 Daniel Sijmons
Daniel Sijmons Andrew J. Guy1,2
Andrew J. Guy1,2 Anna K. Walduck
Anna K. Walduck Paul A. Ramsland
Paul A. Ramsland- 1School of Science, RMIT University, Melbourne, VIC, Australia
- 2ZiP Diagnostics, Collingwood, VIC, Australia
- 3Department of Immunology, Monash University, Melbourne, VIC, Australia
- 4Department of Surgery, Austin Health, University of Melbourne, Heidelberg, VIC, Australia
Helicobacter pylori is an important human pathogen that infects half the human population and can lead to significant clinical outcomes such as acute and chronic gastritis, duodenal ulcer, and gastric adenocarcinoma. To establish infection, H. pylori employs several mechanisms to overcome the innate and adaptive immune systems. H. pylori can modulate interleukin (IL) secretion and innate immune cell function by the action of several virulence factors such as VacA, CagA and the type IV secretion system. Additionally, H. pylori can modulate local dendritic cells (DC) negatively impacting the function of these cells, reducing the secretion of immune signaling molecules, and influencing the differentiation of CD4+ T helper cells causing a bias to Th1 type cells. Furthermore, the lipopolysaccharide (LPS) of H. pylori displays a high degree of phase variation and contains human blood group carbohydrate determinants such as the Lewis system antigens, which are proposed to be involved in molecular mimicry of the host. Lastly, the H. pylori group of outer membrane proteins such as BabA play an important role in attachment and interaction with host Lewis and other carbohydrate antigens. This review examines the various mechanisms that H. pylori utilises to evade the innate immune system as well as discussing how the structure of the H. pylori LPS plays a role in immune evasion.
Introduction
Helicobacter pylori is a gram-negative, spiral shaped bacterium that infects the stomach of up to half the world’s population. The prevalence is dependent on country and can vary between 20% to 80% (1). H. pylori also has a high rate of clinically asymptomatic infection, with some studies reporting a prevalence as high as 67% in entirely asymptomatic populations (2, 3). However, chronic infection with H. pylori can result in severe clinical conditions, and colonisation induces chronic inflammation in all infected individuals (4). These conditions are thought to be caused by damage to the gastric epithelium and prolonged inflammation associated with chronic infection and immune action (3).
Antibiotics are the mainstay of treatment for confirmed infections, and quadruple antibiotic therapy is currently recommended (5). H. pylori strains are however increasingly found to be resistant and treatment failure is not uncommon (6). The outcome of H. pylori infection is strongly influenced by both the bacterial genotype as well as host polymorphisms and is driven by an array of virulence factors possessed by H. pylori (7). Approximately 15% of cases result in symptomatic gastric disease including acute gastritis, chronic gastritis, and peptic ulcer (8). Furthermore, 1-2% of infections result in malignant neoplastic diseases such as mucosal-associated lymphoid tissue (MALT) lymphoma and gastric adenocarcinoma (8). As a result of this H. pylori is classified as a type one carcinogen by the World Health Organisation (WHO) (9). The severity of H. pylori disease, and the increasing incidence of antibiotic resistance means there is an urgent need for new therapies and vaccines.
H. pylori employs several mechanisms to evade innate immunity allowing the bacterium to establish chronic infection. Firstly, H. pylori occupies a unique infective niche in the hostile conditions of the human stomach, using the secretion of the enzyme urease to survive the natural barrier of the stomach’s acidity, coupled with the motility via its unipolar flagella (10). The motility of H. pylori allows it to maintain position and reside in the gastric mucous gel layer where it triggers inflammation. The net effect of H. pylori-induced inflammation is damage to the epithelial barrier, which presumably releases nutrients to promote bacterial growth.
Despite chronic infection, inflammatory responses to H. pylori infection remain relatively controlled in most cases due to a series of mechanisms that manipulate the host response, promoting persistent infection. H. pylori is able to adhere to mucins such as MUC5AC (11), as well as interacting with Lewis system antigens expressed on gastric epithelial cells (12), this allows it to maintain its position in the mucus gel layer and crypt isthmus and prevent its removal by the stomachs churning movement (10). Secondly, H. pylori can interact with and manipulate interleukin (IL) secretion of local dendritic cells (DC), negatively regulating the functions of these cells and suppressing cytokine release (13), influencing the differentiation of CD4+ T helper cells towards Th1 type cells (14). Finally, the lipopolysaccharide (LPS) of H. pylori displays a high degree of phase variation and contains human blood group carbohydrate determinants such as the Lewis system antigens (15–17). This review examines the various mechanisms H. pylori utilises to evade the innate immune system as well as discussing how the structure of the H. pylori LPS plays a prominent role in immune evasion.
Overview of Innate Immunity in the Context of Bacterial Infection
The innate immune system is the first line of defence against the many potential pathogens humans are exposed to daily (18). Epithelial surfaces and mucous membranes provide a physical barrier between internal organs and the external environment (19). The innate immune system also employs various antimicrobial peptides within the mucosal layer which have the ability to kill or inhibit microorganisms and viruses in the area (20). These initial defences are supported by more complex innate immune mechanisms. For example, pathogen-associated molecular patterns (PAMP) can be discerned from host molecular patterns by innate and adaptive immune cells allowing the triggering of a more extensive immune response (21). PAMP frequently include molecules such as glycans and glycoconjugates which may be expressed in structures like the LPS of a gram-negative bacterium (22). Detection of these structures are commonly associated with two classes of proteins known as toll-like receptors (TLR) and nucleotide-binding oligomerization domain-like receptors (NLR), as well as complement components circulating in the blood (23). The TLR expressed in humans are particularly well characterised, with TLR2, TLR4 and TLR5 commonly associated with the detection of lipopolysaccharide structures such as a bacterial LPS or flagella, whereas TLR3, TLR7, TLR8 and TLR9 are associated with the detection of foreign nucleic acids (24). Nod-Like Receptors recognise components of the bacterial cell wall, their activation triggers the release of pro-inflammatory cytokines and have synergistic effects with TLR in immune system signaling pathways (25).
Pathogen recognition triggers several supporting immune functions such as the activation of proinflammatory effector molecules. Inflammation is an important part of the immune response and inflammasomes are a system of innate immune receptors that activate the cystine protease caspase-1 and induce inflammation in response to the presence of infectious microbes (26). Additionally, inflammasomes contribute to the activation of proinflammatory cytokines IL-1β and IL-18, which induce an influx of innate immune cells into a potentially infected area, resulting in the secretion of further inflammatory cytokines, including IL-8, IL-6, IL-12, IL-17 and TNF-α, allowing for improved clearance of infection (26–28). Recognition of infectious bacteria often results in the engulfment of the bacterium by phagocytes including macrophages and monocytes. Engulfed bacteria are encased in an intracellular vesicle, the phagosome, where the phagocytic cells concentrate molecules which are deadly to bacterial cells, including antimicrobial substances, reactive oxygen species and reactive nitrogen intermediates. As well as immediate destruction, the engulfment, or phagocytosis of bacteria can also function as a form of antigen presentation for the activation of the adaptive immune system (29). A specialised type of phagocyte known as the dendritic cell (DC) plays a primary role in the activation of the adaptive immune system. Once a DC engulfs a bacterium, it processes the bacterial cell and presents its antigens in complex with major histocompatibility molecules (MHC) to lymphocytes to induce T cell and B cell maturation and differentiation, triggering the adaptive immune response (30).
Immune Response Against H. pylori
Various innate immune mechanisms are activated by the colonisation of H. pylori. Toll-like receptors (TLR) 2, 4 and 5 can recognise PAMP of H. pylori (31), triggering immune pathways that activate NF-ĸB and IL-8 inducing a local proinflammatory response (32). H. pylori are extracellular and generally non-invasive, but secretion of urease and the VacA toxin trigger local inflammatory responses. The most researched virulence factor is the type IV secretion system (TIVSS) encoded by the Cag pathogenicity island carried by type I strains (33). After bacterial adherence the TIVSS delivers the cytotoxin CagA to the cytoplasm where it initiates intracellular signaling cascades that activate NF-κB (34), contributing to the local inflammatory response. The combined effect of this inflammation results in erosion of the epithelium, releasing nutrients for the bacteria to grow and colonise (35). Furthermore, intracellular pathogen recognising molecules such as NOD1 can bind H. pylori peptidoglycans that are introduced into epithelial cells via the TIVSS and trigger the release of other anti-microbial proteins in response, restricting bacterial growth and initiating additional immune action (36). While additional macrophages may be stimulated, H. pylori is able to neutralise macrophage nitric oxide production reducing the immune action of the cell (37). Once H. pylori has overcome the initial epithelial response, persistent infection drives an adaptive immune response triggered by dendritic cells. The local response consists of humoral and T cell (particularly Th1 and Th17) activation. Overall there is an influx of plasma cells, lymphocytes, neutrophils and other immune cells into the gastric mucosa during H. pylori infection, however this rarely results in complete clearance of the bacterium, and over time a significant population of regulatory T cells become established (38). This control of inflammation by H. pylori probably explains why many H. pylori infected individuals remain asymptomatic.
The stomach has relatively few polymeric immunoglobulin receptors (pIgR), the receptor responsible for IgA transportation, in comparison to the rest of the gastrointestinal tract (39). This is altered in chronic H. pylori infection with upregulation of the pIgR caused by raised γ-interferon levels associated with prolonged inflammation (40). Upregulation of pIgR does not result in a corresponding increase in local secretory IgA levels, with monomeric non-secretory IgA predominating in the stomach of those infected with H. pylori (41). In contrast, secretory IgA is commonly observed in response to intestinal commensals and pathogens, suggesting a different mode of action of pIgR in the stomach. Systemic H. pylori specific IgG is also produced in adults experiencing chronic infection (42).
Role of H. pylori Virulence Factors in Induction of Innate Immune Responses and Immune Evasion
The natural immune response against H. pylori does not effectively clear infection and a combination of immune evasion techniques and bacterial factors leads to a persistent infection (Figure 1 and Table 1). Initial Type 1 inflammatory responses in most infected persons become biased over time towards Th2 and Treg that acts in a kind of damage control measure, which does not clear infection but reduces damage to the host (59–61).

Figure 1 Strategies used by H. pylori to evade innate immune mechanisms. H. pylori uses a number of mechanisms to evade the innate immune system. Various components of the innate immune system present barriers to or respond to H. pylori infection (left). H. pylori is able to overcome these by mechanisms described (right). Created with BioRender.com.

Table 1 Effect of H. pylori virulence factors on the immune response.
An important feature of H. pylori is its ability to overcome natural barriers preventing most microbes from colonising the human stomach (62, 63). Early studies concluded that H. pylori flagellin evades TLR5 recognition and recombinant H. pylori flagellin was considerably less stimulatory than Salmonella flagellin for example (64). Furthermore, a series of residues within the CD0 domain of H. pylori flagella protein FlaA has recently been found to enable evasion of TLR5 (Figure 1), these are speculated to be a result of point mutations not present in flagella proteins from other bacterium (65). However, a number of studies have now reported that H. pylori activates both TLR2 and 5 on epithelial cells, but not TLR4 (66). A recent report revealed that TLR5 activation is via interaction with the CagY protein which forms part of the TIVSS (67).
CagA is one of the most studied and most important virulence factors possessed by H. pylori. Contact and adherence of H. pylori to host gastric epithelial cells upregulates the expression of both CagA and VacA, as well as inducing the synthesis and assembly of the TIVSS (Figure 2) (71). Both CagA and the TIVSS are encoded by the cag PAI which is an approximately 40 kb chromosomal DNA region present in the most virulent strains of H. pylori. The TIVSS is a pilus-based “molecular syringe” structure that translocates CagA protein into host epithelial cells. The Type IV pilus binds epithelial cells via interaction with β1 integrin on the basolateral side (72). The bacteria gain access to the basolateral side through secretion of the HtrA enzyme which breaks down tight junctions (33).
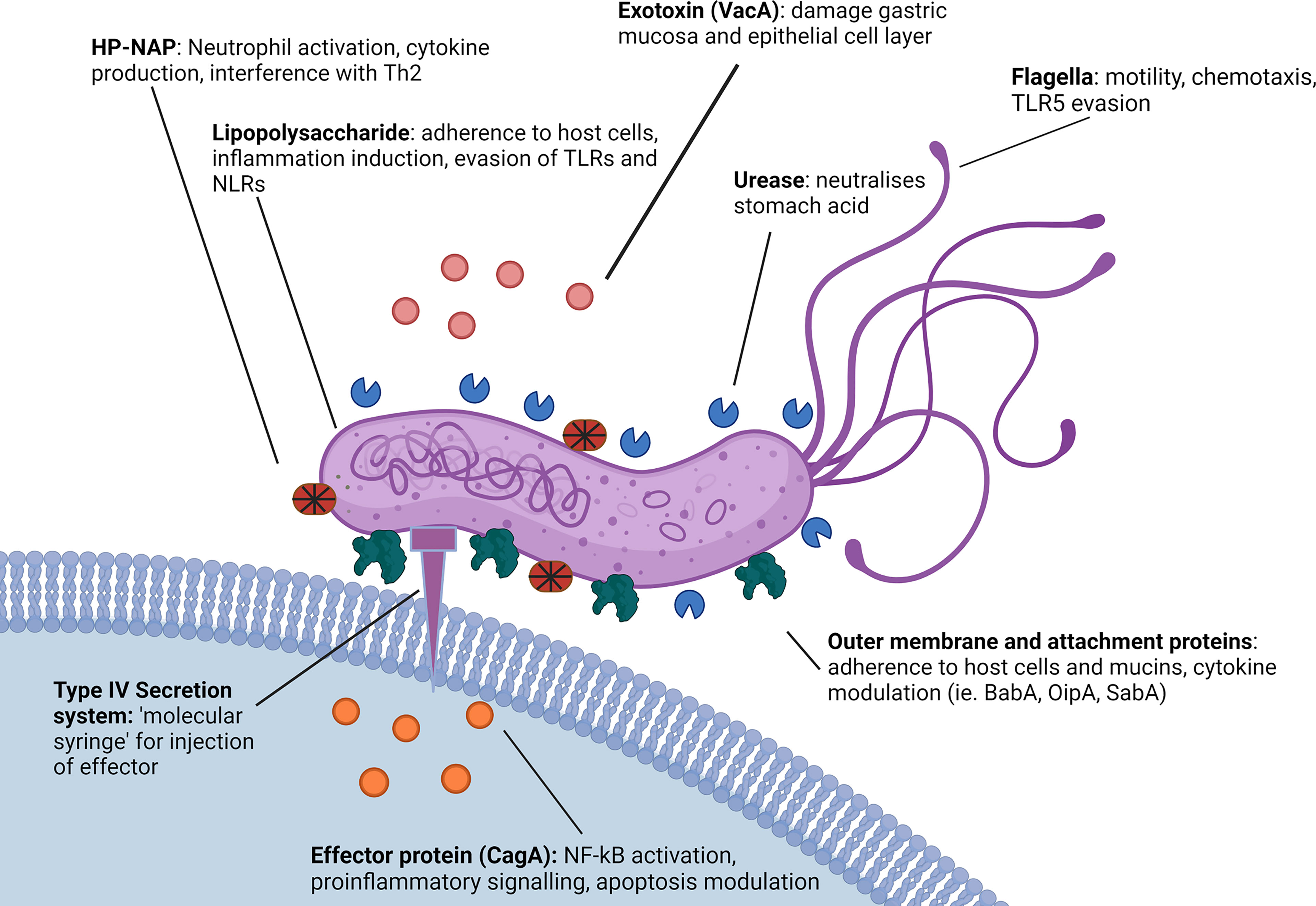
Figure 2 Role of H. pylori virulence factors in innate immune evasion. H. pylori utilises a number of virulence factors to enable immune evasion. H. pylori possesses a less immunogenic LPS in comparison to other gram-negative microbes and both the LPS and several H. pylori virulence proteins such as SabA and OipA demonstrate variable expression patterns affecting the immune response (68, 69). Additionally, H. pylori expresses proteins such as HP-NAP, VacA and CagA that actively modulate cytokine and inflammatory signaling, as well as DC and macrophage function (56, 70). H. pylori flagella protein FlaA also evades detection by TLR5 via a series of complex point mutations (65). Created with BioRender.com.
Once in the cytoplasm, translocated CagA associates with Src and c-Abl family kinases which phosphorylate the CagA carboxyl-terminal EPIYA motif in the inner host cell membrane. Phosphorylated CagA then interferes with host cell signaling pathways, disrupting cell-cell junctions (73). In addition, CagA targets and causes the ubiquitination of the upstream kinase TAK1 (74). The downstream effects of CagA induce signaling cascades that activate NF-κB and downstream proinflammatory signaling (IL-8 secretion), cytoskeletal rearrangements, and changes to tight junctions (31, 33, 75) and inducing apoptosis (70, 76). Higher levels of CagA were shown to cause nuclear translocation of the AP-1 transcription factor and nuclear factor of activated T-cells (NFAT) (77). Furthermore, CagA is directly related to H. pylori carcinogenesis, enhancing DNA double strand breaks, and disabling homologous recombination-mediated DNA repair as well as stimulating Hippo signaling via the inhibition of PAR1b-mediated BRCA1 phosphorylation (78). Additionally, recent work has indicated that CagA initiates the oncogenic YAP pathway, further contributing to the association between CagA and gastric cancer risk (79, 80).
Infection with CagA positive H. pylori strains is one of the strongest risk factors for the development of gastric cancer (75). In addition, multiple H. pylori outer membrane proteins are associated with proinflammatory cytokine induction (Figure 2), these include OipA and BabA, which can induce IL-6, IL-8 and IL-11 (44, 45, 81). Like many bacteria, H. pylori have also been shown to release outer membrane vesicles (OMV) which contain various components, notably including portions of the H. pylori LPS as well as Hop family proteins including BabA and SabA along with other major virulence factors such as VacA and CagA (Figure 2) (74, 82). Interestingly, it appears that H. pylori is also able to regulate cytokine release from host epithelial cells via small noncoding RNAs that have been detected in OMV. These OMV have been shown to suppress IL-8 stimulation from AGS cells in cell culture, presumably by targeting host cell mRNAs, effectively reducing the overall immunostimulatory response to H. pylori LPS and outer membrane proteins (83). Consequently, OMV may play a role in immune modulation and disease progression. Regulation of cytokine release by H. pylori has also been investigated in relation to inflammasomes. H. pylori can modulate NLRP3, a regulator important for inflammasome activation, by inducing secretion of IL-10 in THP-1 monocytic leukemia cell lines, and through the action of miRNAs such as has-miR-223-3p (84). Furthermore, H. pylori infection was reported to suppress the secretion of mature IL-1β from human THP1 monocytes, in spite of upregulation of pro-IL-1β, potentially promoting bacterial persistence (85). Interestingly, cytokine expression has been seen to differ between children and adults, with Il-1β, IL17A, IL-23, IL2, IL-12p70 and IFN-γ upregulated in adults and downregulated in children, and IL-6, TGF-β1, IL-10, TNF-α and IL-1a downregulated in adults and upregulated in children. As a result of these differences, children display a Treg primary response as opposed to the Th1 and Th17 response seen in adults (86–88).
The virulence factor HP-NAP also plays a major role in inducing and modulating host inflammation, recruiting neutrophils to the site of infection and causing a large increase in the neutrophil secretion of reactive nitrogen species, as well as stimulating the release of IL-12 and IL-23 from both neutrophils and monocytes (89). In vitro studies using cloned CD4+ T cells from allergen induced T cell lines have shown HP-NAP acts as a Th2 agonist, redirecting Th2 responses into Th1 to drive further inflammation through cytotoxic Th1 responses characterised by increased TNF-α and interferon-γ (Figure 2) (56).
H. pylori is able to adhere to both the secreted gastric mucin MUC5AC (90) and gastric epithelial cells via several surface proteins, such as the blood group binding adhesin (BabA) and the sialic acid binding adhesin (SabA). These surface proteins interact with carbohydrate determinants such as the host Lewis system antigens, again providing a mechanism by which H. pylori prevents its removal by peristalsis as well as gastric shedding (Figure 2) (46).
H. pylori also expresses Lewis system antigens within the LPS, that is thought to represent a form of molecular mimicry which subverts the host immune response. Indeed, there is also evidence suggesting the expression of Lewis determinants is involved in H. pylori interacting with the DC calcium dependent (C-type) lectin DC-SIGN (Table 1) (91). Upon phagocytosis by an innate immune cell, H. pylori can also avoid destruction by interfering with phagosome maturation (Figure 2) (92). Another method of immune evasion used by H. pylori involves the interaction with DC (Figure 1). While H. pylori inhabits a unique niche in the stomach where few cells have access, there has been strong evidence of DC interaction, with DC maturation and function heavily affected in mouse models (93). Furthermore, there is evidence of H. pylori having the capability to infect DC in cell culture (48). H. pylori is capable of modulating IL-12 secretion by DC affecting the maturation and differentiation of CD4+ T cells and causing Th1 bias in mice (94). In-vitro studies have supported this, with H. pylori demonstrating the ability to modulate expression of IL-12 and TNF-α in cultured DC (95).
In addition to manipulating DC responses, other virulence factors including VacA (96) along with γ-glutamyltranspeptidase (GGT) (97) and CagA (98) have been reported to interfere with the T cell response to H. pylori, causing a characteristic hyposensitivity of CD4+ T cells and suppressing mucosal effector T cells (99). Notably, VacA has been found to have an influence on both DC cytokine expression and T cell differentiation, with VacA positive H. pylori strains showing the ability to supress IL-23 secretion by DC resulting in increased proliferation of Th17 cells. VacA plays additional roles in immune mediation including showing the ability to impair lysosomes and autophagosomes, a function with which VacA shares a synergistic effect with CagA, impairing the ability of both macrophages and gastric epithelial cells to remove internalised or invading bacteria. Specifically, H. pylori is able to inhibit the functions of lysosomal enzymes acid phosphatase, N-acetyl-β-d-glucosaminidase, and cathepsin D, prevent autolysosomal acidification, interfere with retrograde trafficking of mannose-6-phosphate receptors and inhibit autophagosome formation to promote intracellular survival of the bacterium (100).
It is likely H. pylori is highly resistant to destruction by phagocytes, although this has only been seen in-vitro (101). Even though H. pylori is frequently engulfed by phagocytes in vitro, it is proficient at neutralising and resisting the reactive oxygen and nitrate species released by monocytes and macrophages. This allows the bacteria to continue to replicate inside the phagosome, eventually inducing apoptosis of the host cell (49, 101). H. pylori possessing the cag pathogenicity island (cagPI) are even more resistant to phagocytosis than other strains (102), with isogenic mutants of H. pylori lacking cagPI far more readily taken up by phagocytes (49). The mechanism for this appears to be the ability of H. pylori to activate the reverse transsulfuration pathway to induce cystathionine γ-lyase (CTH) in host macrophages, promoting bacterial growth and enhancing bacterial survival in macrophages. H. pylori does this via the CagA-dependant induction of the PI3K/AKT1 pathway, the CagA-independent induction of the MTOR pathway and activation of SP1, increasing macrophage production of CTH (103) Furthermore, it has been shown that H. pylori is able to upregulate the metabolism of macrophage-associated polyamines, impairing M1 macrophage function (104).
Antibody Activity and Humoral Immune Evasion by H. pylori
The mechanisms of humoral immune evasion by H. pylori are somewhat less clear. Most immunocompetent individuals who are infected with H. pylori develop a specific IgG and IgA response, in many cases high antibody titres to the pathogen, however, this often is not enough to clear infection (53, 105). Furthermore, it has been reported that the type of antibody produced correlates with the outcome of infection, with patients experiencing gastritis and duodenal ulcers having greater titres of IgG, and patients suffering from gastric cancers often having greater titres of IgA (106). Other studies have suggested that a weaker overall antibody response is linked to the development of gastric cancers, and patients who had a weak but still detectable antibody response to H. pylori had a higher rate of gastric cancer than those with a stronger antibody response (107–109).
H. pylori LPS and Lewis Antigens
H. pylori both interacts with host Lewis system antigens and displays Lewis antigens in its LPS. The expression of Lewis antigens by H. pylori is associated with immune evasion and a reduced immunogenicity of its LPS when compared to other bacteria (110, 111). The reduced immunogenicity of H. pylori LPS is related to its structure, much like that of other gram-negative bacterium, the H. pylori LPS contains 3 domains: a hydrophobic lipid A domain on the outer bacterial membrane, a core oligosaccharide and a repetitive oligosaccharide termed the O chain (111). The O chain of the H. pylori LPS is the main location where Lewis system antigens are displayed by the organism (112). The H. pylori O antigen is typically composed of a Gal-GlcNAc backbone chain which is divided into two types based on its linkage (Figure 3). Type 1 chains are composed of Galβ1-3GlcNAc, which forms the core saccharide for Lea, sialyl-Lea and Leb. Type 2 chains are composed of Galβ1-4GlcNAc or LacNAc, which forms the core saccharide for Lex, sialyl-Lex and Ley (63). In humans, Lewis system antigens like ABO blood group antigens are expressed in fluids and tissue including the gastric mucosa and endothelium. Lea and Leb are commonly expressed on various cell types from red blood cells to gastric epithelial cells, with Leb in particular associated with various pathologies (113). In the human stomach, Lea and Leb are predominantly expressed on the surface and foveolar epithelia, whereas Lex and Ley are predominantly expressed in the mucus as well as the chief and parietal cells of the gastric glands. More specifically, non-secretory cells in the surface and foveolar epithelia express Lea while Leb and Ley are expressed in secretory cells (114). Clinical isolates of H. pylori typically have a poly-LacNAc with several α-L-fucose residues forming internal Lex determinants with terminal Lex and Ley determinants, while other strains have been described as displaying Lea, Leb and sialyl-Lex as well as group A, B and H-1 determinants (111). As many as 90% of clinical isolates of H. pylori contain Lewis antigens in the O antigen portion of the LPS. This expression has been found to be relatively stable after subculturing using methods such as immunoblot. However, it is possible for cultured H. pylori strains to lose Lewis antigen expression over time (68, 115).
H. pylori LPS Structure, Phase Variation, and Diversity
As described above, the O antigen portion of the H. pylori LPS is associated with the expression of various Lewis antigens in addition to playing a role in the adhesion and immune evasion of the bacterium (116). The O antigen chain is comprised of a glucan group (saccharide composed of glucose), a D-glycerol-D-manno-heptan (DD-heptan) group and a highly conserved trisaccharide (trio) (115). Variability in the O antigen is derived from phase variation, whereby H. pylori uses an on/off system to regulate its biosynthetic genes, including the fucosyltransferase genes FutA, FutB, and FutC (117), allowing the bacterium to adjust the carbohydrate expression of the O antigen with the changing environment of the stomach and gastric mucosa (16). Additionally, phase variation of the H. pylori LPS allows for a greater range of phenotypes and gives H. pylori the ability to modify the expression of Lewis system antigens in its LPS. Additionally, it has been indicated that the H. pylori LPS induces lower biological responses when compared to other gram-negative organisms such as E. coli and Salmonella spp., with in vivo mouse and rabbit studies showing 500 to 1000 fold lower mitogenic and pyrogenic responses (118).
The genes encoding the glycotransferases responsible for the construction of this LPS vary between different regional strains, with the common European strain of H. pylori G27 possessing the trisaccharide fucosyltransferase (HP0102), heptan transferase (HP1283) and GlcNAc transferase (HP1578), the last of which is responsible for initiating synthesis of Lewis system antigens onto a heptan motif (111). In addition to this, a comparison between the European model strain G27 and East Asian strains found that the East Asian strains lacked the genes encoding for heptan transferase and GlcNAc transferase (119). Furthermore, East Asian strains instead express additional copies of other genes, HP1105 and JHP0562, which may act as GlcNAc transferases as well as Gal transferase in place of HP1578 (119). This was identified as an area of interest due to the higher rates of gastric cancer in East Asia and the potential to further characterise the role of the H. pylori LPS heptan in pathogenesis (119). Figure 3 illustrates an abbreviated version of the established structure of the O antigen structure from the mouse-adapted H. pylori SS1 strain including the trio, attachment site and the Lewis antigenic determinants.
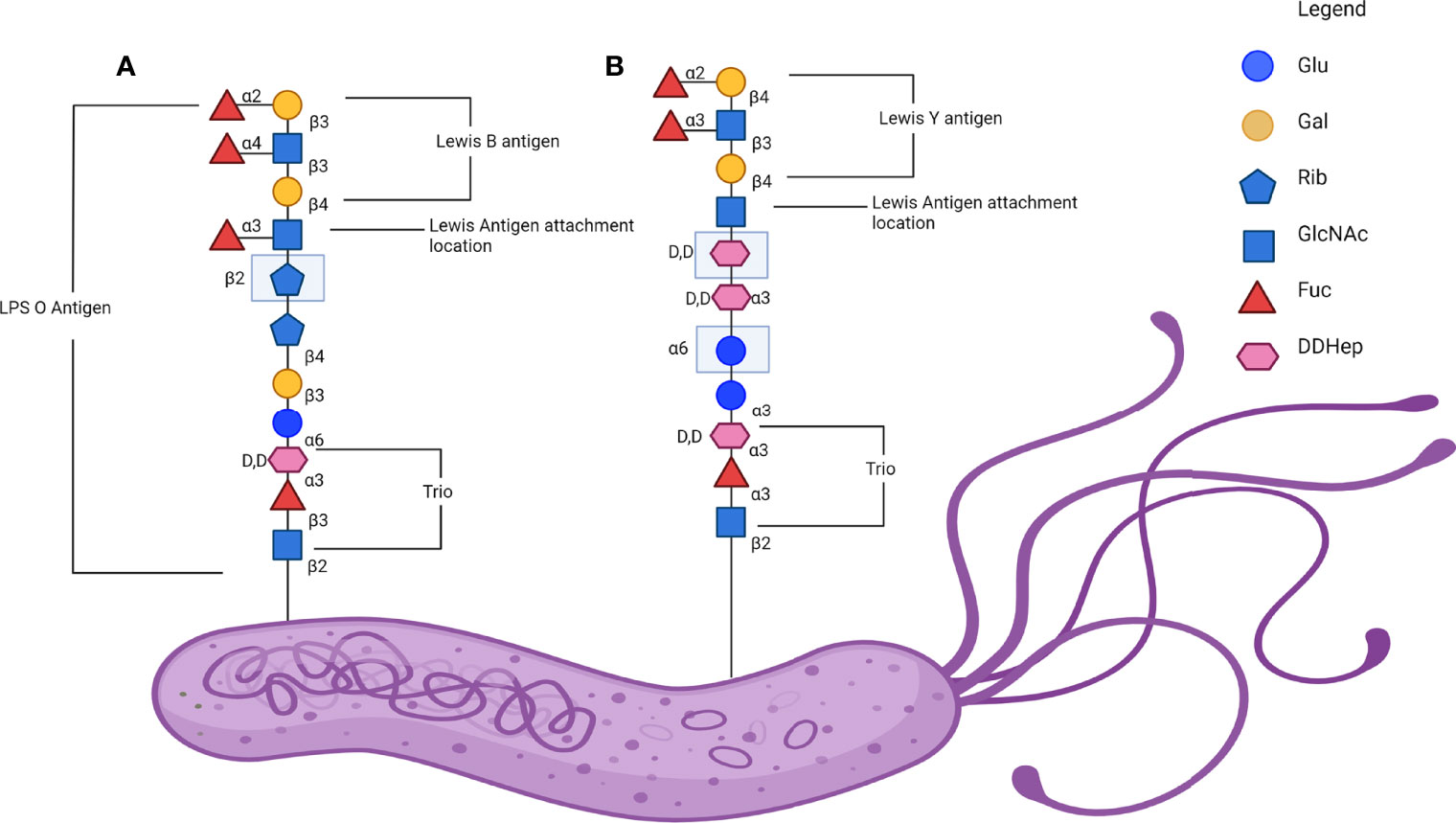
Figure 3 Lewis antigen expression on H. pylori lipopolysaccharide (LPS) O antigen. (A) A representative linear glycan chain of the H. pylori SS1 strain O antigen with the Leb in comparison to (B) a representative linear glycan chain of H. pylori 26695 and G27 O antigen with the Ley antigen attached. Adapted from the established structure in Li et al., 2018. Note that the exact structure and the Lewis determinate expressed is variable and dependant on isolate (115). Created with BioRender.com.
H. pylori LPS Structural Diversity in Infection and Immune Evasion
The H. pylori LPS is generally considered less immunogenic in comparison to enterobacterial LPS, with early studies showing significant reductions in pyrogenicity, mitogenicity and toxicity, as well as a reduced cytokine and chemokine response (118, 120, 121). Previous studies have shown that synthesised partial H. pylori LPS structures, specifically lipid A compounds and Kdo-lipid A compounds, can modulate cytokine production by host cells. All lipid A and Kdo-lipid A H. pylori LPS structures synthesised either failed to induce, or induced very low levels of IL-1β, IL-6 and IL-8, and conversely stimulated high levels of cytokines IL-12 and IL-18 in heparinised human peripheral whole blood (122, 123). IL-12 induction by H. pylori is generally linked to MyD88 expression in macrophages (124). Furthermore, H. pylori modifies the lipid A portion of the LPS by via dephosphorylation of the 1- and 4-’positions of the lipid A backbone. Mutations to the lpxE/F machinery required for this modification have shown an increased susceptibility of H. pylori to the antimicrobial peptide polymyxin B, as well as reducing the ability of H. pylori to colonise mice (125). Knockout studies on H. pylori genes HP0044 and HP1275 demonstrated the production of a truncated H. pylori LPS missing fucose residues found in both the trio and Lewis antigen portions of the H. pylori LPS O antigen, resulting in the loss of a significant portion of the H. pylori LPS. This in turn affected the growth of the bacterium, increased its susceptibility to both the detergent SDS, promoted bacterial autoaggregation, increased surface hydrophobicity and affected bacterial virulence and OMV protein sorting (126). Notably, phase variation of H. pylori fucosyltransferases, and consequently the glycosylation pattern of the LPS has previously been identified to occur in high frequencies, with in-vitro studies demonstrating the expression of Lex can vary at a frequency of 0.2-0.5% resulting in differing LPS variants in the same population. Variation of Lewis antigen expression, particularly between Lex and Ley has been suggested to be influenced by relative pH in liquid medium. The function for this has not been fully identified, but is hypothesised to aid in bacterial persistence (127). On a molecular level the variation in H. pylori fucosylation has previously been attributed to slipped-strand mispairing as a result of differing numbers of polyC repeats in fucosyltransferase genes futA, futB and futC. However, a recent study has described a role for small RNAs (sRNA) in the regulation of the H. pylori LPS biosynthesis. The sRNA RepG modulate HP0102 and TlpB, providing post-transcriptional regulation to LPS biosynthesis potentially indicating further means by which H. pylori is able to adapt to host immune response (128).
Role of H. pylori Adhesins in Immune Evasion
In addition to H. pylori commonly expressing Lewis determinants in its LPS, H. pylori also binds to host Lewis determinants, assisting in the attachment of the bacterium to the gastric epithelium (129, 130). Binding is facilitated by a group of outer membrane proteins, including BabA and SabA.
BabA
H. pylori binds to the various Lewis system antigens expressed on the surface of gastric epithelial cells, including Leb and sialyl-Lex (131). Chronic inflammation also leads to upregulation of Lex and Ley in the host, further enhancing bacterial colonisation (131). Furthermore, H. pylori has a range of adhesive proteins that have been identified and examined. The first identified was BabA, a protein that commonly binds the Leb determinant as well as mucin proteins and the H1 blood group antigen (132). Specifically, BabA binds the lacto series of glycans containing a terminal fucose molecule with an α1,2 linkage attached to a Galβ1-3GlcNAc core (133). This includes H1 blood group antigen as well as other H neoglycoconjugates and Leb. However, BabA binds to Leb with as much as a two-fold increase in affinity in comparison to H neoglycoconjugates (134). The BabA encoding gene(s) can be found in three separate loci on the H. pylori genome with the binding pattern of the protein differing based on the specific loci expressed (135). For example, strains containing locus A BabA (BabA2) bind the classically associated Leb whereas strains with locus B BabA (BabA1) do not bind to Leb (136). Furthermore, some strains may contain more than one BabA encoding gene allowing expression of proteins with varying binding characteristics (137). BabA in general is a highly polymorphic protein with variable expression, which may be lost during laboratory cultivation. Additionally about one quarter of strains isolated from chronically infected hosts tend to lose BabA expression (69, 138). This suggests that BabA is a specific adhesin that is advantageous in the colonisation of a human host that is unnecessary on solid media (137). There has been evidence of numerous binding molecules for BabA in addition to the Leb molecules expressed by gastric epithelia. Several salivary proteins have also been identified to interact with BabA, such as the mucin MUC5B, the agglutinin glycoprotein-340 and the proline-rich glycoprotein containing the Fucα1-2Galβ motif. In addition to this, fucose-containing oligosaccharides present in secretory IgA also play a role in binding BabA, however this is not universal to all secretory IgA (132). It has been observed that BabA presence in a H. pylori strain is associated with expression of VacA, CagA and OipA among other common but variable virulence factors, particularly in the outcome of gastric metaplasia. An in-vitro study found that Leb-positive AGS cells expressed increased levels of mRNA for the cytokines CCL5 and IL-8 as well as precancer related factors CDX2 and MUC2 in the presence of wild type H. pylori, however this was not the case for H. pylori mutants with either BabA genes or TIVSS deleted (139).
SabA
SabA is an additional adhesion molecule associated with the binding of Lewis system determinants by H. pylori. More specifically, SabA binds sialylated molecules such as sialyl-Lex and some gangliosides characterised by Neu5Acα3-neolactohexaosylceramide and Neu5Acα3-neolactooctaosylceramide molecules (58). The gastric inflammation caused by H. pylori infection is essential for changes in glycosylation patterns within the gastric mucosa, promoting the expression of sialyl-Lex as well as sialyl-Lea, which in turn increases the adhesive properties of SabA (140). Similar to BabA, the expression of SabA is highly variable and is often subject to phase variation. It has been suggested that H. pylori may have multiple means of modulating the expression of SabA, with one such method being an acid-responsive ArsRS two-component signal transduction system, and an alternative involving the slipped-strand mispairing of SabA alleles during chromosomal replication in various H. pylori subpopulations (141). Additionally, SabA is commonly subject to homologous recombination and gene conversion due to changing environmental pressures (142). Deactivation of the SabA encoding gene may further assist in bacterial escape as cytotoxic activity by neutrophils is in part triggered by SabA binding to gangliosides. Neutrophil action has been shown to be defective against SabA deleted H. pylori cells (58).
OipA
OipA is also a member of the Hop family of outer membrane proteins possessed by H. pylori. There is currently no available crystal structure for OipA, limiting detailed studies of its binding (143). OipA is known to be closely associated with the cag PAI and CagA, with the oipA locus approximately 100 kb from the cag PAI. OipA is regulated by slipped-strand mispairing and displays on/off states which are closely associated with the expression of CagA. Alleles of oipA have been demonstrated in up to 96% of cag PAI positive H. pylori strains, solidifying the relationship of the two virulence factors (144). With the exception of CagA, OipA is typically independent of other H. pylori virulence factors. OipA is associated with the induction of IL-8 from host epithelial cells, inhibition of apoptosis and enhanced adhesion to gastric cells in vitro (144). Induction of IL-8 and its subsequent role in inducing gastritis has been associated with a synergistic effect from H. pylori strains possessing both the cag PAI and a functional oipA gene (145). In contrast oipA “off” strains can down-regulate anti-apoptotic processes in AGS cells, more regularly inducing apoptosis on infection than oipA “on” strains in-vitro (146).
Conclusions
H. pylori is a very successful global pathogen and has adopted a range of adaptions to evade the innate immune system. These mechanisms include both active systems, such as the neutralisation of reactive oxygen and nitrogen species released by macrophages, the modulation of cytokine secretion and the maturation of dendritic cells, and more passive systems such as the variability and the uniquely low immunogenicity of its LPS (15, 53). In addition, the expression of Lewis antigens in the LPS of H. pylori gives the bacterium the ability to mimic host antigens and thereby hide from the immune system (147). Notably, several of the immune evasion mechanisms H. pylori employs are yet to be fully explained. For example, knowledge of H. pylori activation and modulation of innate immunity outside of interactions with dendritic cells and DC-SIGN is surprisingly limited. It is also known that outer membrane proteins such as BabA, SabA and OipA play a role in the induction of inflammatory cytokines, although the molecular mechanism is still unclear (44, 144). Further study into ways to circumvent the methods H. pylori uses for immune evasion could allow for improved treatment and vaccination options. Additionally, blocking the activity of H. pylori adhesins to prevent attachment in the gastric mucosal gel layer and to epithelial cells has potential for new treatment options in an environment of increased antibiotic resistance.
Author Contributions
DS, prepared figures and wrote the manuscript; AG, critically reviewed and commented on the manuscript; AW, critically reviewed and commented on the manuscript; PR, critically reviewed and commented on the manuscript. All authors conceived and discussed the topic of the review. All authors read and approved the final manuscript.
Funding
DS is supported by a Research Training Program Stipend Scholarship from the Australian Government, Department of Education and Training.
Conflict of Interest
Author AG was employed by company ZiP Diagnostics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Venerito M, Link A, Rokkas T, Malfertheiner P. Review: Gastric Cancer—Clinical Aspects. Helicobacter (2019) 24(S1):e12643. doi: 10.1111/hel.12643
2. Mungazi SG, Chihaka OB, Muguti GI. Prevalence of Helicobacter Pylori in Asymptomatic Patients at Surgical Outpatient Department: Harare Hospitals. Ann Med Surg (Lond) (2018) 35:153–7. doi: 10.1016/j.amsu.2018.09.040
3. Lim SH, Kwon J-W, Kim N, Kim GH, Kang JM, Park MJ, et al. Prevalence and Risk Factors of Helicobacter Pylori Infection in Korea: Nationwide Multicenter Study Over 13 Years. BMC Gastroenterol (2013) 13:104. doi: 10.1186/1471-230X-13-104
4. Dunne C, Dolan B, Clyne M. Factors That Mediate Colonization of the Human Stomach by Helicobacter Pylori. World J Gastroenterol (2014) 20(19):5610–24. doi: 10.3748/wjg.v20.i19.5610
5. Malfertheiner P, Megraud F, O’Morain CA, Gisbert JP, Kuipers EJ, Axon AT, et al. Management of Helicobacter Pylori Infection-the Maastricht V/Florence Consensus Report. Gut (2017) 66(1):6–30. doi: 10.1136/gutjnl-2016-312288
6. Tshibangu-Kabamba E, Yamaoka Y. Helicobacter Pylori Infection and Antibiotic Resistance - From Biology to Clinical Implications. Nat Rev Gastroenterol Hepatol (2021) 18(9):613–29. doi: 10.1038/s41575-021-00449-x
7. Roszczenko-Jasińska P, Wojtyś MI, Jagusztyn-Krynicka EK. Helicobacter Pylori Treatment in the Post-Antibiotics Era-Searching for New Drug Targets. Appl Microbiol Biotechnol (2020) 104(23):9891–905. doi: 10.1007/s00253-020-10945-w
8. Testerman TL, Morris J. Beyond the Stomach: An Updated View of Helicobacter Pylori Pathogenesis, Diagnosis, and Treatment. World J Gastroenterol (2014) 20(36):12781–808. doi: 10.3748/wjg.v20.i36.12781
9. Parkin DM, Bray F, Ferlay J, Pisani P. Global Cancer Statistics, 2002. CA Cancer J Clin (2005) 55(2):74–108. doi: 10.3322/canjclin.55.2.74
10. Kusters JG, van Vliet AHM, Kuipers EJ. Pathogenesis of Helicobacter Pylori Infection. Clin Microbiol Rev (2006) 19(3):449–90. doi: 10.1128/CMR.00054-05
11. Van den Brink GR, Tytgat KM, van der Hulst RW, van der Loos CM, Einerhand AW, Büller HA, et al. H Pylori Colocalises With MUC5AC in the Human Stomach. Gut (2000) 46(5):601–7. doi: 10.1136/gut.46.5.601
12. Doohan D, Rezkitha YAA, Waskito LA, Yamaoka Y, Miftahussurur M. Helicobacter Pylori BabA-SabA Key Roles in the Adherence Phase: The Synergic Mechanism for Successful Colonization and Disease Development. Toxins (Basel) (2021) 13(7):485. doi: 10.3390/toxins13070485
13. Tanaka H, Yoshida M, Nishiumi S, Ohnishi N, Kobayashi K, Yamamoto K, et al. The CagA Protein of Helicobacter Pylori Suppresses the Functions of Dendritic Cell in Mice. Arch Biochem Biophys (2010) 498(1):35–42. doi: 10.1016/j.abb.2010.03.021
14. Kim JM, Kim JS, Yoo DY, Ko SH, Kim N, Kim H, et al. Stimulation of Dendritic Cells With Helicobacter Pylori Vacuolating Cytotoxin Negatively Regulates Their Maturation via the Restoration of E2F1. Clin Exp Immunol (2011) 166(1):34–45. doi: 10.1111/j.1365-2249.2011.04447.x
15. Pece S, Giuliani G, Di Leo A, Fumarola D, Antonaci S, Jirillo E. Role of Lipopolysaccharide and Related Cytokines in Helicobacter Pylori Infection. Recenti Prog Med (1997) 88(5):237–41.
16. Appelmelk BJ, Martino MC, Veenhof E, Monteiro MA, Maaskant JJ, Negrini R, et al. Phase Variation in H Type I and Lewis a Epitopes of Helicobacter Pylori Lipopolysaccharide. Infect Immun (2000) 68(10):5928–32. doi: 10.1128/iai.68.10.5928-5932.2000
17. Perepelov AV, Senchenkova SN, Knirel YA. Variations in the Expression of Terminal Oligosaccharide Units and Glycosylation of Poly(N-Acetyllactosamine) Chain in the Helicobacter Pylori Lipopolysaccharide Upon Colonization of Rhesus Macaques. Biochem (Moscow) (2020) 85(2):234–40. doi: 10.1134/S0006297920020108
18. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Innate Immunity. In: Molecular Biology of the Cell. New York: Garland Science (2002).
19. Muñoz-Carrillo JL, Rodríguez F, Gutierrez O, Moreno Garcìa M, Cordero J. Physiology and Pathology of Innate Immune Response Against Pathogens. In: Rezaei N, editor. Physiology and Pathology of Immunology. London: IntechOpen. (2017) p. 99–134. doi: 10.5772/intechopen.70556
20. Pasupuleti M, Schmidtchen A, Malmsten M. Antimicrobial Peptides: Key Components of the Innate Immune System. Crit Rev Biotechnol (2012) 32(2):143–71. doi: 10.3109/07388551.2011.594423
21. Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, et al. Glycans in the Immune System and The Altered Glycan Theory of Autoimmunity: A Critical Review. J Autoimmun (2015) 57:1–13. doi: 10.1016/j.jaut.2014.12.002
22. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
23. Medzhitov R. Recognition of Microorganisms and Activation of the Immune Response. Nature (2007) 449(7164):819–26. doi: 10.1038/nature06246
24. Diacovich L, Gorvel J-P. Bacterial Manipulation of Innate Immunity to Promote Infection. Nat Rev Microbiol (2010) 8(2):117–28. doi: 10.1038/nrmicro2295
25. Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-Like Proteins in Immunity, Inflammation and Disease. Nat Immunol (2006) 7(12):1250–7. doi: 10.1038/ni1412
26. Guo H, Callaway JB, Ting JPY. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat Med (2015) 21(7):677–87. doi: 10.1038/nm.3893
27. Luo Y, Zheng SG. Hall of Fame Among Pro-Inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front Immunol (2016) 7:604(604). doi: 10.3389/fimmu.2016.00604
28. Tran LS, Chonwerawong M, Ferrero RL. Regulation and Functions of Inflammasome-Mediated Cytokines in Helicobacter Pylori Infection. Microbes Infect (2017) 19(9-10):449–58. doi: 10.1016/j.micinf.2017.06.005
29. Ramachandra L, Simmons D, Harding CV. MHC Molecules and Microbial Antigen Processing in Phagosomes. Curr Opin Immunol (2009) 21(1):98–104. doi: 10.1016/j.coi.2009.01.001
30. Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, et al. Brucella Control of Dendritic Cell Maturation Is Dependent on the TIR-Containing Protein Btp1. PloS Pathog (2008) 4(2):e21–e. doi: 10.1371/journal.ppat.0040021
31. Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 Responds to Peptidoglycan Delivered by the Helicobacter Pylori Cag Pathogenicity Island. Nat Immunol (2004) 5(11):1166–74. doi: 10.1038/ni1131
32. Crabtree JE, Kersulyte D, Li SD, Lindley IJ, Berg DE. Modulation of Helicobacter Pylori Induced Interleukin-8 Synthesis in Gastric Epithelial Cells Mediated by Cag PAI Encoded VirD4 Homologue. J Clin Pathol (1999) 52(9):653–7. doi: 10.1136/jcp.52.9.653
33. Backert S, Haas R, Gerhard M, Naumann M. The Helicobacter Pylori Type IV Secretion System Encoded by the Cag Pathogenicity Island: Architecture, Function, and Signaling. In: Backert S, Grohmann E, editors. Type IV Secretion in Gram-Negative and Gram-Positive Bacteria. Cham: Springer International Publishing (2017). p. 187–220.
34. Tafreshi M, Guan J, Gorrell RJ, Chew N, Xin Y, Deswaerte V, et al. Helicobacter Pylori Type IV Secretion System and Its Adhesin Subunit, CagL, Mediate Potent Inflammatory Responses in Primary Human Endothelial Cells. Front Cell Infect Microbiol (2018) 8:22. doi: 10.3389/fcimb.2018.00022
35. Huang Y, Wang Q-l, Cheng D-d, Xu W-t, Lu N-h. Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter Pylori. Front Cell Infect Microbiol (2016) 6:159(159). doi: 10.3389/fcimb.2016.00159
36. Grubman A, Kaparakis M, Viala J, Allison C, Badea L, Karrar A, et al. The Innate Immune Molecule, NOD1, Regulates Direct Killing of Helicobacter Pylori by Antimicrobial Peptides. Cell Microbiol (2010) 12(5):626–39. doi: 10.1111/j.1462-5822.2009.01421.x
37. Lamarque D, Moran AP, Szepes Z, Delchier JC, Whittle BJ. Cytotoxicity Associated With Induction of Nitric Oxide Synthase in Rat Duodenal Epithelial Cells In Vivo by Lipopolysaccharide of Helicobacter Pylori: Inhibition by Superoxide Dismutase. Br J Pharmacol (2000) 130(7):1531–8. doi: 10.1038/sj.bjp.0703468
38. O’Keeffe J, Moran AP. Conventional, Regulatory, and Unconventional T Cells in the Immunologic Response to Helicobacter Pylori. Helicobacter (2008) 13(1):1–19. doi: 10.1111/j.1523-5378.2008.00559.x
39. Kaneko T, Ota H, Hayama M, Akamatsu T, Katsuyama T. Helicobacter Pylori Infection Produces Expression of a Secretory Component in Gastric Mucous Cells. Virchows Archiv (2000) 437(5):514–20. doi: 10.1007/s004280000285
40. Gorrell RJ, Wijburg OLC, Pedersen JS, Walduck AK, Kwok T, Strugnell RA, et al. Contribution of Secretory Antibodies to Intestinal Mucosal Immunity Against Helicobacter Pylori. Infect Immun (2013) 81(10):3880–93. doi: 10.1128/IAI.01424-12
41. Moyat M, Velin D. Immune Responses to Helicobacter Pylori Infection. World J Gastroenterol (2014) 20(19):5583–93. doi: 10.3748/wjg.v20.i19.5583
42. Blanchard TG, Nedrud JG, Czinn SJ. Local and Systemic Antibody Responses in Humans With Helicobacter Pylori Infection. Can J Gastroenterol (1999) 13(7):591–4. doi: 10.1155/1999/142457
43. van Kooyk Y, Geijtenbeek TB. DC-SIGN: Escape Mechanism for Pathogens. Nat Rev Immunol (2003) 3(9):697–709. doi: 10.1038/nri1182
44. Sugimoto M, Ohno T, Graham DY, Yamaoka Y. Helicobacter Pylori Outer Membrane Proteins on Gastric Mucosal Interleukin 6 and 11 Expression in Mongolian Gerbils. J Gastroenterol Hepatol (2011) 26(11):1677–84. doi: 10.1111/j.1440-1746.2011.06817.x
45. Lamb A, Chen L-F. Role of the Helicobacter Pylori-Induced Inflammatory Response in the Development of Gastric Cancer. J Cell Biochem (2013) 114(3):491–7. doi: 10.1002/jcb.24389
46. Yamaoka Y. Roles of Helicobacter Pylori BabA in Gastroduodenal Pathogenesis. World J Gastroenterol (2008) 14(27):4265–72. doi: 10.3748/wjg.14.4265
47. Semper RP, Mejías-Luque R, Groß C, Anderl F, Müller A, Vieth M, et al. Helicobacter Pylori–Induced IL-1β Secretion in Innate Immune Cells Is Regulated by the NLRP3 Inflammasome and Requires the Cag Pathogenicity Island. J Immunol (2014) 193(7):3566–76. doi: 10.4049/jimmunol.1400362
48. Mitchell P, Germain C, Fiori PL, Khamri W, Foster GR, Ghosh S, et al. Chronic Exposure to Helicobacter Pylori Impairs Dendritic Cell Function and Inhibits Th1 Development. Infect Immun (2007) 75(2):810–9. doi: 10.1128/iai.00228-06
49. Ramarao N, Meyer TF. Helicobacter Pylori Resists Phagocytosis by Macrophages: Quantitative Assessment by Confocal Microscopy and Fluorescence-Activated Cell Sorting. Infect Immun (2001) 69(4):2604–11. doi: 10.1128/IAI.69.4.2604-2611.2001
50. Ottemann KM, Lowenthal AC. Helicobacter Pylori Uses Motility for Initial Colonization and to Attain Robust Infection. Infect Immun (2002) 70(4):1984–90. doi: 10.1128/iai.70.4.1984-1990.2002
51. Tsuda M, Karita M, Morshed MG, Okita K, Nakazawa T. A Urease-Negative Mutant of Helicobacter Pylori Constructed by Allelic Exchange Mutagenesis Lacks the Ability to Colonize the Nude Mouse Stomach. Infect Immun (1994) 62(8):3586–9. doi: 10.1128/iai.62.8.3586-3589.1994
52. Fan X, Gunasena H, Cheng Z, Espejo R, Crowe SE, Ernst PB, et al. Helicobacter Pylori Urease Binds to Class II MHC on Gastric Epithelial Cells and Induces Their Apoptosis. J Immunol (2000) 165(4):1918–24. doi: 10.4049/jimmunol.165.4.1918
53. Lina TT, Alzahrani S, Gonzalez J, Pinchuk IV, Beswick EJ, Reyes VE. Immune Evasion Strategies Used by Helicobacter Pylori. World J Gastroenterol (2014) 20(36):12753–66. doi: 10.3748/wjg.v20.i36.12753
54. Junaid M, Linn AK, Javadi MB, Al-Gubare S, Ali N, Katzenmeier G. Vacuolating Cytotoxin A (VacA) - A Multi-Talented Pore-Forming Toxin From Helicobacter Pylori. Toxicon (2016) 118:27–35. doi: 10.1016/j.toxicon.2016.04.037
55. Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Blixt O, et al. Structural Basis for Distinct Ligand-Binding and Targeting Properties of the Receptors DC-SIGN and DC-SIGNR. Nat Struct Mol Biol (2004) 11(7):591–8. doi: 10.1038/nsmb784
56. D’Elios MM, Amedei A, Cappon A, Del Prete G, de Bernard M. The Neutrophil-Activating Protein of Helicobacter Pylori (HP-NAP) as an Immune Modulating Agent. FEMS Immunol Med Microbiol (2007) 50(2):157–64. doi: 10.1111/j.1574-695X.2007.00258.x
57. Amedei A, Cappon A, Codolo G, Cabrelle A, Polenghi A, Benagiano M, et al. The Neutrophil-Activating Protein of Helicobacter Pylori Promotes Th1 Immune Responses. J Clin Invest (2006) 116(4):1092–101. doi: 10.1172/jci27177
58. Benktander J, Barone A, Johansson MM, Teneberg S. Helicobacter Pylori SabA Binding Gangliosides of Human Stomach. Virulence (2018) 9(1):738–51. doi: 10.1080/21505594.2018.1440171
59. Blaser N, Backert S, Pachathundikandi SK. Immune Cell Signaling by Helicobacter Pylori: Impact on Gastric Pathology. Adv Exp Med Biol (2019) 1149:77–106. doi: 10.1007/5584_2019_360
60. Ren Z, Pang G, Clancy R, Li LC, Lee CS, Batey R, et al. Shift of the Gastric T-Cell Response in Gastric Carcinoma. J Gastroenterol Hepatol (2001) 16(2):142–8. doi: 10.1046/j.1440-1746.2001.02385.x
61. Wang S-K, Zhu H-F, He B-S, Zhang Z-Y, Chen Z-T, Wang Z-Z, et al. CagA+ H Pylori Infection is Associated With Polarization of T Helper Cell Immune Responses in Gastric Carcinogenesis. World J Gastroenterol (2007) 13(21):2923–31. doi: 10.3748/wjg.v13.i21.2923
62. Olivera-Severo D, Uberti AF, Marques MS, Pinto MT, Gomez-Lazaro M, Figueiredo C, et al. A New Role for Helicobacter Pylori Urease: Contributions to Angiogenesis. Front Microbiol (2017) 8:1883(1883). doi: 10.3389/fmicb.2017.01883
63. Sheu B-S, Yang H-B, Yeh Y-C, Wu J-J. Helicobacter Pylori Colonization of the Human Gastric Epithelium: A Bug’s First Step is a Novel Target for Us. J Gastroenterol Hepatol (2010) 25(1):26–32. doi: 10.1111/j.1440-1746.2009.06141.x
64. Sanders CJ, Yu Y, Moore DA 3rd, Williams IR, Gewirtz AT. Humoral Immune Response to Flagellin Requires T Cells and Activation of Innate Immunity. J Immunol (2006) 177(5):2810–8. doi: 10.4049/jimmunol.177.5.2810
65. Forstnerič V, Ivičak-Kocjan K, Plaper T, Jerala R, Benčina M. The Role of the C-Terminal D0 Domain of Flagellin in Activation of Toll Like Receptor 5. PloS Pathog (2017) 13(8):e1006574–e. doi: 10.1371/journal.ppat.1006574
66. Smith SM. Role of Toll-Like Receptors in Helicobacter Pylori Infection and Immunity. World J Gastrointest Pathophysiol (2014) 5(3):133–46. doi: 10.4291/wjgp.v5.i3.133
67. Tegtmeyer N, Neddermann M, Lind J, Pachathundikandi SK, Sharafutdinov I, Gutiérrez-Escobar AJ, et al. Toll-Like Receptor 5 Activation by the CagY Repeat Domains of Helicobacter Pylori. Cell Rep (2020) 32(11):108159. doi: 10.1016/j.celrep.2020.108159
68. Nilsson C, Skoglund A, Moran AP, Annuk H, Engstrand L, Normark S. Lipopolysaccharide Diversity Evolving in Helicobacter Pylori Communities Through Genetic Modifications in Fucosyltransferases. PloS One (2008) 3(11):e3811–e. doi: 10.1371/journal.pone.0003811
69. Solnick JV, Hansen LM, Salama NR, Boonjakuakul JK, Syvanen M. Modification of Helicobacter Pylori Outer Membrane Protein Expression During Experimental Infection of Rhesus Macaques. Proc Natl Acad Sci USA (2004) 101(7):2106–11. doi: 10.1073/pnas.0308573100
70. Sharndama HC, Mba IE. Helicobacter Pylori: An Up-to-Date Overview on the Virulence and Pathogenesis Mechanisms. Braz J Microbiol (2022) 53(1):33–50. doi: 10.1007/s42770-021-00675-0
71. Raghwan, Chowdhury R. Host Cell Contact Induces Fur-Dependent Expression of Virulence Factors CagA and VacA in Helicobacter Pylori. Helicobacter (2014) 19(1):17–25. doi: 10.1111/hel.12087
72. Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter Exploits Integrin for Type IV Secretion and Kinase Activation. Nature (2007) 449(7164):862–6. doi: 10.1038/nature06187
73. Prashar A, Capurro MI, Jones NL. Under the Radar: Strategies Used by Helicobacter Pylori to Evade Host Responses. Annu Rev Physiol (2022) 84(1):485–506. doi: 10.1146/annurev-physiol-061121-035930
74. Lamb A, Yang X-D, Tsang Y-HN, Li J-D, Higashi H, Hatakeyama M, et al. Helicobacter Pylori CagA Activates NF-kappaB by Targeting TAK1 for TRAF6-Mediated Lys 63 Ubiquitination. EMBO Rep (2009) 10(11):1242–9. doi: 10.1038/embor.2009.210
75. Hatakeyama M. Structure and Function of Helicobacter Pylori CagA, the First-Identified Bacterial Protein Involved in Human Cancer. Proc Jpn Acad Ser B Phys Biol Sci (2017) 93(4):196–219. doi: 10.2183/pjab.93.013
76. Baj J, Forma A, Sitarz M, Portincasa P, Garruti G, Krasowska D, et al. Helicobacter Pylori Virulence Factors-Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells (2020) 10(1):27. doi: 10.3390/cells10010027
77. Backert S, Naumann M. What a Disorder: Proinflammatory Signaling Pathways Induced by Helicobacter Pylori. Trends Microbiol (2010) 18(11):479–86. doi: 10.1016/j.tim.2010.08.003
78. Imai S, Ooki T, Murata-Kamiya N, Komura D, Tahmina K, Wu W, et al. Helicobacter Pylori CagA Elicits BRCAness to Induce Genome Instability That may Underlie Bacterial Gastric Carcinogenesis. Cell Host Microbe (2021) 29(6):941–58.e10. doi: 10.1016/j.chom.2021.04.006
79. Li N, Feng Y, Hu Y, He C, Xie C, Ouyang Y, et al. Helicobacter Pylori CagA Promotes Epithelial Mesenchymal Transition in Gastric Carcinogenesis via Triggering Oncogenic YAP Pathway. J Exp Clin Cancer Res (2018) 37(1):280. doi: 10.1186/s13046-018-0962-5
80. Cheok YY, Lee CYQ, Cheong HC, Vadivelu J, Looi CY, Abdullah S, et al. An Overview of Helicobacter Pylori Survival Tactics in the Hostile Human Stomach Environment. Microorganisms (2021) 9(12):2502. doi: 10.3390/microorganisms9122502
81. Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 Proinflammatory Outer Membrane Protein (Oipa) of Helicobacter Pylori. Proc Natl Acad Sci USA (2000) 97(13):7533–8. doi: 10.1073/pnas.130079797
82. Chmiela M, Walczak N, Rudnicka K. Helicobacter Pylori Outer Membrane Vesicles Involvement in the Infection Development and Helicobacter Pylori-Related Diseases. J BioMed Sci (2018) 25(1):78–. doi: 10.1186/s12929-018-0480-y
83. Zhang H, Zhang Y, Song Z, Li R, Ruan H, Liu Q, et al. sncRNAs Packaged by Helicobacter Pylori Outer Membrane Vesicles Attenuate IL-8 Secretion in Human Cells. Int J Med Microbiol (2020) 310(1):151356. doi: 10.1016/j.ijmm.2019.151356
84. Pachathundikandi SK, Backert S. Helicobacter Pylori Controls NLRP3 Expression by Regulating hsa-miR-223-3p and IL-10 in Cultured and Primary Human Immune Cells. Innate Immun (2017) 24(1):11–23. doi: 10.1177/1753425917738043
85. Pachathundikandi SK, Blaser N, Bruns H, Backert S. Helicobacter Pylori Avoids the Critical Activation of NLRP3 Inflammasome-Mediated Production of Oncogenic Mature IL-1β in Human Immune Cells. Cancers (Basel) (2020) 12(4):803. doi: 10.3390/cancers12040803
86. Altobelli A, Bauer M, Velez K, Cover TL, Müller A. Helicobacter Pylori VacA Targets Myeloid Cells in the Gastric Lamina Propria To Promote Peripherally Induced Regulatory T-Cell Differentiation and Persistent Infection. mBio (2019) 10(2):e00261–19, 1–13. doi: 10.1128/mBio.00261-19
87. Araújo GRL, Marques HS, Santos MLC, da Silva FAF, da Brito BB, Correa Santos GL, et al. Helicobacter Pylori Infection: How Does Age Influence the Inflammatory Pattern? World J Gastroenterol (2022) 28(4):402–11. doi: 10.3748/wjg.v28.i4.402
88. Freire de Melo F, Rocha AM, Rocha GA, Pedroso SH, de Assis Batista S, Fonseca de Castro LP, et al. A Regulatory Instead of an IL-17 T Response Predominates in Helicobacter Pylori-Associated Gastritis in Children. Microbes Infect (2012) 14(4):341–7. doi: 10.1016/j.micinf.2011.11.008
89. de Bernard M, D’Elios MM. The Immune Modulating Activity of the Helicobacter Pylori HP-NAP: Friend or Foe? Toxicon (2010) 56(7):1186–92. doi: 10.1016/j.toxicon.2009.09.020
90. Lindén S, Nordman H, Hedenbro J, Hurtig M, Borén T, Carlstedt I. Strain- and Blood Group-Dependent Binding of Helicobacter Pylori to Human Gastric MUC5AC Glycoforms. Gastroenterology (2002) 123(6):1923–30. doi: 10.1053/gast.2002.37076
91. Miszczyk E, Rudnicka K, Moran AP, Fol M, Kowalewicz-Kulbat M, Druszczyńska M, et al. Interaction of Helicobacter Pylori With C-Type Lectin Dendritic Cell-Specific ICAM Grabbing Nonintegrin. J BioMed Biotechnol (2012) 2012:206463. doi: 10.1155/2012/206463
92. Abadi ATB. Strategies Used by Helicobacter Pylori to Establish Persistent Infection. World J Gastroenterol (2017) 23(16):2870–82. doi: 10.3748/wjg.v23.i16.2870
93. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, Reuter S, et al. DC-Derived IL-18 Drives Treg Differentiation, Murine Helicobacter Pylori-Specific Immune Tolerance, and Asthma Protection. J Clin Invest (2012) 122(3):1082–96. doi: 10.1172/JCI61029
94. Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth H-P, et al. Helicobacter Pylori Modulates the T Helper Cell 1/T Helper Cell 2 Balance Through Phase-Variable Interaction Between Lipopolysaccharide and DC-SIGN. J Exp Med (2004) 200(8):979–90. doi: 10.1084/jem.20041061
95. Sarajlic M, Neuper T, Vetter J, Schaller S, Klicznik MM, Gratz IK, et al. H. Pylorimodulates DC Functions via T4SS/Tnfα/P38-Dependent SOCS3 Expression. Cell Communication Signaling (2020) 18(1):160. doi: 10.1186/s12964-020-00655-1
96. Cover TL, Vaughn SG, Cao P, Blaser MJ. Potentiation of Helicobacter Pylori Vacuolating Toxin Activity by Nicotine and Other Weak Bases. J Infect Dis (1992) 166(5):1073–8. doi: 10.1093/infdis/166.5.1073
97. Chevalier C, Thiberge JM, Ferrero RL, Labigne A. Essential Role of Helicobacter Pylori Gamma-Glutamyltranspeptidase for the Colonization of the Gastric Mucosa of Mice. Mol Microbiol (1999) 31(5):1359–72. doi: 10.1046/j.1365-2958.1999.01271.x
98. Yokoyama K, Higashi H, Ishikawa S, Fujii Y, Kondo S, Kato H, et al. Functional Antagonism Between Helicobacter Pylori CagA and Vacuolating Toxin VacA in Control of the NFAT Signaling Pathway in Gastric Epithelial Cells. Proc Natl Acad Sci USA (2005) 102(27):9661–6. doi: 10.1073/pnas.0502529102
99. Ling SSM, Khoo LHB, Hwang L-A, Yeoh KG, Ho B. Instrumental Role of Helicobacter Pylori γ-Glutamyl Transpeptidase in VacA-Dependent Vacuolation in Gastric Epithelial Cells. PloS One (2015) 10(6):e0131460. doi: 10.1371/journal.pone.0131460
100. Zhang L, Hu W, Cho CH, Chan FK, Yu J, Fitzgerald JR, et al. Reduced Lysosomal Clearance of Autophagosomes Promotes Survival and Colonization of Helicobacter Pylori. J Pathol (2018) 244(4):432–44. doi: 10.1002/path.5033
101. Lekmeechai S, Su Y-C, Brant M, Alvarado-Kristensson M, Vallström A, Obi I, et al. Helicobacter Pylori Outer Membrane Vesicles Protect the Pathogen From Reactive Oxygen Species of the Respiratory Burst. Front Microbiol (2018) 9:1837. doi: 10.3389/fmicb.2018.01837
102. Borlace GN, Butler RN, Brooks DA. Monocyte and Macrophage Killing of Helicobacter Pylori: Relationship to Bacterial Virulence Factors. Helicobacter (2008) 13(5):380–7. doi: 10.1111/j.1523-5378.2008.00625.x
103. Gobert AP, Latour YL, Asim M, Finley JL, Verriere TG, Barry DP, et al. Bacterial Pathogens Hijack the Innate Immune Response by Activation of the Reverse Transsulfuration Pathway. mBio (2019) 10(5): e02174–19, 1–18. doi: 10.1128/mBio.02174-19
104. Latour YL, Gobert AP, Wilson KT. The Role of Polyamines in the Regulation of Macrophage Polarization and Function. Amino Acids (2020) 52(2):151–60. doi: 10.1007/s00726-019-02719-0
105. Darwin PE, Sztein MB, Zheng QX, James SP, Fantry GT. Immune Evasion by Helicobacter Pylori: Gastric Spiral Bacteria Lack Surface Immunoglobulin Deposition and Reactivity With Homologous Antibodies. Helicobacter (1996) 1(1):20–7. doi: 10.1111/j.1523-5378.1996.tb00004.x
106. Manojlovic N, Nikolic L, Pilcevic D, Josifovski J, Babic D. Systemic Humoral Anti-Helicobacter Pylori Immune Response in Patients With Gastric Malignancies and Benign Gastroduodenal Disease. Hepatogastroenterology (2004) 51(55):282–4.
107. Yamaji Y, Mitsushima T, Ikuma H, Okamoto M, Yoshida H, Kawabe T, et al. Weak Response of Helicobacter Pylori Antibody is High Risk for Gastric Cancer: A Cross-Sectional Study of 10,234 Endoscoped Japanese. Scand J Gastroenterol (2002) 37(2):148–53. doi: 10.1080/003655202753416795
108. Toyoshima O, Nishizawa T, Sakitani K, Yamakawa T, Takahashi Y, Yamamichi N, et al. Serum Anti-Helicobacter Pylori Antibody Titer and Its Association With Gastric Nodularity, Atrophy, and Age: A Cross-Sectional Study. World J Gastroenterol (2018) 24(35):4061–8. doi: 10.3748/wjg.v24.i35.4061
109. Kishikawa H, Kimura K, Takarabe S, Kaida S, Nishida J. Helicobacter Pylori Antibody Titer and Gastric Cancer Screening. Dis Markers (2015) 2015:156719–. doi: 10.1155/2015/156719
110. Nielsen H, Birkholz S, Andersen LP, Moran AP. Neutrophil Activation by Helicobacter Pylori Lipopolysaccharides. J Infect Dis (1994) 170(1):135–9. doi: 10.1093/infdis/170.1.135
111. Li H, Liao T, Debowski AW, Tang H, Nilsson H-O, Stubbs KA, et al. Lipopolysaccharide Structure and Biosynthesis in Helicobacter Pylori. Helicobacter (2016) 21(6):445–61. doi: 10.1111/hel.12301
112. Sherburne R, Taylor DE. Helicobacter Pylori Expresses a Complex Surface Carbohydrate, Lewis X. Infect Immun (1995) 63(12):4564–8. doi: 10.1128/iai.63.12.4564-4568.1995
113. Xue J, Li L, Li F, Li N, Li T, Li C. Expression of Lewis (B) Blood Group Antigen Interferes With Oral Dienogest Therapy Among Women With Adenomyosis. J Reprod Immunol (2020) 137:103079. doi: 10.1016/j.jri.2019.103079
114. de Mattos LC. Structural Diversity and Biological Importance of ABO, H, Lewis and Secretor Histo-Blood Group Carbohydrates. Rev Bras Hematol Hemoter (2016) 38(4):331–40. doi: 10.1016/j.bjhh.2016.07.005
115. Li H, Tang H, Debowski AW, Stubbs KA, Marshall BJ, Benghezal M. Lipopolysaccharide Structural Differences Between Western and Asian Helicobacter Pylori Strains. Toxins (Basel) (2018) 10(9):364. doi: 10.3390/toxins10090364
116. Moran A. The Role of Lipopolysaccharide in Helicobacter Pylori Pathogenesis. Alimentary Pharmacol Ther (1996) 10(Sup1):39–50. doi: 10.1046/j.1365-2036.1996.22164004.x
117. Nilsson C, Skoglund A, Moran AP, Annuk H, Engstrand L, Normark S. An Enzymatic Ruler Modulates Lewis Antigen Glycosylation of Helicobacter Pylori LPS During Persistent Infection. Proc Natl Acad Sci USA (2006) 103(8):2863–8. doi: 10.1073/pnas.0511119103
118. Muotiala A, Helander IM, Pyhälä L, Kosunen TU, Moran AP. Low Biological Activity of Helicobacter Pylori Lipopolysaccharide. Infect Immun (1992) 60(4):1714–6. doi: 10.1128/iai.60.4.1714-1716.1992
119. Li H, Marceau M, Yang T, Liao T, Tang X, Hu R, et al. East-Asian Helicobacter Pylori Strains Synthesize Heptan-Deficient Lipopolysaccharide. PloS Genet (2019) 15(11):e1008497–e. doi: 10.1371/journal.pgen.1008497
120. Pece S, Fumarola D, Giuliani G, Jirillo E, Moran AP. Activity in the Limulus Amebocyte Lysate Assay and Induction of Tumor Necrosis Factor-α by Diverse Helicobacter Pylori Lipopolysaccharide Preparations. J Endotoxin Res (1995) 2(6):455–62. doi: 10.1177/096805199600200609
121. Moran AP. Lipopolysaccharide in Bacterial Chronic Infection: Insights From Helicobacter Pylori Lipopolysaccharide and Lipid a. Int J Med Microbiol (2007) 297(5):307–19. doi: 10.1016/j.ijmm.2007.03.008
122. Shimoyama A, Saeki A, Tanimura N, Tsutsui H, Miyake K, Suda Y, et al. Chemical Synthesis of Helicobacter Pylori Lipopolysaccharide Partial Structures and Their Selective Proinflammatory Responses. Chem A Eur J (2011) 17(51):14464–74. doi: 10.1002/chem.201003581
123. Fujimoto Y, Shimoyama A, Suda Y, Fukase K. Synthesis and Immunomodulatory Activities of Helicobacter Pylori Lipophilic Terminus of Lipopolysaccharide Including Lipid a. Carbohydr Res (2012) 356:37–43. doi: 10.1016/j.carres.2012.03.013
124. Obonyo M, Sabet M, Cole SP, Ebmeyer J, Uematsu S, Akira S, et al. Deficiencies of Myeloid Differentiation Factor 88, Toll-Like Receptor 2 (TLR2), or TLR4 Produce Specific Defects in Macrophage Cytokine Secretion Induced by Helicobacter Pylori. Infect Immun (2007) 75(5):2408–14. doi: 10.1128/IAI.01794-06
125. Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. Helicobacter Pylori Versus the Host: Remodeling of the Bacterial Outer Membrane Is Required for Survival in the Gastric Mucosa. PloS Pathog (2011) 7(12):e1002454–e. doi: 10.1371/journal.ppat.1002454
126. Liu A-N, Teng K-W, Chew Y, Wang P-C, Nguyen TTH, Kao M-C. The Effects of HP0044 and HP1275 Knockout Mutations on the Structure and Function of Lipopolysaccharide in Helicobacter Pylori Strain 26695. Biomedicines (2022) 10(1):145. doi: 10.3390/biomedicines10010145
127. Moran AP, Knirel YA, Senchenkova SN, Widmalm G, Hynes SO, Jansson PE. Phenotypic Variation in Molecular Mimicry Between Helicobacter Pylori Lipopolysaccharides and Human Gastric Epithelial Cell Surface Glycoforms. Acid-Induced Phase Variation in Lewis(x) and Lewis(y) Expression by H. Pylori Lipopolysaccharides. J Biol Chem (2002) 277(8):5785–95. doi: 10.1074/jbc.M108574200
128. Pernitzsch SR, Alzheimer M, Bremer BU, Robbe-Saule M, De Reuse H, Sharma CM. Small RNA Mediated Gradual Control of Lipopolysaccharide Biosynthesis Affects Antibiotic Resistance in Helicobacter Pylori. Nat Commun (2021) 12(1):4433–. doi: 10.1038/s41467-021-24689-2
129. Monteiro MA, Appelmelk BJ, Rasko DA, Moran AP, Hynes SO, MacLean LL, et al. Lipopolysaccharide Structures of Helicobacter Pylori Genomic Strains 26695 and J99, Mouse Model H. Pylori Sydney Strain, H. Pylori P466 Carrying Sialyl Lewis X, and H. Pylori UA915 Expressing Lewis B Classification of H. Pylori Lipopolysaccharides Into Glycotype Families. Eur J Biochem (2000) 267(2):305–20. doi: 10.1046/j.1432-1327.2000.01007.x
130. Lozniewski A, Haristoy X, Rasko DA, Hatier R, Plénat F, Taylor DE, et al. Influence of Lewis Antigen Expression by Helicobacter Pylori on Bacterial Internalization by Gastric Epithelial Cells. Infect Immun (2003) 71(5):2902–6. doi: 10.1128/iai.71.5.2902-2906.2003
131. Gonciarz W, Walencka M, Moran AP, Hinc K, Obuchowski M, Chmiela M. Upregulation of MUC5AC Production and Deposition of LEWIS Determinants by HELICOBACTER PYLORI Facilitate Gastric Tissue Colonization and the Maintenance of Infection. J Biomed Sci (2019) 26(1):23. doi: 10.1186/s12929-019-0515-z
132. Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter Pylori to Human Gastric Epithelium Mediated by Blood Group Antigens. Science (1993) 262(5141):1892. doi: 10.1126/science.8018146
133. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter Pylori Adhesin Binding Fucosylated Histo-Blood Group Antigens Revealed by Retagging. Science (1998) 279(5349):373–7. doi: 10.1126/science.279.5349.373
134. Moonens K, Gideonsson P, Subedi S, Bugaytsova J, Romaõ E, Mendez M, et al. Structural Insights Into Polymorphic ABO Glycan Binding by Helicobacter Pylori. Cell Host Microbe (2016) 19(1):55–66. doi: 10.1016/j.chom.2015.12.004
135. Matteo MJ, Armitano RI, Romeo M, Wonaga A, Olmos M, Catalano M. Helicobacter Pylori Bab Genes During Chronic Colonization. Int J Mol Epidemiol Genet (2011) 2(3):286–91.
136. Bäckström A, Lundberg C, Kersulyte D, Berg DE, Borén T, Arnqvist A. Metastability of Helicobacter Pylori Bab Adhesin Genes and Dynamics in Lewis B Antigen Binding. Proc Natl Acad Sci USA (2004) 101(48):16923–8. doi: 10.1073/pnas.0404817101
137. Ansari S, Yamaoka Y. Helicobacter Pylori BabA in Adaptation for Gastric Colonization. World J Gastroenterol (2017) 23(23):4158–69. doi: 10.3748/wjg.v23.i23.4158
138. Styer CM, Hansen LM, Cooke CL, Gundersen AM, Choi SS, Berg DE, et al. Expression of the BabA Adhesin During Experimental Infection With Helicobacter Pylori. Infect Immun (2010) 78(4):1593–600. doi: 10.1128/IAI.01297-09
139. Ishijima N, Suzuki M, Ashida H, Ichikawa Y, Kanegae Y, Saito I, et al. BabA-Mediated Adherence Is a Potentiator of the Helicobacter Pylori Type IV Secretion System Activity. J Biol Chem (2011) 286(28):25256–64. doi: 10.1074/jbc.M111.233601
140. Mahdavi J, Sondén B, Hurtig M, Olfat FO, Forsberg L, Roche N, et al. Helicobacter Pylori SabA Adhesin in Persistent Infection and Chronic Inflammation. Science (2002) 297(5581):573–8. doi: 10.1126/science.1069076
141. Goodwin AC, Weinberger DM, Ford CB, Nelson JC, Snider JD, Hall JD, et al. Expression of the Helicobacter Pylori Adhesin SabA Is Controlled via Phase Variation and the ArsRS Signal Transduction System. Microbiology (2008) 154(Pt 8):2231–40. doi: 10.1099/mic.0.2007/016055-0
142. Talarico S, Whitefield SE, Fero J, Haas R, Salama NR. Regulation of Helicobacter Pylori Adherence by Gene Conversion. Mol Microbiol (2012) 84(6):1050–61. doi: 10.1111/j.1365-2958.2012.08073.x
143. Xu C, Soyfoo DM, Wu Y, Xu S. Virulence of Helicobacter Pylori Outer Membrane Proteins: An Updated Review. Eur J Clin Microbiol Infect Dis (2020) 39(10):1821–30. doi: 10.1007/s10096-020-03948-y
144. Horridge DN, Begley AA, Kim J, Aravindan N, Fan K, Forsyth MH. Outer Inflammatory Protein a (OipA) of Helicobacter Pylori is Regulated by Host Cell Contact and Mediates CagA Translocation and Interleukin-8 Response Only in the Presence of a Functional Cag Pathogenicity Island Type IV Secretion System. Pathog Dis (2017) 75(8):ftx113. doi: 10.1093/femspd/ftx113
145. Farzi N, Yadegar A, Aghdaei HA, Yamaoka Y, Zali MR. Genetic Diversity and Functional Analysis of oipA Gene in Association With Other Virulence Factors Among Helicobacter Pylori Isolates From Iranian Patients With Different Gastric Diseases. Infect Genet Evol (2018) 60:26–34. doi: 10.1016/j.meegid.2018.02.017
146. Al-Maleki AR, Loke MF, Lui SY, Ramli NSK, Khosravi Y, Ng CG, et al. Helicobacter Pylori Outer Inflammatory Protein A (OipA) Suppresses Apoptosis of AGS Gastric Cells. Vitro Cell Microbiol (2017) 19(12):e12771. doi: 10.1111/cmi.12771
Keywords: H. pylori, innate immunity, lipopolysaccharide, dendritic cells, Lewis system antigens, molecular mimicry, adhesion, inflammation
Citation: Sijmons D, Guy AJ, Walduck AK and Ramsland PA (2022) Helicobacter pylori and the Role of Lipopolysaccharide Variation in Innate Immune Evasion. Front. Immunol. 13:868225. doi: 10.3389/fimmu.2022.868225
Received: 02 February 2022; Accepted: 04 April 2022;
Published: 13 May 2022.
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Peter Kraiczy, Goethe University Frankfurt, GermanyMarco De Zuani, Wellcome Sanger Institute (WT), United Kingdom
Copyright © 2022 Sijmons, Guy, Walduck and Ramsland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul A. Ramsland, cGF1bC5yYW1zbGFuZEBybWl0LmVkdS5hdQ==