 Weiran Shen1
Weiran Shen1 Li Ou
Li Ou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Immunol. , 27 October 2022
Sec. Vaccines and Molecular Therapeutics
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1001263
This article is part of the Research Topic Immunogenicity and toxicity of AAV gene therapy View all 8 articles
Recombinant Adeno-associated virus (rAAV) is one of the main delivery vectors for gene therapy. To assess immunogenicity, toxicity, and features of AAV gene therapy in clinical settings, a meta-analysis of 255 clinical trials was performed. A total of 7,289 patients are planned to be dosed. AAV2 was the most dominantly used serotype (29.8%, n=72), and 8.3% (n=20) of trials used engineered capsids. 38.7% (n=91) of trials employed neutralizing antibody assays for patient enrollment, while 15.3% (n=36) used ELISA-based total antibody assays. However, there was high variability in the eligibility criteria with cut-off tiers ranging from 1:1 to 1:1,600. To address potential immunogenicity, 46.3% (n=118) of trials applied immunosuppressants (prophylactic or reactive), while 32.7% (n=18) of CNS and 37.5% (n=24) of ocular-directed trials employed immunosuppressants, possibly due to the immune-privileged status of CNS and retina. There were a total of 11 patient deaths across 8 trials, and 18 out of 30 clinical holds were due to toxicity findings in clinical studies. 30.6% (n=78) of trials had treatment-emergent serious adverse events (TESAEs), with hepatotoxicity and thrombotic microangiopathy (systemic delivery) and neurotoxicity (CNS delivery) being the most prominent. Additionally, the durability of gene therapy may be impacted by two distinct decline mechanisms: 1) rapid decline presumably due to immune responses; or 2) gradual decline due to vector dilution. The durability varied significantly depending on disease indication, dose, serotypes, and patient individuals. Most CNS (90.0%) and muscle trials (73.3%) achieved durable transgene expression, while only 43.6% of ocular trials had sustained clinical outcomes. The rAAV production system can affect rAAV quality and thus immunogenicity and toxicity. Out of 186 trials that have disclosed production system information, 63.0% (n=126) of trials used the transient transfection of the HEK293/HEK293T system, while 18.0% (n=36) applied the baculovirus/Sf9 (rBac/Sf9) system. There were no significant differences in TESAEs and durability between AAV generated by rBac/Sf9 and HEK293/HEK293T systems. In summary, rAAV immunogenicity and toxicity poses significant challenges for clinical development of rAAV gene therapies, and it warrants collaborative efforts to standardize monitoring/measurement methods, design novel strategies to overcome immune responses, and openly share relevant information.
Since the first clinical study using recombinant AAV (rAAV) in cystic fibrosis in the 1990s (1), great progress has been made in understanding its virology, production, safety, efficacy, and translatability (2). In the past 10 years, rAAV has been widely applied in treating rare diseases affecting liver, brain, muscle, heart, eye, and other tissues (3). Long-lasting transgene expression has been achieved in multiple trials, and a total of six therapies have received marketing approval from US Food and Drug Administration (Luxturna, Zolgensma) and European Medicines Agency (Glybera, Luxturna, Zolgenmsa, Upstaza, Roctavian) (4). However, the loss of efficacy is not uncommon in clinical trials, which may result from various reasons, including immune response against AAV (4–7). Moreover, immune responses against rAAV vectors include pre-existing antibodies, complement system activation, and T cell immune responses (8–12). Understanding the complex mechanisms of immune activation is difficult, and the data interpretation is further complicated by individual and species differences, and non-standardized outcome measurements in these studies. Serum prevalence studies suggested a significant portion of the human population had high levels of rAAV neutralizing antibodies (13). Pre-existing antibodies can significantly reduce the transduction efficiency (14). Antibodies bind to vector capsid and stimulate lymphocytes to secrete cytokines, activating adaptive immune responses (9). Thus, during patient enrollment in clinical studies (especially for systemic administration), pre-existing antibody levels need to be considered. After vector dosing, transduced antigen presenting cells (APCs) present capsid peptides by MHC-I or MHC-II system, and activate plasmacytoid and conventional dendritic cells (10, 11, 15). Also, rAAV genome is hypomethylated in CpG sequences (16), which activates toll-like receptor 9 (TLR9) (17). TLR9 -dependent and -independent signaling triggers MyD88 phosphorylation, which is a critical step in activation of cytotoxic T cells (CTLs) reactive to viral capsids (17–19). MyD88 also plays an important role in B cell activation and regulates the Th1-dependent antibody production against rAAV (20). A Hemophilia B trial suggests that CpG introduced by codon optimization might be a trigger for silenced transgene expression (21). A statistical study tried to correlate CpG content in genome sequences of clinical used products with efficacy. It also showed that lower CpG content may reduce the overall immune responses and be helpful to maintain the efficacy (22). Further suggestions are made on methods to calculate and mitigate CpG risk (23, 24). Interestingly, it is still unclear how much unmethylated CpG is a safe range. It is meaningful to develop a translatable model and test whether modification of CpG in gene of interest (GOI) is enough, or reducing CpG number in ITR and regulatory elements are also essential. Recent studies also reported that AAV may activate the complement system, causing platelet reduction, acute thrombotic microangiopathy (TMA) (25, 26). To reduce the potential immune responses and toxicity, immunosuppressants are selectively applied across different clinical trials (27). In this study, we performed a meta-analysis of 255 clinical trials using AAV delivery over the past 25 years. The immunosuppressant usage, antibody screening, adverse events, manufacturing systems, and durability of efficacy was evaluated. Results from this study will highlight the trend and new directions in this field, provide a basis for future clinical study design, and uncover crucial details for addressing immune responses against AAV.
This meta-analysis was performed under the guidance of the 2020 PRISMA (28). Clinical trials using AAV delivery were extracted from the U.S National Library of Medicine database (ClinicalTrials.gov), and the cut-off date was June 1, 2022. Observational studies and long-term follow-up studies were not included because AAV administration was not involved. Additionally, the search was broadened to (1) company press releases (2), company official documents (SEC and IPO filings), (3) presentations in research conferences, (4) publications in journal articles, and (5) company websites. Since a lot of information was not available in the database, reports on previous preclinical studies using the same test article were also reviewed. Disease category, disease indication, gene delivered, trial start date, phase, identifier, serotype, dose, sponsor, route of administration (ROA), immunosuppression regimen, antibody screening, manufacturing system, adverse events, efficacy endpoints, and trial status were collected. Missing information was marked as “Undisclosed” or “N/A”. Notably, peer-reviewed resources and information from non-reviewed resources (e.g., websites, databases) were equally used in this study. All diseases were categorized as central nervous system (CNS), liver, lysosomal storage disorders (LSD), ocular, muscle, and other (including oncology, autoimmune, virology, cardiac, metabolic, pulmonary). All raw data is available in Supplementary Table 1.
A previous study reviewed a total of 149 AAV clinical trials from clinical trial databases using the cut-of date as Jan 1, 2020 (3). In that study, phases, serotypes, ROA, promoters used, and sponsors were analyzed. In this study, the cut-off date was expanded to June 1, 2022, and the sources included clinical trial databases, publications, and company press releases, resulting in analysis of 255 trials. The process of data collection was summarized using a PRISMA flowchart (Supplementary Figure 1). A total of 65 trials are in Phase I, 123 in Phase I/II, 25 in Phase II, 6 in Phase II/III, 22 in Phase III, and one in Phase IV (Figure 1A). There is a single trial (GNT0004 sponsored by Genethon) combining Phase I, II, and III studies together. A total of 245 trials report study status, with 79 ‘completed’, 81 ‘recruiting’, 50 ‘active but not recruiting’. There are also 18 trials ‘terminated’, 6 ‘unknown’, and 2 ‘withdrawn’. In terms of trial number, ocular indications constitute the highest portion (25.1%), followed by CNS (21.6%) and liver (15.3%) (Figure 1B). Across 255 trials, more than 7,289 patients are planned to be dosed. A total of 3,192 patients have been enrolled or dosed, while the largest study (NCT04704921) is still actively enrolling patients, targeting a total of 465 people. A total of 94 trials are designed to enroll at least 20 patients. There are 56 trials (22.0%) sponsored by universities, hospitals, or other academic institutes, while the rest (78.0%) are sponsored by industry. The organizations that sponsored the most trials are Spark Therapeutics, Novartis, Nationwide Children’s Hospital, REGENX Bio (n=10, 9, 8, and 8, respectively). In terms of ROA, 92 trials apply systematic injection, 160 trials employ local administration, and 3 trials do not disclose the ROA information (Figure 1C). CNS targeting is the most complicated: intraputaminal (n=18), intracerebral (n=12), intrathecal (n=11), intracisternal magna (ICM, n=9), intrastriatal (n=5), intracerebral (n=4), intraventricular (n=3), 2 intrathalamic (n=2), and intranigral (n=1). Ocular indications are all dosed through local administration, with 37 trials by subretinal, 24 by intravitreal, and 2 by suprachoroidal dosing. For muscle targeting, 22 trials employ systematic dosing while 10 trials apply intramuscular injection. The trial number has significantly increased over the past two decades (Figure 1D). The first AAV trial was for treating cystic fibrosis using tgAAV2-CFTR (1), and the first trial using AAV delivery for in vivo gene editing was for treating Mucopolysaccharidosis type I by zinc finger nucleases (NCT02702115) (29). There are four approved AAV gene therapy drugs: Glybera (30), Zolgensma (31), Luxturna (32), and Upstaza (33) and one conditional approval Roctavian (34) in the USA and/or EU.
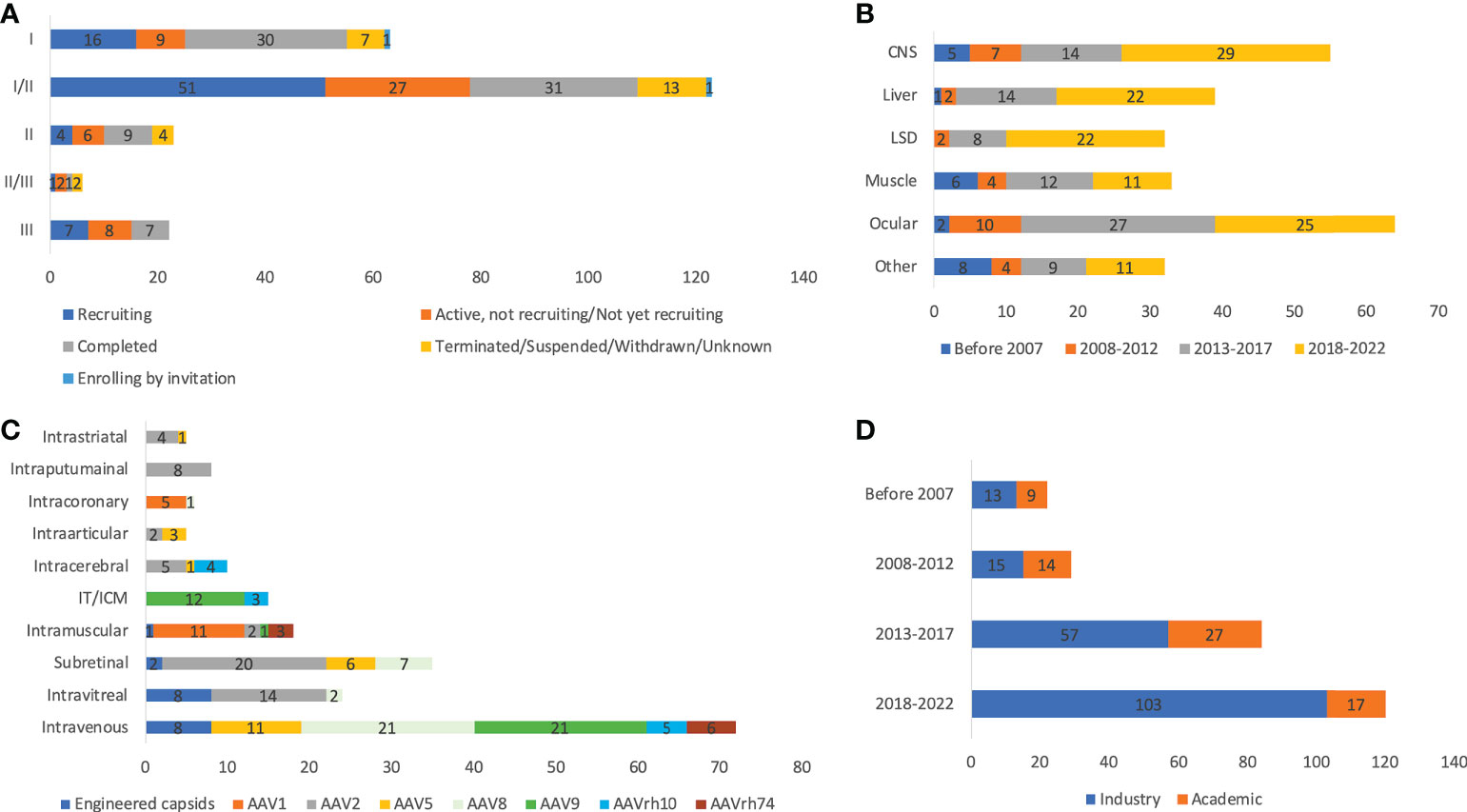
Figure 1 Basic information of 255 AAV clinical trials by status and phases (A), disease indications (B), ROAs and serotypes (C), and sponsors (D). ROA and serotypes associated with <10 trials are not included in the figure. IT and ICM were combined due to the high similarity. Unknown status suggests that study has passed its completion date and status has not been verified in more than two years.
A total of 182 trials disclosed dosing information, and the doses vary largely depending on ROA. For systematic dosing of liver indications, the range is between 5E11 and 1E14 vg/kg. Since vector titers are quantified by each sponsor independently using different primers, probes and assays, it is challenging to perform direct comparison across trials. With this in mind, 14 trials include at least one cohort with a dose higher than 1E14 vg/kg, 6 out of them are targeting muscle indications, and all of them are associated with serious adverse events. Another 6 trials are associated with Zolgensma, which uses self-complementary AAV9 and have been approved as a commercialized product (35). The doses for ocular indications range from 2E8 to 5E11 vg/eye, while 2E10 to 1.1E14 vg for CNS indications. The highest doses for LSD trials are 1.1E14 vg/kg by intravenous injection, 1E14 vg in total by local injection (intraputaminal, intrastriatal, intracerebral), or 1.1E11 vg/g of the brain by ICM injection. A total of 242 trials disclosed serotype information, with 72 of them using AAV2. Other common serotypes are AAV9 (n=36), AAV8 (n=27), AAV5 (n=22), AAV1 (n=17), AAVrh10 (n=13), and AAVrh74 (n=11). There are 20 trials using novel engineered capsids instead of wildtype serotypes (Table 1). Based on disclosed information, 20 trials (8.3%) use self-complementary AAV (scAAV), while others use the single-stranded AAV (ssAAV).
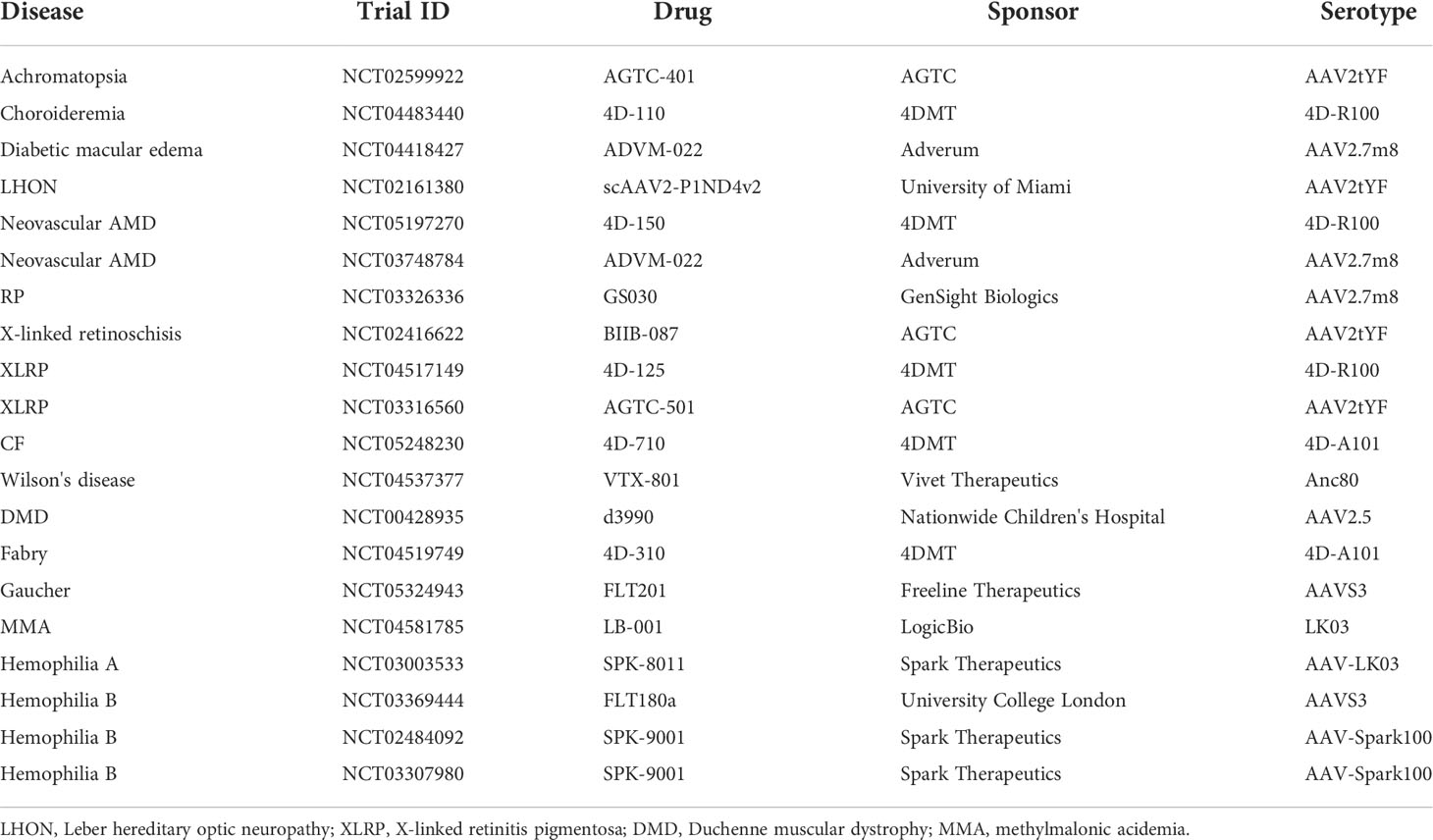
Table 1 Clinical trials using engineered AAV capsids.
As shown in Figure 2, 46.6% (n=118) of trials applied immunosuppressants (prophylactic or reactive). Most trials used corticosteroids, while Sirolimus and Rituximab® were also frequently used. The use of immunosuppressants also depends on the target tissue rAAV delivered to. Only 33.9% of CNS and 37.5% of ocular-directed trials employed immunosuppressants, possibly due to the immune-privileged status of CNS and retina. In contrast, most muscle, liver, and LSD trials employed immunosuppressants (54.5%-78.1%). In terms of ROA, there was also a high variability. 68.5% of trials using intravenous delivery employed immunosuppressants, similarly, 68.4% of trials using CSF delivery (intrathecal/ICM) used immunosuppressants. Other local CNS injections (intraputaminal, intrastriatal, intracerebral) had at most 30% immunosuppressant usage. As time went by, immunosuppressant usage became more frequent from 18.2% before 2007 to 44.6% between 2018 and 2022. This could be explained by a better understanding of AAV immunology through clinical observations over the years. Among all serotypes, AAVrh74 trials were the most frequent user of immunosuppressants (72.7%, 8/11), followed by AAV9 (72.2%, 26/36), AAV8 (53.1%, 17/32) and AAV5 (50.0%, 11/22). Only 24.3% (17/70) of AAV2 trials employed immunosuppressant, which could be partially explained by the fact that AAV2 was most frequently chosen for ocular diseases.
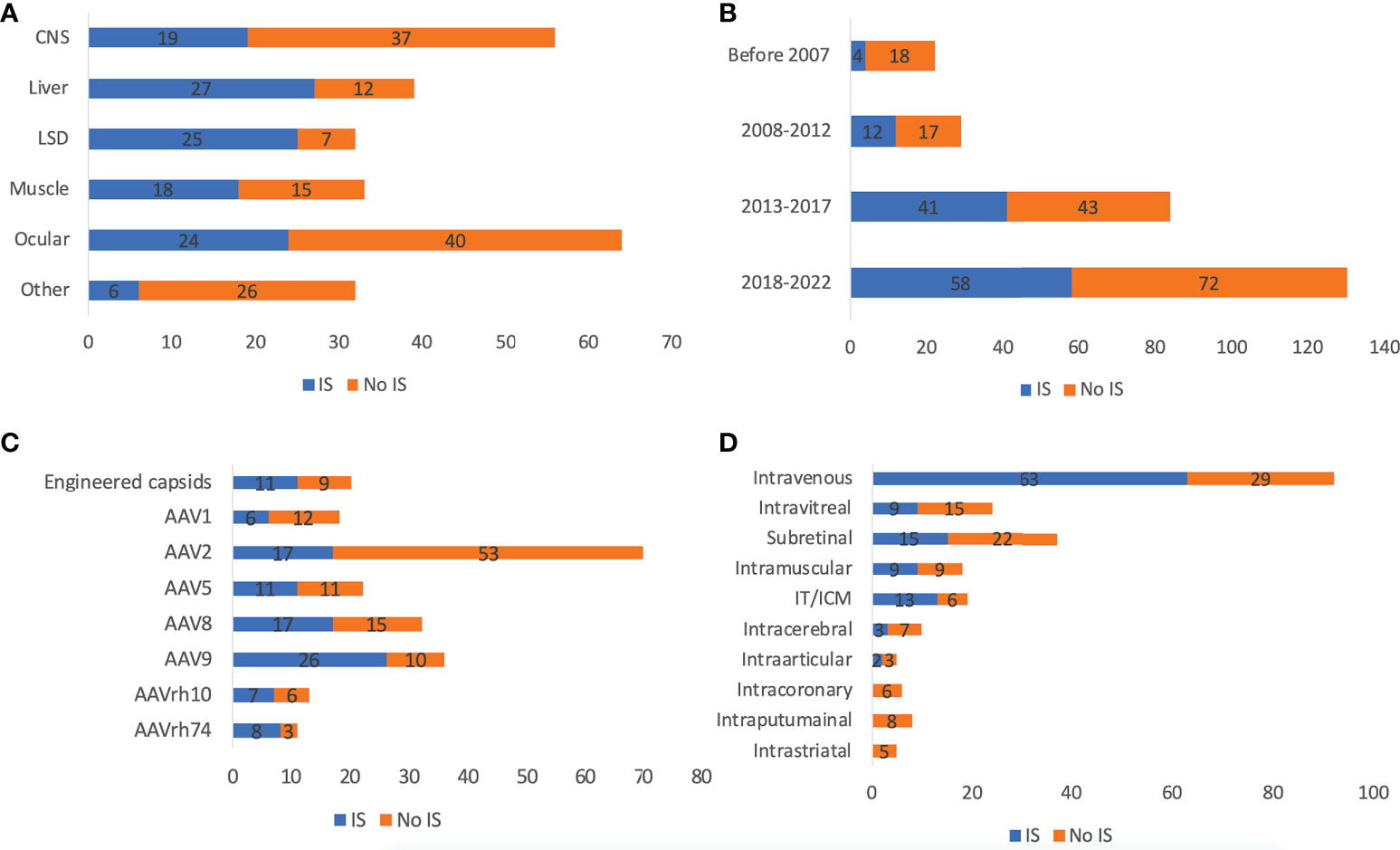
Figure 2 Immunosuppressants usage of 255 AAV clinical trials by disease indications (A), timeframe (B), serotypes (C), and ROAs (D). ROA and serotypes associated with <10 trials are not included in the figure. IT and ICM were combined due to the high similarity.
There was not much fluctuation in usage of antibody screening for patient enrollment over time, from 56.5% before 2007 to 58.5% between 2018 and 2022 (Figure 3B). There was an increase in the usage of total antibody assay (TAB) from 4.3% before 2007 to 15.0% between 2018 and 2022. This could be partially explained by the ease of use and higher reliability of TAB than neutralizing antibody assay (NAB). The antibody screening varied greatly by disease indications (Figure 3A). CNS and ocular trials were relatively low: 53.6% and 15.6%, respectively. In contrast, liver, LSD, and muscle trials were generally high: 87.5%, 78.8%, and 78.1%, respectively. Another interesting finding is that while NAB was more frequently used in other disease categories (60-100% of those used NAB or TAB), TAB was more frequently used in the CNS trials (59.3%). Among all serotypes, 86.1% of AAV9 trials used antibody screening, followed by AAV8 (67.7%) (Figure 3C). In contrast, only 33.8% of AAV2 trials had antibody screening, which could be explained by the frequent usage of AAV2 in ocular diseases. Only 55.0% of trials using engineered capsids had antibody screening. While NAB was more frequently chosen for other serotypes, TAB was the predominant choice for AAV9 (67.9%). In terms of ROA, 86.2% of intravenous delivery employed antibody screening, followed by intraarticular (80%), intracoronary (66.7%), IT/ICM (66.7%), and intramuscular (62.5%) (Figure 3D). In contrast, intravitreal (20.8%), subretinal (10.8%), and intracerebral (20.0%) were low. In these disease categories, NAB was the dominant choice except for intramuscular (NAB 22.2% vs TAB 77.8%). The titer threshold for excluding patients varied significantly among trials: NAB (1:1 to 1:1,200), TAB (1:5 to 1:1,600). What makes it more difficult to perform cross-trial comparisons is that the methods used for these assays varied significantly (36). In addition, the in vitro biological activity of the vectors may also contribute to the wide range of NAB thresholds.
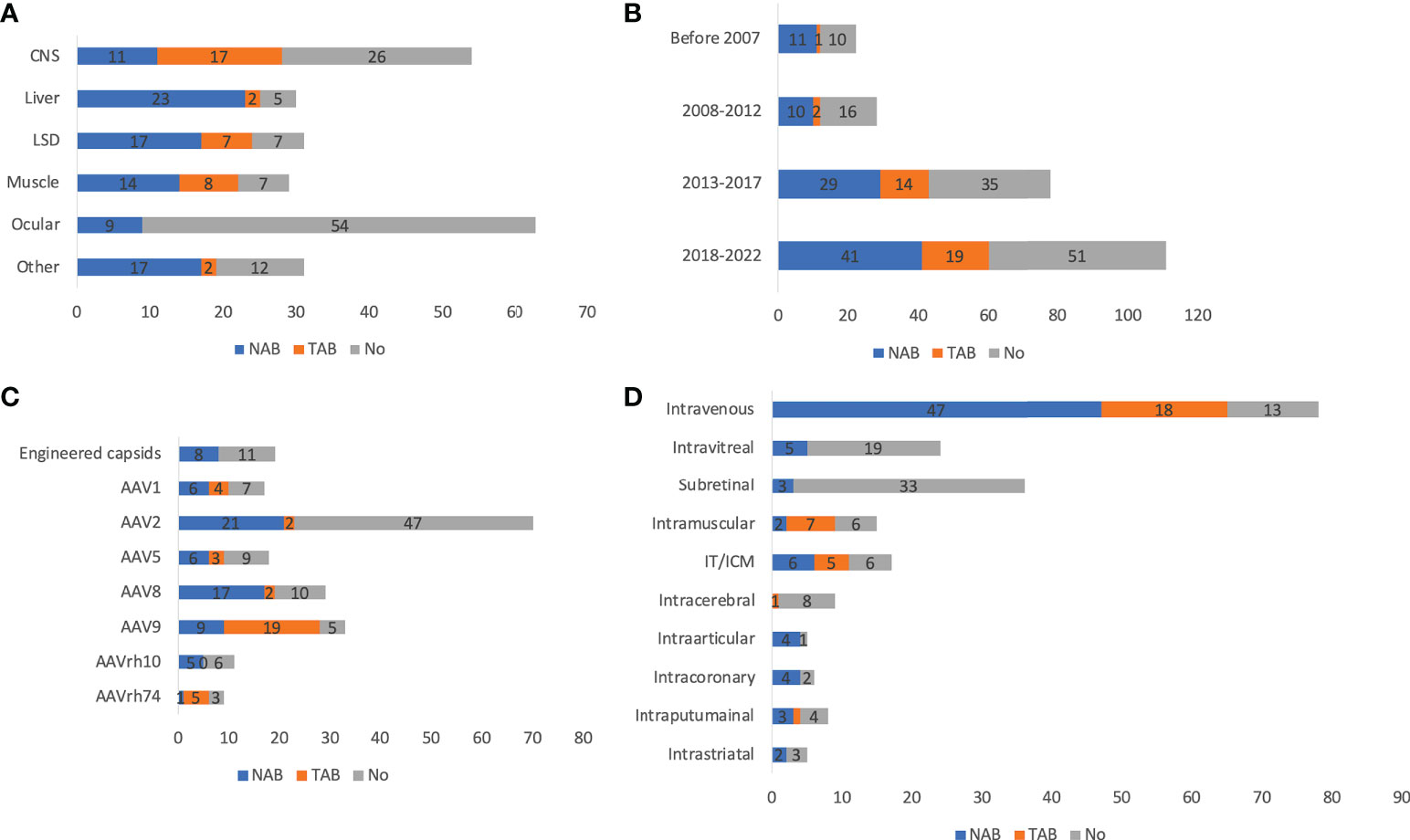
Figure 3 Antibody assays of 255 AAV clinical trials by disease indications (A), timeframe (B), serotypes (C), and ROAs (D). ROA and serotypes associated with <10 trials are not included in the figure. IT and ICM were combined due to the high similarity. Trials that indicated usage of antibody assays but did not specify types of assays are not included.
There were a total of 11 reported patient deaths across 8 trials, with the Audentes trial for X-linked myotubular myopathy having the highest number of patient deaths (n=4), potentially due to hepatotoxicity. The disease indication, ROA, serotype, and production system of these 8 trials are summarized in Table 2. It is worthwhile to mention that whether these patient deaths are vector-related is unclear in most cases. Due to the small sample size and lack of detailed reports, it is difficult to make conclusions on patient deaths in AAV clinical trials. In addition, it was recently reported that two patient deaths due to hepatotoxicity after receiving a commercialized gene therapy drug, Zolgensma (44). There were 30 clinical holds, 18 of which were due to adverse events observed in the respective clinical trials. Others were due to CMC issues, toxicity findings in preclinical studies or other relevant clinical studies, and delivery devices. The most prominent TESAEs were hepatotoxicity (45), TMA (46), and neurotoxicity (41), while DRG toxicity attracted much attention recently (39, 47). Among all disease categories, LSD seemed to be the safest, with the least risk of TESAEs (20.0%), while CNS had the highest risk (42.6%) (Figure 4A). Among all ROAs, IT/ICM seemed to be the safest (5.3%), while local CNS injections had relatively high risk (37.5-50.0%). Interestingly, intravitreal had a lower risk of TESAEs than subretinal (17.4% vs 38.9%), which could be explained by the invasiveness of subretinal injections (Figure 4B). In terms of serotypes, AAV1 and AAV5 seemed to be safer, with a risk of TESAEs at 17.6% and 19.0%, respectively (Figure 4C).
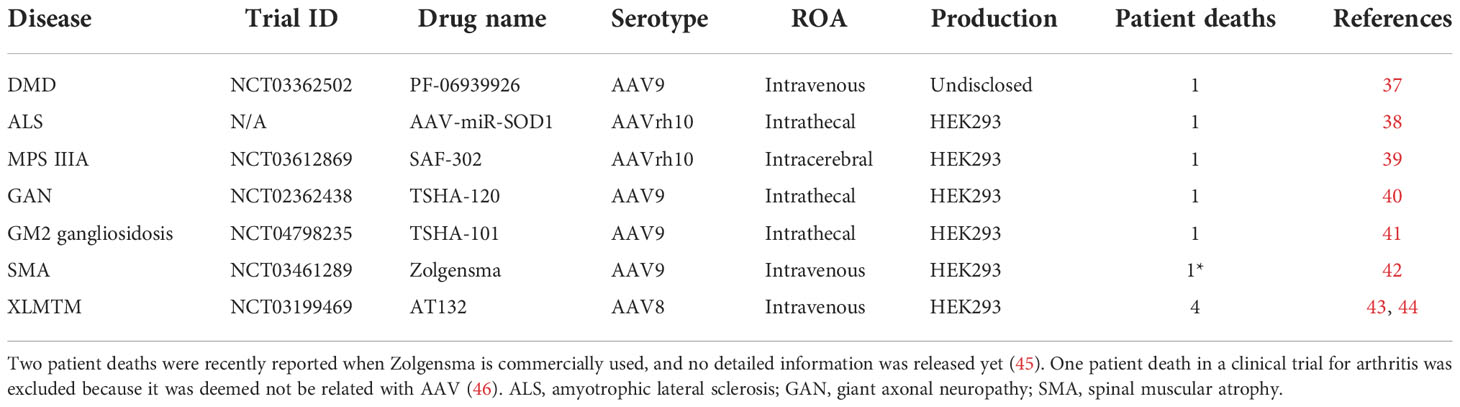
Table 2 Patient deaths that occurred in AAV-related clinical trials.
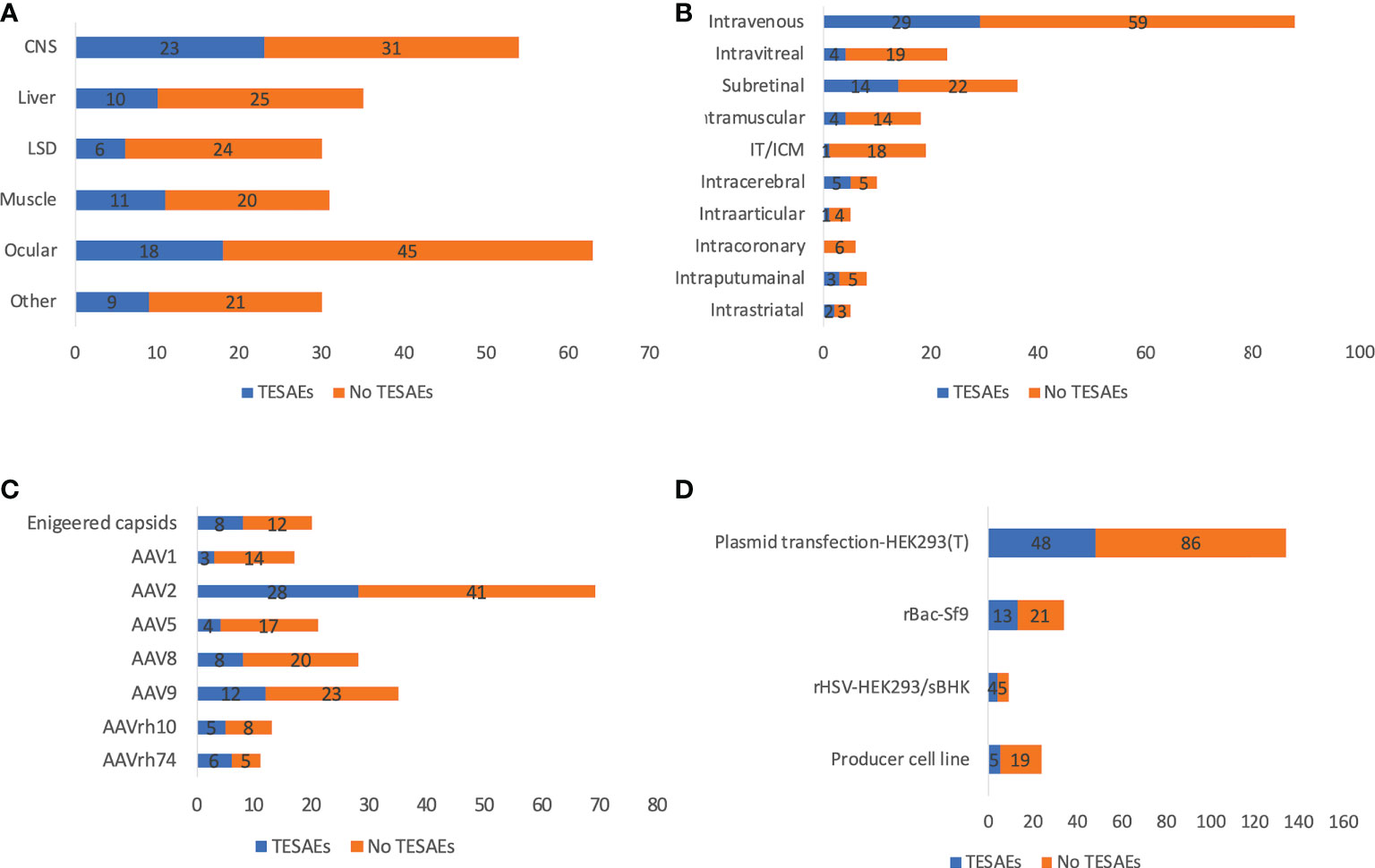
Figure 4 Reported TESAEs of 255 AAV clinical trials by disease indications (A), ROAs (B), serotypes (C), and manufacturing systems (D). ROA and serotypes associated with <10 trials are not included in the figure. IT and ICM were combined due to the high similarity. Trials that initiated recently are not included for analysis.
Long-term clinical data from AAV trials showed incidences of declining transgene expression over time, which may be due to immune response and vector dilution. Declines in transgene expression occur either rapidly or gradually, indicating two distinct mechanisms (presumably acute immune response vs vector dilution). In this study, ‘durable’ was defined as sustained transgene expression or clinical efficacy in all patients during the latest follow-up visit, otherwise it was defined as ‘decline’. Notably, sometimes it is difficult to distinguish lack of initial efficacy and declining transgene expression because some trials observed no efficacy in the first follow-up. Up to 90.0% of CNS and 73.3% of muscle diseases (only those that had publicly available durability data counted) achieved durable transgene expression, in spite of the limited follow-up time and relatively small sample size (Figure 5A). In contrast, only 47.4% of liver trials achieved durability, which can be explained by higher risk of immunogenicity due to systemic administration and active cell divisions of hepatocytes. Only 43.6% of ocular trials achieved durable efficacy, which is counterintuitive because the retina is usually seen as an immune-privileged compartment (48). In general, there was significant inter-patient variability, possibly due to differences in age, genetic background, disease progression, or immune system. This highlights the need of careful patient selection in designing clinical trials. Further, there was no clear correlation between dose and durability even within individual trials. For example, in one trial (RGX-314 for wet AMD), lower doses achieved better durability. This may be because although high dose leads to higher initial transgene expression for decline to begin with, it also causes higher risk of immune response. It is expected to be necessary to identify an optimal balance between high dose and low immunogenicity. Trials using engineered capsids (LK03 for hemophilia A, AAV-Spark100 and AAVS3 for hemophilia B) seemed to achieve better durability even with relatively low doses. However, in general, there is no clear pattern in terms of durability between different serotypes (Figure 5C). One may expect subretinal injections would achieve better durability because AAV is injected into an immune-privileged compartment (49). However, there was no evidence supporting it (intravitreal 64.3% vs subretinal 36.4%) (Figure 5D).
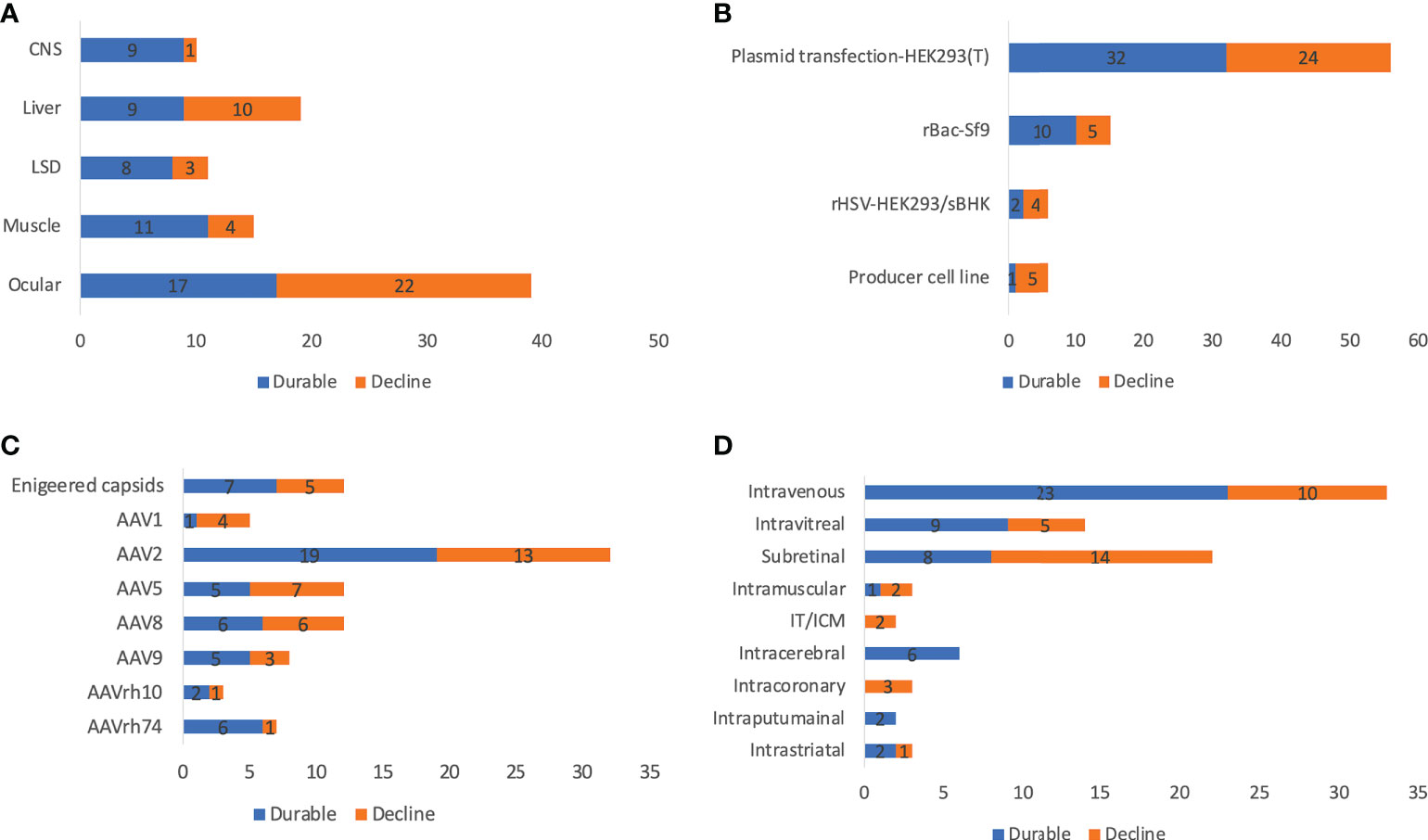
Figure 5 Reported durability of 255 AAV clinical trials by disease indications (A), manufacturing systems (B), serotypes (C), and ROAs (D). ROA and serotypes associated with <10 trials are not included in the figure. IT and ICM were combined due to the high similarity. Trials that initiated recently or did not report efficacy and transgene expression data are not included for analysis.
Plasmid transfection in HEK293 or HEK293T cells became the predominant AAV production system since 2007, while the producer cell line was more frequently used before 2007. There is an increase in the rBac-Sf9 system from 5.6% before 2007 to 20.2% between 2018 and 2022 (Figure 6A). The pros and cons of each system and system used by different companies or institutes are summarized in Table 3. The rBac/Sf9 system has advantages in yield, empty/full ratio, and scalability over other systems (50, 51). Yet, one study showed that AAV produced by the rBac/Sf9 system had a higher degree of truncated and unresolved species than those generated by the HEK293 system (52). Another study showed that the rBac/Sf9 system may lead to difference in post-translational modifications, host cell protein impurity profile, and methylation status of capsid proteins, as well as reduced potency compared to the HEK293 system (53). However, the differences observed in this study was not substantial, and the vectors generated from the two systems were not of similar quality. Moreover, it was reported that by modulating ratio of VP1/2/3 capsid composition in the rBac/Sf9 system, the potency of AAV vectors could be significantly improved to be comparable to the HEK293 system (54). All of these may have implications for AAV immunogenicity and toxicity in clinical studies. Nonetheless, no difference in risk of TESAEs were found between trials using rBac/Sf9 (38.2%) or HEK293 (35.8%) (Figure 4D). Also, a total of 9 patient deaths occurred in 134 AAV trials using the HEK293 system, while there is one individual death for the producer cell line system (n=24) (Figure 6B). The impact of the production system on durability of transgene expression was also assessed. HEK293 and rBac-Sf9 systems had comparable results: 58.2% and 66.7% of trials achieved durable efficacy, respectively (Figure 5B). Interestingly, producer cell line and rHSV systems had only 16.7% and 33.3% of trials achieving durability. Admittedly, the sample size is small.
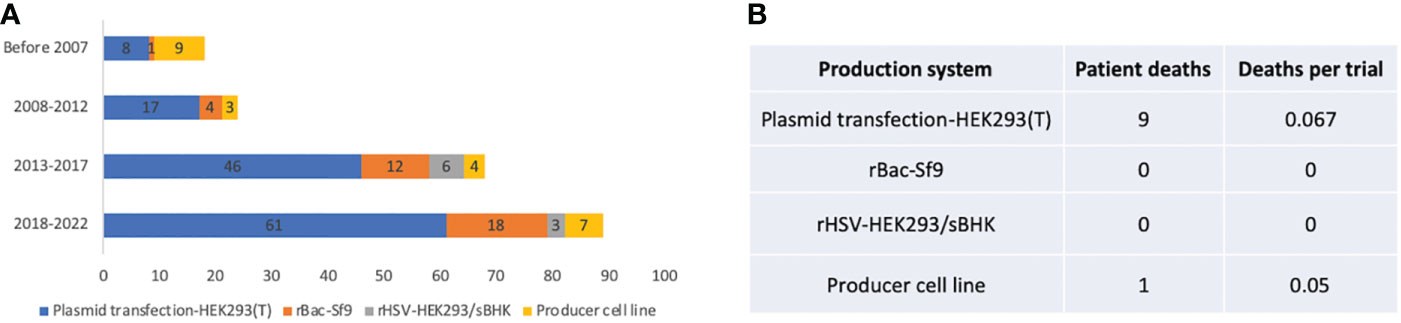
Figure 6 Manufacturing systems of 255 AAV clinical trials. (A) System usage in different time periods. (B) Patient deaths in trials using different manufacturing systems. Trials that did not report manufacturing systems are not included for analysis.
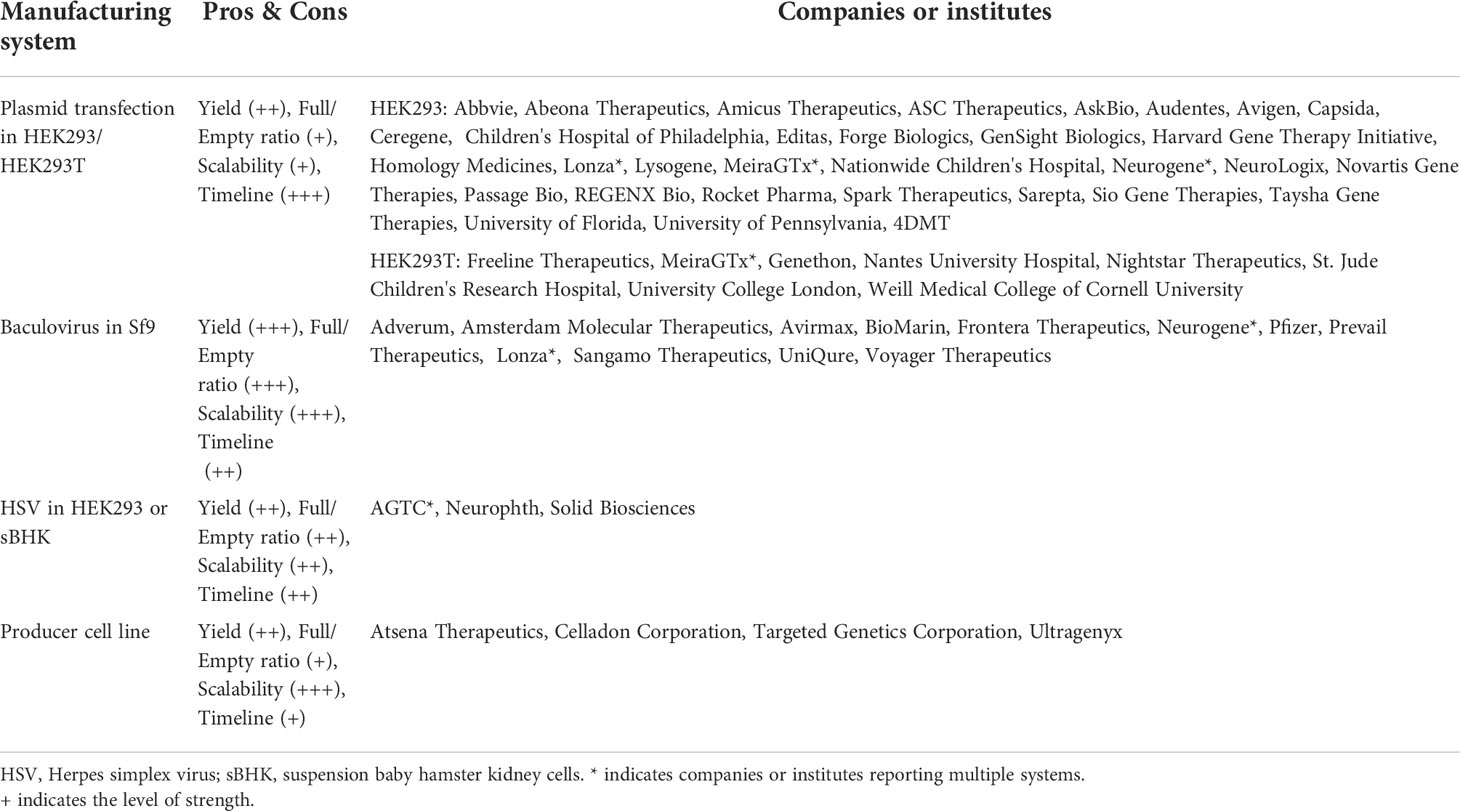
Table 3 Comparison of AAV manufacturing systems.
Immune responses against AAV pose significant challenges for gene therapy development, impacting patient enrollment, toxicity profile, and durability of efficacy. Systematic analysis of currently available clinical data is an important initial step to overcome immune responses against AAV, which could be empowered by accumulating data and more open data-sharing. This study analyzed multiple aspects of 255 AAV clinical trials, providing a knowledge basis for future preclinical and clinical study design. Previous studies have shown that pre-existing antibodies could significantly inhibit AAV transduction (55) and activate memory cells (56). Therefore, pre-existing antibodies exclude a large portion of patients from receiving potentially life-saving treatments due to the relatively high prevalence among human population. It becomes a bigger issue when targeting diseases like Parkinson’s disease because as people age, they would have a higher likelihood to be exposed to AAV. One approach is to use wildtype or engineered capsids that have less seropositive prevalence in human population (53, 54). Another option is using local administration, which is expected to have a reduced risk of interactions between AAV and antibodies (55). Other methods include plasmapheresis (57, 58), saline flushing (59), IgG proteases (60), capsid engineering (61–63), and lipid nanoparticle encapsulated rapamycin (64), most of which are under preclinical development. In clinical settings, a more frequently applied method is antibody screening during patient enrollment. NAB directly measures antibodies that neutralize AAV vectors and inhibit transduction. However, as a cell-based assay, NAB has a higher variability due to differences in cell line maintenance, report gene choices, and multiplicity of infections used (36). Other drawbacks of NAB include the omission of the contribution of non-neutralizing antibodies to immune responses and logistic difficulty when compared with TAB. Possibly due to the aforementioned reasons, this study observed an increased percentage of TAB usage over time. Another key aspect of immune response against AAV in clinical setting is toxicity, evidenced by multiple incidences of hepatotoxicity (44–46, 65), TMA (25, 26), MRI abnormality (66), and DRG toxicity (39, 47). Considering the complexity in chemistry manufacturing and controls (CMC) of recombinant virus products, it is not feasible to directly compare across products from different sponsors. However, a significant number of trials showed a positive correlation between dose and efficacy, meanwhile high doses may lead to a higher risk of complement activation (25, 26), liver enzyme elevations (67), and other adverse effects (66). In many cases, liver enzyme elevations and complement activation can be alleviated by immunosuppressants (67), but not always (46). DRG toxicity has also been reported to be dose-dependent in multiple large animal studies (68). Fortunately, this was less frequently observed in clinical studies, expect two trials for amyotrophic lateral sclerosis (39) and giant axonal neuropathy (41). One potential reason could be species differences. Notably, researchers have developed a potential strategy to reduce the DRG toxicity by silencing the transgene expression in DRG via microRNA (69).
One of the advantages for AAV gene therapy is seen as relatively long-lasting transgene expression, which is achieved mainly through episomal AAV (70, 71). Previous studies showed sustained efficacy for 3 years or longer periods (72, 73). However, many clinical studies observed reduced transgene expression over time, for instance, in some trials for Hemophilia A. In addition to vector dilution, the durability of efficacy can also be impacted by CTL-mediated elimination of transgene-expressing cells (74) and promoter silencing (75). This situation is further exacerbated by the fact that repeated dosing is challenging due to immune responses against AAV (8). In this study, there is high variability in durability across trials, depending on multiple factors, including disease indications, serotypes, ROAs, doses, AAV genome sequences, and genetic background of individual patients. It has been shown that muscle gene transfer could achieve sustained efficacy through inducing immune tolerance by regulatory T cells (76–78). In this study, 73.3% muscular trials achieved durable transgene expression (Figure 5A). However, although there is also clinical evidence of induction of immune tolerance through liver gene transfer (79), only 47.4% of liver trials achieved durable efficacy. This may be partially explained by the more rapid hepatocyte turnover and vector dilution, as well as more antigen presentation due to systemic administration. CNS and retina are usually seen as ‘immune-privileged’ compartments, and gene transfer to CNS and retina are expected to achieve more durable efficacy. Although 90% of CNS achieved durable transgene expression, only 43.6% of ocular trials observed sustained efficacy. This highlighted the complexity in designing ocular trials and the need to carefully select dose, ROA, and patients (genetic background, disease progression, age). Overall, the durability of AAV gene therapies is reasonably good, although careful patient selection and optimized design are warranted.
In addition to immune responses, AAV toxicity can result from potential insertional mutagenesis. rAAV exists mainly as an episome, but still be able to integrate into host chromosome DNA (80). It was estimated that intravenous administration may deliver >100 vector copies per hepatocyte on average (81). With the robust metabolism and strong regeneration activity of hepatocytes, there are concerns about vector genome insertion and oncogenesis. Tumorigenesis events are documented in rodent studies but shown to be less frequent in large animals (71, 82–86), indicating a species dependence. The integration risk also seems to be serotype-dependent. One mouse study using AAV9 observed hepatocellular carcinoma (87), while a similar study using AAV5 did not (88). In addition, the risk also depends on promoter choice as shown in a murine study (86). Currently, only one patient developed a liver tumor (NCT03569891) and finally showed not to be related with AAV dosing (89). Improving the vector potency and specificity by engineering may significantly decrease the required dose, which reduces the risk of genome insertion. Also, exploring the genome design to accelerate the circularization kinetics may help reduce the exposure of the linear genome to double strand breaks, which may reduce the insertion events. In light of this, the FDA recently organized a Cellular, Tissue, and Gene Therapies Advisory Committee (CTGAC) meeting to address AAV toxicity issues, including AAV integration risk. In summary, the insertional mutagenesis risk of AAV is relatively low in the context of currently available clinical data, but it should be monitored in the long-term follow up studies as recommended by the FDA.
Compared with small or large molecule drugs, another crucial challenge for gene therapy is translatability. Remarkable efficacy has been observed in many small animal studies, while the translatability to large animal and clinical trials are limited. For instance, an engineered capsid selected from a C57BL/6J mice strain penetrates the blood brain barrier efficiently (90). However, the receptor for this selected capsid, PHP.B, only exists in some mouse strains but not others or NHPs (91). There is a high demand for designing and screening better capsids to enter target cells or tissue more efficiently and specifically. However, due to the complexity and differences of blood circulation, cell and tissue types, cell surface protein patterns related with attachment and binding, many novel capsids work well only in certain animal models (91, 92). Understanding the species differences and developing new models for mimicking the specific questions to solve will enhance the translatability in the future. Another example is the vector penetration for ocular indications by intravitreal injection. Human eye structure has the inner limiting membrane, which brings extra physical barriers than mouse species. In addition, the cross-species translatability issue also pose significant challenges for predicting immune responses in clinical studies from preclinical data. This holds especially true for T cell responses as expansion of capsid-specific CD8+ T cells was observed in humans (93), but not mice (94), dogs (95), and NHPs (96). Thus, it is critical to develop suitable animal models to recapitulate immune responses against AAV in humans.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
WS and LO designed the study, collected data, performed data analysis, and wrote and revised the manuscript. SL revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors would like to thank Drs. Jingmin Zhou and Patty Biezonski from Genemagic Biosciences for reviewing the manuscript.
LO is an employee of Genemagic Biosciences, WS is an employee of Obio Technologies, and SL is an employee of Avirmax.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1001263/full#supplementary-material
1. Wagner JA, Reynolds T, Moran ML, Moss RB, Wine JJ, Flotte TR, et al. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet (1998) 351(9117):1702–3. doi: 10.1016/S0140-6736(05)77740-0
2. Wang D, Tai PW, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discovery (2019) 18(5):358–78. doi: 10.1038/s41573-019-0012-9
3. Kuzmin DA, Shutova MV, Johnston NR, Smith OP, Fedorin VV, Kukushkin YS. The clinical landscape for AAV gene therapies. Nat Rev Drug Discov (2021) 20(3):173–5. doi: 10.1038/d41573-021-00017-7
4. Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia b. New Engl J Med (2014) 371(21):1994–2004. doi: 10.1056/NEJMoa1407309
5. Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat Med (2006) 12(3):342–7. doi: 10.1038/nm1358
6. Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia a. New Engl J Med (2020) 382(1):29–40.
7. George LA, Sullivan SK, Giermasz A, Rasko JE, Samelson-Jones BJ, Ducore J, et al. Hemophilia b gene therapy with a high-specific-activity factor IX variant. New Engl J Med (2017) 377(23):2215–27.
8. Muhuri M, Maeda Y, Ma H, Ram S, Fitzgerald KA, Tai PW, et al. Overcoming innate immune barriers that impede AAV gene therapy vectors. J Clin Invest (2021) 131(1):e143780.
10. Mingozzi F, A High K. Immune responses to AAV in clinical trials. Curr Gene Ther (2011) 11(4):321–30.
11. Vandamme C, Adjali O, Mingozzi F. Unraveling the complex story of immune responses to AAV vectors trial after trial. Hum Gene Ther (2017) 28(11):1061–74. doi: 10.1089/hum.2017.150
12. Ronzitti G, Gross DA, Mingozzi F. Human immune responses to adeno-associated virus (AAV) vectors. Front Immunol (2020) 11:670. doi: 10.3389/fimmu.2020.00670
13. Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis (2009) 199(3):381–90. doi: 10.1086/595830
14. Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, et al. Effects of transient immunosuppression on adeno associated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood (2006) 108(10):3321–8. doi: 10.1182/blood-2006-04-017913
15. Perrin GQ, Zolotukhin I, Sherman A, Biswas M, De Jong YP, Terhorst C, et al. Dynamics of antigen presentation to transgene product-specific CD4+ T cells and of treg induction upon hepatic AAV gene transfer. Mol Therapy-Methods Clin Dev (2016) 3:16083. doi: 10.1038/mtm.2016.83
16. Tóth R, Mészáros I, Hüser D, Forró B, Marton S, Olasz F, et al. Methylation status of the adeno-associated virus type 2 (AAV2). Viruses (2019) 11(1):38.
17. Zhu J, Huang X, Yang Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest (2009) 119(8):2388–98. doi: 10.1172/JCI37607
18. Ashley SN, Somanathan S, Giles AR, Wilson JM. TLR9 signaling mediates adaptive immunity following systemic AAV gene therapy. Cell Immunol (2019) 346:103997. doi: 10.1016/j.cellimm.2019.103997
19. Rogers GL, Suzuki M, Zolotukhin I, Markusic DM, Morel LM, Lee B, et al. Unique roles of TLR9-and MyD88-dependent and-independent pathways in adaptive immune responses to AAV-mediated gene transfer. J innate Immun (2015) 7(3):302–14. doi: 10.1159/000369273
20. Sudres M, Ciré S, Vasseur V, Brault L, Da Rocha S, Boisgérault F, et al. MyD88 signaling in b cells regulates the production of Th1-dependent antibodies to AAV. Mol Ther (2012) 20(8):1571–81. doi: 10.1038/mt.2012.101
21. Konkle BA, Walsh CE, Escobar MA, Josephson NC, Young G, von Drygalski A, et al. BAX 335 hemophilia b gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood (2021) 137(6):763–74. doi: 10.1182/blood.2019004625
22. Wright JF. Quantification of CpG motifs in rAAV genomes: Avoiding the toll. Mol Ther (2020) 28(8):1756–8. doi: 10.1016/j.ymthe.2020.07.006
23. Wright JF. Codon modification and PAMPs in clinical AAV vectors. Tortoise Hare? Mol Ther (2020) 28(3):701–3. doi: 10.1016/j.ymthe.2020.01.026
24. Hamilton BA, Wright JF. Challenges posed by immune responses to AAV vectors: addressing root causes. Front Immunol (2021) 12:1654. doi: 10.3389/fimmu.2021.675897
25. Available at: https://www.fiercebiotech.com/biotech/pfizer-s-dmd-gene-therapy-looks-good-data-refresh-but-safety-concerns-persist.
26. Available at: https://www.fiercebiotech.com/biotech/fda-lifts-clinical-hold-solid-bio-gene-therapy-trial.
27. Li X, Wei X, Lin J, Ou L. A versatile toolkit for overcoming AAV immunity. Front Immunol (2022) 13:5093. doi: 10.3389/fimmu.2022.991832
28. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Systematic Rev (2021) 10(1):1–11.
29. Ou L, DeKelver RC, Rohde M, Tom S, Radeke R, Martin SJS, et al. ZFN-mediated in vivo genome editing corrects murine hurler syndrome. Mol Ther (2019) 27(1):178–87. doi: 10.1016/j.ymthe.2018.10.018
30. Gaudet D, Méthot J, Déry S, Brisson D, Essiembre C, Tremblay G, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther (2013) 20(4):361–9. doi: 10.1038/gt.2012.43
31. Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. New Engl J Med (2017) 377(18):1713–22. doi: 10.1056/NEJMoa1706198
32. Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr., Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for leber's congenital amaurosis. New Engl J Med (2008) 358(21):2240–8. doi: 10.1056/NEJMoa0802315
33. Tai CH, Lee NC, Chien YH, Byrne BJ, Muramatsu SI, Tseng SH, et al. Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol Ther (2022) 30(2):509–18. doi: 10.1016/j.ymthe.2021.11.005
34. Ozelo MC, Mahlangu J, Pasi KJ, Giermasz A, Leavitt AD, Laffan M, et al. Valoctocogene roxaparvovec gene therapy for hemophilia a. New Engl J Med (2022) 386(11):1013–25. doi: 10.1056/NEJMoa2113708
35. Mercuri E, Muntoni F, Baranello G, Masson R, Boespflug-Tanguy O, Bruno C, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy type 1 (STR1VE-EU): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol (2021) 20(10):832–41. doi: 10.1016/S1474-4422(21)00251-9
36. Weber T. Anti-AAV antibodies in AAV gene therapy: current challenges and possible solutions. Front Immunol (2021) 12:658399. doi: 10.3389/fimmu.2021.658399
37. Available at: https://www.fiercebiotech.com/biotech/pfizer-reports-death-patient-duchenne-trial-halts-enrolment.
38. Mueller C, Berry JD, McKenna-Yasek DM, Gernoux G, Owegi MA, Pothier LM, et al. SOD1 suppression with adeno-associated virus and MicroRNA in familial ALS. New Engl J Med (2020) 383(2):151–8. doi: 10.1056/NEJMoa2005056
39. Philippidis A. Profoundly saddened” lysogene discloses child's death in phase II/III trial. Hum Gene Ther (2020) 31(21-22):1141–3. doi: 10.1089/hum.2020.29139.bfs
40. Bonnemann CG. AAV related immunological safety and toxicity: Preliminary clinical observations in the GAN and MTM1 trials. In: Presented at: Virtual workshop on systemic immunogenicity considerations of AAV-mediated gene therapy. (Baltimore, Maryland: NIH, NCATS) (2020).
41. Available at: https://www.fiercebiotech.com/biotech/taysha-mulls-tweaks-to-gene-therapy-trial-after-subject-dies.
42. Available at: https://www.fiercepharma.com/marketing/zolgensma-s-european-trial-death-unrelated-to-gene-therapy-as-novartis-touts-new.
43. Wilson JM, Flotte TR. Moving forward after two deaths in a gene therapy trial of myotubular myopathy: To learn from these tragedies, the scientific community must commit to full transparency and cooperation. Genet Eng Biotechnol News (2020) 40(8):14–6. doi: 10.1089/gen.40.08.05
44. Shieh PB, Bönnemann CG, Müller-Felber W, Blaschek A, Dowling JJ, Kuntz NL, et al. Re:“Moving forward after two deaths in a gene therapy trial of myotubular myopathy” by Wilson Flotte Hum Gene Ther 31(15-16) (2020) 787. doi: 10.1089/hum.2020.217
45. Available at: https://www.fiercepharma.com/pharma/two-deaths-after-novartis-zolgensma-bring-gene-therapys-liver-safety-spotlight-again.
46. Available at: https://www.fiercebiotech.com/biotech/press-release-fdaprobing-death-targeted-genetics-gene-therapy-trial.
47. Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, et al. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther (2018) 29(3):285–98. doi: 10.1089/hum.2018.015
48. Ramlogan-Steel CA, Murali A, Andrzejewski S, Dhungel B, Steel JC, Layton CJ. Gene therapy and the adeno-associated virus in the treatment of genetic and acquired ophthalmic diseases in humans: trials, future directions and safety considerations. Clin Exp Ophthalmol (2019) 47(4):521–36. doi: 10.1111/ceo.13416
49. Amato A, Arrigo A, Aragona E, Manitto MP, Saladino A, Bandello F, et al. Gene therapy in inherited retinal diseases: an update on current state of the art. Front Med (2021) 8. doi: 10.3389/fmed.2021.750586
50. Sandro Q, Relizani K, Benchaouir R. Aav production using baculovirus expression vector system. In Viral Vectors Gene Ther (2019) pp:91–99). doi: 10.1007/978-1-4939-9065-8_5
51. Clément N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Therapy-Methods Clin Dev (2016) 3:16002. doi: 10.1038/mtm.2016.2
52. Tran NT, Lecomte E, Saleun S, Namkung S, Robin C, Weber K, et al. Human and insect cell-produced recombinant adeno-associated viruses show differences in genome heterogeneity. Hum Gene Ther (2022) 33(7-8):371–88. doi: 10.1089/hum.2022.050
53. Rumachik NG, Malaker SA, Poweleit N, Maynard LH, Adams CM, Leib RD, et al. Methods matter: standard production platforms for recombinant AAV produce chemically and functionally distinct vectors. Mol Therapy-Methods Clin Dev (2020) 18:98–118. doi: 10.1016/j.omtm.2020.05.018
54. Kondratov O, Marsic D, Crosson SM, Mendez-Gomez HR, Moskalenko O, Mietzsch M, et al. Direct head-to-head evaluation of recombinant adeno-associated viral vectors manufactured in human versus insect cells. Mol Ther (2017) 25(12):2661–75. doi: 10.1016/j.ymthe.2017.08.003
55. Ertl HC. T Cell-mediated immune responses to AAV and AAV vectors. Front Immunol (2021) 12:666666. doi: 10.3389/fimmu.2021.666666
56. Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther (2020) 28(3):723–46. doi: 10.1016/j.ymthe.2019.12.010
57. Chicoine LG, Montgomery CL, Bremer WG, Shontz KM, Griffin DA, Heller KN, et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther (2014) 22(2):338–47. doi: 10.1038/mt.2013.244
58. Monteilhet V, Saheb S, Boutin S, Leborgne C, Veron P, Montus MF, et al. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther (2011) 19(11):2084–91. doi: 10.1038/mt.2011.108
59. Mimuro J, Mizukami H, Hishikawa S, Ikemoto T, Ishiwata A, Sakata A, et al. Minimizing the inhibitory effect of neutralizing antibody for efficient gene expression in the liver with adeno-associated virus 8 vectors. Mol Ther J Am Soc Gene Ther (2013) 21(2):318–23. doi: 10.1038/mt.2012.258
60. Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight (2020) 5(19):e139881. doi: 10.1172/jci.insight.139881
61. Tse LV, Klinc KA, Madigan VJ, Castellanos Rivera RM, Wells LF, Havlik LP, et al. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc Natl Acad Sci U S A (2017) 114(24):E4812–E21. doi: 10.1073/pnas.1704766114
62. Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum igg and neutralizing factors against adeno-associated virus (Aav) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using aav vectors. Hum Gene Ther (2010) 21(6):704–12. doi: 10.1089/hum.2009.182
63. Halbert CL, Rutledge EA, Allen JM, Russell DW, Miller AD. Repeat transduction in the mouse lung by using adeno-associated virus vectors with different serotypes. J Virol (2000) 74(3):1524–32. doi: 10.1128/jvi.74.3.1524-1532.2000
64. Ilyinskii PO, Michaud AM, Roy CJ, Rizzo GL, Elkins SL, Capela T, et al. Enhancement of liver-directed transgene expression at initial and repeat doses of AAV vectors admixed with ImmTOR nanoparticles. Sci Adv (2021) 7(9):eabd0321. doi: 10.1126/sciadv.abd0321
65. Available at: https://www.fiercepharma.com/pharma/two-deaths-after-novartis-zolgensma-bring-gene-therapys-liver-safety-spotlight-again.
66. Sondhi D, Kaminsky SM, Hackett NR, Pagovich OE, Rosenberg JB, De BP, et al. Slowing late infantile batten disease by direct brain parenchymal administration of a rh. 10 adeno-associated virus expressing CLN2. Sci Trans Med (2020) 12(572):eabb5413.
67. Chand D, Mohr F, McMillan H, Tukov FF, Montgomery K, Kleyn A, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol (2021) 74(3):560–6. doi: 10.1016/j.jhep.2020.11.001
68. Hordeaux J, Buza EL, Dyer C, Goode T, Mitchell TW, Richman L, et al. Adeno-associated virus-induced dorsal root ganglion pathology. Hum Gene Ther (2020) 31(15-16):808–18. doi: 10.1089/hum.2020.167
69. Hordeaux J, Buza EL, Jeffrey B, Song C, Jahan T, Yuan Y, et al. MicroRNA-mediated inhibition of transgene expression reduces dorsal root ganglion toxicity by AAV vectors in primates. Sci Trans Med (2020) 12(569):eaba9188. doi: 10.1126/scitranslmed.aba9188
70. Duan D, Sharma P, Yang J, Yue Y, Dudus L, Zhang Y, et al. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol (1998) 72(11):8568–77. doi: 10.1128/JVI.72.11.8568-8577.1998
71. Nakai H, Montini E, Fuess S, Storm TA, Grompe M, Kay MA. AAV serotype 2 vectors preferentially integrate into active genes in mice. Nat Genet (2003) 34(3):297–302. doi: 10.1038/ng1179
72. Herzog RW. Encouraging and unsettling findings in long-term follow-up of AAV gene transfer. Mol Ther (2020) 28(2):341–2. doi: 10.1016/j.ymthe.2020.01.007
73. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther (2021) 29(2):464–88. doi: 10.1016/j.ymthe.2020.12.007
74. Scallan CD, Jiang H, Liu T, Patarroyo-White S, Sommer JM, Zhou S, et al. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood (2006) 107(5):1810–7. doi: 10.1182/blood-2005-08-3229
75. Gray SJ, Foti SB, Schwartz JW, Bachaboina L, Taylor-Blake B, Coleman J, et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther (2011) 22(9):1143–53. doi: 10.1089/hum.2010.245
76. Mingozzi F, Liu YL, Dobrzynski E, Kaufhold A, Liu JH, Wang Y, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest (2003) 111(9):1347–56. doi: 10.1172/JCI200316887
77. Brantly ML, Chulay JD, Wang L, Mueller C, Humphries M, Spencer LT, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci (2009) 106(38):16363–8. doi: 10.1073/pnas.0904514106
78. Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing α1-antitrypsin: interim results. Hum Gene Ther (2011) 22(10):1239–47. doi: 10.1089/hum.2011.053
79. Ferreira V, Twisk J, Kwikkers K, Aronica E, Brisson D, Methot J, et al. Immune responses to intramuscular administration of alipogene tiparvovec (AAV1-LPLS447X) in a phase II clinical trial of lipoprotein lipase deficiency gene therapy. Hum Gene Ther (2014) 25(3):180–8. doi: 10.1089/hum.2013.169
80. Hanlon KS, Kleinstiver BP, Garcia SP, Zaborowski MP, Volak A, Spirig SE, et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat Commun (2019) 10(1):1–11. doi: 10.1038/s41467-019-12449-2
81. Kishimoto TK, Samulski RJ. Addressing high dose AAV toxicity–’one and done’or ‘slower and lower’? Expert Opin Biol Ther (2022), 22:1–5. doi: 10.1080/14712598.2022.2060737
82. Russell DW. AAV vectors, insertional mutagenesis, and cancer. Mol Ther (2007) 15(10):1740–3. doi: 10.1038/sj.mt.6300299
83. Sabatino DE, Chair FDB, Chandler RJ, Crystal RG, Davidson BL, Dolmetsch R, et al. Evaluating the state of the science for adeno-associated virus (AAV) integration: An integrated perspective. Mol Ther (2022) 30(8):2646–63. doi: 10.1016/j.ymthe.2022.06.004
84. Nguyen GN, Everett JK, Kafle S, Roche AM, Raymond HE, Leiby J, et al. A long-term study of AAV gene therapy in dogs with hemophilia a identifies clonal expansions of transduced liver cells. Nat Biotechnol (2021) 39(1):47–55. doi: 10.1038/s41587-020-0741-7
85. Dalwadi DA, Calabria A, Tiyaboonchai A, Posey J, Naugler WE, Montini E, et al. AAV integration in human hepatocytes. Mol Ther (2021) 29(10):2898–909. doi: 10.1016/j.ymthe.2021.08.031
86. Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest (2015) 125(2):870–80. doi: 10.1172/JCI79213
87. Li Y, Miller CA, Shea LK, Jiang X, Guzman MA, Chandler RJ, et al. Enhanced efficacy and increased long-term toxicity of CNS-directed, AAV-based combination therapy for krabbe disease. Mol Ther (2021) 29(2):691–701. doi: 10.1016/j.ymthe.2020.12.031
88. Hawkins-Salsbury JA, Shea L, Jiang X, Hunter DA, Guzman AM, Reddy AS, et al. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J Neurosci (2015) 35(16):6495–505. doi: 10.1523/JNEUROSCI.4199-14.2015
89. de Jong YP, Herzog RW. Liver gene therapy and hepatocellular carcinoma: A complex web. Mol Ther (2021) 29(4):1353–4. doi: 10.1016/j.ymthe.2021.03.009
90. Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol (2016) 34(2):204–9. doi: 10.1038/nbt.3440
91. Huang Q, Chan KY, Tobey IG, Chan YA, Poterba T, Boutros CL, et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP. b capsids. PLoS One (2019) 14(11):e0225206. doi: 10.1371/journal.pone.0225206
92. Hordeaux J, Yuan Y, Clark PM, Wang Q, Martino RA, Sims JJ, et al. The GPI-linked protein LY6A drives AAV-PHP. b transport across the blood-brain barrier. Mol Ther (2019) 27(5):912–21.
93. Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, Edmonson SA, Hui DJ, Sabatino DE, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. blood. J Am Soc Hematol (2007) 110(7):2334–41. doi: 10.1182/blood-2007-03-080093
94. Martino AT, Basner-Tschakarjan E, Markusic DM, Finn JD, Hinderer C, Zhou S, et al. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. blood. J Am Soc Hematol (2013) 121(12):2224–33. doi: 10.1182/blood-2012-10-460733
95. Sun J, Shao W, Chen X, Merricks EP, Wimsey L, Abajas YL, et al. An observational study from long-term AAV re-administration in two hemophilia dogs. Mol Therapy-Methods Clin Dev (2018) 10:257–67. doi: 10.1016/j.omtm.2018.07.011
Keywords: rAAV, clinical trials, immunogenicity, toxicity, neutralizing antibodies, immunosuppressants, capsids, gene therapy
Citation: Shen W, Liu S and Ou L (2022) rAAV immunogenicity, toxicity, and durability in 255 clinical trials: A meta-analysis. Front. Immunol. 13:1001263. doi: 10.3389/fimmu.2022.1001263
Received: 23 July 2022; Accepted: 18 October 2022;
Published: 27 October 2022.
Edited by:
Arun Srivastava, University of Florida, United StatesReviewed by:
Hiroaki Mizukami, Jichi Medical University, JapanCopyright © 2022 Shen, Liu and Ou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Ou, b3V4eHgwNDVAdW1uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.