 Ye Guo
Ye Guo Hanfei Guo
Hanfei Guo Yongfei Zhang
Yongfei Zhang Jiuwei Cui
Jiuwei Cui
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 July 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.908894
This article is part of the Research Topic Emerging Technologies and Biology for Tumor Microenvironment and Tumor-immune Interplay View all 13 articles
Alterations in the anaplastic lymphoma kinase (ALK) gene play a key role in the development of various human tumors, and targeted therapy has transformed the treatment paradigm for these oncogene-driven tumors. However, primary or acquired resistance remains a challenge. ALK gene variants (such as gene rearrangements and mutations) also play a key role in the tumor immune microenvironment. Immunotherapy targeting the ALK gene has potential clinical applications. Here, we review the results of recent studies on the immunological relevance of ALK-altered tumors, which provides important insights into the development of tumor immunotherapies targeting this large class of tumors.
Over the past few decades, the anaplastic lymphoma kinase (ALK) gene has been widely known for its role in human tumorigenesis (1). Various rearrangements (fusions), mutations, amplification, and alternative splicing of the ALK gene have been found in anaplastic large cell lymphoma (ALCL), inflammatory myofibroblastoma (IMT), non-small cell lung cancer (NSCLC), and other human tumors (2–4) (Table 1). Currently, ALK gene variants are considered drug targets for these tumors. However, primary or acquired resistance to tyrosine kinase inhibitors is almost unavoidable (5). Although immunotherapy in recent years has provided new hope for patients with a variety of tumors with poor treatment efficacy, the response of these patients with ALK gene abnormalities to immunotherapy has not been clarified. A large retrospective study showed that patients with at least one oncogenic driver alteration (RET, ROS1, EGFR, or ALK) are less likely to benefit from immune checkpoint inhibitor (ICI) monotherapy (6). Until recently, several preclinical and clinical studies suggested that ALK rearrangement may be involved in innate and adaptive immunity through various pathways and is associated with T cell activation, cytokine release, and tumor immune escape (7). In addition, chimeric antigen receptor (CAR-T) therapies and tumor vaccines targeting ALK rearrangements are under development.
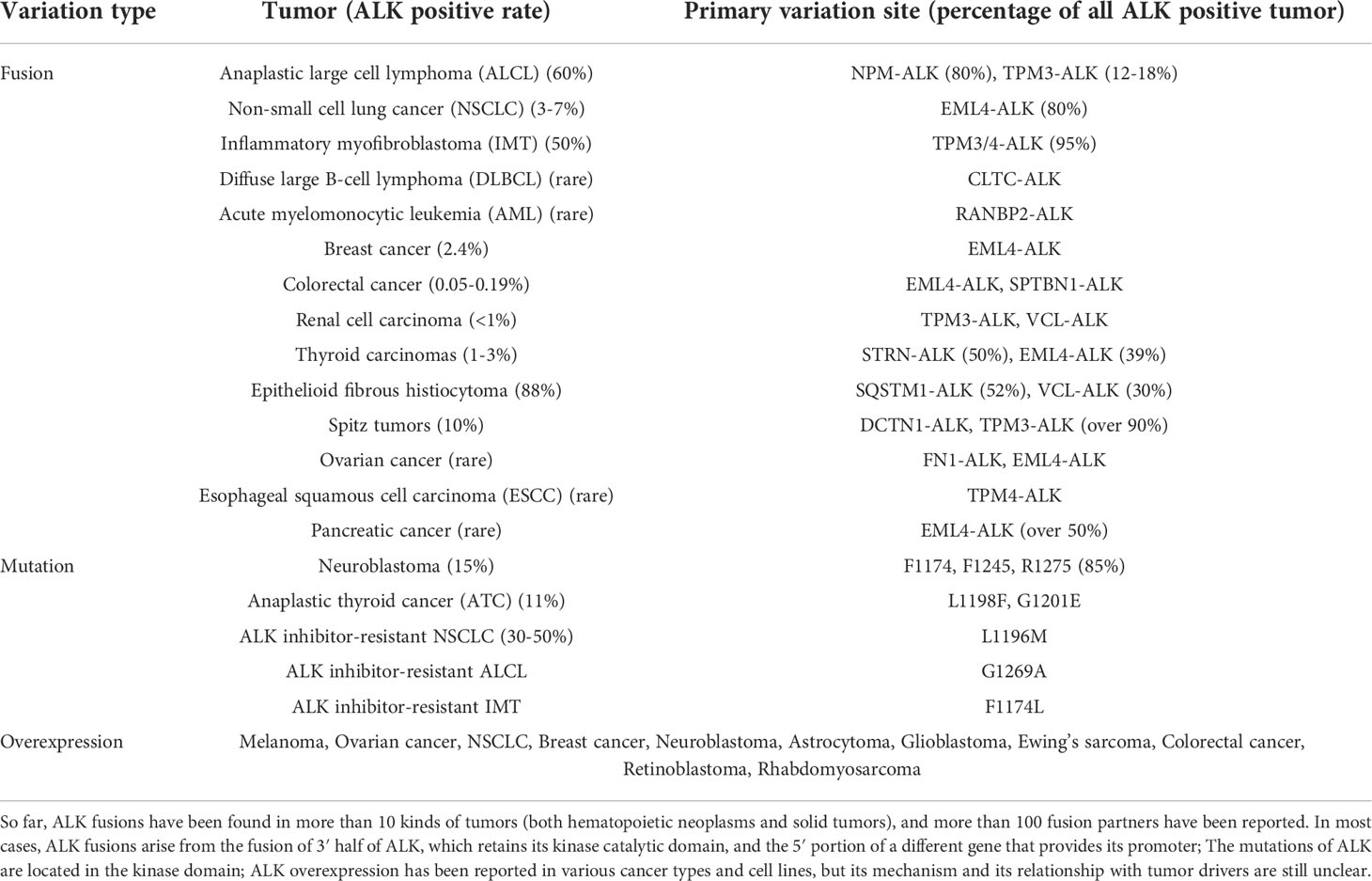
Table 1 Summary of ALK variants.
Therefore, clarifying whether preferred targeted therapy, immunotherapy, or targeted combination immunotherapy is the optimal clinical treatment strategy for such patients is important. Hence, this topic will be the focus of future research in the field of ALK-altered tumor immunotherapy. This article reviews the progress on the knowledge of ALK gene variants in the field of immunotherapy to better understand the mechanism of ALK in the human immune response and may provide new treatment strategies for patients with ALK gene variants.
ALK, consisting of 1,620 amino acids, is a member of the insulin receptor tyrosine kinase (RTK) superfamily, and its gene is located on chromosome 2p23 (8). ALK plays an important role in the growth and development of the mammalian nervous system; however, its expression decreases significantly after birth and remains at a low level in adulthood (9). The tissue expression of ALK in human adults is restricted to the brain, with minimal expression in the lung, colon, small intestine, and testis, as indicated by the expression data of the human protein atlas and several immunohistochemical studies (10). When somatic variations occur, ALK is expressed in tissues that do not originally express ALK, and as such the cells are abnormally activated, resulting in uncontrolled cell proliferation and tumor formation (2, 11). Because ALK expression is restricted to the nervous system, a highly immune-privileged organ, the ALK protein is a potential antigen for the immune system. Similarly, tumor-specific ALK fusions or mutants may also be recognized as neoantigens in the body. Thus, ALK-altered cancer cells may potentially trigger antibody responses in patients. ALK is also involved in innate immunity against microbial pathogens (12, 13). Preclinical and clinical studies have shown that upregulation of immune-related molecules, such as programmed cell death ligand-1 (PD-L1), is commonly observed in ALK-altered tumors (14, 15).
The mechanisms by which ALK-altered tumors lead to immune resistance may include affecting T cell immune responses, regulating cytokine secretion, activating immunosuppressive cells, and upregulating the expression of heterogeneous immune checkpoints (Figure 1).
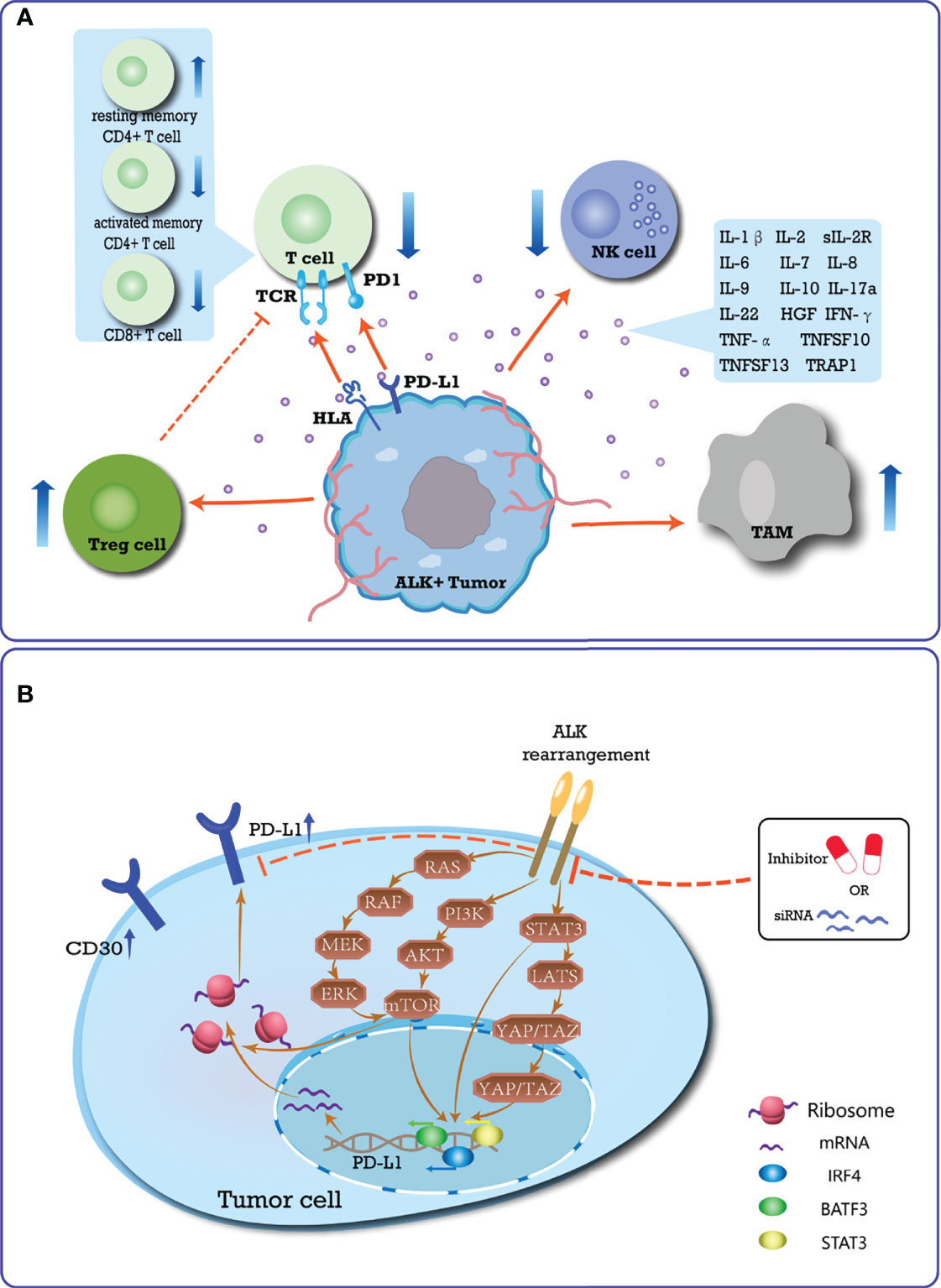
Figure 1 Summary of the immune-suppressive microenvironment induced by ALK-rearrangement. (A) Schematic diagram of the special immune TME of ALK-positive tumor. In ALK-positive tumors, CD30 is expressed continuously, and TCR signaling is inhibited. In the TME, the types of T cells changed, that is, the number of resting memory CD4+ T cells increased, while CD8+ T cells and activated memory CD4+ T cells were lacking. A variety of immunosuppressive cytokines are up-regulated, thereby inhibiting the killing ability of T cells and NK cells to tumor cells, and promoting the function of immunosuppressive cells. The special TME accumulates more Treg cells and TAM cells to promote immune evasion; (B) Mechanism of ALK rearrangement upregulating PD-L1 expression, which plays an essential role in mediating the process of PD-L1 expression. ALK-rearranged protein can activate STAT3, PI3K-AKT-mTOR, and MEK-ERK signaling networks, which upregulate PD-L1 expression through transcription factors acting on the promoter region of PD-L1 gene. Activated mTOR can also recruit PD-L1 transcripts to active polysomes at the post-transcriptional level. The JAK-STAT3-LATS-YAP/TAZ-PD-L1 signaling pathway has gradually been shown to play an important role in mediating ALK-induced upregulation of PD-L1 in multiple cancer cell lines. Conversely, blocking the activation of the ALK pathway inhibits the expression of PD-L1.
In ALK-positive ALCL patients, CD30 is continuously expressed in tumor cells. Compared with CD30- tumors, CD30+ tumors are characterized by downregulation of molecules involved in T cell differentiation/activation (including CD28, CD52, and CD69) and T cell receptor (TCR) signaling (16). CD3 and TCR are negatively expressed in more than 75% of cases, and CD8 expression is rare in T cells (17). In addition, two immunogenic ALK epitopes (P280-89 and p375-86) were identified to elicit cytotoxic T cell (CTL) responses in vitro, in vivo, and in human peripheral blood lymphocytes (PBLs) (18). The anti-ALK CTL generated from the PBL of healthy donors induces an antigen-specific HLA-A2.1 restricted response, which can effectively kill endogenous ALK-expressing tumor targets. Subsequent studies using a mouse model of vaccination identified that, in healthy donors, CD8+ T cells mainly show a naive phenotype, whereas effector and memory CD8+ T cells are detected in ALK-positive ALCL patients (19). ALK-specific CD4+ T cells are detected in HLA-preselected ALCL patients using ALK-derived peptides (20). Recent studies have shown that the in vitro transduction of normal human CD4+ T lymphocytes by NPM-ALK leads to immortalization and malignant transformation (21). Moreover, tumor cells have the morphology and immunophenotype of primary anaplastic large cell lymphoma (21). In ALK+ NSCLC patients, Jin et al. (22) found that tumors are characterized by enriched resting memory CD4+ T cells (P<0.001), as well as a lack of CD8+ T cells (P<0.01), and activated memory CD4+ T cells (P=0.001).
Various pro-inflammatory cytokines and their receptors are significantly upregulated in ALK-positive tumors, including IL-1β, IL-2, soluble IL-2 receptor (sIL-2R), IL-6, IL-7, IL-8, IL-9, IL-10, IL-17a, IL-22, interferon (IFN)-γ, TNF-α, TNFSF10, TNFSF13, hepatocyte growth factor (HGF), CD30, and TRAP1 (23–26). IL-9 and IL-22 activate oncogenic signaling via the JAK3-STAT3 pathway, and neutralizing antibodies against them may inhibit the survival and clonogenicity of ALK+ ALCL cells (27, 28). Furthermore, NPM-ALK promotes the expression of other immunosuppressive signals through the activation of STAT3, including IL-10 and transforming growth factor β (TGFβ) (29). Compared with ALK- ALCL, ALK+ ALCL patients are enriched for the expression of signatures of HIF1-α target genes, IL10-induced genes, and H-ras/K-ras induced genes (30).
Upregulation of IL6 and IL10 expression in ALK+ tumors reduce the antigen-presenting activity of dendritic cells in the TME and inhibits the function of T and NK cells (31, 32), resulting in ALK+ tumors responding to T cells and innate immunity negative effects. Upregulation of CSF1 and CCL18 expression in ALK+ tumors increase M2 tumor−associated macrophages (TAMs) in the TME that contribute to immune evasion (33–35). Previous studies have identified that ALK-mediated activation of TMEM173 (transmembrane protein 173, also known as STING) in macrophages and monocytes is related to the pathogenesis of sepsis caused by infection, and has the potential to activate macrophages and monocytes (12, 36). Recently, Jan et al. compared the immune gene expression profiles and the levels of specific immune cell populations in ALK+ and ALK- lung adenocarcinoma patients. In ALK+ tumors, the proportion of regulatory T cells was significantly increased (P < 0.0005) (35). Further analysis revealed that ALK+ tumors recruit CXCR4+ Tregs by upregulating CXCL12 and CCL22 (35, 37, 38). These studies all showed that ALK variants can activate immune suppressive cells, presenting a challenge to immune-related treatment of patients with ALK+ tumors.
Mutant ALK upregulates the expression of PD-L1, which may potentially confer an immunosuppressive TME, contributing to tolerance and immune evasion in cancer (39, 40). Marzec et al. (29) showed that, in an ALK+ ALCL cell model, NPM-ALK activates the transcription of STAT3 on the PD-L1 promoter. Using CRISPR/Cas9 library screening, Zhang et al. determined that PD-L1 induction is dependent on the NPM-ALK oncoprotein activation of STAT3, as well as a signalosome containing GRB2/SOS1, which activates the MEK-ERK and PI3K-AKT signaling pathways. These signaling networks ultimately induce PD-L1 expression through the action of the transcription factors IRF4 and BATF3 on the enhancer region of the PD-L1 gene (41). A recent clinical study conducted by the MD Anderson Cancer Center of 95 patients with ALCL showed that the positive rate of PD-L1 in ALK+ ALCL patients is higher than that in ALK- cases (76% and 42%, respectively) (42). The same phenomenon was observed in patients with ALK+ NSCLC. Both in vitro and in vivo experiments have shown that the expression level of PD-L1 is positively associated with the presence of EML4-ALK in NSCLC specimens (43–46). EML4-ALK modulates PD-L1 expression via common downstream signaling pathways mediated by PI3K-AKT-mTOR, MEK-ERK, and STAT3 (44, 47, 48). Activated mTOR recruits PD-L1 transcripts to active polysomes at the post-transcriptional level, thereby increasing the level of PD-L1 protein without significantly increasing the mRNA levels (49, 50). STAT3 increases PD-L1 transcription by directly binding to the promoter region of the CD274 gene (located at the 9p24.1 locus) (47). Recently, Nouri et al. (51) identified, through the kinome-wide screen of Hippo pathway regulators, that YAP/TAZ are critical in mediating ALK-induced upregulation of PD-L1 in multiple cancer cell lines. Moreover, ALK may cause enhanced immune evasion and tumorigenesis through the JAK-STAT3-LATS-YAP/TAZ-PD-L1 signaling pathway. Importantly, ALK inhibitors and ALK siRNAs effectively inhibit ALK fusion-induced PD-L1 expression in NSCLC cell models. These results confirmed the effect of ALK on PDL1 expression in NSCLC (44, 52).
Various preclinical and clinical efforts are underway to identify mechanisms related to the interaction of the ALK gene with the tumor immune microenvironment. ICIs targeting programmed cell death ligand-1 (PD-1), PD-L1, and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) are currently the most advanced immunotherapies and have transformed the treatment paradigm for a variety of tumors, including lung cancer. However, there is no firm conclusion regarding the therapeutic effect of ICIs in patients with ALK-altered tumors. Research on tumor vaccines and chimeric antigen receptor T-Cell (CAR-T cell) therapy targeting ALK are also underway (Table 2).
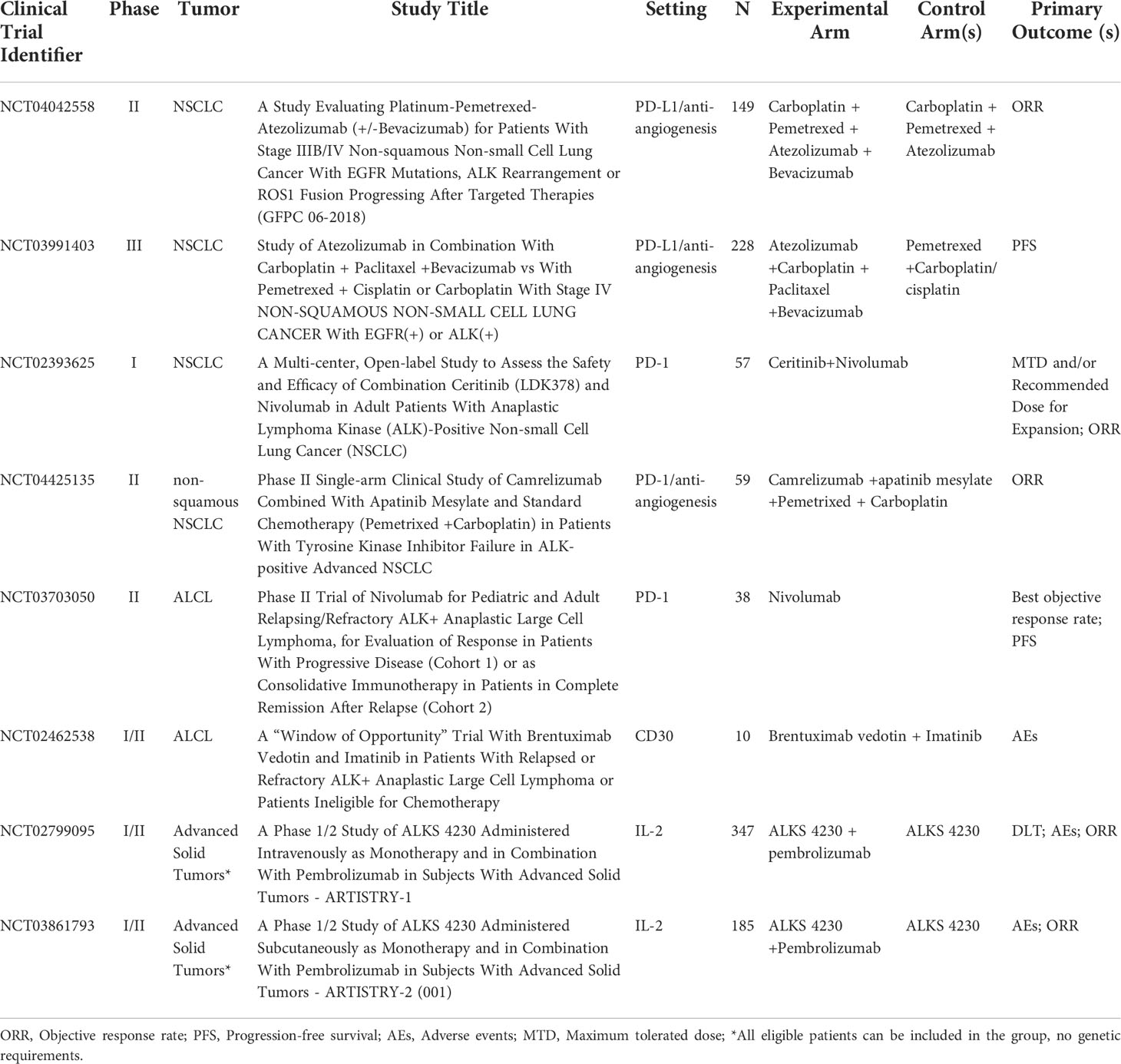
Table 2 Summary of ongoing trials with immunotherapy in ALK+ tumors (source: www.clinicaltrials.gov, last accessed: 30 Mar 2022).
In recent years, ICIs have shown remarkable therapeutic effects in various tumors. Moreover, as mentioned above, ALK variants induce the upregulation of PD-L1 expression in ALK-positive tumors. Based on these findings and in vitro drug trials, some scholars have speculated that anti-PD-1/PD-L1 therapy may be a promising option for NSCLC patients with upregulated PD-L1 carrying the EML4-ALK fusion gene (53). However, whether the high expression of PD-L1 affects the prognosis of ALK+ patients remains inconclusive, and further research is needed (42, 54).
Data from prior randomized studies indicate that immunotherapies are less effective in patients with ALK+ tumors than in those with wild-type tumors, regardless of PD-L1 expression level (55, 56). In a global “real world” study, Mazieres et al. (6) retrospectively analyzed ALK+ NSCLC patients from 10 countries and found that the objective response rate is 0% using ICI monotherapy. The proportion of ALK+ patients who experienced rapid progression within 2 months was 45.5%, which was much higher than that of patients with the wild-type gene. More recently, a multicenter retrospective study showed limited activity in patients with stage III unresectable NSCLC with driver genomic alterations treated with durvalumab (PD-L1 inhibitor) after chemoradiotherapy, especially in the ALK rearrangement subgroup. The median progression-free survival (PFS) was not reached (11.3-NR) in the KRAS-mutation vs. 8.1 month in the EGFR-mutation vs. 7.8 month in the BRAF-mutation/ALK rearrangement (P = 0.02) (57). Therefore, current research on ALK-positive patients has mainly focused on ALK inhibitor resistance (58). For patients with NSCLC, the ATLANTIC trial established an independent cohort of EGFR+/ALK+ patients to evaluate durvalumab as a third line or later treatment. The proportion of patients who achieved a response was generally lower in the cohort of patients with EGFR+/ALK+ NSCLC than in those with EGFR−/ALK− NSCLC. Nevertheless, the proportion of EGFR+/ALK+ patients with at least 25% of tumor cells expressing PD-L1 who achieved an objective response was not substantially lower than that in EGFR−/ALK− patients (12.2% vs 16.4%) (59). Recently, there was a report of a case of a 48-year-old man with ALK+ NSCLC who displayed a complete response for 16 months to nivolumab (PD-1 inhibitor) therapy in a third line setting after ceritinib (second-generation ALK inhibitor) and platin-based chemotherapy (60). Another case report showed that patients with ALK+ ALCL (PD-L1 positive) who were refractory to chemotherapy and ALK inhibitors demonstrated prolonged responses to nivolumab (61, 62). Further clinical trials are needed to verify the effectiveness of ICIs in patients with ALK+ ALCL.
Some studies have analyzed the reasons for the poor effects of ICIs. A majority of ALK-positive NSCLCs lack concurrent PD-L1 expression and high levels of CD8+ tumor infiltrating lymphocytes (TILs) (63). The combined analyses of PD-L1 and CD8+ TILs show a remarkably higher proportion of PD-L1-/TIL- tumors and a lower proportion of PD-L1+/TIL+ tumors in ALK+ groups than in wild-type patients (P = 0.001), suggesting an uninflamed phenotype with immunological ignorance (22). Although a significant number of PD-1 positive CD8+ T cells were found in the ALK-positive tumor bed in early lung adenocarcinoma (64), these PD-1 expressing CD8+ T cells were functionally impaired (65) and did not express interferon-γ mRNA, which could upregulate PD-L1 expression in tumor cells (66, 67). These results indicate that the ALK-positive TME suppresses the immune function of CD8+ TILs through a PD-1/PD-L1 independent mechanism, which might lead to the inability of ALK-positive tumors to respond to PD-1/PD-L1-based immunotherapy (64). Tumor mutational burden (TMB) is an effective marker for predicting the efficacy of ICI treatment. The median TMB of ALK-positive tumor samples is only 2.29 mutations/Mb (ranging from 0.76 to 16.79 mutations/Mb) (68). The TMB (in mutations/Mb) of NSCLC patients with alteration in ALK is significantly lower than in those without (2.1 vs 7.0 mutations/Mb; P < 0.001) (69). These results suggest that the limited benefits of ICI monotherapy are attributable to the low levels of functional CD8+ TILs and TMB.
A preclinical study showed that in vitro application of ceritinib combined with a PD-L1 inhibitor in the treatment of ALK-rearranged NSCLC promotes lymphocyte proliferation and activation, inhibits PD-L1 expression, and enhances lymphocyte cytotoxicity and cell death. In the in vivo xenograft model, tumor volumes treated with a combination of ceritinib and a PD-L1 inhibitor (91.9%) are significantly smaller than those treated with ceritinib (84.9%) or PD-L1 (20.0%) alone (70). Some clinical trials have explored the use of ICIs in combination with ALK inhibitors (71, 72). The primary study was a phase 1/2 study (CheckMate 370) on the safety and tolerability of nivolumab plus crizotinib (first-generation ALK inhibitor) as a first-line treatment for patients with advanced ALK+ NSCLC. The high proportion (38%) of severe hepatotoxicity caused the trial to close prematurely and fail (73). Another phase Ib study evaluated the safety and preliminary antitumor activity of crizotinib plus pembrolizumab (PD-1 inhibitor) as a first-line therapy in patients with ALK+ NSCLC. Although this combination showed antitumor activity, the incidence of dose-limiting toxicities is high, especially with a higher frequency of severe transaminase level increase. Because the study was terminated early, the recommended phase II dose could not be determined (74). Therefore, for a well-designed trial, selecting a suitable combination of partner and treatment population is extremely important. Felip et al. (75) presented the results of a phase Ib trial examining ceritinib plus nivolumab in previously treated or treatment-naive ALK+ NSCLC. This combination appears to elicit activity, and high PD-L1 expression may be enriched in patients more likely to respond. Based on more toxicity findings, especially rash, a protocol amendment to switch to sequential treatment is being investigated in which ceritinib is administered as monotherapy for two cycles before combining it with nivolumab. Two additional phase Ib studies presented at ASCO meeting show promising efficacy and acceptable safety profile of this sequential therapy. In previously treated ALK+ NSCLC, the combination of avelumab (anti-PD-L1) and lorlatinib (third-generation ALK inhibitor) showed no dose-limiting toxicity (76). In treatment-naive ALK+ NSCLC, alectinib (second-generation ALK inhibitor) should be administered 1 week prior to combination with atezolizumab (PD-L1 inhibitor). The objective response rate was 81% (95% CI 58.1–94.6), with a median PFS of 21.7 months and a median DOR of 20.3 months (77). In addition, Chalmers et al. presented a phase I trial of a combination of ipilimumab (a CTLA-4 inhibitor) and crizotinib in ALK+ NSCLC. The median PFS and overall survival (OS) were prolonged, but owing to the small number of enrolled cases (three cases), continued observation was necessary (78). Although a particularly large advantage in ORR was not observed in most combination therapies, given the long-term benefits of ICIs treatment, it remains to be seen whether PFS and OS outcomes can be prolonged in the future.
In the IMpower130 study, for ALK inhibitor-pretreated patients with ALK-sensitizing alterations, atezolizumab plus chemotherapy did not show improved overall survival versus chemotherapy alone (79). However, data from the IMpower150 study showed that the addition of atezolizumab to bevacizumab (angiogenesis inhibitor) plus chemotherapy resulted in significant improvements in PFS and OS (80). In IMpower150, the median PFS for patients with EGFR+/ALK+ status in the atezolizumab plus bevacizumab and chemotherapy (ABCP) group was 9.7 months compared with the PFS of 6.1 months in the bevacizumab plus chemotherapy (BCP) group (HR 0.59, 95% CI, 0.37–0.94). OS data were immature (not reached vs. 17.5 months; HR, 0.54; 95% 0.29–1.03). The 6- and 12-month PFS rates in the ABCP group were 65% and 37%, respectively, compared to 53% and 21% in the BCP group (80, 81). Therefore, after ALK inhibitor resistance, ABCP may be the first choice for patients with ALK+ NSCLC who are still capable of tolerating intensive therapy. The combination of ICIs and anti-vascular endothelial growth factor (VEGF) agents has significantly improved clinical outcomes in a variety of tumors compared with standard treatments (82). Multiple studies have further analyzed the synergistic mechanism between angiogenic factors such as VEGF and PD-(L)1 inhibitors, which is attributed to VEGF-mediated immunosuppression in the TME (83, 84). In addition to inducing vascular abnormalities, angiogenic factors also suppress antigen presentation and immune effector cells or augment the immunosuppressive activity of regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages (85–88). In the PI3K/AKT/mTOR pathway, ALK signaling promotes VEGF expression in tumors, which might enhance the sensitivity of ALK+ patients to bevacizumab (89). In ALK+ patients, CD8+ T cell tumor infiltration decreases (84) and regulatory T cells increase (90) after ALK inhibitor treatment, which induces a lower response rate to ICIs. In several clinical biomarker studies, the combination of bevacizumab and atezolizumab has been proven to overcome ICIs resistance by reversing VEGF-mediated immunosuppression and promoting CD8+ TIL in tumors (91–93). There are also reports that bevacizumab combined with targeted therapy can overcome ALK inhibitor resistance (94, 95). A recent study showed that VEGFR2 inhibition, a promising treatment strategy for oncogene-driven NSCLC, not only inhibits tumor angiogenesis but also exerts direct antiproliferative effects on cancer cells (96). In summary, it can be inferred that ICIs combined with anti-angiogenesis may be a promising treatment method.
Owing to the characteristics of ALK expression in the body, it has long been considered a potential tumor-associated antigen (TAA) (97). There are immunogenic regions located in the ALK kinase domain that can trigger specific T cell responses restricted by HLA alleles (98, 99). These findings provide a basis for peptide vaccine immunotherapy for ALK-driven tumors.
Using an ALK+ ALCL mouse model, Chiarle et al. showed that DNA vaccines with plasmids encoding a part of the ALK cytoplasmic domain elicit ALK-specific interferon-gamma responses and CD8+ T cell-mediated cytotoxicity. The combination of chemotherapy and ALK DNA vaccination significantly enhances the survival of mice challenged with ALK+ lymphomas (100). In mouse models of ALK+ NSCLC, this ALK DNA vaccine induced strong systemic and intratumoral immune responses, significantly reducing tumor growth and extending the survival of treated mice. The combination of this vaccine and ALK TKI is also effective and significantly delayed tumor relapse after TKI treatment. In addition, immunotherapies, such as anti-PD-1/PD-L1 or anti-CTLA, can be used to enhance the benefits of ALK TKI and ALK vaccine combination therapy (101). Another ALK vaccine is based on ALK-overlapping peptides in splenocytes from ALK-vaccinated mice. The vaccine significantly delayed the progression of primary lung tumors in EML4-ALK transgenic mice (102). One of the technologies under study is the use of stabilized multilamellar lipid vesicles with cross-linked lipid bilayers containing an antigenic ALK variant. They can deliver antigens alone in the presence of adjuvants to form an efficient vaccine for ALK-positive glioblastomas (103). Recently, an in vitro test applied a novel anti-epidermal growth factor vaccine (anti-EGF VacAbs) in ALK+ NSCLC cell lines. The anti-EGF VacAbs target the B-cells to generate antibodies that neutralize circulating EGF, thus preventing its binding to EGFR. They potentiate the antitumor effects of ALK-TKIs, significantly enhancing the blockade of downstream oncogenic activation pathways, and delaying the emergence of resistance (104). These experimental results provide a powerful strategy for the treatment of ALK-driven tumors. With the continuous progress in its research, ALK vaccines will soon enter clinical trials.
T cells engineered to express chimeric antigen receptors (CARs) have demonstrated significant activity against many tumors, and CAR-T cells have recently joined a rapidly growing repertoire of immunotherapeutics. Because ALK fusion protein is mainly expressed inside the cell, CAR-T therapy targeting ALK is currently mainly tested in neuroblastoma. It has been found that T cells expressing a CAR incorporating the single-chain variable fragment against the ALK extracellular domain lyse ALK-positive neuroblastoma cell lines. However, CAR functionality is regulated by target antigen and CAR density, and low expression of either contributes to the limited anti-tumor efficacy of ALK CAR-T (105, 106). More specific immunotherapies targeting ALCL surface markers include anti-CD30 CAR-T cells. CD30-specific CAR-T cells have been tested in mouse models and clinical trials have been initiated (107). In one case report, a patient with relapsed ALK+ ALCL achieved remission after CD30-specific CAR-T cell treatment (108). Another trial under investigation is the induction of an immunologic response in a tumor patient using mature dendritic cells transfected with a nucleic acid composition encoding NPM-ALK as a tumor antigen and loaded with a corresponding tumor antigen composition (103).
With the revolutionary breakthroughs in the field of TCR therapy in recent years, an increasing number of ALK epitopes/peptides may become suitable targets for directed immunotherapy (109, 110). An ongoing study is screening for autologous or allogeneic T cell receptor-transgenic T cells to test against ALK+/- patient-derived and cancer cell lines using in vitro and in vivo models to assess the potential utility of cytotoxic TCR-directed immunotherapies (111).
In summary, ALK variants play an important role in a variety of tumors, including both hematological and solid tumors. The development and application of ALK inhibitors have made outstanding contributions to the treatment of ALK+ tumor patients, and it is still the main choice for first-line treatment (112). However, to date, resistance to ALK inhibitors has proven unavoidable in all cases (113). For TKIs resistant patients, the exploration of immunotherapy is currently a promising treatment direction. According to the special immunosuppressive microenvironment of ALK+ tumors, there are still huge challenges in the development and application of immunotherapeutic interventions. Based on the results of current clinical studies, ICIs monotherapy is not the preferred treatment option for TKI-resistant patients. We urgently need to explore better combined treatment options to change tumor immunosuppression to control tumors (114), such as immunotherapy combined with targeted therapy or anti-angiogenesis therapy. Nevertheless, there are still many obstacles in the process of exploration, including the understanding of the specific effects of ALK on the immune microenvironment and development of novel immunotherapy methods. Numerous studies are exploring new treatments and ways to optimize the application of immunotherapy, which may lead to greater survival benefits for the patients (Table 2).
YG carried out the primary literature search, drafted and revised the manuscript. HG and YZ helped modify the manuscript and participated in discussions. JC conceived and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (No. 81874052) and Jilin Scientific and Technological Development Program (CN) (No. 20190303146SF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer (2008) 8(1):11–23. doi: 10.1038/nrc2291
2. Hallberg B, Palmer RH. The role of the alk receptor in cancer biology. Ann Oncol (2016) 27 Suppl 3:iii4–iii15. doi: 10.1093/annonc/mdw301
3. Cao Z, Gao Q, Fu M, Ni N, Pei Y, Ou WB. Anaplastic lymphoma kinase fusions: Roles in cancer and therapeutic perspectives. Oncol Lett (2019) 17(2):2020–30. doi: 10.3892/ol.2018.9856
4. Hallberg B, Palmer RH. Mechanistic insight into alk receptor tyrosine kinase in human cancer biology. Nat Rev Cancer (2013) 13(10):685–700. doi: 10.1038/nrc3580
5. Cameron LB, Hitchen N, Chandran E, Morris T, Manser R, Solomon BJ, et al. Targeted therapy for advanced anaplastic lymphoma kinase (Alk)-rearranged non-small cell lung cancer. Cochrane Database Syst Rev (2022) 1:CD013453. doi: 10.1002/14651858.CD013453.pub2
6. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the immunotarget registry. Ann Oncol (2019) 30(8):1321–8. doi: 10.1093/annonc/mdz167
7. Sankar K, Nagrath S, Ramnath N. Immunotherapy for alk-rearranged non-small cell lung cancer: Challenges inform promising approaches. Cancers (2021) 13(6):1467. doi: 10.3390/cancers13061476
8. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell (2010) 141(7):1117–34. doi: 10.1016/j.cell.2010.06.011
9. Mao R, Zhang X, Kong Y, Wu S, Huo HQ, Kong Y, et al. Transcriptome regulation by oncogenic alk pathway in mammalian cortical development revealed by single-cell rna sequencing. Cereb Cortex (2021) 31(8):3911–24. doi: 10.1093/cercor/bhab058
10. Roskoski R Jr. Anaplastic lymphoma kinase (Alk): Structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res (2013) 68(1):68–94. doi: 10.1016/j.phrs.2012.11.007
11. Reshetnyak AV, Rossi P, Myasnikov AG, Sowaileh M, Mohanty J, Nourse A, et al. Mechanism for the activation of the anaplastic lymphoma kinase receptor. Nature (2021) 600(7887):153–7. doi: 10.1038/s41586-021-04140-8
12. Zeng L, Kang R, Zhu S, Wang X, Cao L, Wang H, et al. Alk is a therapeutic target for lethal sepsis. Sci Transl Med (2017) 9(412):eaan5689. doi: 10.1126/scitranslmed.aan5689
13. Damm-Welk C, Siddiqi F, Fischer M, Hero B, Narayanan V, Camidge DR, et al. Anti-alk antibodies in patients with alk-positive malignancies not expressing npm-alk. J Cancer (2016) 7(11):1383–7. doi: 10.7150/jca.15238
14. Glorieux C, Xia X, Huang P. The role of oncogenes and redox signaling in the regulation of pd-L1 in cancer. Cancers (2021) 13(17):4426. doi: 10.3390/cancers13174426
15. Wang L, Lui VWY. Emerging roles of alk in immunity and insights for immunotherapy. Cancers (2020) 12(2):4426. doi: 10.3390/cancers12020426
16. Bisig B, de Reynies A, Bonnet C, Sujobert P, Rickman DS, Marafioti T, et al. Cd30-positive peripheral T-cell lymphomas share molecular and phenotypic features. Haematologica (2013) 98(8):1250–8. doi: 10.3324/haematol.2012.081935
17. Malcolm TI, Villarese P, Fairbairn CJ, Lamant L, Trinquand A, Hook CE, et al. Anaplastic Large cell lymphoma arises in thymocytes and requires transient tcr expression for thymic egress. Nat Commun (2016) 7:10087. doi: 10.1038/ncomms10087
18. Passoni L, Scardino A, Bertazzoli C, Gallo B, Coluccia AM, Lemonnier FA, et al. Alk as a novel lymphoma-associated tumor antigen: Identification of 2 hla-A2.1-Restricted Cd8+ T-cell epitopes. Blood (2002) 99(6):2100–6. doi: 10.1182/blood.v99.6.2100
19. Passoni L, Gallo B, Biganzoli E, Stefanoni R, Massimino M, Di Nicola M, et al. In vivo T-cell immune response against anaplastic lymphoma kinase in patients with anaplastic Large cell lymphomas. Haematologica (2006) 91(1):48–55.
20. Ait-Tahar K, Barnardo MC, Pulford K. Cd4 T-helper responses to the anaplastic lymphoma kinase (Alk) protein in patients with alk-positive anaplastic Large-cell lymphoma. Cancer Res (2007) 67(5):1898–901. doi: 10.1158/0008-5472.CAN-06-4427
21. Congras A, Hoareau-Aveilla C, Caillet N, Tosolini M, Villarese P, Cieslak A, et al. Alk-transformed mature T lymphocytes restore early thymus progenitor features. J Clin Invest (2020) 130(12):6395–408. doi: 10.1172/JCI134990
22. Jin R, Liu C, Zheng S, Wang X, Feng X, Li H, et al. Molecular heterogeneity of anti-Pd-1/Pd-L1 immunotherapy efficacy is correlated with tumor immune microenvironment in East Asian patients with non-small cell lung cancer. Cancer Biol Med (2020) 17(3):768–81. doi: 10.20892/j.issn.2095-3941.2020.0121
23. Janik JE, Morris JC, Pittaluga S, McDonald K, Raffeld M, Jaffe ES, et al. Elevated serum-soluble interleukin-2 receptor levels in patients with anaplastic Large cell lymphoma. Blood (2004) 104(10):3355–7. doi: 10.1182/blood-2003-11-3922
24. Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N, et al. Mir-135b mediates npm-Alk-Driven oncogenicity and renders il-17-Producing immunophenotype to anaplastic Large cell lymphoma. Blood (2011) 118(26):6881–92. doi: 10.1182/blood-2011-05-354654
25. Yu L, Yan LL, Yang SJ. Sarcomatoid variant of alk- anaplastic Large cell lymphoma involving multiple lymph nodes and both lungs with production of proinflammatory cytokines: Report of a case and review of literature. Int J Clin Exp Pathol (2014) 7(8):4806–16.
26. Knorr F, Damm-Welk C, Ruf S, Singh VK, Zimmermann M, Reiter A, et al. Blood cytokine concentrations in pediatric patients with anaplastic lymphoma kinase-positive anaplastic Large cell lymphoma. Haematologica (2018) 103(3):477–85. doi: 10.3324/haematol.2017.177972
27. Qiu L, Lai R, Lin Q, Lau E, Thomazy DM, Calame D, et al. Autocrine release of interleukin-9 promotes Jak3-dependent survival of alk+ anaplastic Large-cell lymphoma cells. Blood (2006) 108(7):2407–15. doi: 10.1182/blood-2006-04-020305
28. Bard JD, Gelebart P, Anand M, Amin HM, Lai R. Aberrant expression of il-22 receptor 1 and autocrine il-22 stimulation contribute to tumorigenicity in alk+ anaplastic Large cell lymphoma. Leukemia (2008) 22(8):1595–603. doi: 10.1038/leu.2008.129
29. Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase Npm/Alk induces through Stat3 expression of immunosuppressive protein Cd274 (Pd-L1, B7-H1). Proc Natl Acad Sci USA (2008) 105(52):20852–7. doi: 10.1073/pnas.0810958105
30. Iqbal J, Wright G, Wang C, Rosenwald A, Gascoyne RD, Weisenburger DD, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood (2014) 123(19):2915–23. doi: 10.1182/blood-2013-11-536359
31. Liu C, Yang L, Xu H, Zheng S, Wang Z, Wang S, et al. Systematic analysis of il-6 as a predictive biomarker and desensitizer of immunotherapy responses in patients with non-small cell lung cancer. BMC Med (2022) 20(1):187. doi: 10.1186/s12916-022-02356-7
32. Trinchieri G. Interleukin-10 production by effector T cells: Th1 cells show self control. J Exp Med (2007) 204(2):239–43. doi: 10.1084/jem.20070104
33. Li Z, Wang YJ, Zhou J, Umakoshi M, Goto A. The prognostic role of M2 tumor-associated macrophages in non-Small-Cell lung cancer. Histol Histopathol (2022), 18474. doi: 10.14670/HH-18-474
34. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (Csf1r) inhibitors in cancer therapy. J Immunother Cancer (2017) 5(1):53. doi: 10.1186/s40425-017-0257-y
35. Budczies J, Kirchner M, Kluck K, Kazdal D, Glade J, Allgauer M, et al. Deciphering the immunosuppressive tumor microenvironment in alk- and egfr-positive lung adenocarcinoma. Cancer Immunol Immunother (2022) 71(2):251–65. doi: 10.1007/s00262-021-02981-w
36. Zhang B, Wei W, Qiu J. Alk is required for Nlrp3 inflammasome activation in macrophages. Biochem Biophys Res Commun (2018) 501(1):246–52. doi: 10.1016/j.bbrc.2018.04.226
37. Martinenaite E, Munir Ahmad S, Hansen M, Met O, Westergaard MW, Larsen SK, et al. Ccl22-specific T cells: Modulating the immunosuppressive tumor microenvironment. Oncoimmunology (2016) 5(11):e1238541. doi: 10.1080/2162402X.2016.1238541
38. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi: 10.1038/nri1806
39. Panjwani PK, Charu V, DeLisser M, Molina-Kirsch H, Natkunam Y, Zhao S. Programmed death-1 ligands pd-L1 and pd-L2 show distinctive and restricted patterns of expression in lymphoma subtypes. Hum Pathol (2018) 71:91–9. doi: 10.1016/j.humpath.2017.10.029
40. Andorsky DJ, Yamada RE, Said J, Pinkus GS, Betting DJ, Timmerman JM. Programmed death ligand 1 is expressed by non-Hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin Cancer Res (2011) 17(13):4232–44. doi: 10.1158/1078-0432.CCR-10-2660
41. Zhang JP, Song Z, Wang HB, Lang L, Yang YZ, Xiao W, et al. A novel model of controlling pd-L1 expression in alk(+) anaplastic Large cell lymphoma revealed by crispr screening. Blood (2019) 134(2):171–85. doi: 10.1182/blood.2019001043
42. Shen J, Li S, Medeiros LJ, Lin P, Wang SA, Tang G, et al. Pd-L1 expression is associated with alk positivity and Stat3 activation, but not outcome in patients with systemic anaplastic Large cell lymphoma. Mod Pathol (2020) 33(3):324–33. doi: 10.1038/s41379-019-0336-3
43. D’Incecco A, Andreozzi M, Ludovini V, Rossi E, Capodanno A, Landi L, et al. Pd-1 and pd-L1 expression in molecularly selected non-Small-Cell lung cancer patients. Br J Cancer (2015) 112(1):95–102. doi: 10.1038/bjc.2014.555
44. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of pd-L1 expression by the Eml4-alk oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin Cancer Res (2015) 21(17):4014–21. doi: 10.1158/1078-0432.CCR-15-0016
45. Yoneshima Y, Ijichi K, Anai S, Ota K, Otsubo K, Iwama E, et al. Pd-L1 expression in lung adenocarcinoma harboring egfr mutations or alk rearrangements. Lung Cancer (2018) 118:36–40. doi: 10.1016/j.lungcan.2018.01.024
46. Ma L, Lv J, Dong Y, Zhang X, Li X, Zhang H, et al. Pd-L1 expression and its regulation in lung adenocarcinoma with alk translocation. Interdiscip Sci (2019) 11(2):266–72. doi: 10.1007/s12539-019-00331-0
47. Koh J, Jang JY, Keam B, Kim S, Kim MY, Go H, et al. Eml4-alk enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma Via hypoxia-inducible factor (Hif)-1alpha and Stat3. Oncoimmunology (2016) 5(3):e1108514. doi: 10.1080/2162402X.2015.1108514
48. Hu ZY, Huang WY, Zhang L, Huang B, Chen SC, Li XL. Expression of akt and p-akt protein in lung adenocarcinoma and its correlation with pd-L1 protein and prognosis. Ann Transl Med (2020) 8(18):1172. doi: 10.21037/atm-20-5865
49. Sumimoto H, Takano A, Teramoto K, Daigo Y. Ras-Mitogen-Activated protein kinase signal is required for enhanced pd-L1 expression in human lung cancers. PLoS One (2016) 11(11):e0166626. doi: 10.1371/journal.pone.0166626
50. Lamberti G, Sisi M, Andrini E, Palladini A, Giunchi F, Lollini PL, et al. The mechanisms of pd-L1 regulation in non-Small-Cell lung cancer (Nsclc): Which are the involved players? Cancers (2020) 12(11):3129. doi: 10.3390/cancers12113129
51. Nouri K, Azad T, Lightbody E, Khanal P, Nicol CJ, Yang X. A kinome-wide screen using a nanoluc lats luminescent biosensor identifies alk as a novel regulator of the hippo pathway in tumorigenesis and immune evasion. FASEB J (2019) 33(11):12487–99. doi: 10.1096/fj.201901343R
52. Kim SJ, Kim S, Kim DW, Kim M, Keam B, Kim TM, et al. Alterations in pd-L1 expression associated with acquisition of resistance to alk inhibitors in alk-rearranged lung cancer. Cancer Res Treat (2019) 51(3):1231–40. doi: 10.4143/crt.2018.486
53. Hong S, Chen N, Fang W, Zhan J, Liu Q, Kang S, et al. Upregulation of pd-L1 by Eml4-alk fusion protein mediates the immune escape in alk positive nsclc: Implication for optional anti-Pd-1/Pd-L1 immune therapy for alk-tkis sensitive and resistant nsclc patients. Oncoimmunology (2016) 5(3):e1094598. doi: 10.1080/2162402X.2015.1094598
54. Xie W, Medeiros LJ, Li S, Yin CC, Khoury JD, Xu J. Pd-1/Pd-L1 pathway and its blockade in patients with classic Hodgkin lymphoma and non-Hodgkin Large-cell lymphomas. Curr Hematol Malig Rep (2020) 15(4):372–81. doi: 10.1007/s11899-020-00589-y
55. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-Small-Cell lung cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
56. Peters S, Gettinger S, Johnson ML, Janne PA, Garassino MC, Christoph D, et al. Phase ii trial of atezolizumab as first-line or subsequent therapy for patients with programmed death-ligand 1-selected advanced non-Small-Cell lung cancer (Birch). J Clin Oncol (2017) 35(24):2781–9. doi: 10.1200/JCO.2016.71.9476
57. Riudavets M, Auclin E, Mosteiro M, Dempsey N, Majem M, Lobefaro R, et al. Durvalumab consolidation in patients with unresectable stage III non-small cell lung cancer with driver genomic alterations. Eur J Cancer (2022) 167:142–8. doi: 10.1016/j.ejca.2022.02.014
58. Xin Yu J, Hodge JP, Oliva C, Neftelinov ST, Hubbard-Lucey VM, Tang J. Trends in clinical development for pd-1/Pd-L1 inhibitors. Nat Rev Drug Discov (2020) 19(3):163–4. doi: 10.1038/d41573-019-00182-w
59. Garassino MC, Cho BC, Kim JH, Mazières J, Vansteenkiste J, Lena H, et al. Durvalumab as third-line or later treatment for advanced non-Small-Cell lung cancer (Atlantic): An open-label, single-arm, phase 2 study. Lancet Oncol (2018) 19(4):521–36. doi: 10.1016/s1470-2045(18)30144-x
60. Baldacci S, Gregoire V, Patrucco E, Chiarle R, Jamme P, Wasielewski E, et al. Complete and prolonged response to anti-Pd1 therapy in an alk rearranged lung adenocarcinoma. Lung Cancer (2020) 146:366–9. doi: 10.1016/j.lungcan.2020.05.008
61. Hebart H, Lang P, Woessmann W. Nivolumab for refractory anaplastic Large cell lymphoma: A case report. Ann Intern Med (2016) 165(8):607–8. doi: 10.7326/L16-0037
62. Rigaud C, Abbou S, Minard-Colin V, Geoerger B, Scoazec JY, Vassal G, et al. Efficacy of nivolumab in a patient with systemic refractory alk+ anaplastic Large cell lymphoma. Pediatr Blood Cancer (2018) 65(4):10.1002/pbc.26902. doi: 10.1002/pbc.26902
63. Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, et al. Egfr mutations and alk rearrangements are associated with low response rates to pd-1 pathway blockade in non-small cell lung cancer: A retrospective analysis. Clin Cancer Res (2016) 22(18):4585–93. doi: 10.1158/1078-0432.CCR-15-3101
64. Zeng C, Gao Y, Xiong J, Lu J, Yang J, Wang X, et al. Tumor-infiltrating Cd8(+) T cells in alk-positive lung cancer are functionally impaired despite the absence of pd-L1 on tumor cells. Lung Cancer (2020) 150:139–44. doi: 10.1016/j.lungcan.2020.10.009
65. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific Cd8 T cells infiltrating the tumor express high levels of pd-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792
66. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16(5):275–87. doi: 10.1038/nrc.2016.36
67. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating pd-L1 and pd-L2 expression. Cell Rep (2017) 19(6):1189–201. doi: 10.1016/j.celrep.2017.04.031
68. Liu S, Huang T, Liu M, He W, Zhao Y, Yang L, et al. The genomic characteristics of alk fusion positive tumors in Chinese nsclc patients. Front Oncol (2020) 10:726. doi: 10.3389/fonc.2020.00726
69. Singal G, Miller PG, Agarwala V, Li G, Kaushik G, Backenroth D, et al. Association of patient characteristics and tumor genomics with clinical outcomes among patients with non-small cell lung cancer using a clinicogenomic database. JAMA (2019) 321(14):1391–9. doi: 10.1001/jama.2019.3241
70. Du P, Hu T, An Z, Li P, Liu L. In vitro and in vivo synergistic efficacy of ceritinib combined with programmed cell death ligand-1 inhibitor in anaplastic lymphoma kinase-rearranged non-Small-Cell lung cancer. Cancer Sci (2020) 111(6):1887–98. doi: 10.1111/cas.14397
71. Moya-Horno I, Viteri S, Karachaliou N, Rosell R. Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (Nsclc). Ther Adv Med Oncol (2018) 10:1758834017745012. doi: 10.1177/1758834017745012
72. Patel M, Jabbour SK, Malhotra J. Alk inhibitors and checkpoint blockade: A cautionary tale of mixing oil with water? J Thorac Dis (2018) 10(Suppl 18):S2198–S201. doi: 10.21037/jtd.2018.06.118
73. Spigel DR, Reynolds C, Waterhouse D, Garon EB, Chandler J, Babu S, et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation - positive advanced non-small cell lung cancer (Checkmate 370). J Thorac Oncol (2018) 13(5):682–8. doi: 10.1016/j.jtho.2018.02.022
74. Patel SP, Pakkala S, Pennell NA, Reckamp KL, Lanzalone S, Polli A, et al. Phase ib study of crizotinib plus pembrolizumab in patients with previously untreated advanced non-small cell lung cancer with alk translocation. Oncologist (2020) 25(7):562–e1012. doi: 10.1634/theoncologist.2020-0034
75. Felip E, de Braud FG, Maur M, Loong HH, Shaw AT, Vansteenkiste JF, et al. Ceritinib plus nivolumab in patients with advanced alk-rearranged non-small cell lung cancer: Results of an open-label, multicenter, phase 1b study. J Thorac Oncol (2020) 15(3):392–403. doi: 10.1016/j.jtho.2019.10.006
76. Shaw A, Lee S-H, Ramalingam S, Bauer T, Boyer M, Costa E, et al. Avelumab (Anti–Pd-L1) in combination with crizotinib or lorlatinib in patients with previously treated advanced nsclc: Phase 1b results from javelin lung 101. J Clin Oncol (2018) 36:9008–. doi: 10.1200/JCO.2018.36.15_suppl.9008
77. Kim D-S, Gadgeel S, Gettinger S, Riely G, Oxnard G, Mekhail T, et al. Safety and clinical activity results from a phase ib study of alectinib plus atezolizumab in alk + advanced nsclc (Ansclc). J Clin Oncol (2018) 36:9009–. doi: 10.1200/JCO.2018.36.15_suppl.9009
78. Chalmers AW, Patel S, Boucher K, Cannon L, Esplin M, Luckart J, et al. Phase I trial of targeted egfr or alk therapy with ipilimumab in metastatic nsclc with long-term follow-up. Target Oncol (2019) 14(4):417–21. doi: 10.1007/s11523-019-00658-0
79. West H, McCleod M, Hussein M, Morabito A, Rittmeyer A, Conter HJ, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (Impower130): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol (2019) 20(7):924–37. doi: 10.1016/S1470-2045(19)30167-6
80. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous nsclc. N Engl J Med (2018) 378(24):2288–301. doi: 10.1056/NEJMoa1716948
81. Dhillon S, Syed YY. Atezolizumab first-line combination therapy: A review in metastatic nonsquamous nsclc. Target Oncol (2019) 14(6):759–68. doi: 10.1007/s11523-019-00686-w
82. Hack SP, Zhu AX, Wang Y. Augmenting anticancer immunity through combined targeting of angiogenic and pd-1/Pd-L1 pathways: Challenges and opportunities. Front Immunol (2020) 11:598877. doi: 10.3389/fimmu.2020.598877
83. Giannone G, Ghisoni E, Genta S, Scotto G, Tuninetti V, Turinetto M, et al. Immuno-metabolism and microenvironment in cancer: Key players for immunotherapy. Int J Mol Sci (2020) 21(12):4414. doi: 10.3390/ijms21124414
84. Gaissmaier L, Christopoulos P. Immune modulation in lung cancer: Current concepts and future strategies. Respiration (2020), 1–27. doi: 10.1159/000510385
85. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat Rev Clin Oncol (2018) 15(5):325–40. doi: 10.1038/nrclinonc.2018.29
86. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat Rev Clin Oncol (2018) 15(5):310–24. doi: 10.1038/nrclinonc.2018.9
87. Chen DS, Hurwitz H. Combinations of bevacizumab with cancer immunotherapy. Cancer J (2018) 24(4):193–204. doi: 10.1097/PPO.0000000000000327
88. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-vegf and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol (2018) 52(Pt 2):117–24. doi: 10.1016/j.semcancer.2017.12.002
89. Karar J, Maity A. Pi3k/Akt/Mtor pathway in angiogenesis. Front Mol Neurosci (2011) 4:51. doi: 10.3389/fnmol.2011.00051
90. Pyo KH, Lim SM, Park CW, Jo HN, Kim JH, Yun MR, et al. Comprehensive analyses of immunodynamics and immunoreactivity in response to treatment in alk-positive non-Small-Cell lung cancer. J Immunother Cancer (2020) 8(2):e000970. doi: 10.1136/jitc-2020-000970
91. McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med (2018) 24(6):749–57. doi: 10.1038/s41591-018-0053-3
92. Reck M, Mok TSK, Nishio M, Jotte RM, Cappuzzo F, Orlandi F, et al. Atezolizumab plus bevacizumab and chemotherapy in non-Small-Cell lung cancer (Impower150): Key subgroup analyses of patients with egfr mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med (2019) 7(5):387–401. doi: 10.1016/S2213-2600(19)30084-0
93. Shigeta K, Datta M, Hato T, Kitahara S, Chen IX, Matsui A, et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology (2020) 71(4):1247–61. doi: 10.1002/hep.30889
94. Nakasuka T, Ichihara E, Makimoto G, Maeda Y, Kiura K. Primary resistance to alectinib was lost after bevacizumab combined chemotherapy in alk-rearranged lung adenocarcinoma. J Thorac Oncol (2019) 14(8):e168–e9. doi: 10.1016/j.jtho.2019.03.009
95. Choudhury NJ, Young RJ, Sellitti M, Miller A, Drilon A. Lorlatinib and bevacizumab activity in alk-rearranged lung cancers after lorlatinib progression. JCO Precis Oncol (2020) 4:PO.20.00271. doi: 10.1200/PO.20.00271
96. Watanabe H, Ichihara E, Kayatani H, Makimoto G, Ninomiya K, Nishii K, et al. Vegfr2 blockade augments the effects of tyrosine kinase inhibitors by inhibiting angiogenesis and oncogenic signaling in oncogene-driven non-Small-Cell lung cancers. Cancer Sci (2021) 112(5):1853–64. doi: 10.1111/cas.14801
97. Passoni L, Gambacorti-Passerini C. Alk a novel lymphoma-associated tumor antigen for vaccination strategies. Leuk Lymphoma (2003) 44(10):1675–81. doi: 10.1080/1042819031000099625
98. Singh VK, Werner S, Schwalm S, Lennerz V, Ruf S, Stadler S, et al. Npm-Alk-Reactive T-cell responses in children and adolescents with npm-alk positive anaplastic Large cell lymphoma. Oncoimmunology (2019) 8(9):e1625688. doi: 10.1080/2162402X.2019.1625688
99. KS V, Werner S, Hackstein H, Lennerz V, Reiter A, Wolfel T, et al. Analysis of nucleophosmin-anaplastic lymphoma kinase (Npm-Alk)-Reactive Cd8(+) T cell responses in children with npm-alk(+) anaplastic Large cell lymphoma. Clin Exp Immunol (2016) 186(1):96–105. doi: 10.1111/cei.12842
100. Chiarle R, Martinengo C, Mastini C, Ambrogio C, D’Escamard V, Forni G, et al. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat Med (2008) 14(6):676–80. doi: 10.1038/nm1769
101. Voena C, Menotti M, Mastini C, Di Giacomo F, Longo DL, Castella B, et al. Efficacy of a cancer vaccine against alk-rearranged lung tumors. Cancer Immunol Res (2015) 3(12):1333–43. doi: 10.1158/2326-6066.CIR-15-0089
102. Blasco RBJCIR. Abstract A021: Development of an alk vaccine to treat alk-rearranged non-small cell lung cancers. Cancer Immunology Research (2016) 4(11 Supplement):A021–A. doi: 10.1158/2326-6066.IMM2016-A021
103. Kalamatianos T, Denekou D, Stranjalis G, Papadimitriou E. Anaplastic lymphoma kinase in glioblastoma: Detection/Diagnostic methods and therapeutic options. Recent Pat Anticancer Drug Discov (2018) 13(2):209–23. doi: 10.2174/1574892813666180115151554
104. Codony-Servat J, Garcia-Roman S, Molina-Vila MA, Bertran-Alamillo J, Viteri S, d’Hondt E, et al. Anti-epidermal growth factor vaccine antibodies increase the antitumor activity of kinase inhibitors in alk and ret rearranged lung cancer cells. Transl Oncol (2021) 14(1):100887. doi: 10.1016/j.tranon.2020.100887
105. Babar Khan M, Chakraborty S, Boockvar JA. Use of chimeric antigen receptor T cells as a potential therapeutic for glioblastoma. Neurosurgery (2017) 80(5):N33–N4. doi: 10.1093/neuros/nyx105
106. Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther (2017) 25(9):2189–201. doi: 10.1016/j.ymthe.2017.06.008
107. Hombach AA, Gorgens A, Chmielewski M, Murke F, Kimpel J, Giebel B, et al. Superior therapeutic index in lymphoma therapy: Cd30(+) Cd34(+) hematopoietic stem cells resist a chimeric antigen receptor T-cell attack. Mol Ther (2016) 24(8):1423–34. doi: 10.1038/mt.2016.82
108. Ramos CA, Ballard B, Zhang H, Dakhova O, Gee AP, Mei Z, et al. Clinical and immunological responses after Cd30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest (2017) 127(9):3462–71. doi: 10.1172/JCI94306
109. Gaissmaier L, Elshiaty M, Christopoulos P. Breaking bottlenecks for the tcr therapy of cancer. Cells (2020) 9(9):2095. doi: 10.3390/cells9092095
110. Stauss HJ, Tran MGB. Tcr gene therapy: Challenges, opportunities, and future directions. Cells (2020) 9(12):2567. doi: 10.3390/cells9122567
111. Heather JM, Spindler MJ, Cobbold M, Gainor JF, Johnson DS, Hata AN. Anaplastic lymphoma kinase fusions as a target for tcr-directed cellular therapies. J Immunol (2020) 204(1 Supplement):239.10–.10.
112. Elsayed M, Christopoulos P. Therapeutic sequencing in alk(+) nsclc. Pharm (Basel) (2021) 14(2):80. doi: 10.3390/ph14020080
113. Smolle E, Taucher V, Lindenmann J, Jost PJ, Pichler M. Current knowledge about mechanisms of drug resistance against alk inhibitors in non-small cell lung cancer. Cancers (2021) 13(4):699. doi: 10.3390/cancers13040699
Keywords: anaplastic lymphoma kinase, tumor microenvironment, immune evasion, immunotherapy, immune checkpoint inhibitors
Citation: Guo Y, Guo H, Zhang Y and Cui J (2022) Anaplastic lymphoma kinase-special immunity and immunotherapy. Front. Immunol. 13:908894. doi: 10.3389/fimmu.2022.908894
Received: 31 March 2022; Accepted: 30 June 2022;
Published: 25 July 2022.
Edited by:
Min Xue, University of California, Riverside, United StatesCopyright © 2022 Guo, Guo, Zhang and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiuwei Cui, Y3VpandAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.