
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol., 04 January 2023
Sec. Primary Immunodeficiencies
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1033513
This article is part of the Research TopicAdvances in primary Immunodeficiencies (Inborn Errors of Immunity) in Central-Eastern Europe: Volume IIView all 20 articles
 Mojca Železnik1
Mojca Železnik1 Aneta Soltirovska Šalamon1
Aneta Soltirovska Šalamon1 Maruša Debeljak2
Maruša Debeljak2 Aleš Goropevšek3
Aleš Goropevšek3 Nataša Šuštar4
Nataša Šuštar4 Damjana Ključevšek5
Damjana Ključevšek5 Alojz Ihan6
Alojz Ihan6 Tadej Avčin7,8*
Tadej Avčin7,8*Aicardi–Goutières syndrome (AGS) is a genetically determined early-onset progressive encephalopathy caused by mutations leading to overexpression of type I interferon (IFN) and resulting in various clinical phenotypes. A gain-of-function (GOF) mutation in the IFIH1 gene is associated with robust production of type I IFN and activation of the Janus kinase (JAK) signal transducer and activator of the transcription (STAT) pathway, which can cause AGS type 7. We detail the clinical case of an infant who initially presented with Pneumocystis jirovecii pneumonia (PCP), had recurrent respiratory infections, and was later treated with a JAK inhibitor, baricitinib, because of a genetically confirmed GOF mutation in the IFIH1 gene. This spectrum of IFIH1 GOF mutations with overlapping features of hyperinflammation and severe opportunistic infection, which mimics combined immunodeficiency (CID), has not been described before. In this case, therapy with baricitinib effectively blocked IFN-α activation and reduced STAT1 signaling but had no effect on the progression of the neurological disease.
Aicardi–Goutières syndrome (AGS) is a genetically heterogeneous disorder originally defined as an early-onset progressive encephalopathy that is characterized by intracranial calcification, white matter abnormalities, cerebral atrophy, cerebrospinal fluid (CSF) lymphocytosis, and inappropriate induction of a type I interferon (IFN)-mediated immune response, belonging to the group of type I interferonopathies (1, 2). Although AGS particularly affects the brain, immune system, and skin in the first year of life, there is a wide spectrum of disease presentations, progression, and outcomes (3). CSF and serum analyses typically exhibit increased type I IFN activity and increased levels of expression of IFN-stimulated genes in peripheral blood, the so-called IFN signature (2, 4).
As more mutations have been identified in different causative genes for AGS, it has become clear that there are significant clinical differences between patients’ phenotypes, and clinical variability has been observed even within the same genotype or family (4). In 2014, the pathogenic variant IFIH1 (IFN induced with helicase C domain-containing protein 1 gene) was identified as the causative gene for AGS type 7 (2, 5) and accounts for only 4% of cases (6).
IFIH1 is an important intracellular sensor for various viruses; by recognizing viral double-stranded RNA, it triggers antiviral IFN responses (7). Mutations of innate immune sensor IFIH1 are thought to be a predisposing factor for autoimmune diseases and can cause a monogenic form of systemic lupus erythematous similar to genetic overproduction of IFN- α, mutations in upstream components of the classical complement pathway, and apoptosis defects. By producing type I IFN, IFIH1 enhances responses to its own RNA, thereby activating the adaptive immune system (7–9). The mutation in IFIH1 may be gain of function (GOF) and can dramatically up-regulate the production of type I IFNs (5, 10, 11). Conversely, loss-of-function (LOF) variants in IFIH1 result in a deficient antiviral defense. Therefore, it causes a primary immunodeficiency, leading to increased susceptibility to common respiratory RNA viruses (10).
The IFIH1 GOF mutations are associated with the activation of the Janus kinase (JAK) signal transducer and activator of the transcription (STAT) pathway (5, 12). Recent evidence suggests that JAK inhibitors may be effective in blocking IFN-mediated inflammatory signaling by decreasing STAT1 phosphorylation in patients with AGS (12–15). In addition, there are some indications of possible effects on neuroinflammation and thus improvement in neurological function (12, 13). The IFN signature is an indicator of IFN signaling and might reflect disease activity (12, 13, 16). Furthermore, studies have shown that phosphorylated STAT1 (pSTAT) in T lymphocytes is greatly reduced during therapy (16).
A male infant was born to unrelated White parents at 38 weeks of gestation by induction of labor because of oligohydramnios and intrauterine growth restriction. Birth weight was 3,050 g, birth length was 50 cm, head circumference was 34.5 cm, and Apgar scores were 9 and 9 at 1 and 5 minutes, respectively. During pregnancy, the mother had confirmed infection with the SARS-CoV-2 virus.
At 13 days of age, the patient was hospitalized for lack of weight gain and dehydration. On admission, he weighed 2,920 g, was afebrile, and had a systolic murmur below the clavicle, respiratory distress, and abnormal neurological signs. A chest radiograph showed interstitial lung infiltrates, and an echocardiogram revealed a patent foramen ovale, ductus arteriosus, and a high level of pulmonary vascular resistance. Blood test analysis revealed leukopenia, neutropenia, thrombocytopenia, and normal inflammatory parameters. He had a negative hemoculture and a nasopharyngeal swab for respiratory viruses. Screenings for congenital infections and autoimmune thrombocytopenia were also negative. Polymerase chain reaction testing for the SARS-CoV-2 virus in the placenta, umbilical blood, and CSF was negative, and the newborn also had no antibodies against SARS-CoV-2.
He had abnormal neurological signs, with lethargy, hypotonia, and an abnormal spontaneous movement pattern. A cranial ultrasound showed subependymal germinolytic cysts and severe vasculopathy. Brain magnetic resonance imaging (MRI) showed diffusely slightly elevated white matter and basal ganglia signal in the T2-weighted sequences (Figure 1); susceptibility-weighted imaging (SWI) showed a hypointensive signal from slightly more prominent vessels in the basal ganglia areas.
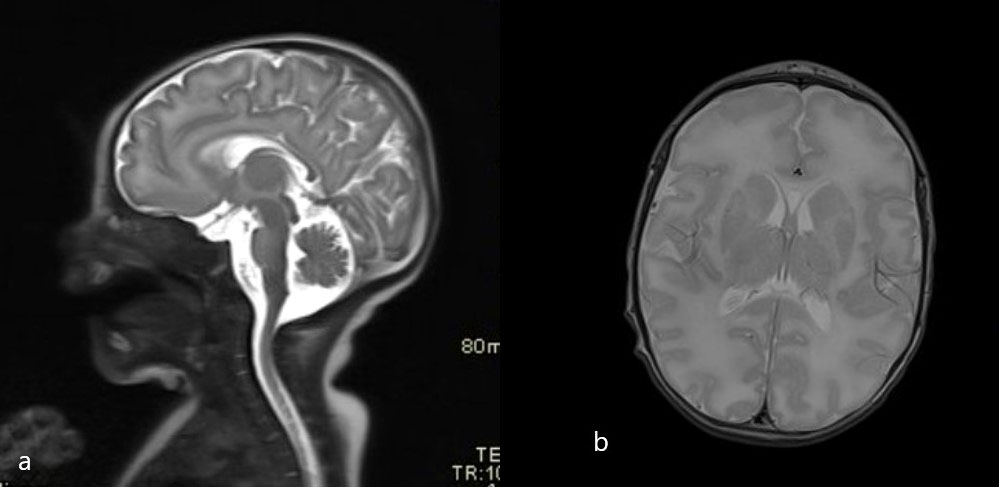
Figure 1 Initial brain MRI scans (T2 sagittal and axial plane). (A) Normal thickness of pons and midbrain. (A, B) Higher T2 signal of the frontal white matter.
Owing to the clinical presentation with abnormal neurological signs and neuroimaging findings, systemic inflammatory disease was suspected. Laboratory testing for interferonopathy was performed in the virology laboratory at the Hôpital Cochin, Paris, France, and showed increased IFN-α concentrations in serum (18 IU/ml) and in CSF (50 IU/ml). Whole-exome sequencing confirmed a heterozygous GOF mutation in exon 11, c.2159G>A (p.Arg720Gln), in the IFIH1 gene (NM_022168.4). The mutation occurred de novo and had previously been reported in six individuals with AGS (5, 17, 18).
The patient had elevated serum immunoglobulins (Ig), with a level of IgG of 6.62 g/l, IgA of 1.32 g/l, and IgM of 1.75 g/l. He was found to have a markedly reduced concentration of B cells, T helper (Th) cells, T cytotoxic cells, and naïve CD4+ T cells. The percentages of Th1 and Th2 cells were also reduced. The percentages of activated T cells (HLA DR+) and Treg cells were markedly increased. The concentration of natural killer (NK) cells and recent thymic emigrants (RTEs) (the marker of thymic T cell production) was normal. In addition, a T-cell proliferation test (CD3/CD28 stimulated T cells) was normal.
At 2.5 months of age, the patient underwent hernioplasty of the right inguinal hernia. After surgery, he presented shortness of breath. A chest radiograph showed interstitial infiltrates on the right side. Laboratory results showed mild elevation of inflammatory parameters, lactate dehydrogenases, and significant positive β-d-glucans. A nasopharyngeal swab was positive for rhinovirus and Pneumocystis jirovecii. P. jirovecii pneumonia (PCP) was confirmed by bronchoscopy, and antibiotic treatment with trimethoprim–sulfamethoxazole was initiated. Because of recurrent mucocutaneous candidiasis, he also received systemic antifungal therapy.
After the resolution of PCP at the age of 3.5 months, the patient was started on the JAK inhibitor baricitinib at a dosage of 0.1 mg/kg/day. Functional studies of STAT signaling (16) before initiation of baricitinib therapy revealed increased total STAT1 expression in monocytes and lymphocytes including CD4+ T cells, and increased levels of pSTAT after stimulation with IFN-α. Follow-up studies after 10 months of therapy with baricitinib revealed persistently increased total STAT1 protein levels, and there was a reduction in the level of pSTAT1 expression after stimulation with IFN-α (Figure 2).
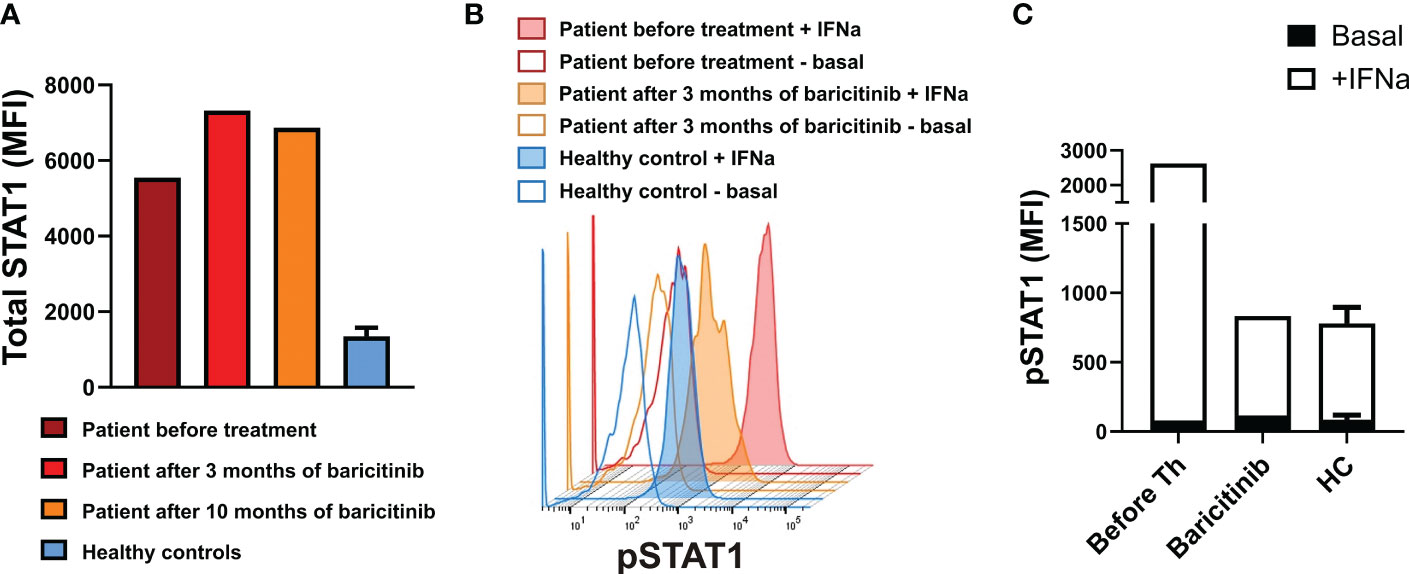
Figure 2 (A): Total STAT1 levels in CD4+ T lymphocytes from the patient on indicated time points and healthy controls. (B) Phosphorylated STAT1 (pSTAT1) levels in unstimulated whole-blood CD4+ T lymphocytes and on interferon (IFN)-α induction. (C) Basal (unstimulated) pSTAT1 levels and pSTAT1 levels on stimulation with IFN-α for 15 minutes in CD4+ T lymphocytes from the patient before treatment and after 3 months of baricitinib, and from healthy controls. Values in (A) and (C) for healthy controls (n=3) are the mean ± SD. MFI, median fluorescence intensity; HC, healthy controls.
At 4.5 months of age, the patient was treated for diarrhea caused by Campylobacter jejuni infection. At 5 months of age, he underwent an emergency right hemicolectomy owing to ischemia of the terminal ileum and ascending colon, and later underwent two relaparotomies owing to dehiscence of the ileotransverse anastomosis.
During the first year, he experienced recurrent viral respiratory infections: rhinovirus, bocavirus, coronavirus, enterovirus, and respiratory syncytial virus were detected. An increase in liver enzymes was noted during respiratory infections, which was partially attenuated by continuing baricitinib therapy during the infections.
After 1 year most immune parameters reached normal levels: concentrations of B cells, T helper cells, T cytotoxic cells, and naive CD4+ T cells and percentage of Th1 and Th2 cells. Only the percentage of Treg cells remained increased.
The neuroimaging results and abnormal neurological signs pointed us to an early diagnosis and initiation of treatment. At a neurological assessment at 3 months of age, the child presented with axial hypotonia and increased muscle tone of the extremities. Treatment with tiagabine and B vitamins was initiated, together with baricitinib. A neurological evaluation at 6 months of age showed decreased growth in head circumference, poor eye contact, and limb spasticity, with occasional dystonia. The patient was unable to hold up his head, and spontaneous movements were poorly expressed, without meaningful coordination. Further neurological follow-ups showed the developmental delay to be severe. A brain MRI scan at 12 months of age showed marked atrophy of the deep and subcortical white matter of both hemispheres, with enlarged ventricles, sulci, and subarachnoid spaces. The brainstem and cerebellar peduncles were also greatly reduced in volume. In the reduced basal nuclei, signs of calcifications were reported (Figure 3). At the age of 1.5 years, global developmental delay was identified, with intellectual disability and without any signs of speech development. Epileptic seizures with twitching in the left arm and short absences had occurred. An electroencephalogram (EEG) showed multifocal epileptiform discharges. Levetiracetam was introduced. Feeding problems became more frequent and, consequently, a percutaneous gastrostoma was introduced. The child and his family were also receiving follow-up treatment from the pediatric palliative care team.
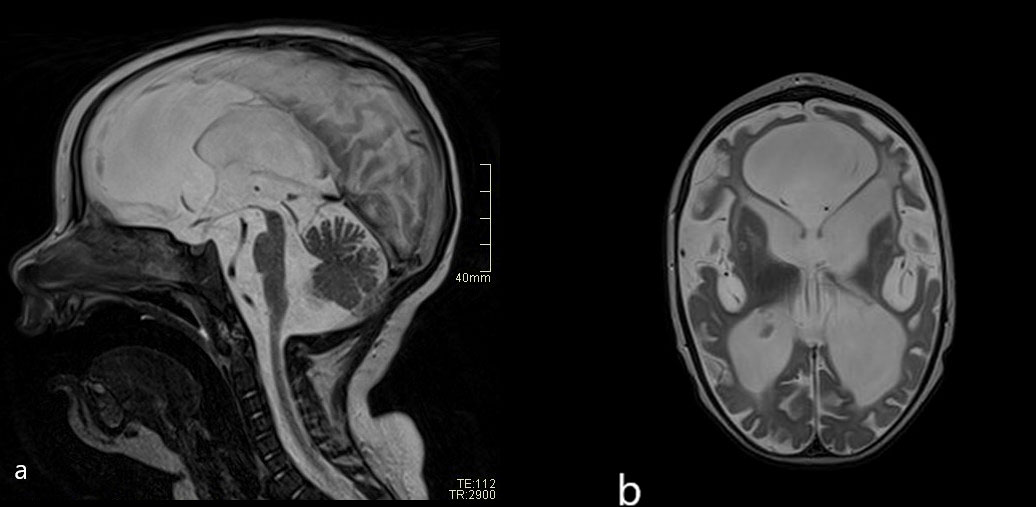
Figure 3 (A) Follow-up brain MRI scans (T2 sagittal and axial plane) after 1 year: severe atrophy of midbrain compared with initial MRI. (B) Severe reduction of brain volume and abnormal signal of the white matter—leucoencephalopathy.
We have detailed the case of an infant with early-diagnosed AGS type 7 with genetically confirmed heterozygous GOF mutation in the IFIH1 gene who initially presented neurological manifestations as well as P. jirovecii pneumonia (PCP) resembling combined immunodeficiency (CID). The patient was later treated with a Janus kinase (JAK) inhibitor, baricitinib, which reduced STAT1 signaling but had no effect on the progression of the neurological disease.
The initial clinical presentation of our patient was suggestive of prenatal onset of the disease and raised suspicion of interferonopathies. In a large case series of AGS patients, only 11.4% of infants had abnormal neurological signs at birth without obvious systemic signs, and others developed symptoms later in life, usually within the first year. Notably, individuals with an IFIH1 mutation had normal development in the first year of life and often did not present until after 1 year (4). Rice et al. reported that clinical presentation at or shortly after birth is associated with a severe AGS phenotype, suggesting prenatal onset of the disease (17).
A heterozygous c.2159G>A (p.Arg720Gln) mutation in the IFIH1 gene (NM_022168.4) has been previously identified in six individuals with a severe AGS phenotype and its functional effect has been demonstrated by IFN scoring (2, 5, 17, 18). Rice et al. reported two patients with intrauterine growth restriction and thrombocytopenia at birth who had severe developmental delay. MRI at 8 and 12 months of age showed cerebral atrophy with calcifications in the basal ganglia and white matter disease. The first patient developed seizures at 13 months of age and died of pneumonia at 2 years of age. The second patient had hypertrophic cardiomyopathy and nephrotic syndrome diagnosed at 7 and 10 months of age, respectively (5). Subsequently, Adang et al. described a male patient with the same mutation, who also presented in the neonatal period with hepatosplenomegaly and severe thrombocytopenia, had profound developmental delay, and developed severe pulmonary hypertension by the age of 2 years (18). Moreover, other studies have reported various amino acid substitutions in IFIH1 mutation-positive patients (3, 11, 18–22). Recent studies suggest that IFN status assessment is a reliable disease marker (13, 17), and a positive correlation has been found between IFN activity in CSF measured within 1 year of disease onset and the degree of subsequent disability (4). In this case, analysis of CSF and serum showed increased IFN type I and an enhanced IFN signature.
P. jirovecii is an opportunistic microbial pathogen that is particularly threatening in immunocompromised individuals and can cause PCP (23). To our knowledge, PCP with a mutation in IFIH1 has not previously been reported. Our patient also had persistent mucocutaneous candidiasis and numerous recurrent respiratory viral infections as clinical signs of immunodeficiency. The deficient antiviral defense has been described only in IFIH1 LOF mutations (10), but even LOF mutations are not associated with opportunistic infections. CID-like presentation with PCP and severe T-cell lymphopenia was previously reported in two patients with stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy (SAVI), which is also a type I interferonopathy and is caused by a GOF mutation in the transmembrane protein 173 gene (TMEM173) encoding STING (24, 25). Based on the disease course in our case, it appears that patients with AGS with the IFIH1 GOF mutation may present similarly to patients with CID, with opportunistic infections associated with autoimmune and hyperinflammatory manifestations.
Previous studies have shown that JAK inhibitors can effectively block the activation of IFNs in patients with AGS (13), especially in patients with IFIH1-related disease (12). IFN signature and STAT1 phosphorylation in T lymphocytes could serve as indicators of response to treatment (12, 13, 16). In our patient, we started treatment with baricitinib, a JAK1 and JAK2 inhibitor, in the first months after birth, using the recommended dosage (13). During treatment, we noted a significant functional improvement, a significant reduction in the IFN signature, and a reduction in pSTAT in peripheral blood T lymphocytes. Studies of STAT signaling in our patient were performed as previously reported (16), including analysis of total STAT1 expression in T cells at different time points without technical repeats.
Some recent studies have reported improvement of neurological functions after JAK inhibitor therapy, even in patients with severe and prolonged disease (13, 26), suggesting that treatment in the earliest stages of the disease could lead to important clinical gains (27). By contrast, our patient experienced severe progression of encephalopathy in spite of early diagnosis and treatment with baricitinib, which was confirmed by neuroimaging at 12 months (28, 29). Our findings indicate that JAK inhibitors effectively blocked the systemic IFN-mediated inflammatory response but had little or no effect on the brain disease. Based on the functional studies of STAT signaling, our data suggest that the measurement of STAT1 phosphorylation can be used as a useful monitoring tool for adjusting the dosage of baricitinib. No common adverse effects were reported (16), and treatment was well tolerated by our patient.
Our case highlights the phenotypic heterogeneity of AGS. PCP and recurrent respiratory infections have not previously been described as part of the clinical spectrum associated with a GOF mutation in the IFIH1 gene. This case shows that AGS with GOF mutation in the IFIH1 gene could mimic CID with opportunistic infections associated with autoimmune and hyperinflammatory manifestations. In our experience, baricitinib effectively blocks IFN-α activation, but even early treatment with a JAK inhibitor failed to halt the progression of neurological manifestations.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
MŽ, ASŠ, NŠ, and TA contributed to patient care and treatment. MŽ, ASŠ, and TA wrote the manuscript and reviewed the literature. DK contributed to interpreting and describing the imaging findings. MD performed genetic evaluation. AG and AI contributed to immunological evaluation and functional testing. All authors contributed to diagnostic procedure, manuscript revision, and read, and approved the submitted version.
This work was partially funded by the Slovenian Research Agency (grant number J3–3061) and University Medical Center Ljubljana (grants number 20210069 and 20220090).
We wish to thank Prof. David Neubauer, University Children’s Hospital Ljubljana, for helpful suggestions in the clinical management of the patient, and Dr. Nuška Pečarič-Meglič and Dr. Tina Vipotnik-Vesnaver, Clinical Institute for Radiology, University Medical Center Ljubljana, for neuroimaging consultation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol (1984) 15:49–54. doi: 10.1002/ana.410150109
2. Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, et al. Aicardi-goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet (2014) 95(1):121–5. doi: 10.1016/j.ajhg.2014.06.007
3. Crow YJ, Zaki MS, Abdel-Hamid M, Boesp O, Cordeiro NJV, Gleeson JG, et al. Mutations in ADAR1 , IFIH1 , and RNASEH2B presenting as spastic paraplegia. Neuropediatrics (2014) 45(6):386–93. doi: 10.1055/s-0034-1389161
4. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte G, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1 , RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1. ADAR, and IFIH1. Am J Med Genet Part A (2015) 167A(2):296–312. doi: 10.1002/ajmg.a.36887
5. Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type i interferon signaling. Nat Genet (2014) 46(5):503–9. doi: 10.1038/ng.2933
6. Al Mutairi F, Alfadhel M, Nashabat M, El-Hattab AW, Ben-Omran T, Hertecant J, et al. Phenotypic and molecular spectrum of aicardi-goutières syndrome: A study of 24 patients. Pediatr Neurol (2018) 78:35–40. doi: 10.1016/j.pediatrneurol.2017.09.002
7. Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity (2014) 40(2):199–212. doi: 10.1016/j.immuni.2013.12.014
8. Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J Immunol (2011) 187(3):1298–303. doi: 10.4049/jimmunol.1100857
9. Gorman JA, Hundhausen C, Errett JS, Stone AE, Allenspach EJ, Ge Y, et al. The A946T variant of the RNA sensor IFIH1 mediates an interferon program that limits viral infection but increases the risk for autoimmunity. Nat Immunol (2017) 18(7):744–52. doi: 10.1038/ni.3766
10. Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci U.S.A. (2017) 114(31):8342–7. doi: 10.1073/pnas.1704259114
11. Amari S, Tsukamoto K, Ishiguro A, Yanagi K, Kaname T, Ito Y. An extremely severe case of aicardi-goutières syndrome 7 with a novel variant in IFIH1. Eur J Med Genet (2020) 63(2):103646. doi: 10.1016/j.ejmg.2019.04.003
12. Kothur K, Bandodkar S, Chu S, Wienholt L, Johnson A, Barclay P, et al. An open-label trial of JAK 1/2 blockade in progressive IFIH1-associated neuroinflammation. Neurology (2018) 90(6):289–91. doi: 10.1212/WNL.0000000000004921
13. Vanderver A, Adang L, Gavazzi F, McDonald K, Helman G. Janus kinase inhibition in the aicardi–goutières syndrome. N Engl J Med (2020) 383(10):986–9. doi: 10.1056/NEJMc2001362
14. Casas-Alba D, Darling A, Caballero E, Mensa-Vilaro A. Efficacy of baricitinib on chronic pericardial effusion in a patient with aicardi–goutieres syndrome. Rheumatology (2021) 61(4):e87–9. doi: 10.1093/rheumatology/keab860
15. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, PJ H, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest (2018) 128(7):3041–52. doi: 10.1172/JCI98814
16. Meesilpavikkai K, Dik WA, Schrijver B, van Helden-Meeuwsen CG, Versnel MA, van Hagen PM, et al. Efficacy of baricitinib in the treatment of chilblains associated with aicardi-goutières syndrome, a type I interferonopathy. Arthritis Rheumatol (2019) 71(5):829–31. doi: 10.1002/art.40805
17. Rice GI, Park S, Gavazzi F, Adang LA, Ayuk LA, Van Eyck L, et al. Genetic and phenotypic spectrum associated with IFIH1 gain-of-function. Hum Mutat (2020) 41(4):837–49. doi: 10.1002/humu.23975
18. Adang LA, Frank DB, Gilani A, Takanohashi A, Ulrick N, Collins A, et al. Aicardi goutières syndrome is associated with pulmonary hypertension. Mol Genet Metab (2018) 125(4):351–8. doi: 10.1016/j.ymgme.2018.09.004
19. Marguet F, Laquerrière A, Goldenberg A, Guerrot AM, Quenez O, Flahaut P, et al. Clinical and pathologic features of aicardi-goutières syndrome due to an IFIH1 mutation: A pediatric case report. Am J Med Genet Part A (2016) 170(5):1317–24. doi: 10.1002/ajmg.a.37577
20. Zheng S, Lee PY, Wang J, Wang S, Huang Q, Huang Y, et al. Interstitial lung disease and psoriasis in a child with aicardi-goutières syndrome. Front Immunol (2020) 11:985. doi: 10.3389/fimmu.2020.00985
21. Van Eyck L, De Somer L, Pombal D, Bornschein S, de Zegher F, Liston A, et al. Gain-of-function mutation in IFIH1 can cause both aicardi-goutières syndrome and systemic lupus erythematosus with IgA-deficiency. Pediatr Rheumatol Online J (2014) 12(Supp1):P309. doi: 10.1186/1546-0096-12-S1-P309
22. Rutsch F, Macdougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, et al. A specific IFIH1 gain-of-function mutation causes singleton-merten syndrome. Am J Hum Genet (2015) 96(2):275–82. doi: 10.1016/j.ajhg.2014.12.014
23. Boillat L, Angelini F, Crucis-Armengaud A, Asner SA, Rochat I. Pneumocystis jirovecii pneumonia in an infant: The tip of the iceberg. Clin Pediatr (Phila) (2019) 58(5):578–81. doi: 10.1177/0009922818825141
24. Saldanha RG, Balka KR, Davidson S, Wainstein BK, Wong M, Macintosh R, et al. A mutation outside the dimerization domain causing atypical STING-associated vasculopathy with onset in infancy. Front Immunol (2018) 9:1–8. doi: 10.3389/fimmu.2018.01535
25. Frémond ML, Hadchouel A, Berteloot L, Melki I, Bresson V, Barnabei L, et al. Overview of STING-associated vasculopathy with onset in infancy (SAVI) among 21 patients. J Allergy Clin Immunol Pract (2021) 9(2):803–18.e11. doi: 10.1016/j.jaip.2020.11.007
26. Tüngler V, Doebler-Neumann M, Salandin M, Kaufmann P, Wolf C, Lucas N, et al. Aicardi-goutières syndrome due to a paternal mosaic IFIH1 mutation. Neurol Genet (2020) 6(1):384. doi: 10.1212/NXG.0000000000000384
27. Crow YJ, Shetty J, Livingston JH. Treatments in aicardi–goutières syndrome. Dev Med Child Neurol (2020) 62(1):42–7. doi: 10.1111/dmcn.14268
28. La Piana R, Uggetti C, Roncarolo F, Vanderver A, Olivieri I, Tonduti D, et al. Neuroradiologic patterns and novel imaging findings in aicardi-goutières syndrome. Neurology (2016) 86(1):28–35. doi: 10.1212/WNL.0000000000002228
Keywords: Aicardi–Goutières syndrome (AGS), IFIH1 gene, interferonopathy, Janus kinase inhibitor, combined immune deficiency
Citation: Železnik M, Soltirovska Šalamon A, Debeljak M, Goropevšek A, Šuštar N, Ključevšek D, Ihan A and Avčin T (2023) Case report: Pneumocystis jirovecii pneumonia in a severe case of Aicardi–Goutières syndrome with an IFIH1 gain-of-function mutation mimicking combined immunodeficiency. Front. Immunol. 13:1033513. doi: 10.3389/fimmu.2022.1033513
Received: 31 August 2022; Accepted: 05 December 2022;
Published: 04 January 2023.
Edited by:
Irina A. Tuzankina, Institute of Immunology and Physiology (RAS), RussiaReviewed by:
Marie-Louise Frémond, Institut Imagine, FranceCopyright © 2023 Železnik, Soltirovska Šalamon, Debeljak, Goropevšek, Šuštar, Ključevšek, Ihan and Avčin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tadej Avčin, dGFkZWouYXZjaW5Aa2Nsai5zaQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.