
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 02 June 2022
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.886540
This article is part of the Research TopicNovel Aspects in Treatment of Skin Disorders in Primary ImmunodeficienciesView all 5 articles
 Ulrich Baumann1
Ulrich Baumann1 Johannes H. Schulte2
Johannes H. Schulte2 Jonathan P. Groß2
Jonathan P. Groß2 Rita Beier3
Rita Beier3 Marius Ludwig2Volker Wahn4Jörg Hofmann5,6Britta Maecker-Kolhoff3Martin Sauer3Petra Kaiser-Labusch7Negin Karimian8
Marius Ludwig2Volker Wahn4Jörg Hofmann5,6Britta Maecker-Kolhoff3Martin Sauer3Petra Kaiser-Labusch7Negin Karimian8 Ulrike Blume-Peytavi8Franziska Ghoreschi8Hagen Ott9
Ulrike Blume-Peytavi8Franziska Ghoreschi8Hagen Ott9 Ludmila Perelygina10Christian Klemann1Oliver Blankenstein11
Ludmila Perelygina10Christian Klemann1Oliver Blankenstein11 Horst von Bernuth4,12,13,14†
Horst von Bernuth4,12,13,14† Renate Krüger4*†
Renate Krüger4*†We report two patients with DNA repair disorders (Artemis deficiency, Ataxia telangiectasia) with destructive skin granulomas, presumably triggered by live-attenuated rubella vaccinations. Both patients showed reduced naïve T cells. Rapid resolution of skin lesions was observed following hematopoietic stem cell transplantation. However, the patient with AT died due to complications of severe hepatic veno-occlusive disease 6 month after HSCT. Dried blood spots obtained after birth were available from this patient and showed absent T-cell receptor excision circles (TRECs). Therefore, newborn screening may help to prevent patients with moderate T-cell deficiency from receiving live-attenuated rubella vaccine potentially causing granulomas.
Vaccine rubella virus (RuV)-induced granuloma (RG) was first described in patients with combined immunodeficiency (CID) in 2014 (1). Initially, patients with DNA double-strand breakage repair disorders were reported. To date, a growing number of patients with inborn errors of immunity (IEI) of different genetic origin and vaccine RuV-induced granulomas have been published (2), also reviewed in (3). Sustained resolution of RG has only been observed after hematopoetic stem cell transplantation (HSCT) (4). However, despite reduced-intensity conditioning, HSCT in patients with DNA repair disorders is complicated by higher transplant-associated morbidity and mortality as compared to other IEI patients (5). In the current issue of Frontiers in Immunology, Perelygina et al. discuss pathomechanisms, clinical characteristics and treatment of vaccine RuV-induced granulomas in a cohort of 28 patients with various IEI with CID (3). HSCT has been shown to be a therapeutic option, but 2 out of 7 patients were lost due to transplantation related mortality. Here, we report two children with DNA repair disorders and destructive skin RG that showed prompt resolution following HSCT. However, one patient, also described in the cohort (Pat. 28), died from HSCT complications, increasing the mortality to 3 out of 7 transplanted patients.
Patient 1: This patient suffered from recurrent diarrhea and chronic unproductive cough beginning in infancy. At school age, he developed a productive cough with bronchiectasis, vitiligo, and retarded growth. Immunological workup at preschool age had shown moderate CD4+ and CD8+ T-cell lymphocytopenia, normal immunoglobulin isotype levels and vaccine-induced antibodies. Next-generation sequencing covering a panel of 104 genes yielded no pathological findings at the age of 6.5 years. Two years later, he suffered from pneumonia due to varicella-zoster virus with a prolonged course of over 2 months. He was subsequently commenced on immunoglobulin substitution. At the age of 7.5 years, granulomatous lesions appeared at the heel and the nose (Figure 1A). Skin biopsy of the granulomatous lesions showed rubella antigen in epitheloid cells, but neither in CD15+ granulocytes nor CD68+ macrophages. No testing for specific rubella strains was performed. The boy had received 2 MMR live vaccinations in his 4th year of life. Topical steroid application was not effective in limiting progression. At age 8.5 years, whole-exome sequencing revealed a homozygous splice variant in DCLRE1C (c.464+1G>A), suggestive for a DNA repair disorder with combined immunodeficiency consistent with Artemis deficiency. As the patient was in a stable condition receiving immunoglobulin treatment, the parents were reluctant to agree to a proposed HSCT. It was due to progressive and disfiguring destruction of the right nostril (Figure 1) that the family eventually accepted HSCT. He subsequently received HSCT from a matched related donor at the age of 10.3 years. Conditioning was performed according to protocol C of the IEWP guideline for hypomorphic Artemis SCID (6) including busulfan (AUC 65 mg*h/l), fludarabine (4 x 45mg/m2/d) and anti-thymocyte globulin (3 x 10mg/kg/d). GvHD prophylaxis consisted of cyclosporin A and mycophenolate mofetil. Engraftment was achieved as expected with a bone marrow graft (leukocytes >1/nl and neutrophils >0.5/nl on day 26, platelets >50/nl on day 34). The post-HSCT course was complicated by an intercurrent secondary von Willebrand Jürgens syndrome and by BK-viremia which was successfully treated with cidofovir. 1.5 years post-HSCT, the patient developed self-limiting autoimmune thrombocytopenia which was more pronounced during viral upper airway infections. Chimerism after HSCT was mixed at day +16 (84% donor) and subsequently always 100% donor. The cutaneous lesions had already started to improve under conditioning and healed largely within two months after HSCT. Even the deep nasal defect partially closed. The lesions before and after HSCT are shown in Figures 1E–G clinical characteristics are summarized in Table 1. Unfortunately, the Guthrie card of patient one was not available for TREC-screening.
Patient 2: A five-year-old microcephalic girl [patient 28 in (3)] was diagnosed with Ataxia telangiectasia (AT) at the age of 3 years following progressive gait disturbances, recurrent viral and bacterial airway infections and failure to thrive (compound heterozygous mutations in ATM: c.2098C>T, p.Gln700*; second mutation probably intronic). She had also suffered from a progressive granulomatous skin lesion on the left upper arm since the age of three years (Figures 1H–J). The immunologic evaluation showed a deficiency of IgA, IgG2, IgG4 and pneumococcal antibodies with normal total IgG and normal antibodies to tetanus-toxoid. Naïve CD4+ T cells were repeatedly markedly reduced with normal lymphocyte, total B- and T-cell counts at initial evaluation (Table 1). Evaluation of the stored dried blood spot (DBS) obtained from the neonatal screening sample showed no TREC copies by using a standardized newborn screening rt-PCR assay (ImmunoIVD, Nacka Strand, Sweden). Prior to the diagnosis of AT the girl had received live vaccines against MMR at the age of 12 and 14 months. Skin biopsy of the lesion at the age of 5 years revealed a granulomatous inflammation, RT-PCR and immunohistochemistry from biopsy material showed rubella vaccine strain RA27/3 (3). The lesion did not improve upon treatment with local corticosteroids or immunoglobulin substitution (initiated due to specific antibody deficiency). HSCT was offered as a therapeutic option once local destructive growth of the lesion (Figure 1J) with increasing pain and pruritus occurred. The parents were counseled by experts for HSCT in IEI from two different hospitals. Limited data on HSCT outcome in patients with AT and risks were intensely discussed. The parents were aware that HSCT would presumably not affect progression of ataxia. They were also given the opportunity to contact parents of AT patients that had been transplanted.
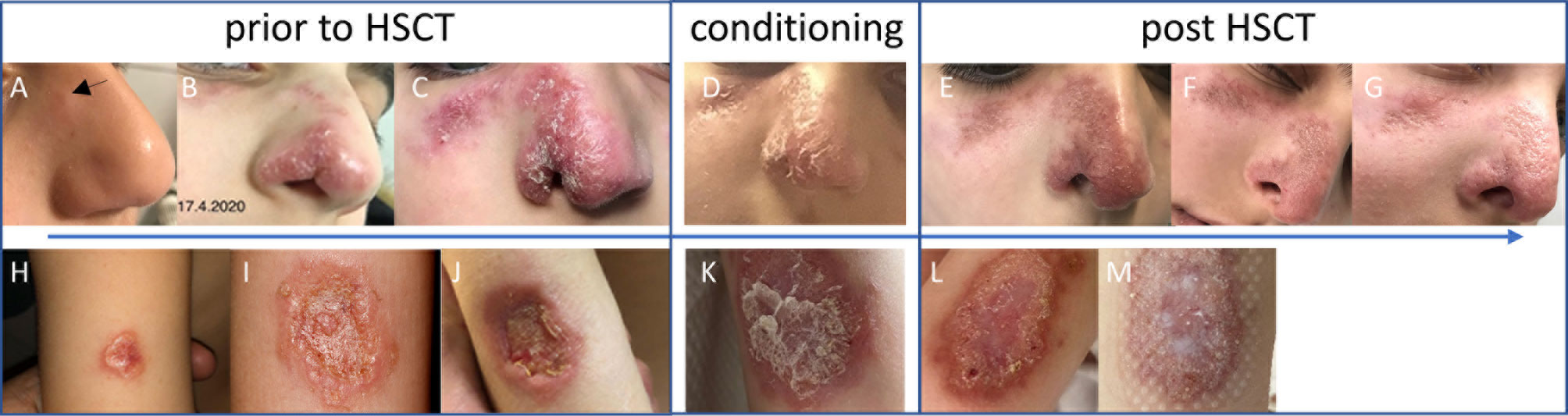
Figure 1 Skin lesion prior to HSCT (A–C, H–J), at conditioning (D, K), and post-HSCT (E–G, L, M) of patient 1 (upper panel) and patient 2 (lower panel). Pictures of patient 1 were taken at age 8.5 (A), 9.3 (B) and 10.1 (C) years, and day +4 (E), +18 (F), and +36 (G) after HSCT. Pictures of patient 2 were taken at age 4.7 (H), 5.6 (I), 5.7 (J) years, at conditioning (K), and at day +5 (L), and +15 (M) after HSCT.
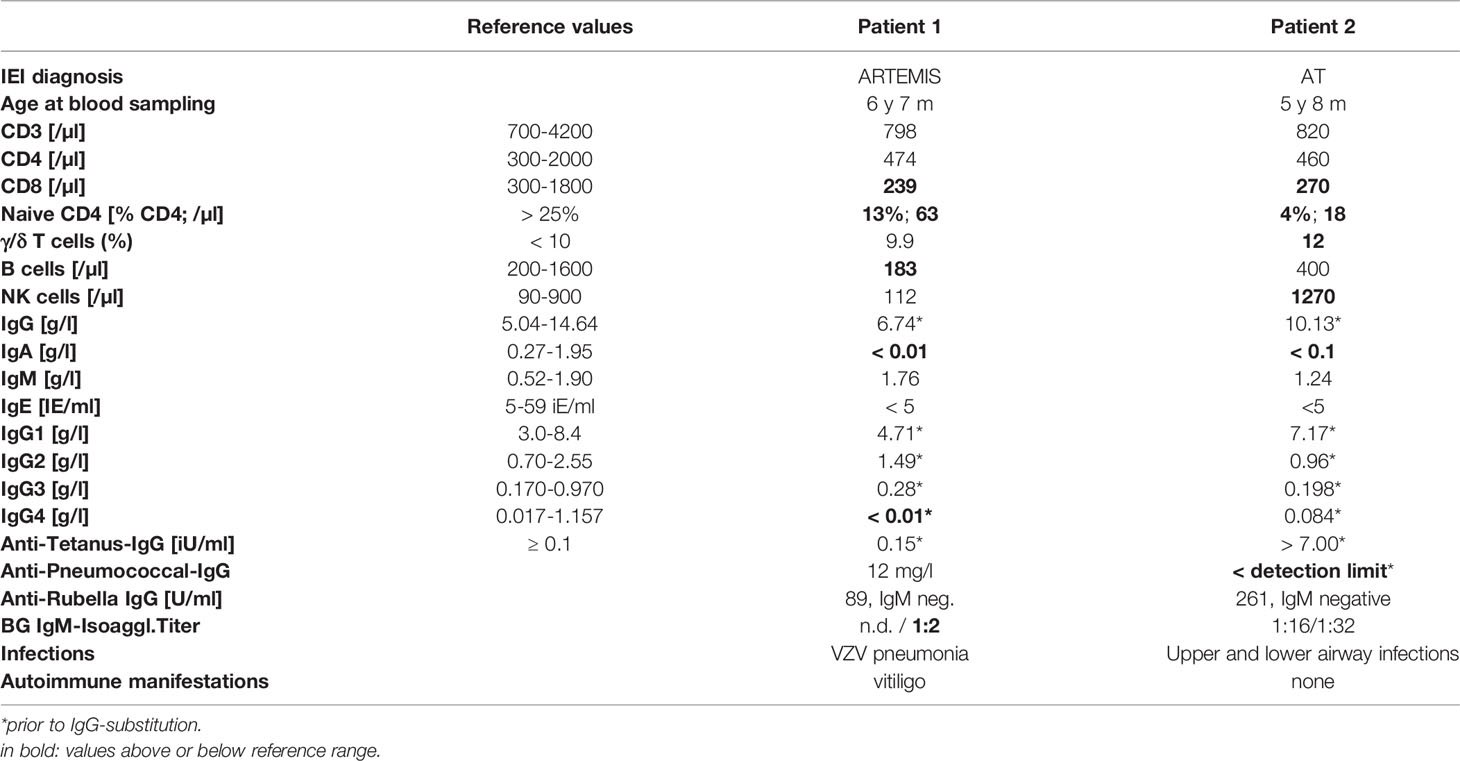
Table 1 Clinical, genetic and immunological characteristics of patient 1 and 2.
HSCT (TCRαβ/CD19-depleted peripheral blood stem cells of a matched (10/10) unrelated donor with add-back of 1 x 106 T cells/kg body weight) was performed at the age of 6 years. To prevent excessive toxicity and to reduce the risk of malignancy in DNA repair disorders, a reduced-intensity conditioning (RIC) was applied consisting of fludarabine (5 x 30 mg/m2/d), cyclophosphamide (4 x 30 mg/kg/d) and rabbit anti-thymocyte globulin (ATG Fresenius® 3 x 20 mg/kg/d) derived from Bakhtiar et al. (7). The skin lesion`s regression was already observed upon treatment with fludarabine and cyclophosphamide prior to HSCT. The girl received standard GvHD-prophylaxis with ATG, mycophenolate mofetil and cyclosporine A. The patient showed timely engraftment with platelets >100/nl on day 8, leucocytes >1/nl on day 12, neutrophils >0.5/nl on day 14, and >1/nl on day 16 after single stimulation with granulocyte-colony stimulating factor on day 15. Whole blood chimerism after HSCT was mixed at day +40 (73% donor) and increased to 100% donor five months post HSCT. She developed a grade III graft-versus-host disease of the skin on day 21, which was treated successfully with topical tacrolimus and corticosteroids as well as systemic corticosteroids. After HSCT, a reactivation of adenovirus in the stool and the blood as well as EBV in the blood was observed with low copy numbers and no severe clinical complications without antiviral therapy. The granuloma resolved with scarring within 10 weeks following HSCT (Figures 1L, M, no pictures were taken after day 15), the first signs of resolution were already seen during conditioning. However, HSCT was complicated by severe and therapy-refractory hepatic veno-occlusive disease (VOD). Sadly, the girl died from complications of VOD 6 months after HSCT [after submission of (3)].
Our report demonstrates that normal ranges for total IgG levels, IgG vaccine responses to protein antigens, total lymphocyte as well as total T- and B-cell counts cannot exclude CID. Delayed diagnosis of CID in patient 1 was complicated by disfiguring rubella-associated skin granuloma. The report underscores the effectiveness of HSCT as, to date, the only known curative treatment of RG in patients with IEI. Granulomas of both patients started to improve within days after initiation of conditioning and largely resolved (with scarring in patient 2) within the first months after HSCT. Even disfiguring lesions in patient 1 healed to a major extent. Resolution of granulomas starting during conditioning and completing by several weeks following HSCT suggests that healing of RG largely depends on patients` depletion of rubella-harboring neutrophils and monocytes/macrophages (see Ref. 3) rather than on B- and T-cell immune reconstitution.
However, patient 2 [patient 28 in (3)] died 6 months after HSCT due to complications of severe hepatic VOD, thus increasing the number of patients in the cohort who died following transplantation to 3 out of 7. Only 7 out of 28 patients in the cohort underwent HSCT, possibly explained by lack of knowledge on treatment of RG in the past. The low number may also reflect concerns about the high morbidity and mortality of HSCT in high-risk patients. No other patient with AT described in the cohort (n=4) underwent HSCT. HSCT in DNA double-strand repair disorders has been reviewed recently (5), indicating particularly poor outcome in patients with AT. Reduced conditioning regimens and adapted protocols for AT appear to improve outcomes (8), the impact on neurologic deterioration in AT patients has to be determined. Other therapeutic options with better outcomes need to be developed.
RG can occur weeks to decades following infection or vaccination [reviewed in (3)], thus it is unclear how many yet unaffected patients with CID/DNA repair disorders will develop RG later in life. Early sensitive and specific risk factors for RG to avoid MMR live-vaccines in infants are yet to be identified. Interestingly, in a cohort of AT, 7 out of 8 patients with granulomas showed IgA deficiency (9). In addition, IgA deficiency has been identified as a surrogate marker for immunodeficiency and mortality in AT patients (10). However, transient IgA deficiency is also common in healthy infants and thus not useful for risk assessment prior to MMR vaccination in patients with AT.
Live vaccines (rotavirus, varicella, measles, rubella, yellow fever, BCG) should be strictly avoided in infants with (S)CID and further defined IEI, since severe vaccine-related infections may occur (11, 12). However, MMR is a crucial live vaccine and avoidance will put children with IEI at risk of severe disease unless IgG substitution is performed. In patients with DNA repair disorders and CID, diagnosis of IEI is often delayed (13), with live vaccines commonly given prior to established IEI diagnosis. Thus, avoidance of rubella vaccination to prevent RG in children at risk is difficult. It is likely, that newborn screening (NBS) for SCID will help to identify some patients with non-SCID IEI at risk for RG as demonstrated for patient 2. However, it remains unclear whether patients identified by NBS as having moderately reduced TREC numbers not requiring HSCT may develop RG upon live vaccination. Further studies are needed to establish immunological parameters that allow the safe use of live vaccines in these infants. Alternatively, the development of a safe, non-live-rubella vaccine, either subunit or mRNA-based, should be considered.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
UB, JS, JG, ML, VW, JH, NK, UB-P, FG, LP, OB, HB and RK contributed patient data. UB, HB and RK wrote the manuscript. All coauthors read and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the United States Centers for Disease Control and Prevention.
Authors JH and HB were employed by Labor Berlin GmbH
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AT, Ataxia telangiectasia; ATG, Anti-thymocyte globulin; CID, Combined immunodeficiency; DBS, Dried blood spot; EBV, Epstein Barr virus; GvHD, Graft versus host disease; HSCT, Hematopoietic stem cell transplantation; IEI, Inborn errors of immunity; MMR, Measles, mumps and rubella; NBS, Newborn screening; RG, Rubella-induced granuloma; RT-PCR, Reverse transcription polymerase chain reaction; SCID, Severe combined immunodeficiency; TREC, T-cell receptor excision circles; TRM, Transplantation related mortality;VOD, Veno-occlusive disease.
1. Bodemer C, Sauvage V, Mahlaoui N, Cheval J, Couderc T, Leclerc-Mercier S, et al. Live Rubella Virus Vaccine Long-Term Persistence as an Antigenic Trigger of Cutaneous Granulomas in Patients With Primary Immunodeficiency. Clin Microbiol Infect (2014) 20:656. doi: 10.1111/1469-0691.12573
2. Buchbinder D, Hauck F, Albert MH, Rack A, Bakhtiar S, Shcherbina A, et al. Rubella Virus-Associated Cutaneous Granulomatous Disease: A Unique Complication in Immune-Deficient Patients, Not Limited to DNA Repair Disorders. J Clin Immunol (2019) 39:81–9. doi: 10.1007/s10875-018-0581-0
3. Perelygina L, Faisthalab R, Abernathy E, Chen M, Hao L, Bercovitch L, et al. Rubella Virus Infected Macrophages and Neutrophils Define Patterns of Granulomatous Inflammation in Inborn and Acquired Errors of Immunity. Front Immunol (2021) 12:796065. doi: 10.3389/fimmu.2021.796065
4. Perelygina L, Icenogle J, Sullivan KE. Rubella Virus-Associated Chronic Inflammation in Primary Immunodeficiency Diseases. Curr Opin Allergy Clin Immunol (2020) 20:574–81. doi: 10.1097/ACI.0000000000000694
5. Wolska-Kuśnierz B, Gennery AR. Hematopoietic Stem Cell Transplantation for DNA Double Strand Breakage Repair Disorders. Front Pediatr (2019) 7:557. doi: 10.3389/fped.2019.00557
6. Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, et al. EBMT/ESID Inborn Errors Working Party Guidelines for Hematopoietic Stem Cell Transplantation for Inborn Errors of Immunity. Bone Marrow Transplant (2021) 56:2052–62. doi: 10.1038/s41409-021-01378-8
7. Bakhtiar S, Woelke S, Huenecke S, Kieslich M, Taylor AM, Schubert R, et al. Pre-Emptive Allogeneic Hematopoietic Stem Cell Transplantation in Ataxia Telangiectasia. Front Immunol (2018) 9:2495. doi: 10.3389/fimmu.2018.02495
8. Ussowicz M, Wawrzyniak-Dzierżek E, Mielcarek-Siedziuk M, Salamonowicz M, Frączkiewicz J, Rybka B, et al. Allogeneic Stem Cell Transplantation After Fanconi Anemia Conditioning in Children With Ataxia-Telangiectasia Results in Stable T Cell Engraftment and Lack of Infections Despite Mixed Chimerism. Biol Blood Marrow Transplant (2018) 24:2245–9. doi: 10.1016/j.bbmt.2018.07.001
9. Woelke S, Valesky E, Bakhtiar S, Pommerening H, Pfeffermann LM, Schubert R, et al. Treatment of Granulomas in Patients With Ataxia Telangiectasia. Front Immunol (2018) 9:2000. doi: 10.3389/fimmu.2018.02000
10. Zielen S, Duecker RP, Woelke S, Donath H, Bakhtiar S, Buecker A, et al. Simple Measurement of IgA Predicts Immunity and Mortality in Ataxia-Telangiectasia. J Clin Immunol (2021) 41:1878–92. doi: 10.1007/s10875-021-01090-8
11. Pöyhönen L, Bustamante J, Casanova J, Jouanguy E, Zhang Q. Life-Threatening Infections Due to Live-Attenuated Vaccines: Early Manifestations of Inborn Errors of Immunity. J Clin Immunol (2019) 39:376–90. doi: 10.1007/s10875-019-00642-3
12. Rosenfeld L, Mas Marques A, Niendorf S, Hofmann J, Gratopp A, Kühl J, et al. Life-Threatening Systemic Rotavirus Infection After Vaccination in Severe Combined Immunodeficiency (SCID). Pediatr Allergy Immunol (2017) 28:841–3. doi: 10.1111/pai.12771
Keywords: inborn errors of immunity, primary immunodeficiency, Ataxia telangiectasia, Artemis deficiency, rubella virus vaccine strain, granuloma formation, stem cell transplantation
Citation: Baumann U, Schulte JH, Groß JP, Beier R, Ludwig M, Wahn V, Hofmann J, Maecker-Kolhoff B, Sauer M, Kaiser-Labusch P, Karimian N, Blume-Peytavi U, Ghoreschi F, Ott H, Perelygina L, Klemann C, Blankenstein O, von Bernuth H and Krüger R (2022) Case Report: Rubella Virus-Induced Cutaneous Granulomas in Two Pediatric Patients With DNA Double Strand Breakage Repair Disorders – Outcome After Hematopoietic Stem Cell Transplantation. Front. Immunol. 13:886540. doi: 10.3389/fimmu.2022.886540
Received: 28 February 2022; Accepted: 25 April 2022;
Published: 02 June 2022.
Edited by:
Mary Slatter, Newcastle University, United KingdomReviewed by:
Kimberly Gilmour, Great Ormond Street Hospital for Children NHS Foundation Trust, United KingdomCopyright © 2022 Baumann, Schulte, Groß, Beier, Ludwig, Wahn, Hofmann, Maecker-Kolhoff, Sauer, Kaiser-Labusch, Karimian, Blume-Peytavi, Ghoreschi, Ott, Perelygina, Klemann, Blankenstein, von Bernuth and Krüger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renate Krüger, ci5rcnVlZ2VyQGNoYXJpdGUuZGU=
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.