Erratum: The efficacy of cancer immunotherapies is compromised by Helicobacter pylori infection
 Paul Oster
Paul Oster Brynn McMillan
Brynn McMillan Dominique Velin
Dominique Velin- Service of Gastroenterology and Hepatology, Centre Hospitalier Universitaire Vaudois, University of Lausanne, Lausanne, Switzerland
Helicobacter pylori infects the gastric mucosa of a large number of humans. Although asymptomatic in the vast majority of cases, H pylori infection can lead to the development of peptic ulcers gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma. Using a variety of mechanisms, H pylori locally suppresses the function of the host immune system to establish chronic infection. Systemic immunomodulation has been observed in both clinical and pre-clinical studies, which have demonstrated that H pylori infection is associated with reduced incidence of inflammatory diseases, such as asthma and Crohn’s disease. The introduction of immunotherapies in the arsenal of anti-cancer drugs has revealed a new facet of H pylori-induced immune suppression. In this review, we will describe the intimate interactions between H pylori and its host, and formulate hypothtyeses describing the detrimental impact of H pylori infection on the efficacy of cancer immunotherapies.
Introduction
Antitumor immunity is a critical concept in oncology that refers to the ability of the immune system to prevent the formation, growth and metastases of tumors (1). Following the recognition of the cellular and genetic alterations that characterize cancer cells, the immune system is capable of generating effector T cell responses that function to eliminate neoplastic cells (2). The generation of tumor-specific immune responses begins when antigen presenting cells (APCs), such as dendritic cells (DCs), capture tumor-specific antigens that have been released by cancer cells and migrate to secondary lymphoid structure(s) which drain the tumor site (2). The processed antigen is then cross-presented by DCs most commonly via major histocompatibility complex (MHC) class I molecules to CD8+ T cells, allowing for their activation, differential expansion and subsequent migration to the tumor site (2, 3). Once at the tumor site, activated CD8+ T cells recognize the tumor-specific antigen present at the surface of the cancer cell through the interaction between the antigenic peptide-MHC class I complex and the T cell receptor (TCR) (2). This interaction induces the release of highly cytotoxic molecules from CD8+ T cells including perforin, granzyme, tumor necrosis factor (TNF) α and interferon (IFN) γ, which leads to the induction of cancer cell death (4). A number of individualistic factors such as tumor and germline genetics, age, and the presence of infectious agents may impact the efficacy of this antitumor response (2). Several additional immune cell populations are implicated in antitumor immunity including CD4+ T cells, natural killer cells (NK cells), T regulatory cells (Treg cells), B cells, various DC subsets, macrophages, monocytes and myeloid-derived suppressor cells (MDSCs).
Cancer immunotherapy is a form of cancer treatment that aims to increase antitumor immunity (5). Currently, there exists six major classes of immunotherapies including lymphocyte promoting cytokines, chimeric antigen receptor (CAR-T) T cells, T cell receptor-engineered T cell (TCR-T), costimulatory receptor agonists, anticancer vaccines and immune checkpoint inhibitors (6, 7). Immune checkpoint receptors, including Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and Programmed cell death 1(PD-1)/Programmed cell death-ligand 1 (PD-L1), are coinhibitory receptors that function in the attenuation of immune cell activation and have recently become a main target in the treatment of cancer (8). Increasing the amount of cytotoxic tumor-infiltrating lymphocytes using vaccination strategies as well as blocking the natural function of the CTLA4 and/or PD-1/PD-L1 immune checkpoints are attractive approaches for anticancer therapies as they enable the host to mount an appropriate immune response to counteract tumor formation, growth and metastasis. Presently, there exist two main cancer immune checkpoint blockade therapies that are approved for clinical use for a number of tumor types, including anti-CTLA4 (ipilimumab) and anti-PD-1/PD-L1 (pembrolizumab, nivolumab/atezolizumab), each of which blocks the attenuation of T cell activity, thereby inducing the expansion of CD8+ T cells and differentially affecting CD4+ T cells (9). Importantly, only a subset of patients respond to these therapies, owing to individualistic factors affecting anticancer immunity as previously described, with additional factors such as the composition of the gut microbiota (2).
Microbiota: A Key Component Influencing the Efficacy of Cancer Immunotherapy
The microbiota represents a highly complex symbiotic relationship between the human body and microorganisms including bacteria, archaea, eukarya and fungi (10, 11). The gut microbiota, referring to the microbial communities along the gastrointestinal tract, functions fundamentally in the maintenance of metabolic homeostasis alongside protecting the body against pathogens (10). Previous studies strongly suggest that the presence of the gut microbiota is in fact necessary in conferring the anticancer effects elicited by anti-CTLA4 immunotherapy (12). Using mice subcutaneously inoculated with cancer cell lines, Vetizou and colleagues demonstrated that mice housed in specific pathogen-free conditions with fully intact microbiotas, had markedly improved tumor control relative to mice housed in germ free conditions, which lack a functional microbiota, following the administration of anti-CTLA4 immunotherapy (12). More particularly, Vetizou et al. demonstrated that the antitumor effects elicited by CTLA4 blockade in mice were dependent on the presence of Burkholderiales Bacteroides fragilis and Bacteroides thetaiotaomicron in the gut microbiota (12). Additional bacteria such as Faecalibacterium prausnitzii, Akkermansia muciniphila, and Bifidobacterium species were also shown to potentiate the efficacy of immune checkpoint inhibitors (13, 14). Importantly, clinical data clearly demonstrate that the intestinal microbiome modulates the anti-cancer immune response (13, 14) and that antibiotics compromise the efficacy of PD-1 blockade therapy (15). Finally, it was recently described that fecal microbiota transplants overcome the resistance to anti–PD-1 therapy in 30% of melanoma patients (16, 17). Collectively, pre-clinical and clinical data demonstrate that the gut microbiota plays an important role in enhancing the anti-cancer immune response following cancer immunotherapies and therefore, is a major research focus for the development of precision medicine and for the improvement of immunotherapy effectiveness.
As described above, some bacterial species can enhance the effectiveness of cancer immunotherapy, whereas other bacterial species have the opposite effect (18). We recently demonstrated that Helicobacter pylori is one of the bacterial species which decreases the efficacy of cancer immunotherapies (19). In this review, we describe Helicobacter pylori and its host interactions, and discuss the different hypotheses explaining the decrease in efficacy of cancer immunotherapies in H pylori infected hosts.
Helicobacter pylori
H pylori are gram-negative microaerophilic bacteria that are largely successful in infecting the stomach mucosa of up to 50% of humans (20). Most commonly colonizing during childhood and persisting throughout life, H pylori has been described to have coevolved with humans for nearly 60,000 years (21). The majority of infected individuals remain asymptomatic, suggesting that H pylori has become well adapted to the acidic environment of the gastric epithelium (20, 21). H pylori infection can be detected by stool antigen test, serology, urea breath test and/or via gastric biopsy (22). H pylori infection takes place mainly in the family circle and is transmitted through ingesting saliva, vomit or fecal matter (23). Upon entering the stomach, the flagella and spiral shape of the bacterium allows for the penetration of the mucus gastric layer (24). Because a minute number of bacteria traverse the gastric epithelial barrier, H pylori is considered a non-invasive bacteria. Most H pylori are living in the mucus, but some organisms adhere to the gastric epithelial cells (25) and even have been detected inside the cells (26). Humans are infected by 104 to 107 H pylori colony forming units (CFU) per gram of gastric mucus (27). Urease and α-carbonic anhydrase of H pylori generate ammonia and HCO32-, which increase the low pH of the gastric juice (28, 29). Outer membrane proteins, such as blood group antigen binding adhesin A (BabA), sialic acid-binding adherence (SabA), adherence-associated lipoprotein A/B (AlpA & AlpB) and H pylori outer membrane protein Z (HopZ) mediate attachment to gastric epithelial cells (30). H pylori effector molecules, such as vacuolating cytotoxin (VacA), cytotoxin- associated gene A (CagA) and components of the type IV secretion system (CagL) modulate the function of gastric epithelial cells, leading to the release of chemokines and nutrients and to the modification of acid secretion (31, 32).
H pylori Infection May Lead to the Development of Gastritis, Peptic Ulcers and Gastric Cancers
H pylori infection can be asymptomatic or symptomatic and may conduct to the development of gastritis, peptic ulcers and gastric cancer (20) (Table 1). Colonization by H pylori induces an inflammatory response, predominantly of the Th1 type. If the infection is not eradicated, chronic gastritis can be life-long (33). Nevertheless, the vast majority of H pylori-positive subjects do not suffer from any symptoms and are unaware of the presence of this inflammation in their stomach. The chronic gastritis generates reactive oxygen species that can give rise to DNA damage and consequently initiate the cancer cascade (chronic gastritis-gastric atrophy-intestinal metaplasia-dysplasia-gastric cancer) (34). The tumorigenic role of H pylori is further supported by data showing that eradication of H pylori reduces the risk of developing H pylori–driven gastric cancer (35, 36). However, depending of the stage of the H pylori-induced diseases, patients might not benefit from the H pylori treatment on the risk to develop gastric cancer (37).
TABLE 1

Table 1 Helicobacterpylori infection promotes or reduces human pathologies.
Peptic ulcers are acid-induced lesions found in the stomach and duodenum, characterized by denuded mucosa (38). Ulcers develop at sites where gastric inflammation is most severe and a variety of host and bacterial factors govern the severity of ulcerations (39).
Chronic inflammation can lead to the destruction of gastric glands and the development of fibrosis and intestinal metaplasia (34). Half of the H pylori-colonized population develops atrophic gastritis and intestinal metaplasia (34). Patients with low acid output show a rapid progression towards atrophy. Moreover, the multifocal extension of intestinal metaplasia and gastric atrophy will increase the risk of gastric cancer development (34). The development of gastric atrophy or cancer is modulated by host and bacterial factors that cause chronic inflammation, endoplasmic reticulum stress, autophagy, and oxidative stress in the gastric epithelium (40). The risk is increased in CagA/VacA-positive strains infected patients (40–42), and also in those with high IL-1 production in response to H pylori infection (40, 43). The VacA gene is present in most all H pylori strains. However, depending on the strain, the toxic activity of VacA may differ. The identified epithelial cells VacA receptors include receptor protein tyrosine phosphatases α (RPTPα) and β (RPTPβ), and low‐density lipoprotein receptor‐related protein‐1 (LRP1) (44). The Type IV secretion system (T4SS) delivers CagA into epithelial cells. This pilus-like structure interacts with β1 integrin at the surface of epithelial cells (30). Injected CagA hijacks multiple intra-cellular signal transduction cascades modulating key pathways involved in the control of apoptosis and self-renewal of the gastric epithelium (45).
Lymphoid tissue is not normally present in the gastric mucosa, but in response to H pylori colonization mucosal associated lymphoid tissue (MALT) nearly always develops (46). Rarely, B lymphoma may develop in those MALT structures. Remarkably, following H pylori eradication a complete remission is observed in approximately 80% of these patients (47).
H pylori Infection Protects From Gastro-Esophageal Reflux Disease or Esophagitis
Only a very small proportion of patients infected with H pylori develop pathologies as described above and paradoxically, studies associate the decrease in the rate of H pylori infection with an increased incidence of several diseases (Table 2). Indeed, H pylori eradication was reported to be associated with gastro-esophageal reflux disease (GERD) or esophagitis (48). The chronic exposure to acid secretion from the stomach of the esophageal mucosa induces GERD. It is hypothesized that the H pylori-induced inflammation in the gastric corpus decreases the acid output and prevents patients from contracting GERD (49). Another detrimental association with H pylori eradication is weight gain (50). Neuroendocrine cells in the gastric epithelium produce hormones that regulate body weight. Among them, Ghrelin promotes weight gain by decreasing energy consumption whilst leptin increases energy consumption. H pylori infection is associated with an increase in plasma leptin levels and a decrease in Ghrelin levels (51). It is believed that the inflammation induced by H pylori colonization interferes with those neuroendocrine cells by altering the epithelial cell turnover (51).
TABLE 2

Table 2 Helicobacter pylori infection reduces the efficacy of cancer immunotherapies.
H pylori Infection Protects From Systemic Inflammatory Diseases
A recently well described positive side effect of H pylori infection is protection against asthma and eosinophilic esophagitis (EoE). Asthma is an immune disease in which allergens trigger a Th2-dependent immune response in the lungs, leading to massive infiltration of eosinophils and mast cells, hyper production of mucus and airway obstruction (52–54). EoE is also an immune disease characterized by esophageal dysfunction, eosinophilic inflammation, a thickened mucosa, muscle layer hyperplasia and collagen deposition (55, 56). Impaired epithelial barrier function and food and aeroallergen hypersensitivity trigger EoE symptoms (57). Incidence of asthma and EoE is growing in developed countries and is commonly explained by the hygiene hypothesis (58, 59). This hypothesis is based on the increased hygiene standard and urbanization that is associated with modern lifestyle. Under these conditions, it is thought that humans are exposed to less pathogens or commensals, especially at a young age, leading to the development of autoimmune/inflammatory diseases later in life (60). Several epidemiological studies in the past decade have inversely correlated asthma, multiple sclerosis and EoE with H pylori infection (61–64). A potential mechanism of protection against asthma came from a recent study demonstrating that H pylori infection, along with VacA and γ-glutamyl transpeptidase (γGT) activity, is able to interfere with dendritic cell (DC) maturation. These immature DCs fail to induce a proinflammatory response but promote Treg cell differentiation. Both immature DCs and Treg cells were shown to be able to migrate to the lungs and exhibit their tolerogenic phenotype, inducing a reduction in immune activation and development of asthma (65). Moreover, immunoregulatory activity of H pylori infection has been shown also in the context of inflammatory bowel diseases (IBD). H pylori DNA was able to reduce colitis induced by Dextran Sulfate Sodium (DSS) in mice (66). NOD-like receptor pyrin domain-containing-3 (NLRP3)-dependent IL-18 production induced by H pylori has been shown to be critical in the reduction of this colitis severity, by H pylori infection in this DSS model (67). Cross-sectional studies also showed a positive correlation between H pylori loss at the population level in developed countries with increased incidence of IBD (68).
H pylori Infection Decreases the Efficacy of Immune Checkpoint Inhibitors
We recently hypothesized that H pylori infection may decrease the effectiveness of ICI therapies. This hypothesis was based on the observation that gut microbiota has been identified as a key driver of ICI efficacy and that as described above H pylori actively suppresses the functioning of the immune system (20). Indeed, H pylori displays major immunomodulators such as CagA, Lipopolysaccharide (LPS), flagelin, VacA, γGGT and urease among others which differentially impact a number of immune cells in the gastric mucosa as well as systemically (20, 69). Noteworthy, H pylori has been shown to modulate key immune cells involved in the generation of the anti-tumor responses. Indeed, H pylori was shown to decrease the activities of Th1 and Th17 cells, DCs, macrophages and Natural killer T cells (NKT cells) and to promote the activities of MDSC and Treg cells, suggesting that H pylori infected hosts might be less responsive to ICI therapies (19).
H pylori Infection Decreases the Effectiveness of Cancer Immunotherapies in Pre-Clinical Models
In order to evaluate whether H pylori decreases ICIs efficacy, we tested whether H pylori reduce the effectiveness of anti-CTLA4 and/or anti-PD-L1 therapy in the MC38 colon adenocarcinoma model. Interestingly, the tumor volumes of H pylori infected mice undergoing treatment were significantly larger than those of non-infected mice (19). Furthermore, we tested whether H pylori reduces the effectiveness of a cancer vaccine in the B16-OVA melanoma model. Mice subcutaneously injected with B16-OVA melanoma cells were intravenously administered with CD8+ T cells specific for OVA and vaccinated with OVA peptide adjuvanted in CpG (70). Remarkably, the tumor volumes of vaccinated infected mice were significantly larger than those of vaccinated non-infected mice, showing that anti-cancer vaccination is less efficient in H pylori infected mice (19). Lastly, by using the colon cancer model induces by the administration of azoxymethane (AOM) and DSS, we evaluated the effect of H pylori infection on immunotherapy in tumors developing in situ (71). Remarkably, the administration of anti-CTLA4 antibody was more efficient to reduce the number of colon tumors in non-infected mice than in infected mice (19). Collectively, we observed that in pre-clinical models, H pylori infection reduces the cancer immunotherapy effectiveness.
H pylori Seropositive Non-Small Cell Lung Cancer Patients Demonstrated Reduced Efficacy of Anti-PD1 Immunotherapy
We performed a retrospective study in two independent cohorts of NSCLC patients to evaluate the correlation between H pylori infection, diagnosed by serology, and cancer immunotherapy efficacy. Out of 60 NSCLC patients in a French cohort, 18 patients were tested H pylori seropositive. Out of 29 NSCLC patients of the second cohort (Canada), 8 patients were tested H pylori seropositive. Remarkably, in the French cohort, a decrease of NSCLC patient survival upon anti-PD-1 therapy were associated with H pylori seropositivity. H pylori seropositive patients demonstrated survival medians of 6.7 months versus 15.4 months for seronegative patients. Additionally, in the second cohort, a decrease of NSCLC patient progression free survival upon anti-PD-1 therapy was observed in H pylori seropositive patients compared to seronegative counterparts. Moreover, a numerical difference for overall survival was also observed in the Canadian cohort, since NSCLC patient survival was 21.7 months for seronegative patients compared to 9.3 months for seropositive patients. Collectively, we observed in two independent cohorts of NSCLC patients that H pylori seropositivity is associated with lower effectiveness of anti-PD-1 immunotherapy (19). A limitation of our data is that the H pylori seropositivity is not synonymous of active H pylori infection, therefore prospective studies detecting past and active H pylori infection are necessary to confirm these results in cancer patients.
H pylori Infection Blocks Tumor Specific Immune Responses Initiated by Cancer Immunotherapies
The triggering of strong tumor specific immune responses are key for the efficacy of cancer immunotherapies. Remarkably, in the tumors of H pylori-infected mice, we detected a decreased number and activation status of tumor-specific T cells. In addition, the vaccine-induced tumor-specific T cells generated in H pylori infected mice displayed lower cytotoxic activities than those generated in non-infected mice. These results show that, in H pylori infected mice, tumor-specific immune responses primed by vaccination are reduced (19).
The functions of DC and more particularly those implicated in the CD8+ T cell priming have been previously been shown to be modulated by the gut microbiota (72, 73). We hypothesized that H pylori infection decreases the abilities of DC to prime tumor-specific immune responses. We indeed observed activation defects of DCs in the tumor draining lymph nodes of infected mice. Secondly, an in vitro co-culture assay showed that splenic DCs of infected mice do not promote efficient tumor specific T cells proliferation. Collectively, it can be speculated that H pylori-induced DC defects (69, 74) are at least partially responsible for the low efficacy of cancer immunotherapies in infected hosts (19).
Remarkably, we detected decrease activation of innate immune responses in infected mice. Indeed, 24 hours post injection of the cancer vaccine, a large decrease of serum inflammatory cytokine levels was detected in infected mice compared to non-infected counterparts. Among other, the level of IFNγ, a cytokine that is critical to sustain anti-tumor responses, was dramatically lower in the serum of infected mice compared to non-infected counterparts. Collectively, since we observed that H pylori infection jeopardizes the activation of the innate immune response, we hypothesized that these defects are key in the reduction of efficacy of cancer immunotherapies in infected hosts (19). Consequently, one might postulate that cancer immunotherapies that do not rely on a strong activation of innate immune responses such as CAR-T/TCR-T could be as effective in patients infected or not by H pylori.
Exploring the H pylori-Dependent Physiological Mechanism(s) Leading to the Decrease of the Tumor-Specific Immune Responses Initiated by Cancer Immunotherapies
We showed that by dampening the cross-presentation activities of DC and the production of inflammatory cytokines, H pylori prevents the triggering of an optimal tumor-specific immune responses. However, we did not identify the H pylori-dependent physiological mechanism(s) leading to these immune defects. Studying the physiological mechanisms which lead to H pylori-induced hypo-responsiveness to ICIs may allow for the identification of new therapeutic approaches that better promote tumor immune responses and/or the selection of more appropriate therapeutic options for patients with cancer. We will discuss the different H pylori-dependent physiological pathways that might lead to decreased efficacy of cancer immunotherapies in H pylori infected hosts.
H pylori Infection Modulates the Composition of the Gut Microbiota
The presence of B. fragilis, B. thetaiotaomicron and Burkholderiales in the gut microbiota, described for the first time by Vetizou et al., was shown to be critical for the efficacy of anti-CTLA4 blockade in mice (12). Later, A. muciniphila, F. prausnitzii, and Bifidobacterium species were also demonstrated to promote responses to immunotherapies (13, 14). More importantly, data collected in humans clearly demonstrate that the gut microbiome also plays critical roles on the responses to anticancer immunotherapies (13, 14, 75) and that antibiotics were shown to be associated with a decrease efficacy of programmed PD-1 blockade therapy (15). Since, it well described that H pylori infection modulates the gut microbiota composition (76), we evaluated whether immune-potentiating bacteria such as B. fragilis, B. thetaiotaomicron and Burkholderiales, A. muciniphila, F. prausnitzii, and Bifidobacterium are present in the gut microbiota of H pylori infected mice. To probe for the role of the gut microbiota the immunosuppression induced by H plyori, three experimental protocols were conducted. Firstly, co-housing experiments were conducted. Mice are coprophagic, hence mice reared in the same cage display very similar fecal microbiota composition. Interesting enough, H pylori infection is not transmitted when infected mice are reared with non-infected mice (19). Using the B16-OVA melanoma model, we observed that in co-housed mice, the tumor volumes of vaccinated infected mice were larger than those of vaccinated non-infected mice (19). Similarly, anti-CTLA4 therapy was less efficient in co-housed infected mice comparted to non-infected counterparts (19). Next, 16S rRNA gene sequencing was perform to determine the composition of the fecal microbiota. As already described by Kienesberger et al. (76), we observed that the composition of the fecal microbiota of non-infected and infected mice differed. Notably, we observed a reduction of Lachnospiraceae and Erysipelotrichaceae genera, and an increased of the Bifidobacterium genus in the fecal microbiota of H pylori infected mice compared to non-infected counterparts. Additionally, non-infected mice demonstrated lower alpha diversity as compared to infected mice. Remarkably enough, H pylori infected individuals compared to non-infected controls also displayed increased alpha diversity (77). Paradoxically, the high Bifidobacterium colonization and high alpha diversity are commonly associated with good responses to cancer immunotherapies in pre-clinical models (72, 78). Lastly, using the B16-OVA melanoma model, we performed fecal transplants. Feces from H pylori infected mice were administered to non-infected mice and cancer immunotherapies them performed. We observed that the transplantation of H pylori infected feces into non-infected mice does not decrease the efficacy of the cancer vaccination to control the tumor growth. Inversely, H pylori infected mice, transplanted with feces of non-infected mice maintained a decreased efficacy of cancer immunotherapies (19), suggesting that FMT will not improve the responses of H pylori infected cancer patients to immunotherapies. Although the fecal microbiota of H pylori infected host differs from non-infected mice, these differences do not play a major role in the low efficacy of cancer immunotherapies in infected hosts (19). However, our data do not address the notion that H pylori-mediated immunosuppression of cancer immunotherapies may rely on the microbiota of the small intestine (79) or stomach (80, 81).
H pylori Infection Modulates Dendritic Cell Activities
It has been described that the gut microbiota promotes the maturation of DCs. The gut microbiota-dependent DC maturation drives potent tumor-specific immune responses and augments the activity of CTLA-4 and/or PD-L1 blockades (12, 78). For exemple, in mice administered with B. fragilis it has been observed that the efficacy of anti-CTLA-4 blockade is increased. This B. fragilis-dependent potentiation effect was associated with an induction of Th1 responses in the lymph nodes draining the tumor and with the maturation of DCs in the tumor bed (12). Additionally, DC activation was shown to be sustained by Bifidobacterium and associated with the induction of very potent tumor-specific CD8+ T cell responses leading to the improvement of the efficacy of PD-L1 blockades (72). Favorable microbiota was also shown to promote the secretion of IL-12 by DCs to recruit, in the tumor beds, CCR9+CXCR3+CD4+ T cells (14). The microbiota has also been shown to modulate the functions of plasmacytoid DCs. Indeed, type I interferon produced by plasmacytoid DCs was shown by modulating the metabolic and epigenomic state of conventional DCs, to increase the priming of T cell responses (82). Importantly, Tanoue K et al. showed that the oral gavage with 11 bacterial strains enhances the therapeutic efficacy of ICIs in a CD8+ T cell dependent manner (83). Though the authors did not identify the cellular and molecular mechanisms by which the 11 strains promote systemic anti-tumor responses, plausible hypotheses include the systemic circulation of bacteria, bacterial antigen, bacteria-loaded DCs, metabolites (induced or produced by the 11 strains) and/or cytokines.
In our context, one might hypothesize that products released by H pylori may directly and/or indirectly stimulate DCs within the tumor microenvironment or in the tumor-draining lymph node to compromise the priming of tumor-specific CD8+ T cells (19).
H pylori infection has, indeed, a major impact on the function of DCs (Figure 1A). In vivo, H pylori was shown to modulate DCs to induce tolerance rather than immunity to H pylori (84, 85) or allergic airway diseases (74, 85). In vitro, Kao et al. showed that H pylori–pulsed bone marrow derived DCs (BM-DCs) produced significantly lower levels of IL-6 and IL-23 compared with DCs pulsed with Escherichia coli or Acinetobacter lwoffii (86). In addition, they showed that H pylori–pulsed BM-DCs preferentially induced Treg cells rather than Th17 cells (86). Remarkably, in vitro and in vivo experiments showed that DCs exposed to live or H pylori extracts are less capable of processing the antigen (74, 87). In addition, Oertli M et al. showed that H pylori mutants lacking γGT or VacA, induced the maturation of BM-DCs more efficiently than wild-type bacteria and showed a reduced ability to inhibit the activation of BM-DCs by LPS of E. Coli (88). They also showed that BM-DCs pulsed with γGT or VacA H pylori mutants displayed reduced ability to induce Treg cells compared to wild-type H pylori (88). These two H pylori virulence determinants expressed by all clinical isolate were also shown to be critically involved in the promotion of H pylori persistence, and protection against allergic airway diseases through tolerogenic reprogramming of DCs (88). Additionally, CagA was shown to dampen DC maturation in mice (89), and to induce tolerogenicity upon activation of the transcription factor STAT3 and IL-10 secretion in human DCs (90). BM-DCs exposed to H pylori extracts produced and secreted large amounts of IL-10, which was dependent on toll-like receptor 2 (TLR2) and Myd88 signaling, but independent on TLR4 (69).
FIGURE 1
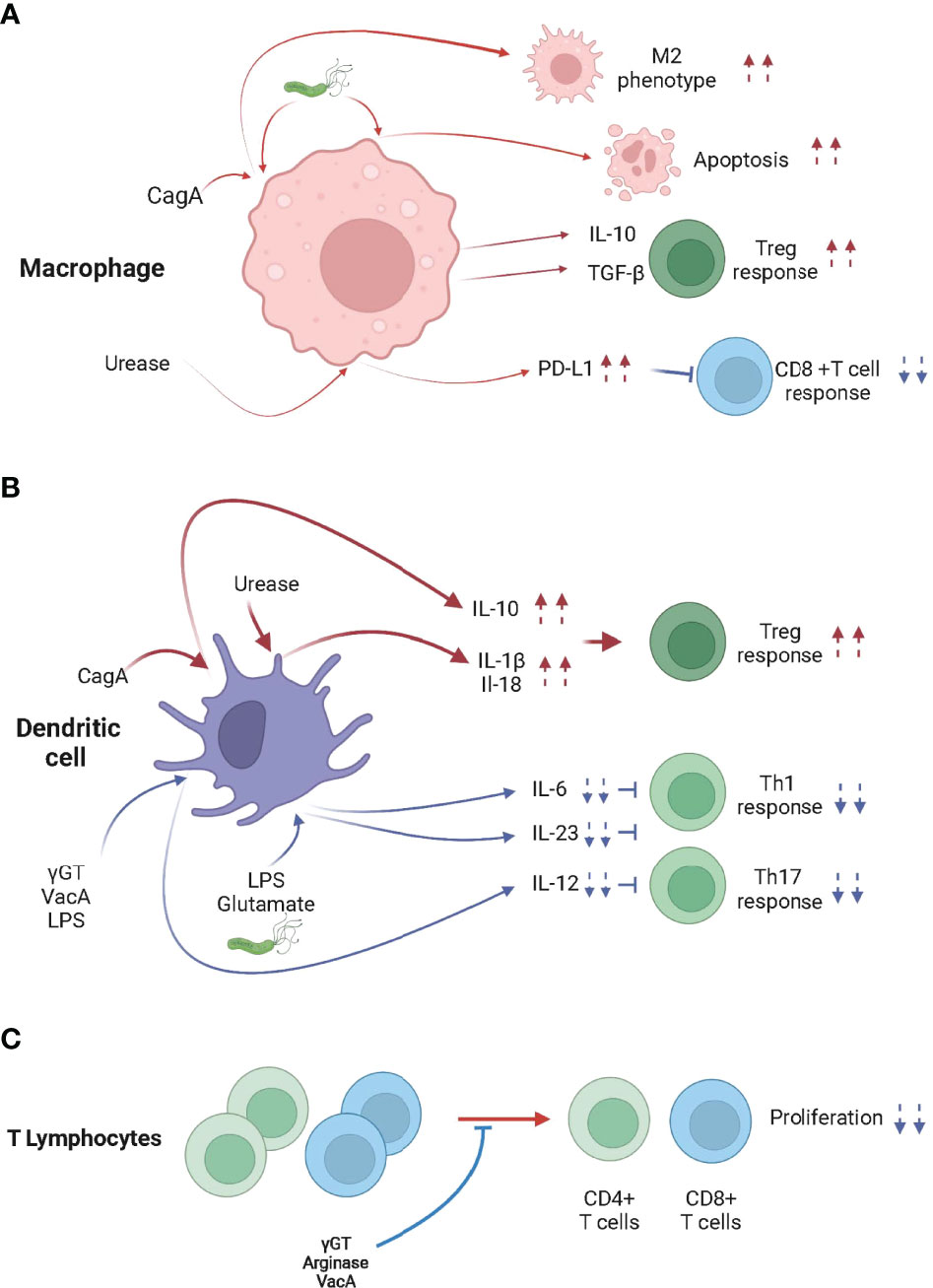
Figure 1 H pylori infection modulates the activity of macrophages, dendritic cells and T lymphocytes. (A) When in contact with H pylori, cytotoxin-associated gene A (CagA) and/or urease, skew macrophages toward an M2 phenotype, which promotes Treg cell differentiation, inhibits CD8+ T cell responses and/or promotes apoptosis. (B) H pylori, CagA, γ-glutamyl transpeptidase (γGT), vacuolating cytotoxin A (VacA), Lipopolysaccharide (LPS), glutamate and urease trigger the differentiation of tolerogenic DCs, leading to the Treg cell-skewed Th response. (C) γGT, VacA and Arginase inhibit the proliferation of CD4+ and CD8+ T cells.
Urease is a secreted and non-secreted protein that has been shown to activate NLRP3 in a TLR2 dependent manner and induce the processing and secretion of the caspase-1-dependent cytokines IL-1β and IL-18 in BM-DCs (91–93). Caspase-1ko, IL-18ko, IL-18Rko, Nlrp3ko or Tlr2ko mice control H pylori infection more effectively than WT animals because they fail to induce Treg cells (85, 93, 94). Importantly, neonatal infection with urease deficient H pylori generated low caspase-1 activation, low NLRP-3 expression, low IL-18 secretion and did not efficiently protect from allergen-induced asthma (94). Taken together, urease, in addition to its acidity buffering role, is key in the H pylori-induced immune tolerance both locally and systemically.
The binding of H pylori LPS to DC-SIGN expressed by DCs resulted in signals which decrease the production of IL-6 and IL-12 and block Th1 cell responses (95, 96). DC-SIGN signaling, by favoring the production of IL-10, a non-inflammatory cytokine, decreases expression of inflammatory cytokines initiated by TLR activation (96). Remarkably, human DCs differentiated from circulating monocytes and infected with H pylori also produced in a DC-SIGN, TLR2 and TLR4 dependent manner IL-10.
Glutamate, produced as the result of γGT enzymatic activity, activates glutamate receptors on DCs, inhibiting cAMP signaling and the release of the proinflammatory cytokine IL-6, favoring the induction of a Treg cell response over Th1 and Th17 responses (97). Recently, it was shown that the metabotropic glutamate receptor 4 (GRM4) suppresses antitumor immunity and antagonizes the treatment of ICIs (98).
One may hypothesize that the DC tolerance induced by H pylori-derived molecules such as γGT, VacA, urease, LPS and/or glutamate may be involved in the H pylori-induced hypo-responsiveness to cancer immunotherapies (Figure 1A). The defects in activation of DC in the tumor draining lymph nodes of infected mice in as little as 24 hours post tumor cell engraftment and the lower cross-presentation activity of splenic DCs of H pylori infected mice support this hypothesis (19).
H pylori Infection Modulates Macrophage Function
It has been reported that macrophages interact with H pylori within the gastric mucosa (99) and that numerous factors released by H pylori, such as urease (100), heat shock protein 60 (Hsp60) (101), and/or LPS (102, 103) were shown to activate macrophages. Remarkably, H pylori impedes the phagocytic killing capacity of macrophages (104) and may promote macrophage apoptosis (105, 106). It has been shown that macrophages infiltrating the gastric mucosa of H pylori infected hosts display the pro-inflammatory (M1) profile (107). Nonetheless, macrophages isolated from the gastric mucosa of H pylori–infected mice also demonstrate anti-inflammatory (M2) characteristics such as the activation of the arginase/ornithine decarboxylase metabolic pathway (106, 108). Remarkably, infected patients also show an increase of M2 markers in their gastric mucosa (109). Moreover, studies have associated the production of IL-10 and TGF-β (99) by macrophages, the typical M2 macrophages cytokines, with H pylori infection (109). H pylori increases the expression of the anti-inflammatory heme oxygenase-1 (HO-1) enzyme in macrophages (110). This process requires CagA phosphorylation and the activation of p38 and NF (erythroid-derived 2)-like 2 (NRF-2). The increased activity of HO-1 reduces the pro-inflammatory M1 markers and promotes of the M2 profile of the infected macrophages, leading to defects in the initiation of innate and adaptive immune responses (Figure 1B). Because tumor associated macrophages tend to be considered M2-skewed macrophages in the majority of established tumors (111), H pylori infection might remotely strengthen the M2 phenotype within tumors and/or tumor draining secondary lymphoid organs, lowering the efficacy of cancer immunotherapies in chronically infected hosts (112).
One may hypothesize that the M2 phenotype induced by H pylori-derived molecules, such as CagA, might be involved in the H pylori-induced hypo-responsiveness to cancer immunotherapies. The significant decrease of the serum levels of inflammatory cytokines in infected mice day one post cancer vaccination supports this hypothesis (19).
H pylori Infection Modulates T Cell Function
Multiple virulence factors of H pylori were shown to directly limit adaptive immune responses by restricting T cell proliferation. This H pylori-dependent inhibition of T cell proliferation may be partly responsible for the decreased efficacy of cancer immunotherapies in H pylori infected hosts.
H pylori arginase, by depleting L-arginine, was shown to inhibit T cell proliferation and reduce the expression of the T cell receptor zeta-chain (113). Similarly, γGT impairs T-cell activation, proliferation and the production of cytokines by specifically depriving glutamine from the extracellular space (114). Endocytosis of the pathogenic factor VacA into activated human primary T cells (115) was also shown to inhibit cell proliferation and the clonal expansion of T cells (116, 117) (Figure 1C).
In addition to the direct effects of H pylori virulence factors on T cell function, H pylori was shown to modulate the function, survival and/or homing of T cells by acting on gastric epithelial cells, macrophages and DCs. Recently, Wu J et al. showed that the effect of urease on macrophages leads to the suppression of cytotoxic CD8+ T cell responses through the induction of PD-L1 (118) (Figure 1B). Similarly, increased expression of PD-L1 in gastric epithelial cells is observed in H pylori infected hosts, leading to suppression of IL-2 synthesis and CD4+ T cell proliferation (119). In addition, it was shown that the high PD-L1 expression (120) and also the production of transforming growth factor beta (TGFβ) expression (121) preferentially promote the maintenance of Foxp3+ Treg cells within the gastric mucosa (Figure 2).
FIGURE 2
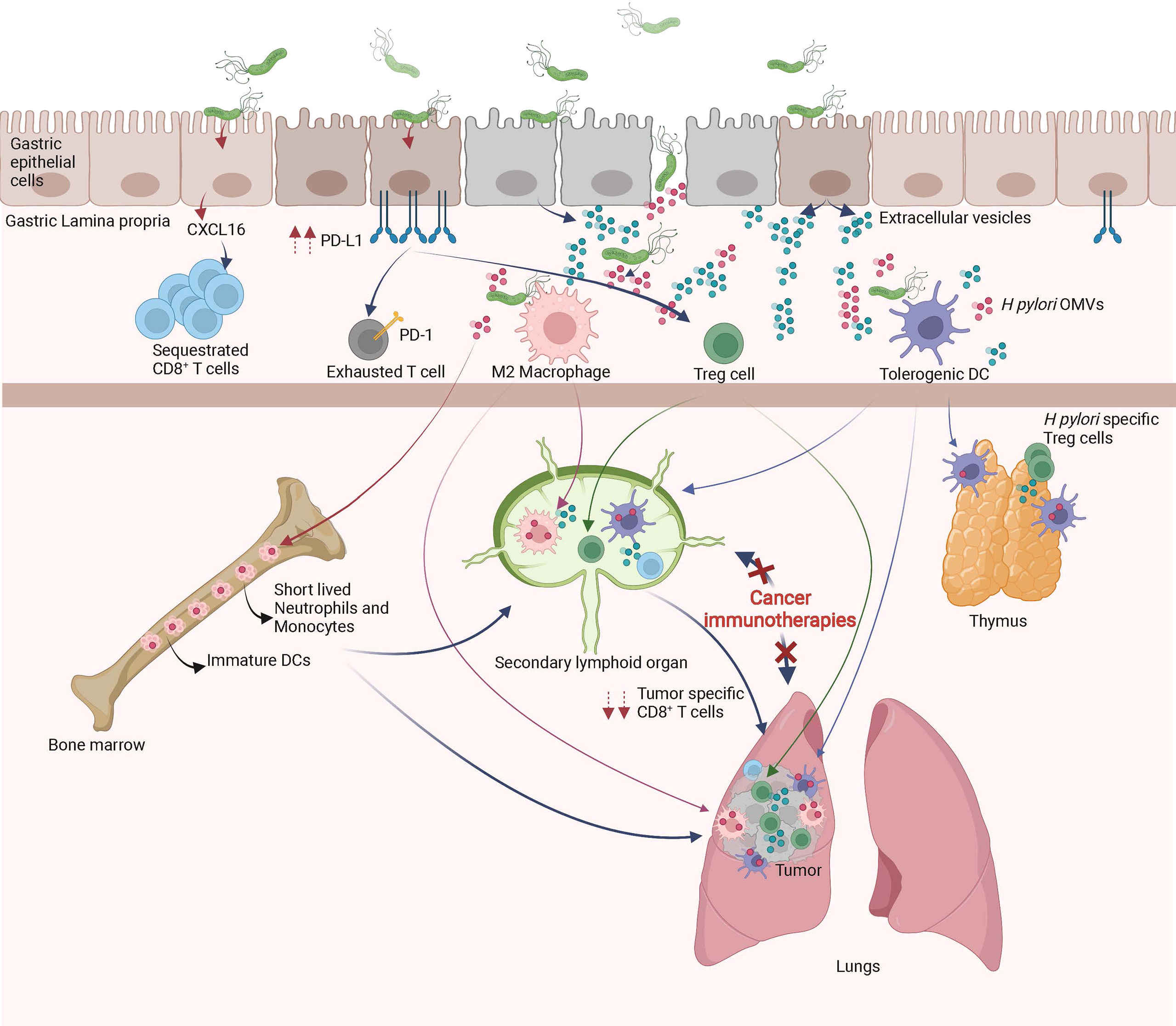
Figure 2 Local and remote effects of H pylori infection on host immune functions. In the gastric mucosa, H pylori promotes the differentiation of tolerogenic DCs, M2 macrophages, Treg cells, inhibits Th1/Th17 responses and/or sequesters CD8+ T cells. The systemic diffusion/migration of extracellular vesicles, outer membrane vesicles (OMVs), Treg cells, tolerogenic DCs and M2 macrophages modulates the function of primary and secondary lymphoid organs, leading to a decrease in tumor specific immune responses triggered by cancer immunotherapies.
As described above, H pylori infection induces tolerogenic properties of DCs, macrophages and gastric epithelial cells, all of which lead to Treg cell differentiation. High numbers of Treg cells is beneficial in the maintenance of chronic infection however, is detrimental for the efficacy of cancer immunotherapies. Indeed, H pylori-induced Treg cells may seed tumor sites and/or secondary lymphoid organs, weakening the initiation and/or effector phase of anti-cancer immune responses initiated by cancer immunotherapies (Figure 2). This hypothesis is supported by previous studies showing that a favorable microbiota in positive responders to CTLA-4 or PD-1 blockade have lower numbers of peripheral blood and tumor Treg cells (122, 123). Importantly, H pylori-induced Treg cells have been shown to reduce the strength of the immune responses initiated in the lungs of rodents suffering from experimental asthma (124).
In addition, chronic H pylori infection may skew anti-cancer immune responses by decreasing the ability of tumor-specific CD8+ T cells to access the tumor bed and/or tumor draining lymph nodes. Indeed, by sustaining chronic immune activation within the gastric mucosa, H pylori may attract immune effectors cells continuously in the gastric mucosa, gastric lamina propria and gastric draining lymph nodes. It has been recently shown that H pylori–induced matrix metallopeptidase-10 promotes the gastric homing of CD8+ T cells through the secretion of CXCL16 (125). One might hypothesize that naïve CD8+ T cells or cancer immunotherapy-primed CD8+ T cells may preferentially home to the gastric mucosa instead of the tumor draining lymphoid organs or tumor bed, respectively (Figure 2). This hypothesis is supported by two recent papers showing that H pylori infection modulates schistosome-induced liver fibrosis or mycobacterial infection (126, 127).
H pylori Infection Modulates Natural Killer T Cells Functions
Natural killer T (NKT) cells recognize, within the context of CD1d, glycolipid antigens. Recent studies have demonstrated that gut microbiota modulate the function of systemic and mucosal iNKT cells (128, 129). Remarkably, a study by Ito et al. showed that cholesteryl α-glucosides of H pylori are recognized by iNKT in the stomach mucosa. The activation of iNKT cells limits H pylori infection by promoting inflammatory responses (130). In addition, it was shown that PI57, a cholesterol-derived lipid from H pylori, protects against the development of allergen-induced airway hyperreactivity (131).
NKT cells display strong anti-tumor responses and thus are a major target in the development of cancer immunotherapies. Following treatment with α-Galactocylceramide, a potent activator of iNKT cells, iNKT cells synthetize multiple cytokines modulating the functions of macrophages, neutrophils, NK cells, DCs, T cells, and B cells. Moreover, activated iNKT cells can directly induce cell death in tumor cells. Mice lacking the iNKT cell subset, compared with wild-type mice, respond to CTLA-4 blockade with markedly increased cure rate and overall survival, indicating that iNKT cells might play a role in efficacy of cancer immunotherapy (132).
Therefore, it may be hypothesized that the function of mucosal and/or systemic iNKT cells are modified by H pylori cholesteryl α-glucosides, leading to the decrease in efficacy of cancer immunotherapy protocols (19).
H pylori Infection Modulates Gastro-Intestinal Permeability
H pylori is known to induce chronic inflammation and directly increase epithelial permeability by redistributing tight junction protein ZO-1 in gastric epithelial cells (20, 133–135) leading to a leaky gut. It has been described that during cancer immunotherapy treatment, the gut barrier is altered (12, 71). Therefore, the leaky gut induced by anti-ICIs and H pylori infection may cause an influx of antigens and immunomodulatory molecules produced by H pylori and other constituents of the gut microbiota in mucosal and systemic tissues. These microbiota derived-molecules modulate the mucosal and systemic immune responses (20, 71, 136) and may potentially decrease cancer immunotherapy effectiveness in H pylori infected hosts (19).
Is the H pylori-Induced Decrease in Efficacy of Cancer Immunotherapies Dependent on the Systemic Diffusion of H pylori Gut Microbiota-Derived Molecules?
A very elegant and highly informative experiment performed by the group of A Macpherson clearly demonstrated that bacterial-derived molecules are detectable in peripheral tissues (137), leading to the conclusion that many metabolic activities of our organism rely on gut microbiota-derived molecules. For instance, molecules produced or modified by the gut microbiota, such as short chain fatty acids (SCFAs; acetate, butyrate and propionate), tryptophan pathway metabolites or bile acids have been shown to diffuse freely throughout the gastro-intestinal epithelial cells and modulate the function of our immune system.
A Role for SCFAs?
SCFAs are the products of the microbial fermentation of nondigestible nutritional fibers. In the human gut, the phylum Firmicutes are the main butyrate producing-bacteria. In particular Faecalibacterium prausnitzii and Clostridium leptum of the family Ruminococcaceae, as well as Eubacterium rectale and Roseburia spp. of the family Lachnospiraceae produce butyrate (138). The SCFA receptors belong the family of G protein-coupled receptors (GPCRs). GPR43, GPR41, GPR109a and GPR164, are expressed by many cells belonging to the gastrointestinal tract and the nervous and immune systems (139). Increased antitumor responses and longer progression-free survival were associated with high fecal SCFA levels, whereas low treatment responses were associated with high systemic SCFA levels (140). Butyrate may inhibit the capacity of DCs to promote tumor-specific T cells, thereby limiting the efficacy of anti-CTLA-4 blockade. Trompette et al. showed that propionate treatment of mice led to modulations of function of hematopoiesis (141). Indeed, the administration of propionate leads to the seeding of the lungs with DCs with impaired ability to promote T helper cell effector functions but with high phagocytic capacity. More specifically, they showed that four days after allergen challenge, the DCs of propionate-treated mice displayed a reduced expression of CD40, PD-L2 and CD86, which is associated with low production of IL-4, IL-5, IL-13, IL-10 and IL-17A by CD4 T cells (141). Taken together, high SCFAs production might account for the observed deficient DC cross presentation activities, low anti-tumor CD8+ T cell cytotoxicity and increased Treg cell number in H pylori infected mice. However, it was recently shown that fecal SCFA levels are decreased in H pylori infected mice compared to the non-infected counterparts, suggesting that CSFAs do not play a role in the reduced efficiency of cancer immunotherapies in H pylori infected hosts (142).
A Role for Tryptophan-Derived Metabolites?
Gut microorganisms such as Escherichia coli, Lactobacillus reuteri and Lactobacillus johnsonii can metabolize tryptophan into indole-3-acetaldehyde and indole acetic acid (IAA), both of which are aryl hydrocarbon receptor (AhR) agonists. These agonists modulate the functions of many cell types such as epithelial cells, immune cells and astrocytes, both locally and systemically (143, 144). Although recent studies show that tryptophan-derived microbial metabolites suppress anti-tumor immunity by activating AhR in tumor-associated macrophages (145), data are lacking to determine whether tryptophan-derived metabolites are implicated in H pylori-induced reduction of cancer immunotherapies (146).
A Role for Bile Acids?
Primary bile acids are produced by the liver and conjugated to glycine (in humans) or taurine (in mice) transported to the gallbladder and the proximal small intestine, after which they are metabolized by gut microbes to generate secondary bile acids. In intestinal epithelial cells, macrophages among other cell types, microbiome-produced deoxycholic acid and lithocholic acid can bind the GPCR TGR5, as well as the nuclear receptor farnesoid X receptor (FXR) (147, 148). DCs and macrophages both express TGR5 and FXR receptors and secondary bile acids, which have been shown to modulate the anti-cancer immune response, promote anti-inflammatory responses in these cells (149) (150). To our knowledge, no data have been published on the modulation of the production of secondary bile acids in H pylori infected hosts. Additional studies are necessary to evaluate whether secondary bile acids play a role in H pylori-induced reduction of cancer immunotherapies.
A Role for Outer Membrane Vesicles or Extracellular Vesicles?
The detection of H pylori specific-cell mediated and humoral immune responses in mucosal and systemic compartments is a clear demonstration that H pylori-derived antigens can gain access to mucosal and systemic secondary lymphoid organs (20).
Derived from the bacterial outer membrane, H pylori secrete outer membrane vesicles (H pylori-OMVs). H pylori-OMVs carry many of molecules expressed at the surface of the bacteria, such as LPS and peptidoglycan. 77% of the bacterial outer membrane proteins were detected by mass spectrometry analysis in H pylori-OMVs. For instance, BabA and SabA adhesins, VacA, CagA, urease subunits, γGT, the protease HtrA, as well as GroEL, catalase are detected in H pylori-OMVs (151). Remarkably, H pylori-OMVs have been shown to deliver peptidoglycan to the cytosol of gastric epithelial cells, activating in a NOD1-dependent manner NF-KB the production of CXCL-2 and IL-8 (152).
In addition, extracellular-vesicles (EVs) are released from pathogen-infected host cells (EVs). Host-derived EVs like OMVs contribute to the local dissemination of pathogenic components. Pathogenic molecules can be transported between cells via EVs and therefore contribute to host-pathogen interactions. EVs may activate and modulate the immune system functions or other cellular signaling pathways. Very interestingly, CagA is present in EVs isolated from the serum of H pylori infected patients (153). Therefore it can be hypothesized that CagA derived from H pylori-infected gastric epithelial cells can diffuse systemically and gain access to distant organs and tissues such primary or secondary lymphoid organs. Thus, the extra-gastric disorders associated with CagA-positive H pylori infection might be induced by the systemic diffusion of CagA-containing exosomes. Although H pylori-OMVs are not detectable in the serum of infected individuals at steady state, one may hypothesize that the anti-ICIs-induced gut barrier dysfunctions might induce a systemic influx of H pylori-OMVs and CagA+-EVs in cancer patients. The H pylori-OMVs and CagA+-EVs may modulate the function of DCs, macrophages, iNKT cells and T cells, leading to decreased efficacy of cancer immunotherapies (Figure 2).
A Role for Systemic Homing of Mucosal-Derived Myeloid and Lymphoid Cells?
In addition to H pylori-OMVs and CagA+-EVs, H pylori crossing the epithelial mucosa promote gastric inflammation. In fact, direct contact (including, in some cases, intracellular penetration) between H pylori, H pylori-derived antigens and stromal cells such as lymphocytes, plasma cells, DCs, macrophages, and granulocytes has been observed in the gastric tissue (154). Occasionally, H pylori have also been detected inside blood capillaries, a finding that is in agreement with the transient detection of H pylori bacteremia (155). Collectively, these observations show that H pylori and H pylori-derived antigens are interacting locally with immune cells.
In a very elegant paper, Morton et al. showed that myeloid and lymphoid cells migrate from the periphery to mucosal tissues and migrate back from the gastro-intestinal mucosa to seed peripheral secondary lymphoid organs (156). Therefore, it might be hypothesized that myeloid cells (monocytes and/or DCs and/or macrophages) alongside B and T lymphocytes educated by H pylori or H pylori-derived antigens within the gastric mucosa migrate to the periphery. Therefore, it can be anticipated that the anti-ICIs-induced gut barrier dysfunctions (12) in H pylori infected hosts, mucosal-derived tolerant myeloid and lymphoid cells massively invade the secondary lymphoid organs and tumor bed, leading to decreased efficacy of cancer immunotherapies (19) (Figure 2).
A Role for the Modulation of Primary Lymphoid Organ Functions?
It has been shown that the complexity of the intestinal microbiota strongly correlates with the size of the pool of bone marrow myeloid cells (157). It was also shown that the gut microbiota, via TLR ligands, regulates the lifespan of inflammatory monocytes and neutrophils, two myeloid cell populations that are key players in inflammatory responses (158). Therefore, one may hypothesize that in H pylori infected hosts, the anti-ICIs-induced gut barrier dysfunctions (12) lead to an influx of H pylori-derived antigens such as LPS, urease, VacA, and CagA into the bone marrow. These H pylori-derived antigens might modulate the number, lifespan and/or activation status of myeloid cells produced by the bone marrow (Figure 2). As a consequence, in H pylori infected hosts, the myeloid cells freshly produced by the bone marrow might be deficient in the priming of tumor-specific CD8+ T cells following of cancer immunotherapy treatment. The significant decrease of serum levels of inflammatory cytokines of H pylori infected mice on day one post cancer vaccination supports this hypothesis (19).
Another hypothesis that could account for H pylori-induced hypo-responsiveness to cancer immunotherapies is the massive neonatal thymic production of H pylori-specific Treg cells (65). Indeed, it has been shown that a trafficking of gut microbiota-derived antigens to the thymus by DCs occurs during the microbiota colonization in early life. This DC-mediated trafficking of microbiota antigens promotes the differentiation of microbiota-specific Treg cells (159). Therefore, during neonatal H pylori infection, the thymus will most probably generate a huge number of H pylori-specific Treg cells. Later in life and in the context of H pylori-induced and anti-ICIs-induced gut leakiness, the H pylori-derived antigens might induce the activation of Treg cells in the tumor draining lymph nodes and/or within the tumor microenvironment and dampen the priming and/or effector phases of cancer immunotherapies (Figure 2).
A Role for the Neonatal Infection?
Upon delivery, neonates leave a sterile environment and are colonized by microorganisms that establish and mold the immune system of the host at both short and long term (128, 160). Depending which microorganism first colonizes the host, the immune system may be heavily impacted. For instance, the neonatal ileum colonization by the segmented filamentous bacteria (SFB) primes a very strong intestinal Th17 response (161). Associated with secretary immunoglobulin A response, Th17 cells control the degree of SFB colonization in young mice as well as other invading pathogens, such as Citrobacter rodentium and Salmonella typhimurium in adult mice. Very importantly, the influence of SFB on the immune system can have effects on peripheral sites. The host can benefit from SFB colonization; indeed, in predisposed non-obese diabetic mice SFB has been shown to delay the development of diabetes. Inversely, by favoring Th17 cell differentiation, SFB colonization may promote the development of arthritis or multiple sclerosis (162). Another example is the neonatal infection by mouse mammary tumor virus (MMTV). MMTV is produced in the mammary glands of mothers, which allows for viral transmission to neonates during suckling. Very interestingly, the MMTV genome encodes a superantigen that will cause the depletion of subsets of CD4+ T cells, creating a hole in the CD4+ T cell repertoire for the lifetime of the infected host (163).
In the case of early-life exposure to H pylori, it has been shown to induce immune tolerance. This immune tolerance is pivotal to prevent inflammation-induced immunopathology and excessive effector T-cell response to infection, allowing high-level of colonization (164). In addition, early-life exposure to H pylori protects from allergic airway disease while the protective effects in mice infected as adults was much weaker (65) Contrastingly, H pylori infection of adult mice is associated with higher degree of gastric inflammation and low-level of colonization (164). Collectively, these data suggest that infection with H pylori during the neonatal period is a key factor in the H pylori-induced local and systemic immune tolerance. Similarly, one may hypothesize that early-life exposure to H pylori may be critical in the H pylori-mediated immunosuppression of cancer immunotherapies (19).
Conclusion
More than 30 years of research on H pylori has generated amazing insights into the biology of H pylori-immune system cross-talks. An array of H pylori-derived factors are known to interact with host cells and signaling pathways, leading to chronic immune cell activation in infected hosts. H pylori-derived molecules such as VacA, γGT, CagA, urease, arginase, Hsp60, LPS, and/or peptitoglycan generate local and systemic pro- and anti-inflammatory cells and molecules which lead to a general state of hypo-responsiveness. On the one hand, H pylori-induced immune tolerance is beneficial to protect individuals from developing severe inflammatory syndrome such as asthma, EoE, IBD and food allergy. However, on the other hand, the immune hypo-responsiveness is most probably the cause of the low efficacy of cancer immunotherapies in H pylori seropositive NSCLC patients (19). In addition, as suggested by Shi et al. (165), the relative resistance of gastric cancers to anti-PD1/PD-L1 blockade therapies may also originate from H pylori-induced hypo-responsiveness.
Studying the physiological mechanisms which lead to H pylori-induced hypo-responsiveness to ICIs may allow for the identification of new therapeutic approaches that better promote tumor immune responses and/or the selection of more appropriate therapeutic options for patients with cancer.
Author Contributions
PO, LV, BMM and DV wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Open access funding provided by University of Lausanne.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work was supported by the Swiss Cancer Research foundation (KFS-4452-02-2018, D.V), the Foundation Emma Muschamp (D.V.) and by the Faculté de Biologie et Médecine de Lausanne (D.V.). Figures created with biorender.com.
References
1. Schaper F, van Spriel AB. Antitumor Immunity Is Controlled by Tetraspanin Proteins. Front Immunol (2018) 9:1185. doi: 10.3389/fimmu.2018.01185
2. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
3. Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. Cd4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity (2017) 47(5):848–61 e5. doi: 10.1016/j.immuni.2017.10.009
4. Menares E, Galvez-Cancino F, Caceres-Morgado P, Ghorani E, Lopez E, Diaz X, et al. Tissue-Resident Memory CD8(+) T Cells Amplify Anti-Tumor Immunity by Triggering Antigen Spreading Through Dendritic Cells. Nat Commun (2019) 10(1):4401. doi: 10.1038/s41467-019-12319-x
5. Liu M, Guo F. Recent Updates on Cancer Immunotherapy. Precis Clin Med (2018) 1(2):65–74. doi: 10.1093/pcmedi/pby011
6. Riley RS, June CH, Langer R, Mitchell MJ. Delivery Technologies for Cancer Immunotherapy. Nat Rev Drug Discovery (2019) 18(3):175–96. doi: 10.1038/s41573-018-0006-z
7. Liu Y, Yan X, Zhang F, Zhang X, Tang F, Han Z, et al. Tcr-T Immunotherapy: The Challenges and Solutions. Front Oncol (2022) 11. doi: 10.3389/fonc.2021.794183
8. Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front Immunol (2018) 9:2306. doi: 10.3389/fimmu.2018.02306
9. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–33.e17. doi: 10.1016/j.cell.2017.07.024
10. Thursby E, Juge N. Introduction to the Human Gut Microbiota. Biochem J (2017) 474(11):1823–36. doi: 10.1042/BCJ20160510
11. Kapitan M, Niemiec MJ, Steimle A, Frick JS, Jacobsen ID. Fungi as Part of the Microbiota and Interactions With Intestinal Bacteria. Curr Top Microbiol Immunol (2019) 422:265–301. doi: 10.1007/82_2018_117
12. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science (2015) 350(6264):1079–84. doi: 10.1126/science.aad1329
13. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut Microbiome Modulates Response to anti-PD-1 Immunotherapy in Melanoma Patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
14. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut Microbiome Influences Efficacy of PD-1-based Immunotherapy Against Epithelial Tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
15. Elkrief A, Derosa L, Kroemer G, Zitvogel L, Routy B. The Negative Impact of Antibiotics on Outcomes in Cancer Patients Treated With Immunotherapy: A New Independent Prognostic Factor? Ann Oncol (2019) 30(10):1572–9. doi: 10.1093/annonc/mdz206
16. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal Microbiota Transplant Promotes Response in Immunotherapy-Refractory Melanoma Patients. Science (2021) 371(6529):602–9. doi: 10.1126/science.abb5920
17. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal Microbiota Transplant Overcomes Resistance to anti-PD-1 Therapy in Melanoma Patients. Science (2021) 371(6529):595–602. doi: 10.1126/science.abf3363
18. Finlay BB, Goldszmid R, Honda K, Trinchieri G, Wargo J, Zitvogel L. Can We Harness the Microbiota to Enhance the Efficacy of Cancer Immunotherapy? Nat Rev Immunol (2020) 20(9):522–8. doi: 10.1038/s41577-020-0374-6
19. Oster P, Vaillant L, Riva E, McMillan B, Begka C, Truntzer C, et al. Helicobacter Pylori Infection Has a Detrimental Impact on the Efficacy of Cancer Immunotherapies. Gut (2022) 71(3):457–66. doi: 10.1136/gutjnl-2020-323392
20. Moyat M, Velin D. Immune Responses to Helicobacter Pylori Infection. World J Gastroenterol WJG (2014) 20(19):5583–93. doi: 10.3748/wjg.v20.i19.5583
21. Atherton JC, Blaser MJ. Coadaptation of Helicobacter Pylori and Humans: Ancient History, Modern Implications. J Clin Invest (2009) 119(9):2475–87. doi: 10.1172/JCI38605
22. Diaconu S, Predescu A, Moldoveanu A, Pop CS, Fierbinţeanu-Braticevici C. Helicobacter Pylori Infection: Old and New. J Med Life (2017) 10(2):112–7.
23. Ding SZ, Du YQ, Lu H, Wang WH, Cheng H, Chen SY, et al. Chinese Consensus Report on Family-Based Helicobacter Pylori Infection Control and Management (2021 Edition). Gut (2021) 71:238–253. doi: 10.1136/gutjnl-2021-325630
24. Schreiber S, Konradt M, Groll C, Scheid P, Hanauer G, Werling HO, et al. The Spatial Orientation of Helicobacter Pylori in the Gastric Mucus. Proc Natl Acad Sci USA (2004) 101(14):5024–9. doi: 10.1073/pnas.0308386101
25. Hazell SL, Lee A, Brady L, Hennessy W. Campylobacter Pyloridis and Gastritis: Association With Intercellular Spaces and Adaptation to an Environment of Mucus as Important Factors in Colonization of the Gastric Epithelium. J Infect Diseases (1986) 153(4):658–63. doi: 10.1093/infdis/153.4.658
26. Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and Interstitial Expression of Helicobacter Pylori Virulence Genes in Gastric Precancerous Intestinal Metaplasia and Adenocarcinoma. J Infect Diseases (2003) 187(8):1165–77. doi: 10.1086/368133
27. Atherton JC, Tham KT, Peek RM Jr., Cover TL, Blaser MJ. Density of Helicobacter Pylori Infection In Vivo as Assessed by Quantitative Culture and Histology. J Infect Diseases (1996) 174(3):552–6. doi: 10.1093/infdis/174.3.552
28. Bauerfeind P, Garner R, Dunn BE, Mobley HL. Synthesis and Activity of Helicobacter Pylori Urease and Catalase at Low Ph. Gut (1997) 40(1):25–30. doi: 10.1136/gut.40.1.25
29. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. The HP0165-HP0166 Two-Component System (ArsRS) Regulates Acid-Induced Expression of HP1186 Alpha-Carbonic Anhydrase in Helicobacter Pylori by Activating the pH-dependent Promoter. J Bacteriol (2007) 189(6):2426–34. doi: 10.1128/JB.01492-06
30. Backert S, Clyne M, Tegtmeyer N. Molecular Mechanisms of Gastric Epithelial Cell Adhesion and Injection of CagA by Helicobacter Pylori. Cell communication Signaling CCS (2011) 9:28. doi: 10.1186/1478-811X-9-28
31. Wiedemann T, Hofbaur S, Tegtmeyer N, Huber S, Sewald N, Wessler S, et al. Helicobacter Pylori CagL Dependent Induction of Gastrin Expression Via a Novel αvβ5-Integrin-Integrin Linked Kinase Signalling Complex. Gut (2012) 61(7):986–96. doi: 10.1136/gutjnl-2011-300525
32. Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter Pylori CagL Activates ADAM17 to Induce Repression of the Gastric H, K-ATPase Alpha Subunit. Gastroenterology (2010) 139(1):239–48. doi: 10.1053/j.gastro.2010.03.036
33. Kuipers EJ, Uyterlinde AM, Peña AS, Roosendaal R, Pals G, Nelis GF, et al. Long-Term Sequelae of Helicobacter Pylori Gastritis. Lancet (1995) 345(8964):1525–8. doi: 10.1016/S0140-6736(95)91084-0
34. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter Pylori Infection. Clin Microbiol Rev (2006) 19(3):449–90. doi: 10.1128/CMR.00054-05
35. Choi IJ, Kook MC, Kim YI, Cho SJ, Lee JY, Kim CG, et al. Helicobacter Pylori Therapy for the Prevention of Metachronous Gastric Cancer. New Engl J Med (2018) 378(12):1085–95. doi: 10.1056/NEJMoa1708423
36. Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, et al. Association Between Helicobacter Pylori Eradication and Gastric Cancer Incidence: A Systematic Review and Meta-Analysis. Gastroenterology (2016) 150(5):1113–24. doi: 10.1053/j.gastro.2016.01.028
37. Chen HN, Wang Z, Li X, Zhou ZG. Helicobacter Pylori Eradication Cannot Reduce the Risk of Gastric Cancer in Patients With Intestinal Metaplasia and Dysplasia: Evidence From a Meta-Analysis. Gastric cancer (2016) 19(1):166–75. doi: 10.1007/s10120-015-0462-7
38. Narayanan M, Reddy KM, Marsicano E. Peptic Ulcer Disease and Helicobacter Pylori Infection. Missouri Med (2018) 115(3):219–24.
39. Alexander SM, Retnakumar RJ, Chouhan D, Devi TNB, Dharmaseelan S, Devadas K, et al. Helicobacter Pylori in Human Stomach: The Inconsistencies in Clinical Outcomes and the Probable Causes. Front Microbiol (2021) 12(2277). doi: 10.3389/fmicb.2021.713955
40. Díaz P, Valenzuela Valderrama M, Bravo J, Quest AFG. Helicobacter Pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front Microbiol (2018) 9:5. doi: 10.3389/fmicb.2018.00005
41. Kuipers EJ, Pérez-Pérez GI, Meuwissen SGM, Blaser MJ. Helicobacter Pylori and Atrophic Gastritis: Importance of the Caga Status. JNCI: J Natl Cancer Institute (1995) 87(23):1777–80. doi: 10.1093/jnci/87.23.1777
42. Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for Gastric Cancer in People With CagA Positive or CagA Negative Helicobacter Pylori Infection. Gut (1997) 40(3):297–301. doi: 10.1136/gut.40.3.297
43. El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, et al. Interleukin-1 Polymorphisms Associated With Increased Risk of Gastric Cancer. Nature (2000) 404(6776):398–402. doi: 10.1038/35006081
44. Chauhan N, Tay ACY, Marshall BJ, Jain U. Helicobacter Pylori VacA, a Distinct Toxin Exerts Diverse Functionalities in Numerous Cells: An Overview. Helicobacter (2019) 24(1):e12544. doi: 10.1111/hel.12544
45. Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M, Nagai T, et al. Helicobacter Pylori Dampens Gut Epithelial Self-Renewal by Inhibiting Apoptosis, A Bacterial Strategy to Enhance Colonization of the Stomach. Cell Host Microbe (2007) 2(4):250–63. doi: 10.1016/j.chom.2007.09.005
46. Kuo SH, Wu MS, Yeh KH, Lin CW, Hsu PN, Chen LT, et al. Novel Insights of Lymphomagenesis of Helicobacter Pylori-Dependent Gastric Mucosa-Associated Lymphoid Tissue Lymphoma. Cancers (2019) 11(4):547. doi: 10.3390/cancers11040547
47. Zullo A, Hassan C, Andriani A, Cristofari F, De Francesco V, Ierardi E, et al. Eradication Therapy for Helicobacter Pylori in Patients With Gastric MALT Lymphoma: A Pooled Data Analysis. Am J Gastroenterol (2009) 104(8):1932–7. doi: 10.1038/ajg.2009.314
48. Labenz J, Blum AL, Bayerdorffer E, Meining A, Stolte M, Borsch G. Curing Helicobacter Pylori Infection in Patients With Duodenal Ulcer may Provoke Reflux Esophagitis. Gastroenterology (1997) 112(5):1442–7. doi: 10.1016/S0016-5085(97)70024-6
49. Scida S, Russo M, Miraglia C, Leandro G, Franzoni L, Meschi T, et al. Relationship Between Helicobacter Pylori Infection and GERD. Acta Bio-medica Atenei Parmensis (2018) 89(8-s):40–3. doi: 10.23750/abm.v89i8-S.7918
50. Nwokolo CU, Freshwater DA, O'Hare P, Randeva HS. Plasma Ghrelin Following Cure of Helicobacter Pylori. Gut (2003) 52(5):637–40. doi: 10.1136/gut.52.5.637
51. Francois F, Roper J, Joseph N, Pei Z, Chhada A, Shak JR, et al. The Effect of H. Pylori Eradication on Meal-Associated Changes in Plasma Ghrelin and Leptin. BMC Gastroenterol (2011) 11:37. doi: 10.1186/1471-230X-11-37
52. Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, et al. Predominant TH2-like Bronchoalveolar T-lymphocyte Population in Atopic Asthma. New Engl J Med (1992) 326(5):298–304. doi: 10.1056/NEJM199201303260504
53. Wang JM, Rambaldi A, Biondi A, Chen ZG, Sanderson CJ, Mantovani A. Recombinant Human Interleukin 5 Is a Selective Eosinophil Chemoattractant. Eur J Immunol (1989) 19(4):701–5. doi: 10.1002/eji.1830190420
54. Renauld JC, Kermouni A, Vink A, Louahed J, Van Snick J. Interleukin-9 and its Receptor: Involvement in Mast Cell Differentiation and T Cell Oncogenesis. J Leukocyte Biol (1995) 57(3):353–60. doi: 10.1002/jlb.57.3.353
55. Rothenberg ME. Molecular, Genetic, and Cellular Bases for Treating Eosinophilic Esophagitis. Gastroenterology (2015) 148(6):1143–57. doi: 10.1053/j.gastro.2015.02.002
56. Simon D, Straumann A, Schoepfer AM, Simon HU. Current Concepts in Eosinophilic Esophagitis. Allergo J Int (2017) 26(7):258–66. doi: 10.1007/s40629-017-0037-8
57. Rothenberg ME. Biology and Treatment of Eosinophilic Esophagitis. Gastroenterology (2009) 137(4):1238–49. doi: 10.1053/j.gastro.2009.07.007
58. Strachan DP. Hay Fever, Hygiene, and Household Size. Bmj (1989) 299(6710):1259–60. doi: 10.1136/bmj.299.6710.1259
59. Giriens B, Yan P, Safroneeva E, Zwahlen M, Reinhard A, Nydegger A, et al. Escalating Incidence of Eosinophilic Esophagitis in Canton of Vaud, Switzerland, 1993-2013: A Population-Based Study. Allergy (2015) 70:1633–9. doi: 10.1111/all.12733
60. Blaser MJ, Falkow S. What Are the Consequences of the Disappearing Human Microbiota? Nat Rev Microbiol (2009) 7(12):887–94. doi: 10.1038/nrmicro2245
61. Chen Y, Blaser MJ. Inverse Associations of Helicobacter Pylori With Asthma and Allergy. Arch Internal Med (2007) 167(8):821–7. doi: 10.1001/archinte.167.8.821
62. Amberbir A, Medhin G, Erku W, Alem A, Simms R, Robinson K, et al. Effects of Helicobacter Pylori, Geohelminth Infection and Selected Commensal Bacteria on the Risk of Allergic Disease and Sensitization in 3-Year-Old Ethiopian Children. Clin Exp Allergy J Br Soc Allergy Clin Immunol (2011) 41(10):1422–30. doi: 10.1111/j.1365-2222.2011.03831.x
63. Reibman J, Marmor M, Filner J, Fernandez-Beros ME, Rogers L, Perez-Perez GI, et al. Asthma is Inversely Associated With Helicobacter Pylori Status in an Urban Population. PloS One (2008) 3(12):e4060. doi: 10.1371/journal.pone.0004060
64. Li W, Minohara M, Su JJ, Matsuoka T, Osoegawa M, Ishizu T, et al. Helicobacter Pylori Infection Is a Potential Protective Factor Against Conventional Multiple Sclerosis in the Japanese Population. J Neuroimmunol (2007) 184(1-2):227–31. doi: 10.1016/j.jneuroim.2006.12.010
65. Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, Taube C, et al. Helicobacter Pylori Infection Prevents Allergic Asthma in Mouse Models Through the Induction of Regulatory T Cells. J Clin Invest (2011) 121(8):3088–93. doi: 10.1172/JCI45041
66. Luther J, Owyang SY, Takeuchi T, Cole TS, Zhang M, Liu M, et al. Helicobacter Pylori DNA Decreases Pro-Inflammatory Cytokine Production by Dendritic Cells and Attenuates Dextran Sodium Sulphate-Induced Colitis. Gut (2011) 60(11):1479–86. doi: 10.1136/gut.2010.220087
67. Engler DB, Leonardi I, Hartung ML, Kyburz A, Spath S, Becher B, et al. Helicobacter Pylori-Specific Protection Against Inflammatory Bowel Disease Requires the NLRP3 Inflammasome and IL-18. Inflammatory Bowel Diseases (2015) 21(4):854–61. doi: 10.1097/MIB.0000000000000318
68. Luther J, Dave M, Higgins PD, Kao JY. Association Between Helicobacter Pylori Infection and Inflammatory Bowel Disease: A Meta-Analysis and Systematic Review of the Literature. Inflammatory Bowel Diseases (2010) 16(6):1077–84. doi: 10.1002/ibd.21116
69. Engler DB, Reuter S, van Wijck Y, Urban S, Kyburz A, Maxeiner J, et al. Effective Treatment of Allergic Airway Inflammation With Helicobacter Pylori Immunomodulators Requires BATF3-dependent Dendritic Cells and IL-10. Proc Natl Acad Sci USA (2014) 111(32):11810–5. doi: 10.1073/pnas.1410579111
70. Murray T, Fuertes Marraco SA, Baumgaertner P, Bordry N, Cagnon L, Donda A, et al. Very Late Antigen-1 Marks Functional Tumor-Resident Cd8 T Cells and Correlates With Survival of Melanoma Patients. Front Immunol (2016) 7:573. doi: 10.3389/fimmu.2016.00573
71. Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-Derived Inosine Modulates Response to Checkpoint Inhibitor Immunotherapy. Science (2020) 369(6510):1481–9. doi: 10.1126/science.abc3421
72. Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates anti-PD-L1 Efficacy. Science (2015) 350(6264):1084–9. doi: 10.1126/science.aac4255
73. Uribe-Herranz M, Bittinger K, Rafail S, Guedan S, Pierini S, Tanes C, et al. Gut Microbiota Modulates Adoptive Cell Therapy Via CD8alpha Dendritic Cells and IL-12. JCI Insight (2018) 3(4):e94952. doi: 10.1172/jci.insight.94952
74. van Wijck Y, de Kleijn S, John-Schuster G, Mertens TCJ, Hiemstra PS, Muller A, et al. Therapeutic Application of an Extract of Helicobacter Pylori Ameliorates the Development of Allergic Airway Disease. J Immunol (2018) 200(5):1570–9. doi: 10.4049/jimmunol.1700987
75. Cheng WY, Wu C-Y, Yu J. The Role of Gut Microbiota in Cancer Treatment: Friend or Foe? Gut (2020) 69(10):1867–76. doi: 10.1136/gutjnl-2020-321153
76. Kienesberger S, Cox LM, Livanos A, Zhang XS, Chung J, Perez-Perez GI, et al. Gastric Helicobacter Pylori Infection Affects Local and Distant Microbial Populations and Host Responses. Cell Rep (2016) 14(6):1395–407. doi: 10.1016/j.celrep.2016.01.017
77. Chen CC, Liou JM, Lee YC, Hong TC, El-Omar EM, Wu MS. The Interplay Between Helicobacter Pylori and Gastrointestinal Microbiota. Gut Microbes (2021) 13(1):1–22. doi: 10.1080/19490976.2021.1909459
78. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell (2018) 33(4):570–80. doi: 10.1016/j.ccell.2018.03.015
79. Roberti MP, Yonekura S, Duong CPM, Picard M, Ferrere G, Tidjani Alou M, et al. Chemotherapy-Induced Ileal Crypt Apoptosis and the Ileal Microbiome Shape Immunosurveillance and Prognosis of Proximal Colon Cancer. Nat Med (2020) 26(6):919–31. doi: 10.1038/s41591-020-0882-8
80. Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative Analysis of Human Gut Microbiota by Barcoded Pyrosequencing. PloS One (2008) 3(7):e2836. doi: 10.1371/journal.pone.0002836
81. Eun CS, Kim BK, Han DS, Kim SY, Kim KM, Choi BY, et al. Differences in Gastric Mucosal Microbiota Profiling in Patients With Chronic Gastritis, Intestinal Metaplasia, and Gastric Cancer Using Pyrosequencing Methods. Helicobacter (2014) 19(6):407–16. doi: 10.1111/hel.12145
82. Schaupp L, Muth S, Rogell L, Kofoed-Branzk M, Melchior F, Lienenklaus S, et al. Microbiota-Induced Type I Interferons Instruct a Poised Basal State of Dendritic Cells. Cell (2020) 181(5):1080–96.e19. doi: 10.1016/j.cell.2020.04.022
83. Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, et al. A Defined Commensal Consortium Elicits CD8 T Cells and Anti-Cancer Immunity. Nature (2019) 565(7741):600–5. doi: 10.1038/s41586-019-0878-z
84. Hitzler I, Oertli M, Becher B, Agger EM, Müller A. Dendritic Cells Prevent Rather Than Promote Immunity Conferred by a Helicobacter Vaccine Using a Mycobacterial Adjuvant. Gastroenterology (2011) 141(1):186–96-96.e1. doi: 10.1053/j.gastro.2011.04.009
85. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-Derived IL-18 Drives Treg Differentiation, Murine Helicobacter Pylori-Specific Immune Tolerance, and Asthma Protection. J Clin Invest (2012) 122(3):1082–96. doi: 10.1172/JCI61029
86. Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, et al. Helicobacter Pylori Immune Escape is Mediated by Dendritic Cell-Induced Treg Skewing and Th17 Suppression in Mice. Gastroenterology (2010) 138(3):1046–54. doi: 10.1053/j.gastro.2009.11.043
87. Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter Pylori Impairs Murine Dendritic Cell Responses to Infection. PloS One (2010) 5(5):e10844. doi: 10.1371/journal.pone.0010844
88. Oertli M, Noben M, Engler DB, Semper RP, Reuter S, Maxeiner J, et al. Helicobacter Pylori Gamma-Glutamyl Transpeptidase and Vacuolating Cytotoxin Promote Gastric Persistence and Immune Tolerance. Proc Natl Acad Sci USA (2013) 110(8):3047–52. doi: 10.1073/pnas.1211248110
89. Tanaka H, Yoshida M, Nishiumi S, Ohnishi N, Kobayashi K, Yamamoto K, et al. The CagA Protein of Helicobacter Pylori Suppresses the Functions of Dendritic Cell in Mice. Arch Biochem Biophysics (2010) 498(1):35–42. doi: 10.1016/j.abb.2010.03.021
90. Kaebisch R, Mejias-Luque R, Prinz C, Gerhard M. Helicobacter Pylori Cytotoxin-Associated Gene A Impairs Human Dendritic Cell Maturation and Function Through IL-10-mediated Activation of STAT3. J Immunol (2014) 192(1):316–23. doi: 10.4049/jimmunol.1302476
91. Semper RP, Mejías-Luque R, Groß C, Anderl F, Müller A, Vieth M, et al. Helicobacter Pylori–Induced IL-1β Secretion in Innate Immune Cells is Regulated by the NLRP3 Inflammasome and Requires the Cag Pathogenicity Island. J Immunol (2014) 193(7):3566–76. doi: 10.4049/jimmunol.1400362
92. Kim DJ, Park JH, Franchi L, Backert S, Núñez G. The Cag Pathogenicity Island and Interaction Between TLR2/NOD2 and NLRP3 Regulate IL-1β Production in Helicobacter Pylori Infected Dendritic Cells. Eur J Immunol (2013) 43(10):2650–8. doi: 10.1002/eji.201243281
93. Hitzler I, Sayi A, Kohler E, Engler DB, Koch KN, Hardt W-D, et al. Caspase-1 Has Both Proinflammatory and Regulatory Properties in Helicobacter Infections, Which are Differentially Mediated by Its Substrates Il-1β and IL-18. J Immunol (2012) 188(8):3594–602. doi: 10.4049/jimmunol.1103212
94. Koch KN, Hartung ML, Urban S, Kyburz A, Bahlmann AS, Lind J, et al. Helicobacter Urease-Induced Activation of the TLR2/NLRP3/IL-18 Axis Protects Against Asthma. J Clin Invest (2015) 125(8):3297–302. doi: 10.1172/JCI79337
95. Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, et al. Helicobacter Pylori Modulates the T Helper Cell 1/T Helper Cell 2 Balance Through Phase-Variable Interaction Between Lipopolysaccharide and DC-SIGN. J Exp Med (2004) 200(8):979–90. doi: 10.1084/jem.20041061
96. Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Geijtenbeek TB. Carbohydrate-Specific Signaling Through the DC-SIGN Signalosome Tailors Immunity to Mycobacterium Tuberculosis, HIV-1 and Helicobacter Pylori. Nat Immunol (2009) 10(10):1081–8. doi: 10.1038/ni.1778
97. Käbisch R, Semper RP, Wüstner S, Gerhard M, Mejías-Luque R. Helicobacter Pylori γ-Glutamyltranspeptidase Induces Tolerogenic Human Dendritic Cells by Activation of Glutamate Receptors. J Immunol (2016) 196(10):4246–52. doi: 10.4049/jimmunol.1501062
98. Wan Z, Sun R, Liu Y-W, Li S, Sun J, Li J, et al. Targeting Metabotropic Glutamate Receptor 4 for Cancer Immunotherapy. Sci Adv (2021) 7(50):eabj4226. doi: 10.1126/sciadv.abj4226
99. Altobelli A, Bauer M, Velez K, Cover TL, Muller A. Helicobacter Pylori VacA Targets Myeloid Cells in the Gastric Lamina Propria to Promote Peripherally Induced Regulatory T-Cell Differentiation and Persistent Infection. mBio (2019) 10(2):e00261–19. doi: 10.1128/mBio.00261-19
100. Gobert AP, Mersey BD, Cheng Y, Blumberg DR, Newton JC, Wilson KT. Cutting Edge: Urease Release by Helicobacter Pylori Stimulates Macrophage Inducible Nitric Oxide Synthase. J Immunol (2002) 168(12):6002–6. doi: 10.4049/jimmunol.168.12.6002
101. Gobert AP, Bambou JC, Werts C, Balloy V, Chignard M, Moran AP, et al. Helicobacter Pylori Heat Shock Protein 60 Mediates Interleukin-6 Production by Macrophages in a Toll-Like Receptor (TLR)-2-, TLR-4-, and Myeloid Differentiation Factor 88-Independent Mechanism. J Biol Chem (2004) 279(1):245–50. doi: 10.1074/jbc.M307858200
102. Pérez-Pérez GI, Shepherd VL, Morrow JD, Blaser MJ. Activation of Human THP-1 Cells and Rat Bone Marrow-Derived Macrophages by Helicobacter Pylori Lipopolysaccharide. Infect Immunity (1995) 63(4):1183–7. doi: 10.1128/iai.63.4.1183-1187.1995
103. Devi S, Rajakumara E, Ahmed N. Induction of Mincle by Helicobacter Pylori and Consequent Anti-Inflammatory Signaling Denote a Bacterial Survival Strategy. Sci Rep (2015) 5:15049. doi: 10.1038/srep15049
104. Allen LA, Schlesinger LS, Kang B. Virulent Strains of Helicobacter Pylori Demonstrate Delayed Phagocytosis and Stimulate Homotypic Phagosome Fusion in Macrophages. J Exp Med (2000) 191(1):115–28. doi: 10.1084/jem.191.1.115
105. Menaker RJ, Ceponis PJ, Jones NL. Helicobacter Pylori Induces Apoptosis of Macrophages in Association With Alterations in the Mitochondrial Pathway. Infect Immunity (2004) 72(5):2889–98. doi: 10.1128/IAI.72.5.2889-2898.2004
106. Chaturvedi R, Asim M, Hoge S, Lewis ND, Singh K, Barry DP, et al. Polyamines Impair Immunity to Helicobacter Pylori by Inhibiting L-Arginine Uptake Required for Nitric Oxide Production. Gastroenterology (2010) 139(5):1686–98, 98.e1-6. doi: 10.1053/j.gastro.2010.06.060
107. Quiding-Järbrink M, Raghavan S, Sundquist M. Enhanced M1 Macrophage Polarization in Human Helicobacter Pylori-Associated Atrophic Gastritis and in Vaccinated Mice. PloS One (2010) 5(11):e15018. doi: 10.1371/journal.pone.0015018
108. Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, et al. Immune Evasion by Helicobacter Pylori is Mediated by Induction of Macrophage Arginase II. J Immunol (2011) 186(6):3632–41. doi: 10.4049/jimmunol.1003431
109. Fehlings M, Drobbe L, Moos V, Renner Viveros P, Hagen J, Beigier-Bompadre M, et al. Comparative Analysis of the Interaction of Helicobacter Pylori With Human Dendritic Cells, Macrophages, and Monocytes. Infect Immunity (2012) 80(8):2724–34. doi: 10.1128/IAI.00381-12
110. Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon Monoxide: From Basic Science to Therapeutic Applications. Physiol Rev (2006) 86(2):583–650. doi: 10.1152/physrev.00011.2005
111. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol (2002) 23(11):549–55. doi: 10.1016/S1471-4906(02)02302-5
112. Duan Z, Luo Y. Targeting Macrophages in Cancer Immunotherapy. Signal Transduction Targeted Ther (2021) 6(1):127. doi: 10.1038/s41392-021-00506-6
113. Zabaleta J, McGee DJ, Zea AH, Hernandez CP, Rodriguez PC, Sierra RA, et al. Helicobacter Pylori Arginase Inhibits T Cell Proliferation and Reduces the Expression of the TCR Zeta-Chain (CD3zeta). J Immunol (2004) 173(1):586–93. doi: 10.4049/jimmunol.173.1.586
114. Wustner S, Mejias-Luque R, Koch MF, Rath E, Vieth M, Sieber SA, et al. Helicobacter Pylori Gamma-Glutamyltranspeptidase Impairs T-lymphocyte Function by Compromising Metabolic Adaption Through Inhibition of cMyc and IRF4 Expression. Cell Microbiol (2015) 17(1):51–61. doi: 10.1111/cmi.12335
115. Sewald X, Jimenez-Soto L, Haas R. PKC-Dependent Endocytosis of the Helicobacter Pylori Vacuolating Cytotoxin in Primary T Lymphocytes. Cell Microbiol (2011) 13(3):482–96. doi: 10.1111/j.1462-5822.2010.01551.x
116. Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of Primary Human T Cell Proliferation by Helicobacter Pylori Vacuolating Toxin (VacA) is Independent of VacA Effects on IL-2 Secretion. Proc Natl Acad Sci USA (2004) 101(20):7727–32. doi: 10.1073/pnas.0401528101
117. Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, et al. The Helicobacter Pylori Vacuolating Toxin Inhibits T Cell Activation by Two Independent Mechanisms. J Exp Med (2003) 198(12):1887–97. doi: 10.1084/jem.20030621
118. Wu J, Zhu X, Guo X, Yang Z, Cai Q, Gu D, et al. Helicobacter Urease Suppresses Cytotoxic CD8+ T-Cell Responses Through Activating Myh9-dependent Induction of PD-L1. Int Immunol (2021) 33(9):491–504. doi: 10.1093/intimm/dxab044
119. Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on Gastric Epithelial Cells: Its Potential Role in Regulating T Cells During Helicobacter Pylori Infection. J Immunol (2006) 176(5):3000–9. doi: 10.4049/jimmunol.176.5.3000
120. Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE. Expression of the Programmed Death Ligand 1, B7-H1, on Gastric Epithelial Cells After Helicobacter Pylori Exposure Promotes Development of CD4+ Cd25+ FoxP3+ Regulatory T Cells. Infect Immunity (2007) 75(9):4334–41. doi: 10.1128/IAI.00553-07
121. Beswick EJ, Pinchuk IV, Earley RB, Schmitt DA, Reyes VE. Role of Gastric Epithelial Cell-Derived Transforming Growth Factor Beta in Reduced CD4+ T Cell Proliferation and Development of Regulatory T Cells During Helicobacter Pylori Infection. Infect Immunity (2011) 79(7):2737–45. doi: 10.1128/IAI.01146-10
122. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The Commensal Microbiome is Associated With anti-PD-1 Efficacy in Metastatic Melanoma Patients. Science (2018) 359(6371):104–8. doi: 10.1126/science.aao3290
123. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline Gut Microbiota Predicts Clinical Response and Colitis in Metastatic Melanoma Patients Treated With Ipilimumab. Ann Oncol (2017) 28(6):1368–79. doi: 10.1093/annonc/mdx108
124. Koch KN, Müller A. Helicobacter Pylori Activates the TLR2/NLRP3/caspase-1/IL-18 Axis to Induce Regulatory T-cells, Establish Persistent Infection and Promote Tolerance to Allergens. Gut Microbes (2015) 6:382–7. doi: 10.1080/19490976.2015.1105427
125. Lv YP, Cheng P, Zhang JY, Mao FY, Teng YS, Liu YG, et al. Helicobacter Pylori-Induced Matrix Metallopeptidase-10 Promotes Gastric Bacterial Colonization and Gastritis. Sci Adv (2019) 5(4):eaau6547. doi: 10.1126/sciadv.aau6547
126. Bhattacharjee S, Mejías-Luque R, Loffredo-Verde E, Toska A, Flossdorf M, Gerhard M, et al. Concomitant Infection of S. Mansoni and H. Pylori Promotes Promiscuity of Antigen-Experienced Cells and Primes the Liver for a Lower Fibrotic Response. Cell Rep (2019) 28(1):231–44.e5. doi: 10.1016/j.celrep.2019.05.108
127. Artola-Borán M, Fallegger A, Priola M, Jeske R, Waterboer T, Dohlman AB, et al. Mycobacterial Infection Aggravates Helicobacter Pylori-Induced Gastric Preneoplastic Pathology by Redirection of De Novo Induced Treg Cells. Cell Rep (2022) 38(6):110359. doi: 10.1016/j.celrep.2022.110359
128. Zeissig S, Blumberg RS. Life at the Beginning: Perturbation of the Microbiota by Antibiotics in Early Life and its Role in Health and Disease. Nat Immunol (2014) 15(4):307–10. doi: 10.1038/ni.2847
129. Wang Y, Cardell SL. The Yin and Yang of Invariant Natural Killer T Cells in Tumor Immunity—Suppression of Tumor Immunity in the Intestine. Front Immunol (2018) 8. doi: 10.3389/fimmu.2017.01945
130. Ito Y, Vela JL, Matsumura F, Hoshino H, Tyznik A, Lee H, et al. Helicobacter Pylori Cholesteryl Alpha-Glucosides Contribute to Its Pathogenicity and Immune Response by Natural Killer T Cells. PloS One (2013) 8(12):e78191. doi: 10.1371/journal.pone.0078191
131. Chang YJ. Innate Lymphoid Cells Mediate Influenza-Induced Airway Hyper-Reactivity Independently of Adaptive Immunity. Nat Immunol (2011) 12:631–8. doi: 10.1038/ni.2045
132. Pilones KA, Kawashima N, Yang AM, Babb JS, Formenti SC, Demaria S. Invariant Natural Killer T Cells Regulate Breast Cancer Response to Radiation and CTLA-4 Blockade. Clin Cancer Res (2009) 15(2):597–606. doi: 10.1158/1078-0432.CCR-08-1277
133. Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the Epithelial Apical-Junctional Complex by Helicobacter Pylori Caga. Science (2003) 300(5624):1430–4. doi: 10.1126/science.1081919
134. Fedwick JP, Lapointe TK, Meddings JB, Sherman PM, Buret AG. Helicobacter Pylori Activates Myosin Light-Chain Kinase to Disrupt Claudin-4 and Claudin-5 and Increase Epithelial Permeability. Infect Immunity (2005) 73(12):7844–52. doi: 10.1128/IAI.73.12.7844-7852.2005
135. Wroblewski LE, Shen L, Ogden S, Romero-Gallo J, Lapierre LA, Israel DA, et al. Helicobacter Pylori Dysregulation of Gastric Epithelial Tight Junctions by Urease-Mediated Myosin II Activation. Gastroenterology (2009) 136(1):236–46. doi: 10.1053/j.gastro.2008.10.011
136. Mu Q, Kirby J, Reilly CM, Luo XM. Leaky Gut as a Danger Signal for Autoimmune Diseases. Front Immunol (2017) 8:598. doi: 10.3389/fimmu.2017.00598
137. Uchimura Y, Fuhrer T, Li H, Lawson MA, Zimmermann M, Yilmaz B, et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity (2018) 49(3):545–59.e5. doi: 10.1016/j.immuni.2018.08.004
138. Louis P, Flint HJ. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ Microbiol (2017) 19(1):29–41. doi: 10.1111/1462-2920.13589
139. Bolognini D, Tobin AB, Milligan G, Moss CE. The Pharmacology and Function of Receptors for Short-Chain Fatty Acids. Mol Pharmacol (2016) 89(3):388–98. doi: 10.1124/mol.115.102301
140. Hayase E, Jenq RR. Role of the Intestinal Microbiome and Microbial-Derived Metabolites in Immune Checkpoint Blockade Immunotherapy of Cancer. Genome Med (2021) 13(1):107. doi: 10.1186/s13073-021-00923-w
141. Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut Microbiota Metabolism of Dietary Fiber Influences Allergic Airway Disease and Hematopoiesis. Nat Med (2014) 20(2):159–66. doi: 10.1038/nm.3444
142. Huang Y, Ding Y, Xu H, Shen C, Chen X, Li C. Effects of Sodium Butyrate Supplementation on Inflammation, Gut Microbiota, and Short-Chain Fatty Acids in Helicobacter Pylori-Infected Mice. Helicobacter (2021) 26(2):e12785. doi: 10.1111/hel.12785
143. Lamas B, Natividad JM, Sokol H. Aryl Hydrocarbon Receptor and Intestinal Immunity. Mucosal Immunol (2018) 11(4):1024–38. doi: 10.1038/s41385-018-0019-2
144. Fung TC, Olson CA, Hsiao EY. Interactions Between the Microbiota, Immune and Nervous Systems in Health and Disease. Nat Neurosci (2017) 20(2):145–55. doi: 10.1038/nn.4476
145. Hezaveh K, Shinde RS, Klötgen A, Halaby MJ, Lamorte S, Ciudad MT, et al. Tryptophan-Derived Microbial Metabolites Activate the Aryl Hydrocarbon Receptor in Tumor-Associated Macrophages to Suppress Anti-Tumor Immunity. Immunity (2022) 55(2):324–40.e8. doi: 10.1016/j.immuni.2022.01.006
146. Gargaro M, Manni G, Scalisi G, Puccetti P, Fallarino F. Tryptophan Metabolites at the Crossroad of Immune-Cell Interaction Via the Aryl Hydrocarbon Receptor: Implications for Tumor Immunotherapy. Int J Mol Sci (2021) 22:4644. doi: 10.3390/ijms22094644
147. Inagaki T, Moschetta A, Lee Y-K, Peng L, Zhao G, Downes M, et al. Regulation of Antibacterial Defense in the Small Intestine by the Nuclear Bile Acid Receptor. Proc Natl Acad Sci USA (2006) 103(10):3920–5. doi: 10.1073/pnas.0509592103
148. Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, et al. TGR5 Activation Inhibits Atherosclerosis by Reducing Macrophage Inflammation and Lipid Loading. Cell Metab (2011) 14(6):747–57. doi: 10.1016/j.cmet.2011.11.006
149. Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile Acids Activated Receptors Regulate Innate Immunity. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01853
150. Phelan JP, Reen FJ, Caparros-Martin JA, O'Connor R, O'Gara F. Rethinking the Bile Acid/Gut Microbiome Axis in Cancer. Oncotarget (2017) 8(70):115736–47. doi: 10.18632/oncotarget.22803
151. Olofsson A, Vallström A, Petzold K, Tegtmeyer N, Schleucher J, Carlsson S, et al. Biochemical and Functional Characterization of Helicobacter Pylori Vesicles. Mol Microbiol (2010) 77(6):1539–55. doi: 10.1111/j.1365-2958.2010.07307.x
152. Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC, et al. Bacterial Membrane Vesicles Deliver Peptidoglycan to NOD1 in Epithelial Cells. Cell Microbiol (2010) 12(3):372–85. doi: 10.1111/j.1462-5822.2009.01404.x
153. Shimoda A, Ueda K, Nishiumi S, Murata-Kamiya N, Mukai SA, Sawada S, et al. Exosomes as Nanocarriers for Systemic Delivery of the Helicobacter Pylori Virulence Factor Caga. Sci Rep (2016) 6:18346. doi: 10.1038/srep18346
154. Mai UE, Perez-Perez GI, Allen JB, Wahl SM, Blaser MJ, Smith PD. Surface Proteins From Helicobacter Pylori Exhibit Chemotactic Activity for Human Leukocytes and are Present in Gastric Mucosa. J Exp Med (1992) 175(2):517–25. doi: 10.1084/jem.175.2.517
155. Ndawula EM, Owen RJ, Mihr G, Borman P, Hurtado A. Helicobacter Pylori Bacteraemia. Eur J Clin Microbiol Infect Diseases (1994) 13(7):621. doi: 10.1007/BF01971319
156. Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. Endoscopic Photoconversion Reveals Unexpectedly Broad Leukocyte Trafficking to and From the Gut. Proc Natl Acad Sci (2014) 111(18):6696–701. doi: 10.1073/pnas.1405634111
157. Balmer ML, Schürch CM, Saito Y, Geuking MB, Li H, Cuenca M, et al. Microbiota-Derived Compounds Drive Steady-State Granulopoiesis Via MyD88/TICAM Signaling. J Immunol (2014) 193(10):5273–83. doi: 10.4049/jimmunol.1400762
158. Hergott CB, Roche AM, Tamashiro E, Clarke TB, Bailey AG, Laughlin A, et al. Peptidoglycan From the Gut Microbiota Governs the Lifespan of Circulating Phagocytes at Homeostasis. Blood (2016) 127(20):2460–71. doi: 10.1182/blood-2015-10-675173
159. Zegarra-Ruiz DF, Kim DV, Norwood K, Kim M, Wu WH, Saldana-Morales FB, et al. Thymic Development of Gut-Microbiota-Specific T Cells. Nature (2021) 594(7863):413–7. doi: 10.1038/s41586-021-03531-1
160. Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, Dejardin F, et al. A Weaning Reaction to Microbiota is Required for Resistance to Immunopathologies in the Adult. Immunity (2019) 50(5):1276–88 e5. doi: 10.1016/j.immuni.2019.02.014
161. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell (2009) 139(3):485–98. doi: 10.1016/j.cell.2009.09.033
162. Flannigan KL, Denning TL. Segmented Filamentous Bacteria-Induced Immune Responses: A Balancing Act Between Host Protection and Autoimmunity. Immunology (2018) 154(4):537–46. doi: 10.1111/imm.12950
163. Velin D, Fotopoulos G, Luthi F, Kraehenbuhl JP. The Nasal-Associated Lymphoid Tissue of Adult Mice Acts as an Entry Site for the Mouse Mammary Tumor Retrovirus. J Exp Med (1997) 185(10):1871–6. doi: 10.1084/jem.185.10.1871
164. Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, et al. Tolerance Rather Than Immunity Protects From Helicobacter Pylori-Induced Gastric Preneoplasia. Gastroenterology (2011) 140(1):199–209. doi: 10.1053/j.gastro.2010.06.047
Keywords: Helicobacter pylori, cancer, immunotherapy; personalized medicine, gut microbiota, immune checkpoint inhibitors
Citation: Oster P, Vaillant L, McMillan B and Velin D (2022) The Efficacy of Cancer Immunotherapies Is Compromised by Helicobacter pylori Infection. Front. Immunol. 13:899161. doi: 10.3389/fimmu.2022.899161
Received: 18 March 2022; Accepted: 26 April 2022;
Published: 23 May 2022.
Edited by:
JoAnn M. Sekiguchi, University of Michigan, United StatesReviewed by:
Xuefeng Gao, Shenzhen University General Hospital, ChinaHolly Algood, Vanderbilt University, China
Copyright © 2022 Oster, Vaillant, McMillan and Velin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dominique Velin, Dominique.velin@chuv.ch