 Xiwen Sun1†
Xiwen Sun1† Chunyu Luo1†
Chunyu Luo1† Ru Tang1
Ru Tang1 Song Mao1
Song Mao1 Ying Zhu1Chonghui Fei2Mengyu Wang3
Ying Zhu1Chonghui Fei2Mengyu Wang3 Shaolin Tan4
Shaolin Tan4 Shiyao Zhang1
Shiyao Zhang1 Jiayao Zhou1
Jiayao Zhou1 Hai Lin1
Hai Lin1 Zhipeng Li1*
Zhipeng Li1* Weitian Zhang1*
Weitian Zhang1*- 1Shanghai Key Laboratory of Sleep Disordered Breathing, Department of Otolaryngology-Head and Neck Surgery, Otolaryngology Institute of Shanghai JiaoTong University, Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Neonatology, Children’s Hospital of Soochow University, Suzhou, China
- 3Department of Ophthalmology, Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 4Jinzhou Medical University Postgraduate Training Base (Department of Otolaryngology-Head and Neck Surgery, Shanghai Sixth People’s Hospital), Shanghai, China
Wiskott–Aldrich syndrome (WAS) is a rare primary immunodeficiency disease with a predisposition towards autoimmunity and lymphoproliferative diseases. Non-Hodgkin lymphoma (NHL) is reported to be the predominant form of malignant tumor in WAS sufferers. Diffuse large B-cell lymphoma (DLBCL) is one of the most common types of NHL while it is uncommon to occur in paranasal sinuses and especially when associated with WAS. In this article, we report a unique case of WAS associated with DLBCL in paranasal sinuses and review the major publications of WAS-related lymphomas that occurred in the head and neck area. This study extends the available therapies for WAS-related lymphomas and emphasizes the significance of recognition for sinonasal lymphomas in WAS patients presenting with sinusitis.
1 Introduction
Wiskott–Aldrich syndrome (WAS) is a rare X-linked disorder affecting 1 in 50,000 and 1 in 250,000 male newborns with disease-causing gene mapped to Xp11.22-p11.23 (1). The gene encodes WAS protein (WASP), whose mutations are associated with combined immunodeficiency, platelet defect, eczema, and increased susceptibility to autoimmune disorders and cancers (2). At the early stage, overwhelming infections and serious bleeding were the leading causes of WAS-related death (3). Afterward, with the modern prophylactic and treatment modalities that have led to a decreased incidence of lethal infections and bleeding, the major life-threatening complications in WAS-affected children became autoimmune disorders and malignant tumors (3, 4). Diffuse large B-cell lymphoma (DLBCL) is one of the most common types of non-Hodgkin lymphoma (NHL) (5). It is reported that NHL is the predominant form of malignancy in patients with WAS while DLBCL is rare, especially in paranasal sinuses (4, 6). Herein, we report a unique case of WAS with sinonasal DLBCL, which had extensively invaded the skull base and caused visual impairment. His medical procedures are reviewed in detail, and the clinical presentation, mechanism, treatment, outcomes, and diagnosis of this type of disease are discussed. Sinonasal malignancy from WAS should be evaluated in patients presenting with sinusitis symptoms. Prompt recognition may avoid unnecessary surgical intervention. However, optic nerve sheath fenestration (ONSF) is urgent when the tumor has caused visual loss. Timely recognition and treatment offer the best prognosis for recovery.
2 Case presentation
A 12-year-old boy diagnosed with WAS was admitted to our hospital in March 2021 with a 3-month history of dizziness, headache, vision loss in both eyes, and intermittent vomiting. In January 2021, the boy felt dizzy and undertook a head CT scan that showed bilateral sinusitis. Then, he was empirically treated with antibiotics and nutritional support. Around March 2021, the boy developed a headache, vomiting, and binocular vision loss. His vital signs were normal when doing a physical examination on admission. However, the vision of his right eye was 0.5 and the left eye had only light perception. Testing of cranial nerves (CNs) revealed that the boy had left abducens nerve paresis and right lower group CN total paresis. The funduscopic examination, perimetry, and F-VEPs showed damage to his left optic nerve (Figure 1). Enhanced MRI showed irregular mass had invaded the nasal cavity, ethmoid sinus, sphenoid sinus, and middle and posterior cranial fossa and compressed the bilateral optic canals, in bilateral paranasal sinuses and nasopharynx areas. The lateral wall of the sinus was destructed. The mass presented with diffuse and slightly longer T1-weighted imaging (T1WI)/T2-weighted imaging (T2WI), slight hyperintensity on fat-suppressed imaging, and significant enhancement. On imaging, the diagnosis was highly likely lymphoma (Figure 2A). The dura mater, cranium, posterior fossa of the trigeminal nerve root, bilateral cavernous sinuses, and internal carotid arteries were eroded to varying degrees. The patient’s medical history was remarkable for thrombocytopenia and recurrent eczema, and he was initially suspected to have WAS shortly after his birth, but it was not until August 2020 that he was diagnosed with WAS through genetic screening. Soon after, ONSF was performed to save his vision. Intraoperatively, the ethmoid tumor was removed to expose the orbital cardboard. The sphenoid plateau was invaded and reduced from the midline to both sides. The optic nerve canal was further completely exposed, and the tumor around the optic canal was removed to decompress the optic nerve (Figure 2B). The final immunohistochemistry findings showed that the malignant cells were positive for CD10, IRF4/MUM1, BCL-6, CD20, and CD79α, and negative for CD3, TIA-1, CD207/langerin, and CD5. The Ki67 was 80% positive. Genetic testing of the malignant cells was positive for BCL-6-related gene translocation but negative for BCL-2- and C-MYC-related genes, and the gene translocations were detected by fluorescence in situ hybridization (FISH) (Figure 3). These results confirmed the diagnosis of DLBCL with germinal center B type. A few days after the surgery, the boy was transferred to the Department of Hematology for further treatment. According to recent follow-up records, the boy underwent chemotherapy and chimeric antigen receptor (CAR) T-cell immunotherapy before the bone marrow transplant. Before chemotherapy and CAR-T therapy, the patient had a low T-cell count (<700/μl) but a compensatory increase in NK cell count (>600/μl), which were resolved after the following treatment. However, no abnormalities were detected in the levels of cytokines including IL-2, IL-4, IL-6, IL-10, TNF-α, IFN-γ, and IL-17A in the patient’s peripheral blood. The condition regimens patient used for CAR-T therapy and allogeneic HSCT are shown in Figure 4. The patient’s response to each therapy was unremarkable, expect on the fifth day of CAR-T therapy, the patient developed red rash on the trunk and limbs, pleural effusion, and decreased blood pressure, which was considered to be related to the cytokine release syndrome induced by allogeneic CAR-T therapy. Tocilizumab and dexamethasone were given, and ulinastatin was added to improve capillary permeability, and norepinephrine was used to increase blood pressure. There was no further deterioration of vision in both eyes after the surgery (Figure 2C) and he remained disease free for 1 year after comprehensive treatment. On the basis of radiographic evidence of tumor shrinkage and the absence of distant metastasis, we considered this patient to be in “partial remission.” The main treatment steps for the patient are shown in Figure 4.
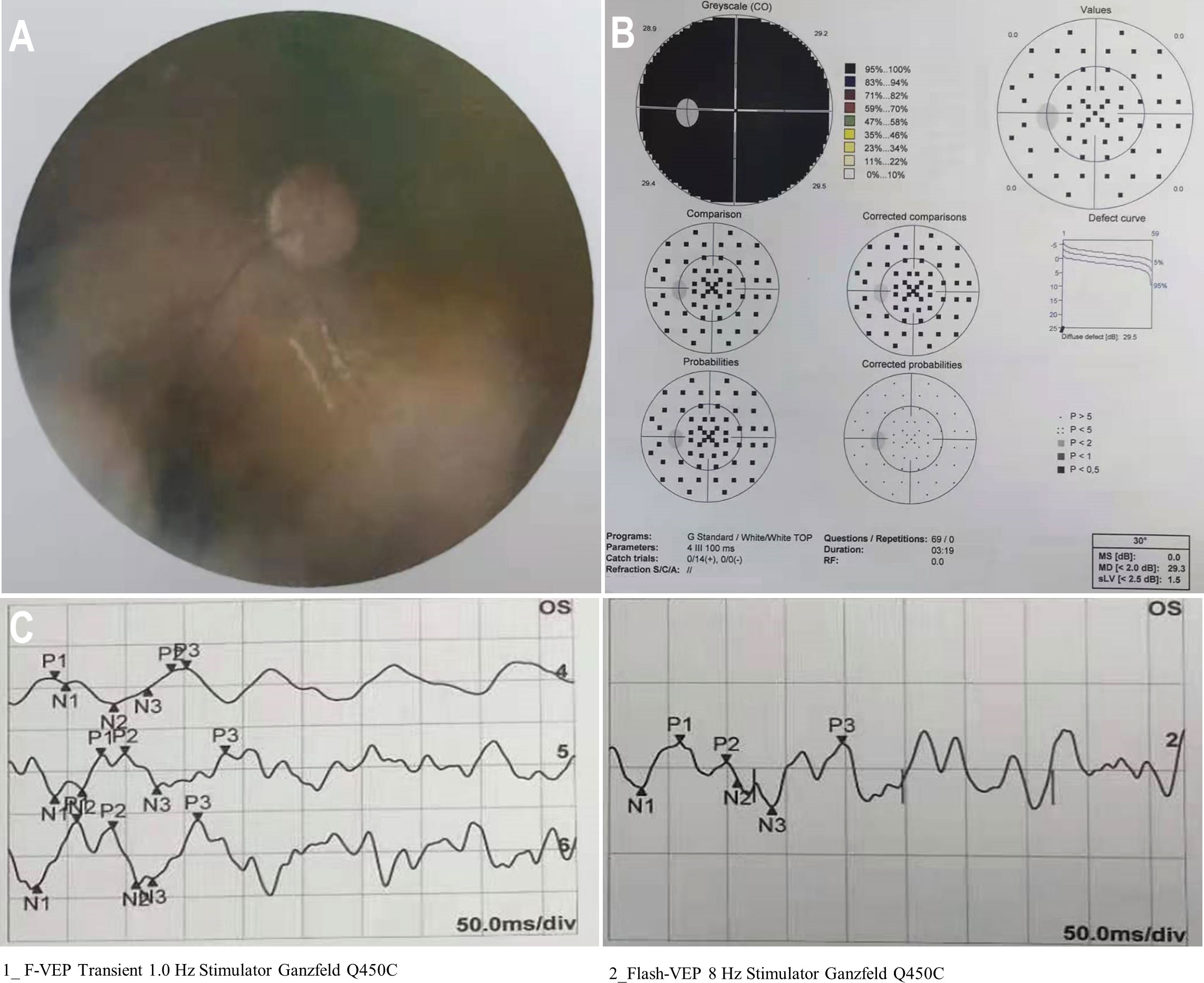
Figure 1 Preoperative condition of the left eye. (A) A funduscopic examination of the left eye revealed a hazy fundus. (B) Automated perimetry showed total visual field defect in the left eye. (C) F-VEPs of the left eye showed a reduced amplitude.
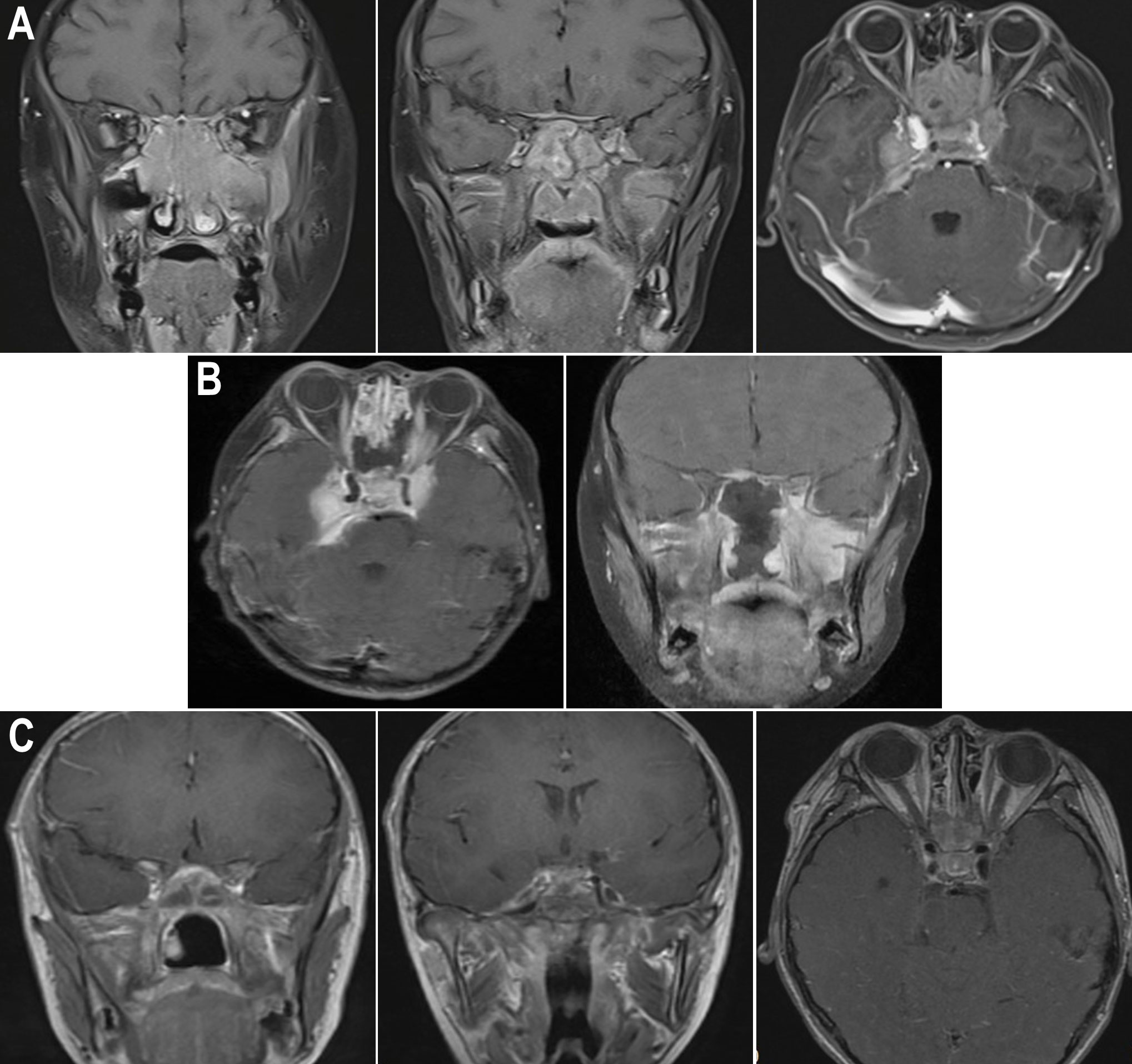
Figure 2 Enhanced nuclear MRI images of the patient during different periods. (A) Pre-op sagittal MRI images showed that the mass invaded the nasal cavity, ethmoid sinus, and sphenoid sinus, and compressed the optic nerve; pre-op axial MRI image revealed that the mass invaded the middle and posterior cranial fossa. (B) Post-op MRI images showed that intraoperative decompression of the optic nerve was performed. (C) Post-op MRI images showed the condition of the optic nerve and intracranial tumor after 1 month of HSCT.
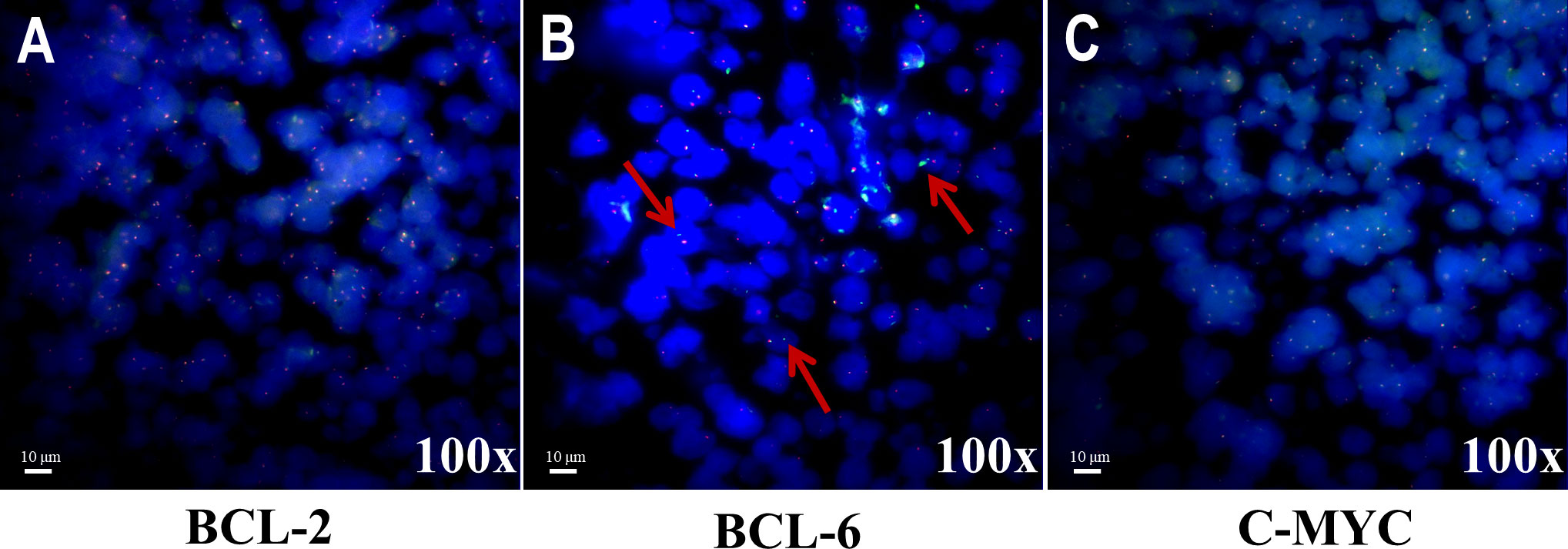
Figure 3 Representative images of translocation statuses by FISH analysis for three genes.(A) Tumor nuclei were negative for translocation of BCL-2. (B) Tumor nuclei were positive for translocation of BCL-6. Multiple tumor nuclei were seen with distinct red and green signals (highlighted by the red arrows). (C) Tumor nuclei were negative for translocation of C-MYC.

Figure 4 Timeline scheme of the major clinical events of the patient.WAS, Wiskott–Aldrich syndrome; ONSF, optic nerve sheath fenestration; COP, cyclophosphamide, vincristine, and prednisolone; VP, vinorelbine and prednisone; TBI, total body irradiation; FC, fluorouracil and carboplatin; CAR-T therapy, chimeric antigen receptor T-cell immunotherapy; CCNU, lomustine; FA, fludarabine; Dex, dexamethasone; Ara-C, cytosine arabinoside; G-CSF, granulocyte colony-stimulating factor; BU, busulfan; CTX, cyclophosphamide; RTX, rituximab; GVHD, graft-versus-host disease; MMF, mycophenolate mofetil; MTX, methotrexate; HSCT, hematopoietic stem-cell transplantation.
3 Discussion
WAS is a serious X-linked recessive primary immunodeficiency disorder characterized by eczema, thrombocytopenia, immune deficiency, and susceptibility to autoimmune diseases or malignancies which was named by the two physicians who described it, Alfred Wiskott (1937) and Robert Aldrich (1954) (1, 7). The boy we mentioned in this report had a single T deletion in exon 10 of the WAS gene (c.1032delT), which led to a frameshift in the Val codon at position 345 (p.Val345fs) (Table 1). Mutations in the WAS gene have various effects on the level of WASP, which, in turn, correlates with the diversity of the disease. Except for classic WAS, the WAS variant also leads to X-linked thrombocytopenia (XLT) or X-linked neutropenia (XLN) which reflect the different clinical phenotypes and clinical outcomes of WAS (3, 7).

Table 1 Genetic variant strongly associated with disease.
The possibility of cancer occurrence in patients with WAS is a significant concern. Approximately 13% of patients with WAS would develop malignant tumors, and the average age at diagnosis is 9.5 years (5). The most frequent malignant tumor is lymphoma, predominantly non-Hodgkin type presenting in extranodal sites induced by EBV infection (5, 7, 8). DLBCL is the most common pathologic type of NHL, which is also the most frequently encountered variant in WAS-affected patients (4). Unfortunately, dozens of cases are poorly characterized concerning the limited molecular detection techniques at early times (9). We located sporadic case reports of extranodal DLBCL in WAS in the published literature, with only one instance occurring in the brain (5), one in the pharyngeal (4), one in the orbit (10), one in the larynx (11), and two cases occurring in the systemic extranodal organs (12, 13) (Table 2). Therefore, the involvement of the nasal cavity and paranasal sinuses is an atypical but probable tumor site presentation that has not been previously reported.
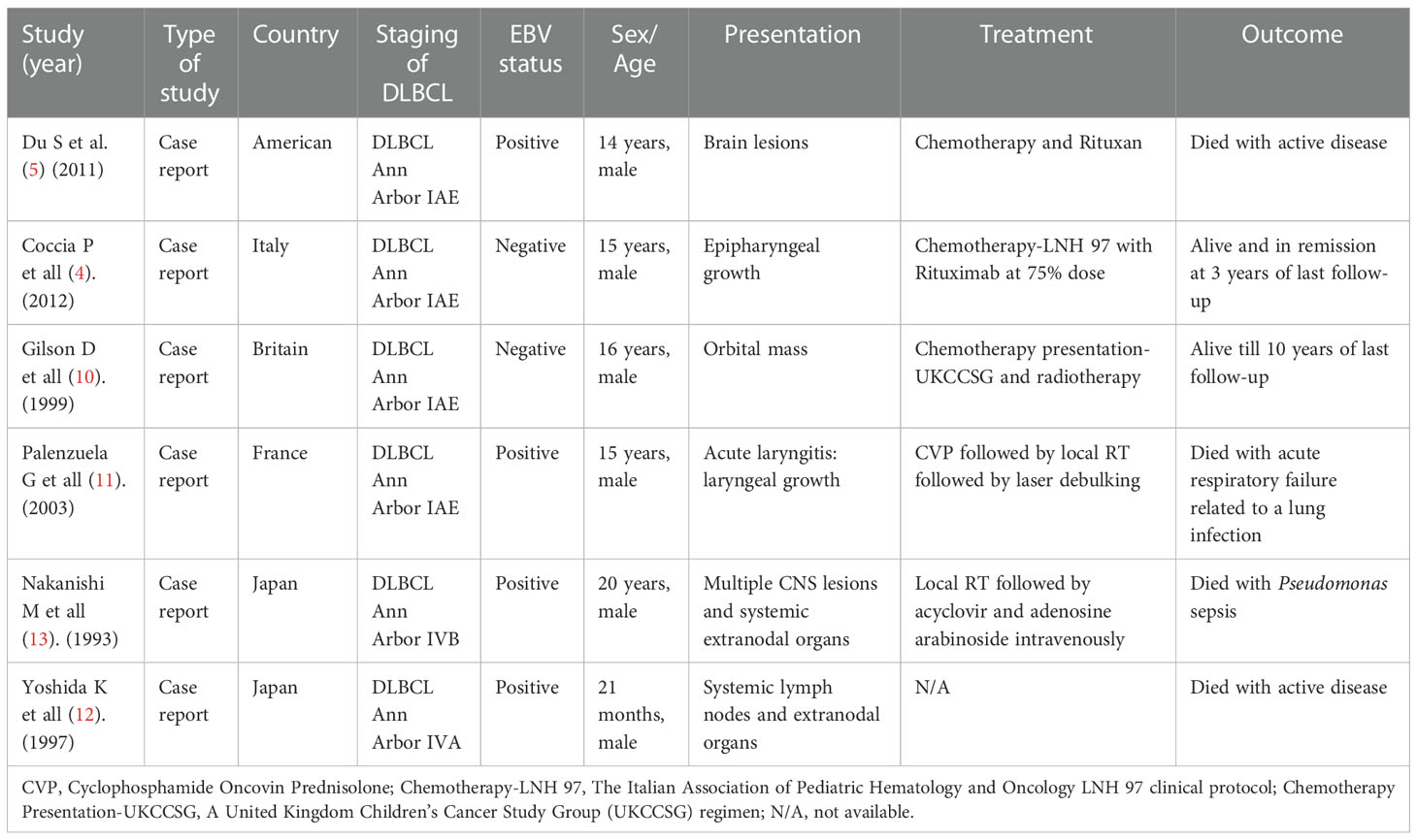
Table 2 Extranodal DLBCL in WAS patients.
Despite the close relationship between WAS and hematolymphoid malignancies, the etiology of lymphoma in patients with WAS is not fully understood (4, 5, 14, 15). Although WASP is considered the main causative factor due to its effect on a variety of cell types and the multiple intracellular and extracellular events that followed, EBV infection may also provide a mechanistic explanation for lymphomagenesis in the majority of WAS-associated lymphoproliferative diseases (7, 12–14, 16). Immunosurveillance by T cells and NK cells, which is generally defective in classical WAS patients, may explain the pathogenesis of DLBCL in WAS patients. Patients with WAS are characterized by defective thymic output and an increased frequency of apoptosis, which may explain the common phenomenon of T-cell lymphopenia in WAS patients. In addition, mutations in the gene encoding WASp cause defects in filamentous actin (F-actin) branching, resulting in the inability of T cells to migrate to secondary lymphoid organs, form stable immune synapses, proliferate, and secrete cytokines (17). WASp-deficient NK cells are unable to rearrange their F-actin cytoskeleton, which is required for NK cell immunological synapse formation and NK cell cytotoxicity (18). Concerns regarding the number and function of T cells and NK cells in patients during treatment are of crucial importance, because they could promptly respond to the treatment effect of the patients, but the monitoring of cytokine levels in peripheral blood of patients often cannot reflect the situation in time, and it may be necessary to conduct multiple tests or use other precise methods to evaluate the functional status of T cells and NK cells. In the case we reported, the patient was EBV-negative. It is more likely that the primary immune defect in WAS increases susceptibility to lymphoma development by several pathways. However, the specific causes of DLBCL in WAS are still unclear.
The prognosis for lymphoma in WAS patients is still dismal due to their immunodeficiency (15). Allogeneic hematopoietic stem-cell transplantation (HSCT) remains the most curative therapy for WAS, with hematopoietic stem-cell (HSC) gene therapy and gene editing developing as optimistic treatment options (19). The most common treatment for DLBCL in WAS is still chemotherapy (4, 15). Surgery is an alternative option when the tumor involved critical blood vessels and nerves or the tumor blocked the respiratory and digestive tract. In this case, endoscopic sinus surgery was performed for his vision salvage and headache relief. Moreover, a pathological biopsy could also be done during the operation, which could determine the nature of the tumor for further treatment strategies. Minimally invasive surgery via nasal endoscopy is more suitable for patients with bleeding tendencies and basic diseases than open surgery.
The sinonasal malignancies initially cause very few symptoms. Moreover, due to the infrequently involved location, tumors in this area are often misdiagnosed as sinusitis at the early stage (20). As a result, such as the case we reported, a high index of suspicion of sinus malignancy in WAS patients with a history of chronic sinusitis in combination with cranial neuralgia or orbital symptoms and signs should be considered. Improved awareness of these symptoms and subsequent early diagnosis are important for the effective treatment of this rare and polymorphic disease.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of Shanghai Sixth People’s Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
WZ, ZL, RT, and XS conceived and designed the study. XS, CL, ZL, SM, ST, CF, and MW were in charge of the data collection and analysis. XS, CL, ZL, WZ, YZ, SZ, JZ, and HL wrote, reviewed, and/or revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Clinical Science and Technology Innovation Project of Municipal Hospital (SHDC12020129) and the National Natural Science Foundation of China (No. 82071014, No.8200951, No.81870700, and No. 82271137).
Acknowledgments
Special thanks to the patient who was willing to share his experience with us.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Binder V, Albert MH, Kabus M, Bertone M, Meindl A, Belohradsky BH. The genotype of the original wiskott phenotype[J]. New Engl J Med (2006) 355(17):1790–3. doi: 10.1056/NEJMoa062520
2. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: A comprehensive review[J]. Ann New York Acad Sci (2013) 1285:26–43. doi: 10.1111/nyas.12049
3. Suri D, Rikhi R, Jindal AK, Rawat A, Sudhakar M, Vignesh P, et al. Wiskott Aldrich syndrome: A multi-institutional experience from India[J]. Front Immunol (2021) 12:627651. doi: 10.3389/fimmu.2021.627651
4. Coccia P, Mastrangelo S, Ruggiero A, Scalzone M, Rosolen A, Maurizi P, et al. Treatment of pharyngeal non-Hodgkin lymphoma in a patient with wiskott-Aldrich syndrome[J]. Pediatr Blood Cancer (2012) 59(2):318–9. doi: 10.1002/pbc.23393
5. Du S, Scuderi R, Malicki DM, Willert J, Bastian J, Weidner N, et al. Hodgkin’s and non-hodgkin’s lymphomas occurring in two brothers with wiskott-Aldrich syndrome and review of the literature[J]. Pediatr Dev Pathol: Off J Soc Pediatr Pathol Paediatric Pathol Soc (2011) 14(1):64–70. doi: 10.2350/10-01-0787-CR.1
6. Lehrich BM, Abiri A, Goshtasbi K, Birkenbeuel J, Yasaka TM, Papagiannopoulos P, et al. Treatment modalities and survival outcomes for sinonasal diffuse Large b-cell Lymphoma[J]. Laryngoscope (2021) 131(11):E2727–35. doi: 10.1002/lary.29584
7. Candotti F. Clinical manifestations and pathophysiological mechanisms of the wiskott-Aldrich Syndrome[J]. J Clin Immunol (2018) 38(1):13–27. doi: 10.1007/s10875-017-0453-z
8. Vignesh P, Suri D, Rawat A, Lau YL, Bhatia A, Das A, et al. Sclerosing cholangitis and intracranial lymphoma in a child with classical wiskott-Aldrich syndrome[J]. Pediatr Blood Cancer (2017) 64(1):106–9. doi: 10.1002/pbc.26196
9. Cotelingam JD, Witebsky FG, Hsu SM, Blaese RM, Jaffe ES. Malignant lymphoma in patients with the wiskott-Aldrich syndrome[J]. Cancer Invest (1985) 3(6):515–22. doi: 10.3109/07357908509039813
10. Gilson D, Taylor RE. Long-term survival following non-hodgkin’s lymphoma arising in wiskott-Aldrich syndrome[J]. Clin Oncol (Royal Coll Radiologists (Great Britain)) (1999) 11(4):283–5. doi: 10.1053/clon.1999.9066
11. Palenzuela G, Bernard F, Gardiner Q, Mondain M. Malignant b cell non-hodgkin’s lymphoma of the larynx in children with wiskott Aldrich syndrome[J]. Int J Pediatr Otorhinolaryngol (2003) 67(9):989–93. doi: 10.1016/S0165-5876(03)00155-1
12. Yoshida K, Minegishi Y, Okawa H, Yata J, Tokoi S, Kitagawa T, et al. Epstein-Barr Virus-associated malignant lymphoma with macroamylasemia and monoclonal gammopathy in a patient with wiskott-Aldrich syndrome[J]. Pediatr Hematol Oncol (1997) 14(1):85–9. doi: 10.3109/08880019709030889
13. Nakanishi M, Kikuta H, Tomizawa K, Kojima K, Ishizaka A, Okano M, et al. Distinct clonotypic Epstein-Barr virus-induced fatal lymphoproliferative disorder in a patient with wiskott-Aldrich syndrome[J]. Cancer (1993) 72(4):1376–81. doi: 10.1002/1097-0142(19930815)72:4<1376::AID-CNCR2820720437>3.0.CO;2-Q
14. Han S-S, Wen K-K, Vyas YM. Deficiency of wiskott-Aldrich syndrome protein has opposing effect on the pro-oncogenic pathway activation in nonmalignant versus malignant lymphocytes[J]. Oncogene (2021) 40(2):345–54. doi: 10.1038/s41388-020-01533-3
15. Senapati J, Devasia AJ, David S, Manipadam MT, Nair S, Jayandharan GR, et al. Diffuse large b cell lymphoma in wiskott-Aldrich syndrome: A case report and review of literature[J]. Indian J Hematol Blood transfusion: An Off J Indian Soc Hematol Blood Transfusion (2014) 30(Suppl 1):309–13. doi: 10.1007/s12288-014-0377-1
16. Rivers E, Thrasher AJ. Wiskott-Aldrich syndrome protein: Emerging mechanisms in immunity[J]. Eur J Immunol (2017) 47(11):1857–66. doi: 10.1002/eji.201646715
17. Mansour R, El-Orfali Y, Saber A, Noun D, Youssef N, Youssef Y, et al. Wiskott-Aldrich syndrome in four male siblings from a consanguineous family from Lebanon[J]. Clin Immunol (Orlando Fla.) (2020) 219:108573. doi: 10.1016/j.clim.2020.108573
18. Jyonouchi S, Gwafila B, Gwalani LA, Ahmad M, Moertel C, Holbert C, et al. Phase I trial of low-dose interleukin 2 therapy in patients with wiskott-Aldrich syndrome[J]. Clin Immunol (Orlando Fla.) (2017) 179:47–53. doi: 10.1016/j.clim.2017.02.001
19. Biffi A. Gene therapy goes the distance in wiskott-Aldrich syndrome[J]. Nat Med (2022) 28(1):24–5. doi: 10.1038/s41591-021-01653-7
Keywords: Wiskott-Aldrich syndrome, diffuse large B cell lymphoma, sinonasal, surgery, case report
Citation: Sun X, Luo C, Tang R, Mao S, Zhu Y, Fei C, Wang M, Tan S, Zhang S, Zhou J, Lin H, Li Z and Zhang W (2023) Sinonasal diffuse large B-cell lymphoma in a patient with Wiskott–Aldrich syndrome: A case report and literature review. Front. Immunol. 13:1062261. doi: 10.3389/fimmu.2022.1062261
Received: 05 October 2022; Accepted: 22 December 2022;
Published: 12 January 2023.
Edited by:
Michel J. Massaad, American University of Beirut, LebanonReviewed by:
Yoji Sasahara, Tohoku University, JapanMarco Antonio Yamazaki-Nakashimada, National Institute of Pediatrics, Mexico
Copyright © 2023 Sun, Luo, Tang, Mao, Zhu, Fei, Wang, Tan, Zhang, Zhou, Lin, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhipeng Li, MzE3NDExNzM0QHFxLmNvbQ==; Weitian Zhang, ZHJ6aGFuZ3d0QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship