 Adrián González-Brito
Adrián González-Brito Mireia Uribe-Herranz*
Mireia Uribe-Herranz*- Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Hospital Clinic de Barcelona, Barcelona, Spain
Adoptive cell therapies, like tumor-infiltrating lymphocytes or chimeric antigen receptor T cells, have become an important immunotherapeutic approach against cancer. One of the main struggles of T cell immunotherapies is how to obtain the most effective T cell phenotype, persistence, and differentiation potential to infuse into patients. Adjusting the T cell ex vivo cell culture conditions is a key factor to increase and improve the efficacy of cellular immunotherapies. In this review, we have summarized the ex vivo impact of short chain fatty acids, a group of gut microbiota derived metabolites, on T cell culture and expansion for immunotherapies. There is a complex gut microbiota-immune system interaction that can affect antitumor immunotherapy efficacy. Indeed, gut microbiota derived metabolites can modulate different biological functions in the immune system local and systemically.
Introduction
According to the World Health Organization, cancer is the second leading cause of death worldwide with almost 10 million deaths in 2020. Cancer immunotherapy has rapidly become one of the cornerstones of cancer therapy along with surgery, radiation therapy and chemotherapy. It works by stimulating the immune system to recognize and kill cancer cells and control tumour growth in a way that prevents damage to the healthy cells. It is agreed that modern cancer immunotherapy began with the publication in 1893 of the first use of Coley’s bacterial toxins to treat tumours (1). Since then, many different types of immunotherapies against cancer have been developed, from stem cell transplants, monoclonal antibodies, and immune checkpoint inhibitors to cellular immunotherapies like cancer vaccines or T-cell transfer therapy. Adoptive cell therapies represent a promising and fast evolving approach for the treatment of cancer and include tumour-infiltrating lymphocytes (TILs) (2) or chimeric antigen receptor (CAR) T cells (3). In the last decade, the CAR T-cell therapy has yielded exceptional efficacy rates for the treatment of several hematologic malignancies (4–8). CAR-T cells are genetically modified T cells that combine, an antigen recognition domain of a specific antibody with an intracellular domain of the CD3-z chain protein into a single chimeric protein region that promotes T-cell cytotoxic activity and proliferation. Hence, CAR-T cells can target the very same antigen expressed in the tumoral cell surface, in a major histocompatibility complex (MHC)-independent manner. There have been ground-breaking responses to treatment but, so far, CAR-T cell therapy is available and effective only for a minority of patients. The main concerns regarding treatment efficacy are CAR-T cell differentiation potential, proliferation, and persistence (9). There are multiple approaches attempting to address these problems from the manipulation of CAR constructs, the selection of T-cell subset populations, pharmacological inhibitors, or the optimization of the CAR T-cell manufacturing process. This last approach will be the focus of this review.
Tuning the ex vivo cell culture conditions is key to determine differentiation status and survival of CAR-T cells. Developing the desired CAR-T profile, undifferentiated, less exhausted, and persistent, is an area of intense research. One of the first options to improve the T-cell fitness is the choice of the T-cell source to manufacture the CAR-T cells. It has been extensively shown that T-cell origin has a critical impact on the CAR-T biological activity and proliferation [reviewed in (10)]. Besides the source of T cells and T-cell subtypes, multiple additional strategies are being tested to tune the ex vivo cell culture conditions; the manufacturing time, the stimulation with cytokines, the T-cell activation, the gene delivery system, and strategies to target or reprogramme the T-cell metabolism.
Currently, protocols to manufacture CAR-T cells are varied and long, taking up to several weeks. In our experience, with a locally developed academic anti-CD19 CAR-T (ARI-0001) for a clinical trial, the expansion takes between 9 and 12 days (11). So far, no standardization has been reached in the field. A key aspect that differs between protocols is the stimulation with cytokines, being the most common and studied interleukin 2 (IL-2), 7 (IL-7), 15 (IL-15) and 21 (IL-21). IL-2 stimulates cell proliferation and maintains viability during the ex vivo expansion (12). However, since stimulation with IL-2 can also induce an exhausted T-cell profile, a well-known alternative is to switch it for IL-7, IL-15, and IL-21, with the aim of promoting naïve and memory cell proliferation and preventing T-cell differentiation (13). Regarding T-cell activation strategies, the most employed are the anti-CD3/anti-CD28 monoclonal antibodies either soluble, bound or in magnetically coated beads, which act as artificial antigen presenting cells. The gene transfer system is another component that can be modified during the CAR-T cell ex vivo manufacturing. Currently, the usual choice is a viral vector because of its high transduction efficacy. The most common system used in the available manufactured CAR-T therapies are the lentivirus, followed by retrovirus. However, viral gene transfer has a major disadvantage. Besides the limitation in gene size (<10 Kb), the insertion of the gene occurs randomly which could potentially lead to oncogenesis through the activation of an oncogene or suppression of a tumor-suppressor gene. To avoid this issue, non-viral systems are being developed such as plasmid-based gene delivery using the transposon/transposase systems (14, 15) or CRISPR/Cas9-based gene editing (16).
Another important area of research is optimizing metabolism at different stages of CAR−T cell production. A low metabolic activity during the manufacturing process correlates with a less differentiated T cell phenotype, longer persistence in vivo and greater antitumoral activity (17). On the other hand, a high metabolic activity has been linked to a more differentiated profile (18). To optimize metabolism, several approaches are being tested during T cell expansion, such as inhibiting glycolysis to limit differentiation and improve T cell function or enhancing mitochondrial metabolism (17, 19–21). Finally, a new area of improvement has been developing for the last 5 years, as few studies are starting to use metabolites of bacterial origin, commonly found in the human body, to modulate T-cell phenotypes ex vivo (Table 1).
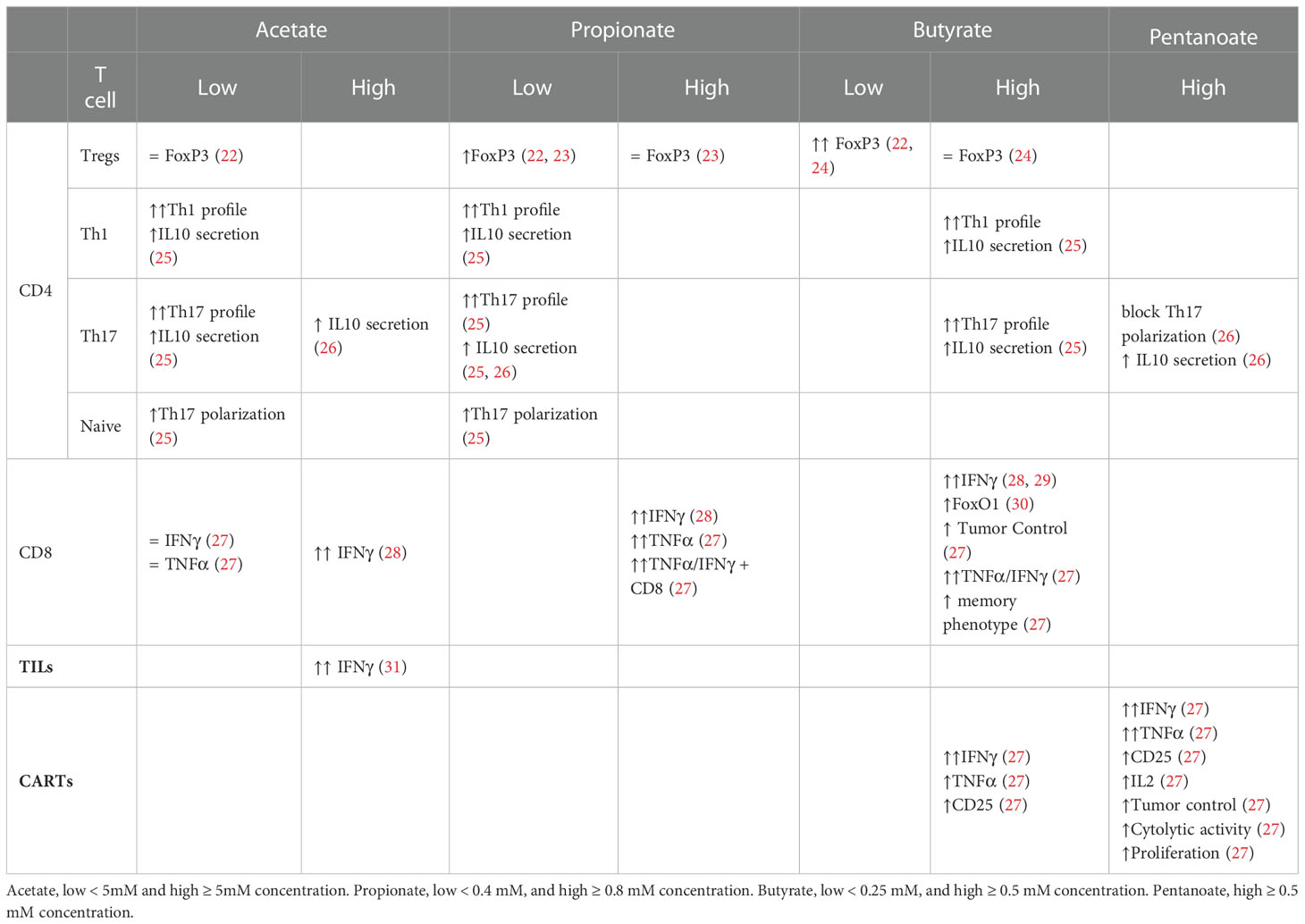
Table 1 ex vivo acetate, propionate, butyrate and pentanoate impact on diverse T cell subsets.
The human body is a complex ecosystem colonized by trillions of bacteria, fungi, yeast, protozoa, and viruses. All these together comprise the commensal microbiota that is developed after birth through vertical transmission and then shaped by environmental factors throughout life. The commensal microbiota and the human host have co-evolved in a mutualistic association, obtaining benefits such as the acquisition of bioactive compounds (32). These metabolites can modulate various biological functions in the immune and nervous systems (33). Beyond the effects on intestinal and local immune physiology, the gut microbiome has systemic effects (34) caused by small molecules such as bacterial-derived metabolites entering the systemic circulation. Several studies have confirmed that gastrointestinal flora impacts the immune system predominantly through bacteria-derived metabolites (35, 36). The gut microbiota has great metabolic capacity, greater than that of the human host. It generates a complex network of metabolic pathways that produce an exceptionally diverse pool of metabolites from modified exogenous dietary components to endogenous compounds generated by the gut microbiota itself (35, 37, 38). For example, in vivo short chain fatty acid (SCFA) can regulate host immunity by facilitating the extrathymic generation of regulatory T (Treg) cells (39), and the function of the colonic Tregs (40); peptidoglycan, an essential molecule of the bacterial wall, can prime the systemic innate immunity by activating neutrophils (41); polysaccharide (PSA) from B. fragilis, a Gram-negative anaerobe, can boost the systemic T helper cell type 1 (Th1) CD4+ T cells (42).
The results from an in vivo study with mice, raised under germ-free conditions, confirmed that the impact of gut microbiota is not restricted to the gastrointestinal tract. It has systemic effects, as these mice had significantly impaired host immune responses to pathogens (43). Moreover, a direct correlation exists between the presence of specific bacteria in the gut microbiota and T-cell development and differentiation (44–47). For instance, colonization of germ-free mice gut with a cocktail of bacteria from Clostridiales clusters IV, XIVa, and XVIII is sufficient to drive Treg differentiation (45). In the cancer context, some specific bacteria have been demonstrated to be involved in the process of initiation and progression of carcinogenesis at epithelial barriers and within sterile tissue (48, 49). In addition, the microbiota has also been implicated in modulating the efficacy and toxicity of cancer therapy, including chemotherapy, radiotherapy, and especially immunotherapy (32, 50–52), including checkpoint blockade approaches targeting the CTLA-4 and PD-1 pathways (53, 54) mainly acting through the local and systemic immune system. There are several studies that support the link between gut microbiota (or bacterial-derived metabolites), Th differentiation and T cells function, and modulation of the antitumor immunity. A high-dietary fiber diet, which is associated with increased gut microbiota diversity and decreased risk of chronic inflammatory diseases, can boost antitumor immunity and increase the infiltration of tumor-killing T cells on melanoma patients (55). In this observational study, fiber-fermenting Ruminococcaceae correlated with the abundance of inducible T cell co-stimulator-expressing TILs on melanoma. In a study investigating four different murine cancer models, inosine, a bacterial purine metabolite produced by Bifidobacterium pseudolongus, promoted Th1 activation and antitumor immunity which improved the antitumor effects (56). In a mouse model of colon carcinoma, a SCFA-rich diet with pectin, a fiber that promotes the growth butyrate-producing bacteria, was associated with increased CD8+ effector T cell function at the tumor site (57). A recent study, showed that the natural polyphenol castalagin improved the intratumoral CD8+/FoxP3+CD4+ ratio in sarcoma tumor-bearing mice, and improved the efficacy of anti-PD-1 immunotherapy through modulation of the gut microbiota, enriching the abundance of Ruminococcaceae and Alistipes bacterial families (58). Another report investigating the immune-checkpoint blockade therapy efficacy, showed that Lactobacillus delbrueckii subsp. bulgaricus produced an extracellular polysaccharide that was able to induce IFN-γ+CCR6+CD8+T cells in Peyer’s patches of tumor-bearing mice, thus enhancing the efficacy of this therapy (59). A study from Wang and colleagues, proved that plasma trimethylamine N-oxide, a microbe-derived metabolite, can activate the endoplasmic stress kinase PERK, boosting the function of IFN-γ+CD8+T cells mediated immunity in triple-negative breast cancer patients (60). Besides gut microbiota, intratumoral bacteria also can have a role in antitumor immunity through T cells. Fusobacterium nucleatum can attenuate T cell-mediated immune responses in rectal and colon cancer cases, as higher amount of F. nucleatum in colorectal carcinoma tissue was associated with lower density of T-cells in tumor tissue (61). All these studies provide strong evidence of a highly dynamic and complex microbiome-immune system interaction that can impact antitumor immunity.
Short Chain Fatty Acids
SCFAs are the main metabolites produced by gut microbiota through bacterial anaerobic fermentation of non-digestible carbohydrates, such as dietary fiber (62). They are fatty acids with fewer than six carbon atoms, the most abundant of which are acetate (two carbons) and propionate (three), which are mainly produced by members of the Bacteroidetes phylum, as well as butyrate (four), predominantly produced by members of the Firmicutes phylum. Acetate derived from colonic bacterial fermentation can flow into the blood compartment and together with endogenous acetate can exert systemic activity upon the immune system. Propionate is a precursor of glucogenesis and has a direct effect on T cell activity locally. Butyrate effects are mainly restricted to the gut, where it is used as energy sourced by the colonocytes and, together with propionate, is processed in the liver (63). The effect of SCFAs on the metabolism of T-cells is yet to be understood but there is abundant data that suggest that SCFAs regulate the adaptive immune system through the modulation of mTOR activity, glucose metabolism, histone acetylation and cytokine gene expression (64). Multiple studies have confirmed that T cells are susceptible to SCFA exposure in vitro, promoting or inhibiting a specific T cell phenotype, and therefore supporting their use in ex vivo T cells expansion.
While research on the effect of microbial metabolites on T cells has primarily been focused on SCFAs, there are also other metabolites that have been investigated. For instance, the inosine, a purine metabolite that can be produced by Bifidobacterium pseudolongum, has been shown to boost the CD4+ Th1 differentiation in the presence of IFN-γ through adenosine A2A receptor (56). Moreover, the bacterial transformation of host bile acids has been demonstrated to directly modulate balance of Th17 and Treg cells (65, 66). Furthermore, tryptophan metabolites are reported to be essential for intestinal immunity and perform their effect on T cells through aryl hydrocarbon receptor signaling (67). Although these bacterial-derived metabolites are promising alternatives to SCFAs, in this manuscript, we will focus on the new and fast developing research field of the T and CAR-T cell ex vivo manufacturing optimization process through the SCFA bacterial derived metabolites. Results summarized in this review, might appear difficult to interpret as the effects of SCFAs are clearly dependent on immunological context and the studies compared often used a wide range of different conditions, from concentrations to exposure time. This is expected, as the field is only just starting to investigate the effect of gut microbial-derived metabolites on immune populations ex vivo. This lack of clarity and consistency reflects the urgent need for more rigorous and systematic protocols to better understand and learn from the microbiome-T cell crosstalk.
The ex vivo effects of SCFAs on T-cells
Naïve CD4+ T cells
The effect of SCFAs in regulating T cell differentiation into effector and IL-10+ regulatory T cells was studied by Park et al. In this study T cells were activated in vitro for 5-6 days in the presence or absence of the SCFAs at different concentrations. The results showed that naïve CD4+ T cells, isolated from murine spleens and lymph nodes (LN), treated with acetate and propionate in vitro, led to differentiation into Th17 cells, which was observed through the increase in IL-17+ and IL-10+ cells as well as the increase in the transcription of the associated genes IL-17A, IL-17F, Rorc, RORα, T-bet, and IFN-g (25) Tables 1, 2.
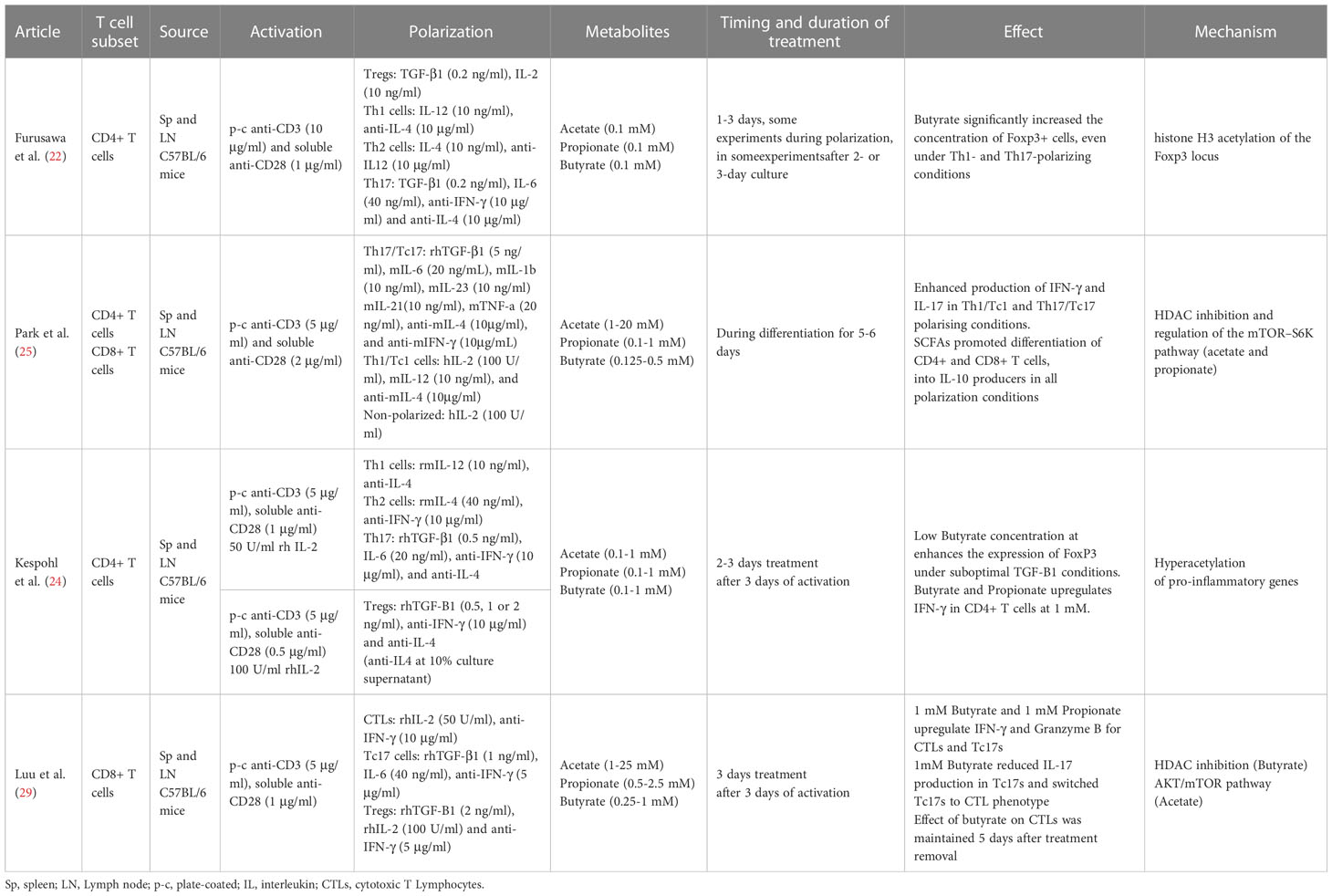
Table 2 Summary of methods and results from ex vivo SCFA effects on murine T cell studies.
Polarized CD4+ T cells
Studies over the last 10 years have shown the ability of SCFAs to modulate and alter polarized CD4+ T cells via epigenetic and metabolic processes. Under tolerogenic conditions, naïve CD4+ T cells supplemented with low or physiogical concentrations of SCFAs seem to play a role in facilitating the activity of T regulatory cells, promoting the production of IL-10 and FoxP3+ Treg polarization (22, 39). Furusawa et al. isolated naïve CD4+ T cells from murine spleen and LNs. After three days of expansion cells were differentiated into Tregs in the presence or absence of acetate, propionate or butyrate, for an additional 2 days. Butyrate significantly increased the concentration of Foxp3+ cells; propionate did so moderately, while acetate showed no effect. The butyrate effect was in part mediated by histone H3 acetylation of the Foxp3 locus (22). Another study provided additional evidence indicating an increase of Foxp3+ CD4+ T cells for lower concentrations of propionate but not for higher ones (23). A third independent study, partially confirmed the enhanced Foxp3 expression in purified murine CD4+ T cells using a suboptimal concentrations of TGF-β1 and low concentrations of butyrate. However, the effect was not observed in the absence or under optimal concentrations of TGF-β1. In any case, higher concetrations of Butyrate did not enhance the expression of Foxp3 and instead, induced the expression of T-bet and IFN-γ via histone acetylation (HDAC inhibition), which is associated with an inhibition of Treg differentiation (24).
Luu and colleagues polarized CD4+ T cells towards Th17 for three days. Supplementation with pentanoate, another SCFA with 5 carbons, during the three-day process, impeded the polarization and inhibited the production of IL-17A through HDAC-inhibitory activity. In this case, and contrary to the results for the shorter SCFAs used in the study from Park et al., pentanoate led to a reduction in the transcription of the Th17 associated genes IL-17A, Rorc, Il21, Stat3, and Tgfb3. The treatment did, however, lead to the increase in the expression of IL-10 in Th17 cells. This increase seems to be due to pentanoate acting as a precursor of acetyl-CoA and activating the mTOR pathway, similarly to the mechanism for acetate. Results showed that Th17 cells treated ex vivo with pentanoate increased the extracellular acidification rate and enhanced their glycolytic activity leading to metabolic and epigenetic reprogramming and the loss of the pathogenic phenotype of Th17 cells in autoimmune disease (26).
Finally, under Th1 CD4+ polarizing conditions in the presence of IL-12, acetate and propionate supplementation potentiated the differentiation into Th1 cells, in a concentration dependent manner (25) Table 1.
Activated CD8+ T cells
Activated CD8+ T cells cultivated in vitro using a SCFA-enriched medium have an increased production of IFN-γ and Granzyme B. SCFAs with shorter chain lengths such as acetate (two carbon chain) require greater concentrations for similar effects to ones with longer chain lengths such as pentanoate (five carbon chain) (28–31). For instance, murine CD8+ T cells polarized to CTLs in culture for three days, were supplemented with acetate for an additional three days at a concentration 25 mM. This led to a 125% increase in IFN-γ cells in comparison to untreated cells. Propionate, which contains a three-carbon chain, seems to require a concentration of approximately 5 mM to achieve similar effects (28). To obtain a comparable effect using butyrate, a SCFA with a four-carbon chain, a concentration of only 1 mM was required (29). In line with this hypothesis, murine CD8+ T expansion under CTL-inducing conditions supplemented with low concentration of acetate showed no effect over TNF-α or IFN-γ expression (27). SCFAs of shorter chain lengths are found in the body at greater concentrations (68). This might explain why T cells may could have evolved to become less sensitive to SCFAs of shorter chain lengths through a variety of mechanisms. In the example above, while acetate caused the increase in IFN-γ via the mTOR pathway (rapamycin, an mTOR inhibitor, led to an abrogation of the effect), butyrate caused it via the inhibition of HDAC activity (29). CD8+ T expansion under CTL-inducing conditions and supplemented with pentanoate, butyrate and, to a lesser extent, propionate showed an increase in the frequencies of TNF-α+ IFN-γ+ CD8+ T cells and secretion of TNF-α by CTLs. The effectiveness of butyrate and pentanoate treated T cells was further tested in a B16-OVA melanoma murine model and showed a better melanoma tumor control. Moreover, pentanoate-treated CD8+ T cells, showed an increase in in vivo persistence in a rag1-deficient mouse model, which was associated to an increase in CD25 expression and continuous IL-2 secretion (27). Qiu et al. tested CD8+ T cells under prolonged glucose restriction in vitro and showed that supplementation of acetate increased the expression of IFN-γ, although Granzyme B levels were not altered. These effects were mediated through histone acylation (31).
Differences in SCFAs concentration in medium lead to differences the expression of cytokines, however, the effect on the length of exposure seems to be less clear. While Luu and colleagues exposed murine activated CD8+ T cells to 1 mM Butyrate for three days (29), He and collaborators exposed activated CD8+ T cells to butyrate at the same concentration for just 12 hours, with the inclusion of IL-12 (28). They both obtained similar results for the proportion of IFN-γ cells, indicating a doubling in relation to untreated cells.
There are other factors to consider beyond concentrations or the duration of treatment, like whether the effect is long-lasting and leads to stable phenotypic change or their memory potential. The first one was explored by treating CTLs with butyrate for two days and then allowing the cells to grow without it for an additional three days. An increase in IFN-γ was still observed after treatment withdrawal compared to the control group, in vitro and in vivo, indicating a phenotypic stable change (29). The relationship between SCFA T cell ex vivo treatment and their memory potential were studied with transgenic gBT-I CD8+T cells treated with butyrate for three days after antigen-specific activation. While butyrate treatment reduced proliferation, it also led to greater responsiveness to IL-15 and a higher expression of transcription factor FoxO1 (30). Expression of FoxO1 is involved in the formation of memory T cells (69–71) while IL-15 promotes memory CD8+ T-cell survival and proliferation (72, 73), suggesting that butyrate may lead to a greater memory potential of the activated CD8+ T cells. This was supported by in vivo experiments that showed that butyrate treated gBT-I CD8+ T cells had a greater expansion upon antigen encounter. The suggested mechanism behind butyrate enhancement of the memory potential of CD8+ T cells might be its impact over glycolytic metabolism, uncoupling the TCA cycle from glycolitic input and favouring oxidative phosphorylation (30) Table 1.
TILs and CAR-T cells
All the studies mentioned above prove that SCFAs can modulate T cell polarization and memory formation ex vivo. There are few reports that show better therapy outcomes polarizing CAR-T cells in vitro before infusion. Interestingly, Th17-polarized human mesothelin-CAR T cells exhibited superior immunity against mesothelioma compared to Th1-polarized mesothelin -CAR T cells (74). In the same line of results, Guedan and colleagues showed that mesothelin-specific CAR Th17/Tc17 cells (that maintained Th17 function with a Th1 bias) had long-lived persistence in vivo and eradicated tumors (75). A third report studying the Th17 polarization effects on CAR-T cells, showed that a Th/Tc17 CAR-T targeting the proto-oncogene Neu, exhibited an increase antitumor immunity and improved early tumor control. The combination of the Th/Tc17 Neu-CAR-T and a STING agonist increased the trafficking, persistence, and tumor control in a murine model of breast cancer (76). Supporting the use of Th17 polarizing conditions for CAR-T cells, Fraietta and colleagues showed that CAR-T cells from chronic lymphocytic leukemia complete responders to CD19-CAR T therapy, had an enhanced transcriptomic profile of STAT3/IL-6 signaling, producing a type-17 signature compared with non-responders (77). Finally, a human mesothelin-CAR polarized towards Th9 cells was able to eliminate advanced human ovarian cancer patient-derived xenograft in humanized NSG mice. Regular expanded CAR-T or high doses of Th1+Tc1 polarized CAR-T cells could not achieve the same results (78). The differentiation status of CART cells plays a crucial role for therapeutic success as well. It is now well stablished that a less differentiated profile, like naïve or central memory T cells which have the capacity to persist and proliferate long-term in vivo lead to a better clinical outcome (77, 79). All the studies mentioned above highlight the potential of SCFAs over the process of expanding tumor-specific lymphocytes and CAR-T cell manufacturing for cancer immunotherapies. In this line, two studies recently published have explored the use of SCFAs on TILs and CAR-T cells.
Murine tumor-infiltrating lymphocytes (TILs) obtained from B16 melanoma and treated ex vivo with acetate showed a significant increase in IFN-γ secretion. This indicates that acetate can promote responsiveness in T cells isolated directly from the tumor microenvironment (31). The effect of butyrate and pentanoate supplementation during CAR-T cell ex vivo expansion was investigated under CTL-inducing conditions. Murine and human CD8+ T cells were used to generate a CAR that recognize receptor tyrosine kinase-like orphan receptor 1 (ROR1). Murine CAR-T cells treated with butyrate and pentanoate enhanced expression of CD25, as well as TNF-α and IFN-γ production. Pentanoate treated cells were further tested in a pancreatic tumor model and showed that tumor volume and weight were significantly reduced in comparison to non-treated cells. Later, the authors developed CAR-T cells from healthy human donor CD8+ T cells using a ROR1-specific CAR. After a two-step 17-day stimulation process, CAR-T cells were treated for four days with pentanoate at different concentrations. CAR-T cells pretreated with pentanoate showed upregulation of CD25 expression, IL-2 secretion, and stronger proliferation, as well as greater cytolytic activity when encountering their target antigen ROR1, together with an increase in IFN-γ and TNF-α production (27). Table 1.
Conclusion
During the last decade multiple studies have confirmed the remarkable effect of gut microbiota and its bacterial derived metabolites upon the immune system. Bacterial T cell polarization and function modulation are two of the most studied aspects of the gut microbiota-immune system axis. These are the basis of several strategies to improve efficacy of immunotherapies or to harness its side effects in clinical trials. According to NIH clinicaltrial.gov site there are 50 clinical trials on cancer immunotherapies that include study or intervention regarding gut microbiota. Nevertheless, the clinical implementation has proven to be difficult. Multiple variables are still largely unknown, like the optimal administration route or how to monitor the evolution once its implemented. Moreover, dosing and administration schedule will be key as it is envisioned as a very dynamic therapeutic process. The studies presented in this review show that microbial metabolites can alter T cell differentiation in vitro, potentially leading to a phenotype that can increase persistence, cytotoxic activity and, ultimately, anti-tumor effect, Tables 2, 3. While some results might appear contradictory, expansion and polarization T cell ex vivo protocols used up to now are inconsistent. Multiple factors are not well defined, for instance the cytokines used in the media culture, the SCFAs concentrations or the duration of the treatment to name just a few examples. Dosing SCFAs might be the most crucial variable to account for, as current available data shows pleiotropic effects of SCFAs upon T cells. Learning from microbiome-T cell crosstalk can help us to develop a more efficient cancer immunotherapy. We foresee the ex vivo use of bacterial-derived metabolites as a new method to improve T cell expansion and polarization, directed to cellular immunotherapies with a special interest on TILs and CAR-T cell production.
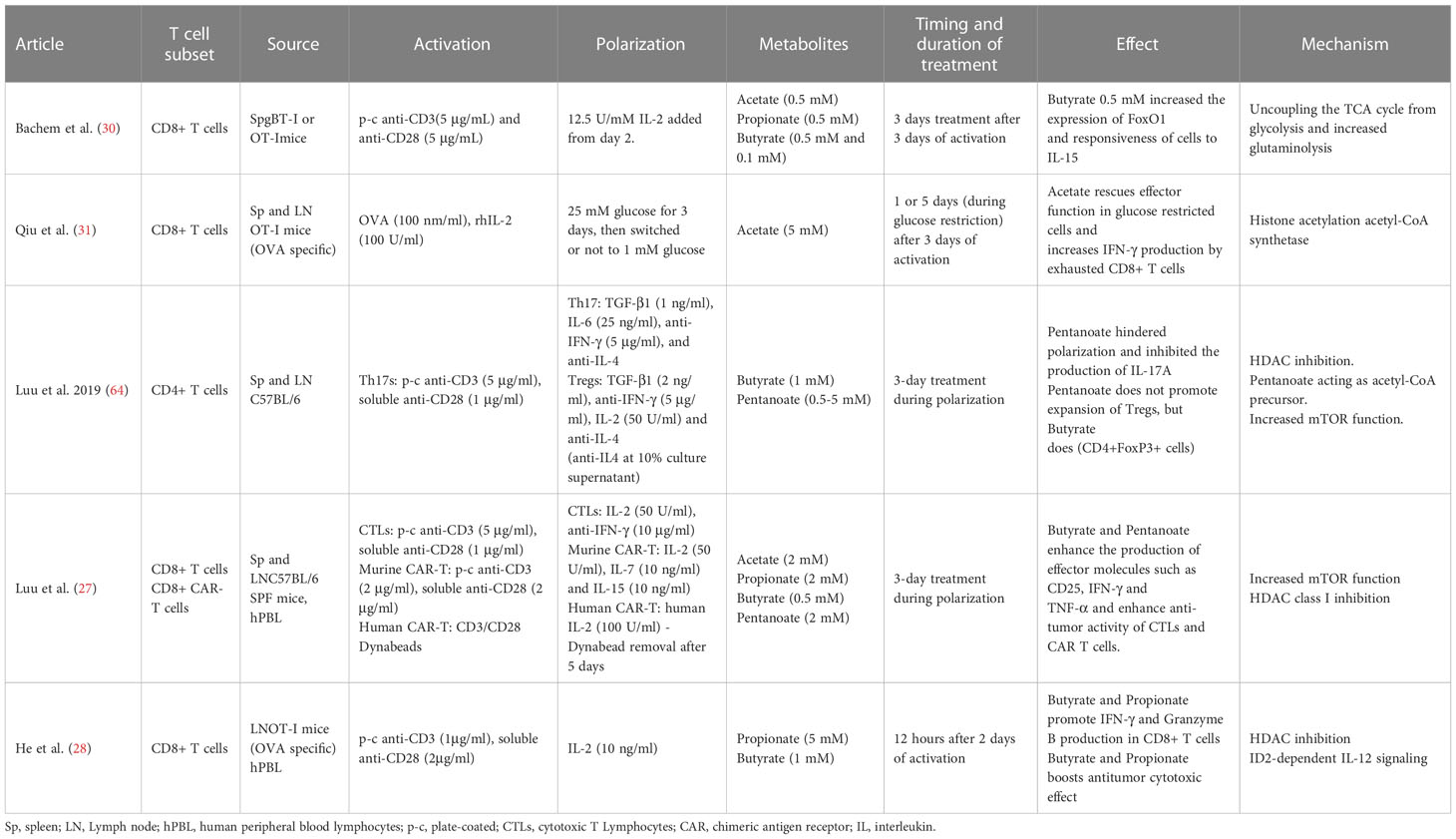
Table 3 Summary of methods and results from ex vivo SCFA effects on murine and human T cell studies.
Author contributions
Conceptualization and Original Draft Preparation, MU-H. Writing—Review and Editing AG-B and MU-H All authors contributed to the article and approved the submitted version.
Funding
MU-H as “Beatriu de Pinos” fellow (2019-BP-00204) has received funding from the Ministry of Business and Knowledge of the Government of Catalonia and the European Union’s Horizon 2020, Marie Sklodowska-Curie COFUND
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. with a report of ten original cases. 1893. Clin Orthop (1991), 3–11.
2. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (2015) 348:62–8. doi: 10.1126/science.aaa4967
3. Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
4. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
5. Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with b-cell NHL. Blood (2016) 127:2980–90. doi: 10.1182/blood-2015-12-686725
6. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
7. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T Cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet Lond Engl (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
8. Ortíz-Maldonado V, Rives S, Castellà M, Alonso-Saladrigues A, Benítez-Ribas D, Caballero-Baños M, et al. CART19-BE-01: A multicenter trial of ARI-0001 cell therapy in patients with CD19+ Relapsed/Refractory malignancies. Mol Ther J Am Soc Gene Ther (2021) 29:636–44. doi: 10.1016/j.ymthe.2020.09.027
9. Porter DL, Hwang W-T, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
10. Bartoló-Ibars A, Uribe-Herranz M, Muñoz-Sánchez G, Arnaldos-Pérez C, Ortiz-Maldonado V, Urbano-Ispizua Á., et al. CAR-T after stem cell transplantation in b-cell lymphoproliferative disorders: Are they really autologous or allogenic cell therapies? Cancers (2021) 13:4664. doi: 10.3390/cancers13184664
11. Castella M, Boronat A, Martín-Ibáñez R, Rodríguez V, Suñé G, Caballero M, et al. Development of a novel anti-CD19 chimeric antigen receptor: A paradigm for an affordable CAR T cell production at academic institutions. Mol Ther Methods Clin Dev (2019) 12:134–44. doi: 10.1016/j.omtm.2018.11.010
12. Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol (2018) 36:411–33. doi: 10.1146/annurev-immunol-042617-053352
13. Dwyer CJ, Knochelmann HM, Smith AS, Wyatt MM, Rangel Rivera GO, Arhontoulis DC, et al. Fueling cancer immunotherapy with common gamma chain cytokines. Front Immunol (2019) 10:263. doi: 10.3389/fimmu.2019.00263
14. Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Dupuy AJ, et al. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol Ther J Am Soc Gene Ther (2003) 8:108–17. doi: 10.1016/s1525-0016(03)00099-6
15. Aronovich EL, McIvor RS, Hackett PB. The sleeping beauty transposon system: A non-viral vector for gene therapy. Hum Mol Genet (2011) 20:R14–20. doi: 10.1093/hmg/ddr140
16. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (2020) 367:eaba7365. doi: 10.1126/science.aba7365
17. Rostamian H, Fallah-Mehrjardi K, Khakpoor-Koosheh M, Pawelek JM, Hadjati J, Brown CE, et al. Metabolic switch to memory CAR T cells: Implications for cancer treatment. Cancer Lett (2021) 500:107–18. doi: 10.1016/j.canlet.2020.12.004
18. Chapman NM, Boothby MR, Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol (2020) 20:55–70. doi: 10.1038/s41577-019-0203-y
19. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123:4479–88. doi: 10.1172/JCI69589
20. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–842.e13. doi: 10.1016/j.cell.2016.09.031
21. MacPherson S, Keyes S, Kilgour MK, Smazynski J, Chan V, Sudderth J, et al. Clinically relevant T cell expansion media activate distinct metabolic programs uncoupled from cellular function. Mol Ther Methods Clin Dev (2022) 24:380–93. doi: 10.1016/j.omtm.2022.02.004
22. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50. doi: 10.1038/nature12721
23. Sumbria D, Berber E, Rouse BT. Supplementing the diet with sodium propionate suppresses the severity of viral immuno-inflammatory lesions. J Virol (2021) 95:e02056–20. doi: 10.1128/JVI.02056-20
24. Kespohl M, Vachharajani N, Luu M, Harb H, Pautz S, Wolff S, et al. The microbial metabolite butyrate induces expression of Th1-associated factors in CD4+ T cells. Front Immunol (2017) 8:1036. doi: 10.3389/fimmu.2017.01036
25. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the MTOR–S6K pathway. Mucosal Immunol (2015) 8:80–93. doi: 10.1038/mi.2014.44
26. Luu M, Pautz S, Kohl V, Singh R, Romero R, Lucas S, et al. The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun (2019) 10:760. doi: 10.1038/s41467-019-08711-2
27. Luu M, Riester Z, Baldrich A, Reichardt N, Yuille S, Busetti A, et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat Commun (2021) 12:4077. doi: 10.1038/s41467-021-24331-1
28. He J, Zhang P, Shen L, Niu L, Tan Y, Chen L, et al. Short-chain fatty acids and their association with signalling pathways in inflammation, glucose and lipid metabolism. Int J Mol Sci (2020) 21:E6356. doi: 10.3390/ijms21176356
29. Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, et al. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci Rep (2018) 8:14430. doi: 10.1038/s41598-018-32860-x
30. Bachem A, Makhlouf C, Binger KJ, de Souza DP, Tull D, Hochheiser K, et al. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. Immunity (2019) 51:285–297.e5. doi: 10.1016/j.immuni.2019.06.002
31. Qiu J, Villa M, Sanin DE, Buck MD, O’Sullivan D, Ching R, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep (2019) 27:2063–2074.e5. doi: 10.1016/j.celrep.2019.04.022
32. Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (2013) 342:971–6. doi: 10.1126/science.1240537
33. Vernocchi P, Del Chierico F, Putignani L. Gut microbiota profiling: Metabolomics based approach to unravel compounds affecting human health. Front Microbiol (2016) 7:1144. doi: 10.3389/fmicb.2016.01144
34. Belkaid Y, Naik S. Compartmentalized and systemic control of tissue immunity by commensals. Nat Immunol (2013) 14:646–53. doi: 10.1038/ni.2604
35. Skelly AN, Sato Y, Kearney S, Honda K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat Rev Immunol (2019) 19:305–23. doi: 10.1038/s41577-019-0144-5
36. Blacher E, Levy M, Tatirovsky E, Elinav E. Microbiome-modulated metabolites at the interface of host immunity. J Immunol Baltim Md 1950 (2017) 198:572–80. doi: 10.4049/jimmunol.1601247
37. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol (2016) 16:341–52. doi: 10.1038/nri.2016.42
38. Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, et al. Gut microbiota functions: Metabolism of nutrients and other food components. Eur J Nutr (2018) 57:1–24. doi: 10.1007/s00394-017-1445-8
39. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
40. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
41. Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med (2010) 16:228–31. doi: 10.1038/nm.2087
42. Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell (2005) 122:107–18. doi: 10.1016/j.cell.2005.05.007
43. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science (2012) 336:1268–73. doi: 10.1126/science.1223490
44. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
45. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of clostridia strains from the human microbiota. Nature (2013) 500:232–6. doi: 10.1038/nature12331
46. Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A (2010) 107:12204–9. doi: 10.1073/pnas.0909122107
47. Round JL, O’Connell RM, Mazmanian SK. Coordination of tolerogenic immune responses by the commensal microbiota. J Autoimmun (2010) 34:J220–5. doi: 10.1016/j.jaut.2009.11.007
48. Zhang H, Sun L. When human cells meet bacteria: Precision medicine for cancers using the microbiota. Am J Cancer Res (2018) 8:1157–75. doi: 10.1158/1538-7445.AM2018-1157
49. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell (2017) 171:1015–1028.e13. doi: 10.1016/j.cell.2017.09.016
50. Uribe-Herranz M, Bittinger K, Rafail S, Guedan S, Pierini S, Tanes C, et al. Gut microbiota modulates adoptive cell therapy via CD8α dendritic cells and IL-12. JCI Insight (2018) 3. doi: 10.1172/jci.insight.94952
51. Smith M, Dai A, Ghilardi G, Amelsberg KV, Devlin SM, Pajarillo R, et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med (2022) 28:713–23. doi: 10.1038/s41591-022-01702-9
52. Uribe-Herranz M, Rafail S, Beghi S, Gil-de-Gómez L, Verginadis I, Bittinger K, et al. Gut microbiota modulate dendritic cell antigen presentation and radiotherapy-induced antitumor immune response. J Clin Invest (2020) 130:466–79. doi: 10.1172/JCI124332
53. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science (2015) 350:1084–9. doi: 10.1126/science.aac4255
54. Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350:1079–84. doi: 10.1126/science.aad1329
55. Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science (2021) 374:1632–40. doi: 10.1126/science.aaz7015
56. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science (2020) 369:1481–9. doi: 10.1126/science.abc3421
57. Zhang S-L, Mao Y-Q, Zhang Z-Y, Li Z-M, Kong C-Y, Chen H-L, et al. Pectin supplement significantly enhanced the anti-PD-1 efficacy in tumor-bearing mice humanized with gut microbiota from patients with colorectal cancer. Theranostics (2021) 11:4155–70. doi: 10.7150/thno.54476
58. Messaoudene M, Pidgeon R, Richard C, Ponce M, Diop K, Benlaifaoui M, et al. A natural polyphenol exerts antitumor activity and circumvents anti-PD-1 resistance through effects on the gut microbiota. Cancer Discovery (2022) 12:1070–87. doi: 10.1158/2159-8290.CD-21-0808
59. Kawanabe-Matsuda H, Takeda K, Nakamura M, Makino S, Karasaki T, Kakimi K, et al. Dietary lactobacillus-derived exopolysaccharide enhances immune-checkpoint blockade therapy. Cancer Discovery (2022) 12:1336–55. doi: 10.1158/2159-8290.CD-21-0929
60. Wang H, Rong X, Zhao G, Zhou Y, Xiao Y, Ma D, et al. The microbial metabolite trimethylamine n-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab (2022) 34:581–594.e8. doi: 10.1016/j.cmet.2022.02.010
61. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-KB, and up-regulating expression of MicroRNA-21. Gastroenterology (2017) 152:851–866.e24. doi: 10.1053/j.gastro.2016.11.018
62. Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol (2020), 11. doi: 10.3389/fendo.2020.00025
63. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54:2325–40. doi: 10.1194/jlr.R036012
64. Luu M, Visekruna A. Short-chain fatty acids: Bacterial messengers modulating the immunometabolism of T cells. Eur J Immunol (2019) 49:842–8. doi: 10.1002/eji.201848009
65. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile acid metabolites control TH17 and treg cell differentiation. Nature (2019) 576:143–8. doi: 10.1038/s41586-019-1785-z
66. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes peripheral treg cell generation. Nature (2020) 581:475–9. doi: 10.1038/s41586-020-2193-0
67. Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med (2009) 206:43–9. doi: 10.1084/jem.20081438
68. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short chain fatty acids in human Large intestine, portal, hepatic and venous blood. Gut (1987) 28:1221–7. doi: 10.1136/gut.28.10.1221
69. Kim MV, Ouyang W, Liao W, Zhang MQ, Li MO. The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity (2013) 39:286–97. doi: 10.1016/j.immuni.2013.07.013
70. Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-Bet-Mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity (2012) 36:374–87. doi: 10.1016/j.immuni.2012.01.015
71. Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M. FoxO1 controls effector-to-Memory transition and maintenance of functional CD8 T cell memory. J Immunol Baltim Md 1950 (2013) 191:187–99. doi: 10.4049/jimmunol.1300331
72. Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, Harty JT. Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest (2015) 125:3477–90. doi: 10.1172/JCI81261
73. Berard M, Brandt K, Bulfone-Paus S, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol Baltim Md 1950 (2003) 170:5018–26. doi: 10.4049/jimmunol.170.10.5018
74. Bowers JS, Nelson MH, Majchrzak K, Bailey SR, Rohrer B, Kaiser ADM, et al. Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight (2017) 2:e90772. doi: 10.1172/jci.insight.90772
75. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
76. Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med (2020) 218:e20200844. doi: 10.1084/jem.20200844
77. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
78. Xue G, Zheng N, Fang J, Jin G, Li X, Dotti G, et al. Adoptive cell therapy with tumor-specific Th9 cells induces viral mimicry to eliminate antigen-Loss-Variant tumor cells. Cancer Cell (2021) 39:1610–1622.e9. doi: 10.1016/j.ccell.2021.09.011
Keywords: immunotherapy, short chain fatty acids, car-t, T cells, ex vivo, gut microbiota
Citation: González-Brito A and Uribe-Herranz M (2023) The potential role of short chain fatty acids improving ex vivo T and CAR-T cell fitness and expansion for cancer immunotherapies. Front. Immunol. 14:1083303. doi: 10.3389/fimmu.2023.1083303
Received: 28 October 2022; Accepted: 04 January 2023;
Published: 20 January 2023.
Edited by:
Vita Golubovskaya, ProMab Biotechnologies, United StatesReviewed by:
Alberto Bravo-Blas, University of Glasgow, United KingdomSagar Bapat, University of California, San Francisco, United States
Copyright © 2023 González-Brito and Uribe-Herranz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mireia Uribe-Herranz, dXJpYmVAcmVjZXJjYS5jbGluaWMuY2F0