 Manmohan Kumar
Manmohan Kumar- 1Immunobiology Laboratory, Department of Zoology, University of Delhi, Delhi, India
- 2Faculty of Life Sciences and Biotechnology, South Asian University, New Delhi, Delhi, India
Toll-like receptors (TLRs) are epitomized as the first line of defense against pathogens. Amongst TLRs, TLR22 is expressed in non-mammalian aquatic vertebrates, including fish. Using headkidney macrophages (HKM) of Clarias gariepinus, we reported the pro-apoptotic and microbicidal role of TLR22 in Aeromonas hydrophila infection. Mitochondria act as a central scaffold in the innate immune system. However, the precise molecular mechanisms underlying TLR22 signaling and mitochondrial involvement in A. hydrophila-pathogenesis remain unexplored in fish. The aim of the present study was to investigate the nexus between TLR22 and mitochondria in pro-apoptotic immune signaling circuitry in A. hydrophila-infected HKM. We report that TLR22-induced mitochondrial-Ca2+ [Ca2+]mt surge is imperative for mtROS production in A. hydrophila-infected HKM. Mitigating mtROS production enhanced intracellular bacterial replication implicating its anti-microbial role in A. hydrophila-pathogenesis. Enhanced mtROS triggers hif1a expression leading to prolonged chop expression. CHOP prompts mitochondrial unfolded protein response (UPRmt) leading to the enhanced expression of mitochondrial fission marker dnml1, implicating mitochondrial fission in A. hydrophila pathogenesis. Inhibition of mitochondrial fission reduced HKM apoptosis and increased the bacterial burden. Additionally, TLR22-mediated alterations in mitochondrial architecture impair mitochondrial function (ΔΨm loss and cytosolic accumulation of cyt c), which in turn activates caspase-9/caspase-3 axis in A. hydrophila-infected HKM. Based on these findings we conclude that TLR22 prompts mtROS generation, which activates the HIF-1α/CHOP signalosome triggering UPRmt-induced mitochondrial fragmentation culminating in caspase-9/-3-mediated HKM apoptosis and bacterial clearance.
Introduction
Oxidative burst is the rapid release of reactive oxygen species (ROS), which plays a crucial role in intracellular redox profile influencing a wide variety of signaling pathways (1). It also represents one of the most proficient defense arsenals in host innate immunity against pathogens (2). Macrophages release copious amounts of ROS and the two well-recognized sources are NADPH oxidase (NOX) and mitochondria (3). The production of mitochondrial ROS (mtROS) is primarily attributed to the oxidation of electron transport chain (ETC) metabolic intermediates (3), and the complex I, complex III of the mitochondrial ETC serve as the major sites for mtROS production (4). mtROS were initially thought of as an unwanted adjunct of oxidative metabolism however, recent studies have suggested that macrophages exploit mtROS as the unswerving antimicrobial agent to combat pathogens (5, 6), implicating mitochondria act as a central hub in innate immunity. Though the role of mtROS in anti-microbial defense has been reported in fish (7) nonetheless the underlying molecular mechanism that activates the process in fish remains nebulous.
Toll-like receptors (TLRs) represent an evolutionarily conserved family of pattern-recognition receptors (PRRs) and represent a cornerstone of the fish innate immune response (8). TLRs recognize pathogens via pathogen-associated molecular patterns (PAMPs) and endogenous danger signals via damage-associated molecular patterns (DAMPs) released by damaged or dying cells (9). Amongst the TLR repertoire, TLR22 has been reported in non-mammalian aquatic animals including fish (10). The role of TLR22 in fish immunity is not well understood and the presence of this receptor in immune and non-immune tissues suggests it to be a multifaceted molecule (11). Previous studies have implicated the role of TLR22 in the fish immune response against microbial infection (12–15). Recently, we reported the anti-bacterial and pro-apoptotic role of TLR22 in fish macrophages (16) but how it contrives innate immune signaling pathways in fish needs to be investigated. Although TLR signaling has been implicated in mtROS production (5), but the primal role of TLR22 has not been reported.
Bacterial infections lead to surfeit consumption of cellular oxygen triggering hypoxia (17). The hypoxia-inducible factor-1 (HIF-1) plays an integral role in the body’s response to hypoxia. It consists of an inducible α subunit (HIF-1α/-2α) and a constitutive β subunit (HIF-1β). HIF-1α is expressed virtually in all innate and adaptive immune cells while the expression of HIF-2α is limited to endothelial cells and certain immune cells (17). Under hypoxic conditions, HIF-1α/-2α dimerizes with HIF-1β and binds with the hypoxia response elements (HREs) in the nucleus (18), initiating the transcription of genes involved in tissue homeostasis and immune response (19). mtROS has been reported as one of the key contributing factors in propagating hypoxia (20) but the exact mechanisms remain poorly defined. Hypoxia has been reported to exert host protective effects in fish by aiding the production of pro-inflammatory cytokines and nitric oxide vital for controlling bacterial burden (21). However, the exact immune signaling mechanisms regulated by HIFs in fish remains elusive.
The fitness of mitochondria is of paramount importance for cellular health and metabolism. The organelle remains dynamically interconnected, undergoing incessant cycles of fission and fusion which is essential for mitochondrial quality control (22). Mitochondrial fission helps in confiscating damaged mitochondria and is triggered by several factors of which dynamin-related protein 1 (Drp1) is important (23). Recent evidence though suggests mtROS is interlinked with mitochondrial fission and fusion (23, 24) but the intricate mechanisms remain unclear. Additionally, there are also reports implicating the importance of mitochondrial dynamics in regulating the outcome of immune response (25) but its role in fish immunity remains elusive.
Unfolded protein response (UPR) is employed to overcome cellular stress and restore proteostasis (26). Pathogenic assault leads to the aggregation of unfolded or misfolded proteins in mitochondria triggering mitochondrial UPR (UPRmt) (27–29). UPRmt triggers the induction of mitochondrial chaperones like Hsp60 to maintain protein homeostasis in the organelle (30). Additionally, it also induces certain genes for mitochondrial biogenesis, mitochondrial fission, and the repair and recovery of damaged mitochondria (30).
Aeromonas hydrophila, a Gram-negative bacterium is responsible for fatal hemorrhagic septicemia, enteritis, red body disease, and motile Aeromonas septicaemia [MAS] in fish (31). In mammals including humans, it is associated with gastroenteritis, septicaemia, wound infections, and extra-intestinal infections (32). The virulence of A. hydrophila has been attributed to its diverse range of virulence factors which poses a difficulty in understanding its pathogenesis. Previous studies have documented A. hydrophila induces apoptosis of fish macrophages, involving extrinsic and intrinsic caspases (33). Nevertheless, the signaling mechanism triggering apoptosis in A. hydrophila-infected cells needs to be investigated.
It is important to note that information on the physiological processes of mitochondria in the innate immune system had been limited to the mammalian system only. To the best of our knowledge, no direct evidence has been yet presented regarding the role of TLR22 and mitochondrial processes in the fish immune system. In fish, headkidney (HK) is a primary immunocompetent organ and serves as a rich source of macrophages (34). In this study, we have studied the role of TLR22 in triggering the mitochondrial response and shaping the immune outcome in A. hydrophila pathogenesis in fish.
Materials and Methods
Animal Care and Maintenance
Catfish (Clarias gariepinus, 120-150 g; 28 ± 2.5 cm) were procured locally and maintained in 50 L tanks under natural photoperiod. The studies were carried out according to the guidelines issued by Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Govt. of India and permitted by Animal Ethics Committee (DU/ZOOL/IAEC-R/2013/33), University of Delhi. Fish were acclimatized for 15 days prior to the experiments and fed with chicken liver ad libitum (33).
HKM Isolation, Infection and Inhibitor Studies
The fish were euthanized using MS 222 (Sigma), headkidney excised aseptically and HKM were isolated using 34/51% percoll gradient as described earlier (33). The HKM were infected with A. hydrophila (MOI 1:50) for 1 h and extracellular bacteria was removed using chloramphenicol (30 µg/mL) as described earlier (16).
The HKM were pre-incubated separately with mPTP inhibitor [Cyclosporin A (CsA), 5 µM, Sigma], mitochondrial Ca2+ uniporter (MCU) inhibitor [Ruthenium Red (RR), 20 µM, Sigma], caspase-9 inhibitor [Z-LEHD-FMK, 7.5 µM, Sigma], DRP-1 inhibitor [Mdivi-1, 25 µM, Sigma], HIF-1α inhibitor [Dimethyl-bisphenol A (di-BPA), 200 µM, Abcam], mtROS inhibitor [YCG063, 10 µM, Calbiochem], caspase-9 inhibitor [Z-LEHD-FMK, 7.5 µM, Biovision], caspase-3 inhibitor [Ac-DEVD-CHO, 10 µM, Sigma], for 1 h and then infected with A. hydrophila as mentioned earlier (16). The inhibitor concentrations used in the study had no adverse effects on the viability of HKM and bacterial growth per se (data not shown).
siRNA Transfection
tlr22 and chop gene was knocked out using specific siRNA (Table 1) using HiPerFect Transfection Reagent (Qiagen) as per manufacturer’s instructions. HKM were transfected with sc-siRNA or specific-siRNA-HiPerFect complex and the HKM were incubated at 30°C for 16 h and then infected with A. hydrophila as described earlier (33). The knockdown of both the genes was confirmed by RT-qPCR (Figure S1).

Table 1 List of siRNAs.
Cloning and Sequencing of dnm1l gene
Degenerate primers were designed using dnm1l homologous sequences of fishes available in the NCBI database. The cDNA was amplified using degenerate primers and the amplified product was eluted using QIA quick gel extraction kit (Qiagen). The amplified product was cloned into pGEM-T EASY vector (Promega) and sequenced (Macrogen). The sequence obtained (Supplementary Table 1) was aligned to nBLAST and submitted to the NCBI database (Accession no. MZ882392).
RT-qPCR
HKM (1 × 107) pre-incubated with inhibitors or transfected with specific siRNA were infected with A. hydrophila and the cultures were terminated at indicated time points. The total RNA was isolated using TRI reagent (MRC) as per the manufacturer’s instructions. The cDNA was prepared using Revert Aid First Strand cDNA synthesis (Thermo Fischer Scientific) from 1 µg of DNase-treated RNA as described earlier (16).
Gene expression studies were performed using ViiA Real-Time PCR system (ABI) and SYBR green PCR Master Mix (ABI) with specific primers (Table 2). The expression of the genes was quantitated by comparative ΔΔCT method and normalized against β-actin (housekeeping gene) as described earlier (16).
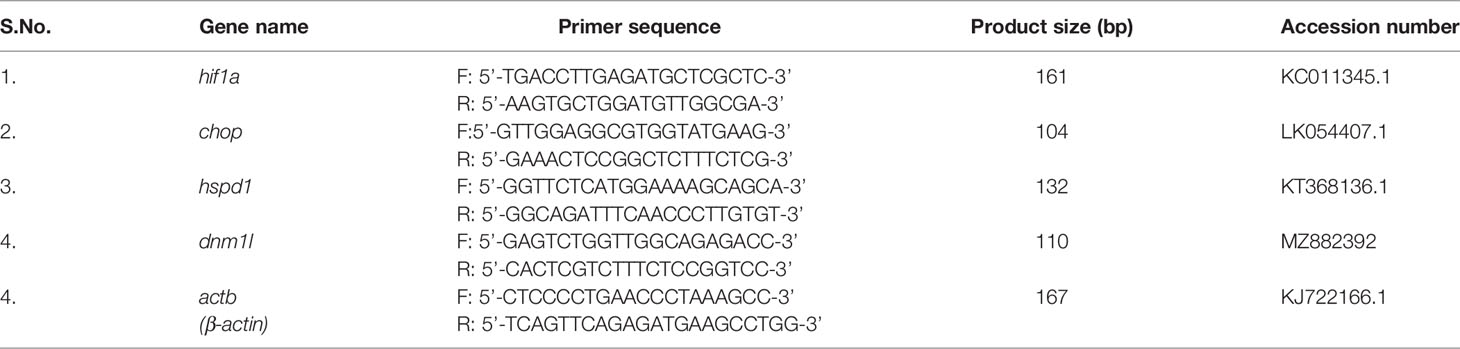
Table 2 List of RT-qPCR primers.
Assessment of Mitochondrial Ca2+ [(Ca2+)mt] Levels
The (Ca2+)mt levels were measured using Rhod-2/AM (Molecular Probes). HKM (2 × 106) were pre-incubated with Ruthenium Red or transfected with tlr22-siRNA and then infected as mentioned earlier. The cells were washed at 1 h p.i. and stained with Rhod-2/AM (50 nM). The excess dye was washed to remove any unbound dye then resuspended in 1× PBS and the changes in fluorescence intensity levels were measured at Ex552 and Em581 using microplate reader (BMG Labtech).
Measurement of Mitochondrial ROS (mtROS) Production
The mtROS levels were determined using MitoSOX™ Red mitochondrial superoxide indicator (Molecular Probes). HKM (2 × 106) were pre-incubated with YCG063, RR or transfected with tlr22-siRNA and then infected with A. hydrophila as described above. At 4 h p.i., the HKM were washed and incubated with MitoSOX (5 µM) at 30 °C for 20 min in dark. The excess dye was washed and the changes in fluorescence intensity levels were measured at Ex510nm and Em580nm using microplate reader (BMG Labtech).
In a parallel study, fluorescence microscopic analysis was also done for monitoring the changes in mtROS levels. For this, HKM were pre-incubated with YCG063, RR or transfected with tlr22-siRNA, infected with A. hydrophila, and then incubated with MitoSOX described above. The nucleus of the cells was stained with DAPI (100 µg/mL, Sigma) for 15 min at 30 °C in dark. The HKM were washed, mounted and observed under fluorescence microscope (×40, Zeiss Imager, Z2).
Apoptosis Assays
Hoechst 33342 Staining
HKM (1 × 106) were pre-incubated with YCG063, CsA, di-BPA, Mdivi-1, Z-LEHD-FMK, Ac-DEVD-CHO or transfected with tlr22-siRNA, chop-siRNA and infected with A. hydrophila as described above. At 24 h p.i., HKM were washed, stained with Hoechst 33342 as described earlier (14) and the slides were visualized under fluorescence microscope (×40, Zeiss Imager, Z2). Hoechst-positive and Hoechst-negative HKM were enumerated and the graph was plotted as % Hoechst-positive HKM.
Caspase Assays
Caspase-9 activity (LEHDase) and caspase-3 activity (DEVDase) were monitored using colorimetric caspase-9 and caspase-3 assay kit (Biovision) respectively following the manufacturer’s instructions and using reagents provided with the assay kits. HKM (1 × 106) pre-incubated with CsA, Mdivi-1, Z-LEHD-FMK, Ac-DEVD-CHO or transfected with tlr22-siRNA were infected with A. hydrophila as mentioned earlier. The HKM were collected at 24 h p.i., washed, re-suspended in chilled lysis buffer (50 μL) and incubated at 4°C for 1 min. Following incubation, the cell lysate was centrifuged at 10, 000 × g for 5 min at 4°C. Supernatant (50 μL) was mixed with 2× reaction buffer supplemented with DTT (10 mM), PMSF (5 mM). Then, 5 μL of substrate (LEHD-pNA for caspase-9 and DEVD-pNA for caspase-3) was added and incubated at 30°C for 5 h. The absorbance was read at A405nm (Epoch2, BioTek) and the relative fold change in the activity of caspase-9 and caspase-3 were calculated.
Confocal Microscopy
The mitochondrial morphology was observed using MitoTracker Green (Molecular Probes). HKM (1 × 106) were infected with A. hydrophila for 24 h p.i., washed and then loaded with MitoTracker Green (50 nM) for 30 min at 30 °C. The nucleus of the HKM was stained with DAPI (1 µg/mL) for 15 min at 30 °C. Excess dye was removed by washing, slide mounted and visualized under fluorescence microscope (×100, Nikon Eclipse Ti2).
Measurement of Cytochrome c (Cyt c) Release
The Cyt c release was studied according to the reported method (35). HKM (1 × 106) pre-incubated with CsA, Mdivi-1 or transfected with tlr22-siRNA were infected with A. hydrophila as described above. The HKM were washed at 24 h p.i., and homogenized in buffer A (50 mM Tris, 1 mM PMSF, 2 mM EDTA, pH 7.5), followed by addition of 2% glucose to remove the impurities and centrifuged at 2,000 × g for 10 min at 4°C. The supernatant was collected to detect the release of Cyt c in cytoplasm and the pellet was re-suspended in buffer B (50 mM Tris, 2 mM EDTA, pH 5) to obtain mitochondrial fraction. The mixture was centrifuged at 5,000 × g for 30 s at 4°C and pellet was re-suspended in TE buffer. The supernatant and pellet were treated with ascorbic acid (500 mg/mL) for 5 min and absorbance was read at A550nm (Epoch2, BioTek).
Determination of Mitochondrial Membrane Potential (Δψm)
The Δψm was studied using Rhodamine 123 (Molecular Probes). HKM (1 × 106) pre-incubated with CsA, Mdivi-1 or transfected with tlr22-siRNA were infected with A. hydrophila as described above. At 24 h p.i., HKM were washed and incubated with Rhodamine 123 (10 µM) for 30 min at 37°C in dark. The unbound dye was removed by washing and changes in fluorescence intensity were measured at Ex511nm and Em534nm (BMG Labtech).
Statistical Analysis
Statistical analysis was performed with IBM SPSS 25. For comparison, one-way ANOVA with Bonferroni post-hoc test was used to compare the means between the groups. *p < 0.05 was considered as level of statistical significance.
Results
TLR22-Induced (Ca2+)mt Flux Prompts Antimicrobial mtROS Generation in A. hydrophila-Infected HKM
Macrophages help in counteracting bacterial infections. Towards this direction, HKM were infected with A. hydrophila and the bacterial load was recorded at indicated time point p.i. We observed time-dependent reduction in the intracellular bacterial load with significant reduction recorded from 4 h p.i. (Figure 1).
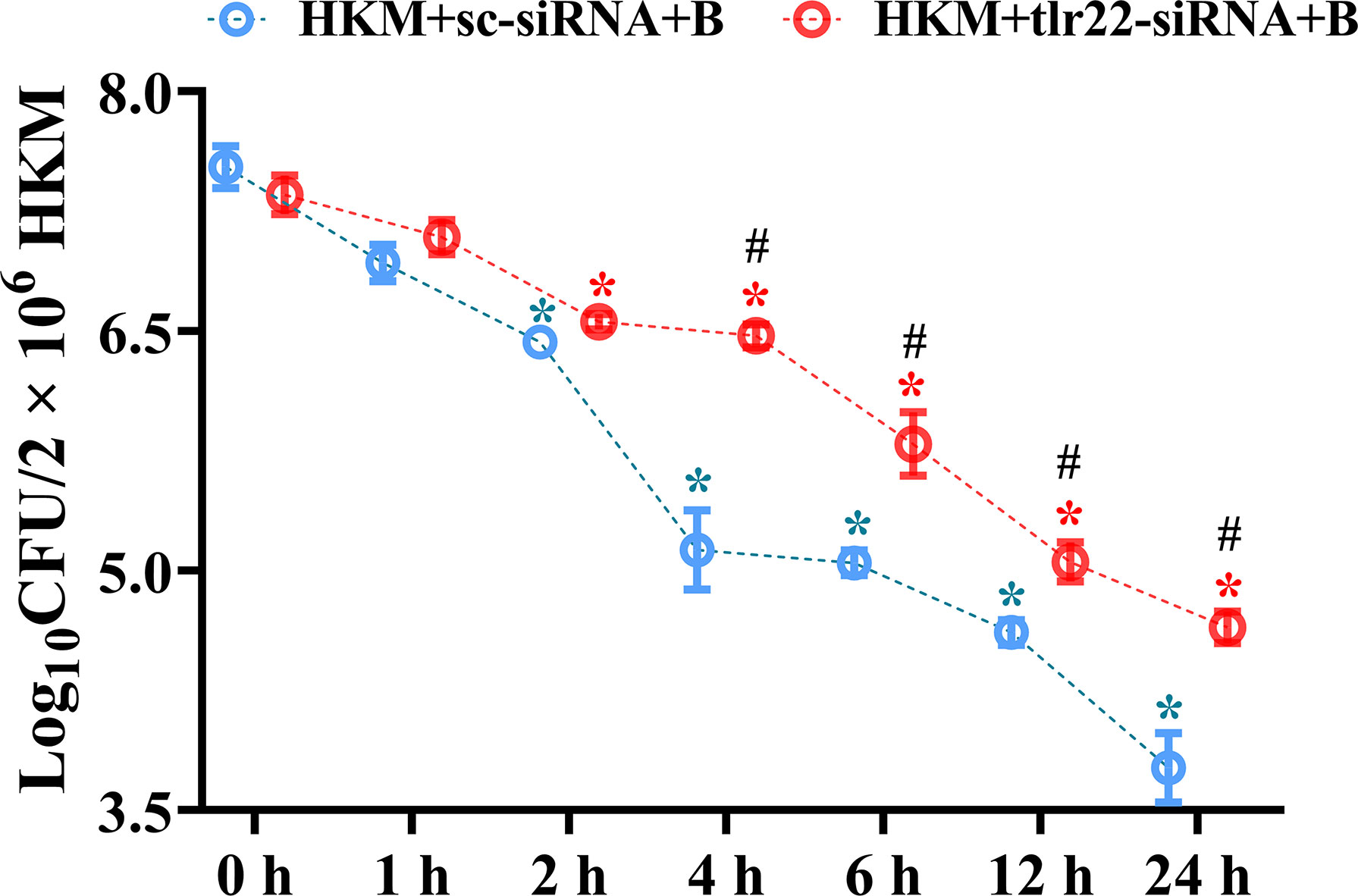
Figure 1 TLR22 signaling restrain A. hydrophila replication in HKM. HKM were transfected with sc-siRNA or tlr22-siRNA, infected with A. hydrophila and bacterial load was enumerated at indicated time points. Data represent mean ± SEM (n=3). Asterisk (*) signifies significant difference as compared to 0 h (*p < 0.05), Hashtag (#) signifies significant difference between the group (#p < 0.05). HKM+sc-siRNA+B, HKM+tlr22-siRNA+B, sc-siRNA and tlr22-siRNA transfected HKM were infected with A. hydrophila.
The next step was to identify the signaling molecules that aid in controlling A. hydrophila replication. Our previous studies implicated the role of TLR22 and mtROS in regulating A. hydrophila replication (16) and HKM apoptosis (36) respectively. Here we aimed to correlate the two molecular events in A. hydrophila pathogenesis. For this, we selected the 4 h time interval because a) significant reduction in intracellular A. hydrophila load was observed from 4 h p.i. and b) maximum mtROS production was noted at 4 h p.i. in A. hydrophila-infected HKM (36). Thus, HKM transfected with tlr22-siRNA were infected with A. hydrophila and the changes in mtROS levels were monitored at 4 h p.i. We observed a significant reduction in mtROS production in tlr22-knockdown HKM (Figures 2A, B) suggesting the role of TLR22 signaling in inducing mtROS production in A. hydrophila-infected HKM. Pre-incubation with the mtROS inhibitor (YCG063) attenuated mtROS production in A. hydrophila-infected HKM (Figures 2A, B). Our results for the first time established the primal role of TLR22 in mtROS production in A. hydrophila infection.
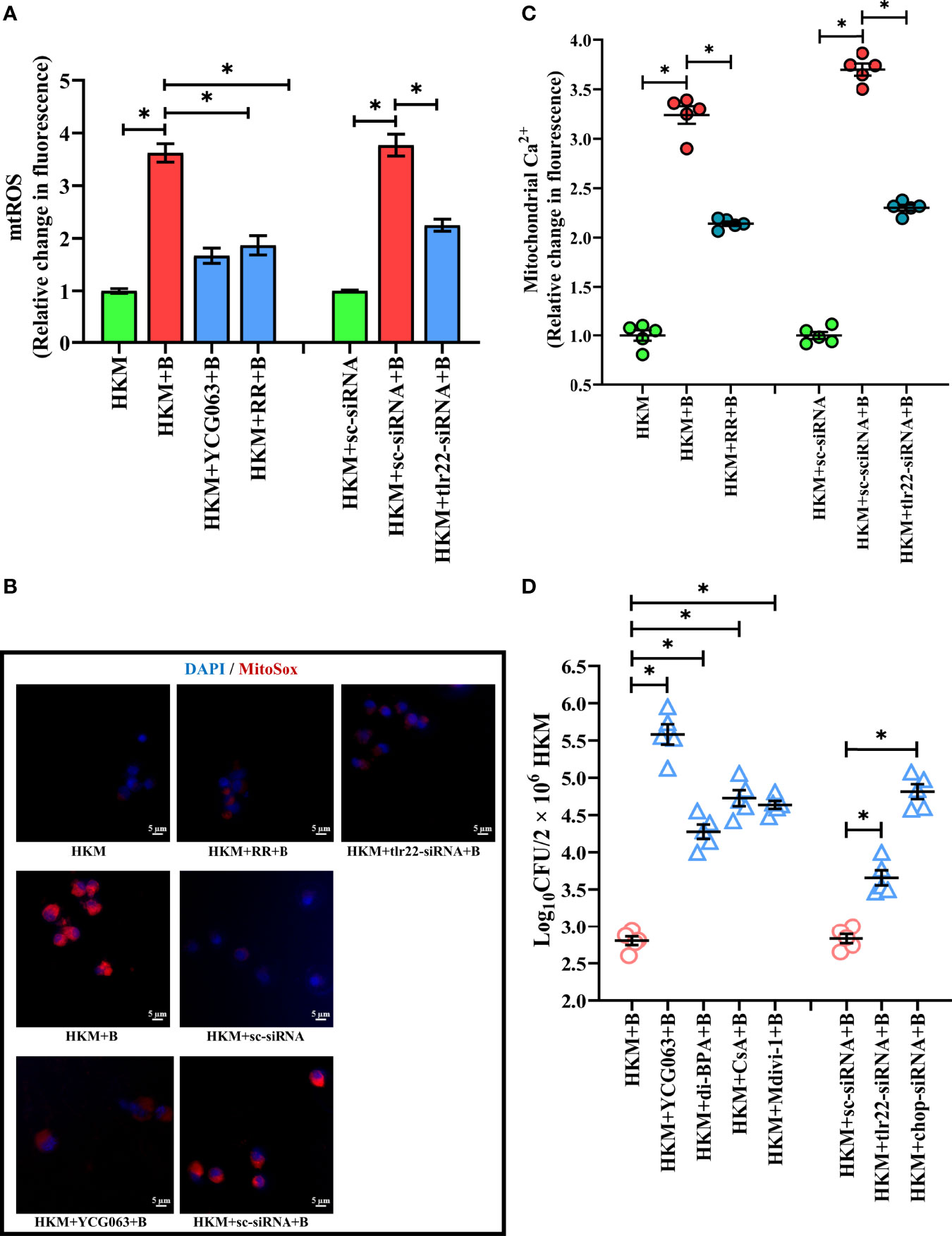
Figure 2 TLR22-induced (Ca2+)mt flux instigates pro-apoptotic mtROS generation in A. hydrophila-infected HKM. HKM pre-incubated with YCG063, RR or transfected with sc-siRNA, tlr22-siRNA were infected with A. hydrophila and at 4 h p.i. (A) changes in mtROS levels were measured, and (B) changes in mtROS levels were visualized under fluorescence microscope. Vertical bars denote mean ± SEM (n=5). Fluorescence microscopic data is representative of three independent experiments. (C) HKM pre-incubated with RR or transfected with sc-siRNA, tlr22-siRNA were infected with A. hydrophila, and changes in (Ca2+)mt were measured using Rhod-2/AM at 1 h p.i. (D) HKM pre-incubated with YCG063, di-BPA, CsA, Mdivi-1 or transfected with sc-siRNA, tlr22-siRNA were infected with A. hydrophila and bacterial load were enumerated at 24 h p.i. Data represent mean ± SEM (n=5). Asterisk (*) signifies significant difference between the indicated group (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+YCG063+B, HKM+RR+B, HKM+di-BPA, HKM+CsA+B, HKM+Mdivi-1, HKM pre-incubated with YCG063, Ruthenium Red, di-BPA, CsA, Mdivi-1 were infected with A. hydrophila; HKM+sc-siRNA, sc-siRNA transfected HKM; HKM+sc-siRNA+B, HKM+tlr22-siRNA+B, sc-siRNA and tlr22-siRNA transfected HKM were infected with A. hydrophila.
Our next step was studying the intermediate molecules that link TLR22 with mtROS generation. Mitochondrial calcium [(Ca2+)mt] flux plays a major role in mtROS production (37). In line with this, we had reported that A. hydrophila infection leads to Ca2+ sequestration in mitochondria at 1 h p.i (33). We hypothesized that TLR22 in (Ca2+)mt flux thereby compounding A. hydrophila pathogenesis. To test this, HKM transfected with tlr22-siRNA, were infected with A. hydrophila and the (Ca2+)mt levels was measured at 1 h p.i., using Rhod-2/AM. The significant reduction in (Ca2+)mt levels in tlr22-knockdown HKM (Figure 2C) suggested that TLR22 signaling positively instigates (Ca2+)mt dynamics in A. hydrophila-infected HKM. Identifying the molecules that aid in (Ca2+)mt flux was the next step and mitochondrial Ca2+ uniporter (MCU) was a rational candidate. Impairment of MCU functioning by ruthenium red (RR) attenuated Ca2+ influx into the mitochondria (Figure 2C) and consequently inhibited mtROS production in A. hydrophila-infected HKM (Figures 2A, B). Collectively our results suggested that TLR22-induced (Ca2+)mt flux through MUP triggers downstream mtROS production in A. hydrophila infected HKM.
We followed this by monitoring the effect of tlr22-induced mtROS on the intracellular A. hydrophila replication. For this, we selected the 24 h time interval and observed that inhibiting the TLR22/mtROS axis significantly increased intracellular A. hydrophila load (Figure 2D). We had previously reported that the inhibition of mtROS production alleviated HKM apoptosis (36) suggesting tlr22-induced mtROS plays anti-bacterial and pro-apoptotic roles in A. hydrophila pathogenesis.
TLR22-mtROS Axis-Induced Hypoxia Instigates Pro-Apoptotic CHOP in A. hydrophila-Infected Macrophages
Bacterial infection induces hypoxia (17). At the outset, we monitored hif1a expression in A. hydrophila-infected HKM and observed maximum expression at 6 h p.i. (Figure S2A) and selected this time point for subsequent studies. TLR signaling has been implicated in HIF-1α activation (38) and we hypothesized the role of TLR22 in the process. To test this, HKM transfected with tlr22-siRNA was infected with A. hydrophila, and hif1a expression was monitored at 6 h p.i. The significant reduction in hif1a expression in tlr22-knockdown HKM (Figure 3A) suggested the role of TLR22 in inducing hypoxia consequent to A. hydrophila infection. HIF-1α inhibitor (dimethyl-BPA) was used as a negative control which effectively repressed hif1α expression in the infected HKM (Figure 3A). Previous studies have implicated the role of mtROS in hypoxia (39). To study this, HKM pre-incubated with YCG063 were infected with A. hydrophila and the hif1a expression was monitored at 6 h p.i. Significant reduction in hif1a expression confirmed the essential role of mtROS on triggering hypoxia in A. hydrophila-infected HKM (Figure 3A).
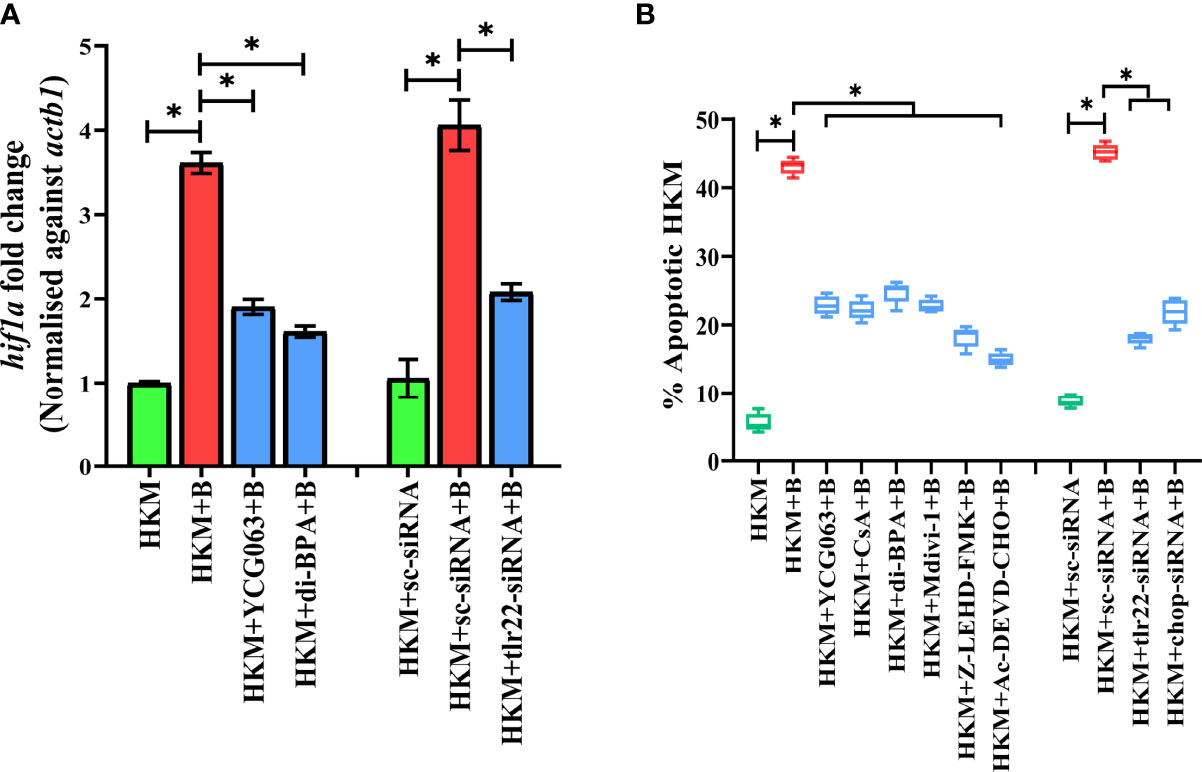
Figure 3 TLR22 induces pro-apoptotic mtROS-dependent HIF-1α activation in A. hydrophila-infected HKM. (A) HKM pre-incubated with YCG063, di-BPA or transfected with sc-siRNA, tlr22-siRNA were infected with A. hydrophila, and the expression of hif1a mRNA studied at 4 h p.i. Vertical bars denote mean ± SEM (n=3). Asterisk (*) signifies significant difference between the indicated group (*p < 0.05). (B) HKM were pre-incubated with YCG063, CsA, di-BPA, Mdivi-1, Z-LEHD-FMK, Ac-DEVD-CHO or transfected with sc-siRNA, tlr22-siRNA, chop-siRNA were infected with A. hydrophila and at 24 h p.i., Hoechst-positive HKM were enumerated at 24 h p.i. Data are presented as box-and-whisker plots (n=5), shows the medians and 25th and 75th percentiles, and the whiskers show 10th and 90th percentiles. HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+YCG063+B, HKM+di-BPA+B; HKM pre-incubated with YCG063 and di-BPA respectively were infected with A. hydrophila; HKM+sc-siRNA, sc-siRNA transfected HKM; HKM+sc-siRNA+B, HKM+tlr22-siRNA+B, sc-siRNA and tlr22-siRNA transfected HKM respectively were infected with A. hydrophila.
The role of hypoxia in containing bacterial growth is well reported (40). Towards this direction, HKM pre-incubated with dimethyl-BPA were infected with A. hydrophila, and bacterial replication was monitored at 24 h p.i. We observed that inhibition of hif1a resulted in the significant increase in intracellular A. hydrophila (Figure 2D). Based on these findings, we suggest that TLR22-induced mtROS triggers hypoxia to counteract intracellular A. hydrophila replication.
Hypoxia triggers apoptosis of macrophages (41). We previously demonstrated the colloquy between CHOP and HKM apoptosis in A. hydrophila infection (36). In this line, we presumed the bactericidal role of HIF-1α in A. hydrophila infection is attributed to the activation of CHOP. At the outset, we monitored chop expression consequent to A. hydrophila infection and observed maximum chop expression at 2 h p.i. (Figure 4A) and thereafter though the levels declined it remained significantly high till 12 h p.i. (Figure 4A). To establish the role of hypoxia on CHOP activation, HKM were pre-treated with di-BPA and chop expression was monitored at indicated time points p.i. Interestingly, di-BPA pre-incubation had little effect on chop expression at early time points (Figure 4A) but repressed its expression at later time points i.e., at 6 h and 12 h p.i. (Figure 4A) suggesting the regulatory role of HIF-1α on prolonged activation of CHOP in A. hydrophila-infected HKM. Also, inhibition of TLR22 signaling attenuated chop expression (Figure 4B). Additionally, silencing of chop by RNAi led to the significant decline in apoptosis of A. hydrophila-infected HKM (Figure 3B) together implicating the intermediary role of CHOP in hypoxia-induced apoptosis of A. hydrophila-infected HKM.
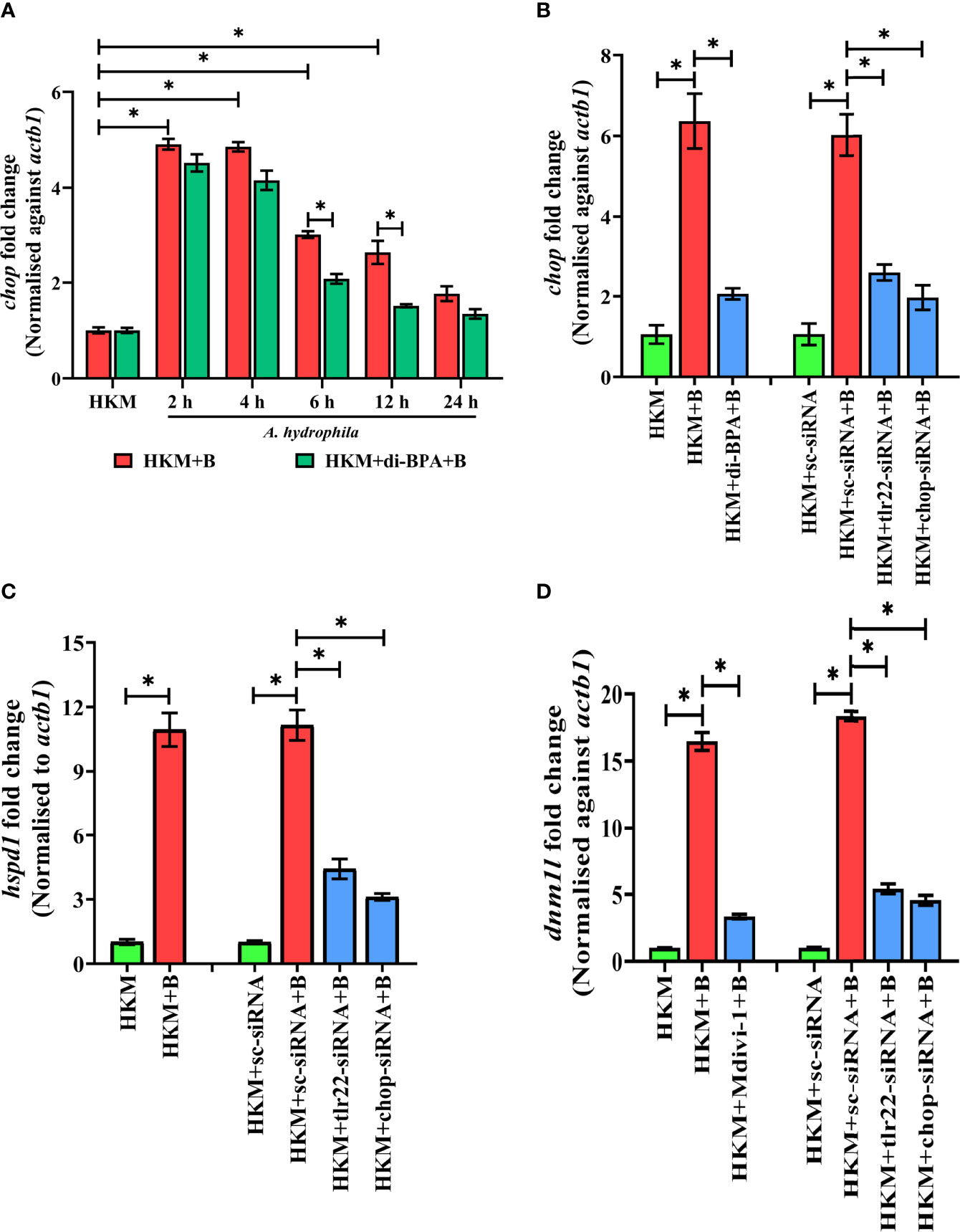
Figure 4 TLR22-induced HIF-1α sustains activation of CHOP triggering UPRmt-mediated mitochondrial fragmentation in A. hydrophila-infected HKM. (A) HKM pre-incubated with di-BPA were infected with A. hydrophila and the expression of chop mRNA studied at indicated time points. HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+di-BPA+B, HKM pre-incubated with di-BPA infected with A. hydrophila. (B) HKM pre-incubated with di-BPA, Mdivi-1 or transfected with sc-siRNA, tlr22-siRNA, chop-siRNA were infected with A. hydrophila and the expression of (B) chop mRNA studied at 6 h p.i., (C) hspd1 mRNA studied at 6 h p.i., and (D) expression of dnm1l mRNA studied at 24 h p.i. Vertical bars denote mean ± SEM (n=3). Asterisk (*) signifies significant difference between the indicated group (*p < 0.05). HKM, uninfected HKM; HKM+B; HKM infected with A. hydrophila; HKM+di-BPA+B, HKM+Mdivi-1+B, HKM pre-incubated with di-BPA, Mdivi-1 infected with A. hydrophila; HKM+sc-siRNA, HKM+sc-siRNA+B, HKM+tlr22-siRNA+B, HKM+chop-siRNA+B, sc-siRNA, tlr22-siRNA, and chop-siRNA transfected HKM were infected with A. hydrophila.
CHOP Activates UPRmt in A. hydrophila-Infected Macrophages
Excessive mtROS induces proteotoxic stress and in turn, mitochondria trigger mitochondrial-UPR (UPRmt) to maintain proteostasis (42, 43). The supra-normal levels of mtROS encouraged us to study UPRmt in A. hydrophila pathogenesis. hspd1 encodes for the mitochondrial chaperone, HSP60 which is a marker for UPRmt (44). At the onset, HKM were infected with A. hydrophila and hspd1 expression monitored at indicated time point p.i. The RT-qPCR data demonstrated maximum fold change in hspd1 expression at 6 h p.i. and was selected for subsequent studies (Figure S2B).
Besides imparting apoptosis, CHOP also plays a role in initiating UPRmt (45). To study the link between chop and UPRmt in A. hydrophila pathogenesis, HKM were transfected with chop-siRNA and hspd1 expression monitored at 6 h p.i. We noticed a significant reduction in hspd1 expression in chop-knockdown HKM (Figure 4C) implicating the role of CHOP in inducing UPRmt in A. hydrophila infection.
UPRmt Induces Mitochondrial Fragmentation in A. hydrophila-Infected Macrophages
UPRmt impacts the mitochondrial architecture (46). In absence of prior information, we assessed whether A. hydrophila infection alters mitochondrial network, and for that, we monitored the expression of dnm1l gene, that encodes for the cytosolic GTPase protein, DRP1 regulating mitochondrial fission (47). We observed a maximum fold increase in dnm1l expression at 24 h p.i. (Figure S3A) and selected this time point for subsequent studies. Mitochondrial fission was further validated by studying the mitochondrial network architecture. Towards that direction, HKM were infected with A. hydrophila then stained with MitoTracker Green dye and visualized at 24 h p.i. by confocal microscopy. Unlike normal mitochondria which form elongated networks; the fragmented mitochondria appear rod-shaped (Figure S3B). The presence of rod-like mitochondria confirmed that A. hydrophila-induced mitochondrial fission in infected HKM. TLR signaling has been reported in regulating mitochondrial network dynamics (48) and therefore we hypothesized the role of TLR22 in the process. In this line, HKM transfected with tlr22-siRNA was infected with A. hydrophila, and dnm1l mRNA expression was monitored at 24 h p.i. The significant reduction in dnm1l mRNA expression in tlr22-knockdown HKM (Figure 4D) clearly indicates the role of TLR22 as a regulator of mitochondrial network architecture consequent to A. hydrophila infection. Together, these results for the first time suggested the occurrence of mitochondrial fragmentation in A. hydrophila infection.
Having established that A. hydrophila infection induces mitochondrial fragmentation, we asked whether inhibition of UPRmt would reverse the mitochondrial fragmentation. CHOP being a regulator for UPRmt response was selected for the study. Thus, HKM transfected with chop-siRNA, were infected with A. hydrophila and the expression of dnm1l was monitored at 24 h p.i. We observed that the silencing of chop resulted in a significant reduction in dnm1l expression (Figure 4D) which implies that alleviating UPRmt restores mitochondrial network architecture in A. hydrophila-infected HKM.
Mitochondrial Fragmentation Induces Mitochondrial Dysfunction Triggering Caspase-9-Mediated HKM Death
Mitochondrial dynamics is crucial in maintaining mitochondrial functioning (49). Depolarization of ΔΨm and release of pro-apoptotic protein such as Cyt c are clear signals of mitochondrial dysfunction (50). To correlate mitochondrial fragmentation with mitochondrial dysfunctioning, HKM were pre-incubated with mitochondrial fission inhibitor, Mdivi-1, then infected with A. hydrophila and the changes in ΔΨm and Cyt c release studied at 24 h p.i. We observed that Mdivi-1 pre-incubation attenuated dnm1l expression (Figure 4D) and ΔΨm (Figure 5A) of A. hydrophila-infected HKM. Consequently, Mdivi-1 inhibited Cyt c release (Figure 5B), repressed the activation of caspase-9/caspase-3 axis (Figures 5C, D), HKM apoptosis (Figure 3B) and increased the number of intracellular A. hydrophila (Figure 2D). These results portray that mitochondrial fragmentation triggers caspase-9/caspase-3-mediated apoptosis of A. hydrophila-infected HKM thereby aiding the clearance of intracellular bacteria.
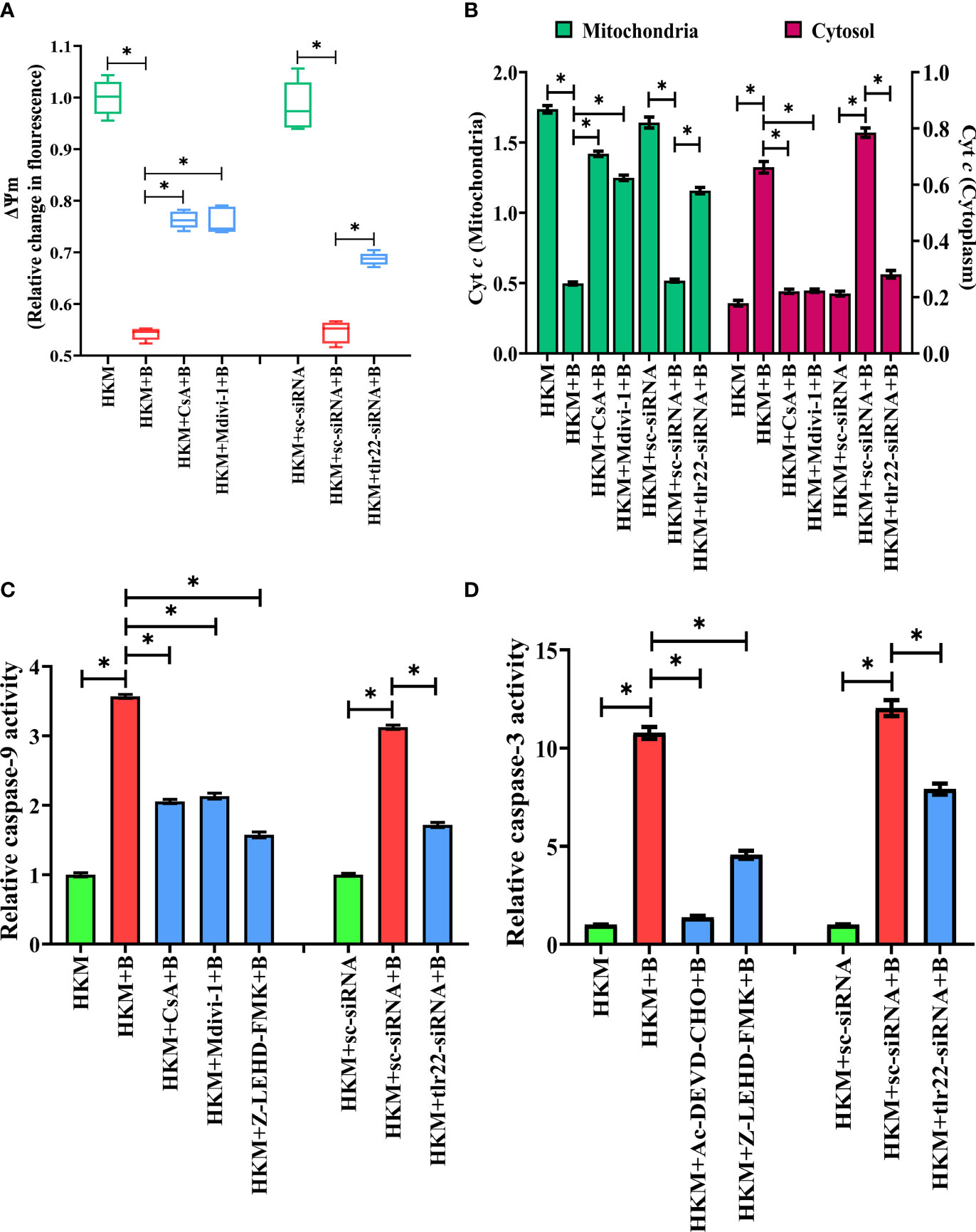
Figure 5 TLR22-induced mitochondrial fragmentation dissipates ψm and triggers Cyt c release actvating caspase-9/caspase-3 axis in A. hydrophila-infected HKM. HKM pre-incubated with CsA, Mdivi-1 or transfected with sc-siRNA, tlr22-siRNA were infected with A. hydrophila and at 24 h p.i., (A) changes in ψm were studied using Rhodamine-123, and (B) changes in Cyt c release was studied. Data are presented as box-and-whisker plots (n=5), shows the median and 25th and 75th percentiles, and the whiskers show 10th and 90th percentiles. Vertical bars denote mean ± SEM (n=5). HKM pre-incubated with CsA, Mdivi-1, Z0LEHD-FMK, Ac-DEVD-CHO or transfected with sc-siRNA, tlr22-siRNA were infected with A. hydrophila and at 24 h p.i., (C) caspase-9 activity and (D) caspase-3 activity were studied. Vertical bars denote mean ± SEM (n=3). Asterisk (*) signifies significant difference between the indicated group (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+CsA+B, HKM+Mdivi-1 +B, HKM+Z-LEHD-FMK, HKM+Ac-DEVD-CHO; HKM pre-incubated with CsA, Mdivi-1, Z-LEHD-FMK, Ac-DEVD-CHO were infected with A. hydrophila; HKM+sc-siRNA, sc-siRNA transfected HKM; HKM+sc-siRNA+B, HKM+tlr22-siRNA+B, HKM+chop-siRNA+B, sc-siRNA, tlr22-siRNA and chop-siRNA transfected HKM were infected with A. hydrophila.
We questioned the role of TLR22 in the activation of caspase-9/caspase-3 axis. To study this, HKM were transfected with tlr22-siRNA and changes in ΔΨm and the translocation of Cyt c were studied in A. hydrophila-infected HKM. We observed that the loss in ΔΨm and Cyt c translocation was repressed in the tlr22-knockdown HKM (Figure 5B). Additionally, RNAi studies also demonstrated that both caspase-9 (Figure 5C) and caspase-3 (Figure 5D) activity to be attenuated in tlr22-knockdown HKM. CsA (mPTP inhibitor) was used as the control which repressed the activation of caspase-9/caspase-3 axis in A. hydrophila-infected HKM (Figures 5C, D). Altogether, our results establish the role of TLR22 in the activation of caspase-9/caspase-3 axis in A. hydrophila-induced HKM apoptosis.
Discussion
During bacterial infection, the host faces a unique set of challenges and relies on efficient TLR signaling to counter the pathogens. However, despite the expanding knowledge of TLR signaling, the innate immune axes regulated by TLRs are not well understood, particularly in fish. TLR22 is an important PRR in non-mammalian aquatic vertebrates, including fish, but its role in immunity remains obscure. Studies have highlighted its contradictory role in fish innate immune system. Some report suggest it triggers inflammatory response (51) whereas other report suggest it acts as an equalizer to suppress excessive inflammation (52). This study highlights the role of TLR22 signaling in mounting mitochondria-mediated innate immune responses in A. hydrophila pathogenesis in fish.
Recent pieces of evidence have highlighted mitochondria as a central hub in the innate immune signaling pathway (53). Nonetheless, reports concerning its involvement in fish innate immunity are lacking. Mitochondria are major sites for ROS production and our previous studies suggested that A. hydrophila induces mtROS production in HKM with pro-apoptotic implications (36). We also reported the pro-apoptotic role of TLR22 in A. hydrophila pathogenesis (16), but the possibility that TLR22 signaling regulates mtROS generation has remained unexplored. Towards that end, we observed a significant reduction in mtROS levels in A. hydrophila-infected HKM in the absence of TLR22 signaling. This finding for the first time established the role of TLR22 in modulating mtROS production thereby impacting bacterial pathogenesis. Previous studies have documented TLR-dependency of mtROS production consequent to microbial infections (5). With the consensus of previous reports, and our own observations we conclude that the ability to trigger mtROS following pathogenic insult is conserved among the members of the TLR superfamily.
The protective role of mtROS in antimicrobial immune defense is well established in mammals (5, 54). We observed an inverse correlation between mtROS levels and intracellular A. hydrophila load and inhibiting mtROS generation increased the bacterial load demonstrating the antimicrobial role of mtROS in fish. To the best of our knowledge, this is the first report on the role of mtROS as a microbicidal factor in fish. Our results are in accord with several previous reports in mammals (5, 54) thereby implicating this event as an evolutionarily conserved innate immune trait. We suggest that TLR22-induced mtROS triggers HKM apoptosis thereby aiding in the removal of infected cells along with pathogens.
The next step was to understand how TLR22 influenced mtROS production in the infected HKM. (Ca2+)mt overload is suggested as one of the major causes for enhancing mtROS production under pathophysiological conditions (55). Mitochondria play a paramount role in regulating the spatiotemporal patterns of Ca2+ signaling thus controlling cell survival and death (56). It can uptake (Ca2+)C through the MCU complex and our previous studies also revealed that A. hydrophila triggers (Ca2+)C influx through MCU (33, 36). To correlate this with TLR22, we used the RNAi approach and measured (Ca2+)mt levels. The marked decline in (Ca2+)mt levels in tlr22-knockdown HKM implicated TLR22 in initiating (Ca2+)C influx inside mitochondria. At present, we do not know how TLR22 influences (Ca2+)mt influx. It has been suggested that TLRs per se do not modulate MCU activity, they rather control Ca2+ mobilization from intracellular stores (57) which subsequently influence MCU activity and prompts (Ca2+)mt influx (58). This highlights the possibility that TLR22 might be playing an indirect role in augmenting (Ca2+)mt levels in A. hydrophila-infected HKM. It has also been observed that following TLR2/4 ligation the mitochondrial adaptor protein ECSIT interacts with TRAF6 to up-regulate mtROS production in macrophages (5). Additionally, the mitochondrial transcription factor A (TFAM) is reported to play a role in triggering mtROS production in response to TLR4 signaling. The conservancy in TLR signaling makes it an interesting proposition to study whether similar cascades of events are initiated following TLR22 activation in A. hydrophila-infected HKM.
The link between (Ca2+)C influx into mitochondria and mtROS production in A. hydrophila pathogenesis has been established (33, 36). RyR and IP3R are Ca2+ channels, accountable for the efflux of (Ca2+)ER to the cytosol (59). Our previous studies suggested that inhibition of IP3R and RyR significantly attenuated (Ca2+)mt uptake and HKM apoptosis (33, 36). The influx of Ca2+ is essential for the normal functioning of mitochondria unless there is an overload that induces structural-functional alterations with pro-apoptotic implications (37). Based on cumulative evidences, we suggest that consequent to A. hydrophila infection TLR22 signaling initiates the influx of (Ca2+)C released by ER into mitochondria to buffer the (Ca2+)C levels and protect the HKM till it reaches a critical threshold whose transgression leads to structural-functional alterations in the organelle. Finding this critical threshold will be important in deciding Ca2+ concentration regulated by TLR22 in different sub-cellular compartments for triggering apoptosis in bacterial infection.
We were intrigued by how mtROS signals HKM apoptosis. Bacterial infections trigger hypoxia-induced host cell apoptosis and HIF-1α plays an important role in the process (17, 40). We hypothesized the role of mtROS in HIF-1α expression. Towards this direction, we recorded significant upregulation in hif1a expression in A. hydrophila-infected HKM. Using pharmacological inhibitors and RNAi studies, we further noted that inhibiting TLR22 signaling or mtROS generation attenuated hif1a expression indicating the role of TLR22/mtROS axis in HIF-1α activation in A. hydrophila-infected HKM. Our results are in consonance with previous reports which suggested mtROS is an essential intermediate in HIF-1α activation and TLR signaling plays a primal role in initiating the chain of events (39, 60).
The next step was to study the role of HIF-1α in regulating A. hydrophila pathogenesis. The significant reduction in HKM apoptosis and concomitant increase in bacterial burden implicated the anti-bacterial role of HIF-1α in A. hydrophila infection. Various reports are in line with our observations which suggested hosts deficient in HIF-1α are susceptible to bacterial infection (18, 40). The mechanism underlying HIF-1α signaling in fish is not well understood. CHOP has been implicated in HIF-1α signaling (61). Previously, we had also reported the role of CHOP in the apoptosis of A. hydrophila-infected HKM (36). At the outset, we questioned whether TLR22 influences CHOP expression, and our RNAi results clearly suggested CHOP activation downstream of TLR22 signaling. Our next step was establishing the link between HIF-1α and CHOP. We observed that inhibition of HIF-1α interfered with prolonged expression of chop implicating the role of HIF-1α in sustaining CHOP activation in A. hydrophila infection. These results suggested that CHOP is an intermediary molecule in the pro-apoptotic TLR22/HIF-1α axis in A. hydrophila pathogenesis.
Exposure to pathogens causes mitochondrial dysfunction and UPRmt activation (28) and to the best of our knowledge, there are no such precedents in A. hydrophila pathogenesis. Importantly, UPRmt has also not been reported in fish. Hence, our first aim was to study UPRmt in A. hydrophila-infected HKM. Hspd1 is considered as a marker for UPRmt (62, 63) and we noticed TLR22-mediated hspd1 expression in A. hydrophila infection. Our findings assume significant importance because a) it is the first report on UPRmt in fish and b) reflects the involvement of TLR22 signaling in UPRmt thereby impacting microbial pathogenesis. Next, we investigated how UPRmt is regulated by TLR22. CHOP playing a key role in UPRmt activation (46) was our prime target. We did observe that CHOP silencing attenuated UPRmt activation in the infected HKM. CHOP binding sites have been reported in UPRmt elements triggering their transcriptional activation (63). To this, we concluded that consequent to A. hydrophila infection TLR22 signaling converges at HIF-1α which in turn induced CHOP-mediated activation of UPRmt in infected HKM. Future studies aimed at identifying other molecules that influence UPRmt activation will help in understanding the role of mitochondrial proteostasis in regulating microbial pathogenesis in fish.
Next, we questioned the implications of the UPRmt activation in A. hydrophila pathogenesis. Activation of UPRmt is a primary retort to defend mitochondrial homeostasis (42) but prolonged UPRmt impacts mitochondrial homeostasis triggering cell death (64). Drp1 plays a critical role in mitochondrial fission (23) and we observed significant upregulation in dnm1l expression and fragmented mitochondrial network in the A. hydrophila-infected HKM. Based on these findings, we suggest that A. hydrophila infection affects mitochondrial homeostasis by favoring mitochondrial fission. This finding is in line with previous reports documenting bacteria-induced mitochondrial fission in mammalian cells (65). Various studies have reported TLR-mediated switching of mitochondrial morphology to fission from fusion (48). We observed that silencing of TLR22 attenuated the expression of dnm1l suggesting TLR22 functions as mitochondrial-fission regulator in A. hydrophila pathogenesis.
Though the biological importance of mitochondrial fission is unclear, reports suggested it is a prerequisite in screening the damaged mitochondria for mitophagic culling (66). Previously, we had reported failure of the autophagic machinery in A. hydrophila infection leading to accretion of damaged mitochondrial fragments (36). This prompted us to investigate the consequences of mitochondrial fragmentation in A. hydrophila-infected HKM. Mitochondrial fission leads to a loss in Δψm and the opening of mPTP thereby releasing pro-apoptotic cyt c (67, 68). We observed that inhibiting TLR22 signaling and mitochondrial fission reinstated Δψm and inhibited cyt c release thereby attenuating HKM apoptosis. These results are in consonance with a previous report documenting mitochondrial fission triggers cyt c release (67). Additionally, the intracellular survival of A. hydrophila was also significantly increased on inhibiting mitochondrial fission, highlighting the relevance of mitochondrial network homeostasis in A. hydrophila pathogenesis. Similar reports support our finding which suggested that mitochondrial fission directs cells towards apoptosis and regulates bacterial pathogenesis (69, 70).
Cyt c is a central molecule in the activation of caspase-9/caspase-3 axis (71). Our earlier reports demonstrated the activation of caspase-9 consequent to A. hydrophila infection (33). Here we aimed to establish the link between TLR22 and caspase-9/caspase-3 axis. We noted that the caspase-9/caspase-3 axis was repressed in tlr22-knockdown HKM suggesting TLR22 signaling culminates in caspase-9/caspase-3-mediated apoptosis of A. hydrophila-infected HKM. Collectively, our findings implicate the pro-apoptotic role of mitochondrial network dynamics in TLR22-mediated apoptosis of A. hydrophila-infected HKM.
To conclude, our findings established the primal role of TLR22 in utilizing mitochondria-derived immune signaling cues integral in controlling the onset and pathogenesis of A. hydrophila infection. We propose that TLR22 serves as a conduit linking A. hydrophila infection with the influx of (Ca2+)C into the mitochondria, entailing mtROS production. Enhanced mtROS induces HIF-1α favoring the sustained activation of CHOP which triggers UPRmt-induced mitochondrial fission and organelle dysfunctioning. Compromised autophagy leads to the accumulation of dysfunctional mitochondria which releases Cyt c triggering downstream caspase-9/caspase-3-mediated HKM apoptosis and the clearance of A. hydrophila (Figure 6). Thus, it will be exciting to extrapolate this current understanding and investigate how innate immune pathways administrated by TLR22 and mitochondria can be translated into active therapeutics to boost the immune system of fish against A. hydrophila and control motile aeromonad septicaemia and ulcerative diseases.
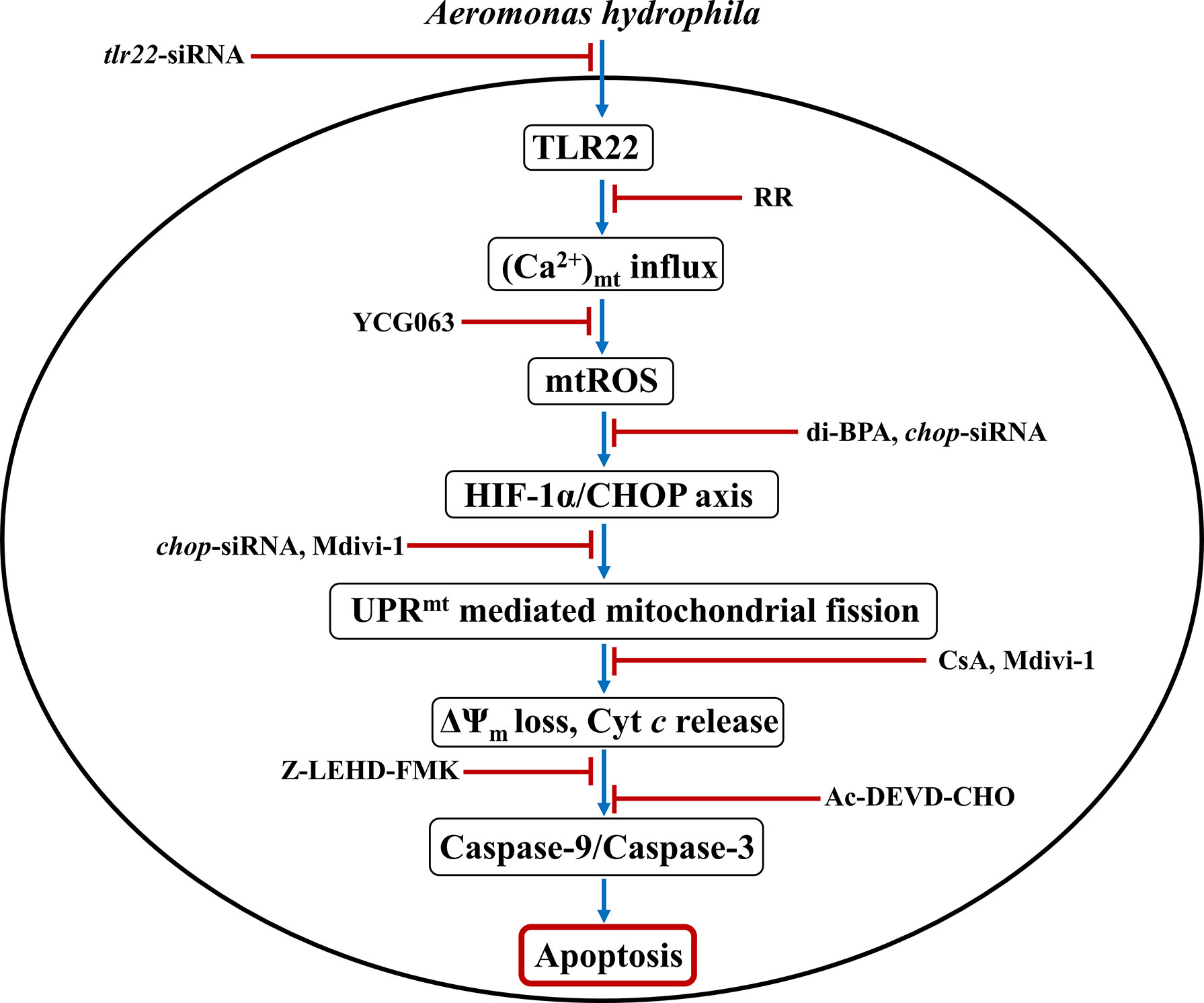
Figure 6 Overview of the study. TLR22-induced Ca2+ influx into mitochondria triggers mtROS generation leading to downstream activation of HIF-1α/CHOP axis. Activated CHOP instigates UPRmt-mediated fragmentation of the mitochondrial network leading to mitochondrial dysfunction which activates caspase-9/caspase-3 axis-mediated apoptosis of A. hydrophila-infected HKM.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, MZ882392.
Ethics Statement
The studies were carried out according to the guidelines issued by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Govt. of India, and permitted by the Animal Ethics Committee (DU/ZOOL/IAEC-R/2013/33), University of Delhi.
Author Contributions
MK: Conceptualization, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review and editing, Formal analysis. SS: Investigation, Methodology, Writing – review and editing. JK: Investigation, Writing – review and editing. MH: Methodology. UH: Methodology. SM: Conceptualization, Resources, Supervision, Writing – original draft, Writing – review and editing. All authors contributed to the article and approved the submitted version.
Funding
MK, SS, and JK were supported by U.G.C.-NET fellowship (Government of India). MH and UH were supported by fellowship provided by South Asian University, India.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank S. Barik, Cleveland State University for helpful discussion and critically analyzing the manuscript. We thank Milind Dongardive, Central Instrumentation Facility, South Asian University, India for the technical help while performing RT-qPCR studies. The authors are particularly grateful to colleagues who helped immensely by providing reagents and support for carrying out the work on conditions of anonymity.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.931021/full#supplementary-material
Supplementary Figure 1 | Transfection of tlr22-siRNA and chop-siRNA attenuates tlr22 and chop gene expression in HKM. HKM transfected with sc-siRNA, tlr22-siRNA or chop-siRNA and tlr22, chop expression was studied using RT-qPCR. Vertical bars represent mean ± S.E (n=3). Asterisk (*) signifies significant difference between indicated groups (*p<0.05). HKM+sc-siRNA, HKM transfected with sc-siRNA; HKM+tlr22-siRNA, HKM transfected with tlr22-siRNA; HKM+chop-siRNA, HKM transfected with chop-siRNA.
Supplementary Figure 2 | A. hydrophila infection triggers hif1a and hspd1 mRNA expression in HKM. HKM were infected with A. hydrophila and at indicated time points (A) hif1a mRNA expression, and (B) hspd1 mRNA expression were studied. Vertical bars denote mean ± SEM (n=3). Asterisk (*) signifies significant difference between the indicated group (*p<0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila.
Supplementary Figure 3 | A. hydrophila induces mitochondrial fragmentation in infected HKM. HKM were infected with A. hydrophila and at indicated time points (A) dnm1l mRNA expression was studied. (B) HKM were infected with A. hydrophila and morphology of mitochondrial network studied at 24 h p.i. HKM were washed, stained with MitoTracker green and DAPI, mounted and visualized under microscope (Scale – 2 µm).
References
1. Circu ML, Aw TY. Reactive Oxygen Species, Cellular Redox Systems, and Apoptosis. Free Radic Biol Med (2010) 48(6):749–62. doi: 10.1016/j.freeradbiomed.2009.12.022
2. Dickson KB, Zhou J. Role of Reactive Oxygen Species and Iron in Host Defense Against Infection. Front Biosci (Landmark Ed) (2020) 25:1600–16. doi: 10.2741/4869
3. Dunn JD, Alvarez LA, Zhang X, Soldati T. Reactive Oxygen Species and Mitochondria: A Nexus of Cellular Homeostasis. Redox Biol (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
4. Scialò F, Fernández-Ayala DJ, Sanz A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front Physiol (2017) 8:428. doi: 10.3389/fphys.2017.00428
5. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR Signalling Augments Macrophage Bactericidal Activity Through Mitochondrial ROS. Nature (2011) 472(7344):476–80. doi: 10.1038/nature09973
6. Stocks CJ, Schembri MA, Sweet MJ, Kapetanovic R. For When Bacterial Infections Persist: Toll-Like Receptor-Inducible Direct Antimicrobial Pathways in Macrophages. J Leukoc Biol (2018) 103(1):35–51. doi: 10.1002/JLB.4RI0917-358R
7. Roca FJ, Ramakrishnan L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria via Mitochondrial Reactive Oxygen Species. Cell (2013) 153(3):521–34. doi: 10.1016/j.cell.2013.03.022
8. Tang R, Wang S, Han P, Zhang Q, Zhang S, Xing X, et al. Toll-Like Receptor (TLR) 2 and TLR13 From the Endangered Primitive-Ray Finned Fish Dabry’s Sturgeon (Acipenser Dabryanus) and Their Expression Profiling Upon Immune Stimulation. Aquac Rep (2020) 16:100247. doi: 10.1016/j.aqrep.2019.100247
9. Goulopoulou S, McCarthy CG, Webb RC. Toll-Like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol Rev (2016) 68(1):142–67. doi: 10.1124/pr.114.010090
10. Roach JC, Glusman G, Rowen L, Kaur A, Purcell MK, Smith KD, et al. The Evolution of Vertebrate Toll-Like Receptors. Proc Natl Acad Sci USA (2005) 102(27):9577–82. doi: 10.1073/pnas.0502272102
11. Gong Y, Feng S, Li S, Zhang Y, Zhao Z, Hu M, et al. Genome-Wide Characterization of Toll-Like Receptor Gene Family in Common Carp (Cyprinus Carpio) and Their Involvement in Host Immune Response to Aeromonas Hydrophila Infection. Comp Biochem Physiol Part D Genomics Proteomics (2017) 24:89–98. doi: 10.1016/j.cbd.2017.08.003
12. Xing J, Zhou X, Tang X, Sheng X, Zhan WJF, Immunology S. Characterization of Toll-Like Receptor 22 in Turbot (Scophthalmus Maximus). Fish Shellfish Immunol (2017) 66:156–62. doi: 10.1016/j.fsi.2017.05.025
13. Li H, Yang G, Ma F, Li T, Yang H, Rombout JH, et al. Molecular Characterization of a Fish-Specific Toll-Like Receptor 22 (TLR22) Gene From Common Carp (Cyprinus Carpio L.): Evolutionary Relationship and Induced Expression Upon Immune Stimulants. Fish Shellfish Immunol (2017) 63:74–86. doi: 10.1016/j.fsi.2017.02.009
14. Wang RH, Li W, Fan YD, Liu QL, Zeng LB, Xiao TY. Tlr22 Structure and Expression Characteristic of Barbel Chub, Squaliobarbus Curriculus Provides Insights Into Antiviral Immunity Against Infection With Grass Carp Reovirus. Fish Shellfish Immunol (2017) 66:120–8. doi: 10.1016/j.fsi.2017.04.018
15. Du X, Wu J, Li Y, Xia P, Li D, Yang X, et al. Multiple Subtypes of TLR22 Molecule From Schizothorax Prenanti Present the Functional Diversity in Ligand Recognition and Signal Activation. Fish Shellfish Immunol (2019) 93:986–96. doi: 10.1016/j.fsi.2019.08.042
16. Kumar M, Kumar J, Sharma S, Hussain MA, Shelly A, Das B, et al. TLR22-Mediated Activation of TNF-α-Caspase-1/IL-1β Inflammatory Axis Leads to Apoptosis of Aeromonas Hydrophila-Infected Macrophages. Mol Immunol (2021) 137:114–23. doi: 10.1016/j.molimm.2021.06.25
17. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF Transcription Factors, Inflammation, and Immunity. Immunity (2014) 41(4):518–28. doi: 10.1016/j.immuni.2014.09.008
18. Dengler VL, Galbraith MD, Espinosa JM. Transcriptional Regulation by Hypoxia Inducible Factors. Crit Rev Biochem Mol Biol (2014) 49(1):1–15. doi: 10.3109/10409238.2013.838205
19. Corrado C, Fontana S. Hypoxia and HIF Signaling: One Axis With Divergent Effects. Int J Mol Sci (2020) 21(16):5611. doi: 10.3390/ijms21165611
20. Klimova T, Chandel NS. Mitochondrial Complex III Regulates Hypoxic Activation of HIF. Cell Death Differ (2008) 15(4):660–6. doi: 10.1038/sj.cdd.4402307
21. Ogryzko NV, Lewis A, Wilson HL, Meijer AH, Renshaw SA, Elks PM. Hif-1α–Induced Expression of Il-1β Protects Against Mycobacterial Infection in Zebrafish. J Immunol (2019) 202(2):494–502. doi: 10.4049/jimmunol.1801139
22. Mishra P, Chan DC. Mitochondrial Dynamics and Inheritance During Cell Division, Development and Disease. Nat Rev Mol Cell Biol (2014) 15(10):634–46. doi: 10.1038/nrm3877
23. Song M, Mihara K, Chen Y, Scorrano L, Dorn GW II. Mitochondrial Fission and Fusion Factors Reciprocally Orchestrate Mitophagic Culling in Mouse Hearts and Cultured Fibroblasts. Cell Metab (2015) 21(2):273–86. doi: 10.1016/j.cmet.2014.12.011
24. Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab (2015) 22(2):207–18. doi: 10.106/j.cmet.2015.06.006
25. Stavru F, Palmer AE, Wang C, Youle RJ, Cossart P. Atypical Mitochondrial Fission Upon Bacterial Infection. Proc Natl Acad Sci USA (2013) 110(40):16003–8. doi: 10.1073/pnas.1315784110
26. Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell (2018) 69(2):169–81. doi: 10.1016/j.molcel.2017.06.017
27. Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science (2012) 337(6094):587–90. doi: 10.1126/science.1223560
28. Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-Regulated Innate Immunity Provides Resistance to Pathogen Infection. Nature (2014) 516(7531):414–7. doi: 10.1038/nture13818
29. Liu Y, Samuel BS, Breen PC, Ruvkun G. Caenorhabditis Elegans Pathways That Surveil and Defend Mitochondria. Nature (2014) 508(7496):406–10. doi: 10.1038/nature13204
30. Naresh NU, Haynes CM. Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb Perspect Biol (2019) 11(6):a033944. doi: 10.1101/cshperspect.a033944
31. Abdelhamed H, Ibrahim I, Baumgartner W, Lawrence ML, Karsi A. Characterization of Histopathological and Ultrastructural Changes in Channel Catfish Experimentally Infected With Virulent Aeromonas Hydrophila. Front Microbiol (2017) 8:1519. doi: 10.3389/fmicb.2017.01519
32. Galindo CL, Fadl AA, Sha J, Gutierrez C, Popov VL, Boldogh I, et al. Aeromonas Hydrophila Cytotoxic Enterotoxin Activates Mitogen-Activated Protein Kinases and Induces Apoptosis in Murine Macrophages and Human Intestinal Epithelial Cells. J Biol Chem (2004) 279(36):37597–612. doi: 10.1074/jbc.M404641200
33. Banerjee C, Singh A, Das TK, Raman R, Shrivastava A, Mazumder S. Ameliorating ER-Stress Attenuates Aeromonas Hydrophila-Induced Mitochondrial Dysfunctioning and Caspase Mediated HKM Apoptosis in Clarias Batrachus. Sci Rep (2014) 4(1):1–13. doi: 10.1038/srep05820
34. Sørensen K, Sveinbjørnsson B, Dalmo R, Smedsrød B, Bertheussen K. Isolation, Cultivation and Characterization of Head Kidney Macrophages From Atlantic Cod, Gadus Morhua L. J Fish Dis (1997) 20(2):93–107. doi: 10.1046/j.1365-2761.1997.d01-112.x
35. Barbu EM, Shirazi F, McGrath DM, Albert N, Sidman RL, Pasqualini R, et al. An Antimicrobial Peptidomimetic Induces Mucorales Cell Death Through Mitochondria-Mediated Apoptosis. PloS One (2013) 8(10):e76981. doi: 10.1371/journal.pone.0076981
36. Kumar M, Shelly A, Dahiya P, Ray A, Mazumder S. Aeromonas Hydrophila Inhibits Autophagy Triggering Cytosolic Translocation of mtDNA Which Activates the Pro-Apoptotic Caspase-1/IL-1β-Nitric Oxide Axis in Headkidney Macrophages. Virulence (2022) 13(1):60–76. doi: 10.1080/21505594.2021.2018767
37. Brookes PS, Yoon Y, Robotham JL, Anders M, Sheu SS. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am J Physiol Cell Physiol (2004) 287(4):C817-33. doi: 10.1152/ajpcell.00139.2004
38. Jantsch J, Wiese M, Schödel J, Castiglione K, Gläsner J, Kolbe S, et al. Toll-Like Receptor Activation and Hypoxia Use Distinct Signaling Pathways to Stabilize Hypoxia-Inducible Factor 1α (HIF1A) and Result in Differential HIF1A-Dependent Gene Expression. J Leukoc Biol (2011) 90(3):551–62. doi: 10.1189/jlb.1210683
39. Patten DA, Lafleur VN, Robitaille GA, Chan DA, Giaccia AJ, Richard DE. Hypoxia-Inducible Factor-1 Activation in Nonhypoxic Conditions: The Essential Role of Mitochondrial-Derived Reactive Oxygen Species. J Mol Cell Biol (2010) 21(18):3247–57. doi: 10.1091/mbc.E10-01-0025
40. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1α Expression Regulates the Bactericidal Capacity of Phagocytes. J Clin Investig (2005) 115(7):1806–15. doi: 10.1172/JCI23865
41. Díaz-Bulnes P, Saiz ML, López-Larrea C, Rodríguez RM. Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Front Immunol (2020) 10:2951. doi: 10.3389/fimmu.2019.02951
42. Fiorese CJ, Haynes CM. Integrating the UPRmt Into the Mitochondrial Maintenance Network. Crit Rev Biochem Mol Biol (2017) 52(3):304–13. doi: 10.1080/10409238.2017.1291577
43. Anderson NS, Haynes CM. Folding the Mitochondrial UPR Into the Integrated Stress Response. Trends Cell Biol (2020) 30(6):428–39. doi: 10.1016/j.tcb.2020.03.001
44. Melber A, Haynes CM. UPRmt Regulation and Output: A Stress Response Mediated by Mitochondrial-Nuclear Communication. Cell Res (2018) 28(3):281–95. doi: 10.1038/cr.2018.16
45. Papa L, Germain D. SirT3 Regulates the Mitochondrial Unfolded Protein Response. Mol Cell Biol (2014) 34(4):699–710. doi: 10.1128/MCB.01337-13
46. Jovaisaite V, Auwerx J. The Mitochondrial Unfolded Protein Response—Synchronizing Genomes. Curr Opin Cell Biol (2015) 33:74–81. doi: 10.1016/j.ceb.2014.12.003
47. Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT. Adaptor Proteins MiD49 and MiD51 can Act Independently of Mff and Fis1 in Drp1 Recruitment and are Specific for Mitochondrial Fission. J Biol Chem (2013) 288(38):27584–93. doi: 10.1074/jbc.M113.479873
48. Gao Z, Li Y, Wang F, Huang T, Fan K, Zhang Y, et al. Mitochondrial Dynamics Controls Anti-Tumour Innate Immunity by Regulating CHIP-IRF1 Axis Stability. Nat Commun (2017) 8(1):1–13. doi: 10.1038/s41467-017-01919-0
49. Detmer SA, Chan DC. Functions and Dysfunctions of Mitochondrial Dynamics. Nat Rev Mol Cell Biol (2007) 8(11):870–9. doi: 10.1038/nrm2275
50. Wang C, Youle RJ. The Role of Mitochondria in Apoptosis. Annu Rev Genet (2009) 43:95–118. doi: 10.5483/bmbrep.2008.41.1.011
51. Xu D, Cui K, Li Q, Zhu S, Zhang J, Gao S, et al. Docosahexaenoic Acid Alleviates Palmitic Acid-Induced Inflammation of Macrophages via TLR22-MAPK-Pparγ/Nrf2 Pathway in Large Yellow Croaker (Larimichthys Crocea). Antioxidants (2022) 11(4):682. doi: 10.3390/antiox11040682
52. Ding X, Liang Y, Peng W, Li R, Lin H, Zhang Y, et al. Intracellular TLR22 Acts as an Inflammation Equalizer via Suppression of NF-κb and Selective Activation of MAPK Pathway in Fish. Fish Shellfish Immunol (2018) 72:646–57. doi: 10.1016/j.fsi.2017.11.042
53. Banoth B, Cassel SL. Mitochondria in Innate Immune Signaling. Transl Res (2018) 202:52–68. doi: 10.1016/j.trsl.2018.07.014
54. Li B, Wan Z, Wang Z, Zuo J, Xu Y, Han X, et al. TLR2 Signaling Pathway Combats Streptococcus Uberis Infection by Inducing Mitochondrial Reactive Oxygen Species Production. Cells (2020) 9(2):494. doi: 10.3390/cells9020494
55. Feno S, Butera G, Vecellio Reane D, Rizzuto R, Raffaello A. Crosstalk Between Calcium and ROS in Pathophysiological Conditions. Oxid Med Cell Longev (2019). 2019:9324018. doi: 10.1155/2019/9324018
56. Cloonan SM, Choi AM. Mitochondria: Sensors and Mediators of Innate Immune Receptor Signaling. Curr Opin Microbiol (2013) 16(3):327–38. doi: 10.1016/j.mib.2013.05.005
57. Shintani Y, Drexler HC, Kioka H, Terracciano CM, Coppen SR, Imamura H, et al. Toll-Like Receptor 9 Protects non-Immune Cells From Stress by Modulating Mitochondrial ATP Synthesis Through the Inhibition of SERCA 2. EMBO Rep (2014) 15(4):438–45. doi: 10.1002/embr.201337945
58. Harrington JL, Murphy E. The Mitochondrial Calcium Uniporter: Mice can Live and Die Without it. J Mol Cell Cardiol (2015) 78:46–53. doi: 10.1016/j.yjmcc.2014.10.013
59. Hajnóczky G, Davies E, Madesh M. Calcium Signaling and Apoptosis. Biochem Biophys Res Commun (2003) 304(3):445–54. doi: 10.1016/s0006-291x(03)00616-8
60. Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting Edge: Essential Role of Hypoxia Inducible Factor-1α in Development of Lipopolysaccharide-Induced Sepsis. J Immunol (2007) 178(12):7516–9. doi: 10.4049/jimmunol.178.12.7516
61. Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, et al. HIF-1α Triggers ER Stress and CHOP-Mediated Apoptosis in Alveolar Epithelial Cells, a Key Event in Pulmonary Fibrosis. Sci Rep (2018) 8(1):1–14. doi: 10.1038/s41598-018-36063-2
62. Münch C, Harper JW. Mitochondrial Unfolded Protein Response Controls Matrix Pre-RNA Processing and Translation. Nature (2016) 534(7609):710–3. doi: 10.1038/nature18302
63. Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of Genes Activated by the Mitochondrial Unfolded Protein Response (mtUPR) and Cognate Promoter Elements. PloS One (2007) 2(9):e874. doi: 10.1371/journal.pone.0000874
64. Deng J, Wang P, Chen X, Cheng H, Liu J, Fushimi K, et al. FUS Interacts With ATP Synthase Beta Subunit and Induces Mitochondrial Unfolded Protein Response in Cellular and Animal Models. Proc Natl Acad Sci USA (2018) 115(41):E9678–86. doi: 10.1073/pnas.1806655115
65. Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. Listeria Monocytogenes Transiently Alters Mitochondrial Dynamics During Infection. Proc Natl Acad Sci USA (2011) 108(9):3612–7. doi: 10.1073/pnas.1100126108
66. Wang Q, Unwalla H, Rahman I. Dysregulation of Mitochondrial Complexes and Dynamics by Chronic Cigarette Smoke Exposure in MitoQC Reporter Mice. Mitochondrion (2022). 63:43-50. doi: 10.1016/j.mito.2022.01.003
67. Duan C, Kuang L, Hong C, Xiang X, Liu J, Li Q, et al. Mitochondrial Drp1 Recognizes and Induces Excessive mPTP Opening After Hypoxia Through BAX-PiC and LRRK2-Hk2. Cell Death Dis (2021) 12(11):1–10. doi: 10.1038/s41419-021-04343-x
68. Ma K, Chen G, Li W, Kepp O, Zhu Y, Chen Q, et al. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front Cell Dev Biol (2020) 8:467. doi: 10.3389/fcell.2020.00467
69. Sheridan C, Martin SJ. Mitochondrial Fission/Fusion Dynamics and Apoptosis. Mitochondrion (2010) 10(6):640–8. doi: 10.1016/j.mito.2010.08.005
70. Gao F, Reynolds MB, Passalacqua KD, Sexton JZ, Abuaita BH, O’Riordan MX. The Mitochondrial Fission Regulator DRP1 Controls Post-Transcriptional Regulation of TNF-α. Front Cell Infect Microbiol (2021) 10:593805. doi: 10.3389/fcimb.2020.593805
Keywords: A. hydrophila, TLR22, mtROS, UPRmt, mitochondrial fission, apoptosis
Citation: Kumar M, Sharma S, Haque M, Kumar J, Hathi UPS and Mazumder S (2022) TLR22-Induced Pro-Apoptotic mtROS Abets UPRmt-Mediated Mitochondrial Fission in Aeromonas hydrophila-Infected Headkidney Macrophages of Clarias gariepinus. Front. Immunol. 13:931021. doi: 10.3389/fimmu.2022.931021
Received: 28 April 2022; Accepted: 09 June 2022;
Published: 04 July 2022.
Edited by:
Yihong Chen, South China Normal University, ChinaReviewed by:
Usha Kumari, Banaras Hindu University, IndiaMrinal Samanta, Central Institute of Freshwater Aquaculture (ICAR), India
Copyright © 2022 Kumar, Sharma, Haque, Kumar, Hathi and Mazumder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shibnath Mazumder, shibnath@sau.int