 Imran J. Anwar
Imran J. Anwar Isabel F. DeLaura
Isabel F. DeLaura Qimeng Gao
Qimeng Gao Joseph Ladowski
Joseph Ladowski Annette M. Jackson
Annette M. Jackson Jean Kwun
Jean Kwun Stuart J. Knechtle*
Stuart J. Knechtle*- Duke Transplant Center, Department of Surgery, Duke University School of Medicine, Durham, NC, United States
Despite dramatic improvement in kidney transplantation outcomes over the last decades due to advent of modern immunosuppressive agents, long-term outcomes remain poor. Antibody-mediated rejection (ABMR), a B cell driven process, accounts for the majority of chronic graft failures. There are currently no FDA-approved regimens for ABMR; however, several clinical trials are currently on-going. In this review, we present current mechanisms of B cell response in kidney transplantation, the clinical impact of sensitization and ABMR, the B cell response under current immunosuppressive regimens, and ongoing clinical trials for ABMR and desensitization treatment.
Introduction
Although the role of alloantibody in kidney transplant rejection has long been acknowledged (1), efforts to elucidate and target mechanisms of rejections have focused primarily on T cell as opposed to B cell responses, partially due to the obvious role of cellular immunity in early graft rejection (2, 3). T cell-centric research in transplantation has produced immunosuppressive regimens that successfully target T cell activation and proliferation, dramatically improving short-term transplantation outcomes. Subsequently, rates and severity of acute and T cell-mediated rejection (TCMR) have decreased over the last 5-6 decades. Currently, 1-year survival rates following kidney transplantation are at an all-time high of 98.11% for living donor and 94.88% for deceased donor transplants, based on the 2020 Scientific Registry of Transplant Recipients (SRTR) report (4). However, long-term graft survival has not seen such dramatic improvements (5). Unsurprisingly, transplantation elicits both T and B cell immune responses, and while TCMR can be usually successfully treated, indolent antibody-mediated rejection (ABMR) has become the dominant mode of allograft injury and contributor to decreased survival (6, 7). Calcineurin inhibitors (CNIs), steroid treatments, and T cell depletion reliably reverse TCMR; however, these therapies are not effective in reversing ABMR. Therefore, a mechanistic understanding of the B cell immune response to transplantation is necessary to develop effective therapeutics and prolong graft survival.
Post-transplant B cell immune responses involve several unique populations. The downstream effector cells, plasma cells (PC), play a key role in producing immunoglobulin products (8, 9). They may be generated following germinal center (GC) formation, or extrafollicularly from memory B cells following an anamnestic response (10). Given the complexity of B cell response, treatment of ABMR will likely require targeting of multiple processes, such as B cell activation and proliferation, plasma cell differentiation, antibody production, and complement activation, in contrast to the singular target of TCMR treatments: the T cell. In this review, we present current mechanisms of B cell response in kidney transplantation, the clinical impact of sensitization and ABMR, B cell response under current immunosuppressive regimens (Table 1), and ongoing ABMR and desensitization treatment clinical trials.
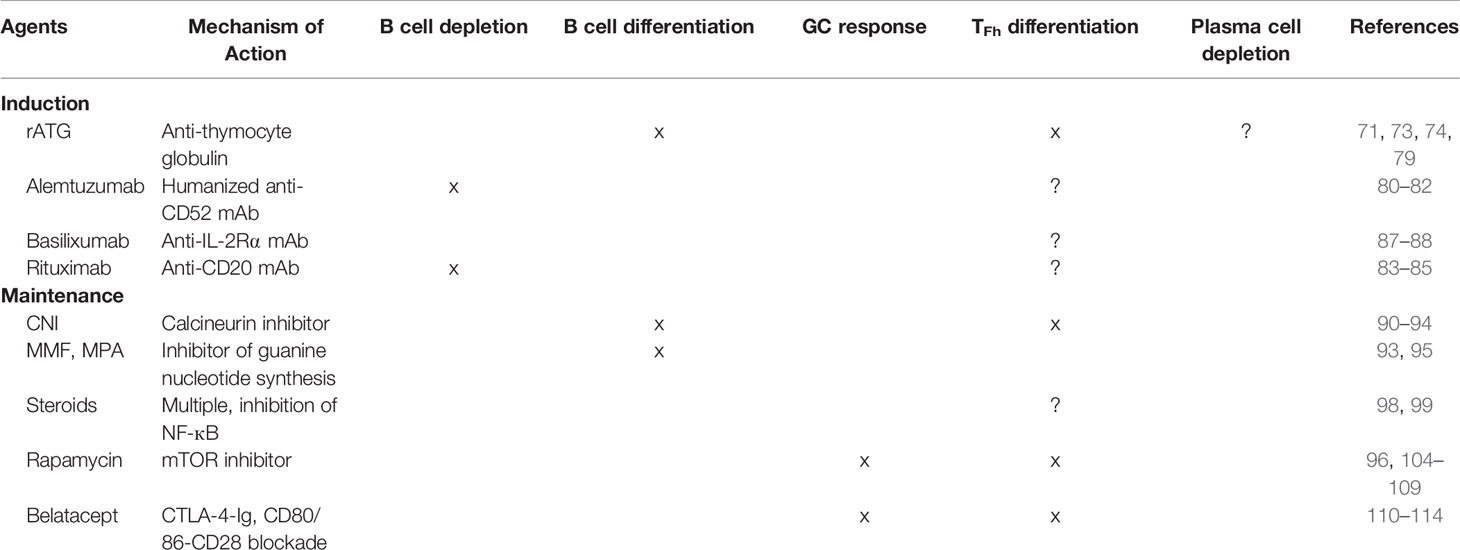
Table 1 B cell response under current immunosuppressive regimens.
Physiology of the B Cell Response
B cell development and maturation has been expertly reviewed elsewhere; thus, we aim to briefly review the lineage and critical steps in B cells maturation as they pertain to allotransplantation and the resultant T cell-dependent antigen activation (43–45). B cell development can be divided into three periods: B cell receptor (BCR) recombination, B cell activation and affinity maturation, and terminal differentiation.
The birth of a B cell occurs in the primary lymphoid tissue (either bone marrow or fetal liver) with the development of a pre-pro-B cell from a common lymphoid progenitor cell. The bone marrow serves as the site of BCR development. A series of functional rearrangements of the heavy chain V, D, J and light chain V and J genes, supported by the survival factor IL-7 which is produced by the surrounding stromal cells, results in a BCR with reactivity to a broad array of foreign antigens (46). BCRs that are autoreactive are negatively selected against through either receptor editing of the Ig light chain or cellular apoptosis. The surviving immature B cells are able to exit the bone marrow as transitional B cells to complete maturation in the spleen (47).
The mature naïve B cell then circulates through peripheral blood and lymph channels in search of a cognate antigen (48). In the context of transplantation, this is most likely alloantigen derived from donor human leukocyte antigen (HLA). The BCR/antigen interaction commonly occurs in the secondary lymphoid tissues (spleen, lymph nodes, and Peyer’s patches) due to the persistent homing of B cells to these tissues. A critical component of this homing and migration is the chemokine CXCL13 which is produced by the T follicular helper (Tfh), follicular dendritic, and stromal cells and serves as the ligand for CXCR5. Specialized antigen presentation cells (APCs) within lymphoid follicles display antigen to the BCR.
A B cell is triggered to proliferate in response to a sufficiently strong co-receptor (e.g. toll-like receptor) stimulation or highly multivalent antigen that successfully activate a BCR signal. These responses are considered T-independent. The surrounding APCs and stromal tissue produce survival factors such as the TNF cytokines, B cell activating factor (BAFF), and a proliferation-inducing ligand (APRIL), which are thought to be essential to B cell survival in T-independent B cell proliferation (49).
B cells that respond to a low valency antigen will commonly progress down a T-dependent pathway and receive secondary signaling and cytokine stimulation from CD4+ Tfh cells. Following initiation of the T-dependent response, the B cell ultimately becomes one of the following three: short-lived plasma cells, memory B cells, or GC B cells. As expected, there are a significant number of cytokines involved in the B cell differentiation and proliferation pathway that are expertly reviewed elsewhere (50). A few cytokines noteworthy to this discussion are IL-4, IL-10, and IL-21— cytokines responsible for promoting B cell proliferation, class switch recombination, and differentiation. The factors associated with the B cell fate are under investigation; antigen affinity, BCR isotype, and exposure time to antigen may play a role (51–53). Given the role of long-lived antibody production in transplantation, GC B cells are of particular relevance to this review.
The GC is histologically organized into light and dark zones containing B cells in differing stages of cell division. Within the GC, B cells proliferate in large part due to the B-Tfh cell interaction consisting of multiple surface protein interactions and Tfh secreted cytokines. Following requisite activation, B cells undergo proliferation and somatic hypermutation, the process by which the variable region of the BCR is altered to produce Ig of varying affinity. Ideally a balance is struck in which the BCRs with the highest affinity for antigen, but not reactive to self-antigen, are selected. Disruption of this balance can lead to the development of auto-immunity. Selected GC cells will ultimately undergo class switch recombination from IgM to IgG, IgA, or Ig. Proteins important to activation and differentiation include the B cell class II HLA complex, CD40, ICOS ligand, and CD80/86 and the T cell TCR, CD40L, ICOS, and CD28 (54, 55). From a cellular standpoint, there is also growing evidence to suggest that certain subsets of Tfh cells also predispose the B cell to IgG and IgM production, the isotypes associated with complement fixation (56–59). Research into the role of these Tfh subsets in the context of allotransplantation is underway. Finally, as mentioned previously, IL-21 is a cytokine produced by Tfh cells with significant importance in B cell development, differentiation, and function.
IL-21 is thought to be critical to generation and maintenance of long-lived plasma cells and the differentiation of B cells into plasma cells capable of producing the complement-fixing IgG1, IgG3, and IgM (50), and potentially leads to long-lived plasma cell development through activation of the STAT3 signaling pathway (60–62). B cells that undergo this process terminally differentiate into either memory B cells or long-lived plasma cells. These cell populations are of importance to transplant recipients as their development results in a cellular reservoir capable of producing donor-specific antibody (DSA) for years after exposure. It is unclear how these cells survive for years; however there is likely a combination of external (cellular and molecular niches) and internal (anti-apoptosis and differentiation factors) influences (63).
Robust B cell response following transplantation has been consistently reported and is particularly relevant in the discussion of sensitized recipients or those experiencing ABMR. Below, we address the role of B cell response in these populations.
HLA Sensitization
Sensitized recipients are those previously exposed to foreign HLA from pregnancy, transfusion, or prior transplant, resulting in the formation of HLA class I and class II antibodies. The role of these alloantibodies in transplant rejection was initially characterized in Patel and Terasaki’s landmark study, which defined positive crossmatch as the lysing of donor cells when mixed with recipient serum, and found a correlation between positive crossmatch and early graft loss (1). In the current era, HLA antibodies are detected in patient sera utilizing large HLA coated bead panels, providing greater sensitivity and specificity than earlier cell panels (64). Detection of donor specific HLA antibodies (DSA) in recipient sera is associated with increased ABMR risk and reduced allograft survival (65, 66).
HLA sensitization induced by previous transplants appears consistently as the strongest sensitizing event, while transfusion is associated with limited HLA antibody production in strength and breadth (67, 68). Pregnancy induced sensitization correlates with the number of live births, the HLA mismatch, and homozygosity in the mother’s HLA phenotype (69). A study examining the impact of sensitization route on long-term allograft survival revealed that highly sensitized recipients sensitized via previous transplants lost grafts at higher rates than those sensitized via pregnancy or transfusion (68).
New assays to detect HLA-specific B cell memory cells are advancing our understanding of B cell memory generation in transplant candidates and the potential ABMR risk that has historically been undetected (70, 71). Pregnancy and previous transplants have been shown to elicit HLA specific B memory cell generation, which may outlast serum HLA antibodies and pose an increased risk for post-transplant anamnestic alloantibody responses even when DSA is absent at time of transplantation (70, 72).
Historically, broad HLA sensitization has limited a candidate’s likelihood of finding a compatible donor resulting in prolonged waiting times. New organ allocation schemas in the United States now assign increasing allocation points according to the breadth of the patient’s sensitization (calculated panel reactive antibody, cPRA) and mandate national organ sharing for 100% cPRA candidates, thereby prioritizing the matching of compatible allografts to sensitized patients (73). Furthermore, the new allocation system with a larger donor pool has provided opportunity to transplant some of these highly sensitized candidates across low level DSA or DSA specific to HLA antigens that are expressed at lower levels on cells (HLA-C and -DP) are associated with lower risks of ABMR and limited impact on allograft survival.
Yet despite this prioritization, transplant inequity still remains for the most highly sensitized candidates (cPRA ≥ 99.9%) due to competition for kidney donors with rare or homozygous HLA antigen phenotypes (74, 75). The Eurotransplant Acceptable Mismatch allocation program for highly sensitized candidates is based on selecting donors whose HLA phenotype includes HLA antigens of the recipient’s phenotype as well as acceptable HLA antigens to which the recipient has never been sensitized. Reports by Heidt et al. (76, 77) show that highly sensitized recipients transplanted within this program experience lower rejection rates and allograft survival equal to non-sensitized patients.
Antibody-Mediated Rejection
Antibody-mediated rejection (ABMR), a clinicopathological diagnosis that was first formally defined in 2003 (78), has gained importance in kidney transplantation and is now thought to be the primary driver of late graft failure. In their 2012 landmark study, Sellarés et al. (6) showed that ABMR was responsible for the majority of late graft failures. Several studies have since confirmed the impact of ABMR on short- and long-term outcomes. A meta-analysis demonstrated rates of acute ABMR (less than 1 year) between 3-12% and chronic ABMR between 5-20% following kidney transplantation (79), with rates up to 50% following positive cross-match living donor kidney transplant (80).
The definition and impact of ABMR has substantially evolved since the establishment of the Banff Criteria in 2003, allowing for improved diagnostic sensitivity and predictive value of graft outcome. The 2017 Banff criteria now incorporate C4d deposition and ABMR-related gene transcripts as a surrogate for DSA (81). It is now accepted that ABMR represents a wide spectrum of lesions at different time points following antibody-mediated injuries, ultimately leading to a spectrum of clinical phenotypes (82). Importantly, biopsies classified as no rejection displayed evidence of increased ABMR-related transcripts in patients with elevated DSA titers, highlighting the high prevalence of ABMR and likely under-diagnosis with current diagnostic modalities (83).
In recent years, the relationship between DSA and ABMR has become more clearly defined, and detection and precise characterization and quantification of DSA against HLA have become SOC following widespread use of Luminex-based assays. Lefaucheur et al. (84) convincingly show that patients who undergo transplantation with pre-existing HLA-DSA fare worse compared to patients without HLA-DSA, and that rates of ABMR are directly correlated to pre-transplant levels of HLA-DSA. However, despite higher rates of ABMR in patients with pre-existing DSA, ABMR following de novo DSA production portends to worse outcomes with a more aggressive molecular phenotype (85). Importantly, patients with ABMR in the absence of HLA-DSA have improved survival compared to HLA-DSA positive patients (86) and a distinct transcriptional signature (87).
Contemporary Challenges With ABMR
Diagnosis and treatment of ABMR remains challenging. Subclinical rejection, the presence of rejection in kidney recipients who have neither clinical nor laboratory evidence of rejection, is common with incidence up to 30% in the first 3 months post-transplant (88). Subclinical ABMR portends to poor long-term graft survival; subclinical ABMR at 1-year is independently associated with a 3.5-fold increase in graft loss (89).
Given the invasive and expensive nature of kidney biopsies, several non-invasive alternatives have been pursued in the hopes of accurately diagnosing rejection and ABMR in particular. Donor-derived cell-free DNA, blood-based molecular biomarkers, urinary biomarkers, and tissue molecular diagnostics all constitute promising venues and have been reviewed by Westphal and Manoon (90).
Additionally, it is well described that interpretation of renal allograft biopsy pathology is limited by inter-observer variation and poor inter-observer agreement (91). Advances in machine learning and artificial intelligence have led to the emergence of digital pathology, paving the way for more accurate and reproducible interpretation of renal pathologies (92, 93). The Banff Digital Pathology Working Group (DPWG) was founded in 2019 at the joint Banff/ASHI meeting to promote and support the development and integration of digital pathology into clinical practice and research protocols (94).
Despite the prevalence of ABMR in kidney transplantation, there are currently no FDA-approved treatments for ABMR (95). In part, ABMR treatment has not been optimized due to the constantly evolving definition of ABMR and our incomplete understanding on the relationship between ABMR and DSA. Despite this, several different therapies have been evaluated in the past and have been extensively reviewed elsewhere (96–99). However, most studies of ABMR treatment are confounded by a heterogenous patient population, small sample size, and varying definition and chronicity of ABMR (96, 97). Intravenous Immunoglobulin (IVIg) and plasmapheresis are often considered standard of care (SOC) despite lack of evidence of efficacy or safety (79). Importantly, the current FDA-approved primary endpoints, 1-year graft and patient survivals, are not appropriate given excellent clinical outcomes under modern immunosuppression (>95% survival at one year) (81). Therefore, it is essential to establish validated surrogate endpoints is essential to develop and trial novel ABMR treatments (100).
B Cell Response With Current Immunosuppression
Induction Therapy
Greater than 90% of all kidney transplants include induction agents as part of their immunosuppressive regimen in the United States, with 60% receiving lymphocytes-depleting agents (usually in the form of rabbit Anti-Thymocyte Globulin (rATG)) and 20% receiving non-depletional agents (IL-2 receptor antagonist, Basilixumab) (4, 101). Alemtuzumab, a humanized anti-CD52 mAb, is used in several transplant centers despite lack of FDA approval for use in solid-organ transplantation. Induction agents have been shown to effectively prevent occurrence of early acute rejection and are now standard of care (102).
Rabbit Anti-thymocyte globulin (rATG) is made by immunizing rabbits with human pediatric thymi and purifying the IgG fraction, resulting in a polyclonal agent that bind multiple epitopes on T cells, causing both complement-dependent lysis and apoptosis and sustained T cell depletion (11). However, the effect of rATG on B cell response is less clearly understood and is thought to occur through several distinct mechanisms. First, the human thymus contains a small percentage of B and plasma-cells (103) and rATG contains antibodies against known B and plasma cell markers (11). Zand et al. (12) showed that rATG triggers B cell apoptosis in vitro, confirming that rATG exerts a direct effect on B cells. However, B cell depletion is typically minimal following rATG induction in kidney transplantation, perhaps due to the low dose commonly used (1.5mg/kg/day for 4 to 7 days) (13, 104–107). Second, rATG inhibits B cell differentiation and promotes B cell proliferation in vivo (14). Despite no significant B cell depletion, rATG induction in humans reduces frequency of memory B cells and class-switched B cells, likely secondary to impaired B cell differentiation (13). Finally, given that rATG causes pan-T cell depletion, including depletion of T follicular helper cells, T-cell-dependent activation of allo-reactive B cells is likely disrupted following rATG induction.
Alemtuzumab, a humanized anti-CD52 mAb, causes profound B cell, T cell, NK cell, and monocyte depletion and is thus used in select centers as an induction agent. B cell depletion is immediate and results in suppressed B cell population for 6 months, followed by repopulation that achieve absolute counts exceeding that of pre-transplantation levels (15–17). Naïve B cells are preferentially depleted (suggesting partial resistance of memory B cells to alemtuzumab), followed by repopulation driven by transitional and pregerminal center B cells that promote long-term expansion and dominance of naïve B cells. Furthermore, B-regulatory cells (BRegs) are increased (16). Todeshini et al. (17) reported that alemzutumab induction was associated with DSA production and worse long-term outcomes, suggesting the B-cell depletion following alemtuzumab induction may promote chronic humoral response against the allograft.
Rituximab, a mouse/human chimeric monoclonal antibody against CD20, potently depletes B cells and is often use in the context of highly sensitized kidney transplantation or ABO incompatible kidney transplants, to prevent antibody-mediated rejection given their higher immunologic risk profile (20). Sustained peripheral depletion (<5 CD19+ cells/µL) is readily achieved after a single dose (375 mg/m2 BSA) in less than 3 days and last for up to 12-14 months (21). The repopulated B-cell population is mostly comprised of transitional B cells (22). Lymph node B cells are partially depleted by a single dose with the resultant B cells showing switched memory phenotype (108).
Basilixumab, a chimeric IgG1 monoclonal antibody against the α-peptide chain of the IL-2Rα (also referred to as CD25), prevents IL-2-mediated proliferation of T lymphocytes, a critical step in the cellular immune response (18). Interestingly, peripheral B cells express IL-2Rα, and IL-2 signaling is critically involved in plasma cell differentiation (19). Basilixumab induction causes minimal B-cell depletion but increases the proportion of naïve B cells (15).
Triple Immunosuppression
Triple immunosuppression, consisting of a calcineurin inhibitor (CNI), an antiproliferative, and a glucocorticoid, is the most common maintenance immunosuppression regimen for kidney transplantation, used in over 60% of cases (4).
CNIs, such as tacrolimus and cyclosporine, bind to intracellular protein FKBT 12, thus inhibiting calcineurin, which subsequently prevents the dephosphorylation and activation of nuclear regulator NFAT, resulting in decreased IL2 production and IL2R expression. CNIs are the backbone of many commonly used immunosuppressive regimens, with some studies such as the ELITE-Symphony trial, showing higher rates of allograft survival and lower rates of antibody rejection with tacrolimus over cyclosporine (109). CNIs act directly on T cells and have been found to inhibit ICOS+PD1+ T follicular helper cell (Tfh) differentiation, thus inhibiting B cell proliferation and differentiation (23–25). Specifically, CNI administration attenuates T cell costimulatory ligand (CD40L and ICOS) expression and production of B cell stimulatory cytokines such as IL21, resulting in decreased B cell activation, plasmablast differentiation, and therefore immunoglobulin secretion in autologous T-B cell co-cultures (24, 26). However, as CNIs do not act directly on B cells, CNI treatment does not inhibit immunoglobulin production from B cells cultured with preactivated T cells (26). Accordingly, preclinical in vivo experiments have shown depletion of GC Tfh, decreased expression of IL21, and subsequent prevention of DSA and ABMR with high dose CNI treatment (27).
Mycophenolate mofetil (MMF) and mycophenolic acid (MPA) directly inhibit T- and B-cell proliferation via the inhibition of guanine nucleotide synthesis. MMF also modulates B cell activation and differentiation indirectly through the downregulation of T cell protein CD40L and suppression of cytokine production (26, 28). Furthermore, MMF directly inhibits B cell expansion and plasma cell differentiation when applied to primed B cells; immunoglobulin production is also reduced with MMF treatment (28, 31). However, MMF does not suppress immunoglobulin production from terminally differentiated plasma cells (28).
Glucocorticoids, such as prednisone, modulate both the innate and adaptive immune responses and suppress inflammation via many mechanisms, including induction of anti-inflammatory genes and inhibition of nuclear regulators such as NF-κB (110). Steroid act both directly on B cells, decreasing antibody production and inducing apoptosis, and indirectly via the suppression of dendritic cell and T cell populations (29, 30).
Rapamycin
Rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, is used as an alternative to CNIs in maintenance immunosuppressive regimens. mTOR inhibitors suppress T cell activation and differentiation via blockade of cell-cycle progression (111), inhibition of thymic maturation (112), and downregulation of chemokine signaling (113, 114). Interestingly, rapamycin treatment results in a decrease in Tfh and effector T cells, with a relative increase in CD4+CD25+FOXP3+ regulatory T cells, which are implicated in attenuating memory T cell proliferation (32–34). However, one important advantage of mTOR inhibitors over CNIs is the direct suppression of GC formation, B cell proliferation and immunoglobulin production (31, 35, 36). In hindering GC activation, rapamycin effectively prevents B cell activation and subsequent differentiation to the plasma cell phenotype; however, treatment does not suppress antibody production from already differentiated plasma cells, suggesting that mTOR is necessary for B cell activation and proliferation, but not for memory response (35, 37).
Costimulation Blockade
Belatacept is a CTLA-4-Ig fusion protein that binds CD80 and CD86 on APCs, thus inhibiting binding to CD28, which suppresses T cell activation and subsequent dependent B cell response (38). Specifically, belatacept decreases ICOS+PD1+ Tfh populations, resulting in decreased B cell maturation and plasmablast differentiation in in vitro experiments (39). Preclinical studies in nonhuman primates also demonstrated suppression of Tfh, and thus GC B cell clonal expansion (40). Transplant recipients treated with belatacept had lower levels of circulating Tfh, memory B cells, and plasmablasts compared to those treated with CNIs (39). In addition to suppressing GC activation by interrupting Tfh-B cell cross talk, experiments in murine models have shown that belatacept treatment disrupts established GCs and reduces alloantibody production even when T cell priming has already occurred (41, 42). Accordingly, belatacept treatment inhibits memory alloreactive B cell and thus recall antibody responses in sensitized recipients (115). Clinically, belatacept has been approved for use in kidney transplantation, with similar rates of acute rejection, graft survival, and death compared to CNIs (116, 117). It has also been used as maintenance monotherapy in immunologically low risk (no DSA, cPRA<20%) patients following alemtuzumab and one-year belatacept/rapamycin dual therapy (118). Post-hoc analysis of BENEFIT and BENEFIT-EXT studies revealed that belatacept-treated patients had lower incidence of de novo DSA compared to cyclosporine-treated patients (119).
Blockade of the CD40/CD40L costimulatory pathway has been extensively evaluated in kidney transplantation following reports of prolonged graft survival in NHP with an anti-CD40L antibody either in combination with CTLA4-Ig (120) or as monotherapy (121). Translation of anti-CD40L agents was halted following thromboembolic events due to high fragment crystallizable (Fc) effector function activity and binding to platelets (122–125). Since then, several anti-CD40L antibodies without Fc effector function have been engineered with improved safety profiles. The humoral response is controlled through several distinct mechanisms. CD40 is constitutively expressed on B lymphocytes (126), and several anti-CD40 agents lead to B cell depletion through Fc effector function (40, 127). Furthermore, blockade of the CD40/CD40L pathway disrupts GC responses (128), and was shown to prevent early DSA and ABMR in NHP (40). Importantly, Fc-silent anti-CD40 lacking the ability to deplete B cells still promotes long-term graft survival in NHP, underlying the crucial role of GC disruption in achieving favorable clinical outcomes (129). Several clinical trials evaluating agents blocking the CD40/CD40L pathway are either on-going (NCT05027906, NCT04046549) or recently ended (NCT03663335, NCT02217410) and will provide insight into their efficacy in preventing the humoral response in humans.
Limitation of Current Immunosuppressive Regimens
Belatacept-based maintenance therapy has emerged as an alternative immunosuppressive regimen with a more favorable side effect profile (i.e. less nephrotoxicity and posttransplant malignancies) compared to CNI-based maintenance therapy. Indeed, the BENEFIT trial revealed a 43% reduction in risk of death of graft loss and enhanced renal function at 7 years for belatacept-based regimens compared to CNI-based regimens (130). Nevertheless, the primary limitation of belatacept-based immunosuppression is that it is less effective in preventing early acute cellular rejection compared to CNI-based immunosuppression (131, 132), including when patients previously maintained on CNI-based therapy were switched to belatacept-based therapy (133). Several subsets of mature T cells have been associated with costimulation blockade resistant rejection (CoBRR) such as CD57+PD1-CD4+ T cells (134, 135) and CD28+CD4+ memory T cells (136, 137). Steroid-sparing, belatacept-based immunosuppression following depletional induction as well as addition of rapamycin to a belatacept-based regimen have reduced rates of acute rejection in small uncontrolled single-center clinical trials, suggesting that a belatacept-based regimen could yield similar rates of acute rejection compared to CNI-regimen if properly optimized (138).
Another limitation is that belatacept is given once monthly intravenously, which may limit compliance and contribute to the generation of de novo DSA (6). As a result, belatacept continues to be seldom used in renal transplantation with less than 10% of all adult transplant recipients on maintenance belatacept (4).
Most renal transplant patients in the U.S. are currently managed long-term with tacrolimus, mycophenolate, and often steroids. While effective, especially short-term, this regimen requires drug adherence and that drug levels of tacrolimus be maintained above 5 ng/ml in order to avoid risk of antibody-mediated rejection (139). When tacrolimus dosing level is lowered to less than 5 ng/ml (mostly due to side-effect avoidance, socioeconomic reasons, or non-adherence) ABMR becomes more likely. DSA development in the late post-transplant period, in association with overt or subclinical rejection, is difficult to clear using current therapies, and most of the time results in graft injury. This scenario occurs in 30-40% of patients at some point after their kidney transplant, and better therapeutic options are needed to avoid graft loss. Graft loss implies return to dialysis and need for retransplantation, further stressing the already insufficient supply of donor kidneys. Therefore, a better solution to management of DSA and ABMR is needed to extend the lives and quality of life of at least a third of renal transplant patients, reduce the need for retransplantation, and reduce this source of demand for donor kidneys—now the third largest cause of end-stage renal failure in the U.S. Fortunately, there are several on-going or upcoming clinical trials in the USA that aim at rigorously investigate novel treatments for both desensitization and ABMR (Table 2).
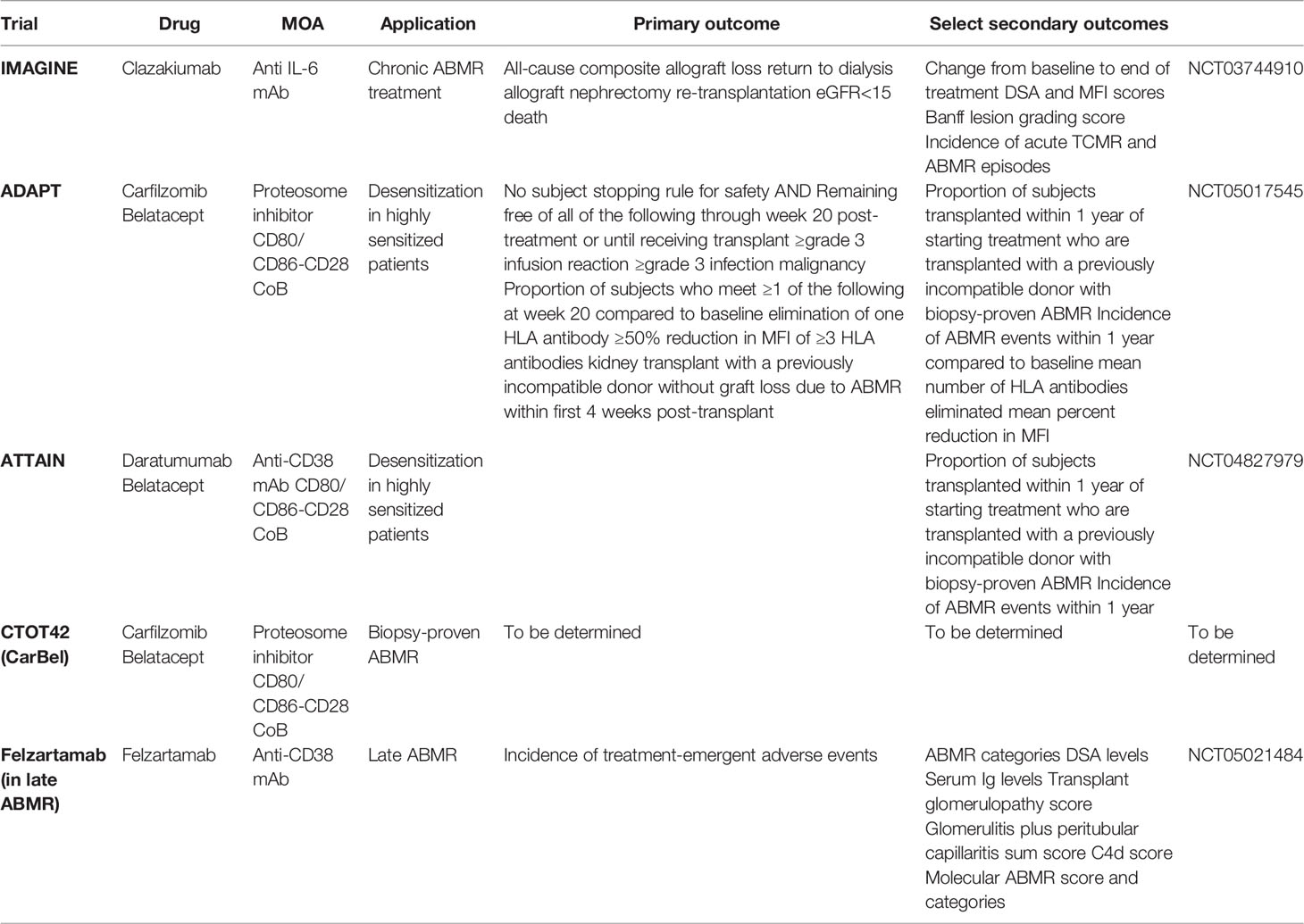
Table 2 Summary of ongoing clinical trials for ABMR and desensitization treatment.
Ongoing and Future Clinical Trials to Address Desensitization and Treatment of ABMR
IMAGINE Trial
The IMAGINE trial (NCT03744910) is an ongoing phase 3 clinical trial to assess the safety and efficacy of clazakizumab for the treatment of chronic ABMR, aiming to enroll 350 kidney transplant patients. Clazakizumab is a humanized monoclonal antibody targeting IL-6 and is administered subcutaneously monthly. IL-6 blockade quiets inflammation and antibody production which may reduce the injury associated with DSA and ABMR (140). A prior phase 2 RCT evaluating clazakizumab in late AMBR showed promising results with early decrease in DSA, slowed eGFR decline, and alleviation—resolution in some patients—of ABMR following clazakizumab treatment (141). The trial was withdrawn due to safety concerns with 25% patients developing serious infectious events, and 10% developing diverticular disease complications. The aforementioned safety events influenced design of the IMAGINE trial which utilized a reduced monthly clazakizumab dose (25 to 12.5mg/kg dose) and stricter exclusion criteria.
The IMAGINE trial has a primary outcome measure of all-cause composite allograft loss, defined as return to dialysis, allograft nephrectomy, re-transplantation, or eGFR <15, or death from any cause. As of this writing, the trial is in active enrollment and randomizes 1:1 to drug vs. placebo. A monthly subcutaneous injection is an attractive route and frequency of drug administration from a patient perspective, and if effective, this treatment would offer a biologic therapy of greater value than currently available. The trial aims to treat a uniform population of patients with chronic ABMR narrowly defined both clinically and by biopsy criteria. One disadvantage of the study design is the long length of time and large number of centers required to achieve target enrollment. Nevertheless, this elegant trial design uses slope of eGFR at one year as a surrogate endpoint for renal allograft survival, and thus represents an advance with respect to adoption of a surrogate endpoint accepted by the FDA in kidney transplant trials (100, 139, 141).
ADAPT and ATTAIN Trials
The ADAPT trial (NCT05017545) is a desensitization trial for adult patients who are highly allosensitized (cPRA >99.9%) and on the wait list for renal transplantation. This single-center study of 15 patients is also mirrored by the ATTAIN study (NCT04827979) which has very similar trial design, but differs in the drug combinations being tested to reduce alloantibody levels. The ADAPT study aims to evaluate the combination of a proteosome inhibitor, carfilzomib, and an anti-CD28 costimulation blocker, Belatacept, to lower alloantibody levels. ATTAIN will use an anti-CD38 mononclonal antibody called daratumumab in place of carfilzomib to target plasma cells, but will also use belatacept. The ADAPT study will maintain belatacept for one year in study patients while a shorter course is given in the ATTAIN study. The primary endpoint of both trials is fulfilling a subject stopping rule, or remaining free of all of the following through week 20 post-treatment initiation or until receiving a transplant, whichever occurs earlier: a. grade 3 or higher infusion reaction, b. grade 3 or higher infections, and c. any malignancy. The study site will grade the severity of adverse events experienced by the study subjects according to the criteria set forth in the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 (Published November 27, 2017). The ADAPT study and the ATTAIN study are largely based on non-human primate results evaluating these therapies for their efficacy and safety in lowering antibody levels and permitting rejection-free renal transplantation subsequently (142–144). While the primary aims of both studies are to assess safety, the secondary goals are to assess efficacy in lowering antibody levels, leading to renal transplants being allocated to study patients with a negative crossmatch test, and to assess how the treatments affect plasma cells in blood and bone marrow of the subjects.
CTOT42, CarBel Trial
The CarBel trial or CTOT42 trial is an NIH-sponsored clinical trial for the treatment of active or chronic active ABMR. This trial, also based largely on the results of pre-clinical work in non-human primates (Schmitz et al., submitted), will evaluate the safety and efficacy of carfilzomib/belatacept dual therapy for the treatment of biopsy-proven ABMR. This multi-center, randomized, prospective clinical trial will compare standard of care treatment consisting of intravenous immune globulin (IVIg) with or without therapeutic plasma exchange (TPE) to combined carfilzomib/belatacept. A total of six doses of carfilzomib will be used along with a year of belatacept, measuring the primary outcome at one year using iBox, a tool developed by the Paris Transplant Group, that has been validated by retrospective data (145). This trial is still in the planning stage with intended initiation in late 2022. The trial includes mechanistic aims to assess the impact of therapy on allo-specific B memory cells and plasma cells derived from blood and bone marrow. Additionally, digital imaging pathology will be used to evaluate artificial intelligence methods to diagnose ABMR and compare such signatures to pathologists’ interpretation of kidney biopsies.
Felzartamab in Late ABMR
Felzartamab, a recombinant fully human monoclonal anti-CD38 monoclonal antibody, will be evaluated for safety and tolerability in a multi-center 12-month randomized placebo-controlled parallel-group trial (NCT05021484) (146). The study hypothesis is that felzartamab will deplete DSA-producing plasma cells thus reducing alloantibody production. Additionally, CD38 is expressed on natural killer (NK) cells, and interference via felzartamab administration may prevent NK cell-triggered tissue injury seen in ABMR. Doberer et al. recently published a case report highlighting the successful reversal of late ABMR using felzartamab, resulting in stabilization of graft function, disappearance of DSA, improved AMBR score on graft histology, and decrease in peripheral and graft NK cells (147).
This trial will enroll 20 kidney transplant recipients with evidence of late ABMR. Participants will be randomized to receive either felzartamab or placebo for 6 months and evaluated for an additional 6 months, including protocol biopsies at 6 months and 1 year. Primary endpoint is assessment of safety and tolerability. Secondary endpoints include pharmacokinetics/pharmacodynamic profiles, serum/urine biomarkers of rejection, biopsy results, and surrogate parameters of allograft dysfunction (eGFR slope and iBOX). While not powered to detect meaningful effect on clinical outcomes of felzartamab compared to placebo, this trial may provide a useful framework for the design of future larger studies.
Conclusion
Given the negative impact of allosensitization in renal transplantation, in particular B cell sensitization and alloantibody production, the development of immunosuppression targeting B cell and plasma cell responses is much needed. Such therapies need to be evaluated not only for efficacy, but also for their impact on B cell responses to infectious agents and thus protective immunity. As current immunosuppressive regimens impair protective immunity leading to increased risk of infection and malignancy, it follows that additional immunosuppression would further heighten these risks. Therefore, careful trial design and collaboration with infectious disease experts is necessary to develop such therapies. Nevertheless, the field of B cell and plasma cell biology has grown exponentially in recent decades, resulting in a plethora of drugs that target specific immune cell populations and that can down-regulate the B cell response. Further applications of these increasingly targeted drugs offer an opportunity to improve survival and graft function outcomes for transplant recipients as we learn how to adapt these strategies in the clinical setting.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was partially supported by the National Institute of Allergy and infectious Diseases of the National Institutes of Health as part of the NHP Transplantation Tolerance Cooperative Study Group under the U19AI131471 (awarded to S.J.K.). Additionally, this work was supported by NIH T32 AI141342-03 (awarded to IJA) and ASTS Presidential Student Mentorship Grant (awarded to IFD).
Conflict of Interest
SJK is a consultant to Novartis, Visterra, and Hansa Biopharma. AMJ is a consultant to Novartis and Hansa Biopharma. SJK and JK has received research materials/grants from Alexion and MorphoSys.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Patel R, Terasaki PI. Significance of the Positive Crossmatch Test in Kidney Transplantation. N Engl J Med (1969) 280:735–9. doi: 10.1056/NEJM196904032801401
2. Billingham RE, Krohn PL, Medawar PB. Effect of Locally Applied Cortisone Acetate on Survival of Skin Homografts in Rabbits. Br Med J (1951) 2:1049–53. doi: 10.1136/bmj.2.4739.1049
3. Benichou G, Yamada Y, Yun SH, Lin C, Fray M, Tocco G. Immune Recognition and Rejection of Allogeneic Skin Grafts. Immunotherapy (2011) 3:757–70. doi: 10.2217/imt.11.2
4. Hart A, Lentine KL, Smith JM, Miller JM, Skeans MA, Prentice M, et al. OPTN/SRTR 2019 Annual Data Report: Kidney. Am J Transplant (2021) 21 Suppl 2:21–137. doi: 10.1111/ajt.16502
5. Hariharan S, Israni AK, Danovitch G. Long-Term Survival After Kidney Transplantation. N Engl J Med (2021) 385:729–43. doi: 10.1056/NEJMra2014530
6. Sellares J, De Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the Causes of Kidney Transplant Failure: The Dominant Role of Antibody-Mediated Rejection and Nonadherence. Am J Transplant (2012) 12:388–99. doi: 10.1111/j.1600-6143.2011.03840.x
7. Halloran PF, Chang J, Famulski K, Hidalgo LG, Salazar ID, Merino Lopez M, et al. Disappearance of T Cell-Mediated Rejection Despite Continued Antibody-Mediated Rejection in Late Kidney Transplant Recipients. J Am Soc Nephrol (2015) 26:1711–20. doi: 10.1681/ASN.2014060588
8. Kwun J, Knechtle SJ. Overcoming Chronic Rejection-Can it B? Transplantation (2009) 88:955–61. doi: 10.1097/TP.0b013e3181b96646
9. Clatworthy MR. Targeting B Cells and Antibody in Transplantation. Am J Transplant (2011) 11:1359–67. doi: 10.1111/j.1600-6143.2011.03554.x
10. Kurosaki T, Kometani K, Ise W. Memory B Cells. Nat Rev Immunol (2015) 15:149–59. doi: 10.1038/nri3802
11. Mohty M. Mechanisms of Action of Antithymocyte Globulin: T-Cell Depletion and Beyond. Leukemia (2007) 21:1387–94. doi: 10.1038/sj.leu.2404683
12. Zand MS, Vo T, Huggins J, Felgar R, Liesveld J, Pellegrin T, et al. Polyclonal Rabbit Antithymocyte Globulin Triggers B-Cell and Plasma Cell Apoptosis by Multiple Pathways. Transplantation (2005) 79:1507–15. doi: 10.1097/01.TP.0000164159.20075.16
13. Gurkan S, Luan Y, Dhillon N, Allam SR, Montague T, Bromberg JS, et al. Immune Reconstitution Following Rabbit Antithymocyte Globulin. Am J Transplant (2010) 10:2132–41. doi: 10.1111/j.1600-6143.2010.03210.x
14. Klaus P, Heine G, Rasche C, Worm M. Low-Dose Anti-Thymocyte Globulin Inhibits Human B-Cell Differentiation Into Antibody-Secreting Cells. Acta Derm Venereol (2015) 95:676–80. doi: 10.2340/00015555-2046
15. Cherukuri A, Salama AD, Carter C, Smalle N, Mccurtin R, Hewitt EW, et al. An Analysis of Lymphocyte Phenotype After Steroid Avoidance With Either Alemtuzumab or Basiliximab Induction in Renal Transplantation. Am J Transplant (2012) 12:919–31. doi: 10.1111/j.1600-6143.2011.03891.x
16. Heidt S, Hester J, Shankar S, Friend PJ, Wood KJ. B Cell Repopulation After Alemtuzumab Induction-Transient Increase in Transitional B Cells and Long-Term Dominance of Naive B Cells. Am J Transplant (2012) 12:1784–92. doi: 10.1111/j.1600-6143.2012.04012.x
17. Todeschini M, Cortinovis M, Perico N, Poli F, Innocente A, Cavinato RA, et al. In Kidney Transplant Patients, Alemtuzumab But Not Basiliximab/Low-Dose Rabbit Anti-Thymocyte Globulin Induces B Cell Depletion and Regeneration, Which Associates With a High Incidence of De Novo Donor-Specific Anti-HLA Antibody Development. J Immunol (2013) 191:2818–28. doi: 10.4049/jimmunol.1203261
18. Mckeage K, Mccormack PL. Basiliximab: A Review of its Use as Induction Therapy in Renal Transplantation. BioDrugs (2010) 24:55–76. doi: 10.2165/11203990-000000000-00000
19. Le Gallou S, Caron G, Delaloy C, Rossille D, Tarte K, Fest T. IL-2 Requirement for Human Plasma Cell Generation: Coupling Differentiation and Proliferation by Enhancing MAPK-ERK Signaling. J Immunol (2012) 189:161–73. doi: 10.4049/jimmunol.1200301
20. Tomita Y, Iwadoh K, Ogawa Y, Miki K, Kato Y, Kai K, et al. Single Fixed Low-Dose Rituximab as Induction Therapy Suppresses De Novo Donor-Specific Anti-HLA Antibody Production in ABO Compatible Living Kidney Transplant Recipients. PLoS One (2019) 14:e0224203. doi: 10.1371/journal.pone.0224203
21. Genberg H, Hansson A, Wernerson A, Wennberg L, Tyden G. Pharmacodynamics of Rituximab in Kidney Allotransplantation. Am J Transplant (2006) 6:2418–28. doi: 10.1111/j.1600-6143.2006.01497.x
22. Kamburova EG, Koenen HJ, Van Den Hoogen MW, Baas MC, Joosten I, Hilbrands LB. Longitudinal Analysis of T and B Cell Phenotype and Function in Renal Transplant Recipients With or Without Rituximab Induction Therapy. PLoS One (2014) 9:e112658. doi: 10.1371/journal.pone.0112658
23. Tsuda K, Yamanaka K, Kitagawa H, Akeda T, Naka M, Niwa K, et al. Calcineurin Inhibitors Suppress Cytokine Production From Memory T Cells and Differentiation of Naive T Cells Into Cytokine-Producing Mature T Cells. PLoS One (2012) 7:e31465. doi: 10.1371/journal.pone.0031465
24. Wallin EF, Hill DL, Linterman MA, Wood KJ. The Calcineurin Inhibitor Tacrolimus Specifically Suppresses Human T Follicular Helper Cells. Front Immunol (2018) 9, 1184 doi: 10.3389/fimmu.2018.01184
25. Kraaijeveld R, Li Y, Yan L, De Leur K, Dieterich M, Peeters AMA, et al. Inhibition of T Helper Cell Differentiation by Tacrolimus or Sirolimus Results in Reduced B-Cell Activation: Effects on T Follicular Helper Cells. Transplant Proc (2019) 51:3463–73. doi: 10.1016/j.transproceed.2019.08.039
26. Heidt S, Roelen DL, Eijsink C, Eikmans M, Van Kooten C, Claas FH, et al. Calcineurin Inhibitors Affect B Cell Antibody Responses Indirectly by Interfering With T Cell Help. Clin Exp Immunol (2010) 159:199–207. doi: 10.1111/j.1365-2249.2009.04051.x
27. Steines L, Poth H, Schuster A, Amann K, Banas B, Bergler T. Disruption of Tfh:B Cell Interactions Prevents Antibody-Mediated Rejection in a Kidney Transplant Model in Rats: Impact of Calcineurin Inhibitor Dose. Front Immunol (2021) 12:657894. doi: 10.3389/fimmu.2021.657894
28. Karnell JL, Karnell F, Stephens GL, Rajan B, Morehouse C, Li Y, et al. Mycophenolic Acid Differentially Impacts B Cell Function Depending on the Stage of Differentiation. J Immunol (2011) 187:3603–12. doi: 10.4049/jimmunol.1003319
29. Strehl C, Ehlers L, Gaber T, Buttgereit F. Glucocorticoids-All-Rounders Tackling the Versatile Players of the Immune System. Front Immunol (2019) 10, 1744 doi: 10.3389/fimmu.2019.01744
30. Taves MD, Ashwell JD. Glucocorticoids in T Cell Development, Differentiation and Function. Nat Rev Immunol (2021) 21:233–43. doi: 10.1038/s41577-020-00464-0
31. Heidt S, Roelen DL, Eijsink C, Van Kooten C, Claas FH, Mulder A. Effects of Immunosuppressive Drugs on Purified Human B Cells: Evidence Supporting the Use of MMF and Rapamycin. Transplantation (2008) 86:1292–300. doi: 10.1097/TP.0b013e3181874a36
32. Coenen JJ, Koenen HJ, Van Rijssen E, Kasran A, Boon L, Hilbrands LB, et al. Rapamycin, Not Cyclosporine, Permits Thymic Generation and Peripheral Preservation of CD4+ CD25+ FoxP3+ T Cells. Bone Marrow Transplant (2007) 39:537–45. doi: 10.1038/sj.bmt.1705628
33. Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory Functions of mTOR Inhibition. Nat Rev Immunol (2009) 9:324–37. doi: 10.1038/nri2546
34. Xie A, Yan H, Fu J, He A, Xiao X, Li XC, et al. T Follicular Helper and Memory Cell Responses and the mTOR Pathway in Murine Heart Transplantation. J Heart Lung Transplant (2020) 39:134–44. doi: 10.1016/j.healun.2019.11.017
35. Ye L, Lee J, Xu L, Mohammed AU, Li W, Hale JS, et al. mTOR Promotes Antiviral Humoral Immunity by Differentially Regulating CD4 Helper T Cell and B Cell Responses. J Virol (2017) 91(4). doi: 10.1128/JVI.01653-16
36. Tuijnenburg P, Aan De Kerk DJ, Jansen MH, Morris B, Lieftink C, Beijersbergen RL, et al. High-Throughput Compound Screen Reveals mTOR Inhibitors as Potential Therapeutics to Reduce (Auto)Antibody Production by Human Plasma Cells. Eur J Immunol (2020) 50:73–85. doi: 10.1002/eji.201948241
37. Traitanon O, Mathew JM, La Monica G, Xu L, Mas V, Gallon L. Differential Effects of Tacrolimus Versus Sirolimus on the Proliferation, Activation and Differentiation of Human B Cells. PLoS One (2015) 10:e0129658. doi: 10.1371/journal.pone.0129658
38. Larsen CP, Pearson TC, Adams AB, Tso P, Shirasugi N, Strobert E, et al. Rational Development of LEA29Y (Belatacept), a High-Affinity Variant of CTLA4-Ig With Potent Immunosuppressive Properties. Am J Transplant (2005) 5:443–53. doi: 10.1111/j.1600-6143.2005.00749.x
39. Leibler C, Thiolat A, Henique C, Samson C, Pilon C, Tamagne M, et al. Control of Humoral Response in Renal Transplantation by Belatacept Depends on a Direct Effect on B Cells and Impaired T Follicular Helper-B Cell Crosstalk. J Am Soc Nephrol (2018) 29:1049–62. doi: 10.1681/ASN.2017060679
40. Kim EJ, Kwun J, Gibby AC, Hong JJ, Farris A, Iwakoshi NN, et al. Costimulation Blockade Alters Germinal Center Responses and Prevents Antibody-Mediated Rejection. Am J Transplant (2014) 14:59–69. doi: 10.1111/ajt.12526
41. Chen J, Yin H, Xu J, Wang Q, Edelblum KL, Sciammas R, et al. Reversing Endogenous Alloreactive B Cell GC Responses With Anti-CD154 or CTLA-4ig. Am J Transplant (2013) 13:2280–92. doi: 10.1111/ajt.12350
42. Young JS, Chen J, Miller ML, Vu V, Tian C, Moon JJ, et al. Delayed Cytotoxic T Lymphocyte-Associated Protein 4-Immunoglobulin Treatment Reverses Ongoing Alloantibody Responses and Rescues Allografts From Acute Rejection. Am J Transplant (2016) 16:2312–23. doi: 10.1111/ajt.13761
43. Klein U, Dalla-Favera R. Germinal Centres: Role in B-Cell Physiology and Malignancy. Nat Rev Immunol (2008) 8:22–33. doi: 10.1038/nri2217
44. Karahan GE, Claas FHJ, Heidt S. B Cell Immunity in Solid Organ Transplantation. Front Immunol (2017) 7:686. doi: 10.3389/fimmu.2016.00686
45. Cyster JG, Allen CDC. B Cell Responses: Cell Interaction Dynamics and Decisions. Cell (2019) 177:524–40. doi: 10.1016/j.cell.2019.03.016
46. Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-Lymphoid Lineage Depends on the Transcription Factor Pax5. Nature (1999) 401:556–62. doi: 10.1038/44076
47. Cariappa A, Chase C, Liu H, Russell P, Pillai S. Naive Recirculating B Cells Mature Simultaneously in the Spleen and Bone Marrow. Blood (2007) 109:2339–45. doi: 10.1182/blood-2006-05-021089
48. Schulz O, Hammerschmidt SI, Moschovakis GL, Forster R. Chemokines and Chemokine Receptors in Lymphoid Tissue Dynamics. Annu Rev Immunol (2016) 34:203–42. doi: 10.1146/annurev-immunol-041015-055649
49. Balazs M, Martin F, Zhou T, Kearney J. Blood Dendritic Cells Interact With Splenic Marginal Zone B Cells to Initiate T-Independent Immune Responses. Immunity (2002) 17:341–52. doi: 10.1016/S1074-7613(02)00389-8
50. Moens L, Tangye SG. Cytokine-Mediated Regulation of Plasma Cell Generation: IL-21 Takes Center Stage. Front Immunol (2014) 5:65. doi: 10.3389/fimmu.2014.00065
51. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 Mediates the Development of T Follicular Helper Cells. Science (2009) 325:1001–5. doi: 10.1126/science.1176676
52. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 Inhibits Germinal Center Formation by Limiting T Follicular Helper Cell Differentiation. Immunity (2012) 36:847–56. doi: 10.1016/j.immuni.2012.02.012
53. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The Generation of Antibody-Secreting Plasma Cells. Nat Rev Immunol (2015) 15:160–71. doi: 10.1038/nri3795
54. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-Controlled T-B Cell Interactions Underlie Germinal Centre Formation. Nature (2008) 455:764–9. doi: 10.1038/nature07345
55. Cannons JL, Qi H, Lu KT, Dutta M, Gomez-Rodriguez J, Cheng J, et al. Optimal Germinal Center Responses Require a Multistage T Cell:B Cell Adhesion Process Involving Integrins, SLAM-Associated Protein, and CD84. Immunity (2010) 32:253–65. doi: 10.1016/j.immuni.2010.01.010
56. Chenouard A, Chesneau M, Bui Nguyen L, Le Bot S, Cadoux M, Dugast E, et al. Renal Operational Tolerance Is Associated With a Defect of Blood Tfh Cells That Exhibit Impaired B Cell Help. Am J Transplant (2017) 17:1490–501. doi: 10.1111/ajt.14142
57. Cano-Romero FL, Laguna Goya R, Utrero-Rico A, Gomez-Massa E, Arroyo-Sanchez D, Suarez-Fernandez P, et al. Longitudinal Profile of Circulating T Follicular Helper Lymphocytes Parallels Anti-HLA Sensitization in Renal Transplant Recipients. Am J Transplant (2019) 19:89–97. doi: 10.1111/ajt.14987
58. La Muraglia G, Wagener ME, Ford ML, Badell IR. Circulating T Follicular Helper Cells are a Biomarker of Humoral Alloreactivity and Predict Donor-Specific Antibody Formation After Transplantation. Am J Transplant (2020) 20:75–87. doi: 10.1111/ajt.15517
59. Louis K, Macedo C, Bailly E, Lau L, Ramaswami B, Marrari M, et al. Coordinated Circulating T Follicular Helper and Activated B Cell Responses Underlie the Onset of Antibody-Mediated Rejection in Kidney Transplantation. J Am Soc Nephrol (2020) 31:2457–74. doi: 10.1681/ASN.2020030320
60. Ozaki K, Spolski R, Feng CG, Qi CF, Cheng J, Sher A, et al. A Critical Role for IL-21 in Regulating Immunoglobulin Production. Science (2002) 298:1630–4. doi: 10.1126/science.1077002
61. Ettinger R, Sims GP, Fairhurst AM, Robbins R, Da Silva YS, Spolski R, et al. IL-21 Induces Differentiation of Human Naive and Memory B Cells Into Antibody-Secreting Plasma Cells. J Immunol (2005) 175:7867–79. doi: 10.4049/jimmunol.175.12.7867
62. Fornek JL, Tygrett LT, Waldschmidt TJ, Poli V, Rickert RC, Kansas GS. Critical Role for Stat3 in T-Dependent Terminal Differentiation of IgG B Cells. Blood (2006) 107:1085–91. doi: 10.1182/blood-2005-07-2871
63. Khodadadi L, Cheng Q, Radbruch A, Hiepe F. The Maintenance of Memory Plasma Cells. Front Immunol (2019) 10:721. doi: 10.3389/fimmu.2019.00721
64. Wehmeier C, Honger G, Schaub S. Caveats of HLA Antibody Detection by Solid-Phase Assays. Transpl Int (2020) 33:18–29. doi: 10.1111/tri.13484
65. Loupy A, Lefaucheur C. Antibody-Mediated Rejection of Solid-Organ Allografts. New Engl J Med (2018) 379:1150–60. doi: 10.1056/NEJMra1802677
66. Tambur AR, Campbell P, Claas FH, Feng S, Gebel HM, Jackson AM, et al. Sensitization in Transplantation: Assessment of Risk (STAR) 2017 Working Group Meeting Report. Am J Transplant (2018) 18:1604–14. doi: 10.1111/ajt.14752
67. Yabu JM, Anderson MW, Kim D, Bradbury BD, Lou CD, Petersen J, et al. Sensitization From Transfusion in Patients Awaiting Primary Kidney Transplant. Nephrol Dial Transplant (2013) 28:2908–18. doi: 10.1093/ndt/gft362
68. Redfield RR, Scalea JR, Zens TJ, Mandelbrot DA, Leverson G, Kaufman DB, et al. The Mode of Sensitization and its Influence on Allograft Outcomes in Highly Sensitized Kidney Transplant Recipients. Nephrol Dialysis Transplant (2016) 31:1746–53. doi: 10.1093/ndt/gfw099
69. Honger G, Fornaro I, Granado C, Tiercy JM, Hosli I, Schaub S. Frequency and Determinants of Pregnancy-Induced Child-Specific Sensitization. Am J Transplant (2013) 13:746–53. doi: 10.1111/ajt.12048
70. Lucia M, Luque S, Crespo E, Melilli E, Cruzado JM, Martorell J, et al. Preformed Circulating HLA-Specific Memory B Cells Predict High Risk of Humoral Rejection in Kidney Transplantation. Kidney Int (2015) 88:874–87. doi: 10.1038/ki.2015.205
71. Karahan GE, Krop J, Wehmeier C, De Vaal YJH, Langerak-Langerak J, Roelen DL, et al. An Easy and Sensitive Method to Profile the Antibody Specificities of HLA-Specific Memory B Cells. Transplantation (2019) 103:716–23. doi: 10.1097/TP.0000000000002516
72. Karahan GE, De Vaal YJ, Roelen DL, Buchli R, Claas FH, Heidt S. Quantification of HLA Class II-Specific Memory B Cells in HLA-Sensitized Individuals. Hum Immunol (2015) 76:129–36. doi: 10.1016/j.humimm.2015.01.014
73. Stewart DE, Wilk AR, Toll AE, Harper AM, Lehman RR, Robinson AM, et al. Measuring and Monitoring Equity in Access to Deceased Donor Kidney Transplantation. Am J Transplant (2018) 18:1924–35. doi: 10.1111/ajt.14922
74. Houp JA, Schillinger KP, Eckstein AJ, Vega RM, Desai NM, Lonze BE, et al. Casting a Smaller Net Into a Bigger Donor Pool: A Single Center's Experience With the New Kidney Allocation System. Hum Immunol (2017) 78:49–53. doi: 10.1016/j.humimm.2016.11.004
75. Schinstock CA, Smith BH, Montgomery RA, Jordan SC, Bentall AJ, Mai M, et al. Managing Highly Sensitized Renal Transplant Candidates in the Era of Kidney Paired Donation and the New Kidney Allocation System: Is There Still a Role for Desensitization? Clin Transplant (2019) 33:e13751. doi: 10.1111/ctr.13751
76. Heidt S, Haasnoot GW, Van Rood JJ, Witvliet MD, Claas FHJ. Kidney Allocation Based on Proven Acceptable Antigens Results in Superior Graft Survival in Highly Sensitized Patients. Kidney Int (2018) 93:491–500. doi: 10.1016/j.kint.2017.07.018
77. Heidt S, Haasnoot GW, Witvliet MD, van der Linden-Van Oevelen MJH, Kamburova EG, Wisse BW, et al. Allocation to Highly Sensitized Patients Based on Acceptable Mismatches Results in Low Rejection Rates Comparable to Nonsensitized Patients. Am J Transplant (2019) 19:2926–33. doi: 10.1111/ajt.15486
78. Racusen LC, Colvin RB, Solez K, Mihatsch MJ, Halloran PF, Campbell PM, et al. Antibody-Mediated Rejection Criteria - an Addition to the Banff 97 Classification of Renal Allograft Rejection. Am J Transplant (2003) 3:708–14. doi: 10.1034/j.1600-6143.2003.00072.x
79. Hart A, Singh D, Brown SJ, Wang JH, Kasiske BL. Incidence, Risk Factors, Treatment, and Consequences of Antibody-Mediated Kidney Transplant Rejection: A Systematic Review. Clin Transplant (2021) 35:e14320. doi: 10.1111/ctr.14320
80. Haririan A, Nogueira J, Kukuruga D, Schweitzer E, Hess J, Gurk-Turner C, et al. Positive Cross-Match Living Donor Kidney Transplantation: Longer-Term Outcomes. Am J Transplant (2009) 9:536–42. doi: 10.1111/j.1600-6143.2008.02524.x
81. Haas M, Loupy A, Lefaucheur C, Roufosse C, Glotz D, Seron D, et al. The Banff 2017 Kidney Meeting Report: Revised Diagnostic Criteria for Chronic Active T Cell-Mediated Rejection, Antibody-Mediated Rejection, and Prospects for Integrative Endpoints for Next-Generation Clinical Trials. Am J Transplant (2018) 18:293–307. doi: 10.1111/ajt.14625
82. Loupy A, Haas M, Roufosse C, Naesens M, Adam B, Afrouzian M, et al. The Banff 2019 Kidney Meeting Report (I): Updates on and Clarification of Criteria for T Cell- and Antibody-Mediated Rejection. Am J Transplant (2020) 20:2318–31. doi: 10.1111/ajt.15898
83. Madill-Thomsen KS, Bohmig GA, Bromberg J, Einecke G, Eskandary F, Gupta G, et al. Donor-Specific Antibody Is Associated With Increased Expression of Rejection Transcripts in Renal Transplant Biopsies Classified as No Rejection. J Am Soc Nephrol (2021) 32:2743–58. doi: 10.1681/ASN.2021040433
84. Lefaucheur C, Loupy A, Hill GS, Andrade J, Nochy D, Antoine C, et al. Preexisting Donor-Specific HLA Antibodies Predict Outcome in Kidney Transplantation. J Am Soc Nephrol (2010) 21:1398–406. doi: 10.1681/ASN.2009101065
85. Aubert O, Loupy A, Hidalgo L, Duong Van Huyen JP, Higgins S, Viglietti D, et al. Antibody-Mediated Rejection Due to Preexisting Versus De Novo Donor-Specific Antibodies in Kidney Allograft Recipients. J Am Soc Nephrol (2017) 28:1912–23. doi: 10.1681/ASN.2016070797
86. Senev A, Coemans M, Lerut E, Van Sandt V, Daniels L, Kuypers D, et al. Histological Picture of Antibody-Mediated Rejection Without Donor-Specific Anti-HLA Antibodies: Clinical Presentation and Implications for Outcome. Am J Transplant (2019) 19:763–80. doi: 10.1111/ajt.15074
87. Callemeyn J, Lerut E, De Loor H, Arijs I, Thaunat O, Koenig A, et al. Transcriptional Changes in Kidney Allografts With Histology of Antibody-Mediated Rejection Without Anti-HLA Donor-Specific Antibodies. J Am Soc Nephrol (2020) 31:2168–83. doi: 10.1681/ASN.2020030306
88. Rush D, Nickerson P, Gough J, Mckenna R, Grimm P, Cheang M, et al. Beneficial Effects of Treatment of Early Subclinical Rejection: A Randomized Study. J Am Soc Nephrol (1998) 9:2129–34. doi: 10.1681/ASN.V9112129
89. Loupy A, Vernerey D, Tinel C, Aubert O, Duong Van Huyen JP, Rabant M, et al. Subclinical Rejection Phenotypes at 1 Year Post-Transplant and Outcome of Kidney Allografts. J Am Soc Nephrol (2015) 26:1721–31. doi: 10.1681/ASN.2014040399
90. Westphal SG, Mannon RB. Emerging Biomarkers in Kidney Transplantation and Challenge of Clinical Implementation. Curr Opin Organ Transplant (2022) 27:15–21. doi: 10.1097/MOT.0000000000000941
91. Furness PN, Taub N, Convergence of European Renal Transplant Pathology Assessment Procedures P. International Variation in the Interpretation of Renal Transplant Biopsies: Report of the CERTPAP Project. Kidney Int (2001) 60:1998–2012. doi: 10.1046/j.1523-1755.2001.00030.x
92. Barisoni L, Lafata KJ, Hewitt SM, Madabhushi A, Balis UGJ. Digital Pathology and Computational Image Analysis in Nephropathology. Nat Rev Nephrol (2020) 16:669–85. doi: 10.1038/s41581-020-0321-6
93. Hermsen M, Smeets B, Hilbrands L, van der Laak J. Artificial Intelligence: Is There a Potential Role in Nephropathology? Nephrol Dial Transplant (2022) 37:438–40. doi: 10.1093/ndt/gfaa181
94. Farris AB, Moghe I, Wu S, Hogan J, Cornell LD, Alexander MP, et al. Banff Digital Pathology Working Group: Going Digital in Transplant Pathology. Am J Transplant (2020) 20:2392–9. doi: 10.1111/ajt.15850
95. Archdeacon P, Chan M, Neuland C, Velidedeoglu E, Meyer J, Tracy L, et al. Summary of FDA Antibody-Mediated Rejection Workshop. Am J Transplant (2011) 11:896–906. doi: 10.1111/j.1600-6143.2011.03525.x
96. Wan SS, Ying TD, Wyburn K, Roberts DM, Wyld M, Chadban SJ. The Treatment of Antibody-Mediated Rejection in Kidney Transplantation: An Updated Systematic Review and Meta-Analysis. Transplantation (2018) 102:557–68. doi: 10.1097/TP.0000000000002049
97. Schinstock CA, Mannon RB, Budde K, Chong AS, Haas M, Knechtle S, et al. Recommended Treatment for Antibody-Mediated Rejection After Kidney Transplantation: The 2019 Expert Consensus From the Transplantion Society Working Group. Transplantation (2020) 104:911–22. doi: 10.1097/TP.0000000000003095
98. Kim MY, Brennan DC. Therapies for Chronic Allograft Rejection. Front Pharmacol (2021) 12:651222. doi: 10.3389/fphar.2021.651222
99. Olaso D, Manook M, Moris D, Knechtle S, Kwun J. Optimal Immunosuppression Strategy in the Sensitized Kidney Transplant Recipient. J Clin Med (2021) 10(16):3656. doi: 10.3390/jcm10163656
100. Anwar IJ, Srinivas TR, Gao Q, Knechtle SJ. Shifting Clinical Trial Endpoints in Kidney Transplantation: The Rise of Composite Endpoints and Machine Learning to Refine Prognostication. Transplantation (2022). doi: 10.1097/TP.0000000000004107
101. Hellemans R, Bosmans JL, Abramowicz D. Induction Therapy for Kidney Transplant Recipients: Do We Still Need Anti-IL2 Receptor Monoclonal Antibodies? Am J Transplant (2017) 17:22–7. doi: 10.1111/ajt.13884
102. Kidney Disease: Improving Global Outcomes Transplant Work, G. KDIGO Clinical Practice Guideline for the Care of Kidney Transplant Recipients. Am J Transplant (2009) 9 Suppl 3:S1–155. doi: 10.1111/j.1600-6143.2009.02834.x
103. Castaneda J, Hidalgo Y, Sauma D, Rosemblatt M, Bono MR, Nunez S. The Multifaceted Roles of B Cells in the Thymus: From Immune Tolerance to Autoimmunity. Front Immunol (2021) 12:766698. doi: 10.3389/fimmu.2021.766698
104. Morelon E, Lefrancois N, Besson C, Prevautel J, Brunet M, Touraine JL, et al. Preferential Increase in Memory and Regulatory Subsets During T-Lymphocyte Immune Reconstitution After Thymoglobulin Induction Therapy With Maintenance Sirolimus vs Cyclosporine. Transpl Immunol (2010) 23:53–8. doi: 10.1016/j.trim.2010.04.004
105. Kho MM, Bouvy AP, Cadogan M, Kraaijeveld R, Baan CC, Weimar W. The Effect of Low and Ultra-Low Dosages Thymoglobulin on Peripheral T, B and NK Cells in Kidney Transplant Recipients. Transpl Immunol (2012) 26:186–90. doi: 10.1016/j.trim.2012.02.003
106. Svachova V, Sekerkova A, Hruba P, Tycova I, Rodova M, Cecrdlova E, et al. Dynamic Changes of B-Cell Compartments in Kidney Transplantation: Lack of Transitional B Cells is Associated With Allograft Rejection. Transpl Int (2016) 29:540–8. doi: 10.1111/tri.12751
107. Alfaro R, Legaz I, Gonzalez-Martinez G, Jimenez-Coll V, Martinez-Banaclocha H, Galian JA, et al. Monitoring of B Cell in Kidney Transplantation: Development of a Novel Clusters Analysis and Role of Transitional B Cells in Transplant Outcome. Diagn (Basel) (2021) 11(4):641. doi: 10.3390/diagnostics11040641
108. Kamburova EG, Koenen HJ, Borgman KJ, Ten Berge IJ, Joosten I, Hilbrands LB. A Single Dose of Rituximab Does Not Deplete B Cells in Secondary Lymphoid Organs But Alters Phenotype and Function. Am J Transplant (2013) 13:1503–11. doi: 10.1111/ajt.12220
109. Ekberg H, Tedesco-Silva H, Demirbas A, Vitko S, Nashan B, Gurkan A, et al. Reduced Exposure to Calcineurin Inhibitors in Renal Transplantation. N Engl J Med (2007) 357:2562–75. doi: 10.1056/NEJMoa067411
110. Rhen T, Cidlowski JA. Antiinflammatory Action of Glucocorticoids–New Mechanisms for Old Drugs. N Engl J Med (2005) 353:1711–23. doi: 10.1056/NEJMra050541
111. Song J, Salek-Ardakani S, So T, Croft M. The Kinases Aurora B and mTOR Regulate the G1-S Cell Cycle Progression of T Lymphocytes. Nat Immunol (2007) 8:64–73. doi: 10.1038/ni1413
112. Damoiseaux JG, Defresne MP, Reutelingsperger CP, Van Breda Vriesman PJ. Cyclosporin-A Differentially Affects Apoptosis During In Vivo Rat Thymocyte Maturation. Scand J Immunol (2002) 56:353–60. doi: 10.1046/j.1365-3083.2002.01110.x
113. Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al. Phosphatidylinositol-3-OH Kinase and Nutrient-Sensing mTOR Pathways Control T Lymphocyte Trafficking. Nat Immunol (2008) 9:513–21. doi: 10.1038/ni.1603
114. Zhang S, Readinger JA, Dubois W, Janka-Junttila M, Robinson R, Pruitt M, et al. Constitutive Reductions in mTOR Alter Cell Size, Immune Cell Development, and Antibody Production. Blood (2011) 117:1228–38. doi: 10.1182/blood-2010-05-287821
115. Chen J, Wang Q, Yin D, Vu V, Sciammas R, Chong AS. Cutting Edge: CTLA-4ig Inhibits Memory B Cell Responses and Promotes Allograft Survival in Sensitized Recipients. J Immunol (2015) 195:4069–73. doi: 10.4049/jimmunol.1500940
116. Durrbach A, Pestana JM, Pearson T, Vincenti F, Garcia VD, Campistol J, et al. A Phase III Study of Belatacept Versus Cyclosporine in Kidney Transplants From Extended Criteria Donors (BENEFIT-EXT Study). Am J Transplant (2010) 10:547–57. doi: 10.1111/j.1600-6143.2010.03016.x
117. Masson P, Henderson L, Chapman JR, Craig JC, Webster AC. Belatacept for Kidney Transplant Recipients. Cochrane Database Syst Rev (2014) 11, CD010699. doi: 10.1002/14651858.CD010699.pub2
118. Kirk AD, Guasch A, Xu H, Cheeseman J, Mead SI, Ghali A, et al. Renal Transplantation Using Belatacept Without Maintenance Steroids or Calcineurin Inhibitors. Am J Transplant (2014) 14:1142–51. doi: 10.1111/ajt.12712
119. Bray RA, Gebel HM, Townsend R, Roberts ME, Polinsky M, Yang L, et al. De Novo Donor-Specific Antibodies in Belatacept-Treated vs Cyclosporine-Treated Kidney-Transplant Recipients: Post Hoc Analyses of the Randomized Phase III BENEFIT and BENEFIT-EXT Studies. Am J Transplant (2018) 18:1783–9. doi: 10.1111/ajt.14721
120. Kirk AD, Harlan DM, Armstrong NN, Davis TA, Dong Y, Gray GS, et al. CTLA4-Ig and Anti-CD40 Ligand Prevent Renal Allograft Rejection in Primates. Proc Natl Acad Sci U S A (1997) 94:8789–94. doi: 10.1073/pnas.94.16.8789
121. Kirk AD, Burkly LC, Batty DS, Baumgartner RE, Berning JD, Buchanan K, et al. Treatment With Humanized Monoclonal Antibody Against CD154 Prevents Acute Renal Allograft Rejection in Nonhuman Primates. Nat Med (1999) 5:686–93. doi: 10.1038/9536
122. Kawai T, Andrews D, Colvin RB, Sachs DH, Cosimi AB. Thromboembolic Complications After Treatment With Monoclonal Antibody Against CD40 Ligand. Nat Med (2000) 6:114. doi: 10.1038/72162
123. Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, et al. A Short Course of BG9588 (Anti-CD40 Ligand Antibody) Improves Serologic Activity and Decreases Hematuria in Patients With Proliferative Lupus Glomerulonephritis. Arthritis Rheum (2003) 48:719–27. doi: 10.1002/art.10856
124. Koyama I, Kawai T, Andrews D, Boskovic S, Nadazdin O, Wee SL, et al. Thrombophilia Associated With Anti-CD154 Monoclonal Antibody Treatment and its Prophylaxis in Nonhuman Primates. Transplantation (2004) 77:460–2. doi: 10.1097/01.TP.0000110291.29370.C0
125. Schroder PM, Fitch ZW, Schmitz R, Choi AY, Kwun J, Knechtle SJ. The Past, Present, and Future of Costimulation Blockade in Organ Transplantation. Curr Opin Organ Transplant (2019) 24:391–401. doi: 10.1097/MOT.0000000000000656
126. Van Kooten C, Banchereau J. CD40-CD40 Ligand. J Leukoc Biol (2000) 67:2–17. doi: 10.1002/jlb.67.1.2
127. Imai A, Suzuki T, Sugitani A, Itoh T, Ueki S, Aoyagi T, et al. A Novel Fully Human Anti-CD40 Monoclonal Antibody, 4D11, for Kidney Transplantation in Cynomolgus Monkeys. Transplantation (2007) 84:1020–8. doi: 10.1097/01.tp.0000286058.79448.c7
128. Ristov J, Espie P, Ulrich P, Sickert D, Flandre T, Dimitrova M, et al. Characterization of the In Vitro and In Vivo Properties of CFZ533, a Blocking and non-Depleting Anti-CD40 Monoclonal Antibody. Am J Transplant (2018) 18:2895–904. doi: 10.1111/ajt.14872
129. Cordoba F, Wieczorek G, Audet M, Roth L, Schneider MA, Kunkler A, et al. A Novel, Blocking, Fc-Silent Anti-CD40 Monoclonal Antibody Prolongs Nonhuman Primate Renal Allograft Survival in the Absence of B Cell Depletion. Am J Transplant (2015) 15:2825–36. doi: 10.1111/ajt.13377
130. Vincenti F, Rostaing L, Grinyo J, Rice K, Steinberg S, Gaite L, et al. Belatacept and Long-Term Outcomes in Kidney Transplantation. N Engl J Med (2016) 374:333–43. doi: 10.1056/NEJMoa1506027
131. Vincenti F, Larsen C, Durrbach A, Wekerle T, Nashan B, Blancho G, et al. Costimulation Blockade With Belatacept in Renal Transplantation. N Engl J Med (2005) 353:770–81. doi: 10.1056/NEJMoa050085
132. Vincenti F, Charpentier B, Vanrenterghem Y, Rostaing L, Bresnahan B, Darji P, et al. A Phase III Study of Belatacept-Based Immunosuppression Regimens Versus Cyclosporine in Renal Transplant Recipients (BENEFIT Study). Am J Transplant (2010) 10:535–46. doi: 10.1111/j.1600-6143.2009.03005.x
133. Rostaing L, Massari P, Garcia VD, Mancilla-Urrea E, Nainan G, Del Carmen Rial M, et al. Switching From Calcineurin Inhibitor-Based Regimens to a Belatacept-Based Regimen in Renal Transplant Recipients: A Randomized Phase II Study. Clin J Am Soc Nephrol (2011) 6:430–9. doi: 10.2215/CJN.05840710
134. Espinosa J, Herr F, Tharp G, Bosinger S, Song M, Farris Iii AB, et al. CD57+ CD4 T Cells Underlie Belatacept-Resistant Allograft Rejection. Am J Transplant (2016) 16:1102–12. doi: 10.1111/ajt.13613
135. Shaw BI, Espinosa JR, Stempora L, Miller A, Adams B, Kirk AD. Functional Characteristics and Phenotypic Plasticity of CD57 + PD1 – CD4 T Cells and Their Relationship With Transplant Immunosuppression. J Immunol (2021) 206:1668–76. doi: 10.4049/jimmunol.2000736
136. Cortes-Cerisuelo M, Laurie SJ, Mathews DV, Winterberg PD, Larsen CP, Adams AB, et al. Increased Pretransplant Frequency of CD28(+) CD4(+) TEM Predicts Belatacept-Resistant Rejection in Human Renal Transplant Recipients. Am J Transplant (2017) 17:2350–62. doi: 10.1111/ajt.14350
137. Mathews DV, Wakwe WC, Kim SC, Lowe MC, Breeden C, Roberts ME, et al. Belatacept-Resistant Rejection Is Associated With CD28(+) Memory CD8 T Cells. Am J Transplant (2017) 17:2285–99. doi: 10.1111/ajt.14349
138. Kirk AD, Adams AB, Durrbach A, Ford ML, Hildeman DA, Larsen CP, et al. Optimization of De Novo Belatacept-Based Immunosuppression Administered to Renal Transplant Recipients. Am J Transplant (2021) 21:1691–8. doi: 10.1111/ajt.16386
139. Nickerson PW. What Have We Learned About How to Prevent and Treat Antibody-Mediated Rejection in Kidney Transplantation? Am J Transplant (2020) 20 Suppl 4:12–22. doi: 10.1111/ajt.15859
140. Nickerson P, Böhmig G, Chadban S, Kumar D, Mannon R, Gelder T, et al. Clazakizumab for the Treatment of Chronic Active Antibody-Mediated Rejection in Kidney Transplant Recipients: Phase 3 IMAGINE Study Design. Am J Transplant (2021) 21(suppl 3).
141. Doberer K, Duerr M, Halloran PF, Eskandary F, Budde K, Regele H, et al. A Randomized Clinical Trial of Anti-IL-6 Antibody Clazakizumab in Late Antibody-Mediated Kidney Transplant Rejection. J Am Soc Nephrol (2021) 32:708–22. doi: 10.1681/ASN.2020071106
142. Kwun J, Burghuber C, Manook M, Ezekian B, Park J, Yoon J, et al. Successful Desensitization With Proteasome Inhibition and Costimulation Blockade in Sensitized Nonhuman Primates. Blood Adv (2017) 1:2115–9. doi: 10.1182/bloodadvances.2017010991
143. Burghuber CK, Manook M, Ezekian B, Gibby AC, Leopardi FV, Song M, et al. Dual Targeting: Combining Costimulation Blockade and Bortezomib to Permit Kidney Transplantation in Sensitized Recipients. Am J Transplant (2019) 19:724–36. doi: 10.1111/ajt.15067
144. Ezekian B, Schroder PM, Mulvihill MS, Barbas A, Collins B, Freischlag K, et al. Pretransplant Desensitization With Costimulation Blockade and Proteasome Inhibitor Reduces DSA and Delays Antibody-Mediated Rejection in Highly Sensitized Nonhuman Primate Kidney Transplant Recipients. J Am Soc Nephrol (2019) 30:2399–411. doi: 10.1681/ASN.2019030304
145. Loupy A, Aubert O, Orandi BJ, Naesens M, Bouatou Y, Raynaud M, et al. Prediction System for Risk of Allograft Loss in Patients Receiving Kidney Transplants: International Derivation and Validation Study. BMJ (2019) 366:l4923. doi: 10.1136/bmj.l4923
146. Mayer KA, Budde K, Halloran PF, Doberer K, Rostaing L, Eskandary F, et al. Safety, Tolerability, and Efficacy of Monoclonal CD38 Antibody Felzartamab in Late Antibody-Mediated Renal Allograft Rejection: Study Protocol for a Phase 2 Trial. Trials (2022) 23:270. doi: 10.1186/s13063-022-06198-9
Keywords: B cell, antibody-mediated rejection, clinical trial, sensitization, kidney transplantation, immunosuppression
Citation: Anwar IJ, DeLaura IF, Gao Q, Ladowski J, Jackson AM, Kwun J and Knechtle SJ (2022) Harnessing the B Cell Response in Kidney Transplantation – Current State and Future Directions. Front. Immunol. 13:903068. doi: 10.3389/fimmu.2022.903068
Received: 23 March 2022; Accepted: 25 April 2022;
Published: 09 June 2022.
Edited by:
Mayara Garcia De Mattos Barbosa, University of Michigan, United StatesReviewed by:
Georg Böhmig, Medical University of Vienna, AustriaYoshiko Matsuda, National Center for Child Health and Development (NCCHD), Japan
Noble Johan, Centre Hospitalier Universitaire de Grenoble, France
Copyright © 2022 Anwar, DeLaura, Gao, Ladowski, Jackson, Kwun and Knechtle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart J. Knechtle, c3R1YXJ0LmtuZWNodGxlQGR1a2UuZWR1