 Zehua Sun
Zehua Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 April 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.869825
This article is part of the Research Topic Deleterious and Beneficial Humoral Immune Response in Viral Diseases: Two Sides of the Same Coin View all 9 articles
Phage display is a well-established technology for in vitro selection of monoclonal antibodies (mAb), and more than 12 antibodies isolated from phage displayed libraries of different formats have been approved for therapy. We have constructed a large size (10^11) human antibody VH domain library based on thermo-stable, aggregation-resistant scaffolds. This diversity was obtained by grafting naturally occurring CDR2s and CDR3s from healthy donors with optimized primers into the VH library. This phage-displayed library was used for bio-panning against various antigens. So far, panels of binders have been isolated against different viral and tumor targets, including the SARS-CoV-2 RBD, HIV-1 ENV protein, mesothelin and FLT3. In the present study, we discuss domain library construction, characterize novel VH binders against human CD22 and PD-L1, and define our design process for antibody domain drug conjugation (DDC) as tumoricidal reagents. Our study provides examples for the potential applications of antibody domains derived from library screens in therapeutics and provides key information for large size human antibody domain library construction.
Phage display is a well-established technology, widely used for displaying antibody domains or peptides on the surface of bacteriophage and subsequently used in different methods for in vitro selection (1, 2). Antibody or antibody domain (Fab, ScFv and VH/VL) library construction combined with phage display facilitates high-throughput screening, and serves as the crucial stage in the retrievals of biologically relevant binders, since multiple factors such as diversity and size affect the quality of a certain library. At the onset of the SARS-CoV-2/COVID19 pandemic in 2020, we isolated a panel of potent neutralizing binders from our phage-displayed libraries, within two weeks of obtaining the spike receptor binding domain (RBD) sequence, highlighting the importance of a high quality phage-displayed antibody libraries (3–5).
Antibody domains have multiple applications, such as designing as Chimeric Antigen Receptors (CARs) or antibody drug conjugations (ADCs) (6, 7). The first FDA approved bivalent variable-domain-only immunoglobulin fragment, Caplacizumab, was used as inhibitor to block the interaction between Von Willebrand factor multimers and platelets (8). The first instance of CAR T employing a human VH domain targeting the CD33 antigen for the treatment of acute myeloid leukemia was reported in 2008 (6). Various single domain binders were reported as neutralizers against SARS-CoV-2 and other infectious diseases (9, 10). Antibody domain drug conjugates (DDC) are yet to be reported.
Cluster of differentiation-22 (CD22) is a B-cell-specific transmembrane glycoprotein, which has emerged as an attractive target for monoclonal antibody (mAb) based therapy of B cell malignancies (11). ADC therapy against CD22 is a promising therapeutic approach to B cell malignancies which conjugates mAbs with cytotoxic agents (12). Active selection agents recognizing only one antigen on leukemia cells can cause antigen-loss relapse, which is a potential limitation of potent CD22-targeted immunotherapies (13). Multispecific duoCAR-T cells that target CD19, CD20, and CD22 show a promising effect to prevent antigen loss mediated relapse or the down regulation of target antigen in patients with B cell malignancies (7). Blockade of the immune checkpoint such as programmed cell death 1 (PD-1)/programmed death-ligand 1 (PD-L1) augments anti-tumor immunity and induces durable responses in patients bearing up with solid cancers (14). But the combination of ADC therapies against PD-L1 and CD22 on clinical efficacy in B cell malignancies are sparse (15, 16).
We constructed an initial human VH domain phage-displayed library in 2008 (17, 18). Subsequently, we have been continuously optimizing the construction of human VH domain libraries at multiple levels, including primer optimization to increase the diversity, scaffold screening and characterization to reduce toxicity, as well as to lower the risk of aggregation in purified binders. In the present study, we discuss the construction of a large-size human antibody VH domain library, and characterize the quality of the library by bio-panning against two different antigens: TGFβ1 and mesothelin. Further, one PD-L1 specific VH domain and one CD22 specific VH domain were selected for domain drug conjugate (DDC) design, as an example of the application of human antibody VH domains.
There are two considerations for using primary human PBMCs for the direct amplification of naturally occurring full length antibody variable heavy-chain (VH) gene repertoires and selection of scaffolds for library construction: (1) to minimize the workload of humanization; and (2) to avoid introducing immunogenic epitopes. We and others have reported that several VH sub-families such as VH1-69, VH3-7, VH3-15, VH3-23, VH4-34 and VH6-1 showed high frequency in the memory compartment of the immune repertoire obtained from healthy donors (4, 19–22). We collected human PBMCs from 10 healthy donors. Full length natural occurring VH genes were amplified by conserved region primers to construct a small size (10^8) VH domain library. Primers were designed according to the alignments from international ImMunoGeneTics information system® (IMGT, http://www.imgt.org), and are displayed in Table 1 (23, 24). We randomly sequenced 100 clones and no repeats were observed. The dominant sub-families in our VH library (Figure 1A) were consistent with the previous reports mentioned above.

Table 1 Primers for amplification of natural occurring full length VH gene repertoires.
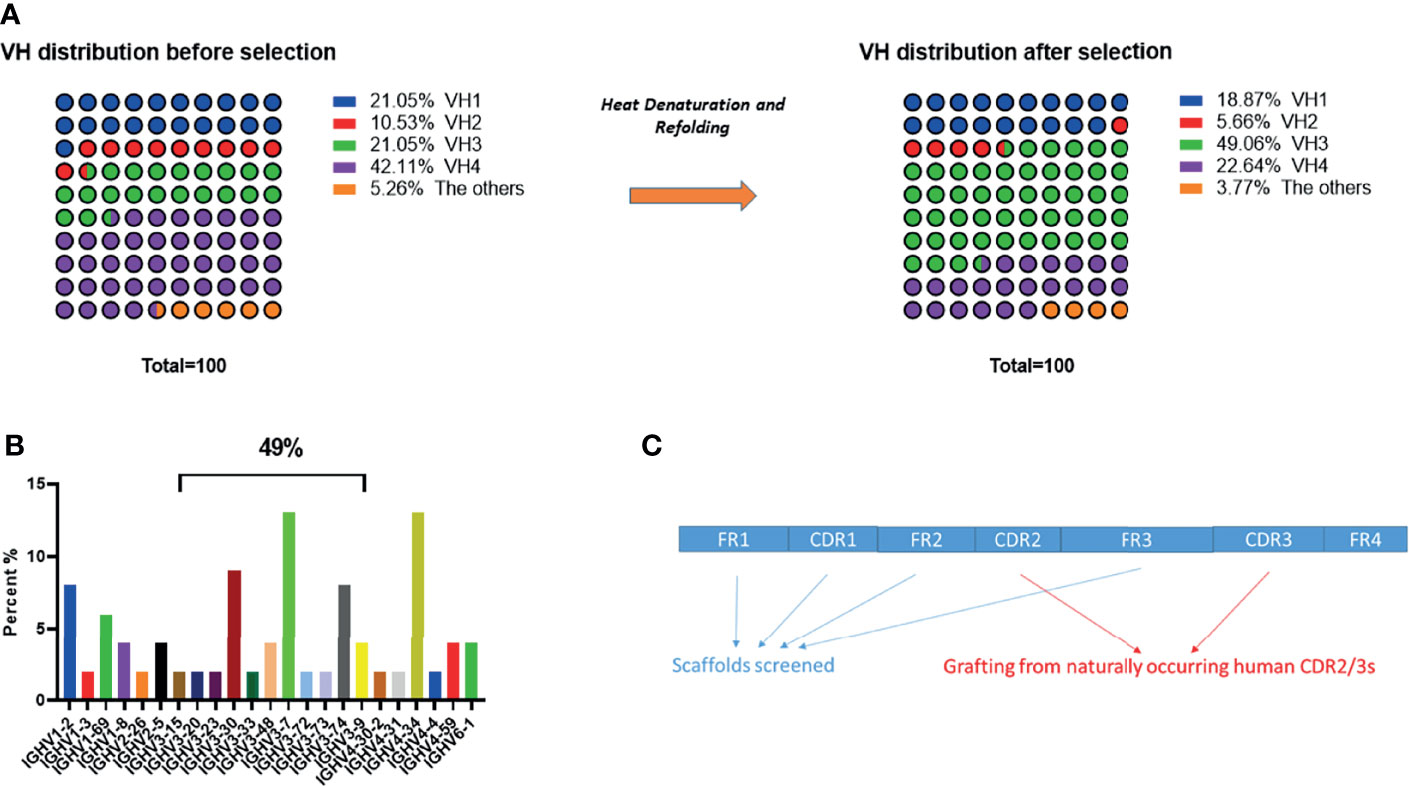
Figure 1 VH segments analysis and grafting of CDR2/3s. (A) General frequency distribution of VH gene segments before and after heat denaturation and refolding screening. (B) Detailed distribution of enriched VH gene segments after heat denaturation and refolding screening. (C) Schematic view of CDR2/3 grafting.
We identified relatively stable VH scaffolds from a small VH phage-displayed library (10^8) by a heat denaturation and refolding strategy (4, 25). After 3 rounds of panning, output clones were enriched according to plate titration. We randomly sequenced 100 clones, and VH genes in VH3 family were largely enriched, composing up to 49% of sequenced genes (Figure 1B) (4, 18). We then sequenced more clones and evaluated the aggregation of the most enriched clones by size exclusion chromatography (SEC) and dynamic light scattering (DLS). The five most enriched clones (1 clone from VH 3-7, 2 clones from VH 3-30 and 2 clones from VH 4-34) were finally selected for construction of a large size VH domain library.
After selection of aggregation resistant scaffolds, we constructed a large size human VH domain library by grafting naturally occurring CDR2s and CDR3s from human PBMCs (from 50 healthy donors) (Figure 1C) (4, 18). Grafting of human CDR3 is an important step, since studies have verified that CDR3 of the VH domain is sufficient to distinguish between a variety of haptens and protein antigens with surprisingly high affinities (26). We optimized the primers for grafting naturally occurring human CDR3 (Table 2), and observed that the optimized set of primers largely increased the diversity of the library constructed. The size of the final VH phage-displayed library constructed (after grafting) was estimated to be 1.3 × 1011, with no repeated sequences found in 100 random sequenced clones, indicating a good diversity. 80% of the clones were productive.

Table 2 Primers for CDR2 and CDR3 gene grafting.
This library was panned against human recombinant Transforming Growth Factor-β1 (TGFβ1), and a panel of binders was obtained with half maximal effective concentration (EC50) ranging from 0.5 nM to 100 nM (Figure 2A). We performed 3 rounds of panning, which will be sufficient for enrichment of antigen-specific binders. The yield of both antibody VH domains ranged from 4 to 8 mg, from 100 ml 2x YT medium culture. To test aggregation, we incubated these binders at 3-5 mg/ml at 37°C for 24 hours and 7 days, and measured the aggregation by DLS (Figure 2C). 83% of binders showed aggregation resistance (aggregation percent < 5%) after 24 hours incubation while 67% of binders showed aggregation resistance after 7 days incubation. We kept incubating of aggregation resistant binders for another 7 days at 37°C, and all the binders showed durable aggregation resistance. We panned the library against another antigen, human recombinant mesothelin (MSLN) (by 3 rounds of panning), and created another panel of binders showing EC50s ranging from 1 nM to 200 nM (Figure 2B), and measured the aggregation of binders by DLS after incubation at 37°C for 24 hours. 75% of binders (9 out of 12) showed aggregation less than 5%. The 9 aggregation resistant binders were incubated for another 13 days at 37°C, and 5 of the candidates showed persistent aggregation resistance, with aggregation accounting for less than 5% of the binder profile (Figure 2D).
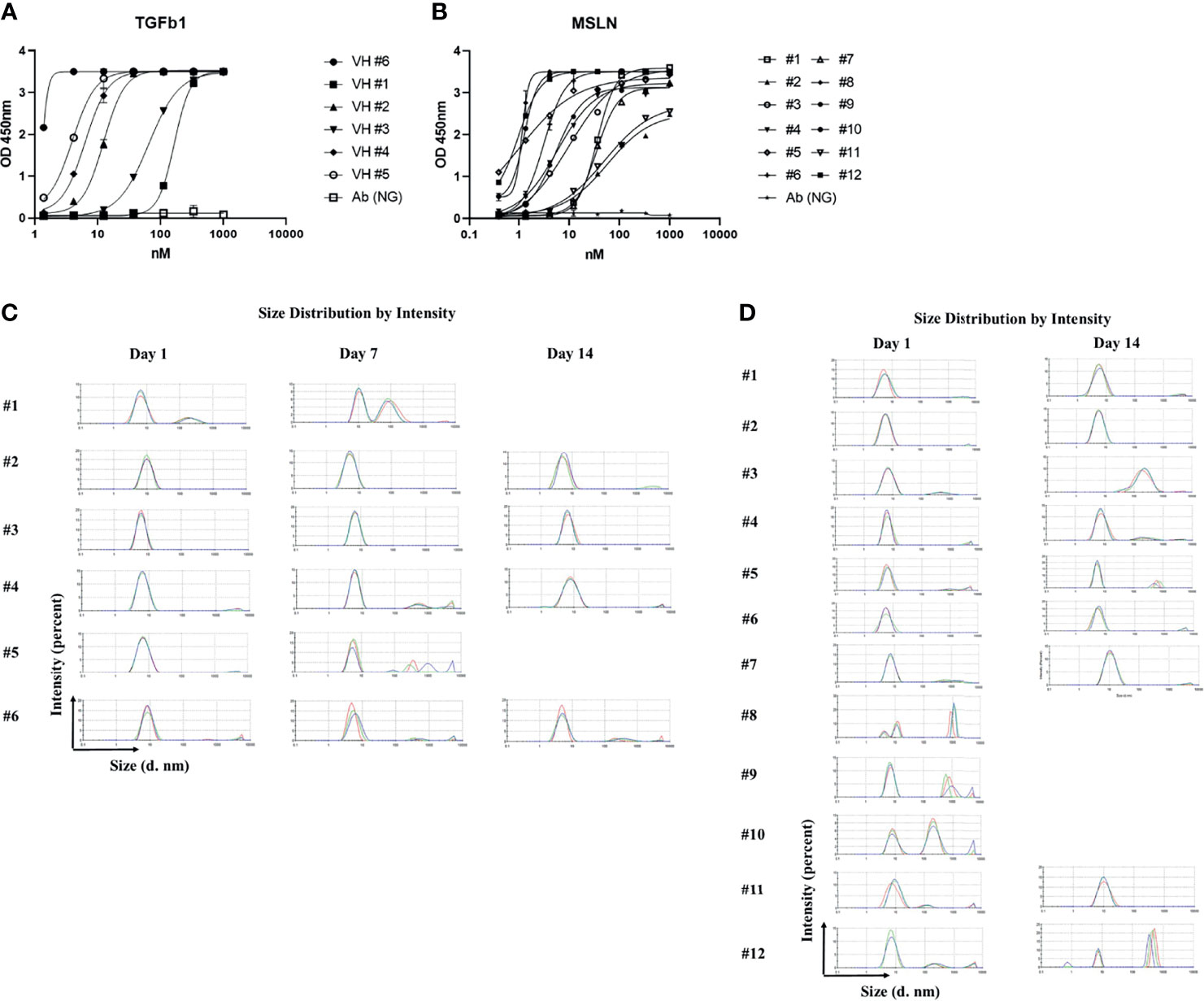
Figure 2 Characterization of constructed library by bio-panning against TGFb1 and MSLN. (A) ELISA of binders against TGFb1; (B) ELISA of binders against MSLN; (C) Dynamic light scattering analysis for evaluation of the aggregation propensity of the TGFb1 specific VH domains; 3 different colors represent 3 different repeats; (D) Dynamic light scattering analysis for evaluation of the aggregation propensity of the MSLN specific VH domains, 3 different colors represent 3 different repeats.
To date, there are several anti-human PD-L1 drugs approved by FDA (27). Most of them function in blocking the PD1/PD-L1 binding interface. We quickly (only 2 rounds of panning) isolated one binder (nominated 1-16) with specific binding to PD-L1. Epitope mapping demonstrated that binders 1-16, targeted the membrane proximal domain of PD-L1, which is far away from known PD1 binding epitopes (Figure 3D). We further modified the CDR3 of 1-16 by grafting and mutagenesis, and acquired one variant of 1-16, nominated 1-16-3, which showed improved binding and aggregation resistance. Surface Plasmon Resonance (SPR) was further performed to measure the equilibrium dissociation constant (KD) of parental domain 1-16 and variant 1-16-3 to human recombinant PD-L1 (Table 3). After conversion to VH-Fc format to further increase the avidity, 1-16-3 showed an EC50 less than 10nM. PD-L1 positive CHO-K1 cells and PD-L1 negative CHO-K1 cells were used for confirming the cell specific binding for both VH 1-16 and VH 1-16-3 by flow cytometry (Figure 3A). The maturation of 1-16 to 1-16-3 did not block the cell specific binding nor shift the epitope since they competed to each other in a competition ELISA (Figure 3C). VH 1-16-3 has an improvement of aggregation resistance after incubation at 37°C for both 24 hours and 7 days by DLS (Figure 3B). In both VH and VH-Fc format, 1-16-3 shows an increase in binding to PD-L1 (Figure 3C). Both 1-16 and 1-16-3 cannot neutralize PD-L1 in a PD-1/PD-L1 Blockade Bioassay (data not shown), indicating the PD-L1 non-neutralization epitope is targeted by 1-16-3 (Figure 3D).
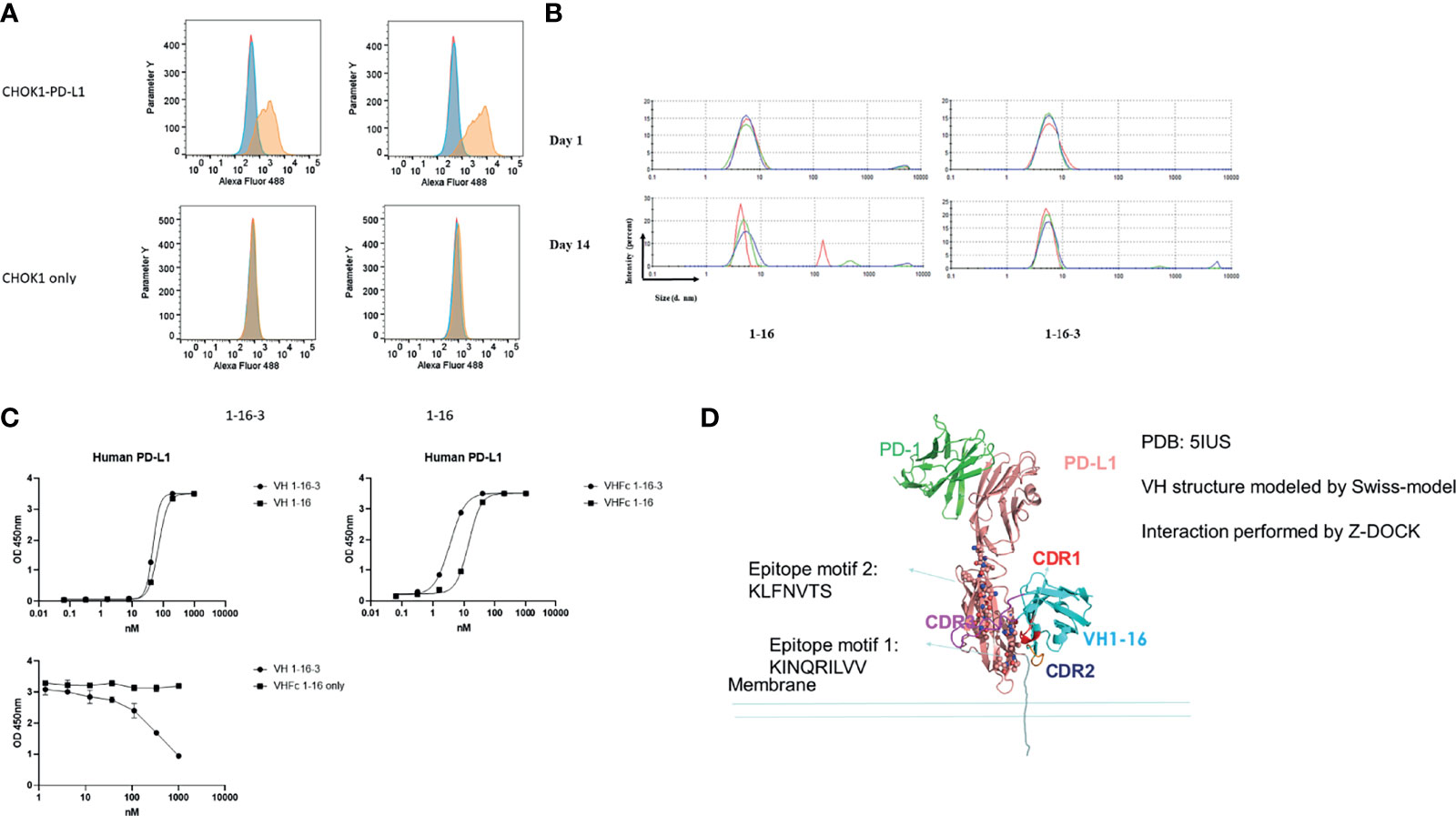
Figure 3 Characterization of VH domains 1-16 and 1-16-3. (A) Flow cytometry analysis for binding of VH 1-16 and 1-16-3 to PD-L1 expressed on the surface of CHOK1 cells; blue peaks represent cells stained with AF488 conjugated secondary antibody only, red peaks represent cells stained with isotype control and then AF488 conjugated secondary antibody, yellow peaks represent cells stained with VH domains and then AF488 conjugated secondary antibody; (B) Dynamic light scattering analysis for evaluation of the aggregation propensity of domain s domains 1-16 and 1-16-3; Curves with different color are different repeats; (C) ELISA of 1-16 and 1-16-3 against PD-L1 with or without human IgG1 Fc, and their competition; (D) Structural modelling of PD-L1 with VH 1-16. Epitopes were labeled in the figure.

Table 3 SPR of PD-L1 specific VH domains 1-16 and 1-16 derived 1-16-3.
CD22, which is overexpressed in B cell malignancies, is a target of great interest. VH-Fc 1-16-3 was conjugated with MMAE for a DDC study exploring killing of CD22 positive leukemia cells. For control purposes, we identified two human VH domains (E1-2 and G10) that specifically bind human recombinant CD22. Cell specific binding of both domains were measured on two different B cell lines (Raji and Bjab) and one T cell line (Jurkat) by flow cytometry. The previously identified IgG1 antibody m971 served as positive control (28, 29). Both E1-2 and G10 can bind to Raji (CD22+) and Bjab (CD22+) cells, but cannot bind to Jurkat cells (CD22-) (Figure 4A). We measured the aggregation of both E1-2 and G10 by DLS. G10 aggregated after incubation 37°C for 24 hours (Figure 4B). In contrast to G10, VH E1-2 showed less than 5% aggregation after incubation at 37°C for 24 hours by DLS (Figure 4B), and an EC50 below 10 nM by ELISA (Figure 4C). The equilibrium dissociation constant (KD) of VH domain E1-2 was measured by SPR (Table 4). After conversion to VH-Fc format, the EC50 of VH-Fc E1-2 was reduced, most likely due to the effect of the orientation of VH domains by human Fc protein (Figure 4D). We also conjugated VH-Fc E1-2 to MMAE for DDC study. VH-Fc E1-2 showed moderate killing of CD22 positive tumor cells, while VH-Fc 1-16-3 showed approximately 10-fold greater potency in comparison to VH-Fc E1-2. VH-Fc 1-16-3 DDC and VH-Fc E1-2 DDC worked efficiently in combination (with equivalent input) in CD22 positive tumor cells (Raji and Bjab) as well (Figures 4E–G). These data indicate that combination of PD-L1 therapy and CD22 therapy may achieve a superior effect to either alone. More extensive studies are required, but we present here an example of the potential applications of antibody VH domains as a therapeutic.
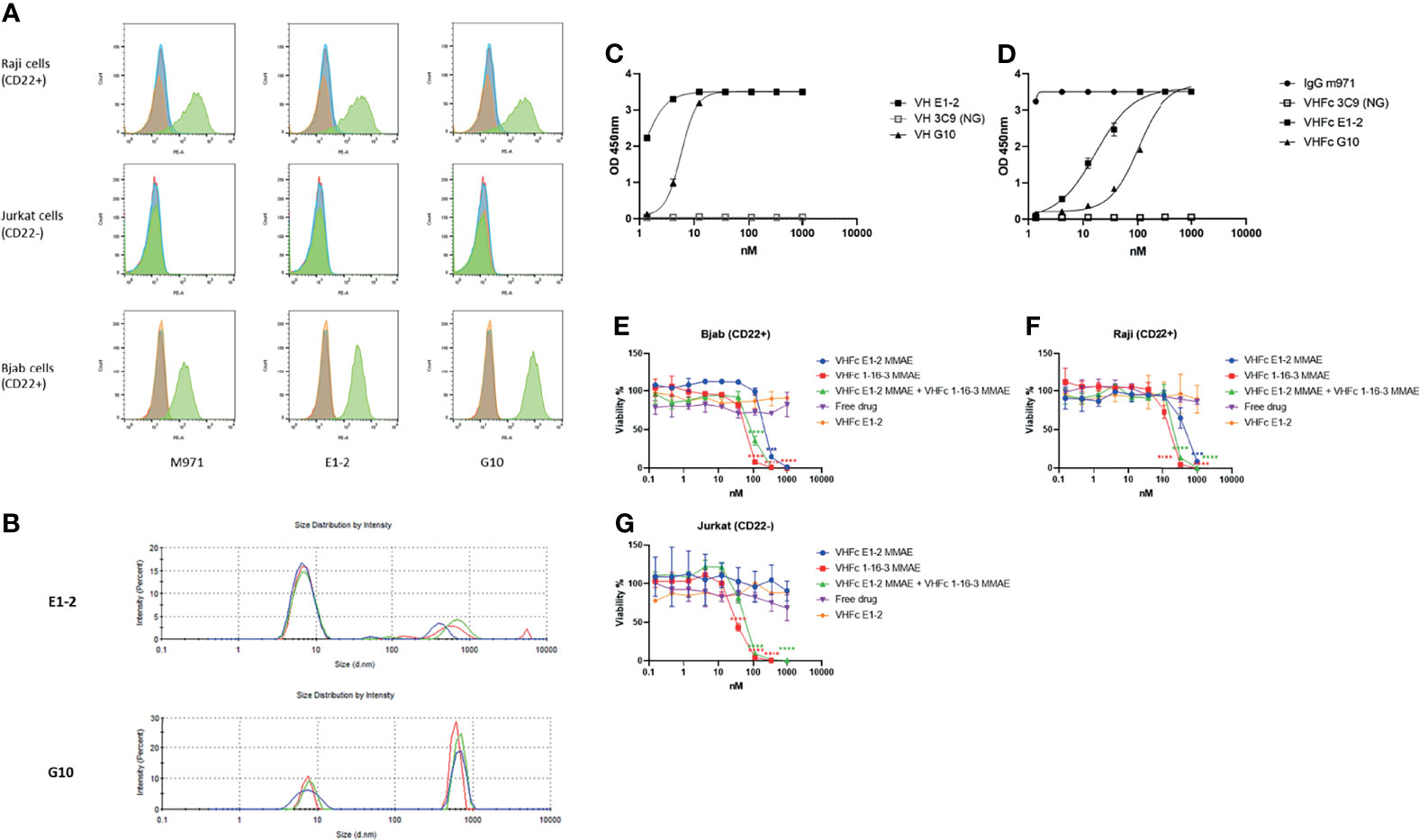
Figure 4 Characterization of VH domains E1-2 and DDC design an example of the potential application. (A) Flow cytometry analysis for binding of domains to CD22 expressed on the surface of Raji cells and Bjab cells (Jurkat cells as negative control); yellow peaks represent unstained cells, blue peaks represent cells stained with PE conjugated secondary antibody only, red peaks represent cells stained with isotype control and then PE conjugated secondary antibody, green peaks represent cells stained with VH domains or m971 (positive control), and then PE conjugated secondary antibody; (B) Dynamic light scattering analysis for evaluation of the aggregation propensity of domains E1-2 and G10 after 24 hours incubation at 37°C; (C, D) ELISA of E1-2 and G10 against CD22 with or without human IgG1 Fc; (E–G) 1-16-3 and E1-2 based antibody domain drug conjugates in killing CD22 positive leukemia cells. Significance was tested using one-way ANOVA, followed by the tukey’s multiple post hoc test. ****P < 0.0001; ***P < 0.001 versus antibody isotype control (VH-Fc E1-2) at each concentration.

Table 4 SPR of CD22 specific VH domain E1-2.
Construction of large size recombinant human domain libraries of high diversity and high aggregation resistance benefits target-specific binder selection, and addresses challenging barriers to finding effective therapeutics (9, 30). In the current study, we demonstrated our experience in construction of a large size VH domain library by optimizing the primers for CDR2/3 grafting into aggregation resistant scaffolds. CDR1 remained the same in scaffolds for several considerations: (1) CDR1 usually contributes less to antigen binding (31–33); (2) CDR2 and CDR3 grafting are sufficient for the target diversity of the library (26, 34, 35); (3) since the scaffolds selected are aggregation resistant, we intended to keep the original sequences unaltered as possible so as to minimize the risk of aggregation in subsequent binders isolated.
One common reason for antibody therapeutics failure in clinical trials is aggregation (36, 37), an essential bottleneck in the generation and production of antibody-based reagents for human therapeutics, especially smaller antibody formats that lack the inter-domain stabilization of their larger counterparts (25, 38, 39). We analyzed the binders we isolated by panning against TGFβ1 and MSLN, and to our experience, the output of binders from the improved library are superior to previous libraries we generated. Our results set an example for illustration of construction a large size VH domain library of high diversity and improved aggregation resistance.
1-16-3 is an aggregation resistant VH domain that targets human PD-L1. Different from the anti-PD-L1 antibodies currently available (40), the epitope of VH 1-16-3 is far away from the epitopes recognized by the natural PD1 ligand. This uncommon characteristic makes it possible for combination artorial applications with PD1/PD-L1 blockage drugs in the market. VH 1-16-3 showed a KD of 48 nM by SPR, while VH-Fc 1-16-3 showed an EC50 less than 10 nM by ELISA. E1-2 is a CD22 specific VH domain with KD of 38 nM and to our knowledge it is the first identified antibody VH domain specific for CD22. MMAE conjugated antibody domains are just one example of the application of domains as DDCs. These identified antibody VH domains of good biophysical properties that can be used for CAR-T constructs and other bi-specific antibody design, such as bispecific T cell engagers (BiTEs) (41, 42) and bispecific NK cell engagers (BiKEs) (43, 44).
Human antibody VH domains offer distinct advantages: (1) no need for further time-consuming humanization; (2) flexible administration such as lung delivery via inhalation for rapid deployment in response to SARS-CoV-2 (9, 45). Rapid progress has been made over the past few decades toward the development of therapeutic proteins (30). Human antibody VH domain is one of the fruition of such rapid progress of potent therapeutic proteins, and provides an exciting paradigm for the next generation of protein based therapeutics with their engineering potential and application advantages.
In summary, we demonstrated our strategy for construction of a large size human antibody VH domain library and briefly characterized the output binders from the current library. We characterized one PD-L1 specific VH domain 1-16-3 and one CD22 specific VH domain E 1-2 as DDCs in vitro. Our study provided key information for scientists who have interest in large size human domain library construction.
Fresh human normal peripheral blood mononuclear cells (PBMCs) (200 million cells/vial, CAT# SER-PBMC-200) were purchased from Zen-Bio, Inc. NC 27709. Consent and waiver was signed for purchasing purpose. Donor ages are ranging from 20 to 40 years old with unknown gender information. Fresh cells were directly used for mRNA extraction as previously described (4, 18).
Full length human PD-L1 and CD22 sequences were synthesized by IDT (Coralville, Iowa) with sequence obtained from Uniprot (https://www.uniprot.org/uniprot/Q13421). The target sequences were cloned into an expression plasmid containing a CMV promotor, woodchuck posttranscriptional regulatory elements, and a His tag. Proteins were purified by Ni-NTA (GE Healthcare) chromatography. Target specific VH domains were cloned in pComb3x vector and purified from Escherichia coli HB2151 bacterial culture at 30°C in 2x YT medium (Y1003, Sigma-Aldrich) for 16 h with stimulation by 1 mM IPTG. Cells were lysed by Polymyxin B (Sigma-Aldrich). Lysate were spun down and supernatant was loaded over Ni-NTA (GE Healthcare). For conversion to Fc-fusion, the VH gene was re-amplified and re-cloned into pSectaq vector containing human Fc. VH-Fc proteins were expressed in the Expi293 expression system (Thermo Fisher Scientific) and purified with protein A resin (GenScript). Buffer replacement in protein purification used Column PD 10 desalting column (GE Healthcare). All protein purity was estimated as >95% by SDS-PAGE and protein concentration was measured spectrophotometrically (NanoVue, GE Healthcare). Further details can be found in our previous publication (4).
For ELISA assays, antigen protein was coated on a 96-well plate (Costar) at 50 ng/well in PBS overnight at 4°C. For the soluble VH binding assay, horseradish peroxidase (HRP)-conjugated mouse anti-FLAG tag antibody (A8592, Sigma-Aldrich) was used to detect VH binding. For detection of human Fc protein, HRP-goat anti-human IgG Fc secondary antibody (A18817, Thermo Fisher Scientific) was used. For the competition ELISA, 200 nM of VH-Fc 1-16 was incubated with serially diluted VH 1-16-3 proteins, and the mixtures were added to antigen-coated wells. After washing, competition was detected by HRP-goat anti-human IgG Fc secondary antibody (A18817, Thermo Fisher Scientific). All colors were developed by 3,3′,5,5′-tetramethylbenzidine (TMB, Sigma) and stopped by 1 M H2SO4 followed by recording absorbance at 450 nm by iMark™ Microplate Absorbance Reader (Bio-Rad Laboratories). Experiments were performed in duplicate and the error bars denote ± 1 SD.
The kinetics of the antibody fragments were determined using a Biacore X100 (GE Healthcare). Human PD-L1/CD22 was (10 mg/mL) was immobilized onto a CM5 sensor chip (GE Healthcare, BR100012) by amine coupling. The antibody fragments diluted in HBS-EP buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant P20, pH 7.4) were injected over an immobilized surface (200 - 400 RU) for 180 sec at a rate of 20 µL/min, followed by dissociation for 600 sec. After each sample injection, the surface was regenerated by injection of regeneration solution (10 mM Glycine/10% Glycerol pH 2.0). The kinetic values, ka, kd, and KD were calculated using the BiacoreX100 Evaluation Software (GE Healthcare).
A large phage-displayed human VH domain library was constructed by grafting of naturally occurring heavy-chain CDR2 s and CDR3 s (from PBMC pool of >30 healthy donors) to VH framework scaffolds. CDR2 s and CDR3 s were PCR amplified with the primers in Table 2. After library construction, 100 dandom clones were sequenced and no repeats were observe, indicating a good diversity of the library. The library was preabsorbed on streptavidin-M280-Dynabeads in phosphate-buffered saline (PBS) for 1 h at room temperature (RT) and incubated with 50 nM biotinylated PD-L1 or CD22 for 2 h at RT with gentle agitation. Phage particles binding to biotinylated antigen were separated from the phage library using streptavidin-M280-Dynabeads and a magnetic separator (Dynal). After washing 20 times with 1 ml of PBS containing 0.1% Tween-20 and another 20 times with 1 ml of PBS, bound phage particles were eluted by 100 mM triethanolamine followed by neutralization with 1 M, pH7.5 Tris-HCl. For the second round of panning, 10 nM (2 nM for the third round) of biotinylated protein was used as antigen. After the third round of panning, 96 individual clones were screened for binding to target protein by phage ELISA. Panels of VHs were selected, sequenced and characterized.
PD-L1 positive aAPC/CHO-K1 (Cat # J1205) cells and PD-L1 negative aAPC/CHO-K1 (Cat # J1191) were purchased from Promega. Bjab cells were purchased from (ABC-TC514S, AcceGen, New Jersey, USA). Raji cells (Cat# CCL-86) and Jurkat cells (TIB-152) were purchased from American Type Culture Collection (ATCC). Both T cells and B cells were maintained in ATCC-formulated RPMI-1640 Medium (ATCC 30-2001) with 10% fetal bovine serum (ATCC 30-2020). To confirm the cell surface binding of isolated antibody binders, cells were treated with VH 1-16, VH 1-16-3, E1-2, G10 or m971 for 1 h at 4°C and then stained with anti-DYKDDDDK Antibody, FITC (130-127-934, Miltenyi Biotec), or anti-DYKDDDDK (flag tag) Antibody, PE(130-101-577, Miltenyi Biotec), or PE anti-human IgG Fc Antibody (410707, BioLegend) for 0.5 h at 4°C. After each incubation with antibodies, cells were washed with cold PBS. Data were acquired using the flow cytometry BD LSR II (San Jose, CA) and analyzed by FlowJo 10.7.1.
Monomethyl auristatin E (MMAE) was conjugated to VH-Fc via the cross-linker OSu-Glu-VC-PAB (SET0100, Levena Biopharma, USA) with a molar ratio mAb : OSu-Glu-VC-PAB-MMAE of 1:10. The conjugation was performed in buffer composed of 50 mM potassium phosphate, 50 mM sodium chloride, and 2 mM EDTA (pH 6.5) with the reaction run for 18-24 h at room temperature. The reaction was stopped by adding 50 mM sodium succinate (pH 5.0) followed by buffer replacement by using Column PD 10 desalting column (GE Healthcare). The protein was washed and concentrated using 30 kDa Amicon centrifugal filter unit (Millipore Sigma CAT#UFC8030).
Cell viability was measured using CellTiter-Glo or LDH-Glo (G7570, J2380, Promega). Briefly, CD22 positive or negative cells were plated into 96-wells, allowing attachment and growth for 24 hr, then triplicate wells were treated with DDCs, naked antibodies, free drugs, or DDCs plus competitor antibodies. Three to five days later, when untreated control wells were 70 to 90% confluent, reagent was added to the plates according to the supplier’s instructions. Wells treated identically but wells without cells were used to subtract background. Fluorescence (ex: 570 nm, Em: 585 nm) was measured using a CLARIOstar microplate reader (BMG Labtech) and data analyzed using GraphPad Prism 8.0.1 software. Significance was tested using one-way ANOVA, followed by the Tukey’s multiple post hoc test. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05 between the indicated groups. PD-L1 neutralization was done in a PD-1/PD-L1 Blockade Bioassay (J1250, Promega).
VH domains were buffer-changed to DPBS and filtered through a 0.22 μm filter. The concentration was adjusted to 5 mg/mL; 500 μL samples were incubated at 37°C. DLS measurement was performed on Zetasizer Nano ZS ZEN3600 (Malvern Instruments Limited, Westborough, MA) to determine the size distributions of protein particles.
Epitope mapping was performed by PEPperPRINT (Heidelberg, Germany). Briefly, a conformational PD-L1 peptide microarray was pre-stained with the secondary and control antibodies to investigate background interactions with the antigen-derived peptides that could interfere with the main assays. Subsequent incubation of conformational mesothelin peptide microarrays with human VH-Fc antibodies 1-16 at concentrations of 1 µg/ml, 10 µg/ml and 100 µg/ml in incubation buffer was followed by staining with the secondary and control antibodies. Read-out was performed with an Innopsys InnoScan 710-IR Microarray Scanner at scanning gains of 50/40 (red/green). The additional HA control peptides framing the peptide microarrays were simultaneously stained with the control antibody as internal quality control to confirm assay performance and peptide microarray integrity.
PD-L1 structure was extracted from the PD1/PD-L1 complex (PDB ID:5IUS). VH1-structure was modeled by Swiss-model (46) followed by energy minimization. Z-DOCK program (https://zdock.umassmed.edu/) is used for docking VH1-16 onto PD-L1 (47). The antibody binding and blocking regions on PD-L1 were selected based on the experimental epitope mapping results. Z-DOCK outputed top 10 optimal poses, which are visually scrutinized for compatibility at the interaction interface and are avoiding of side chain clashes. We choose the most favorable pose as the binding model for further analysis. The structural figures are prepared by PyMol (48).
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
ZS designed the primers and constructed library, identified and characterized antibodies. ZS produced PD-L1 recombinant proteins and WL produced CD22 proteins and studied the modeling of epitope of antibody. ZS characterized DDCs. ZS wrote the first draft of the article. ZS and RO interpreted data. ZS, WL, JM, RO, and DD discussed the results and further revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the University of Pittsburgh Medical Center.
ZS, JM, and DD are co-inventors of a patent, filed on March 12 by the University of Pittsburgh, related to MSLN specific antibodies described in this paper. Binders against CD22 and PD-L1 are in invention disclosures in the University of Pittsburgh. Sequences of binders will be provided upon request and collaboration.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the members of our center (Center of Antibody Therapeutics, PITT) for their helpful discussions.
1. Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, et al. Efficient Construction of a Large Nonimmune Phage Antibody Library: The Production of High-Affinity Human Single-Chain Antibodies to Protein Antigens. Proc Natl Acad Sci USA (1998) 95(11):6157–62. doi: 10.1073/pnas.95.11.6157
2. Van Blarcom T, Lindquist K, Melton Z, Cheung WL, Wagstrom C, McDonough D, et al. Productive Common Light Chain Libraries Yield Diverse Panels of High Affinity Bispecific Antibodies. MAbs (2018) 10(2):256–68. doi: 10.1080/19420862.2017.1406570
3. Li W, Schafer A, Kulkarni SS, Liu X, Martinez DR, Chen C, et al. High Potency of a Bivalent Human VH Domain in SARS-CoV-2 Animal Models. Cell (2020) 183(2):429–41.e16. doi: 10.1016/j.cell.2020.09.007
4. Sun Z, Chen C, Li W, Martinez DR, Drelich A, Baek DS, et al. Potent Neutralization of SARS-CoV-2 by Human Antibody Heavy-Chain Variable Domains Isolated From a Large Library With a New Stable Scaffold. MAbs (2020) 12(1):1778435. doi: 10.1080/19420862.2020.1778435
5. Li W, Chen C, Drelich A, Martinez DR, Gralinski LE, Sun Z, et al. Rapid Identification of a Human Antibody With High Prophylactic and Therapeutic Efficacy in Three Animal Models of SARS-CoV-2 Infection. Proc Natl Acad Sci USA (2020) 117(47):29832–8. doi: 10.1073/pnas.2010197117
6. Schneider D, Xiong Y, Hu P, Wu D, Chen W, Ying T, et al. A Unique Human Immunoglobulin Heavy Chain Variable Domain-Only CD33 CAR for the Treatment of Acute Myeloid Leukemia. Front Oncol (2018) 8:539. doi: 10.3389/fonc.2018.00539
7. Schneider D, Xiong Y, Wu D, Hu P, Alabanza L, Steimle B, et al. Trispecific CD19-CD20-CD22-Targeting duoCAR-T Cells Eliminate Antigen-Heterogeneous B Cell Tumors in Preclinical Models. Sci Transl Med (2021) 13(586). doi: 10.1126/scitranslmed.abc6401
8. Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N Engl J Med (2019) 380(4):335–46. doi: 10.1056/NEJMoa1806311
9. Bracken CJ, Lim SA, Solomon P, Rettko NJ, Nguyen DP, Zha BS, et al. Bi-Paratopic and Multivalent VH Domains Block ACE2 Binding and Neutralize SARS-CoV-2. Nat Chem Biol (2021) 17(1):113–21. doi: 10.1038/s41589-020-00679-1
10. Anthony-Gonda K, Bardhi A, Ray A, Flerin N, Li M, Chen W, et al. Multispecific Anti-HIV duoCAR-T Cells Display Broad In Vitro Antiviral Activity and Potent In Vivo Elimination of HIV-Infected Cells in a Humanized Mouse Model. Sci Transl Med (2019) 11(504). doi: 10.1126/scitranslmed.aav5685
11. Sullivan-Chang L, O'Donnell RT, Tuscano JM. Targeting CD22 in B-Cell Malignancies: Current Status and Clinical Outlook. BioDrugs (2013) 27(4):293–304. doi: 10.1007/s40259-013-0016-7
12. Polson AG, Williams M, Gray AM, Fuji RN, Poon KA, McBride J, et al. Anti-CD22-MCC-DM1: An Antibody-Drug Conjugate With a Stable Linker for the Treatment of Non-Hodgkin's Lymphoma. Leukemia (2010) 24(9):1566–73. doi: 10.1038/leu.2010.141
13. Ruella M, Maus MV. Catch Me If You Can: Leukemia Escape After CD19-Directed T Cell Immunotherapies. Comput Struct Biotechnol J (2016) 14:357–62. doi: 10.1016/j.csbj.2016.09.003
14. McClanahan F, Hanna B, Miller S, Clear AJ, Lichter P, Gribben JG, et al. PD-L1 Checkpoint Blockade Prevents Immune Dysfunction and Leukemia Development in a Mouse Model of Chronic Lymphocytic Leukemia. Blood (2015) 126(2):203–11. doi: 10.1182/blood-2015-01-622936
15. Brusa D, Serra S, Coscia M, Rossi D, D'Arena G, Laurenti L, et al. The PD-1/PD-L1 Axis Contributes to T-Cell Dysfunction in Chronic Lymphocytic Leukemia. Haematologica (2013) 98(6):953–63. doi: 10.3324/haematol.2012.077537
16. Blaeschke F, Stenger D, Apfelbeck A, Cadilha BL, Benmebarek MR, Mahdawi J, et al. Augmenting Anti-CD19 and Anti-CD22 CAR T-Cell Function Using PD-1-CD28 Checkpoint Fusion Proteins. Blood Cancer J (2021) 11(6):108. doi: 10.1038/s41408-021-00499-z
17. Chen W, Zhu Z, Feng Y, Dimitrov DS. Human Domain Antibodies to Conserved Sterically Restricted Regions on Gp120 as Exceptionally Potent Cross-Reactive HIV-1 Neutralizers. Proc Natl Acad Sci USA (2008) 105(44):17121–6. doi: 10.1073/pnas.0805297105
18. Chen W, Zhu Z, Xiao X, Dimitrov DS. Construction of a Human Antibody Domain (VH) Library. Methods Mol Biol (2009) 525:81–99, xiii. doi: 10.1007/978-1-59745-554-1_4
19. Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, et al. A Fully Synthetic Human Fab Antibody Library Based on Fixed VH/VL Framework Pairings With Favorable Biophysical Properties. MAbs (2013) 5(3):445–70. doi: 10.4161/mabs.24218
20. Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise Determination of the Diversity of a Combinatorial Antibody Library Gives Insight Into the Human Immunoglobulin Repertoire. Proc Natl Acad Sci USA (2009) 106(48):20216–21. doi: 10.1073/pnas.0909775106
21. DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, et al. In-Depth Determination and Analysis of the Human Paired Heavy- and Light-Chain Antibody Repertoire. Nat Med (2015) 21(1):86–91. doi: 10.1038/nm.3743
22. Wu Y, Li C, Xia S, Tian X, Kong Y, Wang Z, et al. Identification of Human Single-Domain Antibodies Against SARS-CoV-2. Cell Host Microbe (2020) 27(6):891–898 e5. doi: 10.1016/j.chom.2020.04.023
23. Lefranc MP. IMGT, the International ImMunoGeneTics Information System. Cold Spring Harb Protoc (2011) 2011(6):595–603. doi: 10.1101/pdb.top115
24. Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, et al. IMGT, the International ImMunoGeneTics Information System. Nucleic Acids Res (2009) 37(Database issue):D1006–12. doi: 10.1093/nar/gkn838
25. Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, et al. General Strategy for the Generation of Human Antibody Variable Domains With Increased Aggregation Resistance. Proc Natl Acad Sci USA (2012) 109(27):10879–84. doi: 10.1073/pnas.1202866109
26. Xu JL, Davis MM. Diversity in the CDR3 Region of V(H) is Sufficient for Most Antibody Specificities. Immunity (2000) 13(1):37–45. doi: 10.1016/S1074-7613(00)00006-6
27. Twomey JD, Zhang B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J (2021) 23(2):39. doi: 10.1208/s12248-021-00574-0
28. Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T Cells With Dual Targeting of CD19 and CD22 in Adult Patients With Recurrent or Refractory B Cell Malignancies: A Phase 1 Trial. Nat Med (2021) 27(8):1419–31. doi: 10.1038/s41591-021-01436-0
29. Haso W, Lee DW, Shah NN, Stetler-Stevenson M, Yuan CM, Pastan IH, et al. Anti-CD22-Chimeric Antigen Receptors Targeting B-Cell Precursor Acute Lymphoblastic Leukemia. Blood (2013) 121(7):1165–74. doi: 10.1182/blood-2012-06-438002
30. Dimitrov DS. Therapeutic Proteins. Methods Mol Biol (2012) 899:1–26. doi: 10.1007/978-1-61779-921-1_1
31. Ruiz-Zamora RA, Guillaume S, Al-Hilaly YK, Al-Garawi Z, Rodriguez-Alvarez FJ, Zavala-Padilla G, et al. The CDR1 and Other Regions of Immunoglobulin Light Chains are Hot Spots for Amyloid Aggregation. Sci Rep (2019) 9(1):3123. doi: 10.1038/s41598-019-39781-3
32. Arbabi-Ghahroudi M, To R, Gaudette N, Hirama T, Ding W, MacKenzie R, et al. Aggregation-Resistant VHs Selected by In Vitro Evolution Tend to Have Disulfide-Bonded Loops and Acidic Isoelectric Points. Protein Eng Des Sel (2009) 22(2):59–66. doi: 10.1093/protein/gzn071
33. Li W, Prabakaran P, Chen W, Zhu Z, Feng Y, Dimitrov DS. Antibody Aggregation: Insights From Sequence and Structure. Antibodies (Basel) (2016) 5(3). doi: 10.3390/antib5030019
34. D'Angelo S, Ferrara F, Naranjo L, Erasmus MF, Hraber P, Bradbury ARM. Many Routes to an Antibody Heavy-Chain CDR3: Necessary, Yet Insufficient, for Specific Binding. Front Immunol (2018) 9:395. doi: 10.3389/fimmu.2018.00395
35. Erasmus MF, D'Angelo S, Ferrara F, Naranjo L, Teixeira AA, Buonpane R, et al. A Single Donor is Sufficient to Produce a Highly Functional In Vitro Antibody Library. Commun Biol (2021) 4(1):350. doi: 10.1038/s42003-021-01881-0
36. Horning JS. Investigation of Monoclonal Antibody Therapy With Conventional and High-Dose Treatment: Current Clinical Trials. Semin Hematol (2000) 37(4 Suppl 7):27–33. doi: 10.1016/S0037-1963(00)90057-X
37. Glennie MJ, Johnson PW. Clinical Trials of Antibody Therapy. Immunol Today (2000) 21(8):403–10. doi: 10.1016/S0167-5699(00)01669-8
38. Holliger P, Hudson PJ. Engineered Antibody Fragments and the Rise of Single Domains. Nat Biotechnol (2005) 23(9):1126–36. doi: 10.1038/nbt1142
39. Lowe D, Dudgeon K, Rouet R, Schofield P, Jermutus L, Christ D. Aggregation, Stability, and Formulation of Human Antibody Therapeutics. Adv Protein Chem Struct Biol (2011) 84:41–61. doi: 10.1016/B978-0-12-386483-3.00004-5
40. Akinleye A, Rasool Z. Immune Checkpoint Inhibitors of PD-L1 as Cancer Therapeutics. J Hematol Oncol (2019) 12(1):92. doi: 10.1186/s13045-019-0779-5
41. Aldoss I, Bargou RC, Nagorsen D, Friberg GR, Baeuerle PA, Forman SJ. Redirecting T Cells to Eradicate B-Cell Acute Lymphoblastic Leukemia: Bispecific T-Cell Engagers and Chimeric Antigen Receptors. Leukemia (2017) 31(4):777–87. doi: 10.1038/leu.2016.391
42. Huehls AM, Coupet TA, Sentman CL. Bispecific T-Cell Engagers for Cancer Immunotherapy. Immunol Cell Biol (2015) 93(3):290–6. doi: 10.1038/icb.2014.93
43. Schmohl JU, Gleason MK, Dougherty PR, Miller JS, Vallera DA. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-Cell Activity Against CD133+ Colorectal Cancer Cells. Target Oncol (2016) 11(3):353–61. doi: 10.1007/s11523-015-0391-8
44. Li W, Wu Y, Kong D, Yang H, Wang Y, Shao J, et al. One-Domain CD4 Fused to Human Anti-CD16 Antibody Domain Mediates Effective Killing of HIV-1-Infected Cells. Sci Rep (2017) 7(1):9130. doi: 10.1038/s41598-017-07966-3
45. Salvatori P. The Rationale of Ethanol Inhalation for Disinfection of the Respiratory Tract in SARS-CoV-2-Positive Asymptomatic Subjects. Pan Afr Med J (2021) 40:201. doi: 10.11604/pamj.2021.40.201.31211
46. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. SWISS-MODEL: Modelling Protein Tertiary and Quaternary Structure Using Evolutionary Information. Nucleic Acids Res (2014) 42(Web Server issue):W252–8. doi: 10.1093/nar/gku340
47. Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK Server: Interactive Docking Prediction of Protein-Protein Complexes and Symmetric Multimers. Bioinformatics (2014) 30(12):1771–3. doi: 10.1093/bioinformatics/btu097
Keywords: antibody VH domains, CD22, PD-L1, library construction, antibody domain drug conjugations (DDC)
Citation: Sun Z, Li W, Mellors JW, Orentas R and Dimitrov DS (2022) Construction of a Large Size Human Immunoglobulin Heavy Chain Variable (VH) Domain Library, Isolation and Characterization of Novel Human Antibody VH Domains Targeting PD-L1 and CD22. Front. Immunol. 13:869825. doi: 10.3389/fimmu.2022.869825
Received: 05 February 2022; Accepted: 16 March 2022;
Published: 06 April 2022.
Edited by:
Yuejin Liang, University of Texas Medical Branch at Galveston, United StatesReviewed by:
Sami Hamdoun, Johannes Gutenberg University Mainz, GermanyCopyright © 2022 Sun, Li, Mellors, Orentas and Dimitrov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zehua Sun, zes20@pitt.edu; Dimiter S. Dimitrov, DSD116@pitt.edu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.