 Yosuke Asano
Yosuke Asano Yoshinori Matsumoto
Yoshinori Matsumoto Jun Wada
Jun Wada Robert Rottapel2,3,4,5,6
Robert Rottapel2,3,4,5,6
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 23 February 2023
Sec. Autoimmune and Autoinflammatory Disorders : Autoimmune Disorders
Volume 14 - 2023 | https://doi.org/10.3389/fimmu.2023.1120710
This article is part of the Research TopicProtein Ubiquitylation and OsteoimmunologyView all articles
Ubiquitin-mediated proteasomal degradation is a post-transcriptional protein modification that is comprised of various components including the 76-amino acid protein ubiquitin (Ub), Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), ubiquitin ligase (E3), deubiquitinating enzyme (DUB) and proteasome. We and others have recently provided genetic evidence showing that E3-ubiquitin ligases are associated with bone metabolism, the immune system and inflammation through ubiquitylation and subsequent degradation of their substrates. Dysregulation of the E3-ubiquitin ligase RNF146-mediated degradation of the adaptor protein 3BP2 (SH3 domain-binding protein 2) causes cherubism, an autosomal dominant disorder associated with severe inflammatory craniofacial dysmorphia syndrome in children. In this review, on the basis of our discoveries in cherubism, we summarize new insights into the roles of E3-ubiquitin ligases in the development of human disorders caused by an abnormal osteoimmune system by highlighting recent genetic evidence obtained in both human and animal model studies.
Ubiquitin-mediated proteasomal degradation is a post-transcriptional protein modification that is comprised of various components including the 76-amino acid protein ubiquitin (Ub), Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), ubiquitin ligase (E3), deubiquitinating enzyme (DUB) and proteasome. E1 activates ubiquitin and forms an E1-ubiquitin intermediate, and ubiquitin is transferred from E1 to E2, leading to the formation of an E2-ubiquitin intermediate (1–3). Then E3 recognizes its substrate proteins and the E2-ubiquitin intermediate, resulting in the formation of a protein complex and transference of the activated ubiquitin from E2 to most often a lysine residue in the substrates (4). Ubiquitin has seven lysine residues, including K6, K11, K27, K29, K33, K48 and K63, that are used as attachment sites for subsequent Ub proteins (1, 2, 4), while K48-linked chains are the most abundant linkages for polyubiquitylation. This process is repeated to form a polyubiquitin chain, and ubiquitin-tagged proteins are recognized and degraded into small fragments by the 26S proteasome. Protein ubiquitylation is reversed by DUBs that hydrolyze the peptide bonds linking the substrates to ubiquitin (1, 2, 5) (Figure 1, top). More than 600 E3-ubiquitin ligases have been identified in humans (6) and they are classified into four major groups on the basis of their structures: HECT (homologous to E6-AP carboxyl terminus) type, RING (really interesting new gene)-finger type, U-box type and RBR (RING-between RING-RING) type (7, 8).
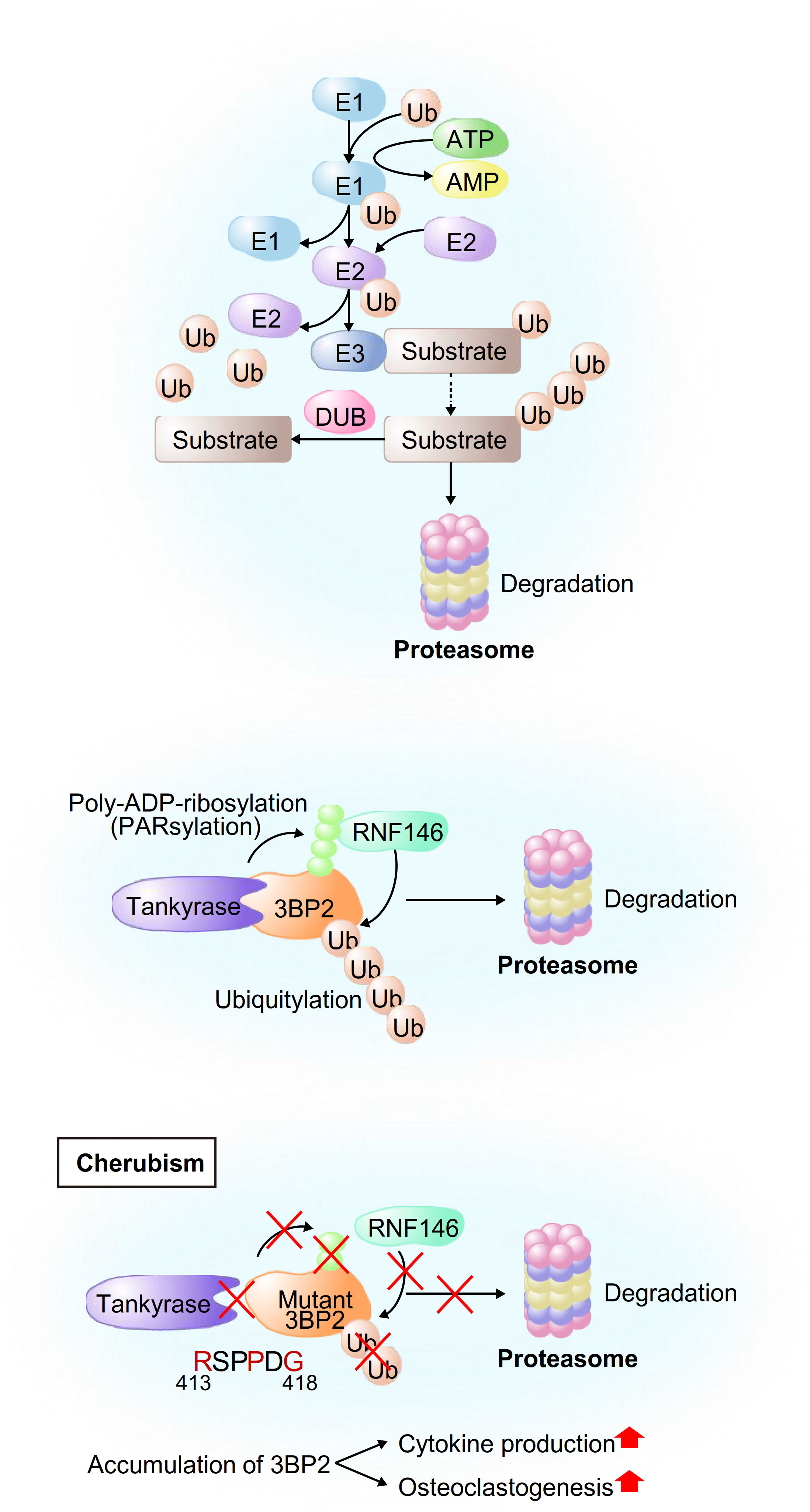
Figure 1 Schematic model of ubiquitylation of general proteins and 3BP2. (Upper) Schematic model of proteasomal degradation mediated by E3-ubiquitin ligases. First, E1 activates ubiquitin and forms an E1-ubiquitin intermediate. Ubiquitin is then transferred from E1 to E2, leading to the formation of an E2-ubiquitin intermediate. Finally, E3 recognizes its substrate proteins and the E2-ubiquitin intermediate, resulting in the formation of a protein complex and transference of the activated ubiquitin from E2 to the substrates. (Middle and Bottom) Schematic model of the development of cherubism. Tankyrase-mediated PARsylation of 3BP2 creates a recognition site for RNF146, leading to ubiquitylation and subsequent proteasomal degradation of 3BP2 (middle). Cherubism mutations uncouple 3BP2 from Tankyrase, which results in impairment of RNF146-mediated ubiquitylation and subsequent stabilization of 3BP2 in macrophages, leading to activation of cytokine production and osteoclastogenesis (bottom).
We have extensively investigated the roles of E3-ubiquitin ligases in bone metabolism, the immune system and inflammation through ubiquitylation and subsequent degradation of their substrates. The adaptor protein 3BP2 (SH3 domain-binding protein 2) nucleates a signaling complex including ABL, SRC, VAV and SYK and enforces an open active configuration of these proteins, leading to their kinase activation. Gain-of-function missense mutations in the SH3BP2 gene cause cherubism, an autosomal dominant disorder associated with severe inflammatory craniofacial dysmorphia syndrome in children (9, 10). Prof. Robert Rottapel’s group at the University of Toronto have provided a mechanistic understanding of cherubism by showing that impairment of ubiquitylation of the cherubism mutant 3BP2 leads to accumulation of 3BP2 and subsequent activation of osteoclastogenesis and cytokine production in macrophages (11, 12).
On the basis of our discoveries in cherubism, we have provided further genetic and mechanistic evidence showing that E3-ubiquitin ligase-mediated protein degradation of their substrates is associated with various human disorders caused by an abnormal osteoimmune system.
In this review, we summarize new insights into the roles of E3-ubiquitin ligases in the development of human disorders caused by an abnormal osteoimmune system by highlighting recently reported genetic studies that have provided mechanistic evidence.
Recent studies have provided genetic evidence linking HECT-domain E3-ubiquitin ligases, which contain an N-terminal C2 domain, a WW domain and a C-terminal catalytic HECT domain, to the pathogenesis of human disorders. ITCH was originally identified in the mouse agouti locus, in which mutations lead to characteristic coat color changes (13). In humans, Lohr et al. reported the first pathogenic mutation in the ITCH gene that resulted in ITCH deficiency in ten Amish children with multisystem autoimmune disease and developmental abnormalities (14). After various examinations that failed to reveal a diagnosis, they performed single-nucleotide polymorphism autozygosity mapping and identified a large homozygous block in chromosome 20q11. They further found a pathogenic truncating homozygous mutation in the ITCH gene in all of the affected children. Characteristic clinical features were dysmorphic faces, multiple organomegaly of the lung, liver and gut with inflammatory cell infiltration and delayed motor development, indicating that human ITCH deficiency leads to a complex phenotype affecting physical growth, craniofacial morphology, muscle development and immune function. Three children died due to respiratory failure caused by cellular nonspecific interstitial pneumonitis. Mouse studies and in vitro studies have provided mechanistic evidence of these clinical features. Mutations of Itch cause a fatal autoimmune disease characterized by histiocyte and lymphocyte infiltration of the lungs, liver, kidney and heart in mice that shows a phenotypic similarity to human diseases (15). Both antigen processing and T-cell anergy are abnormal (16), and multiple organs are infiltrated by lymphocytes, particularly autoreactive B cells, leading to fatal lung disease early in life. Ubiquitylation of the TCR (T-cell receptor) results in its downregulation (17), and downstream signaling is also altered by ubiquitylation of JUNB, which may inhibit IL-2 production and T-cell proliferation (15). Itch deficiency results in loss of tolerance to self-antigens and autoreactive T/B cells (18). ITCH is also a negative regulator of the BCR (B-cell receptor) signaling pathway possibly through ubiquitylation and regulation of components of the mTORC1 complex that are key contributors to glycolysis in B cells and maintenance of germinal centers (19). Itch-deficient B cells have been shown to exhibit increased survival and proliferation through activation of mTORC1-mediated glycolysis (19). Detailed studies on immunological function, B-cell regulation and autoimmunity are currently underway. In addition to immune cells, ITCH is expressed in various tissues including the gut, pancreas, nerves and lymphoid tissues. ITCH might regulate NOD2 signaling (20), which could explain the bowel inflammation observed in these patients with ITCH deficiency since NOD is associated with activation of the innate immune system and its mutations have been identified in patients with Crohn’s disease (21). Craniofacial dysmorphic phenotype is associated with other osteoimmunological roles of ITCH in nonhematopoietic cells, possibly cells of mesenchymal origin. ITCH thus has critical functions in the maintenance of homeostasis in different physiologic states (Figure 2).
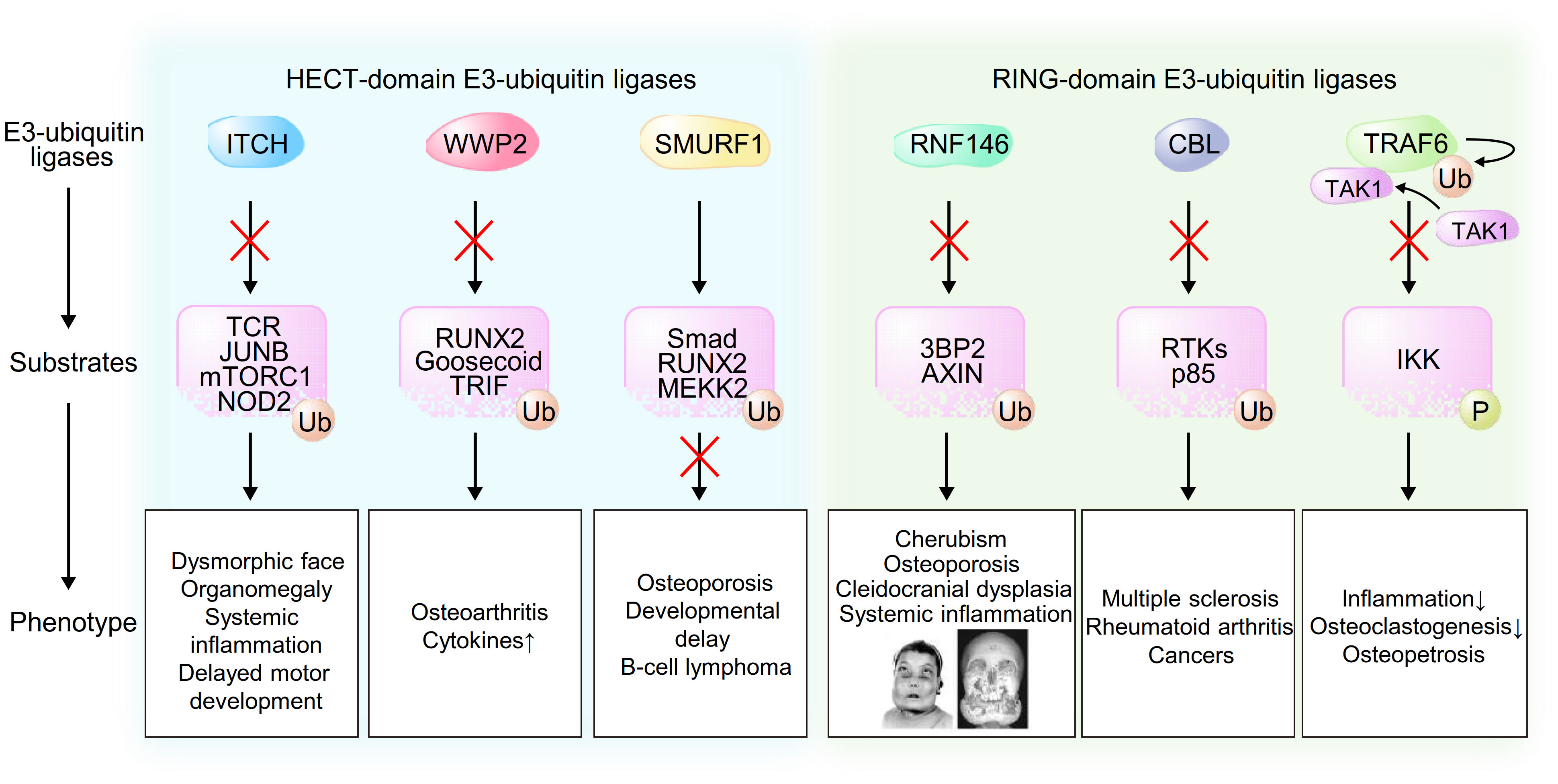
Figure 2 Schematic model of the associations between E3-ubiquitin ligases, their substrates and phenotype caused by dysregulation of these pathways. Pictures of an individual reproduced from Ueki et al. Nat Genet. 28(2):125-126, 2001 (9).
WWP2, which was originally identified as a protein binding to atrophin-1 from yeast two-hybrid screening and in vitro binding analysis and was named atrophin-1 interacting protein 2 (AIP2) (22), might be involved in the pathogenesis of several human disorders on the basis of results of previous in vivo studies. Gao et al. reported that WWP2 was identified from 187 genetic variants as a susceptible gene in osteoarthritis (OA) from a GWAS (genome-wide association study) (23). WWP2 is abundantly expressed in articular cartilage, and WWP2 expression level is decreased in human OA cartilage (24). Mice lacking WWP2 exhibit aggravated spontaneous and surgically induced OA since WWP2 protects cartilage through ubiquitylation and degradation of its substrate RUNX2 (runt-related transcription factor 2), which induces ADAMTS5 and subsequent cartilage degradation (24). WWP2 might also be associated with the development of congenital craniofacial anomalies (CFA) since WWP2-deficient mice display craniofacial malformation (25). In this regard, WWP2 interacts with and ubiquitylates the paired-like homeobox transcription factor Gsc (Goosecoid), leading to its transcriptional activation of Sox6, which plays an important role in craniofacial development. In addition to skeletal formation, WWP2 is associated with the innate immune system through regulation of the Toll-like receptor (TLR) signaling pathway. Upon TLR3 activation by viral double-stranded RNA, WWP2 mediates K48-linked ubiquitylation and degradation of TRIF (TIR-domain-containing adapter-inducing interferon-β), which is required for TLR3-mediated NF-κB and IRF3 (interferon regulatory factor 3) activation, leading to induction of proinflammatory cytokines and type I interferons (26). These findings indicate that loss-of-function mutations in the WWP2 are associated with human skeletal and inflammatory disorders (Figure 2).
A number of in vivo and in vitro studies have shown associations of SMURF1 (Smad Ubiquitin Regulatory Factor-1) with osteoblast function and response to BMPs (bone morphogenetic proteins) (27, 28), and genetic variants in the genes encoding SMURF1-related proteins have been implicated in the risk of osteoporosis by hypothesis-free GWAS (29). Al−Rawi et al. reported the first case of a microduplication in the SMURF1 gene in a 10-year-old girl suffering from two leg fractures with osteoporosis, severe developmental delay, infantile seizures and B-cell lymphoma (30). During the process of osteoblastogenesis, BMPs bind to homomeric type II receptor, which phosphorylates a glycine-serine-rich domain in homomeric type I receptor, leading to induction of signal transduction. The phosphorylated complex of receptor-regulated SMADs (R-Smad; Smad1/5/8) and Co-Smad (Smad4) translocates into the nucleus and binds to the promoter regions of target genes including the osteoblastogenic master transcription factor RUNX2 and Osterix, leading to osteoblast differentiation and maturation. Additionally, the binding of BMPs to the receptors leads to phosphorylation of mitogen-activated protein kinase kinase kinase 2 (MEKK2) and subsequent activation of the c-Jun N-terminal kinase (JNK) signaling pathway, leading to osteoblast activation and increased response to BMPs (31). In bone metabolism, SMURF1 inhibits R-Smad/Co-Smad complex nuclear translocation and directly ubiquitylates and degrades RUNX2, leading to suppression of osteoblastogenesis. Additionally, SMURF1 ubiquitylates and regulates MEKK2, resulting in inhibition of JNK activation and subsequent suppression of osteoblast activity and response to BMPs (28, 30). Consistent with these molecular mechanisms, clinical features in the reported child with osteoporosis and bone fractures showed phenotypic similarity to those observed in mice with Smurf1 mutations. In addition to its roles in skeletal development, SMURF1 may function as an oncoprotein through regulation of the levels of the tumor suppressor RhoB, leading to promotion of tumor metastasis and initiation (32), which may provide the reason why B-cell lymphoma developed in the girl (Figure 2).
CBL (Casitas B-lineage lymphoma proto-oncogene) and CBL-B, a member of the CBL family of proteins including CBL, CBL-B and CBL-C, are RING-domain E3-ubiquitin ligases that ubiquitylate receptor tyrosine kinases (RTKs) for degradation as enzymes (33) and activate several signaling pathways through protein-protein interaction as adaptor proteins (34, 35). A previous GWAS and a previous EWAS (epigenome-wide association study) showed a genetic link between CBL and several human inflammatory disorders such as MS (multiple sclerosis) (36) and RA (rheumatoid arthritis) (37). CBL-B has been reported to ubiquitylate the regulatory p85 subunit of PI3K that results in inhibition of the recruitment of PI3K to CD28 upon activation of the costimulatory pathway rather than its degradation, leading to inhibition of T-cell activation (38). The pathogenic single nucleotide polymorphism of CBL-B in patients with MS is associated with reduction of CBL-B expression levels and induction of CD4+ T-cell proliferation mediated by type I IFNs (39). Additionally, loss-of-function mutations in the CBL gene are associated with the development of cancers since CBL has tumor suppressor functions through both ubiquitylation and degradation of RTKs and inhibition of the PI3K signaling pathway (37), indicating that CBL or CBL-B mutations might be involved in the pathogenesis of autoreactive inflammatory disorders and cancers in humans (Figure 2).
In addition to CBL family proteins, several genetic studies have revealed multiple risk alleles for autoantibody-positive RA within the MHC region, a PTPN22 missense allele and risk alleles in other loci. Raychaudhuri et al. systematically examined 370 SNPs from 179 independent loci with p<0.001 in a published meta-analysis of an RA GWAS of 3,393 cases and 12,462 controls and identified TRAF6/RAG1 as one of the true RA risk alleles (40). TRAF6 is a member of the TNF receptor associated factor (TRAF) protein family in which each protein contains an N-terminal RING domain, zinc-finger motifs, a central coiled-coil region and a highly conserved C-terminal domain and mediates signaling from the TNF receptor superfamily and Toll/IL-1 family. In the IL-1 signaling pathway, TRAF6 induces K-63-linked auto-ubiquitylation after its oligomerization, which results in the recruitment and activation of TAK1 (transforming growth factor β-activated kinase 1), leading to phosphorylation of IKK (IκB kinase) and subsequent activation of NF-κB and cytokine production. In addition to cytokine production, RANKL-induced signaling in macrophages and osteoclastogenesis are also controlled by TRAF6 since TRAF6-deficient mice display osteopetrosis due to an osteoclast defect (41) (Figure 2).
In addition to the roles of these RING-domain E3-ubiquitin ligases in the development of human disorders, we have uncovered another genetic link between RNF146 (Ring Finger Protein 146) and the human hereditary syndrome cherubism.
We have investigated the roles of the RING-domain E3-ubiquitin ligase RNF146, which contains a WWE domain and a RING domain (42, 43), in the osteoimmune system. RNF146 recognizes poly-ADP-ribosylated (PARsylated) proteins that are catalyzed by Tankyrase, a member of the PARP (Poly(ADP-ribose) polymerase) family, through its WWE domain, resulting in its structural change and subsequent activation of the RING domain. Then RNF146 induces K48-linked polyubiquitylation and subsequent degradation of its substrates by the 26S proteasome (43, 44). 3BP2 (SH3 domain-binding protein 2), which was originally identified as a binding protein for the ABL kinase (Abelson murine leukemia viral oncogene homolog 1) (45, 46), is one of the identified substrates that are regulated by RNF146-mediated ubiquitylation (Figure 1, middle). It has been reported that single missense mutations in the SH3BP2 gene cause cherubism, a rare hereditary syndrome associated with severe craniofacial developmental defects in children (9, 47). Prof. Robert Rottapel’s group at the University of Toronto reported that cherubism mutations uncouple 3BP2 from Tankyrase, which results in impairment of RNF146-mediated ubiquitylation and subsequent stabilization of 3BP2 in macrophages, leading to activation of SYK and SRC kinases and hyperosteoclastogenesis (11, 12) (Figure 1, bottom). In addition to the roles of 3BP2 in osteoclastogenesis, 3BP2 is also required for osteoblastogenesis since Sh3bp2-/- mice display osteoporosis due to defective osteoblastogenesis (48). We have shown that 3BP2-mediated ABL kinase activation potentiates the formation of a transcriptional complex of the osteoblastogenic master transcription factor RUNX2 and TAZ (transcriptional co-activator with PDZ-binding motif), leading to phosphorylation and activation of RUNX2 and subsequent enhancement of osteoblastogenesis (49). ABL and TAZ reciprocally stabilize each other through suppression of the respective E3-ubiquitin ligases SMURF1 and β-TrCP. Similarly, we showed that ABL-mediated RUNX2 phosphorylation is also required for breast cancer invasion through an increase of MMP13 transcripts that is independent of TAZ-mediated RUNX2 activation (50).
On the basis of our discoveries in cherubism, we have further investigated the genetic link between RNF146 and the osteoimmune system through generation of conditional knockout mice in which endogenous Rnf146 is deleted in myeloid cells (Rnf146fl/fl LysM-Cre (+)) or osteoblasts (Rnf146fl/fl Osterix-Cre (+)). We showed that RANKL represses RNF146 transcripts through activation of NF-κB and subsequent inhibition of the RNF146 promoter (51). Repression of RNF146 by RANKL results in stabilization of its substrates 3BP2 and AXIN1, which triggers SRC activation and β-catenin attenuation, respectively, both of which are required to execute the osteoclast developmental program (11, 52–54). Consistently, we showed that dysfunction of the RNF146-mediated 3BP2 degradation program in Rnf146fl/fl LysM-Cre (+) mice results in enhanced osteoclastogenesis and bone loss. Additionally, depletion of RNF146 leads to hypersensitivity to LPS-induced TNF-α production in vivo, indicating that RNF146 acts as an inhibitory switch controlling osteoclastogenesis and cytokine production that could be a control point underlying the pathogenesis of chronic inflammatory diseases (51). In addition to the roles of RNF146 in osteoclastogenesis and cytokine production, we have provided further genetic evidence showing that mice lacking RNF146 in osteoblasts show phenotypic similarities to cleidocranial dysplasia (CCD) (55), an autosomal dominant human disorder characterized by abnormal bone development mainly due to defective intramembranous bone formation by osteoblasts (56, 57). We showed that loss of RNF146 in osteoblasts stabilizes AXIN1, which results in inhibition of Wnt3a-induced β-catenin activation and reduced Fgf18 expression. FGF18 induces TAZ expression required for osteoblast proliferation and differentiation through activation of TEAD and RUNX2 transcription factors, respectively. These findings indicate that dysfunction of RNF146 could be the pathogenesis of CCD in addition to known mutations in a single allele of RUNX2 (55, 58–63) (Figure 2).
In this review, we have summarized the roles of E3-ubiquitin ligases in the development of human disorders caused by an abnormal osteoimmune system. On the basis of our discoveries in cherubism, we and others have further investigated and provided evidence that links E3-ubiquitin ligases to autoimmune/autoinflammatory disorders. We have shown that mice lacking Tankyrase in myeloid cells develop severe systemic inflammation with elevated inflammatory cytokine production through the impairment of RNF146-medaited 3BP2 degradation (64). An increased level of 3BP2 in macrophages results in tyrosine phosphorylation and activation of TLR2, and TLR2 (Y647) phosphorylation within the TIR domain by SRC and SYK is essential for TLR2 stabilization and signaling. In the myeloid cell lineage, 3BP2 is also required for G protein-coupled receptor-mediated neutrophil functions (65) and adhesion and migration of mast cells (66). In addition to the innate immune system, 3BP2 is required for B cell proliferation and cell survival following cross-linking of the BCR through SYK phosphorylation (67). 3BP2 is also part of a signaling complex of the TCR with LCK, ZAP-70 and VAV that is induced by CD28 co-stimulation, leading to proliferation and differentiation of T cells since 3BP2-deficient CD8+ T cells exhibit a proliferation defect (68). In an animal model of rheumatoid arthritis (RA), 3BP2 deficiency reduced induction of arthritis and bone erosion through reduction of autoantibody production (69), while an increased level of 3BP2 caused exacerbation of bone loss with increased osteoclast formation in an arthritis mouse model (70), indicating that a high expression level of 3BP2 might be a pathogenesis of RA. In addition to 3BP2, the chondrogenic master transcription factor SOX9 is ubiquitylated by another E3-ligase that is mediated by Tankyrase, while Tankyrase inhibitors ameliorate osteoarthritis in mice (71).
These studies have thus shown that dysregulation of E3-ligases-mediated ubiquitylation causes abnormalities in skeletal formation and the osteoimmune system that are associated with a pathogenesis of human autoimmune/autoinflammatory disorders (Figure 2). E3-ligases and their substrates could be therapeutic targets for these disorders, and some of them are in preclinical or clinical trials, especially for cancer (72, 73). On the contrary, our recent study showed that RNF146-deficient myeloid cells are highly active for the production of inflammatory cytokines through elevation of 3BP2, indicating that tissue-specific inhibitors or enhancers of E3-ubiquitin ligases or their substrates should be established to avoid unexpected side effects (74). Additionally, these genetic studies have partially uncovered the association between E3-ligases and inflammatory human disorders. Further studies in humans are thus required in order to provide new genetic evidence of other ligases, including NEDD4-2 (75), HUWE1 (76), c-MIR (77), Cullin3 (78), FBW7 (79–82), MARCH1 (83–88), MURF1 (89–92), RNF90 (93–95), SAG (96), Hrd1 (97), Peli1 (98, 99), TRIM (100–102) and MYCBP2 (103), that have been shown to be associated with the osteoimmune system in previous in vitro and in vivo mouse studies.
YM: conceptualization and guidance. YA and YM: writing the original draft. JW and RR: proofreading. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Zeng R, Xiong Y, Lin Z, Panayi AC, Sun Y, Cao F, et al. E3 ubiquitin ligases: Potential therapeutic targets for skeletal pathology and degeneration. Stem Cells Int (2022) 2022:6948367. doi: 10.1155/2022/6948367
2. Abdalla O, Mascarenhas B, Cheng HM. Death of a protein: The role of E3 ubiquitin ligases in circadian rhythms of mice and flies. Int J Mol Sci (2022) 23(18):10569. doi: 10.3390/ijms231810569
3. Kleiger G, Mayor T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol (2014) 24(6):352–9. doi: 10.1016/j.tcb.2013.12.003
4. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol (2016) 18(6):579–86. doi: 10.1038/ncb3358
5. Guo YC, Zhang SW, Yuan Q. Deubiquitinating enzymes and bone remodeling. Stem Cells Int (2018) 2018:3712083. doi: 10.1155/2018/3712083
6. Li X, Elmira E, Rohondia S, Wang J, Liu J, Dou QP. A patent review of the ubiquitin ligase system: 2015-2018. Expert Opin Ther Pat (2018) 28(12):919–37. doi: 10.1080/13543776.2018.1549229
7. Wang D, Ma L, Wang B, Liu J, Wei W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev (2017) 36(4):683–702. doi: 10.1007/s10555-017-9703-z
8. Yang Q, Zhao J, Chen D, Wang Y. E3 ubiquitin ligases: Styles, structures and functions. Mol Biomed (2021) 2(1):23. doi: 10.1186/s43556-021-00043-2
9. Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, et al. Mutations in the gene encoding c-abl-binding protein SH3BP2 cause cherubism. Nat Genet (2001) 28(2):125–6. doi: 10.1038/88832
10. Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, et al. Increased myeloid cell responses to m-CSF and RANKL cause bone loss and inflammation in SH3BP2 "cherubism" mice. Cell (2007) 128(1):71–83. doi: 10.1016/j.cell.2006.10.047
11. Levaot N, Voytyuk O, Dimitriou I, Sircoulomb F, Chandrakumar A, Deckert M, et al. Loss of tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell (2011) 147(6):1324–39. doi: 10.1016/j.cell.2011.10.045
12. Guettler S, LaRose J, Petsalaki E, Gish G, Scotter A, Pawson T, et al. Structural basis and sequence rules for substrate recognition by tankyrase explain the basis for cherubism disease. Cell (2011) 147(6):1340–54. doi: 10.1016/j.cell.2011.10.046
13. Perry WL, Hustad CM, Swing DA, O'Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet (1998) 18(2):143–6. doi: 10.1038/ng0298-143
14. Lohr NJ, Molleston JP, Strauss KA, Torres-Martinez W, Sherman EA, Squires RH, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet (2010) 86(3):447–53. doi: 10.1016/j.ajhg.2010.01.028
15. Matesic LE, Haines DC, Copeland NG, Jenkins NA. Itch genetically interacts with Notch1 in a mouse autoimmune disease model. Hum Mol Genet (2006) 15(24):3485–97. doi: 10.1093/hmg/ddl425
16. Zhang M, Veselits M, O'Neill S, Hou P, Reddi AL, Berlin I, et al. Ubiquitinylation of ig beta dictates the endocytic fate of the b cell antigen receptor. J Immunol (2007) 179(7):4435–43. doi: 10.4049/jimmunol.179.7.4435
17. Matesic LE, Copeland NG, Jenkins NA. Itchy mice: The identification of a new pathway for the development of autoimmunity. Curr Top Microbiol Immunol (2008) 321:185–200. doi: 10.1007/978-3-540-75203-5_9
18. Parravicini V, Field AC, Tomlinson PD, Basson MA, Zamoyska R. Itch-/- alphabeta and gammadelta T cells independently contribute to autoimmunity in itchy mice. Blood (2008) 111(8):4273–7282. doi: 10.1182/blood-2007-10-115667
19. Moser EK, Roof J, Dybas JM, Spruce LA, Seeholzer SH, Cancro MP, et al. The E3 ubiquitin ligase itch restricts antigen-driven b cell responses. J Exp Med (2019) 216(9):2170–83. doi: 10.1084/jem.20181953
20. Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW. ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol (2009) 19(15):1255–63. doi: 10.1016/j.cub.2009.06.038
21. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to crohn's disease. Nature (2001) 411(6837):603–6. doi: 10.1038/35079114
22. Wood JD, Yuan J, Margolis RL, Colomer V, Duan K, Kushi J, et al. Atrophin-1, the DRPLA gene product, interacts with two families of WW domain-containing proteins. Mol Cell Neurosci (1998) 11(3):149–60. doi: 10.1006/mcne.1998.0677
23. Gao F, Yao Y, Zhang Y, Tian J. Integrating genome-wide association studies with pathway analysis and gene expression analysis highlights novel osteoarthritis risk pathways and genes. Front Genet (2019) 10:827. doi: 10.3389/fgene.2019.00827
24. Mokuda S, Nakamichi R, Matsuzaki T, Ito Y, Sato T, Miyata K, et al. Wwp2 maintains cartilage homeostasis through regulation of Adamts5. Nat Commun (2019) 10(1):2429. doi: 10.1038/s41467-019-10177-1
25. Zou W, Chen X, Shim JH, Huang Z, Brady N, Hu D, et al. The E3 ubiquitin ligase Wwp2 regulates craniofacial development through mono-ubiquitylation of goosecoid. Nat Cell Biol (2011) 13(1):59–65. doi: 10.1038/ncb2134
26. Yang Y, Liao B, Wang S, Yan B, Jin Y, Shu HB, et al. E3 ligase WWP2 negatively regulates TLR3-mediated innate immune response by targeting TRIF for ubiquitination and degradation. Proc Natl Acad Sci USA (2013) 110(13):5115–20. doi: 10.1073/pnas.1220271110
27. Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H, Du K, et al. A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol (2007) 27(16):5776–89. doi: 10.1128/MCB.00218-07
28. Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell (2005) 121(1):101–13. doi: 10.1016/j.cell.2005.01.035
29. Wu S, Liu Y, Zhang L, Han Y, Lin Y, Deng HW. Genome-wide approaches for identifying genetic risk factors for osteoporosis. Genome Med (2013) 5(5):44. doi: 10.1186/gm448
30. Al-Rawi R, Al-Beshri A, Mikhail FM, McCormick K. Fragile bones secondary to SMURF1 gene duplication. Calcif Tissue Int (2020) 106(5):567–73. doi: 10.1007/s00223-020-00668-5
31. Wu M, Chen G, Li YP. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res (2016) 4:16009. doi: 10.1038/boneres.2016.9
32. Wang M, Guo L, Wu Q, Zeng T, Lin Q, Qiao Y, et al. ATR/Chk1/Smurf1 pathway determines cell fate after DNA damage by controlling RhoB abundance. Nat Commun (2014) 5:4901. doi: 10.1038/ncomms5901
33. Swaminathan G, Tsygankov AY. The cbl family proteins: Ring leaders in regulation of cell signaling. J Cell Physiol (2006) 209(1):21–43. doi: 10.1002/jcp.20694
34. Thien CB, Langdon WY. C-cbl and cbl-b ubiquitin ligases: Substrate diversity and the negative regulation of signalling responses. Biochem J (2005) 391(Pt 2):153–66. doi: 10.1042/BJ20050892
35. Thien CB, Langdon WY. Cbl: Many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol (2001) 2(4):294–307. doi: 10.1038/35067100
36. Julia A, Absher D, Lopez-Lasanta M, Palau N, Pluma A, Waite Jones L, et al. Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in b cells. Hum Mol Genet (2017) 26(14):2803–11. doi: 10.1093/hmg/ddx177
37. Liyasova MS, Ma K, Lipkowitz S. Molecular pathways: Cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res (2015) 21(8):1789–94. doi: 10.1158/1078-0432.CCR-13-2490
38. Fang D, Liu YC. Proteolysis-independent regulation of PI3K by cbl-b-mediated ubiquitination in T cells. Nat Immunol (2001) 2(9):870–5. doi: 10.1038/ni0901-870
39. Sturner KH, Borgmeyer U, Schulze C, Pless O, Martin R. A multiple sclerosis-associated variant of CBLB links genetic risk with type I IFN function. J Immunol (2014) 193(9):4439–47. doi: 10.4049/jimmunol.1303077
40. Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, et al. Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet (2009) 41(12):1313–8. doi: 10.1038/ng.479
41. Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev (1999) 13(8):1015–24. doi: 10.1101/gad.13.8.1015
42. He F, Tsuda K, Takahashi M, Kuwasako K, Terada T, Shirouzu M, et al. Structural insight into the interaction of ADP-ribose with the PARP WWE domains. FEBS Lett (2012) 586(21):3858–64. doi: 10.1016/j.febslet.2012.09.009
43. Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, et al. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev (2012) 26(3):235–40. doi: 10.1101/gad.182618.111
44. DaRosa PA, Wang Z, Jiang X, Pruneda JN, Cong F, Klevit RE, et al. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature (2015) 517(7533):223–6. doi: 10.1038/nature13826
45. Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science (1993) 259(5098):1157–61. doi: 10.1126/science.8438166
46. Khatri A, Wang J, Pendergast AM. Multifunctional abl kinases in health and disease. J Cell Sci (2016) 129(1):9–16. doi: 10.1242/jcs.175521
47. Jones WA, Gerrie J, Pritchard J. Cherubism–familial fibrous dysplasia of the jaws. J Bone Joint Surg Br volume (1950) 32-B(3):334–47. doi: 10.1302/0301-620X.32B3.334
48. Levaot N, Simoncic PD, Dimitriou ID, Scotter A, La Rose J, Ng AH, et al. 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J Clin Invest (2011) 121(8):3244–57. doi: 10.1172/JCI45843
49. Matsumoto Y, La Rose J, Kent OA, Wagner MJ, Narimatsu M, Levy AD, et al. Reciprocal stabilization of ABL and TAZ regulates osteoblastogenesis through transcription factor RUNX2. J Clin Invest (2016) 126(12):4482–96. doi: 10.1172/JCI87802
50. He F, Matsumoto Y, Asano Y, Yamamura Y, Katsuyama T, La Rose J, et al. RUNX2 phosphorylation by tyrosine kinase ABL promotes breast cancer invasion. Front Oncol (2021) 11:665273. doi: 10.3389/fonc.2021.665273
51. Matsumoto Y, Larose J, Kent OA, Lim M, Changoor A, Zhang L, et al. RANKL coordinates multiple osteoclastogenic pathways by regulating expression of ubiquitin ligase RNF146. J Clin Invest (2017) 127(4):1303–15. doi: 10.1172/JCI90527
52. Albers J, Keller J, Baranowsky A, Beil FT, Catala-Lehnen P, Schulze J, et al. Canonical wnt signaling inhibits osteoclastogenesis independent of osteoprotegerin. J Cell Biol (2013) 200(4):537–49. doi: 10.1083/jcb.201207142
53. Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, et al. TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol (2012) 188(6):2612–21. doi: 10.4049/jimmunol.1102836
54. Wei W, Zeve D, Suh JM, Wang X, Du Y, Zerwekh JE, et al. Biphasic and dosage-dependent regulation of osteoclastogenesis by beta-catenin. Mol Cell Biol (2011) 31(23):4706–19. doi: 10.1128/MCB.05980-11
55. Matsumoto Y, La Rose J, Lim M, Adissu HA, Law N, Mao X, et al. Ubiquitin ligase RNF146 coordinates bone dynamics and energy metabolism. J Clin Invest (2017) 127(7):2612–25. doi: 10.1172/JCI92233
56. Mundlos S. Cleidocranial dysplasia: Clinical and molecular genetics. J Med Genet (1999) 36(3):177–82.
58. Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. (1997) 89(5):773–9. doi: 10.1016/S0092-8674(00)80260-3
59. Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, et al. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet (1997) 16(3):307–10. doi: 10.1038/ng0797-307
60. Kobayashi T, Kronenberg HM. Overview of skeletal development. Methods Mol Biol (2014) 1130:3–12. doi: 10.1007/978-1-62703-989-5_1
61. Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell (1997) 89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3
62. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell (1997) 89(5):755–64. doi: 10.1016/s0092-8674(00)80258-5
63. Ott CE, Leschik G, Trotier F, Brueton L, Brunner HG, Brussel W, et al. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia. Hum Mutat (2010) 31(8):E1587–93. doi: 10.1002/humu.21298
64. Matsumoto Y, Dimitriou ID, La Rose J, Lim M, Camilleri S, Law N, et al. Tankyrase represses autoinflammation through the attenuation of TLR2 signaling. J Clin Invest (2022) 132(7):e140869. doi: 10.1172/JCI140869
65. Strathmann MP, Simon MI. G Alpha 12 and G alpha 13 subunits define a fourth class of G protein alpha subunits. Proc Natl Acad Sci USA (1991) 88(13):5582–6. doi: 10.1073/pnas.88.13.5582
66. Navines-Ferrer A, Ainsua-Enrich E, Serrano-Candelas E, Sayos J, Martin M. Myo1f, an unconventional long-tailed myosin, is a new partner for the adaptor 3BP2 involved in mast cell migration. Front Immunol (2019) 10:1058. doi: 10.3389/fimmu.2019.01058
67. Chen G, Dimitriou ID, La Rose J, Ilangumaran S, Yeh WC, Doody G, et al. The 3BP2 adapter protein is required for optimal b-cell activation and thymus-independent type 2 humoral response. Mol Cell Biol (2007) 27(8):3109–22. doi: 10.1128/MCB.01014-06
68. Dimitriou ID, Lee K, Akpan I, Lind EF, Barr VA, Ohashi PS, et al. Timed regulation of 3BP2 induction is critical for sustaining CD8(+) T cell expansion and differentiation. Cell Rep (2018) 24(5):1123–35. doi: 10.1016/j.celrep.2018.06.075
69. Mukai T, Gallant R, Ishida S, Kittaka M, Yoshitaka T, Fox DA, et al. Loss of SH3 domain-binding protein 2 function suppresses bone destruction in tumor necrosis factor-driven and collagen-induced arthritis in mice. Arthritis Rheumatol (2015) 67(3):656–67. doi: 10.1002/art.38975
70. Mukai T, Ishida S, Ishikawa R, Yoshitaka T, Kittaka M, Gallant R, et al. SH3BP2 cherubism mutation potentiates TNF-alpha-induced osteoclastogenesis via NFATc1 and TNF-alpha-mediated inflammatory bone loss. J Bone Miner Res (2014) 29(12):2618–35. doi: 10.1002/jbmr.2295
71. Kim S, Han S, Kim Y, Kim HS, Gu YR, Kang D, et al. Tankyrase inhibition preserves osteoarthritic cartilage by coordinating cartilage matrix anabolism via effects on SOX9 PARylation. Nat Commun (2019) 10(1):4898. doi: 10.1038/s41467-019-12910-2
72. LaPlante G, Zhang W. Targeting the ubiquitin-proteasome system for cancer therapeutics by small-molecule inhibitors. Cancers (Basel) (2021) 13(12):3079. doi: 10.3390/cancers13123079
73. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther (2020) 5(1):11. doi: 10.1038/s41392-020-0107-0
74. Asano Y, Matsumoto Y, He F, Katsuyama T, Katsuyama T, Tsuji S, et al. Pharmacologic inhibition of PARP5, but not that of PARP1 or 2, promotes cytokine production and osteoclastogenesis through different pathways . Clin Exp Rheumatol (2023). doi: 10.55563/clinexprheumatol/qf55h8
75. Lu L, Yakoumatos L, Ren J, Duan X, Zhou H, Gu Z, et al. JAK3 restrains inflammatory responses and protects against periodontal disease through Wnt3a signaling. FASEB J (2020) 34(7):9120–40. doi: 10.1096/fj.201902697RR
76. Guo Y, Li L, Xu T, Guo X, Wang C, Li Y, et al. HUWE1 mediates inflammasome activation and promotes host defense against bacterial infection. J Clin Invest (2020) 130(12):6301–16. doi: 10.1172/JCI138234
77. Toyomoto M, Ishido S, Miyasaka N, Sugimoto H, Kohsaka H. Anti-arthritic effect of E3 ubiquitin ligase, c-MIR, expression in the joints. Int Immunol (2011) 23(3):177–83. doi: 10.1093/intimm/dxq470
78. Meyer SJ, Boser A, Korn MA, Koller C, Bertocci B, Reimann L, et al. Cullin 3 is crucial for pro-b cell proliferation, interacts with CD22, and controls CD22 internalization on b cells. J Immunol (2020) 204(12):3360–74. doi: 10.4049/jimmunol.1900925
79. Takeishi S, Nakayama KI. Role of Fbxw7 in the maintenance of normal stem cells and cancer-initiating cells. Br J Cancer (2014) 111(6):1054–9. doi: 10.1038/bjc.2014.259
80. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer (2008) 8(2):83–93. doi: 10.1038/nrc2290
81. Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, et al. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol (2011) 13(4):351–60. doi: 10.1038/ncb2183
82. Petsouki E, Gerakopoulos V, Szeto N, Chang W, Humphrey MB, Tsiokas L. FBW7 couples structural integrity with functional output of primary cilia. Commun Biol (2021) 4(1):1066. doi: 10.1038/s42003-021-02504-4
83. Ohmura-Hoshino M, Goto E, Matsuki Y, Aoki M, Mito M, Uematsu M, et al. A novel family of membrane-bound E3 ubiquitin ligases. J Biochem (2006) 140(2):147–54. doi: 10.1093/jb/mvj160
84. Santamaria P, Gehrz RC, Bryan MK, Barbosa JJ. Involvement of class II MHC molecules in the LPS-induction of IL-1/TNF secretions by human monocytes. quantitative differences at the polymorphic level. J Immunol (1989) 143(3):913–22.
85. Tze LE, Horikawa K, Domaschenz H, Howard DR, Roots CM, Rigby RJ, et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J Exp Med (2011) 208(1):149–65. doi: 10.1084/jem.20092203
86. Thibodeau J, Bourgeois-Daigneault MC, Huppe G, Tremblay J, Aumont A, Houde M, et al. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. Eur J Immunol (2008) 38(5):1225–30. doi: 10.1002/eji.200737902
87. Hunt D, Wilson JE, Weih KA, Ishido S, Harton JA, Roche PA, et al. Francisella tularensis elicits IL-10 via a PGE(2)-inducible factor, to drive macrophage MARCH1 expression and class II down-regulation. PloS One (2012) 7(5):e37330. doi: 10.1371/journal.pone.0037330
88. Galbas T, Raymond M, Sabourin A, Bourgeois-Daigneault MC, Guimont-Desrochers F, Yun TJ, et al. MARCH1 E3 ubiquitin ligase dampens the innate inflammatory response by modulating monocyte functions in mice. J Immunol (2017) 198(2):852–61. doi: 10.4049/jimmunol.1601168
89. Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol (2001) 306(4):717–26. doi: 10.1006/jmbi.2001.4448
90. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab (2014) 307(6):E469–84. doi: 10.1152/ajpendo.00204.2014
91. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. (2001) 294(5547):1704–8. doi: 10.1126/science.1065874
92. Liu Z, Liu H, He J, Lin P, Tong Q, Yang J. Myeloma cells shift osteoblastogenesis to adipogenesis by inhibiting the ubiquitin ligase MURF1 in mesenchymal stem cells. Sci Signal (2020) 13(633):eaay8203. doi: 10.1126/scisignal.aay8203
93. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity (2008) 29(4):538–50. doi: 10.1016/j.immuni.2008.09.003
94. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
95. Yang B, Liu Y, Cui Y, Song D, Zhang G, Ma S, et al. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PloS Pathog (2020) 16(3):e1008387. doi: 10.1371/journal.ppat.1008387
96. Xiong X, Mathewson ND, Li H, Tan M, Fujiwara H, Li H, et al. SAG/RBX2 E3 ubiquitin ligase differentially regulates inflammatory responses of myeloid cell subsets. Front Immunol (2018) 9:2882. doi: 10.3389/fimmu.2018.02882
97. Yang Y, Kong S, Zhang Y, Melo-Cardenas J, Gao B, Zhang Y, et al. The endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls a critical checkpoint in b cell development in mice. J Biol Chem (2018) 293(33):12934–44. doi: 10.1074/jbc.RA117.001267
98. Xiao Y, Jin J, Chang M, Chang JH, Hu H, Zhou X, et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med (2013) 19(5):595–602. doi: 10.1038/nm.3111
99. Huang X, Hao S, Liu J, Huang Y, Liu M, Xiao C, et al. The ubiquitin ligase Peli1 inhibits ICOS and thereby tfh-mediated immunity. Cell Mol Immunol (2021) 18(4):969–78. doi: 10.1038/s41423-021-00660-5
100. Ozato K, Shin DM, Chang TH, Morse HC 3rd. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol (2008) 8(11):849–60. doi: 10.1038/nri2413
101. Hu MM, Xie XQ, Yang Q, Liao CY, Ye W, Lin H, et al. TRIM38 negatively regulates TLR3/4-mediated innate immune and inflammatory responses by two sequential and distinct mechanisms. J Immunol (2015) 195(9):4415–25. doi: 10.4049/jimmunol.1500859
102. Zhao L, Hao Y, Song Z, Fan Y, Li S. TRIM37 negatively regulates inflammatory responses induced by virus infection via controlling TRAF6 ubiquitination. Biochem Biophys Res Commun (2021) 556:87–92. doi: 10.1016/j.bbrc.2021.03.147
Keywords: E3-ubiquitin ligases, ubiquitylation, proteasomal degradation, osteoimmunology, cherubism
Citation: Asano Y, Matsumoto Y, Wada J and Rottapel R (2023) E3-ubiquitin ligases and recent progress in osteoimmunology. Front. Immunol. 14:1120710. doi: 10.3389/fimmu.2023.1120710
Received: 10 December 2022; Accepted: 10 February 2023;
Published: 23 February 2023.
Edited by:
Uday Kishore, United Arab Emirates University, United Arab EmiratesReviewed by:
Åsa Andersson, Halmstad University, SwedenCopyright © 2023 Asano, Matsumoto, Wada and Rottapel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yoshinori Matsumoto, eW1hdHN1bW90b0Bva2F5YW1hLXUuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.