
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 January 2023
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1053136
This article is part of the Research Topic Targeting Signalling Pathways in Inflammatory Diseases View all 17 articles
 Sizhu Gong
Sizhu Gong Fang Deng*
Fang Deng*Emotional disorders, including depression and anxiety, contribute considerably to morbidity across the world. Depression is a serious condition and is projected to be the top contributor to the global burden of disease by 2030. The role of the renin-angiotensin system (RAS) in hypertension and emotional disorders is well established. Evidence points to an association between elevated RAS activity and depression and anxiety, partly through the induction of neuroinflammation, stress, and oxidative stress. Therefore, blocking the RAS provides a theoretical basis for future treatment of anxiety and depression. The evidence for the positive effects of RAS blockers on depression and anxiety is reviewed, aiming to provide a promising target for novel anxiolytic and antidepressant medications and/or for improving the efficacy of currently available medications used for the treatment of anxiety and depression, which independent of blood pressure management.
Emotional disorders, including depression and anxiety, contribute considerably to morbidity across the world (1). Anxiety disorders are the most common class of disorders listed in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V), which are complex interactions between biological, psychological, temperamental, and environmental factors (2). As a group, anxiety disorders represent a heterogeneous group of illnesses that are characterized by excessive fear and anxiety, hypervigilance, and related behavioral disturbances (3). Furthermore, anxiety is one of the most common comorbid disorders with major depressive disorder (MDD) (4, 5). A large psychiatric cohort study has reported that depression preceded anxiety in 18% of such comorbid cases, while in 57% of the cases anxiety preceded depression (5). Comorbid anxiety and many core depression symptoms may be caused by hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis combined with amygdala dysfunction (4). The amygdala is a key element of anxiety circuitry and produces behavioral responses associated with fear and anxiety by integrating information from sensory inputs in the cortex and thalamus (6). Similarly, some neuroimaging studies have reported enhanced amygdala glucose metabolism and activation in patients with depression (7), and that depression-associated anxiety is accompanied by an increase in amygdala volume (8). First-degree relatives of individuals with anxiety have an increased risk of developing anxiety disorders or depression (9). Importantly, however, only 40–70% of patients with depression respond to pharmacological treatment and therapies often have a delayed onset (10), and anxiety associated with depression often leads to reduced responses and decreased compliance with pharmacotherapy (6). Consequently, emotional disorders are a serious public health issue, and identifying novel targets for their treatment is worthy of further attention.
The role of the renin-angiotensin system (RAS) in hypertension and emotional disorders is well established. Evidence points to an association between elevated RAS activity and depression and anxiety, partly through the induction of neuroinflammation, stress, and oxidative stress (11). Importantly, blocking RAS can have anti-inflammatory and anti-oxidative stress effects, providing a theoretical basis for future treatment of anxiety and depression. Captopril and enalapril (angiotensin-converting enzyme inhibitors; ACEIs) may rapidly improve depressive moods in hypertensive patients (12). This has sparked significant interest in RAS targets. The evidence for the positive effects of RAS blockers on depression and anxiety is reviewed here to evaluate a promising target for novel anxiolytic and antidepressant medications. Furthermore, this knowledge may aid the improvement of the efficacy of currently available medications used for the treatment of anxiety and depression, which are independent of blood pressure management.
RAS-blockers or RAS inhibitors are classes of medications that block the renin-angiotensin axis, primarily inhibiting angiotensin (Ang) II activity. Examples include ACEIs and selective Ang II type 1 receptor blockers (ARBs) (13). The most common ACEIs include captopril, enalapril, lisinopril, perindopril, ramipril, and imidapril. The most common ARBs include losartan, irbesartan, candesartan, telmisartan, and valsartan. As effective first-line antihypertensive medications, ACEIs and ARBs have been shown to minimize the risk of cardiovascular and renal events as well as mortality (14). These classes of drugs target Ang II, however, differences in their mechanisms of action impact their effects on other pathways and receptors, which may have therapeutic implications. For example, ACEIs inhibit RAS activation by preventing the conversion of Ang I to Ang II, resulting in reduced activation of both Ang II type 1 (AT1/AT1) receptors and Ang II type 2 (AT2/AT2) receptors (15). Moreover, ACEIs prevent the degradation of Ang-(1–7) by angiotensin-converting enzyme (ACE), thereby the level increased due to a build-up caused by the lack of degradation of Ang-(1–7) (15). Additionally, ACEIs block the degradation of bradykinin, leading to activation of the β-2 receptor and promotion of nitric oxide (NO) release with vasodilatory and tissue-protective effects (13). One study showed that ACEIs rapidly ameliorate depressive behaviors via the bradykinin-dependent activation of the target of the rapamycin complex (10). Unlike ACEIs, ARBs block RAS by antagonizing the binding of Ang II to the AT1 receptor and activating the AT2 receptors, thus producing insufficient Ang II to elevate Ang-(1–7) level (16). High levels of Ang-(1–7) reduce anxiety and depression behaviors, providing positive benefits (see below). RAS blockers shift the balance to increase circulating levels of Ang-(1–7), this may contribute to shunt the ACE/Ang II/AT1 pathway toward the ACE2/Ang-(1–7)/MasR pathway providing beneficial effects on mood disorders (17).
Although RAS is widely acknowledged as a cardiovascular circulation hormonal system, it is found in a variety of organs, including the brain. The RAS is composed of two pathways that are mutually antagonistic that maintain the balance through angiotensin-converting enzyme 2 (ACE2): the classical pathway angiotensin-converting enzyme/angiotensin II/angiotensin II type 1 receptor (ACE/Ang II/AT1R) and the non-classical pathway angiotensin-converting enzyme 2/angiotensin- (1–7)/Mas receptor (ACE2/Ang-(1–7)/MasR).
The classical pathway contains renin, angiotensin (Ang) II, angiotensin-converting enzyme (ACE), angiotensin II type 1 receptor (AT1R/AT1R), and angiotensin II type 2 receptor (AT2R/AT2R). Renin is an aspartyl protease typically produced in the juxtaglomerular cells of the kidney, and it cleaves angiotensinogen (ATN, an inactive peptide formed and secreted by the liver) to produce angiotensin I (Ang I). Ang I has few physiological effects and produces Ang II as a substrate for ACE. Ang II exerts several physiological effects: constriction of blood vessels, stimulation of aldosterone secretion, and release of catecholamines. Ang II acts by binding to the AT1R and AT2R. When Ang II activates the AT1 receptor, it causes neurotoxicity, such as vasoconstriction, pro-inflammatory, apoptotic, and anti-diuresis. Furthermore, increased circulating levels of Ang II disrupt blood-brain barrier (BBB) integrity, allowing circulating Ang II to access the brain parenchyma and trigger the AT1R directly, producing oxidative stress and brain inflammation (18). AT2R is activated by Ang II and may counterbalance AT1R neurotoxic effects and determine a neuroprotective role in RAS activation, such as vasodilation, diuresis, anti-fibrosis, antihypertensive, and cognitive improvement. AT2R activation is important in blunting the negative effects of AT1R, such as neuroinflammation and oxidative stress (19). However, evidence shows that AT1 receptors predominate in adult tissues and AT2 receptors predominated in the developing brain (20).
The non-classical axis is neuroprotective and composes angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas receptor axis (ACE2/Ang-(1–7)/MasR). The non-classical axis exerts neuroprotective effects, such as promoting the release of NO and promoting anti-inflammatory, anti-fibrotic, and vasodilatation effects. In the brain, all components of the ACE2/Ang-(1–7)/MasR axis are expressed. ACE2 correlates with AT1R and Ang II levels and ACE2 overexpression results in the downregulation of AT1R and increases the expression of AT2R and MasR. As a homologous enzyme of ACE, ACE2 is found in the hippocampus and cerebral cortex that cleaves Ang II to produce Ang-(1–7), which activates the Mas receptor and produces an inverse regulation of the ACE/Ang II/AT1 pathway (21, 22). Ang-(1–7) generated in the rat hippocampus has been reported (23). As for the Mas receptor, it was a G protein-coupled receptor specific for Ang-(1–7), which is expressed in brains and other different organs, including the hippocampus, amygdala, and cortex (21).
In addition to the systemic RAS, all the components of the RAS independently exist in the brain involving the pathophysiology of depression and anxiety. Hyperactivation of the ACE/Ang II/AT1R classical pathway accelerate the disease process via activated AT1R, while AT2R plays a protective role (24). We will discuss the evidence and mechanisms of RAS involvement in depression and anxiety in this part (Figure 1A) (Table 1).
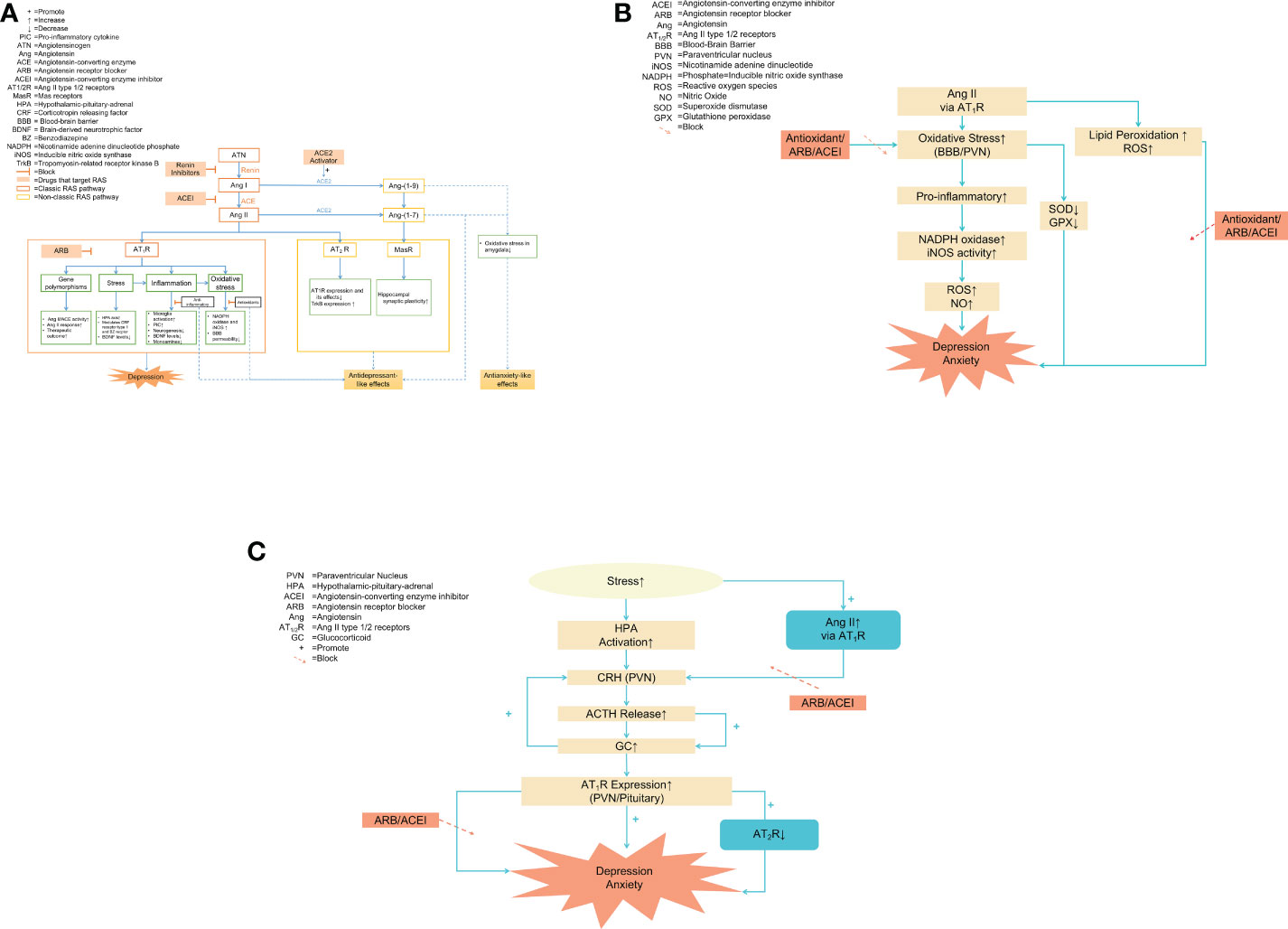
Figure 1 (A) The overview of RAS and the role of the RAS in the pathophysiology of anxiety and depression. Notes: The blue dashed line pointed to the positive effects, such as antidepressant-like and anxiolytic effects. Two key regulatory pathways: the classical axis ACE/Ang II/AT1 receptor pathway and the non-classical pathway ACE2/Ang-(1–7)/Mas receptor pathway. Under physiological conditions, the two pathways regulated each other and maintained a dynamic balance. Ang II aggravated oxidative stress and inflammation response by activating AT1R to upregulate the ACE/Ang II/AT1R pathway, promoting the development of emotional disorders. Ang II activated non-classical pathways by activating AT2R and MasR, producing antidepressant-like and anxiolytic effects. Thus, the beneficial effects of RAS blockers may be due to inhibiting oxidative stress and inflammation by directly targeting Ang II and its AT1 receptor. Other potential targets of anxiolytic drugs include renin, ACE2, AT2 receptors, and Mas receptors. (B) RAS and oxidative stress in depression and anxiety Notes: The pro-inflammatory effects of Ang II were largely mediated by increased oxidative stress. During inflammation, Ang II activated AT1R to promote the oxidative stress process by increasing NADPH oxidase and iNOS activity in the BBB and PVN, resulting in an accumulation of ROS and NO and ultimately aggravating emotional disorders. Antioxidant enzyme activity was decreased during oxidative stress, which was prevented by RAS blockers and antioxidants. Furthermore, Ang II directly increased ROS generation and triggered lipid peroxidation, inhibited by RAS inhibitors and antioxidants. (C) HPA and RAS. Notes: The type of stress caused HPA axis activation and increased the downstream hormones such as CRH, ACTH, and GC, eventually promoting anxiety and depression. Stress increased Ang II levels, which in turn raised the expression of CRH mRNA, induced the release of ACTH and GC, and enhanced the stimulatory effects of CRH through AT1R located in the pituitary and PVN. The absence of AT2 receptor transcription enhanced the AT1 receptor expression in brain areas and was involved in regulating the HPA axis, which was associated with anxiety and depression. Pretreatment with RAS blockers attenuated neuroendocrine responses, preventing the development of stress-induced anxiety/depression-like behaviors. (C) RAS and oxidative stress in depression and anxiety. Notes: The pro-inflammatory effects of Ang II were largely mediated by increased oxidative stress. During inflammation, Ang II activated AT1R to promote the oxidative stress process by increasing NADPH oxidase and iNOS activity in the BBB and PVN, resulting in an accumulation of ROS and NO and ultimately aggravating emotional disorders. Antioxidant enzyme activity was decreased during oxidative stress, which was prevented by RAS blockers and antioxidants. Furthermore, Ang II directly increased ROS generation and triggered lipid peroxidation, inhibited by RAS inhibitors and antioxidants.
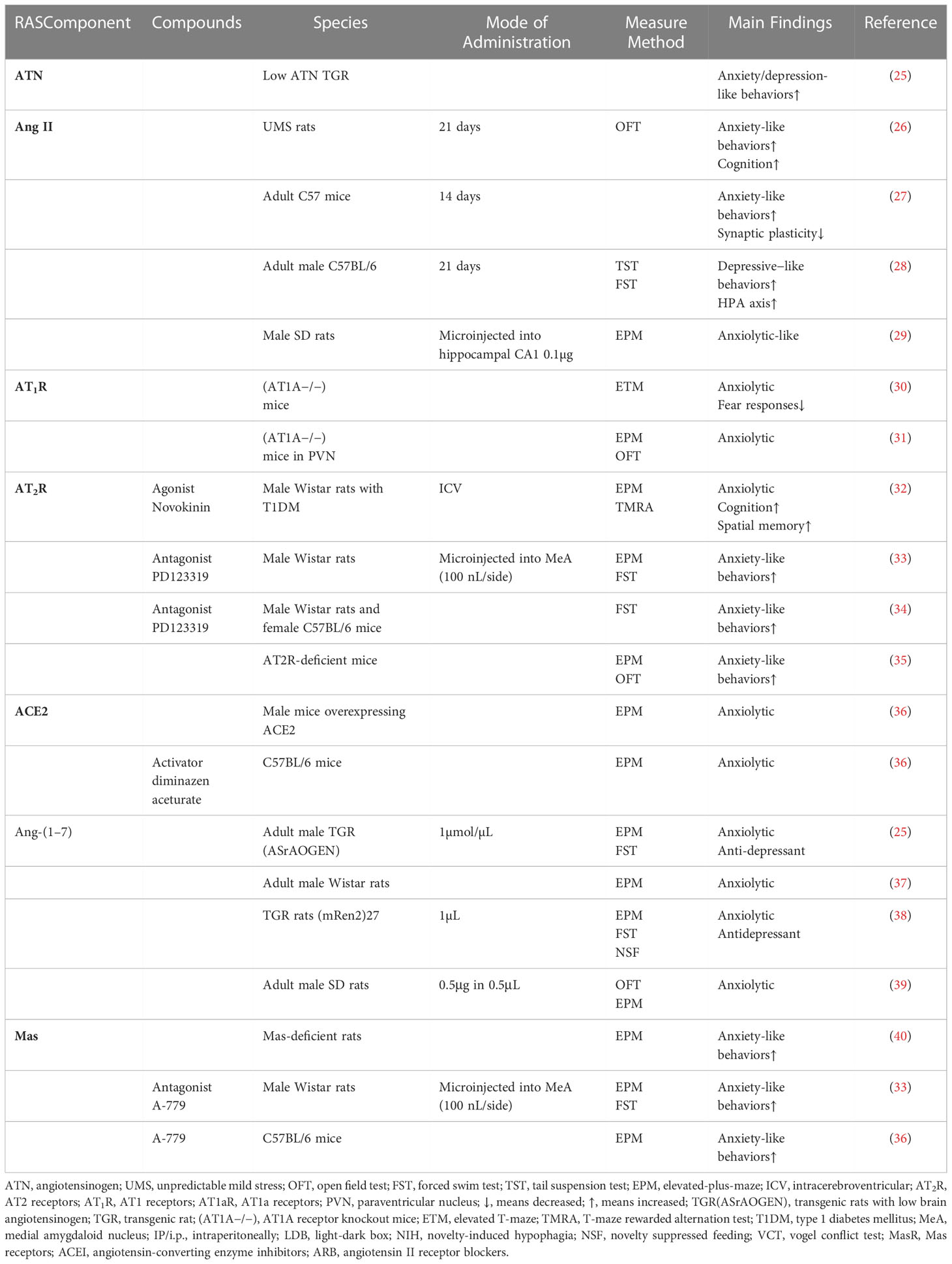
Table 1 The relationship between RAS components and depression/anxiety.
Angiotensinogen is the glycoprotein precursor of angiotensin II. Voigt et al. were the first time to describe transgenic rats with low brain angiotensinogen behavioral phenotype as characterized by increased anxiety-related behaviors (41). Subsequently, studies showed low angiotensinogen concentration in the brain leads to anxiety-like behaviors accompanied by a depression-like state (25).
Ang II was previously discovered as a pro-hypertensive factor present in areas of the brain associated with cardiovascular and has recently been found to be associated with motor activity, anxiety, learning, and memory (20). Additionally, increased Ang II level is significantly associated with depression, anxiety, hyperactivity of the HPA axis, and stress (28, 42). For instance, treatment with Ang II for 14 consecutive days had significant anxious-like behaviors and bidirectional synaptic plasticity impairment, and increase expression of GABAARα1 (γ-aminobutyric acid A receptor) (27). Administered Ang II for 3 weeks induced cognitive impairment and anxiety-like behaviors as shown by spending less time in the four center squares in the open field tests (OFT) (26). Telmisartan and imipramine reversed chronic Ang II infusion-induced behavioral changes, including changes in TST and forced swimming test (FST) (28). Losartan microinjects into the hippocampus CA1 region showed an anxiolytic-like effect in bilateral olfactory bulbectomy rats (OBX, rat model of depression), indicating the involvement of Ang II in the pathogenesis of anxiety by activating AT1R (42). Whereas microinjections Ang II (0.1,0.5,1.0µg) into the CA1 hippocampal area, at a dose of 0.1µg shows some anxiolytic effects manifested as an increasing number of entries into the open arms in the elevated plus maze (EPM) (29). The results are inconsistent with previous studies, but there may be anxiolytic and anxiety effects of Ang II in a dose-related U-shaped manner.
Furthermore, hyperactivation of AT1a receptors is associated with promoting anxiety-like behaviors in the brain (31). Deletion of AT1a receptors (AT1a−/−) from the paraventricular nucleus (PVN) attenuated anxiety-like behaviors in rodents as manifested by increased time spent in the open arms of the EPM (31). Further, (AT1a−/−)mice reduced flight behavior in the elevated T-maze test (a model of anxiety and panic) and diminished fear responses despite threat levels (30).
Chronically infused intracerebroventricular (i.c.v.) AT2 receptors agonist evokes anxiolytic-like effects (32). Treatment with selective AT2 receptor antagonist PD123319 decreased the open arms exploration in EPM and changed the pattern of swimming during the FST (33). AT2 receptor-deficient mice increased anxiety-like behaviors, which can be reversed by captopril, and show no depression-like behaviors compared to wild-type mice, providing a theoretical basis for ACEIs for the treatment of emotional disorders (35). Recently, it was reported that the modulatory role of the AT2 receptor in the development of depressive-like behavior (43). Administration of AT2R antagonist PD123319 into the prefrontal cortex reversed the antidepressant effect of losartan (34), indicating AT2R has positive effects on depression and anxiety.
Walther was the first to discover that the ACE2/Ang-(1–7)/MasR pathway is associated with the development of anxiety, demonstrating that upregulating ACE2 significantly improved anxiety-like behaviors (40). ACE2 is essential for maintaining the balance between ACE2/Ang-(1–7)/Mas receptor and the ACE/Ang II/AT1R pathway. The main function of ACE2 is to inhibit ACE activity by decreasing Ang II bioavailability and increasing Ang-(1–7) levels. Thus, the overexpression of ACE2 not only is related to the upregulation of AT2R and Mas receptors but also to the downregulation of AT1R and ACE (44). Elevated ACE2 activity decreases anxiety-like behaviors and inhibits stress-induced activation of the HPA axis in male mice (45). However, in female mice, increasing ACE2 expression only produces anxiolysis without reversing HPA axis activity (45). Consistent with the effect, central administration of diminazen aceturate to mice, an ACE2 activator reduces anxiety-like behaviors in EPM (36).
Ang-(1–7) is associated with reduced depressive and anxious behaviors as a selective non-competitive antagonist of Ang II at type 1 Ang II receptors (46). Overexpression of circulation Ang-(1–7) produced anxiolytic-like effects have been found in transgenic rats (37), which were manifested by increasing the percentage of time spent and frequency of entries in the open arms and decreasing stretching in closed arms of the EPM (38, 47). In addition, overexpression of Ang-(1–7) reverses the increase in heart rate associated with emotional stress and demonstrates less anxious behaviors in transgenic rats (48). Low angiotensinogen levels in the brain lead to anxiety-like behaviors and depression-like behaviors, while intracerebroventricular administration of Ang-(1–7), selective serotonin reuptake inhibitor fluoxetine, enalapril (ACEI) attenuated behavioral changes in transgenic hypertensive rats, as shown by spending a lower percentage of time in the open arms of EPM and decreasing immobility time in FST (25, 38).
Centrally injecting the MasR antagonist reverses the ACE2 and Ang-(1–7)-induced anxiolytic effects, indicating the anxiolytic effects of ACE2/Ang-(1–7)/MasR pathway due to activate Mas receptors (36, 47). Pre-treatment with A779 (a selective Mas receptor antagonist) enhances the anxiety-like effects (38), showing decreases open arms exploration in the EPM and changes the pattern of swimming during the forced swim test (33). MasR-deficient mice influence hippocampal synaptic plasticity and exhibit increased anxiety behaviors in EPM (40). Taken together, the upregulation of the ACE/Ang II/AT1R pathway accelerates the process of the emotional disorder and the non-classical axis ACE2/Ang-(1–7)/MasR has neuroprotective effects on emotional disorders.
ACEIs and ARBs have been shown to have protective and potential therapeutic benefits in mood disorders (49)(Table 2). Saavedra et al. proposed that Ang II expression is associated with mood disorders and that reducing brain Ang II levels might decrease anxiety and depression in animal models (57). For example, treatment with captopril had significant antidepressant activity as shown by the forced swim-induced behavioral despair (immobility) test in mice. In addition, it reversed escape deficits in the learned helplessness model (55, 58). Studies in rodents injected with losartan systemically and locally to the anterior prefrontal cortex and medial amygdaloid nucleus have shown antidepressant effects as evidenced by decreased immobility time in the FST (33, 34).
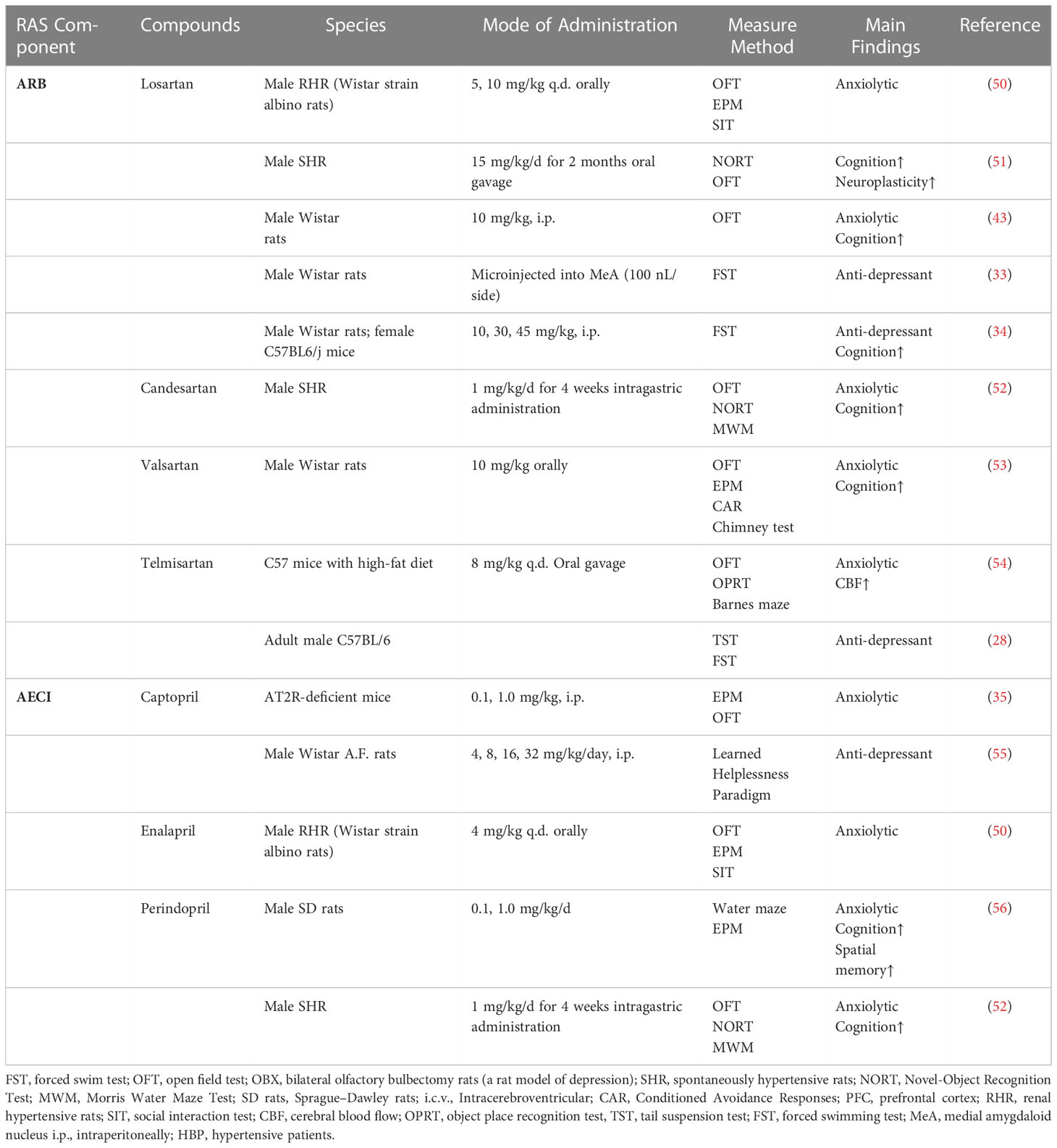
Table 2 Anxiolytic/anti-depressant effect of RAS blockers.
Chronic administration of the ACEI perindopril has anxiolytic effects on rats (56). Chronic cerebral hypoperfusion induces ACE/Ang II/AT1R overexpression in the hippocampus and causes anxiety. Candesartan and perindopril attenuate anxiety-like behavior and improve memory impairment by downregulating the ACE/Ang II/AT1R pathway and upregulating the ACE2/Ang-(1–7)/MasR pathway in the hippocampus (52). Ang II-induced rats spent significantly less time in the open arms of the elevated plus maze (EPM), this effect was abolished by the administration of valsartan and losartan (53, 59). Losartan effectively attenuated hyperactivity and anxiogenic behaviors in mice as seen in the EPM, social-interaction tests, and open field tests (OFT) (51) (50). Moreover, telmisartan treatment prevented diet-induced anxiety-like behaviors in behavioral tests (54).
The main RAS-related gene polymorphisms are found in the angiotensinogen gene, ACE, and the angiotensin 1 receptor gene, which have alleles associated with high levels of Ang II, high ACE activity, and elevated Ang II response, respectively. The ACE insertion/deletion (I/D) polymorphism determines functional variations of the ACE gene that significantly influence ACE plasma concentrations, which account for 30–40% of the variation in plasma ACE levels (60). Baghai et al. investigated the genetic association between 35 single-nucleotide polymorphisms (SNPs) and an I/D polymorphism in the ACE gene. They reported that carrying the T-allele is correlated with higher ACE serum activity, in which the highest ACE activities are found in patients homozygous for the T-allele, and the lowest is noted in patients homozygous for the A-allele (61). This research indicates that enhanced ACE activity is associated with depressive symptoms and increased susceptibility to affective disorders (61). In addition, a study based on the Iranian population showed that high serum ACE activity is associated with the pathogenesis of depression (62). The GG genotype of the A2350G polymorphism is associated with MDD and exhibits significantly higher serum ACE activity than AA or AG. Furthermore, certain variants of the ACE gene, such as the D allele, are more frequently noted in patients with affective disorders and associated with a higher risk of affective disorders (63). The D allele may be associated with the severity of depression in DD genotypes carriers of ACE I/D polymorphism (64).
In fact, in individuals with depression, the ACE I/D polymorphism is significantly associated with HPA axis hyperactivity. Patients with depression carrying the D/D variant of the ACE gene show considerably greater activation of the HPA axis (65). Cortisol secretion is increased in patients carrying homozygous T-alleles, showing higher HPA axis activity (61). In addition, the I/D polymorphism of the ACE gene is associated with both late-life depression and cortisol secretion (66).
Polymorphisms of the RAS-related gene variants have also been associated with a higher risk of depression (67). Saab et al. collected buccal cells from 132 patients with major depression and their first-degree relatives (case controls) in Lebanon (67). Their study showed that the angiotensin receptor type 1 (A1166C) CC genotype is more common in patients with depression, indicating that the CC genotype is significantly associated with depression (p=0.036) (67). A population-based cohort study found the ACE gene (rs1799752) is associated with the incidence of major depression in older individuals in followed up for over 12 years (68). Moreover, variation in the angiotensin II type 1 receptor has been linked with depression diagnosis and frontotemporal brain volumes (69). Two haplotype-tagging SNPs, rs10935724 (p=0.0487) and rs12721331(p=0.0082) showed statistically significant changes in frequency between diagnostic cohorts (69).
Clinically, the ACE I/D polymorphism seems to influence the therapeutic outcome in patients with depression, including the onset of action of antidepressant pharmacotherapies and the responses to selective serotonin reuptake inhibitors (SSRIs) (70), which plays an important role in the individualized treatment of depression. To investigate the impact of ACE2 gene variants on the antidepressant efficacy of SSRIs, a randomized, controlled trial was completed, involving 200 patients with newly diagnosed depression who underwent fluoxetine or sertraline for 6 weeks, along with ACE2 allele genotyping (71). The result showed that the patients with GA and AA genotypes respond significantly better to sertraline and confirm the role of G8790A in response to some SSRIs (71). Elevated levels of substance P are associated with mood symptoms, such as depression (72) and treatment with substance P receptor antagonists has antidepressant properties (73). DD allele carriers possessing higher ACE activity can promote the degradation of substance P, which may be related to having a positive impact on antidepressant treatment efficacy (63, 72). Another study conducted a survey among 313 patients with depression receiving various antidepressant treatments and found patients with the D/D and I/D genotypes have shorter hospitalization durations and better treatment outcomes than those with the I/I genotype (74). Surprisingly, when the 313 patients were classified by sex, the ACE I/D polymorphism only influences the therapeutic outcome in women with major depression, not men. This may be due to the sex-dependent influence of the ACE I/D polymorphism on therapeutic outcomes in antidepressant therapies through the influence of gonadal hormones (74). Based on this study, the D allele has a beneficial effect on the onset of therapy for depression and can be used as a predictor of faster onset of different antidepressant treatments, but the I-allele seemed to have a delayed effect on therapy (74). Another study enrolled 273 patients with MDD who received various antidepressant treatments and assessed the severity of depression with the Hamilton Depression Scale-17 (HAMD) before and after 4 weeks of therapy (75). That study revealed that patients carrying the D allele respond better to antidepressant treatment than those carrying other genotypes (75). More than 70% of AT1 CC homozygotes have a 50% reduction in the HAMD-17 scale within 4 weeks of antidepressant treatment, implying that patients with a haplotype combining the CC and DD/ID genotypes respond better to treatment than those with a single allele (75). Although the therapeutic outcome in various genotypes is related to pharmaceutical variety, the findings demonstrate that the ACE gene may generate varied antidepressant effects.
Inflammation is triggered by cellular damage caused by infection or injury. The term “neuroinflammation” refers to an immune-related process that occurs within the brain and spinal cord as a result of harm induced by infection, psychological or physical stress, or indirectly as a result of infection emerging in the periphery (76). Subsequently, innate immune cells in the brain (microglia, astrocytes, and oligodendroglia) are activated in response to inflammatory stimuli including the production of cytokines, chemokines, and secondary inflammatory mediators such as prostaglandins (77).
The brain lacks T- and B-cells that are involved in cellular and humoral immunity but contain innate immune cells, including macrophages and dendritic cells (78). Macrophages in the brain referred to as microglia, are the primary immune cells and contain surface membrane receptors recognizing neurotransmitters and hormones (79). Microglia respond to local and systemic inflammatory stimuli by producing pro-inflammatory cytokines (PIC), including interleukin-1α and β (IL-1α and IL-1β), tumor necrosis factor α (TNF-α), and interleukin-6 (IL-6) (79). In addition, microglia produce the anti-inflammatory cytokines IL-10 and transforming growth factor (TGF)-β. Activated microglia also trigger a chain reaction between chemokines, prostaglandins, and NO. In addition to the direct promotion of brain inflammation, chemokines such as CCR2 promote the recruitment of peripheral immune cells into the brain, thereby increasing the effects of inflammation (80).
Appropriate central inflammatory responses are essential to protect the brain from infection and restore homeostasis; however, prolonged inflammation is harmful. Long-term neuroinflammation leads to the activation of peripheral macrophages and central microglia and nerve dysfunction (76). Moreover, excessive responses may lead to decreased levels of brain-derived neurotrophic factors chronic inflammation, and neuronal damage. Sustained activation of the immune response increases inflammation and nitro-oxidative stress, ultimately leading to changes in monoamine levels that increase the risk of many neurological and psychiatric disorders (81). Such a procedure most likely occurs in chronic psychiatric disorders such as depression (82).
In recent years it has become apparent that inflammation is associated with psychiatric disorders. For example, depression is characterized by a chronic low-grade inflammatory state, increased levels of peripheral inflammatory cytokines, and microglial activation (83–85). Clinically, high levels of inflammatory markers are associated with the development of depression (83). In the general population, an elevated C-reactive protein (CRP) level is linked to a higher risk of developing depression (86). Elevated levels of inflammatory cytokines have been observed in both peripheral and cerebrospinal fluid in patients with depression (87). Particularly, elevated levels of circulating pro-inflammatory mediators have been found in patients with treatment-refractory depression (TRD), including TNF-α, IL-6, IL-1β, CRP, and macrophage inflammatory protein-1 (88).
Consequently, it has been hypothesized that medications that suppress levels of pro-inflammatory cytokines might also contribute to treating depression (87, 89). Some antidepressants with known anti-inflammatory effects have been shown to reduce the level of IL-6 and IL-1β in patients with MDD (88, 90). The use of anti-cytokine and anti-tumor necrosis factor drugs (e.g. infliximab, etanercept, and adalimumab) has been associated with significant improvements in depressive symptoms (91). LPS promotes the activation of microglia and induces depressive-like symptoms, while treatment with anti-inflammatory medication alleviates depressive symptoms (92, 93).
In addition to the regulation of blood pressure, the RAS is also an important regulator of the inflammatory states in the nervous system (94). Excessively elevated Ang II levels enhance plasma cytokine levels such as IL-6, interferon-γ (IFN-γ), TNF-α, and IL-1β (95). IL-6 levels increase the most after Ang II infusion, and plasma IFN-γ levels also increases significantly (96). Cytokine expression is controlled at the transcriptional level by pro-inflammatory transcription factors, such as nuclear factor kappa B (NF-κB) and activator protein-1 (AP-1) (97). Ang II induces the differentiation of immune cells and promotes the production of cytokines through NF-κB and/or AP-1, initiating an inflammatory cascade that leads to microglial activation (98). These findings suggest that the RAS has an intimate and complex regulatory role in the immune system.
ARBs have been proven to effectively inhibit inflammation by reducing gene expression of brain pro-inflammatory cytokines (99)(Table 3). Ang II facilitates the production of IL-1β and NO. This effect is reversed by losartan, which inhibits NF-κB and AP-1 (110). Administration of candesartan reduces brain AT1R synthesis and inhibits LPS-induced acute brain inflammation throughout the inflammatory cascade, including decreased production and release to the circulation of centrally acting pro-inflammatory cytokines; reduction of brain pro-inflammatory cytokines, cytokine, and prostanoid receptors; and reduced microglial activation (105, 106). Pretreatment with candesartan (1 mg/kg/d, for 3 d before the LPS treatment) lessens LPS-induced ACTH and corticosterone release and reduces gene expression of cyclooxygenase-2 (COX-2), IL-6, and TNF-α (105). Moreover, candesartan prevents the synthesis and release of the pro-inflammatory hormone aldosterone (108). In the pituitary, candesartan decreases the expression of the genes for IL-6, iNOS, and COX-2 (109). It also lessens the release of inflammatory markers such as TNF-, IL-1, and IL-6 in the circulation (109). AT1 receptor blockades are demonstrated to provide superior neuroprotective properties to ACE inhibition (107). In the rat model of neuroinflammation, candesartan (1 nM) inhibits LPS-induced neuroinflammation more effectively even at lower dosages and increases AT2R and anti-inflammatory IL-10 expression than perindopril (1 μM) (107). Systemic administration of telmisartan directly ameliorates the IL-1β-induced neuronal inflammatory response and inhibits oxidative stress (102). Administration of telmisartan attenuates chronic intermittent hypoxia (CIH)-induced neuronal apoptosis and decreases levels of CD45 (leukocyte common antigen), CRP, and IL-6 in the hippocampus and circulation through inhibiting inflammatory response (103). Intranasal administration of telmisartan (1 mg/kg; two months) significantly reduces glial activation in the brain and ameliorates the synthesis of NO, iNOS, TNF-α, as well as IL1-β (104).
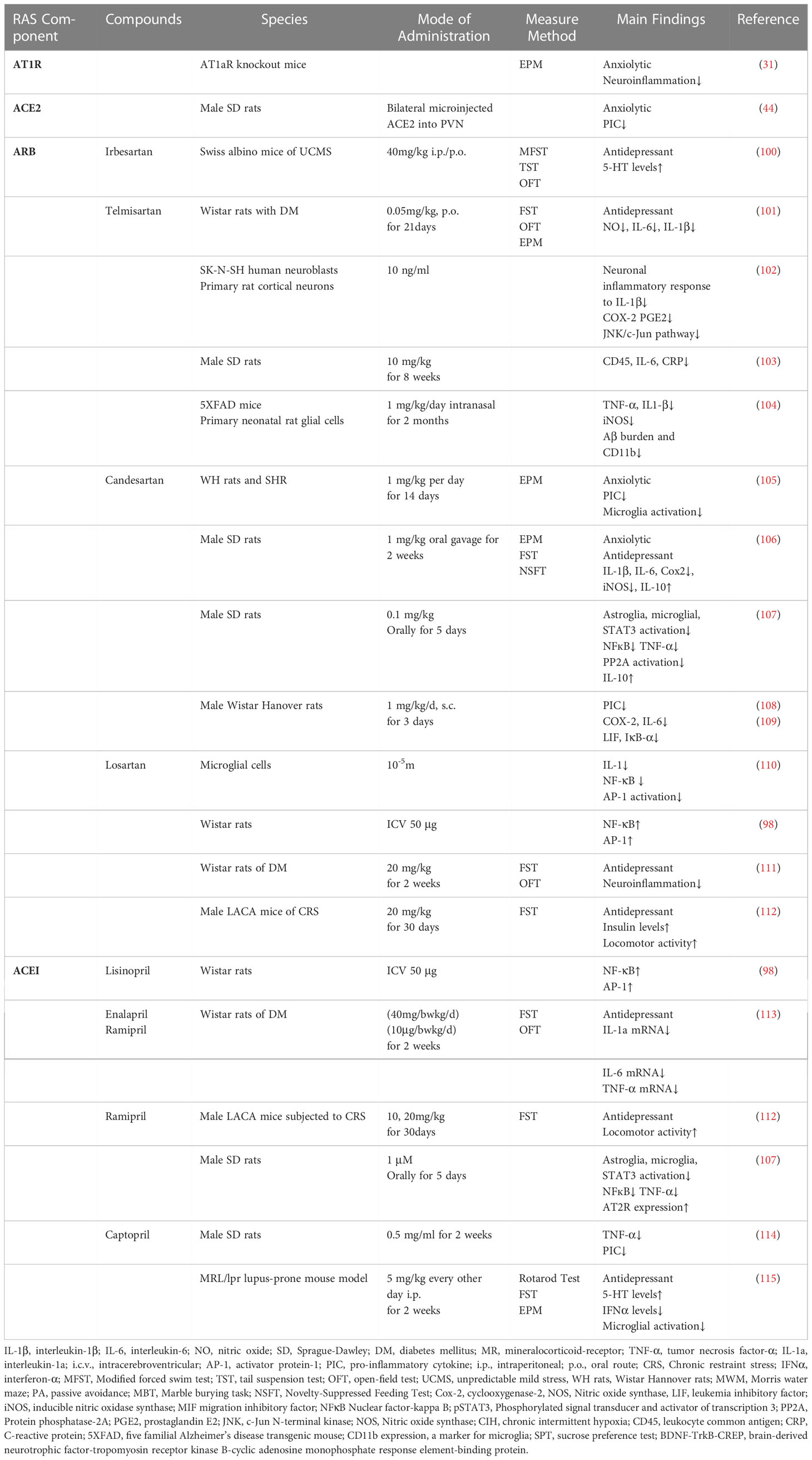
Table 3 RAS blockers inhibit inflammation.
Consequently, it is hypothesized that medications with anti-inflammatory effects might also have antidepressant potential. Losartan and ramipril can reverse depression-like behaviors in restraint-stressed mice and insulin resistance through anti-inflammatory mechanisms (112). Administration of irbesartan reduces the level of inflammatory mediators and reverses Ang II-induced depressive-like behaviors as manifested by decreased immobility times in the modified forced swim test (MFST) and the TST (100). Pretreatment with losartan significantly improves FST performance and prevents LPS-induced anhedonia and anxiety-like behaviors in addition to preventing LPS-induced higher levels of the pro-inflammatory cytokine (TNF, IL-1, and IL-6) (99, 116). A model of diabetes-associated depression rats exhibited depression-like behavior, which can be therapeutically reversed by losartan (20 mg/kg) via altering diabetes-induced neuroinflammatory responses (111). Telmisartan effectively reduces the concentration of pro-inflammatory mediators, including NO, IL-6, and IL-1β, in depressed rats with diabetes (101). Moreover, in a rat model of post-traumatic stress disorder (PTSD), treatment with captopril decreases pro-inflammatory cytokines levels and inhibits microglial activation in the hypothalamus (114). More importantly, the anxiolytic/antidepressant effects of RAS blockers may be mediated by their anti-inflammatory effects, providing new treatment directions.
Oxidative stress occurs when there is an imbalance between the production of reactive oxygen species (ROS) and endogenous antioxidant enzymes. Antioxidant enzymes, such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPX), maintain low levels of ROS in vivo. Excessive ROS generation and exhaustion of anti-oxidative defense-produced pro-inflammatory mediators results in damage to vital macromolecules and induces cellular apoptosis (117). Another major consequence of ROS-derived damage is lipid peroxidation. The brain is particularly susceptible to damage from reactive oxygen species because of its elevated oxygen consumption and lower levels of endogenous antioxidant enzymes (118).
Many studies have highlighted associations between oxidative stress and the development of affective disorders, and that antioxidants can improve symptoms of emotional disorders (119). Increased levels of oxidative stress markers, pro-inflammatory cytokines, and lipid peroxidation are observed in patients with anxiety and depression (120). Depression patients have significantly higher levels of F8-isoprostanes and lower GPX activity, two markers of oxidative stress, compared to healthy controls (121). Moreover, antioxidant treatment improves diabetes-induced depressive-like behaviors, increases levels of antioxidant enzymes CAT and SOD in brain tissue, and reduces oxidative stress in the hippocampus (118). Additionally, the administration of antioxidants shows an anxiolytic effect (122, 123). F2-isoprostanes and oxidized glutathione are positively associated with total Hamilton Anxiety ratings and the severity of anxiety in MDD (124). Moderate treadmill exercises prevent anxiety-like behaviors and the production of oxidative stress markers in the hippocampus, amygdala, and locus coeruleus (125). Importantly, antioxidants have the same effects as treadmill exercises, performing an anxiolytic effect (125), indicating that oxidative stress metabolites play an important role in mood disorders.
RAS overactivation is involved in oxidative stress via increasing Ang II levels and oxidative stress in the central nervous system is associated with depression (126) (Figure 1B). Ang II induces the production of superoxide anion and impairs cerebral microvascular endothelial function in vivo (127). Ang II stimulates inflammatory responses in the microvascular endothelium of the brain through AT1R, allowing more interaction between immune cells and the endodermis, and in turn, leading to disrupted BBB permeability partly via oxidative stress cascades (128). Ang II increases leukocyte adhesion 2.6-fold and BBB permeability 3.8-fold in male mice via oxidative stress-mediated cerebral microvascular inflammation (128). Furthermore, Ang II directly increases the production of ROS and subsequently induces lipid peroxidation and modification (129).
After systemic inflammation in the brain, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and inducible nitric oxide synthase (iNOS) activities rise, resulting in an accumulation of ROS and NO (126). During inflammation, Ang II promotes the oxidative stress process by increasing NADPH oxidase and iNOS activity in the BBB and PVN (126), and ARBs attenuate iNOS activity (126)(Table 4). Moreover, pretreatment with losartan at 3 mg/kg attenuates NO metabolite accumulation in hippocampal and cortical tissues (116). Treatment with telmisartan attenuates CIH-induced neuronal apoptosis in the hippocampus by suppressing NOS activity and inhibiting excessive NO generation (103). In addition, RAS blockers have a positive effect on depression as a comorbidity. Administration of RAS blockers prevents indices of systemic oxidative/nitrosative stress increased in rats with diabetes mellitus by inhibiting oxidative stress (131). Treatment with perindopril reduces severe acute respiratory syndrome-related coronavirus 2 spike protein-induced inflammatory and oxidative stress responses in cells and significantly blunted apoptosis and ROS (130).
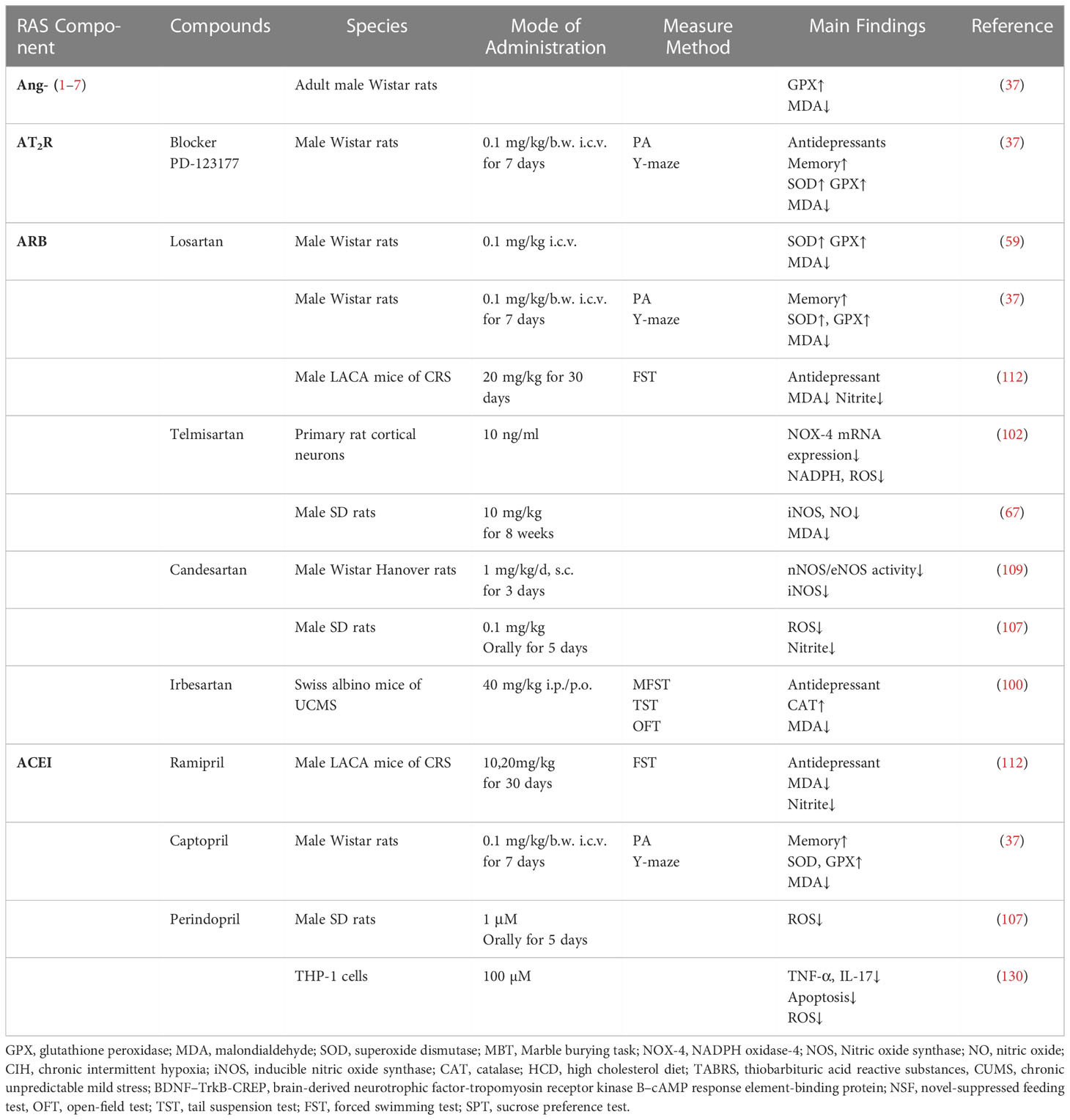
Table 4 RAS blockers inhibit oxidative stress.
The limbic system, comprised primarily of the amygdala and hippocampus, includes widely distributed AT1 receptors and is sensitive to oxidative stress 184). Oxidative stress upregulates angiotensin-1 receptor levels and elevates NF-κB-mediated pro-inflammatory factors levels (IL-6, TNF-α) in these brain areas, leading to anxiety-like behaviors (132, 133). Candesartan significantly inhibits nuclear translocation of NF-κB expression, while also decreasing ROS levels and increasing IL-10 levels in the cortex and hippocampus (133). Captopril and losartan reverse the disruption of BBB permeability and prevent Ang II-induced enhancement of oxidative stress in the hippocampus (59, 134). Moreover, captopril and losartan decrease lipid peroxidation levels, reduce anxiety-like behaviors, and increase antioxidant enzymes including SOD and GPX (59, 134). Furthermore, antioxidant treatment improves Ang II-induced disruption of BBB permeability and prevented anxiety-like behaviors in rats (135). LPS injection raises the concentration of malondialdehyde (MDA), a marker of lipid peroxidation, in hippocampus tissue (99). Losartan prominently increases the activity of antioxidant enzymes and reduces lipid peroxidation, such as MDA (99, 136). Pretreatment with losartan (3 mg/kg) significantly decreases MDA levels and reverses the negative effects of LPS on the activity of CAT and SOD in the hippocampal and cortical tissues (116). Consistent with losartan, telmisartan suppresses CIH-induced lipid peroxidation and overexpression of inflammatory mediators in the hippocampus (103). Irbesartan, alone or in combination with fluoxetine, significantly decreases the levels of thiobarbituric acid reactive substances, CAT, and MDA, and reverses the reduction in GSH levels in unpredictable chronic mild stress (UCMS) mice (100). Taken together, RAS blockers improve emotional disorders through anti-oxidative stress effects.
In response to stress stimulation, the subgenual prefrontal cortex is suppressed and the amygdala is activated, which activates the HPA axis (137). As the major stress mediator and crucial regulatory center of the neuroendocrine system, HPA releases corticotropin-releasing factor (CRF) to regulate stress responses (138). When the HPA axis is activated, CRF is released from the paraventricular nucleus(PVN) into the portal circulation, stimulating the pituitary to produce and release adrenocorticotropic hormone (ACTH). ACTH further stimulates the glucocorticoid (GC) hormone cortisol synthesis and release by the adrenal cortex (139). Interestingly, increased CRF in the cerebrospinal fluid and hyperactivation of the HPA axis have been reported in both anxiety and depression (140, 141).
Corticotrophin-releasing factor or hormone (CRH) is a 41 amino acid neuropeptide that mediated the neuroendocrine, immunological, autonomic, and behavioral responses to stress (142). In various animal models of anxiety disorders, centrally injected CRF induces anxiety-like responses such as sleep disturbances, loss of appetite, and anhedonia (143). CRF antagonists show anxiolytic effects in a variety of animal models (143). In addition, a large literature indicates that stress responses upregulate the transcription and expression of Ang II-related receptors and enhance the expression of central Ang II (144–146). Ang II, as a stress hormone, increases the expression of CRF mRNA through AT1R, which contributes to increasing the expression of CRF receptors and promoting CRF release during stress (147, 148). Ang II stimulates ACTH and GC secretion through pituitary AT1R and/or activates Ang II afferent terminals innervating present in PVN neurons enhancing the stimulatory effect of CRF (149, 150)(Figure 1C).
The various stressors enhance Ang II levels and affect the expression of receptors. Pavel reported that ARBs prevent the response of the HPA axis to isolate stress and reduce the expression of CRH receptors and benzodiazepine receptors, demonstrating that ARBs exert powerful anti-anxiety properties by downregulating CRH receptor type and benzodiazepine receptors in stress models (151, 152). Saavedra et al. pretreated rats for 13 days with candesartan (0.5 mg/kg/day) followed by 24 h of isolation in metabolic cages, and they found that candesartan blocks the stress-induced augments of CRF in the cortical and prevents benzodiazepine receptors from binding in the paraventricular nucleus and cortex (153). Injecting Ang II-induced depressive-like behaviors via microglia activation and activates the HPA axis (28), pretreatment with candesartan (1.0mg/kg/days for 14 days) attenuates the response of the HPA axis to stress and reduces cold restraint stress (placed in plastic restraining devices and maintained at 4°C for 2 h)-induced ulceration of the gastric mucosa eventrally (154). Candesartan increases gastric blood flow by 40-50% and prevents gastric ulcer formation by 70-80% after cold-restraint stress (154). Administration of losartan effectively attenuates the stress-induced fear memory impairment and prevents the development of depression-like behaviors caused by chronic mild unpredictable stress (CMS) (116, 155). Low doses of candesartan completely reverse chronic restraint stress (2 h/21 days in tight plastic tubes)-induced memory deficits (156). Isolation stress increases AT1 receptor binding in the PVN and anterior pituitary. The administration of candesartan is sufficient to block the isolation stress (24h isolation in individual metabolic cages)-induced increased binding of AT1R to PVN and to reduce the HPA response to stress (157)(Table 4).
Systemic administration of angiotensin II receptor antagonist inhibits stress-induced anxiety (158). Ovariectomized rats treated with losartan decreased plasma corticosterone levels (p<0.05) and AT1R mRNA expression in the CA3 region of the hippocampus (159). Administration of losartan improves anxiety responses in stressed rats via blockade of the AT1 receptor within the amygdala under both non-stress and acutely stressed rats (160). In sub-chronic swim stress models, pretreatment with losartan (10 mg/kg) decreases anxiety-like and stress behaviors as manifested by enhancing the tendency to spend more time in the center area in the OFT (43). Chronic stress lead to neuropsychiatric disorders, such as anxiety and depression, the neuroprotective effect of losartan alleviates chronic fatiguing stress-induced anxiety-like behaviors (161). Moreover, pretreatment of losartan reveres the chronic restraint stress-induced increased anxiety-like behaviors and decreased motor activity (162).
Empirical studies in animals show RAS blockers exert antidepressive effects. Treatment with losartan significantly abolishes the increased Ang II level and prevents the development of stress-induced depression-like behaviors in UCMS rats (163). Chronic administration of irbesartan significantly increases swimming and climbing times, decreases immobility times in the MFST, and decreases immobility time in the tail suspension test in UCMS rats (100). Administration with telmisartan for five weeks notably prevents the depression-like behaviors in OFT and sucrose preference test (SPT) in animals under chronic stress (164).
Serotonin (5-HT) is an important neuromodulatory transmitter and decreased 5-HT production may result in mood disturbance, aggression, and other neuropsychological impairment (165). There is an interaction between Ang II and 5-HT, particularly in the hippocampus, and brain Ang II regulates stress-related effects by modulating 5-HT release and synthesis (166, 167). The major serotonin metabolite 5-hydroxyindoleaceticacid (5-HIAA) is significantly elevated in the striatum after Ang II administration, indicating that Ang II increases the 5-HT levels (167). Furthermore, the AT1 receptor antagonist losartan reduces basal levels of 5HIAA (167). However, irbesartan (an ARB) increases time spent in the center of the OFT and elevates the 5-HT levels in UCMS rats (100), suggesting the biphasic response of Ang II on 5-HT synthesis (166). It is speculated that the biphasic response was related to the concentration of Ang II. Ang II stimulated tryptophan (TRP) hydroxylase at high concentrations to increase the synthesis of 5-HT. At low concentrations, an inhibitory effect is found, Ang II inhibits tryptophan hydroxylase resulting in decreasing 5-HT levels (166). Above all, it represents a subtle regulation of serotonin and the RAS system.
Perindopril (1.0 mg/kg/day) and candesartan (10 mg/kg/day) were administered via drinking water for 1 week, and serotonin levels increase in the prefrontal cortex and hippocampus, suggesting that decreased Ang II levels are associated with increased serotonin (168). Systemic administration of captopril increased serotonin levels and decrease depressive-like behaviors (115). In addition, captopril significantly increases the concentration of 5-HT and 5-HIAA in the parabrachial lateral nucleus (169). Administration with telmisartan for five weeks notably prevents the depression-like behaviors in OFT and SPT, and enhances expression of 5-HT transporter in the hippocampus of mice through activation of peroxisome proliferator-activated receptor δ, indicating RAS blockers improve stress-induced depressive symptoms in animals under chronic stress (164). Although these findings support that Ang II regulates stress by altering 5-HT levels in the brain, the association between Ang II and 5-HT in the formation of stress-related behaviors needs further investigation.
Stress generally triggers the autonomic nervous system, which is one of the major neural pathways (170). Depression is characterized by autonomic imbalance, with elevated sympathetic tone and weak parasympathetic tone, or both (171). A study of more than 600 subjects reports that depression and anxiety are shown to be more significantly and positively linked with the activation of the sympathetic nervous system (SNS), and depression is negatively correlated with parasympathetic nervous system (PNS) activation (172). SNS is stimulated in response to stress and increases the concentration of serum catecholamines to maintain body homeostasis (173). Indeed, patients with depression have been reported to have elevated catecholamines in plasma and cerebrospinal fluid (CSF) (174).
Norepinephrine (NE), one of the catecholamine hormones, is a major monoamine neurotransmitter that widely affects multiple brain regions (175). Noradrenergic hyperactivity has been shown to be an important component of the stress response and dysregulation of noradrenergic signaling has been implicated in the pathogenesis of anxiety and depression disorders. In fact, RAS has a complex bidirectional interaction with the autonomic nervous system activity under both physiological and pathophysiological conditions via receptors localized to peripheral and central sites of action (176). In the brain, Ang II increases sympathetic discharge and decreases vagal discharge. As a neuromodulator, Ang II stimulates ganglionic transmission and catecholamine release from adrenal medullary chromaffin cells and potentiated NE release from sympathetic nerve terminals in the periphery (177). In addition, there is some evidence that Ang II inhibits norepinephrine reuptake to promote neurotransmission (177). Administration of angiotensin-converting enzyme inhibitors can attenuate sympathetic neurotransmission and facilitate parasympathetic (175). In addition, Ang-(1–7) inhibits sympathetic tone and facilitates parasympathetic tone effects in experimental animal models, which will become an attractive treatment for autonomic nerve dysfunction (178).
In rat hypothalamic tissue, losartan partially reduces norepinephrine secretion (179). Also, noradrenaline levels are also shown to be significantly reduced in the striatum after chronic candesartan, although the mechanism is unclear (168). Ang II elevates catecholamines in the periphery and central, and pretreatment with Ang II receptor antagonism losartan significantly attenuates neuroendocrine responses, indicating Ang II activates sympathetic-adrenomedullary system activity through AT1R during stress (180). Although these findings support that Ang II regulated stress-related behaviors by altering NE levels, the association between Ang II and NE and sympathetic and parasympathetic in the formation of stress-related behaviors needs further investigation.
In clinical studies, elevated catecholamine activity impaired prefrontal cortex (PFC) function under stress and is associated with PTSD and other anxiety disorders (181). Chronic stress exposure leads to dendritic atrophy in PFC and enhances the noradrenergic NE system in the PFC (181). Cerebrospinal fluid norepinephrine concentrations are significantly higher in patients with chronic PTSD than in healthy subjects (182). Moreover, CSF norepinephrine concentrations are shown to be more significantly and positively link with the severity of PTSD symptoms, rather than plasma norepinephrine concentrations (182). Gold observed significant elevations in twenty-four-hour indices of norepinephrine secretion in both cerebrospinal fluid and plasma in severely depressed patients, compared with the control group (183). Clinical studies find that alpha-1 receptor blockers or alpha-2A receptor agonists can reduce the high concentration of NE release during stress, suggesting targeting the autonomic nervous system can be utilized as a pharmaceutical therapy for stress-related symptoms (181, 184). Taken together, the above finding indicates that RAS is involved in stress and that inhibiting RAS reduced the effects of stress.
Brain-derived neurotrophic factor (BDNF) is a neurotrophic factor bound to its receptor tropomyosin-related receptor kinase B (TrkB). BDNF is associated with the neurobiology of depression and antidepressant effects (185). BDNF plays an important role in neuronal growth, maturation, and survival and it mainly has the following functions (186): (1) increase synaptic plasticity and affect learning and memory. (2) promote neurogenesis, especially in the hippocampus. Further, the expression of BDNF is regulated via cyclic adenosine monophosphate response element-binding protein (CREB), and the phosphorylated cyclic adenosine monophosphate response element-binding protein (pCREB) level in the hippocampus was one of the pathogenesis of depression (187). TrkB signaling is essential for antidepressants, activating TrkB and increasing levels of BDNF (188). Hippocampal biopsies showed that individuals with major depression reveal lower levels of BDNF and its receptor TrkB, and long-term use of antidepressants promotes increased BDNF levels (111). Chauhan et al. revealed that in depressed patients with low BDNF levels, after four weeks of antidepressant treatment, the serum BDNF levels and depressive symptoms significantly improve (189).
BDNF exhibits a negative regulatory effect on brain inflammation along with inflammation and oxidative stress (83, 189). RAS performs an integral effect in mediating BDNF, which is essential in the neurobiology of depression and antidepressant effects (34). Decreased BDNF levels in the hippocampal are associated with depressive-like behaviors in UCMS rats and losartan minimizes depressive-like behaviors via modulating the BDNF pathway (113, 190)(Table 5). Losartan treatment significantly elevates TrkB and p-CREB protein levels and reduces NF-κB protein, IL-6, and TNF-α mRNA levels, and facilitates the BDNF-TrkB-CREB, indicating that the TrkB signal promoted neuronal survival (111). Losartan exerts neuroprotective effects by alleviating neuroinflammatory responses and elevating BDNF levels in astrocyte (111). Oral administration of candesartan ameliorates chronic neuroinflammation-induced behavioral changes and apoptosis by inhibiting Ang II-induced NF-κB inflammatory signaling and enhancing the phosphorylated CREB and BDNF expression level in the cortex and hippocampus regions (133). In the case of AT1R blockade, AT2R activation increases the expression of AT2R mRNA (192). The antidepressant-like effect of losartan may increase the binding of Ang II to AT2R by inhibiting AT1R, finally, increase the surface levels of TrkB and coupling of TRK/FYN in the hippocampus and ventral medial prefrontal cortex (vmPFC) prelimbic aspects (34).
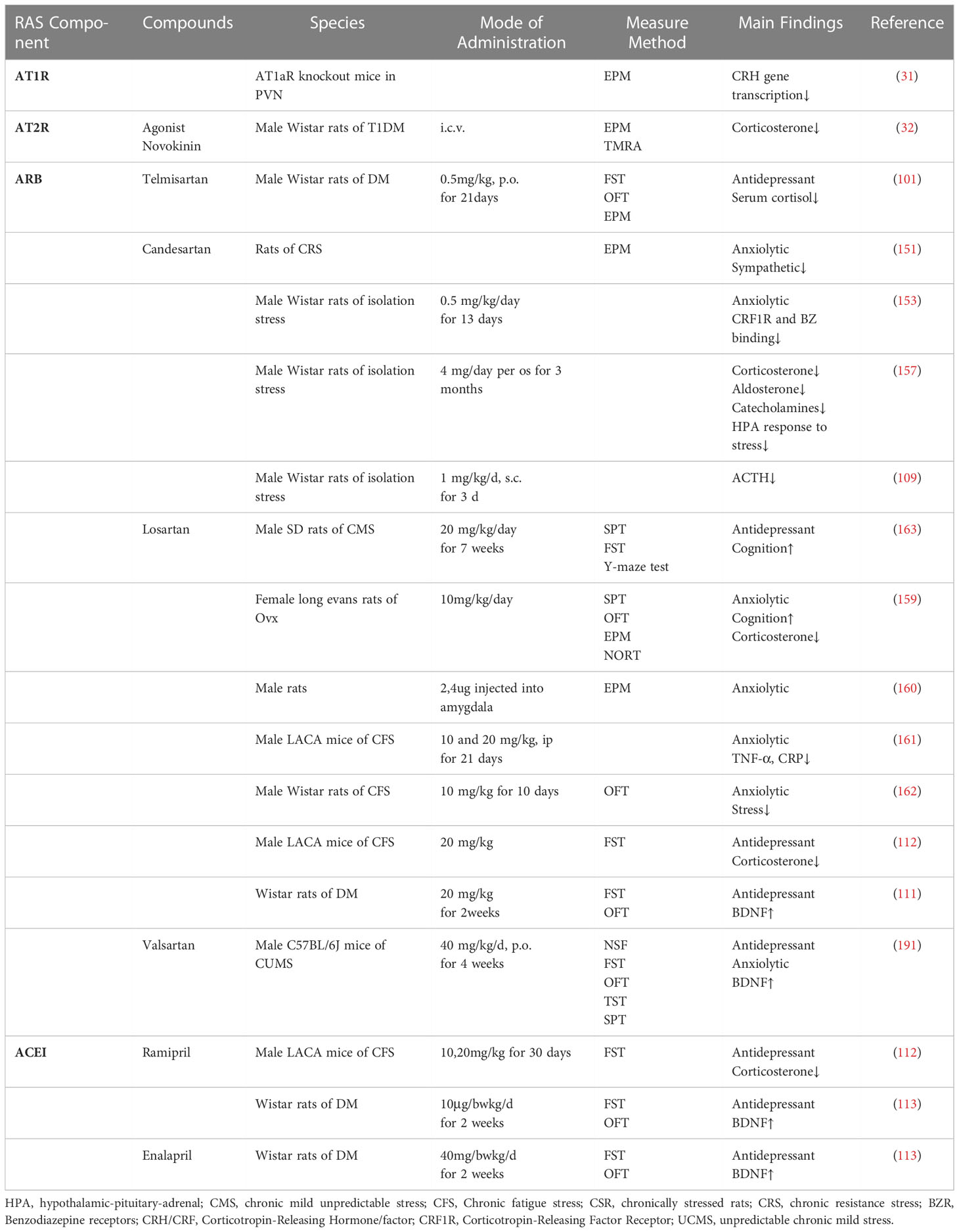
Table 5 RAS blockers reduce stress responses and elevate BDNF levels.
Reduced BDNF levels promote oxidative stress processes that lead to anxiety (120, 132). For example, social stress lowers BDNF and glutathione reductase levels, leading to oxidative stress-induced anxiety/depression-like behaviors in rats (193). Impaired hippocampal neurogenesis and reduced BDNF levels were observed in UCMS mice. Orally administration of valsartan (10–40 mg/kg/day, 4 weeks) promotes the hippocampus neurogenesis and the BDNF expression, exerting an antidepressant-like effect, which may be one of the mechanisms for its antidepressant (191). Importantly, the antidepressant-like property is dose-dependent, with the maximum effect obtained when valsartan is administered at a dose of 40 mg/kg/day (191).
To date, no randomized controlled trial has examined the impact of ACEIs or ARBs on depression. However, case reports and observational studies have demonstrated a bidirectional relationship between antihypertensive medications and depression, indicating mood-improving effects of RAS blockers, whereas other antihypertensive agents did not (Table 6). For example, losartan positively affects individuals with high-trait anxiety and prevents the development of anxiety disorders (205). In hypertensive individuals without a psychiatric history, discontinuation of valsartan (160 mg/day) is accompanied by anxiety symptoms such as palpitations, insomnia, and increased respiratory rate (206). These anxiety symptoms were significantly relieved with recontinuation of valsartan (80 mg/day) (206). A recent neuroimaging study showed that losartan prevents sustained amygdala activation in individuals with high-trait anxiety and leads to increased activation in other brain areas associated with threat processing, such as the insula and putamen (205). A recent study reports a significant association between the presence of an ACE inhibitor/ARB medication and decreased post-traumatic stress disorder symptoms compared with other blood pressure medications, including β-blockers, calcium channel blockers, and diuretics (207).
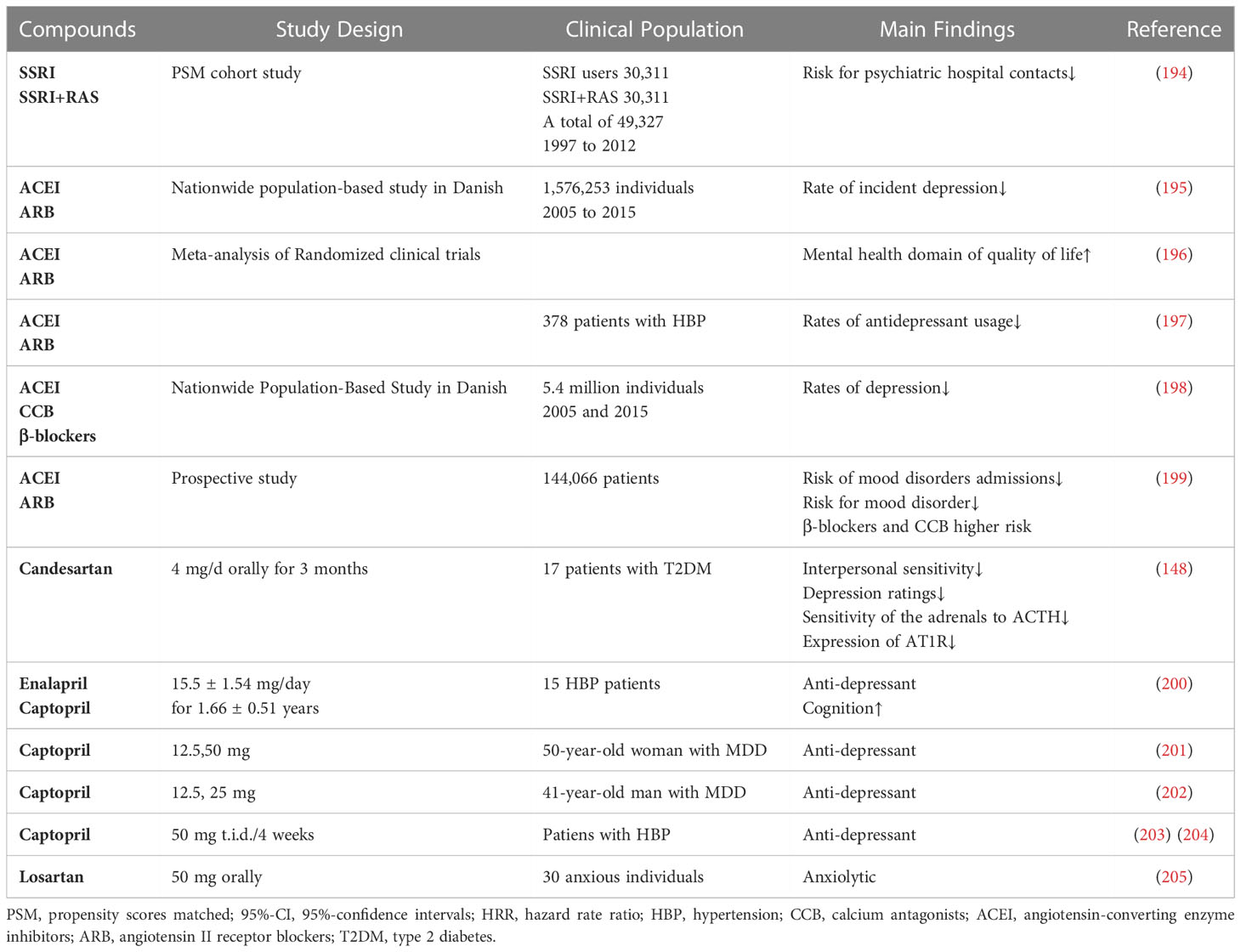
Table 6 Clinical data of RAS blockers.
Moreover, captopril and enalapril partly reverse the significant negative emotional effects of hypertension (12). Several cases have reported that captopril has mood-improving properties and antidepressant effects (202–204). Patients treated with captopril had significantly reduced total Hamilton Depression Scale (HAMD) scores and corrected neuroendocrine dysfunction during one-year follow-up (201). Moreover, captopril (12.5 mg b.i.d.2 weeks) decreases HAMD-21 scores and improves depressive symptomatology in patients with recurrent unipolar major depression (208). Captopril and enalapril improve depressive and anxiety symptoms in patients with hypertension (200). Hertzman reported that ten patients who suffered from hypertension and mood disorders (major depressive disorder or bipolar disorder) had improved mood with a combination of antidepressants and lisinopril, and no serious negative effects were reported by any of these patients while using lisinopril (209). A clinical trial reported that ACE inhibitors reduced the likelihood of depression risk and significantly improved general well-being, work performance, and cognitive function in 625 white men with mild hypertension administered captopril for 6 months (210). Two prospective multicenter randomized trials have shown that captopril has a significant tendency to reduce depressive symptoms compared with other antihypertensive drugs (211). Subsequently, another ACE inhibitor (enalapril) showed a significant improvement in health-related quality of life (HRQoL) compared to selective beta-blockers, although the overall tolerability of the two drugs was similar (212). In a clinical trial that enrolled 387 subjects aged 75+ years with hypertension in Italy, the results showed that the use of ACE inhibitors was associated with significantly better HRQoL among older adults (213). A case-control study enrolled 972 patients with both diabetes and depression, and the results suggest that those given ACEIs show a lower odds ratio for depression (OR 1.3, 95%confidence interval (95%CI):0.8–2.2) compared to beta-blockers (OR 2.6, 95% CI:1.1–7.0) and calcium channel blockers (OR 2.2, 95% CI:1.2–4.2) (214). Patients with type 2 diabetes and depressive symptoms received chronic candesartan administration for at least three months, which significantly improved interpersonal sensitivity and depression ratings and reset the HPA axis by reducing the sensitivity of the adrenals to ACTH and expression of AT1R (148).
In clinical and cohort studies, targeted RAS compounds have neuropsychiatric advantages through their anti-inflammatory properties, especially in depression (215). A Danish nationwide population-based study enrolled 1 576 253 subjects exposed to one of the six drugs(low-dose aspirin, statins, allopurinol, ACEIs, ARBs, and non-aspirin non-steroidal anti-inflammatory drugs) during the exposure period from 2005 to 2015. The incidence of depression decreased in patients taking ACEIs and ARBs, suggesting that these agents may act on inflammation and the stress response system (195). A recent meta-analysis identified 11 randomized controlled trials on the effects of antihypertensives on mental health and reported that ACEI and AT1R blockers have better effects on quality of life, anxiety, and mental well-being than placebo and other antihypertensives (196). Nasr analyzed data from 378 patients whom both suffered from hypertension and depression and found that patients being treated with an ACEI or ARB showed significantly lower rates of antidepressant usage, while beta-blocker and calcium channel blocker usage led to the highest rate of antidepressant usage (197). Consistent with these studies, Boal et al. analyzed cohort data and found that the use of RAS blockers in patients was associated with a low prevalence of depressive symptoms. The risk of affective disorders is reduced with the usage of ACEIs or Ang II receptor blockers, whereas the risk is increased with the administration of beta-blockers or calcium channel blockers (199). A nationwide population-based study showed that 3747190 subjects were exposed to antihypertensive drugs between 2005 to 2015. The findings of this study suggest that patients who continue to use classes of angiotensin agents, calcium antagonists, and β-blockers have significantly decreased rates of depression, whereas those using diuretic medication do not (198). A retrospective cohort study to investigate the distinct effects of antihypertensives on depression enrolled 181,709 patients and found that other antihypertensives may have a negative effect on the risk of depression compared with ARBs (216). Furthermore, 58.6 million patients aged 18–90 years were enrolled to study the influence of antihypertensive drugs on the onset and recurrence of psychiatric disorders (217). This study showed that ARB users exhibited the lowest incidence of psychotic, affective, and anxiety disorders for the first and recurrent diagnoses compared with CCB and β-blockers (217). However, there are limited randomized clinical trials on ACEI- and ARB-targeting medications and depression, which raises the possibility that future clinical research should conduct trials to examine these apparent advantages.
Despite the success demonstrated, a significant limitation is that not all research has revealed that ACEIs and ARBs have positive benefits on mood disorders, and these opposing results provide a new perspective for future clinical therapy. A small double-blind pilot study of eight patients was conducted to assess whether captopril has euphoric effects in the treatment of hypertension and found that the administration of captopril failed to elevate depressive mood (218). This result may be due to the small sample size of this study. A case report shows the dose of captopril is increased from 25 mg t.i.d. to 37.5 mg q.i.d. and the patient becomes intensely dysphoric within 2 days (203). When the captopril dose was raised to 37.5 mg q.i.d, anxiety and dysphoria reappeared. Furthermore, the dose was increased to 50 mg q.i.d., and the patient exhibited incoherent speech and suicidal ideation. It should be noted that although several studies have found that captopril has a positive effect on depression, its anxiolytic effect may be dose-dependent. In future studies, more attention should be paid to the frequency and different dosages of RAS blockers and the negative effects of different doses on mood.
In contrast, some epidemiological studies highlight concerns regarding possible links between the use of RAS blockers and increased risk of suicide, although the underlying mechanisms remain unknown (49). Mamdani et al. carried out a population-based nested case-control study, which included 964 patients and 3856 controls. The study found that ARB exposure is associated with a higher risk of suicide compared with ACEI (adjusted odds ratio, 1.63; 95% CI, 1.33–2.00). The preferred use of ACEI instead of ARB should be explored whenever possible, especially in individuals with severe mental disorders (219). Nonetheless, when a subsequent analysis was performed in 2020, a nationwide population-based propensity score matching study demonstrated that ARB use was not associated with an increased risk of suicide compared to non-ARB use (220). In summary, evidence from epidemiological studies suggests that the relationship between RAS medication use and suicide is inconsistent. Even if a direct cause-and-effect link exists, it is difficult to prove whether higher or lower RAS contributes to increased suicide risk.
Furthermore, most studies investigating the role of RAS in mood disorders have focused on pharmacological compounds that target ACE or AT1 receptors. Recently, several novel pharmacological compounds have been discovered, including ACE2 activators, Mas receptor agonists, AT2 receptor agonists, and renin blockers (221, 222), and further work to understand their roles is required.
A growing body of experimental and clinical data highlights the important role of the RAS in the pathophysiology of mood disorders. In this review, we presented an overview of the RAS, which consisted of two mutually antagonistic pathways that maintain balance through ACE2. The ACE/Ang II/AT1R classical pathway aggravates depression and anxiety by activating AT1R, while the non-classical pathway exerts anxiety/antidepressant effects by activating MasR. Moreover, RAS, mainly Ang II, is involved in the pathological process of depression by promoting inflammation, oxidative stress, and stress responses and reducing BDNF levels. Similarly, agents that inhibit RAS reduce inflammation, oxidative stress, and stress responses and facilitate neurogenesis. This may be the underlying mechanism of RAS blocker treatment for anxiety and depression. The full potential of RAS blockers as antidepressants and anti-anxiety drugs has not yet been elucidated. Hence, in future work, large-scale, randomized, controlled clinical trials are necessary to evaluate the therapeutic efficiency of RAS compounds in emotional disorders. RAS blockers need to be tested as potential therapies for emotional disorders, such as comorbid cardiovascular/cerebrovascular disease and depression. Furthermore, the role of RAS blockers in males and females in emotional disorders and pharmaceutical dosage in men and women should be carefully established.
Thus, RAS blockers may be a promising strategy for the treatment of mood disorders in the future. However, to realize the full therapeutic potential of RAS in mood disorders, further research is required.
SG performed information collection and drafted the manuscript. FD supervised the review and approved the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the National Natural Science Foundation of China [grant number 82071293].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Machado MO, Veronese N, Sanches M, Stubbs B, Koyanagi A, Thompson T, et al. The association of depression and all-cause and cause-specific mortality: an umbrella review of systematic reviews and meta-analyses. BMC Med (2018) 16(1):112. doi: 10.1186/s12916-018-1101-z
2. Clark DA, Beck AT. Cognitive theory and therapy of anxiety and depression: Convergence with neurobiological findings. Trends Cogn Sci (2010) 14(9):418–24. doi: 10.1016/j.tics.2010.06.007
3. Regier DA, Kuhl EA, Kupfer DJ. The DSM-5: Classification and criteria changes. World Psychiatry (2013) 12(2):92–8. doi: 10.1002/wps.20050
4. Young EA, Abelson JL, Cameron OG. Effect of comorbid anxiety disorders on the hypothalamic-pituitary-adrenal axis response to a social stressor in major depression. Biol Psychiatry (2004) 56(2):113–20. doi: 10.1016/j.biopsych.2004.03.017
5. Lamers F, van Oppen P, Comijs HC, Smit JH, Spinhoven P, van Balkom AJLM, et al. Comorbidity patterns of anxiety and depressive disorders in a large cohort study: the Netherlands study of depression and anxiety (NESDA). J Clin Psychiatry (2011) 72(3):341–8. doi: 10.4088/JCP.10m06176blu
6. Babaev O, Piletti Chatain C, Krueger-Burg D. Inhibition in the amygdala anxiety circuitry. Exp Mol Med (2018) 50(4):1–16. doi: 10.1038/s12276-018-0063-8
7. Drevets WC, Price JL, Bardgett ME, Reich T, Todd RD, Raichle ME. Glucose metabolism in the amygdala in depression: relationship to diagnostic subtype and plasma cortisol levels. Pharmacol Biochem Behav (2002) 71(3):431–47. doi: 10.1016/S0091-3057(01)00687-6
8. Frodl T, Meisenzahl E, Zetzsche T, Bottlender R, Born C, Groll C, et al. Enlargement of the amygdala in patients with a first episode of major depression. Biol Psychiatry (2002) 51(9):708–14. doi: 10.1016/S0006-3223(01)01359-2
9. Smoller JW. The genetics of stress-related disorders: PTSD, depression, and anxiety disorders. Neuropsychopharmacology (2016) 41(1):297–319. doi: 10.1038/npp.2015.266
10. Luo H, Wu PF, Cao Y, Jin M, Shen TT, Wang J, et al. Angiotensin-converting enzyme inhibitor rapidly ameliorates depressive-type behaviors via bradykinin-dependent activation of mammalian target of rapamycin complex 1. Biol Psychiatry (2020) 88(5):415–25. doi: 10.1016/j.biopsych.2020.02.005
11. Welcome MO, Mastorakis NE. Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways. Pharmacol Res (2020) 157:104769. doi: 10.1016/j.phrs.2020.104769
12. Karwowska-Polecka W, Halicka D, Jakubów P, Braszko JJ. [The effect of enalapril and captopril on emotional processes in hypertensive patients]. Psychiatr Pol (2002) 36(4):591–601.
13. Robles NR, Cerezo I, Hernandez-Gallego R. Renin-angiotensin system blocking drugs. J Cardiovasc Pharmacol Ther (2014) 19(1):14–33. doi: 10.1177/1074248413501018
14. Turnbull F. Blood pressure lowering treatment trialists’ collaboration. effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. Lancet (2003) 362(9395):1527–35. doi: 10.1016/s0140-6736(03)14739-3
15. Miller AJ, Arnold AC. The renin-angiotensin system in cardiovascular autonomic control: recent developments and clinical implications. Clin Auton Res (2019) 29(2):231–43. doi: 10.1007/s10286-018-0572-5
16. Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7). Hypertension (2002) 40(5):774–9. doi: 10.1161/01.HYP.0000035396.27909.40
17. Yousif MHM, Dhaunsi GS, Makki BM, Qabazard BA, Akhtar S, Benter IF. Characterization of angiotensin-(1-7) effects on the cardiovascular system in an experimental model of type-1 diabetes. Pharmacol Res (2012) 66(3):269–75. doi: 10.1016/j.phrs.2012.05.001
18. Biancardi VC, Stern JE. Compromised blood-brain barrier permeability: novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension: AngII-mediated BBB breakdown. J Physiol (2016) 594(6):1591–600. doi: 10.1113/JP271584
19. Sabuhi R, Ali Q, Asghar M, Al-Zamily NRH, Hussain T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: opposing effects in lean and obese zucker rats. Am J Physiol Renal Physiol (2011) 300(3):F700–706. doi: 10.1152/ajprenal.00616.2010
20. Saavedra JM, Ando H, Armando I, Baiardi G, Bregonzio C, Juorio A, et al. Anti-stress and anti-anxiety effects of centrally acting angiotensin II AT1 receptor antagonists. Regul Pept (2005) 128(3):227–38. doi: 10.1016/j.regpep.2004.12.015
21. Gironacci MM, Vicario A, Cerezo G, Silva MG. The depressor axis of the renin-angiotensin system and brain disorders: a translational approach. Clin Sci (Lond) (2018) 132(10):1021–38. doi: 10.1042/CS20180189
22. Fraga-Silva RA, Pinheiro SVB, Gonçalves ACC, Alenina N, Bader M, Souza Santos RA. The antithrombotic effect of angiotensin-(1-7) involves mas-mediated NO release from platelets. Mol Med (2008) 14(1):28–35. doi: 10.2119/2007-00073.Fraga-Silva
23. Pereira MGAG, Souza LL, Becari C, Duarte DA, Camacho FRB, Oliveira JAC, et al. Angiotensin II-independent angiotensin-(1-7) formation in rat hippocampus: involvement of thimet oligopeptidase. Hypertension (2013) 62(5):879–85. doi: 10.1161/HYPERTENSIONAHA.113.01613
24. Armando I, Terrón JA, Falcón-Neri A, Takeshi I, Häuser W, Inagami T, et al. Increased angiotensin II AT1 receptor expression in paraventricular nucleus and hypothalamic-Pituitary-Adrenal axis stimulation in AT2 receptor gene disrupted mice. Neuroendocrinology (2002) 76(3):137–47. doi: 10.1159/000064525
25. Kangussu LM, Almeida-Santos AF, Bader M, Alenina N, Fontes MAP, Santos RAS, et al. Angiotensin-(1-7) attenuates the anxiety and depression-like behaviors in transgenic rats with low brain angiotensinogen. Behav Brain Res (2013) 257:25–30. doi: 10.1016/j.bbr.2013.09.003
26. Duchemin S, Belanger E, Wu R, Ferland G, Girouard H. Chronic perfusion of angiotensin II causes cognitive dysfunctions and anxiety in mice. Physiol Behav (2013) 109:63–8. doi: 10.1016/j.physbeh.2012.10.005
27. Gao N, Wang H, Xu X, Yang Z, Zhang T. Angiotensin II induces cognitive decline and anxiety-like behavior via disturbing pattern of theta-gamma oscillations. Brain Res Bull (2021), 174:84–91. doi: 10.1016/j.brainresbull.2021.06.002
28. Park HS, You MJ, Yang B, Jang KB, Yoo J, Choi HJ, et al. Chronically infused angiotensin II induces depressive-like behavior via microglia activation. Sci Rep (2020) 10(1):22082. doi: 10.1038/s41598-020-79096-2
29. Belcheva I, Georgiev V, Chobanova M, Hadjiivanova C. Behavioral effects of angiotensin II microinjected into CA1 hippocampal area. Neuropeptides (1997) 31(1):60–4. doi: 10.1016/S0143-4179(97)90021-4
30. Choy KHC, Chavez CA, Yu J, Mayorov DN. The effect of angiotensin AT1A inactivation on innate and learned fear responses in mice and its relationship to blood pressure. Psychoneuroendocrinology (2019), 107:208–16. doi: 10.1016/j.psyneuen.2019.05.004
31. Wang L, Hiller H, Smith JA, de Kloet AD, Krause EG. Angiotensin type 1a receptors in the paraventricular nucleus of the hypothalamus control cardiovascular reactivity and anxiety-like behavior in male mice. Physiol Genomics (2016) 48(9):667–76. doi: 10.1152/physiolgenomics.00029.2016
32. Pechlivanova D, Petrov K, Grozdanov P, Nenchovska Z, Tchekalarova J, Stoynev A. Intracerebroventricular infusion of angiotensin AT2 receptor agonist novokinin aggravates some diabetes-mellitus-induced alterations in wistar rats. Can J Physiol Pharmacol (2018) 96(5):471–8. doi: 10.1139/cjpp-2017-0428
33. Moreno-Santos B, Marchi-Coelho C, Costa-Ferreira W, Crestani CC. Angiotensinergic receptors in the medial amygdaloid nucleus differently modulate behavioral responses in the elevated plus-maze and forced swimming test in rats. Behav Brain Res (2021) 397:112947. doi: 10.1016/j.bbr.2020.112947
34. Diniz CRAF, Casarotto PC, Fred SM, Biojone C, Castrén E, Joca SRL. Antidepressant-like effect of losartan involves TRKB transactivation from angiotensin receptor type 2 (AGTR2) and recruitment of FYN. Neuropharmacology (2018) 135:163–71. doi: 10.1016/j.neuropharm.2018.03.011
35. Okuyama S, Sakagawa T, Chaki S, Imagawa Y, Ichiki T, Inagami T. Anxiety-like behavior in mice lacking the angiotensin II type-2 receptor. Brain Res (1999) 821(1):150–9. doi: 10.1016/S0006-8993(99)01098-7
36. Wang L, de Kloet AD, Pati D, Hiller H, Smith JA, Pioquinto DJ, et al. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central mas receptors. Neuropharmacology (2016) 105:114–23. doi: 10.1016/j.neuropharm.2015.12.026
37. Bild W, Ciobica A. Angiotensin-(1–7) central administration induces anxiolytic-like effects in elevated plus maze and decreased oxidative stress in the amygdala. J Affect Disord (2013) 145(2):165–71. doi: 10.1016/j.jad.2012.07.024
38. Almeida-Santos AF, Kangussu LM, Moreira FA, Santos RAS, Aguiar DC, Campagnole-Santos MJ. Anxiolytic- and antidepressant-like effects of angiotensin-(1–7) in hypertensive transgenic (mRen2)27 rats. Clin Sci (2016) 130(14):1247–55. doi: 10.1042/CS20160116
39. Zhu D, Sun M, Liu Q, Yue Y, Lu J, Lin X, et al. Angiotensin (1–7) through modulation of the NMDAR–nNOS–NO pathway and serotonergic metabolism exerts an anxiolytic-like effect in rats. Behav Brain Res (2020) 390:112671. doi: 10.1016/j.bbr.2020.112671
40. Walther T, Balschun D, Voigt JP, Fink H, Zuschratter W, Birchmeier C, et al. Sustained long term potentiation and anxiety in mice lacking theMas protooncogene. J Biol Chem (1998) 273(19):11867–73. doi: 10.1074/jbc.273.19.11867
41. Voigt JP, Hörtnagl H, Rex A, van Hove L, Bader M, Fink H. Brain angiotensin and anxiety-related behavior: The transgenic rat TGR(ASrAOGEN)680. Brain Res (2005) 1046(1–2):145–56. doi: 10.1016/j.brainres.2005.03.048
42. Tashev R, Ivanova M. Involvement of hippocampal angiotensin 1 receptors in anxiety-like behaviour of olfactory bulbectomized rats. Pharmacol Rep (2018) 70(5):847–52. doi: 10.1016/j.pharep.2018.03.001
43. Ranjbar H, Aghaei I, Moosazadeh M, Shabani M. Angiotensin II type 1 receptor blocker losartan attenuates locomotor, anxiety-like behavior and passive avoidance learning deficits in a sub-chronic stress model. Iranian J Basic Med Sci [Internet] (2018) 21(8):856–62. doi: 10.22038/ijbms.2018.27113.6632
44. Sriramula S, Cardinale JP, Lazartigues E, Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res (2011) 92(3):401–8. doi: 10.1093/cvr/cvr242
45. de Kloet AD, Cahill KM, Scott KA, Krause EG. Overexpression of angiotensin converting enzyme 2 reduces anxiety-like behavior in female mice. Physiol Behav (2020) 224:113002. doi: 10.1016/j.physbeh.2020.113002
46. Mahon JM, Carr RD, Nicol AK, Henderson IW. Angiotensin(1-7) is an antagonist at the type 1 angiotensin II receptor. J Hypertens (1994) 12(12):1377–81.
47. Kangussu LM, Almeida-Santos AF, Moreira FA, Fontes MAP, Santos RAS, Aguiar DC, et al. Reduced anxiety-like behavior in transgenic rats with chronically overproduction of angiotensin-(1–7): Role of the mas receptor. Behav Brain Res (2017) 331:193–8. doi: 10.1016/j.bbr.2017.05.026.
48. Moura Santos D, Ribeiro Marins F, Limborço-Filho M, de Oliveira ML, Hamamoto D, Xavier CH, et al. Chronic overexpression of angiotensin-(1-7) in rats reduces cardiac reactivity to acute stress and dampens anxious behavior. Stress (2017) 20(2):189–96. doi: 10.1080/10253890.2017.1296949
49. Sanches M, Teixeira AL. The renin-angiotensin system, mood, and suicide: Are there associations? World J Psychiatry (2021) 11(9):581–8.
50. Srinivasan J, Suresh B, Ramanathan M. Differential anxiolytic effect of enalapril and losartan in normotensive and renal hypertensive rats. Physiol Behav (2003) 78(4–5):585–91. doi: 10.5498/wjp.v11.i9.581.
51. Coatl-Cuaya H, Tendilla-Beltrán H, de Jesús-Vásquez LM, Garcés-Ramírez L, Gómez-Villalobos M de J, Flores G. Losartan enhances cognitive and structural neuroplasticity impairments in spontaneously hypertensive rats. J Chem Neuroanat (2022) 120:102061. doi: 10.1016/j.jchemneu.2021.102061
52. Feng P, Wu Z, Liu H, Shen Y, Yao X, Li X, et al. Electroacupuncture improved chronic cerebral hypoperfusion-induced anxiety-like behavior and memory impairments in spontaneously hypertensive rats by downregulating the ACE/Ang II/AT1R axis and upregulating the ACE2/Ang-(1-7)/MasR axis. Neural Plast (2020) 2020:9076042. doi: 10.1155/2020/9076042
53. Braszko JJ. Valsartan abolishes most of the memory-improving effects of intracerebroventricular angiotensin II in rats. Clin Exp Hypertens (2005) 27(8):635–49. doi: 10.1080/10641960500298723
54. Huber G, Ogrodnik M, Wenzel J, Stölting I, Huber L, Will O, et al. Telmisartan prevents high-fat diet-induced neurovascular impairments and reduces anxiety-like behavior. J Cereb Blood Flow Metab (2021) 41(9):2356–69. doi: 10.1177/0271678X211003497
55. Martin P, Massol J, Puech AJ. Captopril as an antidepressant? effects on the learned helplessness paradigm in rats. Biol Psychiatry (1990) 27(9):968–74. doi: 10.1016/0006-3223(90)90034-Y
56. Jenkins TA, Chai SY. Effect of chronic angiotensin converting enzyme inhibition on spatial memory and anxiety-like behaviours in rats. Neurobiol Learn Memory (2007) 87(2):218–24. doi: 10.1016/j.nlm.2006.08.010
57. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin ii At1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: Therapeutic implications. Psychoneuroendocrinology (2011) 36(1):1–18. doi: 10.1016/j.psyneuen.2010.10.001
58. Giardina WJ, Ebert DM. Positive effects of captopril in the behavioral despair swim test. Biol Psychiatry (1989) 25(6):697–702. doi: 10.1016/0006-3223(89)90240-0
59. Ciobica A, Hritcu L, Bild W, Padurariu M, Bild V. P.4.b.011 effects of angiotensin II and its specific receptor antagonists on anxiety status and some oxidative stress markers in rat. Eur Neuropsychopharmacol (2011) 21:S538–9. doi: 10.1016/S0924-977X(11)70876-8
60. Jeunemaitre X. [Genetic polymorphisms in the renin-angiotensin system]. Therapie (1998) 53(3):271–7.
61. Baghai TC, Binder EB, Schule C, Salyakina D, Eser D, Lucae S, et al. Polymorphisms in the angiotensin-converting enzyme gene are associated with unipolar depression, ACE activity and hypercortisolism. Mol Psychiatry (2006) 11(11):1003–15. doi: 10.1038/sj.mp.4001884
62. Firouzabadi N, Shafiei M, Bahramali E, Ebrahimi SA, Bakhshandeh H, Tajik N. Association of angiotensin-converting enzyme (ACE) gene polymorphism with elevated serum ACE activity and major depression in an Iranian population. Psychiatry Res (2012) 200(2–3):336–42. doi: 10.1016/j.psychres.2012.05.002
63. Arinami T, Li L, Mitsushio H, Itokawa M, Hamaguchi H, Toru M. An insertion/deletion polymorphism in the angiotensin converting enzyme gene is associated with both brain substance p contents and affective disorders. Biol Psychiatry (1996) 40(11):1122–7. doi: 10.1016/S0006-3223(95)00597-8
64. Ghorbani E, Mohammadi M, Malakouti SK, Mohammadi-Kangarani H, Abdollahi E, Torab M, et al. Association of ACE gene Insertion/Deletion polymorphism with suicidal attempt in an Iranian population. Biochem Genet (2021) 59(1):31–41. doi: 10.1007/s10528-020-09986-7
65. Baghai TC, Schule C, Zwanzger P, Minov C, Zill P, Ella R, et al. Hypothalamic-pituitary-adrenocortical axis dysregulation in patients with major depression is influenced by the insertion/deletion polymorphism in the angiotensin I-converting enzyme gene. Neurosci Lett (2002) 328(3):299–303. doi: 10.1016/S0304-3940(02)00527-X
66. Ancelin ML, Carrière I, Scali J, Ritchie K, Chaudieu I, Ryan J. Angiotensin-converting enzyme gene variants are associated with both cortisol secretion and late-life depression. Transl Psychiatry (2013) 3:e322. doi: 10.1038/tp.2013.95
67. Saab YB, Gard PR, Yeoman MS, Mfarrej B, El-Moalem H, Ingram MJ. Renin–angiotensin-system gene polymorphisms and depression. Prog Neuropsychopharmacol Biol Psychiatry (2007) 31(5):1113–8. doi: 10.1016/j.pnpbp.2007.04.002
68. Zettergren A, Kern S, Gustafson D, Gudmundsson P, Sigström R, Östling S, et al. The ACE gene is associated with late-life major depression and age at dementia onset in a population-based cohort. Am J Geriatr Psychiatry (2017) 25(2):170–7. doi: 10.1016/j.jagp.2016.06.009
69. Taylor WD, Benjamin S, McQuoid DR, Payne ME, Krishnan RR, MacFall JR, et al. AGTR1 gene variation: Association with depression and frontotemporal morphology. Psychiatry Res: Neuroimaging (2012) 202(2):104–9. doi: 10.1016/j.pscychresns.2012.03.007
70. Baghai TC, Schüle C, Zwanzger P, Minov C, Schwarz MJ, de Jonge S, et al. Possible influence of the insertion/deletion polymorphism in the angiotensin I-converting enzyme gene on theraputic outcome in affective disorders. Mol Psychiatry (2001) 6(3):258–9. doi: 10.1038/sj.mp.4000857
71. Firouzabadi N, Farshadfar P, Haghnegahdar M, Alavi-Shoushtari A, Ghanbarinejad V. Impact of ACE2 genetic variant on antidepressant efficacy of SSRIs. Acta Neuropsychiatr (2022) 34(1):30–6. doi: 10.1017/neu.2021.32
72. Skidgel RA, Erdös EG. The broad substrate specificity of human angiotensin I converting enzyme. In: Clinical and experimental hypertension. part a: Theory and practice (1987), vol. 9:(2–3):243–59. Available at: https://www.tandfonline.com/doi/abs/10.3109/10641968709164184.
73. Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, et al. Distinct mechanism for antidepressant activity by blockade of central substance p receptors. Science (1998) 281(5383):1640–5. doi: 10.1126/science.281.5383.1640
74. Baghai TC, Schule C, Zill P, Deiml T, Eser D, Zwanzger P, et al. The angiotensin I converting enzyme insertion/deletion polymorphism influences therapeutic outcome in major depressed women, but not in men. Neurosci Lett (2004) 363(1):38–42. doi: 10.1016/j.neulet.2004.03.052
75. Bondy B, Baghai TC, Zill P, Schule C, Eser D, Deiml T, et al. Genetic variants in the angiotensin I-converting-enzyme (ACE) and angiotensin II receptor (AT1) gene and clinical outcome in depression. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29(6):1094–9. doi: 10.1016/j.pnpbp.2005.03.015
76. Leonard BE. Inflammation and depression: a causal or coincidental link to the pathophysiology? Acta Neuropsychiatr (2018) 30(1):1–16. doi: 10.1017/neu.2016.69
77. Licinio J, Wong ML. Pathways and mechanisms for cytokine signaling of the central nervous system. J Clin Invest (1997) 100(12):2941–7.
78. Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci (2008) 9(1):46–56.
79. Ah M, Cl R. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol (2016) 16(1):22–34.
80. Prinz M, Priller J. Tickets to the brain: role of CCR2 and CX3CR1 in myeloid cell entry in the CNS. J Neuroimmunol (2010) 224(1–2):80–4.
81. Moylan S, Berk M, Dean OM, Samuni Y, Williams LJ, O’Neil A, et al. Oxidative & nitrosative stress in depression: why so much stress? Neurosci Biobehav Rev (2014) 45:46–62.
82. Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res (2007) 32(10):1749–56.
83. Porter GA, O’Connor JC. Brain-derived neurotrophic factor and inflammation in depression: Pathogenic partners in crime? World J Psychiatry (2022) 12(1):77–97.
84. Osimo EF, Baxter LJ, Lewis G, Jones PB, Khandaker GM. Prevalence of low-grade inflammation in depression: a systematic review and meta-analysis of CRP levels. Psychol Med (2019) 49(12):1958–70.
85. Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev (2012) 36(2):764–85.
86. Wium-Andersen MK, Orsted DD, Nordestgaard BG. Elevated c-reactive protein, depression, somatic diseases, and all-cause mortality: a mendelian randomization study. Biol Psychiatry (2014) 76(3):249–57.
87. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol Psychiatry (2009) 65(9):732–41. doi: 10.1016/j.biopsych.2008.11.029
88. Yang C, Wardenaar KJ, Bosker FJ, Li J, Schoevers RA. Inflammatory markers and treatment outcome in treatment resistant depression: A systematic review. J Affect Disord (2019) 257:640–9. doi: 10.1016/j.jad.2019.07.045
89. Strawbridge R, Hodsoll J, Powell TR, Hotopf M, Hatch SL, Breen G, et al. Inflammatory profiles of severe treatment-resistant depression. J Affect Disord (2019) 246:42–51. doi: 10.1016/j.jad.2018.12.037
90. Hannestad J, DellaGioia N, Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacol (2011) 36(12):2452–9. doi: 10.1038/npp.2011.132
91. Kappelmann N, Lewis G, Dantzer R, Jones PB, Khandaker GM. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry (2018) 23(2):335–43. doi: 10.1038/mp.2016.167
92. O’Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry (2009) 14(5):511–22. doi: 10.1038/sj.mp.4002148
93. Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, et al. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry (2014) 19(6):699–709. doi: 10.1038/mp.2013.155
94. Tao SH, Ren XQ, Zhang LJ, Liu MY. Association between common cardiovascular drugs and depression. Chin Med J (Engl) (2021) 134(22):2656–65. doi: 10.1097/CM9.0000000000001875
95. Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL-6 and serum amyloid a synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol (2009) 20(3):604–12. doi: 10.1681/ASN.2008060628
96. Recinos A, LeJeune WS, Sun H, Lee CY, Tieu BC, Lu M, et al. Angiotensin II induces IL-6 expression and the jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis (2007) 194(1):125–33. doi: 10.1016/j.atherosclerosis.2006.10.013
97. Kranzhöfer R, Browatzki M, Schmidt J, Kübler W. Angiotensin II activates the proinflammatory transcription factor nuclear factor-kappaB in human monocytes. Biochem Biophys Res Commun (1999) 257(3):826–8. doi: 10.1006/bbrc.1999.0543
98. Watanabe K, Taniguchi M, Miyoshi M, Shimizu H, Imoto T, Sato K, et al. Effects of central injection of angiotensin-converting-enzyme inhibitor and angiotensin type 1 receptor antagonist on the brain NF-kappaB and AP-1 activities of rats given LPS. Peptides (2006) 27(6):1538–46. doi: 10.1016/j.peptides.2005.11.005
99. Salmani H, Hosseini M, Baghcheghi Y, Moradi-Marjaneh R, Mokhtari-Zaer A. Losartan modulates brain inflammation and improves mood disorders and memory impairment induced by innate immune activation: The role of PPAR-γ activation. Cytokine (2020) 125:154860. doi: 10.1016/j.cyto.2019.154860
100. Ayyub M, Najmi A, Akhtar M. Protective effect of irbesartan an angiotensin (AT1) receptor antagonist in unpredictable chronic mild stress induced depression in mice. Drug Res (Stuttg) (2016) 67(01):59–64. doi: 10.1055/s-0042-118172
101. Aswar U, Chepurwar S, Shintre S, Aswar M. Telmisartan attenuates diabetes induced depression in rats. Pharmacol Rep (2017) 69(2):358–64. doi: 10.1016/j.pharep.2016.12.004
102. Pang T, Wang J, Benicky J, Sánchez-Lemus E, Saavedra JM. Telmisartan directly ameliorates the neuronal inflammatory response to IL-1β partly through the JNK/c-jun and NADPH oxidase pathways. J Neuroinflamm (2012) 9(1):588. doi: 10.1186/1742-2094-9-102
103. Yuan X, Guo X, Deng Y, Zhu D, Shang J, Liu H. Chronic intermittent hypoxia-induced neuronal apoptosis in the hippocampus is attenuated by telmisartan through suppression of iNOS/NO and inhibition of lipid peroxidation and inflammatory responses. Brain Res (2015) 1596:48–57. doi: 10.1016/j.brainres.2014.11.035
104. Torika N, Asraf K, Danon A, Apte RN, Fleisher-Berkovich S. Telmisartan modulates glial activation: In vitro and In vivo studies. PloS One (2016) 11(5):e0155823. doi: 10.1371/journal.pone.0155823
105. Benicky J, Sánchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacol (2011) 36(4):857–70. doi: 10.1038/npp.2010.225
106. Gong X, Hu H, Qiao Y, Xu P, Yang M, Dang R, et al. The involvement of renin-angiotensin system in lipopolysaccharide-induced behavioral changes, neuroinflammation, and disturbed insulin signaling. Front Pharmacol (2019) 10:318. doi: 10.3389/fphar.2019.00318
107. Bhat SA, Goel R, Shukla R, Hanif K. Angiotensin receptor blockade modulates NFκB and STAT3 signaling and inhibits glial activation and neuroinflammation better than angiotensin-converting enzyme inhibition. Mol Neurobiol (2016) 53(10):6950–67. doi: 10.1007/s12035-015-9584-5
108. Sanchez-Lemus E, Murakami Y, Larrayoz-Roldan IM, Moughamian AJ, Pavel J, Nishioku T, et al. Angiotensin II AT1 receptor blockade decreases lipopolysaccharide-induced inflammation in the rat adrenal gland. Endocrinology (2008) 149(10):5177–88. doi: 10.1210/en.2008-0242
109. Sánchez-Lemus E, Benicky J, Pavel J, Saavedra JM. In vivo angiotensin II AT1 receptor blockade selectively inhibits LPS-induced innate immune response and ACTH release in rat pituitary gland. Brain Behav Immun (2009) 23(7):945–57. doi: 10.1016/j.bbi.2009.04.012
110. Miyoshi M, Miyano K, Moriyama N, Taniguchi M, Watanabe T. Angiotensin type 1 receptor antagonist inhibits lipopolysaccharide-induced stimulation of rat microglial cells by suppressing nuclear factor κB and activator protein-1 activation: Lipopolysaccharide-induced stimulation of rat microglial cells. Eur J Neurosci (2008) 27(2):343–51. doi: 10.1111/j.1460-9568.2007.06014.x
111. Lenart L, Balogh DB, Lenart N, Barczi A, Hosszu A, Farkas T, et al. Novel therapeutic potential of angiotensin receptor 1 blockade in a rat model of diabetes-associated depression parallels altered BDNF signalling. Diabetologia (2019) 62(8):1501–13. doi: 10.1007/s00125-019-4888-z
112. Singh B, Mourya A, Sah SP, Kumar A. Protective effect of losartan and ramipril against stress induced insulin resistance and related complications: Anti-inflammatory mechanisms. Eur J Pharmacol (2017) 801:54–61. doi: 10.1016/j.ejphar.2017.02.050
113. Balogh DB, Molnar A, Hosszu A, Lakat T, Hodrea J, Szabo AJ, et al. Antidepressant effect in diabetes-associated depression: A novel potential of RAAS inhibition. Psychoneuroendocrinology (2020) 118:104705. doi: 10.1016/j.psyneuen.2020.104705
114. Xue B, Yu Y, Wei SG, Beltz TG, Guo F, Felder RB, et al. Stress-induced sensitization of angiotensin II hypertension is reversed by blockade of angiotensin-converting enzyme or tumor necrosis factor-α. Am J Hypertens (2019) 32(9):909–17. doi: 10.1093/ajh/hpz075
115. Nocito C, Lubinsky C, Hand M, Khan S, Patel T, Seliga A, et al. Centrally acting angiotensin-converting enzyme inhibitor suppresses type I interferon responses and decreases inflammation in the periphery and the CNS in lupus-prone mice. Front Immunol (2020) 11:573677. doi: 10.3389/fimmu.2020.573677
116. Salmani H, Hosseini M, Beheshti F, Baghcheghi Y, Sadeghnia HR, Soukhtanloo M, et al. Angiotensin receptor blocker, losartan ameliorates neuroinflammation and behavioral consequences of lipopolysaccharide injection. Life Sci (2018) 203:161–70. doi: 10.1016/j.lfs.2018.04.033
117. Bhatt S, Nagappa AN, Patil CR. Role of oxidative stress in depression. Drug Discov Today (2020) 25(7):1270–6. doi: 10.1016/j.drudis.2020.05.001
118. Réus GZ, Dos Santos MAB, Abelaira HM, Titus SE, Carlessi AS, Matias BI, et al. Antioxidant treatment ameliorates experimental diabetes-induced depressive-like behaviour and reduces oxidative stress in brain and pancreas. Diabetes Metab Res Rev (2016) 32(3):278–88. doi: 10.1002/dmrr.2732
119. Salim S. Oxidative stress and psychological disorders. Curr Neuropharmacol (2014) 12(2):140–7. doi: 10.2174/1570159X11666131120230309
120. Vaváková M, Ďuračková Z, Trebatická J. Markers of oxidative stress and neuroprogression in depression disorder. Oxid Med Cell Longevity (2015) 2015:1–12. doi: 10.1155/2015/898393
121. Diniz BS, Mendes-Silva AP, Silva LB, Bertola L, Vieira MC, Ferreira JD, et al. Oxidative stress markers imbalance in late-life depression. J Psychiatr Res (2018) 102:29–33. doi: 10.1016/j.jpsychires.2018.02.023
122. Lee SY, Lee SJ, Han C, Patkar AA, Masand PS, Pae CU. Oxidative/nitrosative stress and antidepressants: Targets for novel antidepressants. Prog Neuropsychopharmacol Biol Psychiatry (2013) 46:224–35. doi: 10.1016/j.pnpbp.2012.09.008
123. Hovatta I, Juhila J, Donner J. Oxidative stress in anxiety and comorbid disorders. Neurosci Res (2010) 68(4):261–75. doi: 10.1016/j.neures.2010.08.007
124. Steenkamp LR, Hough CM, Reus VI, Jain FA, Epel ES, James SJ, et al. Severity of anxiety- but not depression- is associated with oxidative stress in major depressive disorder. J Affect Disord (2017) 219:193–200. doi: 10.1016/j.jad.2017.04.042
125. Salim S, Sarraj N, Taneja M, Saha K, Tejada-Simon MV, Chugh G. Moderate treadmill exercise prevents oxidative stress-induced anxiety-like behavior in rats. Behav Brain Res (2010) 208(2):545–52. doi: 10.1016/j.bbr.2009.12.039
126. Rodriguez-Perez AI, Borrajo A, Rodriguez-Pallares J, Guerra MJ, Labandeira-Garcia JL. Interaction between NADPH-oxidase and rho-kinase in angiotensin II-induced microglial activation: NADPH-oxidase and rho-kinase interaction. Glia (2015) 63(3):466–82. doi: 10.1002/glia.22765
127. Pedreanez A, Arcaya JL, Carrizo E, Mosquera J. Forced swimming test increases superoxide anion positive cells and angiotensin II positive cells in the cerebrum and cerebellum of the rat. Brain Res Bull (2006) 71(1–3):18–22. doi: 10.1016/j.brainresbull.2006.07.018
128. Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience (2010) 171(3):852–8. doi: 10.1016/j.neuroscience.2010.09.029
129. Lee SH, Fujioka S, Takahashi R, Oe T. Angiotensin II-induced oxidative stress in human endothelial cells: Modification of cellular molecules through lipid peroxidation. Chem Res Toxicol (2019) 32(7):1412–22. doi: 10.1021/acs.chemrestox.9b00110
130. Barhoumi T, Alghanem B, Shaibah H, Mansour FA, Alamri HS, Akiel MA, et al. SARS-CoV-2 coronavirus spike protein-induced apoptosis, inflammatory, and oxidative stress responses in THP-1-Like-Macrophages: Potential role of angiotensin-converting enzyme inhibitor (Perindopril). Front Immunol (2021) 12:728896. doi: 10.3389/fimmu.2021.728896
131. Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS. Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: effects of ACEI and ARB. Kidney Int (2002) 61(1):186–94. doi: 10.1046/j.1523-1755.2002.00123.x
132. Salim S, Asghar M, Taneja M, Hovatta I, Chugh G, Vollert C, et al. Potential contribution of oxidative stress and inflammation to anxiety and hypertension. Brain Res (2011), 1404:63–71. doi: 10.1016/j.brainres.2011.06.024
133. Goel R, Bhat SA, Hanif K, Nath C, Shukla R. Angiotensin II receptor blockers attenuate lipopolysaccharide-induced memory impairment by modulation of NF-κB-Mediated BDNF/CREB expression and apoptosis in spontaneously hypertensive rats. Mol Neurobiol (2018) 55(2):1725–39. doi: 10.1007/s12035-017-0450-5
134. Bild W, Hritcu L, Stefanescu C, Ciobica A. Inhibition of central angiotensin II enhances memory function and reduces oxidative stress status in rat hippocampus. Biol Psychiatry (2013) 43:79–88. doi: 10.1016/j.pnpbp.2012.12.009
135. Patki G, Salvi A, Liu H, Atrooz F, Alkadhi I, Kelly M, et al. Tempol treatment reduces anxiety-like behaviors induced by multiple anxiogenic drugs in rats. PloS One (2015) 10(3):e0117498. doi: 10.1371/journal.pone.0117498
136. AlSaad AMS, Alasmari F, Abuohashish HM, Mohany M, Ahmed MM, Al-Rejaie SS. Renin angiotensin system blockage by losartan neutralize hypercholesterolemia-induced inflammatory and oxidative injuries. Redox Rep (2020) 25(1):51–8. doi: 10.1080/13510002.2020.1763714
137. Gold PW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry (2015) 20(1):32–47. doi: 10.1038/mp.2014.163
138. Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A (2012) 109(16):5995–9. doi: 10.1073/pnas.1118355109
139. Juruena MF, Bocharova M, Agustini B, Young AH. Atypical depression and non-atypical depression: Is HPA axis function a biomarker? a systematic review. J Affect Disord (2018) 233:45–67. doi: 10.1016/j.jad.2017.09.052
140. Boyer P. Do anxiety and depression have a common pathophysiological mechanism? Acta Psychiatr Scand Suppl (2000) 406):24–9. doi: 10.1111/j.0065-1591.2000.acp29[dash]04.x
141. Kalin NH. Novel insights into pathological anxiety and anxiety-related disorders. Am J Psychiatry (2020) 177(3):187–9. doi: 10.1176/appi.ajp.2020.20010057
142. Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci (1995) 15(10):6340–50. doi: 10.1523/JNEUROSCI.15-10-06340.1995
143. Holsboer F. The rationale for corticotropin-releasing hormone receptor (CRH-r) antagonists to treat depression and anxiety. J Psychiatr Res (1999) 33(3):181–214. doi: 10.1016/S0022-3956(98)90056-5
144. Yang G, Xi ZX, Wan Y, Wang H, Bi G. Changes in circulating and tissue angiotensin II during acute and chronic stress. Biol Signals (1993) 2(3):166–72. doi: 10.1159/000109488
145. Peng J, Kimura B, Phillips MI. The predominant role of brain angiotensinogen and angiotensin in environmentally induced hypertension. Regul Peptides (2002) 110(1):25–32. doi: 10.1016/S0167-0115(02)00156-8
146. Castren E, Saavedra JM. Repeated stress increases the density of angiotensin II binding sites in rat paraventricular nucleus and subfornical organ. Endocrinology (1988) 122(1):370–2. doi: 10.1210/endo-122-1-370
147. Jezova D, Ochedalski T, Kiss A, Aguilera G. Brain angiotensin II modulates sympathoadrenal and hypothalamic pituitary adrenocortical activation during stress: Neuropeptide y. J Neuroendocrinol (2008) 10(1):67–72. doi: 10.1046/j.1365-2826.1998.00182.x
148. Pavlatou MG, Mastorakos G, Lekakis I, Liatis S, Vamvakou G, Zoumakis E, et al. Chronic administration of an angiotensin II receptor antagonist resets the hypothalamic–pituitary–adrenal (HPA) axis and improves the affect of patients with diabetes mellitus type 2: Preliminary results: Research report. Stress (2008) 11(1):62–72. doi: 10.1080/10253890701476621
149. Veltmar A, Culman J, Qadri F, Rascher W, Unger T. Involvement of adrenergic and angiotensinergic receptors in the paraventricular nucleus in the angiotensin II-induced vasopressin release. J Pharmacol Exp Ther (1992) 263(3):1253–60.
150. Rivier C, Vale W. Effect of angiotensin II on ACTH release in vivo: role of corticotropin-releasing factor. Regul Pept (1983) 7(3):253–8. doi: 10.1016/0167-0115(83)90018-6
151. Pavel J, Benicky J, Murakami Y, Sanchez-Lemus E, Saavedra JM. Peripherally administered angiotensin II AT 1 receptor antagonists are anti-stress compounds in vivo. Ann New York Acad Sci (2008) 1148(1):360–6. doi: 10.1196/annals.1410.006
152. Saavedra JM, Benicky J. Brain and peripheral angiotensin II play a major role in stress. Stress (2007) 10(2):185–93. doi: 10.1080/10253890701350735
153. Saavedra JM, Armando I, Bregonzio C, Juorio A, Macova M, Pavel J, et al. A centrally acting, anxiolytic angiotensin II AT1 receptor antagonist prevents the isolation stress-induced decrease in cortical CRF1 receptor and benzodiazepine binding. Neuropsychopharmacol (2006) 31(6):1123–34. doi: 10.1038/sj.npp.1300921
154. Bregonzio C, Armando I, Ando H, Jezova M, Baiardi G, Saavedra JM. Anti-inflammatory effects of angiotensin II AT1 receptor antagonism prevent stress-induced gastric injury. Am J Physiol Gastrointest Liver Physiol (2003) 285(2):G414–423. doi: 10.1152/ajpgi.00058.2003
155. Costa R, Tamascia ML, Sanches A, Moreira RP, Cunha TS, Nogueira MD. Tactile stimulation of adult rats modulates hormonal responses, depression-like behaviors, and memory impairment induced by chronic mild stress. In: Role of angiotensin II. Behav Brain Res (2020) 379:112250. doi: 10.1016/j.bbr.2019.112250. Available at: https://reader.elsevier.com/reader/sd/pii/S0166432819304784?token=89053D7496A3BE06CDD995A8EE12059DFC1F1D89BA4C836C07522F9DFA6CDC552C257BF51C70A785D342B9A91BC7DD7E&originRegion=us-east-1&originCreation=20220420031144.
156. Braszko JJ, Wincewicz D, Jakubów P. Candesartan prevents impairment of recall caused by repeated stress in rats. Psychopharmacol (Berl) (2013) 225(2):421–8. doi: 10.1007/s00213-012-2829-3
157. Armando I, Carranza A, Nishimura Y, Hoe KL, Barontini M, Terrón JA, et al. Peripheral administration of an angiotensin II AT1 receptor antagonist decreases the hypothalamic-Pituitary-Adrenal response to isolation stress. Endocrinology (2001) 142(9):3880–9. doi: 10.1210/endo.142.9.8366
158. Barnes NM, Costall B, Kelly ME, Murphy DA, Naylor RJ. Anxiolytic-like action of DuP753, a non-peptide angiotensin II receptor antagonist. Neuroreport (1990) 1(1):20–1. doi: 10.1097/00001756-199009000-00006
159. Campos GV, de Souza AMA, Ji H, West CA, Wu X, Lee DL, et al. The angiotensin type 1 receptor antagonist losartan prevents ovariectomy-induced cognitive dysfunction and anxiety-like behavior in long Evans rats. Cell Mol Neurobiol (2020) 40(3):407–20. doi: 10.1007/s10571-019-00744-x
160. Llano López LH, Caif F, García S, Fraile M, Landa AI, Baiardi G, et al. Anxiolytic-like effect of losartan injected into amygdala of the acutely stressed rats. Pharmacol Rep (2012) 64(1):54–63. doi: 10.1016/S1734-1140(12)70730-2
161. Kumar A, Singh B, Mishra J, Sah SP, Pottabathini R. Neuroprotective mechanism of losartan and its interaction with nimesulide against chronic fatigue stress. Inflammopharmacology (2015) 23(6):291–305. doi: 10.1007/s10787-015-0238-z
162. Pechlivanova DM, Stoynev AG, Tchekalarova JD. The effects of chronic losartan pretreatment on restraint stress-induced changes in motor activity, nociception and pentylenetetrazol generalized seizures in rats. Folia Med (2011) 53(2):69–73. doi: 10.2478/v10153-010-0040-z
163. Costa R, Tamascia ML, Sanches A, Moreira RP, Cunha TS, Nogueira MD, et al. Tactile stimulation of adult rats modulates hormonal responses, depression-like behaviors, and memory impairment induced by chronic mild stress: Role of angiotensin II. Behav Brain Res (2020) 379:112250. doi: 10.1016/j.bbr.2019.112250
164. Li Y, Cheng KC, Liu KF, Peng WH, Cheng JT, Niu HS. Telmisartan activates PPARδ to improve symptoms of unpredictable chronic mild stress-induced depression in mice. Sci Rep (2017) 7(1):14021. doi: 10.1038/s41598-017-14265-4
165. McALLISTER-WILLIAMS RH, Ferrier IN, Young AH. Mood and neuropsychological function in depression: the role of corticosteroids and serotonin. Psychol Med (1998) 28(3):573–84. doi: 10.1017/S0033291798006680
166. Nahmod VE, Finkielman S, Benarroch EE, Pirola CJ. Angiotensin regulates release and synthesis of serotonin in brain. Science (1978) 202(4372):1091–3. doi: 10.1126/science.152460
167. Bali A, Jaggi AS. Angiotensin as stress mediator: role of its receptor and interrelationships among other stress mediators and receptors. Pharmacol Res (2013) 76:49–57. doi: 10.1016/j.phrs.2013.07.004
168. Jenkins TA. Effect of angiotensin-related antihypertensives on brain neurotransmitter levels in rats. Neurosci Lett (2008) 444(2):186–9. doi: 10.1016/j.neulet.2008.08.021
169. Tanaka J, Miyakubo H, Kawakami A, Hayashi Y, Nomura M. Involvement of NMDA receptor mechanisms in the modulation of serotonin release in the lateral parabrachial nucleus in the rat. Brain Res Bull (2006) 71(1–3):311–5. doi: 10.1016/j.brainresbull.2006.09.017
170. Stone LB, McCormack CC, Bylsma LM. Cross system autonomic balance and regulation: Associations with depression and anxiety symptoms. Psychophysiology (2020) 57(10):e13636. doi: 10.1111/psyp.13636
171. Streeter CC, Gerbarg PL, Saper RB, Ciraulo DA, Brown RP. Effects of yoga on the autonomic nervous system, gamma-aminobutyric-acid, and allostasis in epilepsy, depression, and post-traumatic stress disorder. Med Hypotheses (2012) 78(5):571–9. doi: 10.1016/j.mehy.2012.01.021
172. Lin KD, Chang LH, Wu YR, Hsu WH, Kuo CH, Tsai JR, et al. Association of depression and parasympathetic activation with glycemic control in type 2 diabetes mellitus. J Diabetes Complications (2022) 36(8):108264. doi: 10.1016/j.jdiacomp.2022.108264
173. Selye H. A syndrome produced by diverse nocuous agents. 1936. J Neuropsychiatry Clin Neurosci (1998) 10(2):230–1. doi: 10.1176/jnp.10.2.230a
174. Grippo AJ, Johnson AK. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress (2009) 12(1):1–21. doi: 10.1080/10253890802046281
175. Vatta MS, Bianciotti LG, Locatelli AS, Papouchado ML, Fernández BE. Monophasic and biphasic effects of angiotensin II and III on norepinephrine uptake and release in rat adrenal medulla. Can J Physiol Pharmacol (1992) 70(6):821–5. doi: 10.1139/y92-110
176. Piano MR, Prasun M. Neurohormone activation. Crit Care Nurs Clin North Am (2003) 15(4):413–21. doi: 10.1016/S0899-5885(02)00096-5
177. Fabiani ME, Sourial M, Thomas WG, Johnston CI, Johnston CI, Frauman AG. Angiotensin II enhances noradrenaline release from sympathetic nerves of the rat prostate via a novel angiotensin receptor: implications for the pathophysiology of benign prostatic hyperplasia. J Endocrinol (2001) 171(1):97–108. doi: 10.1677/joe.0.1710097
178. Machado-Silva A, Passos-Silva D, Santos RA, Sinisterra RD. Therapeutic uses for angiotensin-(1-7). Expert Opin Ther Pat (2016) 26(6):669–78. doi: 10.1080/13543776.2016.1179283
179. Rodriguez-Campos M, Kadarian C, Rodano V, Bianciotti L, Fernandez B, Vatta M. AT-1 receptor and phospholipase c are involved in angiotensin III modulation of hypothalamic noradrenergic transmission. Cell Mol Neurobiol (2000) 20(6):747–62. doi: 10.1023/A:1007059010571
180. Kregel KC, Stauss H, Unger T. Modulation of autonomic nervous system adjustments to heat stress by central ANG II receptor antagonism. Am J Physiol (1994) 266(6 Pt 2):R1985–1991. doi: 10.1152/ajpregu.1994.266.6.R1985
181. Arnsten AFT, Raskind MA, Taylor FB, Connor DF. The effects of stress exposure on prefrontal cortex: Translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress (2015) 1:89–99. doi: 10.1016/j.ynstr.2014.10.002
182. Geracioti TD, Baker DG, Ekhator NN, West SA, Hill KK, Bruce AB, et al. CSF norepinephrine concentrations in posttraumatic stress disorder. Am J Psychiatry (2001) 158(8):1227–30. doi: 10.1176/appi.ajp.158.8.1227
183. Gold PW, Wong ML, Goldstein DS, Gold HK, Ronsaville DS, Esler M, et al. Cardiac implications of increased arterial entry and reversible 24-h central and peripheral norepinephrine levels in melancholia. Proc Natl Acad Sci U S A (2005) 102(23):8303–8. doi: 10.1073/pnas.0503069102
184. Strawn JR, Geracioti TD. Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety (2008) 25(3):260–71. doi: 10.1002/da.20292
185. Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histol Histopathol (2010) 25(2):237–58. doi: 10.14670/HH-25.237
186. Alonso M, Vianna MRM, Depino AM, Mello e Souza T, Pereira P, Szapiro G, et al. BDNF-triggered events in the rat hippocampus are required for both short- and long-term memory formation. Hippocampus (2002) 12(4):551–60. doi: 10.1002/hipo.10035
187. Chen DY, Bambah-Mukku D, Pollonini G, Alberini CM. Glucocorticoid receptors recruit the CaMKIIα-BDNF-CREB pathways to mediate memory consolidation. Nat Neurosci (2012) 15(12):1707–14. doi: 10.1038/nn.3266
188. Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci (2003) 23(1):349–57. doi: 10.1523/JNEUROSCI.23-01-00349.2003
189. Chauhan VS, Khan SA, Kulhari K. Correlation of brain-derived neurotrophic factor with severity of depression and treatment response. Med J Armed Forces India (2020). doi: 10.1016/j.mjafi.2020.09.014
190. Tayyab M, Shahi MH, Farheen S, Mariyath PMM, Khanam N, Hossain MM. Exploring the potential role of sonic hedgehog cell signalling pathway in antidepressant effects of nicotine in chronic unpredictable mild stress rat model. Heliyon (2019) 5(5):e01600. doi: 10.1016/j.heliyon.2019.e01600
191. Ping G, Qian W, Song G, Zhaochun S. Valsartan reverses depressive/anxiety-like behavior and induces hippocampal neurogenesis and expression of BDNF protein in unpredictable chronic mild stress mice. Pharmacol Biochem Behav (2014) 124:5–12. doi: 10.1016/j.pbb.2014.05.006
192. Shibata K, Makino I, Shibaguchi H, Niwa M, Katsuragi T, Furukawa T. Up-regulation of angiotensin type 2 receptor mRNA by angiotensin II in rat cortical cells. Biochem Biophys Res Commun (1997) 239(2):633–7. doi: 10.1006/bbrc.1997.7521
193. Patki G, Solanki N, Atrooz F, Allam F, Salim S. Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res (2013) 1539:73–86. doi: 10.1016/j.brainres.2013.09.033
194. Köhler-Forsberg O, Petersen L, Berk M, Gasse C, Østergaard SD. The effect of combined treatment with SSRIs and renin-angiotensin system (RAS) drugs: A propensity score matched cohort study. Eur Neuropsychopharmacol (2020) 32:120–30. doi: 10.1016/j.euroneuro.2020.01.004
195. Kessing LV, Rytgaard HC, Gerds TA, Berk M, Ekstrøm CT, Andersen PK. New drug candidates for depression - a nationwide population-based study. Acta Psychiatr Scand (2019) 139(1):68–77. doi: 10.1111/acps.12957
196. Brownstein DJ, Salagre E, Köhler C, Stubbs B, Vian J, Pereira C, et al. Blockade of the angiotensin system improves mental health domain of quality of life: A meta-analysis of randomized clinical trials. Aust N Z J Psychiatry (2018) 52(1):24–38. doi: 10.1177/0004867417721654
197. Nasr SJ, Crayton JW, Agarwal B, Wendt B, Kora R. Lower frequency of antidepressant use in patients on renin-Angiotensin-Aldosterone system modifying medications. Cell Mol Neurobiol (2011) 31(4):615–8. doi: 10.1007/s10571-011-9656-7
198. Kessing LV, Rytgaard HC, Ekstrøm CT, Torp-Pedersen C, Berk M, Gerds TA. Antihypertensive drugs and risk of depression: A nationwide population-based study. Hypertension (2020) 76(4):1263–79. doi: 10.1161/HYPERTENSIONAHA.120.15605
199. Boal AH, Smith DJ, McCallum L, Muir S, Touyz RM, Dominiczak AF, et al. Monotherapy with major antihypertensive drug classes and risk of hospital admissions for mood disorders. Hypertension (2016) 68(5):1132–8. doi: 10.1161/HYPERTENSIONAHA.116.08188
200. Braszko JJ, Karwowska-Polecka W, Halicka D, Gard PR. Captopril and enalapril improve cognition and depressed mood in hypertensive patients. J Basic Clin Physiol Pharmacol (2003) 14(4):323–43. doi: 10.1515/JBCPP.2003.14.4.323
201. Germain L, Chouinard G. Captopril treatment of major depression with serial measurements of blood cortisol concentrations. Biol Psychiatry (1989) 25(4):489–93. doi: 10.1016/0006-3223(89)90203-5
202. Germain L, Chouinard G. Treatment of recurrent unipolar major depression with captopril. Biol Psychiatry (1988) 23(6):637–41. doi: 10.1016/0006-3223(88)90010-8
203. Zubenko GS, Nixon RA. Mood-elevating effect of captopril in depressed patients. Am J Psychiatry (1984) 141(1):110–1. doi: 10.1176/ajp.141.1.110
204. Deicken RF. Captopril treatment of depression. Biol Psychiatry (1986) 21(14):1425–8. doi: 10.1016/0006-3223(86)90334-3
205. Reinecke A, Browning M, Klein Breteler J, Kappelmann N, Ressler KJ, Harmer CJ, et al. Angiotensin regulation of amygdala response to threat in high-Trait-Anxiety individuals. Biol Psychiatry Cognit Neurosci Neuroimaging (2018) 3(10):826–35. doi: 10.1016/j.bpsc.2018.05.007
206. Shad MU. Is there an association between anxiety symptoms and valsartan treatment? J Affect Disord (2020) 261:111–2. doi: 10.1016/j.jad.2019.10.004
207. Khoury NM, Marvar PJ, Gillespie CF, Wingo A, Schwartz A, Bradley B, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry (2012) 73(6):849–55. doi: 10.4088/JCP.11m07316
208. Cohen BM, Zubenko GS. Captopril in the treatment of recurrent major depression. J Clin Psychopharmacol (1988) 8(2):143–4. doi: 10.1097/00004714-198804000-00018
209. Hertzman M, Adler LW, Arling B, Kern M. Lisinopril may augment antidepressant response. J Clin Psychopharmacol (2005) 25(6):618–20. doi: 10.1097/01.jcp.0000186736.99523.1d
210. Breckenridge A. Angiotensin converting enzyme inhibitors and quality of life. Am J Hypertens (1991) 4(1 Pt 2):79S–82S. doi: 10.1093/ajh/4.1.79S
211. Hill JF, Bulpitt CJ, Fletcher AE. Angiotensin converting enzyme inhibitors and quality of life: the European trial. J Hypertens Suppl (1985) 3(2):S91–94.
212. Edmonds D, Vetter H, Vetter W. Angiotensin converting enzyme inhibitors in the clinic: quality of life. J Hypertens Suppl (1987) 5(3):S31–35. doi: 10.1097/00004872-198708003-00007
213. Laudisio A, Giovannini S, Finamore P, Gemma A, Bernabei R, Incalzi RA, et al. Use of ACE-inhibitors and quality of life in an older population. J Nutr Health Aging (2018) 22(10):1162–6. doi: 10.1007/s12603-018-1135-0
214. Rathmann W, Haastert B, Roseman JM, Giani G. Cardiovascular drug prescriptions and risk of depression in diabetic patients. J Clin Epidemiol (1999) 52(11):1103–9. doi: 10.1016/S0895-4356(99)00082-7
215. Vian J, Pereira C, Chavarria V, Köhler C, Stubbs B, Quevedo J, et al. The renin-angiotensin system: a possible new target for depression. BMC Med (2017) 15(1):144. doi: 10.1186/s12916-017-0916-3
216. Cao YY, Xiang X, Song J, Tian YH, Wang MY, Wang XW, et al. Distinct effects of antihypertensives on depression in the real-world setting: A retrospective cohort study. J Affect Disord (2019) 259:386–91. doi: 10.1016/j.jad.2019.08.075
217. Colbourne L, Luciano S, Harrison PJ. Onset and recurrence of psychiatric disorders associated with anti-hypertensive drug classes. Transl Psychiatry (2021) 11(1):319. doi: 10.1038/s41398-021-01444-1
218. Callender JS, Hodsman GP, Hutcheson MJ, Lever AF, Robertson JI. Mood changes during captopril therapy for hypertension. a double-blind pilot study. Hypertension (1983) 5(5 Pt 2):III90–93. doi: 10.1161/01.hyp.5.5_pt_2.iii90
219. Mamdani M, Gomes T, Greaves S, Manji S, Juurlink DN, Tadrous M, et al. Association between angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and suicide. JAMA Netw Open (2019) 2(10):e1913304. doi: 10.1001/jamanetworkopen.2019.13304
220. Lin SY, Lin CL, Lin CC, Hsu WH, Lin CD, Wang IK, et al. Association between angiotensin receptor blockers and suicide: nationwide population-based propensity score matching study. J Affect Disord (2020) 276:815–21. doi: 10.1016/j.jad.2020.07.106
221. Vasile S, Hallberg A, Sallander J, Hallberg M, Åqvist J, Gutiérrez-de-Terán H. Evolution of angiotensin peptides and peptidomimetics as angiotensin II receptor type 2 (AT2) receptor agonists. Biomolecules (2020) 10(4):649. doi: 10.3390/biom10040649
Keywords: deprssion, anxiety, renin-angiotensin system, angiotensin II, neuroinflammation
Citation: Gong S and Deng F (2023) Renin-angiotensin system: The underlying mechanisms and promising therapeutical target for depression and anxiety. Front. Immunol. 13:1053136. doi: 10.3389/fimmu.2022.1053136
Received: 25 September 2022; Accepted: 05 December 2022;
Published: 24 January 2023.
Edited by:
Mirza S. Baig, Indian Institute of Technology Indore, IndiaReviewed by:
Milica Milovan Borovcanin, University of Kragujevac, SerbiaCopyright © 2023 Gong and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Deng, ZGVuZ19mYW5nQGpsdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.