 Walter J. Lukiw
Walter J. Lukiw
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Microbiol. , 26 September 2016
Sec. Microbial Symbioses
Volume 7 - 2016 | https://doi.org/10.3389/fmicb.2016.01544
This article is part of the Research Topic Microbiome interplay and control View all 37 articles
The human microbiome consists of ~3.8 × 1013 symbiotic microorganisms that form a highly complex and dynamic ecosystem: the gastrointestinal (GI) tract constitutes the largest repository of the human microbiome by far, and its impact on human neurological health and disease is becoming increasingly appreciated. Bacteroidetes, the largest phylum of Gram-negative bacteria in the GI tract microbiome, while generally beneficial to the host when confined to the GI tract, have potential to secrete a remarkably complex array of pro-inflammatory neurotoxins that include surface lipopolysaccharides (LPSs) and toxic proteolytic peptides. The deleterious effects of these bacterial exudates appear to become more important as GI tract and blood-brain barriers alter or increase their permeability with aging and disease. For example, presence of the unique LPSs of the abundant Bacteroidetes species Bacteroides fragilis (BF-LPS) in the serum represents a major contributing factor to systemic inflammation. BF-LPS is further recognized by TLR2, TLR4, and/or CD14 microglial cell receptors as are the pro-inflammatory 42 amino acid amyloid-beta (Aβ42) peptides that characterize Alzheimer’s disease (AD) brain. Here we provide the first evidence that BF-LPS exposure to human primary brain cells is an exceptionally potent inducer of the pro-inflammatory transcription factor NF-kB (p50/p65) complex, a known trigger in the expression of pathogenic pathways involved in inflammatory neurodegeneration. This ‘Perspectives communication’ will in addition highlight work from recent studies that advance novel and emerging concepts on the potential contribution of microbiome-generated factors, such as BF-LPS, in driving pro-inflammatory degenerative neuropathology in the AD brain.
Homo sapiens and their complex microbiome, consisting chiefly of bacteria with microbial eukaryotes, archaea, fungi, protozoa, viruses, and other microorganisms making up the balance, together compromise the entire metaorganism whose symbiotic associations and host interactions are critical to human health and disease (Hugon et al., 2015; Seksik and Landman, 2015; Youssef et al., 2015). There are approximately 52 recognized divisions of bacteria, however, humans, have co-evolved with – 2 dominant phyla: Bacteroidetes [about 20% of all gastrointestinal (GI) tract bacteria] and Firmicutes (about 80%); with Actinobacteria (about 3%), Proteobacteria (~1%), and Verrucomicrobia (~0.1%) making up significantly smaller fractions. These four major bacterial divisions represent the ‘bacterial-core’ of the human microbiome (Hakansson and Molin, 2011; Zhao et al., 2015; Hug et al., 2016; Sender et al., 2016). About 98% of GI tract microbiota consists of anaerobic bacteria, and Bacteroidetes species, constituting ~30% of all GI tract bacteria are the most abundant Gram-negative bacteria found in the GI tract outnumbering Escherichia coli abundance by at least 100 to 1 (Fathi and Wu, 2016; Foster et al., 2016; Hug et al., 2016; Rogers and Aronoff, 2016; Sampson and Mazmanian, 2016; Sender et al., 2016).
Bacteroidetes species such as Bacteroides fragilis, normal commensals of the GI tract, are thought to be generally beneficial to human health via their production of polysaccharides, volatile fatty acids, cleavage of dietary fibers into digestible short-chain fatty acids, and other nutrients, however, when they escape this environment they can cause substantial inflammatory pathology with significant morbidity and mortality (Hofer, 2014; Khanna and Tosh, 2014; Fathi and Wu, 2016). Dietary intake may have a role in regulating the composition and stoichiometry of the GI tract microbiome; for example Bacteroidetes species have been observed to proliferate in porcine models fed high-fat diets deprived of sufficient dietary fiber (Heinritz et al., 2016). In addition to their lipopolysaccharide (LPS) generation, B. fragilis endotoxins are a leading cause of anaerobic bacteremia and sepsis/systemic inflammatory distress through their generation of the highly pro-inflammatory zinc metalloprotease metalloproteinase B. fragilis toxin (BFT) fragilysin (Choi et al., 2016; Fathi and Wu, 2016). BFT has recently been shown to disrupt epithelial cells of GI tract barriers via cleavage of the synaptic adhesion zonula adherens protein E-cadherin (Seong et al., 2015; Choi et al., 2016; Zhan and Davies, 2016). It is currently not well understood if GI tract barrier-disrupting proteolytic endotoxins such as BFT are able to propagate pathogenic actions via the systemic circulation to further disrupt the blood-brain barrier and transfer LPS, BFT, and other endotoxins into the cerebrovascular circulation to the neural cells and synaptic circuitry of the CNS. B. fragilis has been shown to play a pathological role in neurodevelopment including autism spectrum disorder (ASD) via circulating metabolites (Hofer, 2014). It has also recently been reported that along with BFTs amyloid peptide-dependent changes in synaptic adhesion affect both the function and integrity of synapses, suggesting that the observed deficits in synaptic adhesion in Alzheimer’s disease (AD) play key roles in the progressive disruption of functional signaling throughout neuronal networks (Lin et al., 2014; Seong et al., 2015; Leshchyns’ka and Sytnyk, 2016).
Multiple and highly interactive aspects of increased inflammatory signaling is a consistent and recurrent feature of AD and the major pathological lesions that define AD, including insoluble Aβ42-enriched peptide deposits, neurofibrillary tangles, apoptotic, damaged, and dying neurons, and activated microglia are potent neuropathological stimulants that maintain the brain in a chronic and self-reinforcing inflammatory state (Hill and Lukiw, 2015; Calsolaro and Edison, 2016; Minter et al., 2016; Richards et al., 2016; Varatharaj and Galea, 2016). These progressive and ultimately fatal pro-inflammatory and neurodegenerative processes appear to be further stimulated by aberrant or excessive deregulation of the innate-immune response, and an increasing focus has been placed on pathological contributions by the human microbiome including dietary effects on microbial composition that appear to support pathological functions (Hill and Lukiw, 2015; Zhao et al., 2015; Foster et al., 2016). There is a wealth of accumulating evidence (a) that specific types of microbial LPS and endotoxins (such as BF-LPS and BFT) from enterotoxigenic microbes specifically impact microglial-mediated innate-immune responses, phagocytic, and detoxifying mechanisms and amyloidogenesis characteristic of inflammatory neurodegeneration (Bhattacharjee and Lukiw, 2013; Hill et al., 2014; Clark and Vissel, 2015; Hill and Lukiw, 2015; Lim et al., 2015); and (b) a resurgence of our interest in the importance of GI tract and blood-brain barrier systems that normally exclude these microbiome-sourced toxins from the systemic circulation and CNS but become leaky with age and disease (Montagne et al., 2015; Choi et al., 2016; Minter et al., 2016; Richards et al., 2016; Soenen et al., 2016; van de Haar et al., 2016; Varatharaj and Galea, 2016; Zhan and Davies, 2016; Zhao et al., 2016).
Interestingly, while secreted LPS, proteolytic endotoxins, and amyloid monomers are generally soluble as monomers over time they form into highly insoluble fibrous protein aggregates that are implicated in the progressive degenerative neuropathology of several common, age-related disorders of the human systemic circulation and CNS including systemic inflammation response syndrome, multiple sclerosis (MS), prion disease, and AD (Asti and Gioglio, 2014; Clark and Vissel, 2015; Devier et al., 2015; Richards et al., 2016; Zhao et al., 2016). At the genetic level, virtually all of this inducible inflammatory signaling within the CNS involves NF-kB activation and NF-kB-recognition and binding to target NF-kB DNA sequences as a prelude to the up-regulation of pro-inflammatory gene expression pathways, including the up-regulation of discrete families of pro-inflammatory pathogenic microRNAs that selectively down-regulate their mRNA targets (Lukiw and Bazan, 1998; Lukiw et al., 2008; Devier et al., 2015; Zhao et al., 2016).
Lipopolysaccharides are characteristic components of the outer leaflet of the outer membrane of Gram-negative bacteria shed into the extracellular space that play key roles in host–pathogen interactions of the innate-immune system (Hill and Lukiw, 2015; Zhao et al., 2015; Jiang et al., 2016; Maldonado et al., 2016). While LPSs contain large and hypervariable polysaccharide/oligosaccharide regions, the relatively conserved lipid region (lipid A) is the endotoxic and biologically active moiety that is responsible for septic shock (Jiang et al., 2016; Maldonado et al., 2016). A “canonical” LPS structure is represented by that of LPS from E. coli, that contains one of the most potent neurotoxic lipid A species known, consisting of a 1,4′-biphosphorylated glucosamine disaccharide bearing six fatty acids which are unbranched chains 12–14 methyl(ene) units in length. Other ‘lipid A’ species show variability in the number, length, and composition of the attached fatty acids, as well as variability in the degree of phosphorylation and number and types of substituted phosphate ligands. For instance, BF-LPS lipid A is penta-acylated and mono-phosphorylated and contains branched fatty acids 15–17 methyl(ene) units in length; deviations from the canonical lipid A structure are known to have a profound impact on the host innate-immune responses. LPS activates Toll-like receptors (TLRs), membrane-spanning protein receptors expressed in microglial cells of the innate immune system which recognize common damage- or pathogen-associated molecular-patterns (DAMPS, PAMPs; Gustot et al., 2013; Land, 2015; Maldonado et al., 2016; Minter et al., 2016; Richards et al., 2016; Varatharaj and Galea, 2016). TLRs play key roles in host protection from microbial invasion via the activation of the innate-immune system by sensing structurally conserved DAMPS or PAMPs from microbes or microbial exudates that are distinguishable from, and not innate to, the host organism (Land, 2015; Yu and Ye, 2015). Interestingly, of the 13 currently characterized TLRs the microglial TLR2 and TLR4 are activated by amyloid, LPS, lipoglycans and/or other microbial triggers that subsequently induce cytokine production, inflammation, phagocytosis and innate immune defense responses that directly impact the development of CNS pathology. More specifically, the TLR2 complex can recognize biofilm-associated LPS and amyloids produced by both Bacteroidetes and Firmicutes (Nishimori et al., 2012; Bhattacharjee and Lukiw, 2013; Asti and Gioglio, 2014; Hill and Lukiw, 2015). Interestingly, Aβ42 peptides overproduced in AD that are associated with microglia-mediated inflammatory responses have very recently been shown to activate TLR2 and/or TLR4 (Hill and Lukiw, 2015; Yu and Ye, 2015; Minter et al., 2016). Of further interest is: (a) that microbial amyloids induce pro-inflammatory interleukin IL-17A and IL-22, triggers for NF-kB activation/signaling and cyclooxygenase 2 activation via direct TLR2 activation (Nishimori et al., 2012); and (b) that increased levels of both IL-17A and IL-22 are associated with age-related inflammatory neurodegenerative diseases such as AD (Zhang et al., 2013; Calsolaro and Edison, 2016; Maldonado et al., 2016; Richards et al., 2016). Bacterial endotoxin- and LPS-induced neuro-inflammation has been known for some time to be important in driving the generation of Aβ42 (Lee et al., 2008; Asti and Gioglio, 2014; Hill and Lukiw, 2015; Zhao et al., 2015). In addition to TLR2 and TLR4 at least one additional microglial transmembrane LPS receptor CD14 mediates phagocytosis of both bacterial components and Aβ42 peptides, hence expanding roles for microglia and microglial LPS receptors in the pathophysiology of AD (Lee et al., 2008; Halmer et al., 2015; Jiang et al., 2016). Of further interest is that gram negative bacterial exudates such as BF-LPS are hypervariable in composition, and different Bacteroidetes species appear to generate unique temporal patterns of LPS that exhibit rapid adaptive changes – these include the modulation of LPS synthesis and structure and alterations in DAMP/PAMP recognition features as strategies for host immune system evasion (Land, 2015; Maldonado et al., 2016; Richards et al., 2016).
We hope that this ‘Perspectives’ article has adequately highlighted some recent findings on microbial-derived LPS and proteolytic endotoxins and has engendered interest in the potential contribution of these neurotoxic and pro-inflammatory microbial exudates to amyloidogenesis and age-related inflammatory neurodegeneration. Taken together, these current observations advance seven key areas in our understanding of the role of the microbiome in progressive, age-related inflammatory neurodegeneration: (a) that relatively low, nanomolar amounts of bacterial LPS are extremely potent inducers of the pro-inflammatory transcription factor NF-kB (p50/p65 complex) in human primary human neuronal-glial (HNG) co-cultures of brain cells (Figure 1); (b) that different LPS preparations from different bacterial species appear to exhibit slightly different trends in the induction of an inflammatory response, as quantified by the extent (mean values) of NF-kB activation and DNA-binding (Figure 1); (c) that the proliferation of microbial species such as the BF-LPS and BFT generating Bacteroidetes may be regulated by diet, environment, and lifestyle factors such as dietary fiber intake can impact neurological health, CNS inflammation, and degenerative disease (Hofer, 2014; Khanna and Tosh, 2014; Zhan and Davies, 2016); (d) that LPS transit across compromised GI tract and blood-brain barriers underscore the critical roles of cellular adhesion structures in allowing passage of noxious molecules from the GI tract into the systemic circulation and CNS (Montagne et al., 2015; Soenen et al., 2016; van de Haar et al., 2016); (e) that biophysical, gastrointestinal, and neurobiological barriers which may become more ‘leaky’ with aging again underscore the important role of tight junctions in moderating systemic and CNS inflammation and immune-mediated inflammatory disease (Hill and Lukiw, 2015; Varatharaj and Galea, 2016); (f) that DAMPS and/or PAMPs common to both LPS, endotoxins, and/or amyloid may trigger TRL2, TRL4 and/or CD14 microglial receptors to propagate and sustain inducible AD-relevant inflammatory responses within the CNS (Land, 2015; Zhao and Lukiw, 2015; Varatharaj and Galea, 2016); and (g) that LPS abundance, speciation and complexity in the CSF and/or blood serum may be useful diagnostically for the onset of mild cognitive impairment (MCI), the clinical precursor for the development of AD. Clearly, more research into the intriguing realm of human microbiome-host interaction is warranted, and the study of the complex interactions between each biological niche is certain to shed new light on the still evolving concepts and mechanisms of microbiome interplay and control in human neurological health and disease.
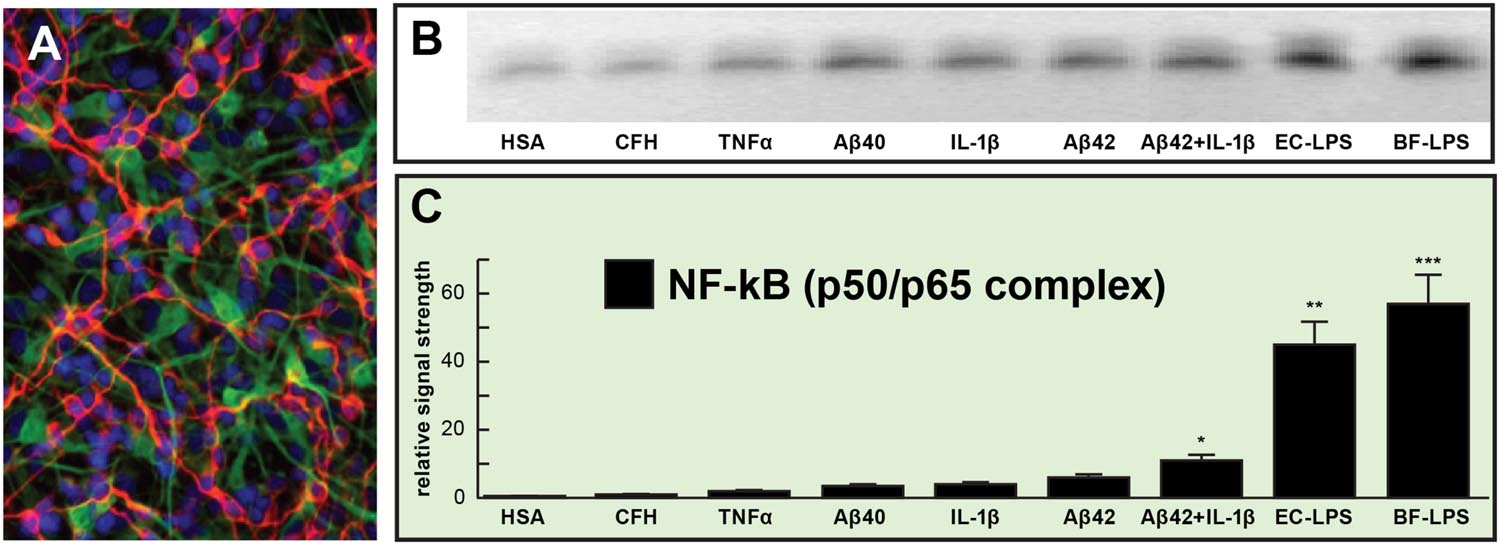
FIGURE 1. Relative induction of NF-kB (p50/p65)-DNA binding in pro-inflammatory factor and lipopolysaccharides (LPS)-treated primary human neuronal-glial (HNG) co-cultures. (A) HNG cells in primary co-culture for 1.5 weeks; HNG cells were stained with a neuron-specific β-tubulin III (red fluorescence λ max~650 nm; anti-βTUBIII antibody, Sigma-Aldrich, St Louis, MO, USA); an antibody against glial fibrillary acidic protein (GFAP; green fluorescence; λ max ~510 nm; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and DAPI nuclear stain (blue fluorescence; λ max~470 nm; Thermo Fisher Scientific, Waltham, MA, USA); 20×; (B) induction of the pro-inflammatory transcription factor NF-kB (p50/p65 activation complex) by various physiologically relevant, pro-inflammatory factors all at equal dosage (25 nM); NF-kB abundance was measured by NF-kB-DNA binding assay (a measure of NF-kB activation and binding to NF-kB-DNA recognition sequences) onto a 36 nucleotide end-labeled double stranded DNA fragment containing the canonical human NF-kB (p50/p65) recognition sequence 5′-GGGGACTTTCCC-3′ as previously described (Lukiw and Bazan, 1998; Lukiw et al., 2008; Devier et al., 2015; Clement et al., 2016); a scrambled control nucleotide containing no such NF-kB recognition sequence showed NF-kB-DNA binding activity (data not shown); (C) data from gel bands in panel (B) quantified in bar graph format; note robust induction of the NF-kB (p50/p65 complex) by Escherichia coli lipopolysaccharide (EC-LPS; LPS from Escherichia coli 0111:B4; Sigma L3012, St Louis, MO, USA) or B. fragilis lipopolysaccharide (BF-LPS; prepared by methods previously published; Eidhin and Mouton, 1993) that was ~45- to ~55-fold higher than that of the control human serum albumen (HSA) protein, and was fivefold to sevenfold higher than the combination of the pro-inflammatory Aβ42 peptide and IL-1β together (at 25 nM each); complex mixtures of microbiome bacterial LPS on NF-kB induction might be expected to be additive or synergistic; HSA = (control) human serum albumen; CFH = complement factor H; TNFα = tumor necrosis factor alpha (cachectin); IL-1β = interleukin 1-beta; Aβ40, Aβ42 = amyloid beta peptide, 40 and 42 amino acids in length; EC-LPS, BF-LPS = E. coli, Bacteroides fragilis lipopolysaccharide; error bars represent one standard error of the mean; N = 4; *p < 0.05; **p < 0.01; ***p < 0.001, ANOVA.
WL and the late Dr. James Hill performed experiments, researched and wrote this paper; the author is sincerely grateful to colleagues and collaborators for helpful discussions, medical artwork and unpublished data; the contributions of these researchers have been recognized in the ‘Acknowledgements’ section.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research work was presented in part at the Society for Neuroscience (SFN) Annual Meeting 17–21 October 2015, Chicago IL, USA and at the Alzheimer Association International Congress 2016 (AAIC 2016) Annual conference 21–27 July 2016 in Toronto CANADA. These studies utilized total nucleic acid and/or cytoplasmic fractions extracted from primary human neuronal-glial (HNG) co-cultures; sincere thanks are extended to Drs. P. Alexandrov, J. G. Cui, F. Culicchia, W. Poon, and Y. Zhao for short post-mortem interval (PMI) human brain tissues or extracts, HNG tissue culture and NF-kB-DNA binding assay and initial data interpretation, and to D. Guillot and A. I. Pogue for expert technical assistance and medical artwork. Thanks are also extended to the many neuropathologists, physicians, and researchers of the US, Canada, and Europe who have provided high quality, short PMI human CNS, or extracted tissue fractions for scientific study. Research on the human microbiome, pro-inflammatory, and pathogenic signaling in the Lukiw laboratory involving the innate-immune response, neuroinflammation and amyloidogenesis in AD and in other neurological diseases was supported through an unrestricted grant to the LSU Eye Center from Research to Prevent Blindness (RPB); the Louisiana Biotechnology Research Network (LBRN) and NIH grants NEI EY006311, NIA AG18031, and NIA AG038834.
Asti, A., and Gioglio, L. (2014). Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J. Alzheimers Dis. 39, 169–179. doi: 10.3233/JAD-131394
Bhattacharjee, S., and Lukiw, W. J. (2013). Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. 7:153. doi: 10.3389/fncel.2013.00153
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Choi, V. M., Herrou, J., Hecht, A. L., Teoh, W. P., Turner, J. R., Crosson, S., et al. (2016). Activation of Bacteroides fragilis toxin by a novel bacterial protease contributes to anaerobic sepsis in mice. Nat. Med. 22, 563–567. doi: 10.1038/nm.4077
Clark, I. A., and Vissel, B. (2015). Amyloid β: one of three danger-associated molecules that are secondary inducers of the proinflammatory cytokines that mediate Alzheimer’s disease. Br. J. Pharmacol. 172, 3714–3727. doi: 10.1111/bph.13181
Clement, C., Hill, J. M., Dua, P., Culicchia, F., and Lukiw, W. J. (2016). Analysis of RNA from Alzheimer’s disease post-mortem brain tissues. Mol. Neurobiol. 53, 1322–1328. doi: 10.1007/s12035-015-9105-6
Devier, D. J., Lovera, J. F., and Lukiw, W. J. (2015). Increase in NF-κB-sensitive miRNA-146a and miRNA-155 in multiple sclerosis (MS) and pro-inflammatory neurodegeneration. Front. Mol. Neurosci. 8:5. doi: 10.3389/fnmol.2015.00005
Eidhin, D. N., and Mouton, C. (1993). A rapid method for preparation of rough and smooth lipopolysaccharide from Bacteroides, Porphyromonas and Prevotella. FEMS Microbiol. Lett. 110, 133–138. doi: 10.1111/j.1574-6968.1993.tb06309.x
Fathi, P., and Wu, S. (2016). Isolation, detection, and characterization of enterotoxigenic Bacteroides fragilis in clinical samples. Open Microbiol. J. 10, 57–63. doi: 10.2174/1874285801610010057
Foster, J. A., Lyte, M., Meyer, E., and Cryan, J. F. (2016). Gut microbiota and brain function: an evolving field in neuroscience. Int. J. Neuropsychopharmacol. 19, yv114. doi: 10.1093/ijnp/pyv114
Gustot, A., Raussens, V., Dehousse, M., Dumoulin, M., Bryant, C. E., Ruysschaert, J. M., et al. (2013). Activation of innate immunity by lysozyme fibrils is critically dependent on cross-β sheet structure. Cell. Mol. Life. Sci. 70, 2999–3012. doi: 10.1007/s00018-012-1245-5
Hakansson, A., and Molin, G. (2011). Gut microbiota and inflammation. Nutrients 3, 637–682. doi: 10.3390/nu3060637
Halmer, R., Davies, L., Liu, Y., Fassbender, K., and Walter, S. (2015). The innate immune receptor CD14 mediates lymphocyte migration in EAE. Cell. Physiol. Biochem. 37, 269–275. doi: 10.1159/000430351
Heinritz, S. N., Weiss, E., Eklund, M., Aumiller, T., Heyer, C. M., Messner, S., et al. (2016). Impact of a high-fat or high-fiber diet on intestinal microbiota and metabolic markers in a pig podel. Nutrients 8:E317. doi: 10.3390/nu8050317
Hill, J. M., Clement, C., Pogue, A. I., Bhattacharjee, S., Zhao, Y., and Lukiw, W. J. (2014). Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front. Aging Neurosci. 6:127. doi: 10.3389/fnagi.2014.00127
Hill, J. M., and Lukiw, W. J. (2015). Microbial-generated amyloids and Alzheimer’s disease (AD). Front. Aging Neurosci. 7:9. doi: 10.3389/fnagi.2015.00009
Hofer, U. (2014). Microbiome: B. fragilis and the brain. Nat. Rev. Microbiol. 12, 76–77. doi: 10.1038/nrmicro3197
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Hugon, P., Dufour, J. C., Colson, P., Fournier, P. E., Sallah, K., and Raoult, D. (2015). A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect. Dis. 15, 1211–1219. doi: 10.1016/S1473-3099(15)00293-5
Jiang, Q., Jin, S., Jiang, Y., Liao, M., Feng, R., Zhang, L., et al. (2016). Alzheimer’s disease variants with the genome-wide significance are significantly enriched in immune pathways and active in immune cells. Mol. Neurobiol. doi: 10.1007/s12035-015-9670-8 [Epub ahead of print].
Khanna, S., and Tosh, P. K. (2014). A clinician’s primer on the role of the microbiome in human health and disease. Mayo Clin. Proc. 89, 107–114. doi: 10.1016/j.mayocp.2013.10.011
Land, W. G. (2015). The role of damage-associated molecular patterns (DAMPs) in human diseases: part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 15, e157–e170.
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., et al. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5:37. doi: 10.1186/1742-2094-5-37
Leshchyns’ka, I., and Sytnyk, V. (2016). Synaptic cell adhesion molecules in Alzheimer’s disease. Neural Plast. 2016, 6427537. doi: 10.1155/2016/6427537
Lim, S. L., Rodriguez-Ortiz, C. J., and Kitazawa, M. (2015). Infection, systemic inflammation, and Alzheimer’s disease. Microbes Infect. 17, 549–556. doi: 10.1016/j.micinf.2015.04.004
Lin, C. S., Chang, C. J., Lu, C. C., Martel, J., Ojcius, D. M., Ko, Y. F., et al. (2014). Impact of the gut microbiota, prebiotics, and probiotics on human health and disease. Biomed. J. 37, 259–268. doi: 10.4103/2319-4170.138314
Lukiw, W. J., and Bazan, N. G. (1998). Strong NF-kB-DNA binding parallels cyclooxygenase-2 gene transcription in aging and in sporadic Alzheimer’s disease superior temporal lobe neocortex. J. Neurosci. Res. 53, 583–592. doi: 10.1002/(SICI)1097-4547(19980901)53:5<583::AID-JNR8>3.0.CO;2-5
Lukiw, W. J., Zhao, Y., and Cui, J. G. (2008). An NF-kB-sensitive microRNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J. Biol. Chem. 283, 31315–31322. doi: 10.1074/jbc.M805371200
Maldonado, R. F., Sá-Correia, I., and Valvano, M. A. (2016). Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 40, 480–493. doi: 10.1093/femsre/fuw007
Minter, M. R., Taylor, J. M., and Crack, P. J. (2016). The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 136, 457–474. doi: 10.1111/jnc.13411
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Nishimori, J. H., Newman, T. N., Oppong, G. O., Rapsinski, G. J., Yen, J. H., Biesecker, S. G., et al. (2012). Microbial amyloids induce interleukin 17A and IL-22 rvia TLR2 activation. Infect. Immun. 80, 4398–4408. doi: 10.1128/IAI.00911-12
Richards, R. I., Robertson, S. A., O’Keefe, L. V., Fornarino, D., Scott, A., Lardelli, M., et al. (2016). The enemy within: innate surveillance-mediated cell death, the common mechanism of neurodegenerative disease. Front Neurosci. 10:193. doi: 10.3389/fnins.2016.00193
Rogers, M. A., and Aronoff, D. M. (2016). The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 22, .e1–.e178. doi: 10.1016/j.cmi.2015.10.003
Sampson, T. R., and Mazmanian, S. K. (2016). Control of brain development, function, and behavior by the microbiome cell host microbe. Cell Host Microbe 17, 565–576. doi: 10.1016/j.chom.2015.04.011
Seksik, P., and Landman, C. (2015). Understanding microbiome data: a primer for clinicians. Dig. Dis. 33(Suppl. 1), 11–16. doi: 10.1159/000437034 [Epub ahead of print]
Sender, R., Fuchs, S., and Milo, R. (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14:e1002533. doi: 10.1371/journal.pbio.1002533
Seong, E., Yuan, L., and Arikkath, J. (2015). Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh. Migr. 9, 202–213. doi: 10.4161/19336918.2014.994919
Soenen, S., Rayner, C. K., Jones, K. L., and Horowitz, M. (2016). The ageing gastrointestinal tract. Curr. Opin. Clin. Nutr. Metab. Care 19, 12–18. doi: 10.1097/MCO.0000000000000238
van de Haar, H. J., Burgmans, S., Jansen, J. F., van Osch, M. J., van Buchem, M. A., Muller, M., et al. (2016). Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology doi: 10.1148/radiol.2016152244 [Epub ahead of print].
Varatharaj, A., and Galea, I. (2016). The blood-brain barrier in systemic inflammation. Brain Behav. Immun. doi: 10.1016/j.bbi.2016.03.010 [Epub ahead of print],
Youssef, N. H., Couger, M. B., McCully, A. L., Criado, A. E., and Elshahed, M. S. (2015). Assessing the global phylum level diversity within the bacterial domain: a review. J. Adv. Res. 6, 269–282. doi: 10.1016/j.jare.2014.10.005
Yu, Y., and Ye, R. D. (2015). Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 35, 71–83. doi: 10.1007/s10571-014-0101-6
Zhan, L. S., and Davies, S. S. (2016). Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med. 8, 46. doi: 10.1186/s13073-016-0296-x
Zhang, J., Ke, K. F., Liu, Z., Qiu, Y. H., and Peng, Y. P. (2013). Th17 cell-mediated neuroinflammation is involved in neurodegeneration of Aβ1-42-induced Alzheimer’s disease. PLoS ONE 8:e75786. doi: 10.1371/journal.pone.0075786
Zhao, Y., Dua, P., and Lukiw, W. J. (2015). Microbial sources of amyloid and relevance to amyloidogenesis and Alzheimer’s disease (AD). J. Alzheimers Dis. Parkinsonism 5, 177.
Zhao, Y., Jaber, V., and Lukiw, W. J. (2016). Over-expressed pathogenic miRNAs in Alzheimer’s disease (AD) and prion disease (PrD) drive deficits in TREM2-mediated Aβ42 peptide clearance. Fron. Aging Neurosci. 8:140. doi: 10.3389/fnagi.2016.00140
Keywords: 42 amino acid amyloid-beta (Aβ42) peptides, Alzheimer’s disease, bacteroidetes, DAMPs and PAMPs, lipopolysaccharides, microbiome, NF-kB, systemic inflammation
Citation: Lukiw WJ (2016) Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease. Front. Microbiol. 7:1544. doi: 10.3389/fmicb.2016.01544
Received: 06 June 2016; Accepted: 15 September 2016;
Published: 26 September 2016.
Edited by:
Christine Moissl-Eichinger, Medical University of Graz, AustriaReviewed by:
Benjamin P. Willing, University of Alberta, CanadaCopyright © 2016 Lukiw. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Walter J. Lukiw, d2x1a2l3QGxzdWhzYy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.