 Qingbo Li1†
Qingbo Li1† Fengxian Tan2,3†
Fengxian Tan2,3† Yuanyuan Wang2,3
Yuanyuan Wang2,3 Xiaohui Liu3
Xiaohui Liu3 Xianbin Kong1*
Xianbin Kong1* Jingyan Meng1*
Jingyan Meng1* Long Yang2,3*
Long Yang2,3* Shan Cen4*
Shan Cen4*- 1College of Traditional Chinese medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2Research Center for Infectious Diseases, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 3School of Integrative Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 4Institute of Medicinal Biotechnology, Chinese Academy of Medical Science, Beijing, China
Various studies are being conducted on oncolytic virotherapy which one of the mechanisms is mediating interferon (IFN) production by it exerts antitumor effects. The antiviral effect of IFN itself has a negative impact on the inhibition of oncolytic virus or tumor eradication. Therefore, it is very critical to understand the mechanism of IFN regulation by oncolytic viruses, and to define its mechanism is of great significance for improving the antitumor effect of oncolytic viruses. This review focuses on the regulatory mechanisms of IFNs by various oncolytic viruses and their combination therapies. In addition, the exerting and the producing pathways of IFNs are briefly summarized, and some current issues are put forward.
1 Introduction
In recent years, research on tumor treatment by oncolytic viruses has been carried out continuously, and the related mechanism studies have been explored step by step, including direct lysis of tumor cells, inhibition of tumor angiogenesis and activation of human immunity (1). Among these mechanisms, IFN, as an active component of the human immune system, always plays an essential role in the treatment of tumors (2–4). Viruses can cause changes in IFN, as can oncolytic viruses. The release of IFN activates the human immune response through various pathways, thereby reversing the immunosuppressive state of the tumor microenvironment, which plays a positive role in tumor treatment.
There is an unstable effect of oncolytic virus therapy when applied to tumor treatment, which is very closely related to the effect of IFN. On the one hand, many tumor cells have an intact IFN pathway, which will have an immune clearance effect on the oncolytic virus (5) and cannot continue to exert therapeutic effect on tumor cells. On the other hand, the IFN secretion caused by oncolytic viruses will recruit more immune cells, and the antiviral effect they play will undoubtedly be the extinction for oncolytic viruses, which eventually leads to making the therapeutic effect greatly reduced.
However, incomplete IFN pathway genes are present in some cells in cancer patients. It has been shown that nearly half of the 85 genes with methylation-dependent down-regulation after immortality are associated with IFN signal transduction (6), the deletion of these genes is more common in glioma, leukemia, and bladder cancer cells (7–9). Besides, pancreatic cancer, gastric cancer, hepatocellular carcinoma, and colon cancer all exhibit low expression of IFN receptors (10–14). In addition to tumor cells, many cancer patients have impaired IFN signaling in immune cells (15). Although these defects allow tumor cells to survive and can accelerate tumor cell proliferation, the absence of the IFN pathway allows the virus to escape from the immune system, thereby avoiding immune clearance and increasing the efficiency of viral tumor lysis (16). Therefore, clarifying the mechanism of action between oncolytic virus and IFN will provide a strategy for combining oncolytic virus with other therapies or modifying oncolytic virus. This will help us to properly deal with the relationship between IFN and oncolytic virus, which is very important to achieve enhanced anti-tumor effects.
In this review, it presents a 5-year review of the mechanisms of action of various oncolytic virus therapies associated with IFN. Beyond that, the mechanisms of IFN production and signaling are briefly introduced. This demonstrates the current level of research on oncolytic virus therapies in order to hopefully provide new ideas for future studies on the mechanisms regulating IFN. Of course, some of these issues need to be noted.
2 IFN generation and signals transmission
2.1 Generation of IFN
IFN was discovered in humans more than 50 years ago for its ability to elicit antiviral responses in cells (17). Currently, IFNs are classified into three types based on their sequences and cellular receptors, which including the IFNs of type I, type II and type III (18, 19). There are some differences in the pathways of production of different types of IFNs.
2.1.1 Generation of type I IFN
The type I IFN family consists of several genetically encoded members, among which IFN-α and IFN-β are well known. In fact, they can be specifically divided into 16 species, which contain 12 IFN-α isoforms, IFN-β, IFN-ϵ, IFN-κ, and IFN-ω (20–27). The production of type I IFN is induced by pathogen-associated molecular patterns (PAMPs). These PAMPs can stimulate Toll-like receptors (TLRs) located on the cell membrane or endosomal membrane (28), or cytosolic pattern recognition receptors, including nucleotide sensors such as retinoid acid-inducible gene I(RIG-I)-like receptors (RLRs) or DNA sensors, to induce IFN production (29).
Firstly, the generation of type I IFNs is dependent on the TLR pathway. TLR recognizes double-stranded RNA and single-stranded RNA or double-stranded DNA, respectively, through TLR3, 7/8, and 9 (30), which activate and mediate IFN regulatory factor (IRF) to generate type I IFN (28, 31, 32). Second, type I IFN production can also occur through a non-TLR-dependent pathway. RIG-I and melanoma differentiation-associated protein 5 (MDA5) recognize endogenous RNA (single- and double-stranded, respectively) (33) and activate IRF3 and IRF7 to generate type I IFN through a mitochondrial antiviral signaling protein (MAVS)-dependent mechanism. In addition, endogenous cytoplasmic double-stranded DNA (dsDNA)-triggered synthesis of cyclic GMP-AMP (cGAMP) activates IFN gene stimulating protein (STING), which induces IRF3 to produce type I IFN (34).
2.1.2 Generation of type II IFN
There exists only one type of type II IFN, that is IFN-γ. Diverse cells in the immune system are the primary source of its secretion, including innate-like lymphocyte populations such as innate lymphocytes (ILC) and natural killer (NK) cells, and also adaptive immune cells consisting of T helper 1 (Th1) cells and CD8 cytotoxic T lymphocytes (CTL) (35).
First of all, in innate lymphocytes, microbial infection or tissue injury activates pattern recognition receptors (PRR) as well as broadly reactive antigen receptors, which induce IFN-γ production. In addition, cytokines which consisting of interleukin (IL)-12 and IL-18 can also lead to IFN-γ production. Second, in adaptive immune cells, T-cell receptor (TCR)-mediated recognition of microbial peptides causes sustained high levels of IFN-γ production in Th1 cells and CTL. However, the mechanism of IFN-γ production differs between the two cell types, with Th1 cells producing IFN-γ associated with major histocompatibility complex (MHC) Class II molecules, whereas CTL production of IFN-γ is associated with MHC Class I molecules (35).
2.1.3 Generation of type III IFN
Type III IFN was discovered later (known as IFN-λ) and it was reported in 2003 for the first time (36, 37). It includes four kinds of IFN-λ isoforms, namely IFN-λ1 or IL-28a, IFN-λ2 or IL-28b, IFN-λ3 or IL-29, and IFN-λ4 (38–40). Similarly, viruses can mediate the expression of type III IFNs in diverse cell types (41–43).
Type III IFN is expressed in various primary human cells of the hematopoietic spectrum (44–48), in parallel with the production of large amounts of type I IFN. Meanwhile, type III IFN is mainly produced by epithelial cells in non-hematopoietic cells (49–51). However, the exact mechanism by which it produces is not clearly explained. It has been claimed that the HSV molecular pattern is distinguished by TLR3 and TLR9 in the endosome as well as melanoma differentiation-associated gene 5 (MDA5) in the cytoplasm, which leads to the activation of nuclear factor κB (NF-κB), IRF3 and IRF7 transcription factors and their subsequent translocation to the nucleus, where they then stimulate IFN-λ gene transcription (52). In this process, the transcriptional mediator Med23 and anchoring protein repeat domain protein 1 (ANKRD1) target IRF7 and IRF3, individually, which promote the gene of type III IFN expression (53, 54).
2.2 Signal transduction of IFN
When IFN acts, it is transduced through different pathways. The three IFNs mainly signal through the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway. There are both similarities and differences. The main differences are that the signaling of the three IFNs is carried out through the binding of different heterodimeric receptor complexes (20) (Figure 1).
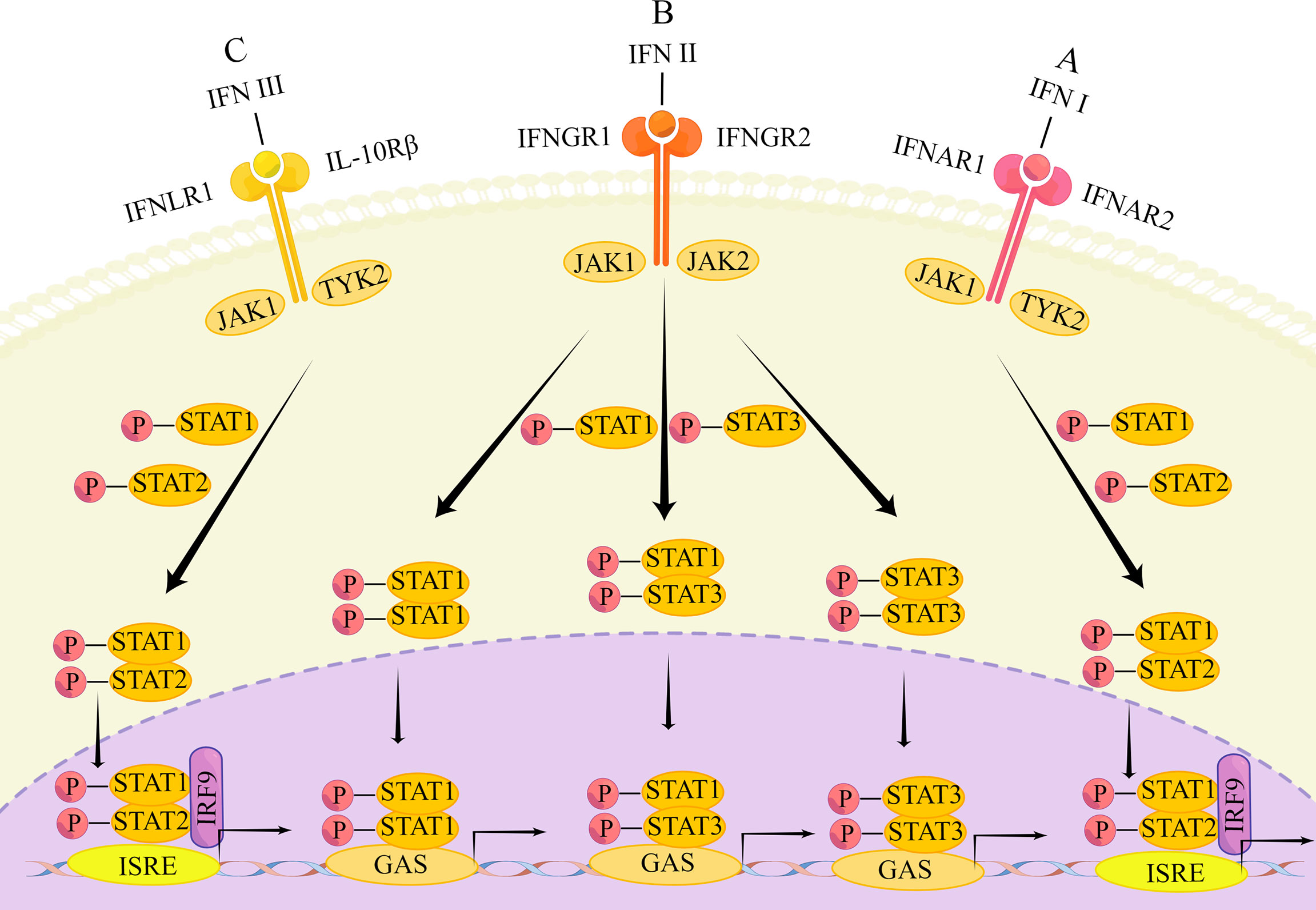
Figure 1 The main transduction pathway of IFN signaling. (A) IFN first binds to the heterodimers of IFNAR1 and IFNAR2, causing phosphorylation of JAK1 and TYK2, followed by phosphorylation of STAT1 and STAT2. Phosphorylated STATs form heterodimers that enter the nucleus and bind to IRF9 to form ISGF3, which subsequently binds to the transcriptional enhancer ISRE, triggering transcriptional induction of ISG. (B) Type II IFNs first bind to IFNGR1 and IFNGR2, causing phosphorylation of STAT1 and STAT2. The phosphorylated STAT forms a heterodimer that enters the nucleus and binds to IRF9 to form ISGF3. ISGF3 subsequently binds to the transcriptional enhancer ISRE, triggering the transcriptional induction of ISG. In addition, phosphorylated STAT1 homodimers, STAT3 homodimers and STAT1-STAT3 heterodimers enter the nucleus. These dimers bind to GAS elements to induce transcription factor production and initiate a second wave of gene expression; (C) Type III IFN binds to IFNLR1 and IL10R2, followed by the same response as the type I IFN signaling cascade.
Type I IFN is bound to a heterodimer of type I interferon α/β receptor 1 (IFNAR1) and IFN-α/β receptor 2 (IFNAR2) (18, 55). Signaling through the JAK/STAT pathway is the most common, which starts with phosphorylation of JAK1 and tyrosine kinase (TYK)2 and leads to phosphorylation of STAT1 as well as STAT2. Phosphorylated STATs are able to form heterodimers. The heterodimers will enter the nucleus and link with IRF9 to create the transcriptional activator IFN-stimulated gene factor (ISGF) 3. Next, ISGF3 integrates with the transcriptional enhancer called IFN-stimulated response element (ISRE), leading to transcriptional induction of ISGs (56–59). Besides this typical pathway, type I IFN can induce the expression of other genes, through STAT1 or STAT3 homodimers as well. For instance, homodimers formed by STAT1 combined with gamma activated sequence (GAS) elements that belong to different genes’ promoters (60). STAT1 and STAT3 are the most frequent, but it has also been shown that in some cell types, STAT-3, -4, -5, and -6 can also be activated by interferon receptor (IFNAR), causing the next series of signaling cascades (61).
Type II IFN is bound to type II interferon gamma receptor 1 (IFNGR1) and IFN-γ receptor 2 (IFNGR2) (18, 55). The JAK/STAT pathway can be activated as well, although not in the same way as the changes in the JAK/STAT pathway caused by type I IFN. In this pathway, type II IFN can signal through ISGF3 as type I IFN does (62), causing the following series of responses. The difference is that the phosphorylation of JAK2 upon binding of type II IFN to its receptor is accompanied by allowing phosphorylation of JAK1 and IFNGR1 (63), which will recruit and phosphorylate STAT1s. Then, the homodimer formed by phosphorylated STAT1 enters the nucleus, unites with the GAS element to induce transcription, inducing the generation of many transcription factors that initiate the second wave of gene expression (64). Furthermore, IFN-γ signaling can activate not only STAT1 homodimers, but also generate STAT3 homodimers and STAT1-STAT3 heterodimers. These still translocated to the nucleus to combined with the GAS element that is in the IRG gene promoter (65, 66). Apart from the above pathway, type II IFN can trigger the expression of MHCII as well, that is by inducing a different piece of genes through the function of the class II, major histocompatibility complex, transactivator (CIITA) (67).
Type III IFN is bound to interferon λ receptor 1 (IFNLR1) and IL-10 receptor 2 (IL10R2) (also known as IL-10 receptor β (IL10Rβ)) (18, 55). The induced signaling pathway is also essentially the same as that of type I IFN (36). Type III IFNs similarly form ISGF3 through phosphorylated STAT1 and STAT2, followed by binding to IRF9, which in turn triggers the expression of ISGs (42). Alternatively, type III IFN can induce the activation of STAT-3, -4, and -5 in certain cell types (68). However, the durability of ISG induction by type III IFN is demonstrated by the fact that ISGs peak later after type III IFN stimulation and persist over time. In contrast, type I IFN only induces ISG expression at an early stage and persists for a relatively short period of time (69).
Aside from the classical pathway of JAK/STAT, the three IFNs can also function in other signaling pathways, including MAPK and PI3-kinase pathways (61, 70). In addition, type I IFN can also activate and signal through the NF-κB pathway, and type II IFN can function through this pathway as well (70). Also, the bioinformatic analysis revealed the presence of NF-κB binding sites in the promoter of the type III IFN gene (68), suggesting the possibility that it also acts in the NF-κB pathway.
3 IFN and tumor treatment
IFN is able to modulate multiple pathways to achieve tumor inhibition or killing. This has been demonstrated in several experimental studies.
Under in vitro conditions, IFN can inhibit tumor growth through various pathways. First, IFN can affect the cell cycle of tumor cells. For example, IFNα can arrest the cell cycle of prostate cancer cells, which is achieved by upregulating endogenous inhibitors of cell cycle protein-dependent protein-dependent kinases, such as p21 (71); it has been discovered that type I IFNs can prolong the cell cycle of human breast cancer cells under in vitro conditions, which can suppress the growth of tumor cells (72). Second, IFN can also induce apoptosis of tumor cells. It has been shown that type I IFNs and Toll-like receptor 3 (TLR3) agonists when combined, are able to upregulate DR ligands, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), thus leading the breast tumor cell lines to be apoptotic (64). Moreover, type I IFN-induced apoptosis was connected with other ISGs, consisting of Fas, Fas ligand (FASLG), protein kinase R (PKR), and 2′-5′-oligoadenosine synthase (OAS) (64).
In the vivo environment, IFNs have been suggested to have a crucial role in tumorigenesis process. For example, in studies of 3-methylcholanthrene (MCA)-induced sarcoma models, it was found that deletion of immune cell type I and/or type II IFN signaling pathways sped up tumorigenesis and development (73, 74). Furthermore, STAT1 is thought to exert antitumor effects in transgenic mouse models of breast cancer through activation of immune and antiproliferative mechanisms (75), which is significant in type three types of IFNs signaling.
4 Molecular mechanisms by which different oncolytic virus therapies affect the IFN pathway
4.1 Vesicular stomatitis lysis virus
VSV is a prototypical member of the genus Blister virus, belonging to the family Rhabdoviridae (76). Recently, it has been extensively studied as an oncolytic agent (77). Among the IFN-related mechanisms, VSV mainly regulates IFN-induced antiviral factors, the expression of classical JAK/STAT, nuclear factor red lineage 2-related factor 2 (Nrf2), and IFN-mediated programmed cell death-ligand 1 (PD-L1) (Figure 2).
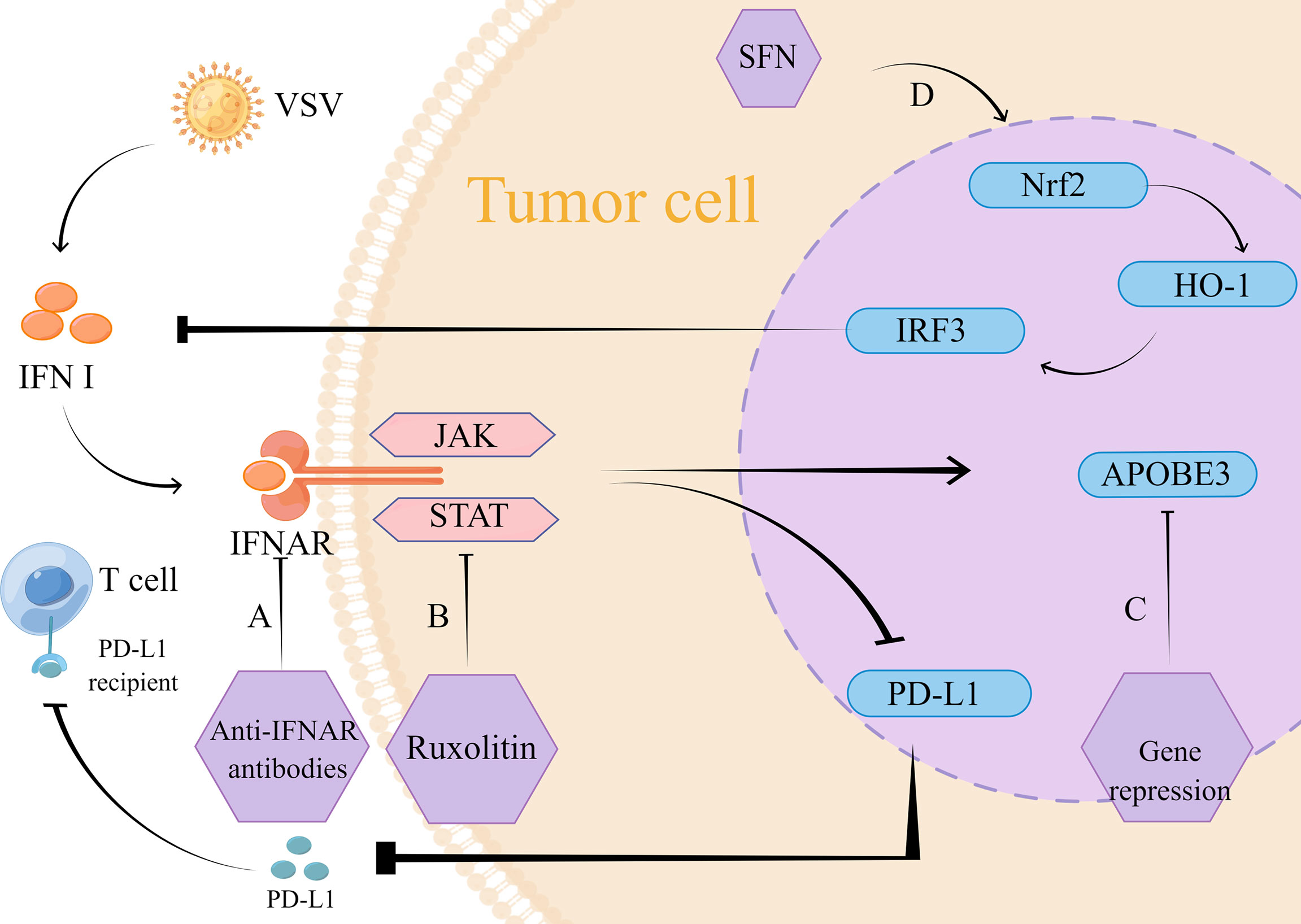
Figure 2 Mechanism of action of VSV therapy. (A) The intervention of monoclonal IFNAR antibody blocks the VSV-mediated IFN inflammatory pathway and reduces type I IFN-induced PD-L1 expression. Its low expression reduced PD-L1 binding to T cells, which thus exerted normal antitumor effects. (B) VSV strongly induced the JAK/STAT pathway, and inhibition of JAK/STAT using lusolidin prevented IFN-mediated antiviral response to VSV immune clearance, which promoted VSV replication and dissemination. Similarly, type I IFN-induced PD-L1 expression was reduced, thus preventing PD-L1 from binding to T cells and providing conditions for T cells to exert normal anti-tumor effects. (C) APOBEC3 gene expression is mediated by type I IFN, targeted inhibition of APOBEC3 gene can reduce IFN-mediated tumor resistance. (D) SFN through Nrf2/HO-1 pathway activates autophagy to inhibit IRF3 activity, which suppresses the type I IFN response. The inhibition of IFN response reduces the restriction of replication of VSVΔ51, so VSV exerts its normal oncolytic effect.
The type I IFN response is considered as an essential pathway for the development of drug resistance to VSV during tumor treatment, and tumor cells with intact or partially intact IFN signaling are resistant to viral replication (78–80). Human A375 as well as mouse B16-OVA melanoma cell lines were reported to be shielded by type I IFN and resistant to tumor lysis by wild-type VSV and VSV-GP (81)., thus preventing VSV from exerting its normal antitumor effects. VSV significantly upregulates the JAK/STAT pathway, which is an important component of the functioning of IFN. Inhibition of this link can effectively improve resistance to VSV therapy. In a preclinical trial in small cell lung cancer, the use of the JAK/STAT inhibitor lusolidin effectively increased viral replication, and the killing effect of VSV-IFN-β on tumor cells was enhanced in vitro conditions. Besides, it turned out that PD-L1 expression was restricted, which was also beneficial for tumor treatment. However, this combination treatment strategy did not significantly improve the survival rate of mice (82), so the safety of this combination therapy is uncertain. Additionally, this pathway was also studied in an animal experiment in melanoma, where tumor sensitivity to VSV-Δ51 was significantly increased under conditions of JAK/STAT pathway inhibition (83).
PD-L1 performs a functional role in regulating the cancer immune clearance cycle by binding to T cell-activated negative regulators, such as programmed cell death-1 (PD-1) and B7.1 (CD80) (84). In order to inhibit the killing effect on tumor cells, the combination of PD-L1 and its receptor inhibits T cells from migrating, meanwhile, the combination keeps down the T cells’ proliferation as well as the release of mediators that have cytotoxic, therefore inhibition of PD-L1 expression is one of the strategies for tumor therapy. Several previous studies have shown that PD-L1 expression can be induced independently of the IFN inflammatory pathway, but is often dependent on the IRF1 pathway, a transcription factor associated with PD-L1 regulation (85–88). In contrast, according to a recent research in melanoma, VSV optimizes PD-L1 upregulation in tumors which is dependent on type I IFN expression, and in-depth studies revealed that VSV-induced type I IFN proceeds in an IFNAR-dependent manner (89). This provides an opportunity to improve the therapeutic use of VSV for tumors. In order to block the viral-mediated IFN inflammatory pathway, the monoclonal IFNAR antibody can be taken into consideration. The paper shows monoclonal IFNAR antibody can reduce PD-L1 expression which is induced by type I IFN, this will promotes immune responses with tumor-specific T cells (89). This research result provides a new target for the treatment of solid tumors.
Among the IFN-induced antiviral factors, the APOBEC cytosine deaminase family is associated with viral resistance (90), which has been demonstrated in retroviruses, herpesviruses, and hepatitis viruses, among others (91–94). In addition to being a viral limiting factor, over expression of APOBEC3 family proteins occurs in several types of cancers, so that APOBEC3 upregulation and the genomic mutations it causes to mediate therapeutic resistance are important for the prognostic profile of cancer (92, 95). In contrast, VSV, a retrovirus, is able to mediate APOBEC3 expression in tumor cells. This expression is dependent on type I IFN upregulation. This research suggests that APOBEC3 is a key gene for type I IFN stimulation and plays an influential part in the build-up of resistance to oncolytic virus therapy (96).
Nrf2 is a transcriptional regulator that maintains redox homeostasis by controlling basal and induced expression of a series of antioxidant enzymes (90). Furthermore, Nrf2 actively regulates autophagy as an essential component of the regulatory network that responses to different types of stress, including protein aggregation, nutritional deficiency, and viral infection (97). Therefore, it may also influence VSV replication and infection. A study on lung cancer and osteosarcoma showed that for drug-resistant lung cancer cells (A549) and osteosarcoma cells (U-2OS), sulforaphane (SFN) inhibited IRF3 activity by activating autophagy through the Nrf2/HO-1 pathway. This inhibited the type I IFN response and promoted VSVΔ51 replication, leading to better tumor lysis. what’s more, it has shown a good safety profile in animal experiments (98).
4.2 Herpes simplex virus-1
Herpes simplex virus 1 (HSV-1) belongs to the subfamily Alphaherpesvirinae (99). Regulation of IFN by HSV and its combination therapies is achieved through multiple pathways, including STAT3-PKR-dependent antiviral responses, IRF3, and TNF-related apoptosis-inducing ligand (TRAIL). Remarkably, there is a possibility as a potential cancer vaccine of it. (Figure 3)
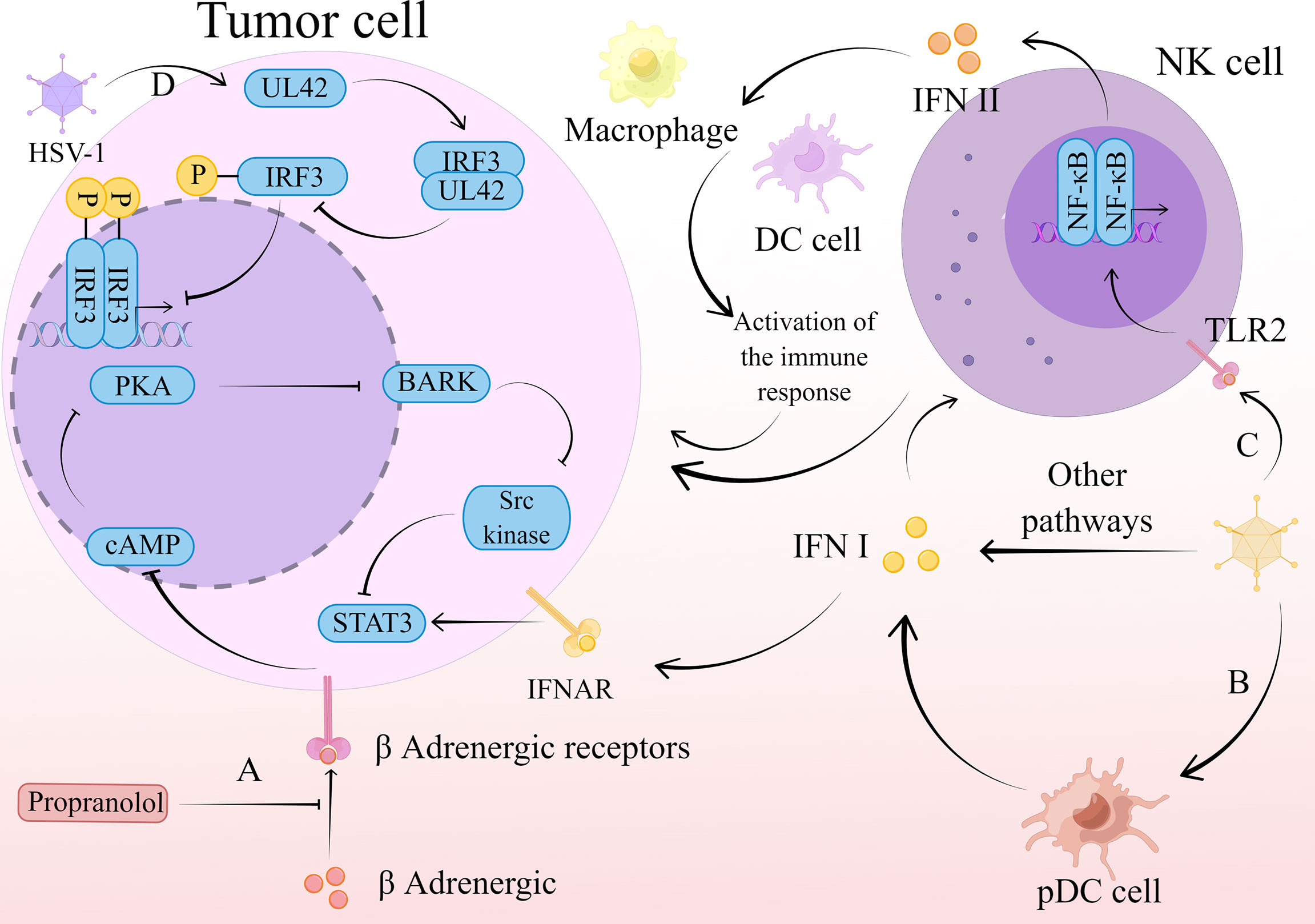
Figure 3 The mechanism of action of HSV-1 therapy. (A) Propranolol blocks β-adrenergic binding to β-adrenergic receptors, leading to a restriction of Gαs-mediated cAMP synthesis. This restriction limits the transient flux of intracellular cAMP to activate PKA production and inhibits its phosphorylation to produce BARK. activation of Src kinase is dependent on BARK, and its inhibition in turn inhibits activation of the transcription factor STAT3. This series of reactions inhibit the type I IFN-mediated antiviral response and promotes the normal replication and propagation of HSV-1, thus exerting an oncolytic effect. (B) HSV-1 stimulates the production of type I IFN by pDC, which activates NK cells to exert a direct killing effect on tumor cells. (C) HSV-1 stimulates the release of type II IFN by NK cells through the TLR2/NF-κB signaling pathway. Type II IFNs released by NK cells recruit macrophages, DC cells, and other immune cells, which act as immune agents to remove tumor cells. (D) The viral polymerase synthesis factor UL42 is able to interact with the host transcription factor IRF3. This interaction inhibits IRF3 phosphorylation and downstream IFN-β gene transcription, thereby suppressing IFN-β expression and thus the antiviral effect. The virus is consequently able to replicate and spread normally, exerting an oncolytic effect.
Similarly, HSV still faces resistance due to innate immunity when applied. The type I IFN antiviral signaling pathway bears the brunt of viral defense in infected cells (100). Previous studies have identified protein kinase R (PKR) as a host antiviral kinase that inhibits cell proliferation and blocks the production of viral proteins, thus preventing viral replication (101). Therefore, inhibition of its expression and activation can attenuate the type I IFN-mediated antiviral response. In contrast, STAT3, as part of the type I IFN signaling pathway, can also inhibit the expression of PKR to limit the type I IFN cascade response (102–105). Similarly, protein kinase A (PKA) can also achieve this effect. Recently, it has been shown that β-blocker pretreatment inhibits the binding of catecholamines to β-adrenergic receptors, leading to a limitation of Gαs-mediated cyclic 3’-5’ adenosine monophosphate (cAMP) synthesis. This limits the transient flux of intracellular cAMP to activate PKA production, whose phosphorylation to produce a variety of target proteins is necessarily affected, including β-adrenergic receptor kinase (BARK). Src kinase can be activated by BARK, thereby suppressing the activation of the important transcription factor STAT3 (106). By inhibiting this series of interferon-related responses, the antitumor efficacy of oncolytic virus T1012G is improved (107).
The IFN gene stimulating factor (STING), which is upstream of IFN production, is similar to STAT. Its mediated IFN gene stimulating factor (STING)- TANK-binding kinase 1 (TBK1)-IRF3-IFN pathway is a central cellular host defense against viral infection (108–110). However, in a preclinical study of pancreatic ductal adenocarcinoma (PDAC), it was shown that cell lines defective in the STING pathway had relatively low susceptibility to being C-REV (a type of HSV) (111). In contrast, those cell lines that capable of responsive STING pathway had relatively higher susceptibility to C-REV. This suggests that there is a correlation between STING pathway activation and resistance to C-REV, and this pathway does have an effect on oncolytic virus replication, However, data analysis revealed that it is not the main pathway affecting C-REV in human pancreatic ductal adenocarcinoma cell lines (111).
Although C-REV has no critical effect on STING, one study found that modification of HSV can act on IRF3 in the STING pathway to exert antitumor effects. Engineered HSV-1ΔN146 containing amino acids 147 to 263 of γ134.5 could efficiently replicate and lyse in malignant cells refractory to the γ134.5 null mutant. ΔN146 activated IRF3 and IFN expression, triggering immunity against the virus and the tumor. Unexpectedly, ΔN146 exposed to exogenous IFN-α was also able to replicate normally, and in a 4T1 tumor model, ΔN146 also exhibited significant inhibition of tumor growth and metastasis. Thus, ΔN146 is able to stimulate the expression of inflammatory cytokines that do not have a serious impact on the replication of the virus in tumor cells. This can ensure to a greater extent that the oncolytic virus works (112). Furthermore, a similar effect can be achieved by causing mutations in the HSV-1 gene through the random insertion of a corruptive 1.2-kbp transposon into the viral genome. This mutation is capable of generating the viral polymerase synthesis factor UL42, which interacts with the host transcription factor IRF3 (113). The above studies suggest that genetic modification of HSV may be a new strategy to avoid immune clearance of HSV and provide a new way to enhance the effect of oncolytic virus therapy, but this may only be applicable to personalized treatment.
One of the pathways mediated by HSV-1 to generate IFN-α/β is through the stimulation of plasmacytoid dendritic cells (pDCs) (114), whose production of type I IFNs can activate NK cells (115). A current study has found that the type I IFN produced by pDCs and activated NK cells are an important link in the anti-myeloma effect of HSV-1, type I IFN has a direct cytotoxic effect on tumor cells and induces IFN to release from NK cells, thereby enhancing the killing effect of NK cells (116). In addition, type I IFNs have direct oncolytic activity against plasmacytoid tumors, where type I IFNs upregulate TRAIL expression, mitochondrial cytochrome c release, while limiting the expression of B-cell leukemia/lymphoma 2 (Bcl-2) and Bcl-XL (117). Regulation of these genes ultimately leads to apoptosis.
HSV not only acts through type I IFNs with NK cells, but also mediates type II IFNs stimulation of NK cells. Recent studies have shown that UV-oHSV2 can stimulate NK cells to secrete IFN-γ, which is achieved through the Toll-like receptor 2 (TLR2)/NF-κB signaling pathway, which activates multiple immune cells to exert anti-tumor responses (118). Furthermore, UV-oHSV2 stimulation promoted the expression of two checkpoint molecules, one located on NK cells, NKG2A, and the other on tumor cells, HLA-E. This finding predicts that anti-NKG2A may further enhance the antitumor effects occurring from UV-oHSV2 stimulation, and that anti-HLA-E treatment also has this possibility (118).
Speaking of IFN-γ, it plays an active role in the induction of apoptosis as well as tumor-infiltrating T cell recruitment. It has been shown that oHSV-1 stimulates tumor cells to secrete IFN-γ, which increases the immune activity of T cells, which is also able to enhance the activity of CD70-specific CAR T cells. This combination of specific T-cell therapy and oHSV-1 enhanced the pro-inflammatory environment and reduced anti-inflammatory factors in vitro, which achieved the goal of promoting tumor extinction. While this immune activation environment, this combined strategy increased the ratio of T cells and natural killer cells in the tumor microenvironment (TME) and decreased the expression of regulatory T cells as well as transforming growth factor-β1 in glioblastoma (GBM) in an in situ xenograft animal model. It undoubtedly brings a new therapeutic strategy for the treatment of GBM (119).
In addition, one study found that HF10 was able to prevent secondary tumors while activating anti-tumor effects. In an animal experiment with mice with squamous cell carcinoma, mice that survived HF10 treatment all showed rejection of tumors upon reactivation. Studies of systemic immunity in mice revealed that a large number of granulocytes and CD8 T cells were accumulated in the spleen at the time of HF10 application, and when co-cultured with SCC-VII cells, splenocytes released type I IFN (IFN-α and IFN-β), IFN-γ, IL-2, IL-12, and TNF-α. This suggests that mice developed anti-tumor immunity and implies that HSV has the potential for an in situ cancer vaccine (120).
4.3 Reovirus
MRV is a virus with double stranded RNA (dsRNA), which belongs to Reoviridae (121). Its mechanisms associated with IFN when treating tumors are mainly related to the PD-1/PD-L1 axis, STAT, and IFNAR signaling.
Similar to VSV, IFN is a key cytokine in reovirus-mediated activation of immune cell populations (122). A research found that type I and type II IFNs are able to promote PD-L1 expression in patient-derived glioma cells in a synergistic manner, while type I IFNs can induce strong expression of PD-L1 with IFN-γ, which undoubtedly has a detrimental effect on tumor treatment (123). PD-L1 binding to PD-1 prevents attack by the host’s own immune system, thereby reducing T-cell activation and proliferation of cytotoxic T lymphocytes. tumor cells evade immune surveillance as a result of T-cell depletion (124, 125). In a vitro experiment, IFN-γ was the cytokine secreted after reovirus treatment of HGG cells (123). The above analysis revealed that reovirus treatment could improve the clinical outcome of brain tumor patients by activating leukocytes, enhancing T-cell infiltration into tumors and upregulating PD-L1, which prepared for later anti-PD-1 therapy. Further studies found that the addition of PD-1 blockers to reovirus enhanced systemic therapy in preclinical glioma models. These results analyze the mechanisms by which reovirus affect IFN-related pathways and provide theoretical support for the development of PD-L1 blockade combined with systemic immunoviral treatment strategies (123).
It has been noted that in a mouse model, IL-15 can be induced by type I IFN in dendritic cells (DC production) and this cytokine can activate NK cells to act (126, 127). NK cells are a type of innate lymphocyte (ILC) that, on the one hand, are able to recognize and kill infected cells as well as tumor cells, on the other hand, have an ability to induce adaptive immunity to function (128, 129).NK component of the cytotoxic machinery is triggered by type I IFN (126, 130, 131). Recent studies have shown that Reovirus are able to activate NK cells in a type I IFN -dependent manner, inducing STAT1 and STAT4 signaling in CD56dim as well as a subset of CD56bright NK cells. However, It is puzzling that MRV is dependent on type I IFN to inhibit IL-15-induced NK cell proliferation, which may be involved with reduced AKT signaling. In in vivo experiments, CD56bright NK cells disappeared from the peripheral circulation for a brief period during the peak of the type I IFN response, which may suggest that they underwent redistribution and migrated to secondary lymphoid tissues. In combination with OV-mediated direct tumor cell killing, CD56 activation and CD56bright NK cells induce a spectrum of activity via antiviral pathways, including NK cell-mediated tumor cell killing and regulation of adaptive NK cells to lymph nodes by transport of IFN-γ-expressing CD56 (132). However, reovirus does not always depend on the type I IFN pathway for its antitumor effects. By comparing IFN-β promoter stimulator-1promoter stimulator-1 knockout (KO) mice with TLR-3 KO mice under reovirus treatment conditions, it was found that Reovirus inhibits the immunosuppressive activity of bone marrow mesenchymal stem cells in a TLR3 manner, but not in an IFN-β promoter stimulator-1 signaling-dependent manner (133).
Alternatively, the type of IFN produced by reovirus-mediated production can impact the tumor’s therapeutic outcome. Studies have shown that MRV infection has a superior stimulatory effect on type III IFN production, but does not show satisfactory performance for type I IFN production. Although activation of STAT1 and STAT2 can be achieved by both type I and type III IFNs, triple-negative breast cancer cell proliferation is only sensitive to type I IFN (134). For this issue, researchers treated triple-negative breast cancer cells with a topoisomerase inhibitor that activated the DNA damage response pathway. This combination promoted the replication of the eutherian virus and enhanced cytotoxicity, achieving effective infection and killing of triple-negative breast cancer cells (134).
Again, the problem of ineffectiveness against IFN pathway-deficient tumor cells has been faced with the application of the eutherian virus. Researchers identified IFN regulatory factor 3, as a crucial transcription factor for IFN-β expression, in transformed human myeloid cells infected with tumor-selective MRV, IFN-α/β receptor (IFNAR) signaling both gradually promoted IFN I secretion from infected cells by enhancing the activation of IFN regulatory factor 3, and also promoted viral replication. However, tumors can interfere with the IFNAR pathway to maintain their own survival, and tumors that do not respond to IFNAR signaling may require other therapeutic strategies to promote adequate type I IFN secretion into the tumor microenvironment. Therefore, the parameters of eutherian virus-induced type I IFN levels need to be further explored (135).
Some viruses from the same ancestor have small genetic differences that cannot be ignored, and their effects on cell signaling and regulation of cytokine secretion may differ dramatically (136–142), which can affect antitumor effects. For example, T3D lab strains have a large variability in the regulation of RIG-I and/or IFN-dependent genes, with the least tumorigenic T3DTD strains inducing significantly higher levels compared to the most tumorigenic T3DPL strains (the difference may be a result of polymorphisms in the dsRNA-binding protein and the PKR antagonist σ3), which is crucial for the selection of the appropriate tumorigenic virus strain (143). This suggests the need to consider minor differences between viruses and to clarify the target of action when selecting combination therapies.
4.4 Newcastle disease virus
NDV belongs to the genus Aviravirus in the family Paramyxoviridae (144). The mechanism has not been extensively studied in terms of IFN-associated tumor lysis, which is associated with both type I and type II IFNs.
Similar to numerous viruses, NDV affects type I IFN (145). It is well known that type I IFN-mediated PD-L1 expression is an unfavorable factor in tumor treatment. Unexpectedly, the inflammatory response and PD-L1 upregulation induced by NDV treatment of tumors can precisely enhance the sensitivity of these tumors to PD-1/PD-L1 blockade. The strategy of intratumoral NDV injection combined with systemic PD-1 or PD-L1 blockade significantly enhances the antitumor immune effect, which provides a theoretical basis for future clinical trials (146). Further analysis revealed that this is mainly evidenced by its upregulation of PD-L1 expression in tumor cells as well as in tumor-infiltrating immune cells, which plays an important role in the development of late adaptive mechanisms of immune resistance to increased immune cell infiltration into tumors (146).
NDV infection also affects changes in type II IFN, as demonstrated in glioblastoma, colorectal and cervical cancers (147–149), and the mechanism of action needs to be further elucidated. In an animal experiment on lung cancer, it was identified that, compared to IL-4, the increase in IFN-γ concentration far exceeded its increase, IFN-γ is one of the cytokines secreted by Th1, under NDV intervention conditions (150). This indicates a shift in cellular distribution from Th2-dominant to Th1-dominant, suggesting that NDV plays a role in regulating humoral immunity and inhibiting tumor growth (148).
Naturally, genetic modification of NDV is one of the strategies to overcome the body’s antiviral reflection (151). It was found that genetic modification of NDV to express influenza virus NS1 protein can improve the susceptibility of GBM to type I NDV-activated cells, resulting in better tumor lysis (152). This viral protein can suppress the host immune response (153, 154), the virus can thus exert its antitumor effects more effectively.
4.5 Vaccinia virus
The virus of VV belongs to the genus Orthopoxvirus (OPXV) (155). Its mediated IFN exerts antitumor effects mainly related to IRF-3, JAK-STAT signaling pathway, and Th1 cells.
In the past decades, some progress has been made in the study of VV for antitumor therapy (156–159). The finding of a recent study that a recombinant VV can induce high levels of IFN while blocking the IFN-mediated antiviral response is undoubtedly an important finding. It was shown that OncoVV-WCL could achieve induction of high levels of type I IFN expression by promoting IRF-3 transcriptional activity, which could enhance the antitumor effects of oncolytic virus. Specifically, blocking the IFN-induced antiviral response is achieved through two pathways. On the one hand, OncoVV-WCL can inhibit the activity of IFN stimulatory response element (ISRE), and on the other hand, inhibition of JAK-STAT signaling pathway by OncoVV-WCL limits ISG expression (160). By these means, the virus can avoid elimination by the antiviral pathway and thus exert a normal lytic effect (160).
Another study on multiple tumors showed that vvDD-IL-23 can promote the expression and release of Th1 chemokines and some anti-tumor factors, which contained IFN-γ, as well as tumor necrosis factor-α (TNF-α), IL-2, perforin, and granzyme B (GzmB). These cytokines keep the ratio of infiltrating activated T cells CD8 and Treg to high levels, which play a therapeutic role in reversing the immunosuppressive state to achieve antitumor (161). In a clinical study, VV also acted through a similar mechanism. It was found that a classical IFN response, including the release of inflammatory cytokines/chemokines, was induced in patients who were lysing virus responders. These factors activate T cells, which can then infiltrate into the tumor to exert antitumor effects (162).
4.6 Other viruses
4.6.1 Measles virus
MV is a type of negative-stranded RNA virus that belongs to the family Paramyxoviridae, genus Morbillivirus (163). It mainly affects type I IFN to exert antitumor effects. A variety of malignant pleural mesothelioma (MPM) cell lines have a defect in antiviral type I IFN response (164), and this defect in type I IFN response is located upstream of the IFNAR. It was thought that type I IFN-deficient tumor cells sensitive to the antitumor effects of MV still participate in part of the type I IFN response though. This is achieved by relocalizing IRF3 and NF-κB in the nucleus, but the resulting ISG expression is minimal, which is very favorable for the oncolytic virus to function. Indeed, type I IFN-deficient tumor cells can induce a response that induces immunogenic death of tumor cells and additionally induces an endoplasmic reticulum stress response, enhancing the antitumor effect. At a deeper genetic level, pure deletion (HD) of all genes of type I IFN in human MPM cells leads to their sensitivity to MV virus, and HD of the type I IFN-encoding gene in MPM occurs frequently together with HD of the CDKN2A gene, it suggests a new therapeutic target (165–167). Another study using a sequential transformation model also identified reduced type 1 IFN pathway function as a significant factor in MV-mediated selectivity of transformed cytolytic tumors (168).
4.6.2 Coxsackie virus
Fewer studies have been conducted on the mechanisms by which coxsackieviruses exert tumor lysis. Recent clinical trials in non-muscle invasive bladder cancer have identified a kind of coxsackievirus, CAVATAK, that upregulates PD-L1 and lymphocyte activation gene-3 (LAG3) among the IFN-inducible genes. In parallel, this virus promotes the release of Th1-related chemokines as well as induces RIG-I. Through these pathways, it induces an inflammatory response, reverses the “cold” tumor state, has an anti-tumor effect, and shows a good biosafety profile (169).
4.6.3 Poliovirus
The neurally attenuated recombinant poliovirus PVSRIPO has also been used in oncology treatment, it has shown good efficacy in clinical trials in glioblastoma (170). Recently, researchers have explored its mechanism of action, and unlike other oncolytic viruses, PVSRIPO is insensitive to both upstream and downstream endogenous intrinsic responses to IFN triggered by MDA5 under in vitro conditions. Although the involvement of PRR inhibited the kinetics of PVSRIPO, PVSRIPO could still be translated in diseased cells and propagate in the cell. This occurrence may be related to the translation strategy of PVSRIPO, which prevents the body’s antiviral immune response while destroying tumor cells, and immune escape occurs (171). This property could sustain activation of the IFN response. This finding suggests that poliovirus has a significant advantage in exerting its oncolytic effect.
5 Discussions and challenges
From the above review, we can understand that a variety of viruses show the ability to activate the immune of body response, which is closely related to the IFN pathway. They reverse the immunosuppressive state in the tumor microenvironment, resulting in the production of various other cytokines and various immune cells and changing cold tumors to hot tumors. Finally, they achieve the suppressive and clearing effect on tumor cells.
For different oncolytic viruses, the pathways affecting IFN are not identical, and almost all IFN-related pathways are included, summarizing that they mainly interact with IFN through the following mechanisms: (1) PD-L1/PD-1; (2) JAK/STAT signaling pathway; (3) APOBEC cytosine deaminase family; (4) Nrf2; (5) TLR2/NF-κB signaling pathway; (6) IRF3. The elucidation of these mechanisms provides a theoretical basis for future combination therapies with various oncolytic viruses, thus providing guidance for targeted enhancement of oncolytic viral therapeutic efficacy.
At the same time, we found that oncolytic viruses are similar to other viruses. When the immunity of body is activated, it will activate the anti-virus response related to IFN, which will make the virus unable to exist in the immune microenvironment for a relatively long time. Although this can improve the biological safety of oncolytic virus therapy, it is difficult to achieve an effective anti-tumor effect for a short time. Through various ways, it can inhibit the immune clearance of oncolytic viruses, better therapeutic effect can be achieved. Here, we focus on IFNs. By summarizing, we found that researchers mainly take the following ways to achieve better anti-tumor effects: (1) Combined drugs target the inhibition sites of oncolytic viruses; (2) Genetic modification of oncolytic virus; (3) Select the appropriate strain. In addition, for tumor types with IFN deficiency, some specific defects in the IFN signaling cascade can be used as potential biomarkers, which may help identify such individual cancer patients and obtain personalized treatment (16).
Of course, there are still some problems in the study of the mechanism of action between oncolytic virus and IFNs. First, most studies are still in the preclinical stage, and their mechanisms and effects in the clinical setting are still unknown. Researchers should accelerate their studies to better benefit oncology patients. Second, due to the complexity of in vivo immunity, the relationship between the efficacy of viral tumor lysis and the IFNs gene has not been fully elucidated. Further studies in this area are expected in the future. In addition, many studies have used the expression level of IFNs as an indicator of antitumor effects, and the specific mechanism of its increased expression level and antitumor effects need to be further elucidated. Notably, there are few studies on type III IFN and oncolytic viruses, and it is hoped that future studies will fill this gap. Finally, the degree of research on various viruses varies greatly. Some viruses are able to activate IFN-induced antitumor immunity while avoiding immune clearance, such as PVSRIPO and HSV-1, etc. Such viruses may have more advantages in antitumor, and research on their oncolytic mechanism can be more widely carried out.
Author contributions
QL and FT: writing, editing, and visualization. YW and XL: reviewing and editing. XK: conceptualization. JM: validation. LY: supervision. SC: funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Scientific research project of Tianjin Education Commission (2021KJ134), National Natural Science Foundation of China (81973728) and Science and Technology Program of Tianjin (21ZYJDJC00070).
Acknowledgments
The figures were drawn by Figdraw (www.figdraw.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yang L, Gu X, Yu J, Ge S, Fan X. Oncolytic virotherapy: From bench to bedside. Front Cell Dev Biol (2021) 9:790150. doi: 10.3389/fcell.2021.790150
2. Borden EC. Progress toward therapeutic application of interferons, 1979-1983. Cancer (1984) 54(11 Suppl):2770–6. doi: 10.1002/1097-0142(19841201)54:2+<2770::aid-cncr2820541425>3.0.co;2-0
3. Gresser I, Maury C, Brouty-Boyé D. Mechanism of the antitumour effect of interferon in mice. Nature (1972) 239(5368):167–8. doi: 10.1038/239167a0
4. Gutterman JU. Cytokine therapeutics: lessons from interferon alpha. Proc Natl Acad Sci USA (1994) 91(4):1198–205. doi: 10.1073/pnas.91.4.1198
5. Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity (2006) 25(3):373–81. doi: 10.1016/j.immuni.2006.08.007
6. Kulaeva OI, Draghici S, Tang L, Kraniak JM, Land SJ, Tainsky MA. Epigenetic silencing of multiple interferon pathway genes after cellular immortalization. Oncogene (2003) 22(26):4118–27. doi: 10.1038/sj.onc.1206594
7. Olopade OI, Jenkins RB, Ransom DT, Malik K, Pomykala H, Nobori T, et al. Molecular analysis of deletions of the short arm of chromosome 9 in human gliomas. Cancer Res (1992) 52(9):2523–9.
8. Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, et al. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci USA (1988) 85(14):5259–63. doi: 10.1073/pnas.85.14.5259
9. Cairns P, Tokino K, Eby Y, Sidransky D. Homozygous deletions of 9p21 in primary human bladder tumors detected by comparative multiplex polymerase chain reaction. Cancer Res (1994) 54(6):1422–4.
10. Kondo M, Nagano H, Sakon M, Yamamoto H, Morimoto O, Arai I, et al. Expression of interferon alpha/beta receptor in human hepatocellular carcinoma. Int J Oncol (2000) 17(1):83–8.
11. Damdinsuren B, Nagano H, Wada H, Noda T, Natsag J, Marubashi S, et al. Interferon alpha receptors are important for antiproliferative effect of interferon-alpha against human hepatocellular carcinoma cells. Hepatol Res Off J Japan Soc Hepatol (2007) 37(1):77–83. doi: 10.1111/j.1872-034X.2007.00007.x
12. Saidi RF, Williams F, Silberberg B, Mittal VK, ReMine SG, Jacobs MJ. Expression of interferon receptors in pancreatic cancer: identification of a novel prognostic factor. Surgery (2006) 139(6):743–8. doi: 10.1016/j.surg.2005.11.010
13. Chen HM, Tanaka N, Mitani Y, Oda E, Nozawa H, Chen JZ, et al. Critical role for constitutive type I interferon signaling in the prevention of cellular transformation. Cancer Sci (2009) 100(3):449–56. doi: 10.1111/j.1349-7006.2008.01051.x
14. Slattery ML, Lundgreen A, Bondurant KL, Wolff RK. Interferon-signaling pathway: associations with colon and rectal cancer risk and subsequent survival. Carcinogenesis (2011) 32(11):1660–7. doi: 10.1093/carcin/bgr189
15. Critchley-Thorne RJ, Simons DL, Yan N, Miyahira AK, Dirbas FM, Johnson DL, et al. Impaired interferon signaling is a common immune defect in human cancer. Proc Natl Acad Sci USA (2009) 106(22):9010–5. doi: 10.1073/pnas.0901329106
16. Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol (2018) 28(6):e2008. doi: 10.1002/rmv.2008
17. Isaacs A, Lindenmann J. Virus interference. i. the interferon. Proc R Soc London Ser B Biol Sci (1957) 147(927):258–67. doi: 10.1098/rspb.1957.0048.
18. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16(3):131–44. doi: 10.1038/nrc.2016.14
19. Sprooten J, Agostinis P, Garg AD. Type I interferons and dendritic cells in cancer immunotherapy. Int Rev Cell Mol Biol (2019) 348:217–62. doi: 10.1016/bs.ircmb.2019.06.001
20. Walter MR. The role of structure in the biology of interferon signaling. Front Immunol (2020) 11:606489. doi: 10.3389/fimmu.2020.606489
21. Pestka S. The human interferon-alpha species and hybrid proteins. Semin Oncol (1997) 24(3 Suppl 9):S9–4-s9-17.
22. Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem (2007) 282(28):20047–51. doi: 10.1074/jbc.R700004200
23. Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol (2004) 22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622
24. Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
25. Hardy MP, Owczarek CM, Jermiin LS, Ejdebäck M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics (2004) 84(2):331–45. doi: 10.1016/j.ygeno.2004.03.003
26. LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J Biol Chem (2001) 276(43):39765–71. doi: 10.1074/jbc.M102502200
27. Hauptmann R, Swetly P. A novel class of human type I interferons. Nucleic Acids Res (1985) 13(13):4739–49. doi: 10.1093/nar/13.13.4739
28. Honda K, Taniguchi T. IRFs: master regulators of signalling by toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol (2006) 6(9):644–58. doi: 10.1038/nri1900
29. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22(2):240–73. doi: 10.1128/cmr.00046-08
30. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
31. Honda K, Taniguchi T. Toll-like receptor signaling and IRF transcription factors. IUBMB Life (2006) 58(5-6):290–5. doi: 10.1080/15216540600702206
32. Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol (2018) 14(4):214–28. doi: 10.1038/nrrheum.2018.31
33. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol (2014) 5:342. doi: 10.3389/fimmu.2014.00342
34. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
35. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol (2018) 18(9):545–58. doi: 10.1038/s41577-018-0029-z
36. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4(1):69–77. doi: 10.1038/ni875
37. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol (2003) 4(1):63–8. doi: 10.1038/ni873
38. Geoffroy K, Bourgeois-Daigneault MC. The pros and cons of interferons for oncolytic virotherapy. Cytokine Growth factor Rev (2020) 56:49–58. doi: 10.1016/j.cytogfr.2020.07.002
39. Onabajo OO, Muchmore B, Prokunina-Olsson L. The IFN-λ4 conundrum: When a good interferon goes bad. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (2019) 39(10):636–41. doi: 10.1089/jir.2019.0044
40. Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis c virus. Nat Genet (2013) 45(2):164–71. doi: 10.1038/ng.2521
41. Durbin RK, Kotenko SV, Durbin JE. Interferon induction and function at the mucosal surface. Immunol Rev (2013) 255(1):25–39. doi: 10.1111/imr.12101
42. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol (2006) 80(9):4501–9. doi: 10.1128/jvi.80.9.4501-4509.2006
43. Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (2010) 30(8):555–64. doi: 10.1089/jir.2010.0078
44. Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol (2004) 34(3):796–805. doi: 10.1002/eji.200324610
45. Osterlund P, Veckman V, Sirén J, Klucher KM, Hiscott J, Matikainen S, et al. Gene expression and antiviral activity of alpha/beta interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J Virol (2005) 79(15):9608–17. doi: 10.1128/jvi.79.15.9608-9617.2005
46. Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, et al. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med (2010) 207(12):2703–17. doi: 10.1084/jem.20092720
47. Hillyer P, Mane VP, Schramm LM, Puig M, Verthelyi D, Chen A, et al. Expression profiles of human interferon-alpha and interferon-lambda subtypes are ligand- and cell-dependent. Immunol Cell Biol (2012) 90(8):774–83. doi: 10.1038/icb.2011.109
48. Melchjorsen J, Sirén J, Julkunen I, Paludan SR, Matikainen S. Induction of cytokine expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J Gen Virol (2006) 87(Pt 5):1099–108. doi: 10.1099/vir.0.81541-0
49. Khaitov MR, Laza-Stanca V, Edwards MR, Walton RP, Rohde G, Contoli M, et al. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy (2009) 64(3):375–86. doi: 10.1111/j.1398-9995.2008.01826.x
50. Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza a infection. J Immunol (Baltimore Md 1950) (2009) 182(3):1296–304. doi: 10.4049/jimmunol.182.3.1296
51. Ioannidis I, Ye F, McNally B, Willette M, Flaño E. Toll-like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol (2013) 87(6):3261–70. doi: 10.1128/jvi.01956-12
52. Yin Y, Favoreel HW. Herpesviruses and the type III interferon system. Virologica Sinica (2021) 36(4):577–87. doi: 10.1007/s12250-020-00330-2
53. Griffiths SJ, Koegl M, Boutell C, Zenner HL, Crump CM, Pica F, et al. A systematic analysis of host factors reveals a Med23-interferon-λ regulatory axis against herpes simplex virus type 1 replication. PloS Pathogens (2013) 9(8):e1003514. doi: 10.1371/journal.ppat.1003514
54. Bilichodmath S, Nair SK, Bilichodmath R, Mangalekar SB. mRNA expression of IFN-λs in the gingival tissue of patients with chronic or aggressive periodontitis: A polymerase chain reaction study. J Periodontol (2018) 89(7):867–74. doi: 10.1002/jper.17-0349
55. de Weerd NA, Nguyen T. The interferons and their receptors–distribution and regulation. Immunol Cell Biol (2012) 90(5):483–91. doi: 10.1038/icb.2012.9
56. Lin FC, Young HA. Interferons: Success in anti-viral immunotherapy. Cytokine Growth factor Rev (2014) 25(4):369–76. doi: 10.1016/j.cytogfr.2014.07.015
57. Zan J, Zhang H, Gu AP, Zhong KL, Lu MY, Bai XX, et al. Yin yang 1 dynamically regulates antiviral innate immune responses during viral infection. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2017) 44(2):607–17. doi: 10.1159/000485116
58. Maney SK, Xu HC, Huang J, Pandyra AA, Ehlting C, Aguilar-Valenzuela R, et al. RAIDD mediates TLR3 and IRF7 driven type I interferon production. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2016) 39(4):1271–80. doi: 10.1159/000447832
59. Cui H, Liu Y, Huang Y. Roles of TRIM32 in corneal epithelial cells after infection with herpes simplex virus. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2017) 43(2):801–11. doi: 10.1159/000481563
60. Li SF, Gong MJ, Zhao FR, Shao JJ, Xie YL, Zhang YG, et al. Type I interferons: Distinct biological activities and current applications for viral infection. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2018) 51(5):2377–96. doi: 10.1159/000495897
61. Platanias LC. Mechanisms of type-i- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. doi: 10.1038/nri1604
62. Matsumoto M, Tanaka N, Harada H, Kimura T, Yokochi T, Kitagawa M, et al. Activation of the transcription factor ISGF3 by interferon-gamma. Biol Chem (1999) 380(6):699–703. doi: 10.1515/bc.1999.087
63. Briscoe J, Rogers NC, Witthuhn BA, Watling D, Harpur AG, Wilks AF, et al. Kinase-negative mutants of JAK1 can sustain interferon-gamma-inducible gene expression but not an antiviral state. EMBO J (1996) 15(4):799–809.
64. Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (1997) 17(3):121–34. doi: 10.1089/jir.1997.17.121
65. Wesoly J, Szweykowska-Kulinska Z, Bluyssen HA. STAT activation and differential complex formation dictate selectivity of interferon responses. Acta Biochim Polonica (2007) 54(1):27–38.
66. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukocyte Biol (2004) 75(2):163–89. doi: 10.1189/jlb.0603252
67. Steimle V, Siegrist CA, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Sci (New York NY) (1994) 265(5168):106–9. doi: 10.1126/science.8016643
68. Dumoutier L, Lejeune D, Hor S, Fickenscher H, Renauld JC. Cloning of a new type II cytokine receptor activating signal transducer and activator of transcription (STAT)1, STAT2 and STAT3. Biochem J (2003) 370(Pt 2):391–6. doi: 10.1042/bj20021935
69. Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, et al. Interferons alpha and lambda inhibit hepatitis c virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology (2006) 131(6):1887–98. doi: 10.1053/j.gastro.2006.09.052
70. Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the jak-STAT pathway and the mitogen-activated protein kinases. J Virol (2007) 81(14):7749–58. doi: 10.1128/jvi.02438-06
71. Levraud JP, Boudinot P, Colin I, Benmansour A, Peyrieras N, Herbomel P, et al. Identification of the zebrafish IFN receptor: implications for the origin of the vertebrate IFN system. J Immunol (Baltimore Md 1950) (2007) 178(7):4385–94. doi: 10.4049/jimmunol.178.7.4385
72. Balkwill F, Watling D, Taylor-Papadimitriou J. Inhibition by lymphoblastoid interferon of growth of cells derived from the human breast. Int J Cancer (1978) 22(3):258–65. doi: 10.1002/ijc.2910220307
73. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6(7):722–9. doi: 10.1038/ni1213
74. Swann JB, Hayakawa Y, Zerafa N, Sheehan KC, Scott B, Schreiber RD, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol (Baltimore Md 1950) (2007) 178(12):7540–9. doi: 10.4049/jimmunol.178.12.7540
75. Koromilas AE, Sexl V. The tumor suppressor function of STAT1 in breast cancer. Jak-stat (2013) 2(2):e23353. doi: 10.4161/jkst.23353
76. Bishnoi S, Tiwari R, Gupta S, Byrareddy SN, Nayak D. Oncotargeting by vesicular stomatitis virus (VSV): Advances in cancer therapy. Viruses (2018) 10(2):90. doi: 10.3390/v10020090
77. Barber GN. VSV-tumor selective replication and protein translation. Oncogene (2005) 24(52):7710–9. doi: 10.1038/sj.onc.1209042
78. Escobar-Zarate D, Liu YP, Suksanpaisan L, Russell SJ, Peng KW. Overcoming cancer cell resistance to VSV oncolysis with JAK1/2 inhibitors. Cancer Gene Ther (2013) 20(10):582–9. doi: 10.1038/cgt.2013.55
79. Patel MR, Jacobson BA, Ji Y, Drees J, Tang S, Xiong K, et al. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget (2015) 6(32):33165–77. doi: 10.18632/oncotarget.5320
80. Dold C, Rodriguez Urbiola C, Wollmann G, Egerer L, Muik A, Bellmann L, et al. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol Ther Oncolytics (2016) 3:16021. doi: 10.1038/mto.2016.21
81. Kimpel J, Urbiola C, Koske I, Tober R, Banki Z, Wollmann G, et al. The oncolytic virus VSV-GP is effective against malignant melanoma. Viruses (2018) 10(3):108. doi: 10.3390/v10030108
82. Patel MR, Dash A, Jacobson BA, Ji Y, Baumann D, Ismail K, et al. JAK/STAT inhibition with ruxolitinib enhances oncolytic virotherapy in non-small cell lung cancer models. Cancer Gene Ther (2019) 26(11-12):411–8. doi: 10.1038/s41417-018-0074-6
83. Nguyen TT, Ramsay L, Ahanfeshar-Adams M, Lajoie M, Schadendorf D, Alain T, et al. Mutations in the IFNγ-JAK-STAT pathway causing resistance to immune checkpoint inhibitors in melanoma increase sensitivity to oncolytic virus treatment. Clin Cancer Res an Off J Am Assoc Cancer Res (2021) 27(12):3432–42. doi: 10.1158/1078-0432.Ccr-20-3365
84. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515(7528):563–7. doi: 10.1038/nature14011
85. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci United States America (2003) 100(9):5336–41. doi: 10.1073/pnas.0931259100
86. Lee SJ, Jang BC, Lee SW, Yang YI, Suh SI, Park YM, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274). FEBS Letters (2006) 580(3):755–62. doi: 10.1016/j.febslet.2005.12.093
87. Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Letters (2017) 184:7–14. doi: 10.1016/j.imlet.2017.02.006
88. Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(12):3643–57. doi: 10.1158/1078-0432.Ccr-18-2402
89. El-Sayes N, Walsh S, Vito A, Reihani A, Ask K, Wan Y, et al. IFNAR blockade synergizes with oncolytic VSV to prevent virus-mediated PD-L1 expression and promote antitumor T cell activity. Mol Ther Oncolytics (2022) 25:16–30. doi: 10.1016/j.omto.2022.03.006
90. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem (2009) 284(20):13291–5. doi: 10.1074/jbc.R900010200
91. Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat Rev Immunol (2004) 4(11):868–77. doi: 10.1038/nri1489
92. Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun (2015) 6:6997. doi: 10.1038/ncomms7997
93. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature (2002) 418(6898):646–50. doi: 10.1038/nature00939
94. Suspène R, Aynaud MM, Koch S, Pasdeloup D, Labetoulle M, Gaertner B, et al. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J Virol (2011) 85(15):7594–602. doi: 10.1128/jvi.00290-11
95. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet (2013) 45(9):970–6. doi: 10.1038/ng.2702
96. Huff AL, Wongthida P, Kottke T, Thompson JM, Driscoll CB, Schuelke M, et al. APOBEC3 mediates resistance to oncolytic viral therapy. Mol Ther Oncolytics (2018) 11:1–13. doi: 10.1016/j.omto.2018.08.003
97. Pajares M, Jiménez-Moreno N, García-Yagüe ÁJ, Escoll M, de Ceballos ML, Van Leuven F, et al. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy (2016) 12(10):1902–16. doi: 10.1080/15548627.2016.1208889
98. Olagnier D, Lababidi RR, Hadj SB, Sze A, Liu Y, Naidu SD, et al. Activation of Nrf2 signaling augments vesicular stomatitis virus oncolysis via autophagy-driven suppression of antiviral immunity. Mol Ther J Am Soc Gene Ther (2017) 25(8):1900–16. doi: 10.1016/j.ymthe.2017.04.022
99. Zhu H, Zheng C. The race between host antiviral innate immunity and the immune evasion strategies of herpes simplex virus 1. Microbiol Mol Biol Rev MMBR (2020) 84(4):e00099–20. doi: 10.1128/mmbr.00099-20
100. Saha D, Wakimoto H, Rabkin SD. Oncolytic herpes simplex virus interactions with the host immune system. Curr Opin Virol (2016) 21:26–34. doi: 10.1016/j.coviro.2016.07.007
101. Kohlhapp FJ, Kaufman HL. Molecular pathways: Mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res an Off J Am Assoc Cancer Res (2016) 22(5):1048–54. doi: 10.1158/1078-0432.Ccr-15-2667
102. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
103. Ho HH, Ivashkiv LB. Role of STAT3 in type I interferon responses. negative regulation of STAT1-dependent inflammatory gene activation. J Biol Chem (2006) 281(20):14111–8. doi: 10.1074/jbc.M511797200
104. Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol (Baltimore Md 1950) (2008) 181(5):3167–76. doi: 10.4049/jimmunol.181.5.3167
105. Tsai MH, Pai LM, Lee CK. Fine-tuning of type I interferon response by STAT3. Front Immunol (2019) 10:1448. doi: 10.3389/fimmu.2019.01448
106. Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2012) 18(5):1201–6. doi: 10.1158/1078-0432.Ccr-11-0641
107. Hu J, Lu R, Zhang Y, Li W, Hu Q, Chen C, et al. β-adrenergic receptor inhibition enhances oncolytic herpes virus propagation through STAT3 activation in gastric cancer. Cell Biosci (2021) 11(1):174. doi: 10.1186/s13578-021-00687-1
108. Ng KW, Marshall EA, Bell JC, Lam WL. cGAS-STING and cancer: Dichotomous roles in tumor immunity and development. Trends Immunol (2018) 39(1):44–54. doi: 10.1016/j.it.2017.07.013
109. Khoo LT, Chen LY. Role of the cGAS-STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep (2018) 19(12):e46935. doi: 10.15252/embr.201846935
110. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455(7213):674–8. doi: 10.1038/nature07317
111. Morimoto D, Matsumura S, Bustos-Villalobos I, Sibal PA, Ichinose T, Naoe Y, et al. C-REV retains high infectivity regardless of the expression levels of cGAS and STING in cultured pancreatic cancer cells. Cells (2021) 10(6):1502. doi: 10.3390/cells10061502
112. Liu X, He B. Selective editing of herpes simplex virus 1 enables interferon induction and viral replication that destroy malignant cells. J Virol (2019) 93(2):e01761–18. doi: 10.1128/jvi.01761-18
113. Chapon M, Parvatiyar K, Aliyari SR, Zhao JS, Cheng G. Comprehensive mutagenesis of herpes simplex virus 1 genome identifies UL42 as an inhibitor of type I interferon induction. J Virol (2019) 93(23):e01446–19. doi: 10.1128/jvi.01446-19
114. Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med (2000) 192(2):219–26. doi: 10.1084/jem.192.2.219
115. Müller L, Aigner P, Stoiber D. Type I interferons and natural killer cell regulation in cancer. Front Immunol (2017) 8:304. doi: 10.3389/fimmu.2017.00304
116. Oku M, Ishino R, Uchida S, Imataki O, Sugimoto N, Todo T, et al. Oncolytic herpes simplex virus type 1 (HSV-1) in combination with lenalidomide for plasma cell neoplasms. Br J Haematol (2021) 192(2):343–53. doi: 10.1111/bjh.17173
117. Chen Q, Gong B, Mahmoud-Ahmed AS, Zhou A, Hsi ED, Hussein M, et al. Apo2L/TRAIL and bcl-2-related proteins regulate type I interferon-induced apoptosis in multiple myeloma. Blood (2001) 98(7):2183–92. doi: 10.1182/blood.v98.7.2183
118. Wang Y, Jin J, Li Y, Zhou Q, Yao R, Wu Z, et al. NK cell tumor therapy modulated by UV-inactivated oncolytic herpes simplex virus type 2 and checkpoint inhibitors. Trans Res J Lab Clin Med (2022) 240:64–86. doi: 10.1016/j.trsl.2021.10.006
119. Zhu G, Zhang J, Zhang Q, Jin G, Su X, Liu S, et al. Enhancement of CD70-specific CAR T treatment by IFN-γ released from oHSV-1-infected glioblastoma. Cancer Immunol Immunother CII (2022). doi: 10.1007/s00262-022-03172-x
120. Esaki S, Goshima F, Ozaki H, Takano G, Hatano Y, Kawakita D, et al. Oncolytic activity of HF10 in head and neck squamous cell carcinomas. Cancer Gene Ther (2020) 27(7-8):585–98. doi: 10.1038/s41417-019-0129-3
121. Bussiere LD, Miller CL. Reovirus and the host integrated stress response: On the frontlines of the battle to survive. Viruses (2021) 13(2):200. doi: 10.3390/v13020200
122. Adair RA, Scott KJ, Fraser S, Errington-Mais F, Pandha H, Coffey M, et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int J Cancer (2013) 132(10):2327–38. doi: 10.1002/ijc.27918
123. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Trans Med (2018) 10(422).eaam7577. doi: 10.1126/scitranslmed.aam7577
124. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-Small-Cell lung cancer. New Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
125. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
126. Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, et al. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol (Baltimore Md 1950) (2002) 169(8):4279–87. doi: 10.4049/jimmunol.169.8.4279
127. Baranek T, Manh TP, Alexandre Y, Maqbool MA, Cabeza JZ, Tomasello E, et al. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe (2012) 12(4):571–84. doi: 10.1016/j.chom.2012.09.002
128. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? the example of natural killer cells. Sci (New York NY) (2011) 331(6013):44–9. doi: 10.1126/science.1198687
129. Vidal SM, Khakoo SI, Biron CA. Natural killer cell responses during viral infections: flexibility and conditioning of innate immunity by experience. Curr Opin Virol (2011) 1(6):497–512. doi: 10.1016/j.coviro.2011.10.017
130. Martinez J, Huang X, Yang Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol (Baltimore Md 1950) (2008) 180(3):1592–7. doi: 10.4049/jimmunol.180.3.1592
131. Sato K, Hida S, Takayanagi H, Yokochi T, Kayagaki N, Takeda K, et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur J Immunol (2001) 31(11):3138–46. doi: 10.1002/1521-4141(200111)31:11<3138::aid-immu3138>3.0.co;2-b
132. Wantoch M, Wilson EB, Droop AP, Phillips SL, Coffey M, El-Sherbiny YM, et al. Oncolytic virus treatment differentially affects the CD56(dim) and CD56(bright) NK cell subsets in vivo and regulates a spectrum of human NK cell activity. Immunology (2022) 166(1):104–20. doi: 10.1111/imm.13453
133. Katayama Y, Tachibana M, Kurisu N, Oya Y, Terasawa Y, Goda H, et al. Oncolytic reovirus inhibits immunosuppressive activity of myeloid-derived suppressor cells in a TLR3-dependent manner. J Immunol (Baltimore Md 1950) (2018) 200(8):2987–99. doi: 10.4049/jimmunol.1700435
134. Rodríguez Stewart RM, Berry JTL, Berger AK, Yoon SB, Hirsch AL, Guberman JA, et al. Enhanced killing of triple-negative breast cancer cells by reassortant reovirus and topoisomerase inhibitors. J Virol (2019) 93(23):e01411–19. doi: 10.1128/jvi.01411-19
135. Oosenbrug T, van den Wollenberg DJM, Duits EW, Hoeben RC, Ressing ME. Induction of robust type I interferon levels by oncolytic reovirus requires both viral replication and interferon-α/β receptor signaling. Hum Gene Ther (2021) 32(19-20):1171–85. doi: 10.1089/hum.2021.140
136. Lanoie D, Boudreault S, Bisaillon M, Lemay G. How many mammalian reovirus proteins are involved in the control of the interferon response? Pathog (Basel Switzerland) (2019) 8(2):83. doi: 10.3390/pathogens8020083
137. Lanoie D, Lemay G. Multiple proteins differing between laboratory stocks of mammalian orthoreoviruses affect both virus sensitivity to interferon and induction of interferon production during infection. Virus Res (2018) 247:40–6. doi: 10.1016/j.virusres.2018.01.009
138. Jacobs BL, Ferguson RE. The Lang strain of reovirus serotype 1 and the Dearing strain of reovirus serotype 3 differ in their sensitivities to beta interferon. J Virol (1991) 65(9):5102–4. doi: 10.1128/jvi.65.9.5102-5104.1991
139. Stuart JD, Holm GH, Boehme KW. Differential delivery of genomic double-stranded RNA causes reovirus strain-specific differences in interferon regulatory factor 3 activation. J Virol (2018) 92(9):e01947–17. doi: 10.1128/jvi.01947-17
140. Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J Neurovirol (2011) 17(4):314–26. doi: 10.1007/s13365-011-0038-1
141. Sherry B, Torres J, Blum MA. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol (1998) 72(2):1314–23. doi: 10.1128/jvi.72.2.1314-1323.1998
142. Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol (2009) 83(5):2178–87. doi: 10.1128/jvi.01787-08
143. Mohamed A, Konda P, Eaton HE, Gujar S, Smiley JR, Shmulevitz M. Closely related reovirus lab strains induce opposite expression of RIG-I/IFN-dependent versus -independent host genes, via mechanisms of slow replication versus polymorphisms in dsRNA binding σ3 respectively. PLoS Pathogens (2020) 16(9):e1008803. doi: 10.1371/journal.ppat.1008803
144. Rahman AU, Habib M, Shabbir MZ. Adaptation of Newcastle disease virus (NDV) in feral birds and their potential role in interspecies transmission. Open Virol J (2018) 12:52–68. doi: 10.2174/1874357901812010052
145. Santos MR, Xavier PLP, Pires PRL, Rochetti AL, Rosim DF, Scagion GP, et al. Oncolytic effect of Newcastle disease virus is attributed to interferon regulation in canine mammary cancer cell lines. Veterinary Comp Oncol (2021) 19(3):593–601. doi: 10.1111/vco.12699
146. Zamarin D, Ricca JM, Sadekova S, Oseledchyk A, Yu Y, Blumenschein WM, et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest (2018) 128(4):1413–28. doi: 10.1172/jci98047
147. Koks CA, Garg AD, Ehrhardt M, Riva M, Vandenberk L, Boon L, et al. Newcastle Disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int J Cancer (2015) 136(5):E313–25. doi: 10.1002/ijc.29202
148. Mozaffari Nejad AS, Fotouhi F, Mehrbod P, Alikhani MY. Antitumor immunity enhancement through Newcastle viral oncolysate in mice model: A promising method to treat tumors. Saudi J Biol Sci (2021) 28(10):5833–40. doi: 10.1016/j.sjbs.2021.06.043
149. Tian L, Liu T, Jiang S, Cao Y, Kang K, Su H, et al. Oncolytic Newcastle disease virus expressing the co-stimulator OX40L as immunopotentiator for colorectal cancer therapy. Gene Ther (2021). doi: 10.1038/s41434-021-00256-8
150. Keshavarz M, Ebrahimzadeh MS, Miri SM, Dianat-Moghadam H, Ghorbanhosseini SS, Mohebbi SR, et al. Oncolytic Newcastle disease virus delivered by mesenchymal stem cells-engineered system enhances the therapeutic effects altering tumor microenvironment. Virol J (2020) 17(1):64. doi: 10.1186/s12985-020-01326-w
151. Zamarin D, Palese P. Oncolytic Newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol (2012) 7(3):347–67. doi: 10.2217/fmb.12.4
152. García-Romero N, Palacín-Aliana I, Esteban-Rubio S, Madurga R, Rius-Rocabert S, Carrión-Navarro J, et al. Newcastle Disease virus (NDV) oncolytic activity in human glioma tumors is dependent on CDKN2A-type I IFN gene cluster codeletion. Cells (2020) 9(6):1405. doi: 10.3390/cells9061405
153. Krug RM. Functions of the influenza a virus NS1 protein in antiviral defense. Curr Opin Virol (2015) 12:1–6. doi: 10.1016/j.coviro.2015.01.007
154. Zamarin D, Martínez-Sobrido L, Kelly K, Mansour M, Sheng G, Vigil A, et al. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol Ther J Am Soc Gene Ther (2009) 17(4):697–706. doi: 10.1038/mt.2008.286
155. El-Jesr M, Teir M, Maluquer de Motes C. Vaccinia virus activation and antagonism of cytosolic DNA sensing. Front Immunol (2020) 11:568412. doi: 10.3389/fimmu.2020.568412
156. Shen Y, Nemunaitis J. Fighting cancer with vaccinia virus: teaching new tricks to an old dog. Mol Ther J Am Soc Gene Ther (2005) 11(2):180–95. doi: 10.1016/j.ymthe.2004.10.015
157. Puhlmann M, Gnant M, Brown CK, Alexander HR, Bartlett DL. Thymidine kinase-deleted vaccinia virus expressing purine nucleoside phosphorylase as a vector for tumor-directed gene therapy. Hum Gene Ther (1999) 10(4):649–57. doi: 10.1089/10430349950018724
158. Guo ZS, Lu B, Guo Z, Giehl E, Feist M, Dai E, et al. Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics. J Immunother Cancer (2019) 7(1):6. doi: 10.1186/s40425-018-0495-7
159. Jia X, Chen Y, Zhao X, Lv C, Yan J. Oncolytic vaccinia virus inhibits human hepatocellular carcinoma MHCC97-h cell proliferation via endoplasmic reticulum stress, autophagy and wnt pathways. J Gene Med (2016) 18(9):211–9. doi: 10.1002/jgm.2893
160. Wang X, Zhou N, Liu T, Jia X, Ye T, Chen K, et al. Oncolytic vaccinia virus expressing white-spotted charr lectin regulates antiviral response in tumor cells and inhibits tumor growth In vitro and in vivo. Mar Drugs (2021) 19(6):292. doi: 10.3390/md19060292
161. Chen L, Chen H, Ye J, Ge Y, Wang H, Dai E, et al. Intratumoral expression of interleukin 23 variants using oncolytic vaccinia virus elicit potent antitumor effects on multiple tumor models via tumor microenvironment modulation. Theranostics (2021) 11(14):6668–81. doi: 10.7150/thno.56494
162. West EJ, Scott KJ, Tidswell E, Bendjama K, Stojkowitz N, Lusky M, et al. Intravenous oncolytic vaccinia virus therapy results in a differential immune response between cancer patients. Cancers (2022) 14(9):2181. doi: 10.3390/cancers14092181
163. Engeland CE, Ungerechts G. Measles virus as an oncolytic immunotherapy. Cancers (2021) 13(3):544. doi: 10.3390/cancers13030544
164. Achard C, Boisgerault N, Delaunay T, Roulois D, Nedellec S, Royer PJ, et al. Sensitivity of human pleural mesothelioma to oncolytic measles virus depends on defects of the type I interferon response. Oncotarget (2015) 6(42):44892–904. doi: 10.18632/oncotarget.6285
165. Delaunay T, Achard C, Boisgerault N, Grard M, Petithomme T, Chatelain C, et al. Frequent homozygous deletions of type I interferon genes in pleural mesothelioma confer sensitivity to oncolytic measles virus. J Thorac Oncol Off Publ Int Assoc Stud Lung Cancer (2020) 15(5):827–42. doi: 10.1016/j.jtho.2019.12.128
166. Yap TA, Aerts JG, Popat S, Fennell DA. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer (2017) 17(8):475–88. doi: 10.1038/nrc.2017.42
167. Jean D, Daubriac J, Le Pimpec-Barthes F, Galateau-Salle F, Jaurand MC. Molecular changes in mesothelioma with an impact on prognosis and treatment. Arch Pathol Lab Med (2012) 136(3):277–93. doi: 10.5858/arpa.2011-0215-RA
168. Aref S, Castleton AZ, Bailey K, Burt R, Dey A, Leongamornlert D, et al. Type 1 interferon responses underlie tumor-selective replication of oncolytic measles virus. Mol Ther J Am Soc Gene Ther (2020) 28(4):1043–55. doi: 10.1016/j.ymthe.2020.01.027
169. Annels NE, Mansfield D, Arif M, Ballesteros-Merino C, Simpson GR, Denyer M, et al. Phase I trial of an ICAM-1-Targeted immunotherapeutic-coxsackievirus A21 (CVA21) as an oncolytic agent against non muscle-invasive bladder cancer. Clin Cancer Res an Off J Am Assoc Cancer Res (2019) 25(19):5818–31. doi: 10.1158/1078-0432.Ccr-18-4022
170. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. New Engl J Med (2018) 379(2):150–61. doi: 10.1056/NEJMoa1716435
Keywords: oncolytic virus, IFN, signaling, oncology, tumor, cancer, immunology
Citation: Li Q, Tan F, Wang Y, Liu X, Kong X, Meng J, Yang L and Cen S (2022) The gamble between oncolytic virus therapy and IFN. Front. Immunol. 13:971674. doi: 10.3389/fimmu.2022.971674
Received: 17 June 2022; Accepted: 08 August 2022;
Published: 25 August 2022.
Edited by:
Jieying Bai, Academy of Military Medical Sciences (AMMS), ChinaReviewed by:
Wenyan Zhang, First Affiliated Hospital of Jilin University, ChinaGuoli Shi, National Institutes of Health (NIH), United States
Copyright © 2022 Li, Tan, Wang, Liu, Kong, Meng, Yang and Cen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianbin Kong, a29uZ3hpYW5iaW52aXBAMTYzLmNvbQ==; Jingyan Meng, bWVuZ2p5QDE2My5jb20=; Long Yang, bG9uZy55YW5nQHRqdXRjbS5lZHUuY24=; Shan Cen, c2hhbmNlbkBpbWIucHVtYy5lZHUuY24=
†These authors have contributed equally to this work