 Jiaxuan Chen1†
Jiaxuan Chen1† Shuzhen Liao1†
Shuzhen Liao1† Huimin Zhou2†
Huimin Zhou2† Lawei Yang1
Lawei Yang1 Fengbiao Guo1
Fengbiao Guo1 Shuxian Chen1
Shuxian Chen1 Aifen Li1
Aifen Li1 Quanren Pan1
Quanren Pan1 Chen Yang1
Chen Yang1 Hua-feng Liu1*
Hua-feng Liu1* Qingjun Pan1*
Qingjun Pan1*- 1Key Laboratory of Prevention and Management of Chronic Kidney Disease of Zhanjiang City, Institute of Nephrology, Affiliated Hospital of Guangdong Medical University, Zhanjiang, China
- 2Zhanjiang Central Hospital, Guangdong Medical University, Zhanjiang, China
Animal models have played a crucial role in the understanding of the mechanisms and treatments of human diseases; however, owing to the large differences in genetic background and disease-specific characteristics, animal models cannot fully simulate the occurrence and progression of human diseases. Recently, humanized immune system mice, based on immunodeficient mice, have been developed that allow for the partial reconstruction of the human immune system and mimic the human in vivo microenvironment. Systemic lupus erythematosus (SLE) is a complex disease characterized by the loss of tolerance to autoantigens, overproduction of autoantibodies, and inflammation in multiple organ systems. The detailed immunological events that trigger the onset of clinical manifestations in patients with SLE are still not well known. Two methods have been adopted for the development of humanized SLE mice. They include transferring peripheral blood mononuclear cells from patients with SLE to immunodeficient mice or transferring human hematopoietic stem cells to immunodeficient mice followed by intraperitoneal injection with pristane to induce lupus. However, there are still several challenges to be overcome, such as how to improve the efficiency of reconstruction of the human B cell immune response, how to extend the lifespan and improve the survival rate of mice to extend the observation period, and how to improve the development of standardized commercialized models and use them. In summary, there are opportunities and challenges for the development of humanized mouse models of SLE, which will provide novel strategies for understanding the mechanisms and treatments of SLE.
Introduction
Systemic lupus erythematosus (SLE) is a typical autoimmune disease characterized by excessive activation of T and B cells, producing a large number of autoantibodies and pro-inflammatory cytokines that result in tissue and organ damage (1). At present, there are few clinically approved traditional therapeutic drugs and biologic therapies for SLE (2, 3). Animal models have made great contributions to the study of SLE pathogenesis and the development of new drugs. Based on the study of spontaneous (4–6) or induced (7–9) lupus-prone mouse model, considerable progress has been made in understanding the pathogenesis of SLE. In these models, disease phenotypes similar to patients with SLE can be observed, including the imbalanced immune responses of T and B cells, the production of a variety of autoantibodies and a large number of pro-inflammatory cytokines, and damage to multiple organs (such as lupus nephritis, etc.) (10). However, the genetic background differences between humans and mice cause the lupus-prone mouse model to have many differences from human SLE, especially when studying the in vivo functions of molecules with poor homology between humans and mice (such as non-coding RNA, etc.) (11–13) and Kv1.3 phenotype, etc. (14). The emergence of humanized mice allows for better studies in vivo, further clarifies the pathogenesis, and improves the success rate of translational medicine research (such as novel drug discovery, etc.) (15–17). At present, there are two main methods of constructing humanized mouse models of SLE, including transferring human peripheral blood mononuclear cells (PBMCs) or peripheral blood lymphocytes (PBLs) from patients with SLE to immunodeficient mice (18, 19), or transferring human hematopoietic stem cells (HSCs) to immunodeficient mice and then injecting intraperitoneally (i. p.) with pristane to induce lupus (20) (Figure 1). For these two humanized SLE mouse models, the PBLs/PBMCs humanized mouse model is widely used, but individual differences in SLE patients often lead to inconsistent model parameters and poor uniformity; the HSCs-pristane humanized mouse model can better reproduce the clinical features of human SLE, but there are very few such studies. The differences between two kinds of humanized SLE mice as shown in Table 1. The above two humanized SLE mouse models provide opportunities to study the pathogenesis and prevention of SLE in vivo, but there are also many challenges.
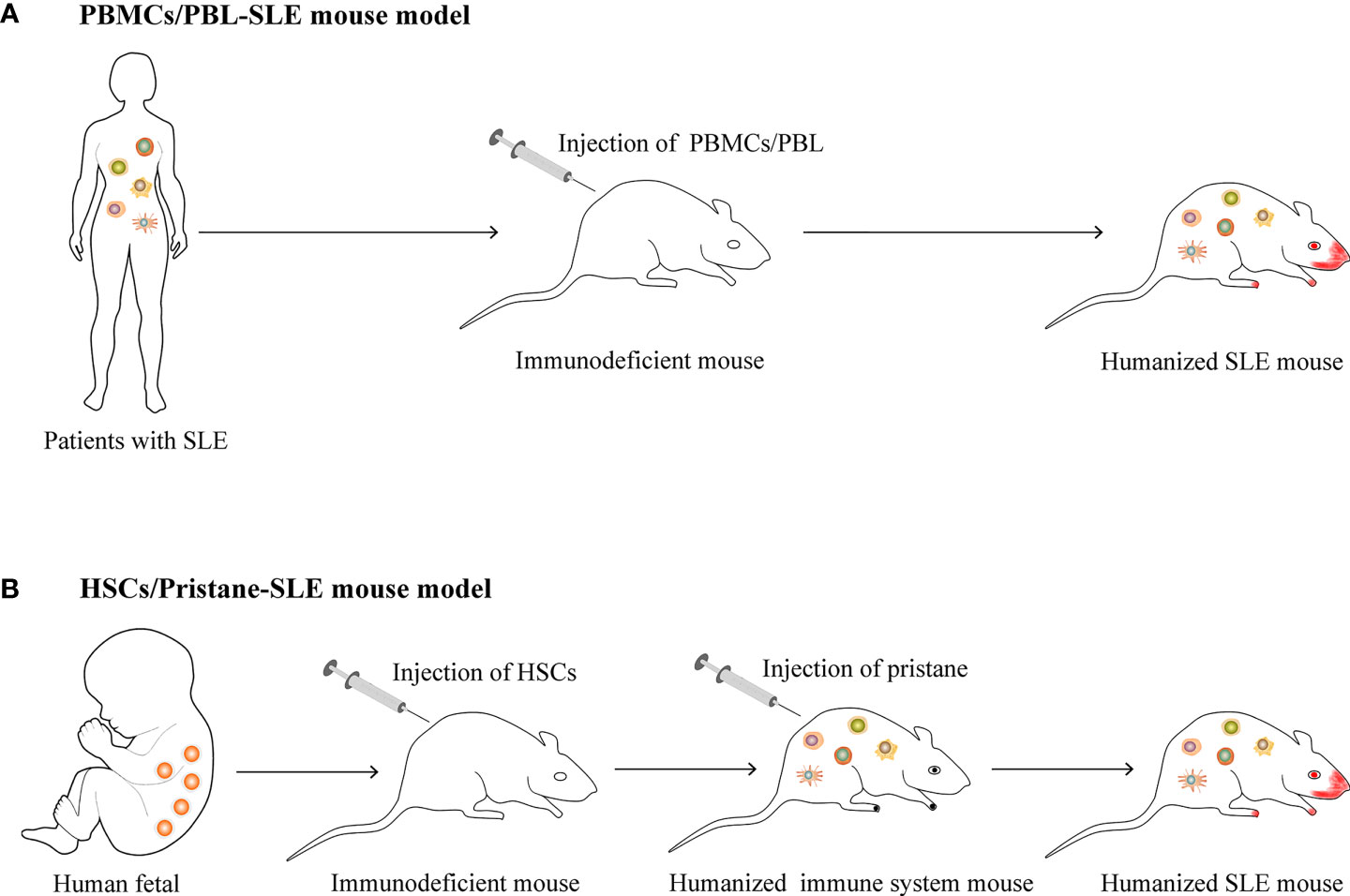
Figure 1 The construction of humanized SLE mouse model. (A) Transferring human peripheral blood mononuclear cells (PBMCs) or peripheral blood lymphocytes (PBLs) from patients with SLE to immunodeficient mice. (B) Transferring human hematopoietic stem cells (HSCs) to immunodeficient mice and then injecting intraperitoneally (i. p.) with pristane to induce lupus.

Table 1 The differences between two kinds of humanized SLE mice.
PBLs/PBMCs Humanized SLE Mouse Model
Development of PBLs/PBMCs Humanized SLE Mouse Model
The main characteristic of humanized mice is the reconstruction of the human immune system in immunodeficient mice. For DKO (BALB-Rag2-/- IL2Rgc-/-) mice (4–5 weeks old) engrafted with PBMCs (0.3–0.5×107) from patients with SLE, the ratio of human CD45+ cells to total PBMCs increased from 5–10% (6–7 weeks old) to 20–80% (8–10 weeks old) (21).
It is known that T and B cells interact to promote the progression of lupus (35). T cells mainly promote the development of SLE through the production of pro-inflammatory cytokines and tissue infiltration (36). Humanized mouse models of SLE constructed by engrafting PBLs/PBMCs from patients with SLE have mainly revealed the presence of human T cells (21). A skewed ratio of CD4 to CD8 (lower frequency of CD4+ and higher CD8+ cells) in the PBMCs of patients with SLE is commonly observed (37, 38). In a humanized mouse model of SLE within 7–8 weeks by engrafting PBMCs (0.3–0.5×107) from patients with SLE, CD3+ cells were found in the CD45+cells of PBMCs from both SLE-DKO and ND-DKO mice. While in SLE-DKO mice, a significantly lower frequency of CD3+CD4+cells (5.5% ± 2.1%) and a higher frequency of CD3+CD8+ cells (79.4 ± 3.6%) was reported; this contrasted with a more typical distribution of CD3+CD4+ (66.2 ± 2.5%) and CD3+CD8+ (16.5 ± 2.1%) cells in the ND-DKO mice (21). In addition, a similar study showed that among T cells, the ratio of CD4+CD8- cells to CD4-CD8+cells were 3:1, and 1:2 at one- and two-months post engraftment, respectively (22). This skewed distribution could also be detected in humanized SLE mice, which supports the hypothesis that this model mimics the characteristics of human SLE.
Previous studies have demonstrated that the effective B cell helper activity of Th17 cells was an important function of pro-inflammatory T cells (39, 40). The increased percentage of human IL-17+ Tfh cells was detected in the spleens of NSG mice (8 weeks old) engrafted with PBMCs (1×107cells/mouse) from patients with active lupus, while this process could be halted by the knockdown of RORγ in human CD4+ T cells (23). In this instance, Th17 could also be detected in humanized SLE mice, and RORγ therapy targeting CD4+ T cells is expected to become a novel strategy.
In SLE, B cells mainly play a role in antibody production, antigen presentation, and cytokine expression (41). NK cells are producers of various cytokines and chemokines (e.g., IFN-g, TNF-a, CCL5, CCL3, and CCL4), which amplify and recruit an inflammatory response through various mechanisms, further contributing to the progression of SLE (42). Myeloid cells (e.g., neutrophils, dendritic cells) have also been reported to be key factors in SLE (43, 44). However, the reconstruction efficiency of B, NK, and myeloid cells in the PBLs/PBMCs humanized SLE mouse model is not satisfactory and remains a huge challenge today. In this mice model, it was found that human CD3+ populations were detected in the CD45+cells in PBMCs of SLE-DKO mice at 3–4 weeks post engraftment, while other human immune cells such as B cells, NK cells, and myeloid cells were rare or undetectable (21). Therefore, improving the reconstruction efficiency of B cells, NK cells, and myeloid cells is a significant challenge to develop this model better.
The success of reconstruction in humanized SLE mice with engraftment of human PBLs/PBMCs can also be assessed based on the expression of human IgG, with successful engraftment indicated by equal to or higher than 200 µg/mL of human IgG in the sera two weeks following PBLs/PBMC administration (28). As previously reported, the average serum level of human IgG was approximately 3000 µg/mL post-PBLs (3×107 cells/mouse) from patients with SLE were injected i.p. into SCID mice (8–10 weeks old) (28). A similar study showed that approximately 500 µg/mL of human IgG in the serum of SCID mice (9–13 weeks old) engrafted with PBMCs (3×107 cells/mouse) from patients with SLE could be detected (29).
The ratio of human CD4/CD8 also can affect the production of IgG in humanized SLE mice. When this ratio increased from a lower ratio (less than 0.5) to a higher ratio (greater than 1.5), serum levels of human IgG could be detected (24). In addition, in vitro activation of human PBMCs also led to ten times higher IgG production in vivo compared with PBMCs without activation (24). Therefore, for the variation in human IgG production in humanized mice, the key effector may be the phenotype and activation status of human PBLs/PBMCs.
For the autoantibodies produced by this PBLs/PBMCs humanized SLE mouse model, several main autoantigens (e.g., dsDNA, Ro, RNP, anti-La, etc.), and subtypes of IgGs (e.g., IgG I, IgG II) can be detected. Human IgG could be detected in the serum of established humanized SLE mice at two weeks and reached maximum levels at two months after the reconstruction of the model with i.p. injection of PBLs (1.5×107 cells/mouse) from patients with SLE into SCID mice (22).
Anti-dsDNA autoantibodies are representative autoantibodies for the diagnosis and modeling of the disease activity of SLE (45). A previous study showed that higher levels of human IgG I and IgG II anti-dsDNA autoantibodies were detected in the serum of humanized SLE mice (10–12 weeks old), after the establishment of this model with i.p. injection of PBMCs (3×107 cells/mouse) from patients with SLE (SLEDAI score 5.88 ± 4.18) into SCID mice (8–10 weeks) (28).
In addition, humanized SLE mice produce autoantibodies against other autoantigens. After the establishment of humanized SLE mice with i.p. injection of 2–5×107 PBMCs from patients with SLE into SCID mice (5–7 weeks old), antibodies against human anti-Ro, anti-RNP, and anti-La in serum could be detected at 4–6 weeks after transplantation (34). DKO mice (4–5 weeks old) were used for engraftment of (0.3–0.5×107 cells/mouse) PBMCs from patients with SLE, and the antibodies of humans (e.g., anti-dsDNA antibody, ANA antibodies, ACL IgG) in serum could be detected at 4–8 weeks post engraftment. Importantly, SLE patients with a high level of antiphospholipid antibodies (>80 GCL) showed high ACL IgG levels in all DKO mice engrafted with their PBMCs. Additionally, all mice had detectable ACL IgG at two to three different times within two to four weeks post engraftment (21). Thus, the engrafted mice showed most of the antibodies in patients with SLE and reflected an accurate phenocopy of certain autoantibodies.
Treatment of PBLs/PBMCs Humanized SLE Mouse Model
Autoantibodies and Pro-Inflammatory Cytokines
In terms of intervention studies, the production of autoantibodies and pro-inflammatory cytokines in humanized SLE mice can also be attenuated or eliminated by drugs or other factors.
The protein annexin A1 (ANX A1) is a modulator of the immune response involving several cell types, and its expression in activated B and T cells is abnormal in autoimmune disease (46–48). In one study, the levels of autoantibodies s (e.g., anti-ssDNA, anti-RNA, anti-histone, and anti-nucleosome IgG), inflammatory cytokines (e.g., IFN-γ and IL-4), and disease symptoms were significantly reduced in anti-ANX A1 antibody-treated humanized SLE mice (12 weeks old NSG mice engrafted with 1×107 PBMCs/mouse from patients with SLE), compared to the humanized SLE mice treated with the isotype control antibody (15).
Myeloid-derived suppressor cells (MDSCs) with immunosuppressive functions are a group of highly heterogeneous populations derived from myeloid progenitors (49). It has been reported that MDSCs have a pathogenic role in promoting the development of autoimmune diseases (50–52). For example, mouse MDSCs can promote the differentiation of Th17 cells (53). However, the role of MDSCs in Th17 differentiation and the pathogenesis of autoimmune diseases in humans is relatively unknown. In a previous study, a humanized SLE mouse model was established by intravenous injection of PBMCs from patients with active SLE into immunodeficient non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice. To investigate the function of MDSCs and Arg-1 in disease progression, the NOD/SCID mice were injected with unaltered PBMCs, MDSC-depleted PBMCs, or unaltered PBMCs plus nor-NOHA (the Arg-1 inhibitor). The study showed that all mice (4–5 weeks old NOD/SCID mice engrafted with 0.5–1×107 PBMCs/mouse from patients with SLE) injected with unaltered PBMCs had detectable human autoantibodies within four to five weeks (30). However, mice receiving MDSC-depleted PBMCs showed significantly less severe symptoms, indicating that MDSCs are necessary for disease progression in vivo. In addition, the deleterious role of MDSCs was possibly dependent on Arg-1, because its inhibitor significantly delayed disease progression in NOD/SCID mice (30). The above research indicates that targeting MDSCs or Arg-1 is expected to alleviate SLE disease progression.
Based on the suppressive activity of complement receptor type 1 on human lymphocytes, the co-crosslinking of this receptor on B cells with the B-cell receptor (BCR) can inhibit the activation and proliferation of B cells, and this receptor may be a novel therapeutic target for negative signal delivery (54, 55). Humanized SLE mice (8 weeks old SCID mice engrafted with 1×107 PBMCs/mouse from patients with SLE) were treated with anti-human DNA-like chimeras, which contained a monoclonal antibody against human inhibitory complement receptor type 1. The results showed that anti-dsDNA antibodies were directly eliminated. The specific clearance of autoreactive B cells not only limited the production of anti-dsDNA IgG, but also limited the activation and proliferation of autoreactive T cells. Additionally, the levels of pro-inflammatory cytokines IL-10 and IFN-γ were also reduced (16, 31). The same study showed that anti-human DNA-like chimeras could prevent the production of anti-dsDNA IgG antibodies (32). Anti-human DNA-like chimeras had an ideal therapeutic effect in humanized SLE mice, and they are expected to enter clinical research as a drug.
Two synthesized peptides (based on the sequence of CDR1 and CDR3 of the pathogenic murine anti-DNA 16/6Id) were reported to be immunodominant T cell epitopes in normal (e.g., BALB/c, SJL) and lupus-prone (NZB×NZW) F1 mice (56–58). Treatment with these peptides improved clinical symptoms and decreased autoantibody production in spontaneous and induced SLE (59–61). Treatment with hCDR1 significantly decreased the serum levels of human anti-dsDNA antibodies and decreased the serum levels of IFN-γ and IL-10, while increasing TGF-β production in humanized SLE mice (8–10 weeks old SCID mice engrafted with 3×107 PBLs/mouse from patients with SLE) (29). However, this treatment did not affect anti-tetanus toxoid antibodies. Therefore, the effect of hCDR1 treatment may be restricted to SLE-associated responses, and the hCDR1 peptide is a potential novel candidate for SLE treatment.
One potential therapeutic strategy for SLE is antisense/ribozyme, which specifically inhibits the expression of the target mRNA without severe side effects (62, 63). In a study, humanized SLE mice (SCID mice engrafted with 0.5×107 PBLs/mouse from patients with SLE) treatment with the chemically modified ribozyme (RZ-I) not only decreased anti-DNA antibody production in these humanized SLE mice but also inhibited IgG deposition in the kidneys of these mice (17). Therefore, a novel therapeutic strategy for SLE may be based on the usefulness of chemically modified ribozymes.
Whether the delivery of IL-2 and TGF-β, which are deficient in SLE, mediated by nanoparticles (NPs) to mouse CD2+ and CD4+ cells, could induce a tolerogenic immune response and then protect mice from a lupus-like disorder was investigated (64, 65). Humanized SLE mice (8–12 weeks old NSG mice engrafted with 1×107 PBMCs/mouse from patients with SLE) treated with T cell-targeted NPs loaded with IL-2/TGF-β showed significantly reduced serum levels of human IgG and improved skin morphology (25). Therefore, NPs may provide a novel therapeutic strategy in vivo for the suppression of proinflammatory responses in SLE and other autoimmune diseases.
In another study, the binding of XmAb5871 (the Fc domain of one anti-human CD19 antibody) with FcγRIIb promoted the engagement of FcγRIIb with the BCR complex (66). This antibody stimulated phosphorylation of the ITIM of FcγRIIb and suppressed BCR-induced calcium mobilization. It also allowed for the proliferation of human B cells, costimulatory molecule expression on B cells from healthy persons and patients with SLE, as well as the proliferation of B cells induced by LPS, IL-4, or B cell-activating factor (BAFF) (67). Another study involved anti-XmAb5871 treatment performed on humanized SLE mice (6–12 weeks old SCID mice engrafted with 1–3×107 PBMCs/mouse from patients with SLE). It was found that anti-XmAb5871 inhibited the activation of B cells and the total human IgG2 level (26). In addition, anti-XmAb5871 substantially inhibited anti-tetanus titer in vivo (26). Thus, anti-XmAb5871 should be considered a novel B cell-targeted immunosuppressive therapeutic strategy for SLE.
AS101 as an immunomodulator can significantly decrease serum levels of human IgG, IgA, IgM (e.g., anti-dsDNA IgG, anti-Sm IgG), and IL-10 in humanized SLE mice (SCID mice engrafted with 1.5×107 PBMCs/mouse from patients with SLE) (27).
In addition, treatment of humanized SLE mice (6–10 weeks old SCID mice engrafted with 1.5×107 PBMCs/mouse from patients with SLE) with an anti-IL-6 monoclonal antibody inconsistently decreased the serum concentration of anti-dsDNA IgG produced by PBMCs from patients with SLE. In contrast, administration of an anti-IL-10 monoclonal antibody consistently decreased autoantibodies produced by SLE PBMCs (33).
Lupus Nephritis
The kidney is one of the most involved organs in SLE (lupus nephritis) (68). Approximately 50% of patients with SLE have clinical renal involvement with lupus nephritis (69), and humanized SLE mice also show similar renal disease. SLE-DKO mice have mild proteinuria at 4–6 weeks after implantation of PBMCs (0.3–0.5×107) from patients with SLE and human IgG deposits in the glomeruli, and the glomeruli were enlarged, showing severe capillary thrombosis and endothelial cell necrosis. Multifocal acute tubular necrosis with hyaline casts was also observed (21). The overall appearance of the kidney was similar to that of a human lupus class IV-G proliferative nephritis. It has also been reported that 1.5×107 PBLs of patients with SLE were injected i.p. into SCID mice. The kidney tissue showed that human IgA and IgG were granular and circularly deposited along the mesangium and capillaries, and proteinuria occurred (14, 22). It can be seen that the humanized mice modeled by the PBMCs of patients with SLE also displayed kidney lesions, which were similar to spontaneous and induced mouse models and are closer to clinical patients.
The intervention of humanized lupus mice can reduce pathological changes in their kidneys. Anti-ANX A1 treatment of humanized SLE mice (12 weeks old NSG mice engrafted with 1×107 PBMCs/mouse from patients with SLE) reduced the proteinuria of the mice, significantly reduced cell infiltration in the kidney, and no immune complex deposition was observed (15). NOD/SCID mice receiving MDSC-depleted PBMCs showed a substantial decrease in proteinuria levels, IL-17A, and human IgG deposition in glomeruli and mesangial cell proliferation (30). The proteinuria level of humanized SLE mice (8–10 weeks old SCID mice engrafted with 3×107 PBLs/mouse from patients with SLE) was significantly reduced after hCDR1 treatment; however, IgG and C3 deposits in the kidney sections were detected in only one (6%) in 17 mice treated with hCDR1 (29). Treatment with RZ-I reduced the level of proteinuria, inhibited the production of anti-DNA, and there was no glomerular IgG, IgM, or IgA deposition in humanized SLE mice (SCID mice engrafted with 0.5×107 PBLs/mouse from patients with SLE) (17). Targeting immunogenic self-DNA-specific Tfh cells through human RORγ knockdown in CD4+ T cells and IL-17 neutralization effectively eliminated the levels of kidney inflammation, IgG deposition, and proteinuria in humanized SLE mice (8 weeks old NSG mice engrafted with 1×107 PBMCs/mouse from patients with SLE) (23). In anti-human DNA-like chimera treatment, this has also been proven to considerably reduce immune complex deposition and improve kidney disease (16, 31, 32). The application of humanized lupus mice has allowed for the increase in attempts to treat lupus nephritis and has guided researchers in the clinical development of new drugs and treatment measures.
Lifespan and Survival Rates
Immunodeficient mice transplanted with PBMCs from patients with SLE generally die spontaneously after four weeks. In contrast, the survival rate of mice modeled with normal human PBMCs was significantly higher than that of lupus patients (21).
It has been reported that specific treatment of humanized SLE mice can improve their survival rate. PBMCs from patients with lupus nephritis were pretreated with Kv1.3-NPs and then transferred 0.8×107 PBMCs into 6–10 weeks old NSG mice. It was found that this pretreatment increased the survival rate of PBMC-humanized mice with lupus nephritis by 66% compared with those in the non-treated PBMCs group (14). Pretreated T cells with NPs loaded with IL-2/TGF-β further improved the survival rate of humanized SLE mice (8–12 weeks old NSG mice engrafted with 1×107 PBMCs/mouse from patients with SLE) compared to those of the non-treated T cell group (25). Treatment with an anti-XmAb5871 antibody inhibited the activation of B cells in humanized SLE mice (6–12 weeks old SCID mice engrafted with 1–3×107 PBMCs/mouse from patients with SLE), and significantly improved the survival rate compared with non-treated mice (26). Therefore, further studies are required to extend the lifespan and improve the survival rate of humanized SLE mice.
In summary, specific intervention for humanized SLE mice can significantly reduce the levels of autoantibodies and pro-inflammatory cytokines, improve renal function, and prolong the life span (Table 2).
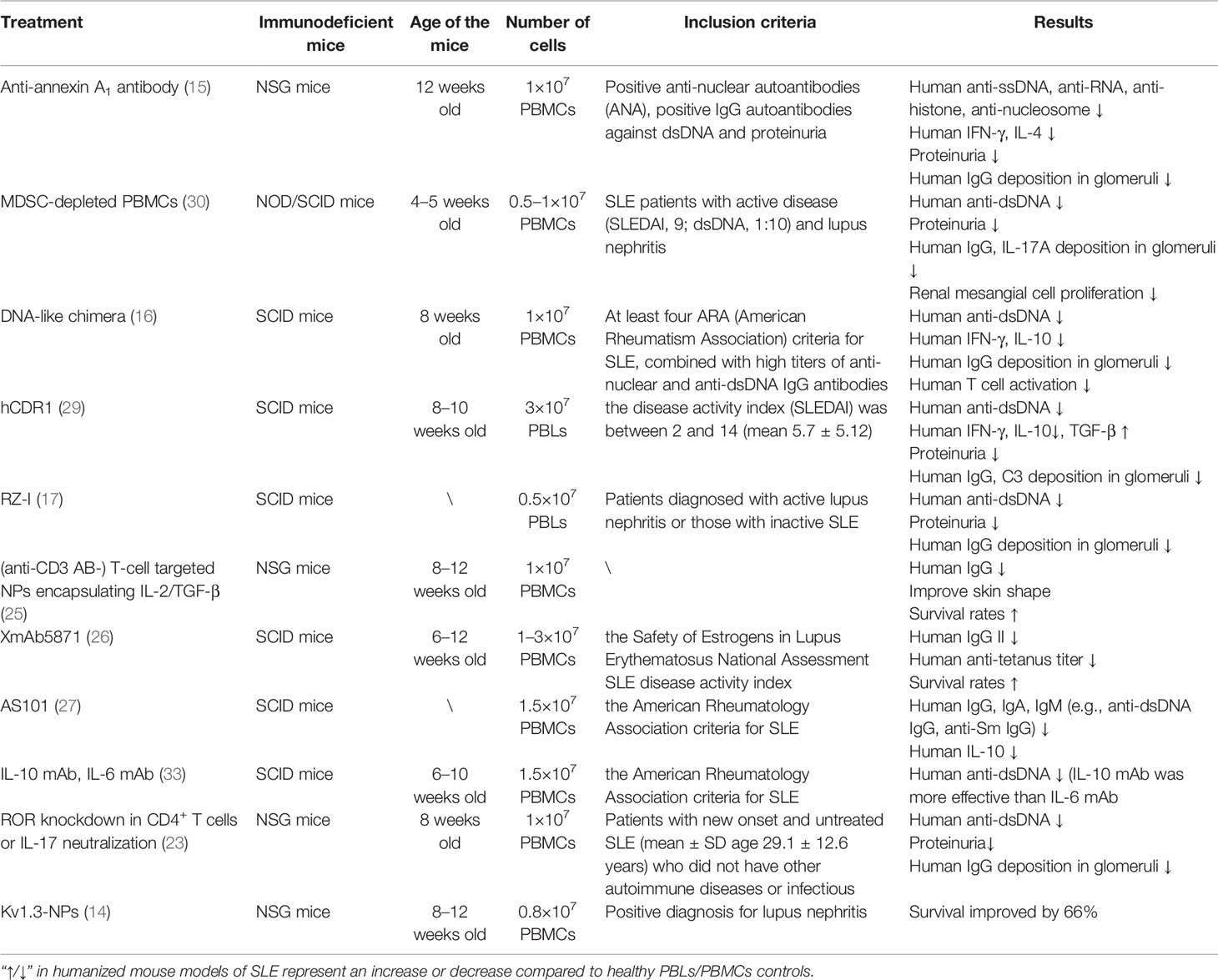
Table 2 The therapeutic effect on humanized SLE mice.
HSCs-Pristane Humanized SLE Mouse Model
In the HSCs-pristane humanized SLE mouse model, NSG mice (within three days after birth) were sublethally irradiated with 1 Gy γ-rays first and then transplanted with human CD34+ HSCs (1×105 cells/mouse) by intra-hepatic injections. The results showed that these humanized mice consistently achieved a good reconstitution of the human immune system, with reconstitution levels in the blood (42.1%), and higher levels in the tissues at 12 weeks, including the spleen (82.8%), mesenteric lymph nodes (97.4%), and liver (89.0%). Subsequently, pristane was injected i.p. into 12–13 weeks old humanized NSG mice and normal NSG mice. Pristane injection induced the hyperactivation of B cells, as shown by the increased expression of CD86, a B cell activation marker. In addition, the percentage and absolute number of CD19+CD20−CD27hiCD38hi plasmablasts/plasma cells in the peripheral blood and spleen of pristane-injected humanized NSG mice. Moreover, a relative expansion in the percentage of CD27+ memory B cells and CD27−IgD− B cell populations and a reduction in the CD27−IgD+ naïve/transitional B cell compartment were found in these pristane-injected humanized mice. Finally, pristane-injected humanized mice showed the activation of both CD4+ and CD8+ T cells, a marked reduction in both CD4+ and CD8+ T cells with a naïve phenotype, and an increased percentage of T cells with an effector memory phenotype in the peripheral blood, spleen, mesenteric lymph nodes, and peritoneal lavage, indicating a systemic proinflammatory condition (20). For the production of autoantibodies and pro-inflammatory cytokines by these pristane-injected humanized mice, the total levels of human IgG and IgM and human anti-nuclear autoantibodies (e.g., anti-dsDNA antibody, anti-histone antibody, anti-RNP70 antibody) were detected. In particular, human anti-dsDNA IgG can be detected as early as four weeks after the injection of pristane and gradually increased to eight weeks. The serum levels of human pro-inflammatory cytokines (e.g., IFN-γ, IL-8, IL-6) also increased significantly in the plasma and peritoneal lavage fluid (20). Lupus nephritis is the most severe manifestation of organ involvement in patients with SLE (70). It is characterized by the deposition of immune complexes in the glomerulus and infiltration of leukocytes, leading to proteinuria (71). In the upper pristane-injected humanized mice, focal to diffuse global glomerular enlargement by mesangial/endocapillary proliferation and increased glomerular cellularity and human CD45+ cells in the glomeruli were reported. All of these were not observed in NSG mice injected with pristine alone (20). For lung injury, the upper pristane-injected humanized mice showed increased multifocal serosal and subpleural inflammation with fibrosis, as well as perivascular interstitial and intra-alveolar mononuclear cell infiltrate (20). For the survival rate, the upper pristane-injected humanized mice showed significantly earlier mortality (median survival at 13 weeks) after pristane injection. NSG mice injected with pristine alone appeared healthy, and there was no mortality during the observation period (20 weeks after pristane injection) (20).
The above HSCs-pristane humanized SLE mouse model provided another strategy for the development of a humanized SLE mouse model. This model is more consistent with the clinical characteristics of SLE patients and reflects the interaction of various immune cells, which is an ideal mouse lupus model. At present, there are few intervention studies based on this model, and more follow-up studies are needed to confirm its stability and clinical value.
The Improvement of Humanized SLE Mouse Model
In the development of humanized SLE mice, PBLs/PBMCs humanized SLE mouse models are widely used, but individual differences in patients with SLE often lead to inconsistent parameters and poor uniformity. This model can better study human T cells, but the effect of human B cell reconstruction is poor, the level of human NK cells is low, and the differentiation of human myeloid cells is lacking.
Regarding the poor reconstruction of human B cells, the reason may be that human T cells proliferate too fast, and the proportion of human B cells decreases as time increases (72). In addition, some reports have shown that certain proteins related to B cell survival showed weak cross-reaction between mice and humans, and there was a lack of signal supporting human B cell survival in mice (73). It has been reported that the lentiviral vector carrying the human IL-7 gene was overexpressed in Rag2-/-γC-/- mice, and the serum level of human IL-7 in mice was maintained at a high level during the observation period of six months. Overexpression of human IL-7 significantly increased the proportion of T and B cells in peripheral blood (74). It has also been reported that the proportion of human B cells can be increased by injecting recombinant human BLyS protein into humanized mice (73).
The low level of human NK cells in humanized mice may be due to the lack of relevant cytokines that support the survival of human NK cells in mice, resulting in a short survival time (72). To solve this problem, a study was conducted involving human IL-15 and Flt3l vectors that were injected into humanized mice, and it was found that the NK cell reconstruction level was significantly improved (75). In addition, the induced human NK cells normally express both activation and inhibition, causing NK cell-dependent liver damage and having the ability to kill target cells in vitro. The above results indicate that the reconstructed human NK cells were functional (75).
Regarding the problem of poor myeloid differentiation, it has been reported that human neutrophils, monocytes, and dendritic cells (DCs) were significantly increased after the injection of human G-CSF into NOG mice (76). Similarly, NOD/SCID mice were injected with human SCF, IL-3, GM-CSF, and TPO for two weeks, and the development of lymphocytes and myeloid cells was significantly improved (77). The injection of human FLT3L in NOD/SCID mice significantly increased the number and function of DCs (78). In addition, Nsg-sgm3 mice were constructed using a transgenic technique to express human SCF, GM-CSF, and IL-3. The results showed that the reconstruction level of myeloid cells was significantly improved, especially in DCs (79).
Another major challenge is that although PBLs/PBMCs humanized SLE mouse model can better simulate the clinical characteristics of patients with SLE, their lifespan and survival rate are significantly lower than those of spontaneous or induced lupus-prone mouse models, which may lead to a narrow period for observation or treatment. One study found immunodeficient mice transplanted with high lupus activity PBMC had a low survival rate and transplanted with low lupus activity PBMC had a high survival rate (21). Therefore, in future research, determining a consistent standard and unifying it is an important direction to better construct a humanized SLE mouse model.
Another HSCs-pristane humanized SLE mouse model irradiates mice before modeling. This can provide more “space” for humanized construction through irradiation or pretreatment with chemical reagents. A previous study compared the efficiency of transplantation with irradiation and found that human immune cells could survive better by pre-radiation 2–3 Gy to NOD/SCID mice before injection of human HSCs (80). It has also been found that mouse NK cells could be knocked out using CD122 or IL-2R antibodies (80). Cl2MDP can knock out mouse macrophages and obtain a better reconstruction of the human immune system (81). This modeling method will theoretically better reproduce the clinical features of human SLE, but there are still few research reports.
Based on the above, the treatment of humanized mice can significantly increase the number of human B, NK, and myeloid cells, and better reconstruct the human immune system (Table 3). However, these interventions have rarely been used in humanized SLE mice. It can be seen that the humanized SLE mouse model still has a long way to go.
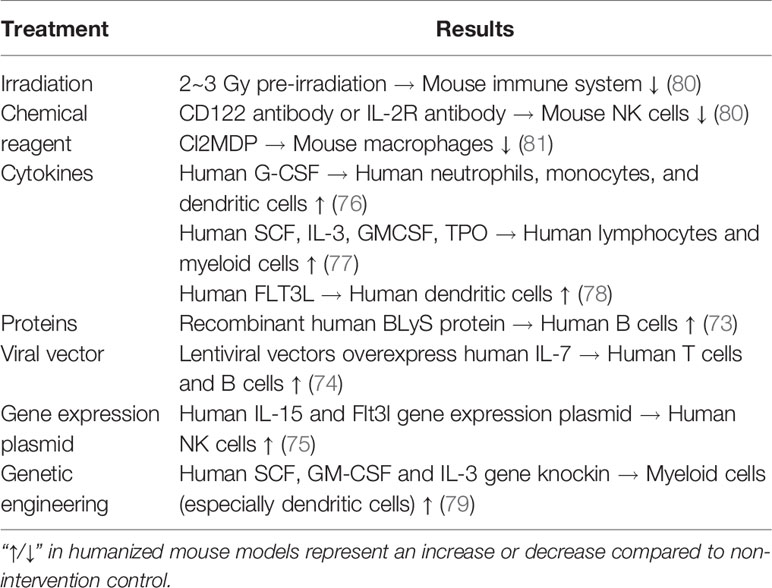
Table 3 The improvement of humanized SLE mice.
Future Perspectives
Currently, the pathogenesis of SLE is still not well known, and the clinically approved traditional therapeutic drugs, as well as biologic therapies for SLE are still very few. The successful development of a humanized SLE mouse model has provided a new path for the study of SLE. However, there are still many challenges to overcome, such as how to better reconstruct the B-cell immune response and how to extend the lifespan and survival rate of mice to extend the period of medical treatment. In summary, to further improve humanized SLE mouse models and develop standardized or even commercialized models, these models can better clarify the pathogenesis of SLE and provide new strategies for the prevention and treatment of SLE, especially the development of new drugs.
Author Contributions
JC, SL, and HZ wrote the manuscript and designed the figures. LY, FG, SC, AL, QRP, CY, H-fL, and QJP revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Natural Science Foundation of China (no. 82070757), the Project of “Dengfeng Plan” and Department of established positions for the Zhujiang Scholar from Guangdong Medical University, and Guangdong Basic and Applied Basic Research Foundation (no. 2019A1515012203), the Zhanjiang City Program for Tackling Key Problems in Science and Technology (no. 2019B01179, no. 2017A01009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tsokos GC. Systemic Lupus Erythematosus. N Engl J Med (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
2. Murphy G, Isenberg DA. New Therapies for Systemic Lupus Erythematosus - Past Imperfect, Future Tense. Nat Rev Rheumatol (2019) 15:403–12. doi: 10.1038/s41584-019-0235-5
3. Li A, Guo F, Pan Q, Chen S, Chen J, Liu HF, et al. Mesenchymal Stem Cell Therapy: Hope for Patients With Systemic Lupus Erythematosus. Front Immunol (2021) 12:728190. doi: 10.3389/fimmu.2021.728190
4. Steinberg AD, Raveché ES, Laskin CA, Smith HR, Santoro T, Miller ML, et al. NIH Conference. Systemic Lupus Erythematosus: Insights From Animal Models. Ann Internal Med (1984) 100:714–27. doi: 10.7326/0003-4819-100-5-714
5. Pan Q, Gong L, Xiao H, Feng Y, Li L, Deng Z, et al. Basophil Activation-Dependent Autoantibody and Interleukin-17 Production Exacerbate Systemic Lupus Erythematosus. Front Immunol (2017) 8:348. doi: 10.3389/fimmu.2017.00348
6. Pan Q, Xiao H, Shi L, He Y, Cai J, Wu J, et al. IgG4 Autoantibodies Attenuate Systemic Lupus Erythematosus Progression by Suppressing Complement Consumption and Inflammatory Cytokine Production. Front Immunol (2020) 11:1047. doi: 10.3389/fimmu.2020.01047
7. Yung R, Chang S, Hemati N, Johnson K, Richardson B. Mechanisms of Drug-Induced Lupus. IV. Comparison of Procainamide and Hydralazine With Analogs In Vitro and In Vivo. Arthritis Rheum (1997) 40:1436–43. doi: 10.1002/art.1780400811
8. Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of Autoimmunity by Pristane and Other Naturally Occurring Hydrocarbons. Trends Immunol (2009) 30:455–64. doi: 10.1016/j.it.2009.06.003
9. Yung RL, Richardson BC. Drug-Induced Lupus. Rheum Dis Clinics North America (1994) 20:61–86. doi: 10.1016/S0889-857X(21)00226-X
10. Cheung YH, Loh C, Pau E, Kim J, Wither J. Insights Into the Genetic Basis and Immunopathogenesis of Systemic Lupus Erythematosus From the Study of Mouse Models. Semin Immunol (2009) 21:372–82. doi: 10.1016/j.smim.2009.10.005
11. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell (2007) 129:1401–14. doi: 10.1016/j.cell.2007.04.040
12. Noviello TMR, Di Liddo A, Ventola GM, Spagnuolo A, D’Aniello S, Ceccarelli M, et al. Detection of Long Non-Coding RNA Homology, a Comparative Study on Alignment and Alignment-Free Metrics. BMC Bioinf (2018) 19:407. doi: 10.1186/s12859-018-2441-6
13. Ma L, Bajic VB, Zhang Z. On the Classification of Long Non-Coding RNAs. RNA Biol (2013) 10:925–33. doi: 10.4161/rna.24604
14. Khodoun M, Chimote AA, Ilyas FZ, Duncan HJ, Moncrieffe H, Kant KS, et al. Targeted Knockdown of Kv1.3 Channels in T Lymphocytes Corrects the Disease Manifestations Associated With Systemic Lupus Erythematosus. Sci Adv (2020) 6:eabd1471. doi: 10.1126/sciadv.abd1471
15. Mihaylova N, Chipinski P, Bradyanova S, Velikova T, Ivanova-Todorova E, Chausheva S, et al. Suppression of Autoreactive T and B Lymphocytes by Anti-Annexin A1 Antibody in a Humanized NSG Murine Model of Systemic Lupus Erythematosus. Clin Exp Immunol (2020) 199:278–93. doi: 10.1111/cei.13399
16. Kerekov NS, Mihaylova NM, Grozdev I, Todorov TA, Nikolova M, Baleva M, et al. Elimination of Autoreactive B Cells in Humanized SCID Mouse Model of SLE. Eur J Immunol (2011) 41:3301–11. doi: 10.1002/eji.201141439
17. Suzuki Y, Funato T, Munakata Y, Sato K, Hirabayashi Y, Ishii T, et al. Chemically Modified Ribozyme to V Gene Inhibits Anti-DNA Production and the Formation of Immune Deposits Caused by Lupus Lymphocytes. J Immunol (Baltimore Md: 1950) (2000) 165:5900–5. doi: 10.4049/jimmunol.165.10.5900
18. Vladutiu AO. The Severe Combined Immunodeficient (SCID) Mouse as a Model for the Study of Autoimmune Diseases. Clin Exp Immunol (1993) 93:1–8. doi: 10.1111/j.1365-2249.1993.tb06488.x
19. Duchosal MA. SCID Mice in the Study of Human Autoimmune Diseases. Springer Semin Immunopathol (1992) 14:159–77. doi: 10.1007/bf00195292
20. Gunawan M, Her Z, Liu M, Tan SY, Chan XY, Tan WWS, et al. A Novel Human Systemic Lupus Erythematosus Model in Humanised Mice. Sci Rep (2017) 7:16642. doi: 10.1038/s41598-017-16999-7
21. Andrade D, Redecha PB, Vukelic M, Qing X, Perino G, Salmon JE, et al. Engraftment of Peripheral Blood Mononuclear Cells From Systemic Lupus Erythematosus and Antiphospholipid Syndrome Patient Donors Into BALB-RAG-2-/- IL-2rγ-/- Mice: A Promising Model for Studying Human Disease. Arthritis Rheum (2011) 63:2764–73. doi: 10.1002/art.30424
22. Duchosal MA, McConahey PJ, Robinson CA, Dixon FJ. Transfer of Human Systemic Lupus Erythematosus in Severe Combined Immunodeficient (SCID) Mice. J Exp Med (1990) 172:985–8. doi: 10.1084/jem.172.3.985
23. Wen Z, Xu L, Xu W, Xiong S. Retinoic Acid Receptor-Related Orphan Nuclear Receptor γt Licenses the Differentiation and Function of a Unique Subset of Follicular Helper T Cells in Response to Immunogenic Self-DNA in Systemic Lupus Erythematosus. Arthritis Rheumatol (2021) 73:1489–500. doi: 10.1002/art.41687
24. Cavacini LA, Kennel M, Lally EV, Posner MR, Quinn A. Human Immunoglobulin Production in Immunodeficient Mice: Enhancement by Immunosuppression of Host and In Vitro Activation of Human Mononuclear Cells. Clin Exp Immunol (1992) 90:135–40. doi: 10.1111/j.1365-2249.1992.tb05845.x
25. Giang S, Horwitz DA, Bickerton S, La Cava A. Nanoparticles Engineered as Artificial Antigen-Presenting Cells Induce Human CD4(+) and CD8(+) Tregs That Are Functional in Humanized Mice. Front Immunol (2021) 12:628059. doi: 10.3389/fimmu.2021.628059
26. Horton HM, Chu SY, Ortiz EC, Pong E, Cemerski S, Leung IW, et al. Antibody-Mediated Coengagement of Fcγriib and B Cell Receptor Complex Suppresses Humoral Immunity in Systemic Lupus Erythematosus. J Immunol (Baltimore Md: 1950) (2011) 186:4223–33. doi: 10.4049/jimmunol.1003412
27. Kalechman Y, Gafter U, Da JP, Albeck M, Alarcon-Segovia D, Sredni B. Delay in the Onset of Systemic Lupus Erythematosus Following Treatment With the Immunomodulator AS101: Association With IL-10 Inhibition and Increase in TNF-Alpha Levels. J Immunol (Baltimore Md: 1950) (1997) 159:2658–67.
28. Sthoeger Z, Zinger H, Dekel B, Arditi F, Reisner Y, Mozes E. Lupus Manifestations in Severe Combined Immunodeficient (SCID) Mice and in Human/Mouse Radiation Chimeras. J Clin Immunol (2003) 23:91–9. doi: 10.1023/a:1022520710570
29. Mauermann N, Sthoeger Z, Zinger H, Mozes E. Amelioration of Lupus Manifestations by a Peptide Based on the Complementarity Determining Region 1 of an Autoantibody in Severe Combined Immunodeficient (SCID) Mice Engrafted With Peripheral Blood Lymphocytes of Systemic Lupus Erythematosus (SLE) Patients. Clin Exp Immunol (2004) 137:513–20. doi: 10.1111/j.1365-2249.2004.02559.x
30. Wu H, Zhen Y, Ma Z, Li H, Yu J, Xu ZG, et al. Arginase-1-Dependent Promotion of TH17 Differentiation and Disease Progression by MDSCs in Systemic Lupus Erythematosus. Sci Trans Med (2016) 8:331ra340. doi: 10.1126/scitranslmed.aae0482
31. Kerekov N, Mihaylova N, Prechl J, Tchorbanov A. Humanized SCID Mice Models of SLE. Curr Pharm Design (2011) 17:1261–6. doi: 10.2174/138161211795703780
32. Nikolova K, Mihaylova N, Voynova E, Kerekov N, Gesheva V, Prechl J, et al. Re-Establishing Tolerance to DNA in Humanized and Murine Models of SLE. Autoimmun Rev (2010) 9:499–502. doi: 10.1016/j.autrev.2010.02.009
33. Llorente L, Zou W, Levy Y, Richaud-Patin Y, Wijdenes J, Alcocer-Varela J, et al. Role of Interleukin 10 in the B Lymphocyte Hyperactivity and Autoantibody Production of Human Systemic Lupus Erythematosus. J Exp Med (1995) 181:839–44. doi: 10.1084/jem.181.3.839
34. Ashany D, Hines J, Gharavi A, Mouradian J, Elkon KB. Analysis of Autoantibody Production in SCID-Systemic Lupus Erythematosus (SLE) Chimeras. Clin Exp Immunol (1992) 88:84–90. doi: 10.1111/j.1365-2249.1992.tb03043.x
35. Lech M, Anders HJ. The Pathogenesis of Lupus Nephritis. J Am Soc Nephrol: JASN (2013) 24:1357–66. doi: 10.1681/asn.2013010026
36. Mak A, Kow NY. The Pathology of T Cells in Systemic Lupus Erythematosus. J Immunol Res (2014) 2014:419029. doi: 10.1155/2014/419029
37. Erkeller-Yüsel F, Hulstaart F, Hannet I, Isenberg D, Lydyard P. Lymphocyte Subsets in a Large Cohort of Patients With Systemic Lupus Erythematosus. Lupus (1993) 2:227–31. doi: 10.1177/096120339300200404
38. Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF. Increase in Activated CD8+ T Lymphocytes Expressing Perforin and Granzyme B Correlates With Disease Activity in Patients With Systemic Lupus Erythematosus. Arthritis Rheum (2005) 52:201–11. doi: 10.1002/art.20745
39. Mitsdoerffer M, Lee Y, Jäger A, Kim HJ, Korn T, Kolls JK, et al. Proinflammatory T Helper Type 17 Cells are Effective B-Cell Helpers. Proc Natl Acad Sci USA (2010) 107:14292–7. doi: 10.1073/pnas.1009234107
40. An N, Chen Y, Wang C, Yang C, Wu ZH, Xue J, et al. Chloroquine Autophagic Inhibition Rebalances Th17/Treg-Mediated Immunity and Ameliorates Systemic Lupus Erythematosus. Cell Physiol Biochem (2017) 44:412–22. doi: 10.1159/000484955
41. Dörner T, Giesecke C, Lipsky PE. Mechanisms of B Cell Autoimmunity in SLE. Arthritis Res Ther (2011) 13:243. doi: 10.1186/ar3433
42. Spada R, Rojas JM, Barber DF. Recent Findings on the Role of Natural Killer Cells in the Pathogenesis of Systemic Lupus Erythematosus. J Leukocyte Biol (2015) 98:479–87. doi: 10.1189/jlb.4RU0315-081RR
43. Wirestam L, Arve S, Linge P, Bengtsson AA. Neutrophils-Important Communicators in Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Front Immunol (2019) 10:2734. doi: 10.3389/fimmu.2019.02734
44. Kaewraemruaen C, Ritprajak P, Hirankarn N. Dendritic Cells as Key Players in Systemic Lupus Erythematosus. Asian Pac J Allergy Immunol (2020) 38:225–32. doi: 10.12932/ap-070919-0639
45. Munoz LE, Gaipl US, Herrmann M. Predictive Value of anti-dsDNA Autoantibodies: Importance of the Assay. Autoimmun Rev (2008) 7:594–7. doi: 10.1016/j.autrev.2008.06.003
46. Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J Immunol Res (2016) 2016:8239258. doi: 10.1155/2016/8239258
47. Vago JP, Nogueira CR, Tavares LP, Soriani FM, Lopes F, Russo RC, et al. Annexin A1 Modulates Natural and Glucocorticoid-Induced Resolution of Inflammation by Enhancing Neutrophil Apoptosis. J Leukocyte Biol (2012) 92:249–58. doi: 10.1189/jlb.0112008
48. Perretti M, D’Acquisto F. Annexin A1 and Glucocorticoids as Effectors of the Resolution of Inflammation. Nat Rev Immunol (2009) 9:62–70. doi: 10.1038/nri2470
49. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
50. Guo C, Hu F, Yi H, Feng Z, Li C, Shi L, et al. Myeloid-Derived Suppressor Cells Have a Proinflammatory Role in the Pathogenesis of Autoimmune Arthritis. Ann Rheum Dis (2016) 75:278–85. doi: 10.1136/annrheumdis-2014-205508
51. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ Myeloid Precursors Migrate to the CNS and Play a Pathogenic Role During Autoimmune Demyelinating Disease. Blood (2009) 113:3190–7. doi: 10.1182/blood-2008-07-168575
52. Yi H, Guo C, Yu X, Zuo D, Wang XY. Mouse CD11b+Gr-1+ Myeloid Cells can Promote Th17 Cell Differentiation and Experimental Autoimmune Encephalomyelitis. J Immunol (Baltimore Md: 1950) (2012) 189:4295–304. doi: 10.4049/jimmunol.1200086
53. Chatterjee S, Das S, Chakraborty P, Manna A, Chatterjee M, Choudhuri SK. Myeloid Derived Suppressor Cells (MDSCs) can Induce the Generation of Th17 Response From Naïve CD4+ T Cells. Immunobiology (2013) 218:718–24. doi: 10.1016/j.imbio.2012.08.271
54. Józsi M, Prechl J, Bajtay Z, Erdei A. Complement Receptor Type 1 (CD35) Mediates Inhibitory Signals in Human B Lymphocytes. J Immunol (Baltimore Md: 1950) (2002) 168:2782–8. doi: 10.4049/jimmunol.168.6.2782
55. Wagner C, Ochmann C, Schoels M, Giese T, Stegmaier S, Richter R, et al. The Complement Receptor 1, CR1 (CD35), Mediates Inhibitory Signals in Human T-Lymphocytes. Mol Immunol (2006) 43:643–51. doi: 10.1016/j.molimm.2005.04.006
56. Waisman A, Ruiz PJ, Israeli E, Eilat E, Könen-Waisman S, Zinger H, et al. Modulation of Murine Systemic Lupus Erythematosus With Peptides Based on Complementarity Determining Regions of a Pathogenic Anti-DNA Monoclonal Antibody. Proc Natl Acad Sci USA (1997) 94:4620–5. doi: 10.1073/pnas.94.9.4620
57. Brosh N, Eilat E, Zinger H, Mozes E. Characterization and Role in Experimental Systemic Lupus Erythematosus of T-Cell Lines Specific to Peptides Based on Complementarity-Determining Region-1 and Complementarity-Determining Region-3 of a Pathogenic Anti-DNA Monoclonal Antibody. Immunology (2000) 99:257–65. doi: 10.1046/j.1365-2567.2000.00957.x
58. Brosh N, Dayan M, Fridkin M, Mozes E. A Peptide Based on the CDR3 of an Anti-DNA Antibody of Experimental SLE Origin is Also a Dominant T-Cell Epitope in (NZBXNZW)F1 Lupus-Prone Mice. Immunol Lett (2000) 72:61–8. doi: 10.1016/s0165-2478(00)00161-9
59. Eilat E, Dayan M, Zinger H, Mozes E. The Mechanism by Which a Peptide Based on Complementarity-Determining Region-1 of a Pathogenic Anti-DNA Auto-Ab Ameliorates Experimental Systemic Lupus Erythematosus. Proc Natl Acad Sci USA (2001) 98:1148–53. doi: 10.1073/pnas.98.3.1148
60. Eilat E, Zinger H, Nyska A, Mozes E. Prevention of Systemic Lupus Erythematosus-Like Disease in (NZBxNZW)F1 Mice by Treating With CDR1- and CDR3-Based Peptides of a Pathogenic Autoantibody. J Clin Immunol (2000) 20:268–78. doi: 10.1023/a:1006663519132
61. Zinger H, Eilat E, Meshorer A, Mozes E. Peptides Based on the Complementarity-Determining Regions of a Pathogenic Autoantibody Mitigate Lupus Manifestations of (NZB X NZW)F1 Mice via Active Suppression. Int Immunol (2003) 15:205–14. doi: 10.1093/intimm/dxg026
62. Stein CA, Cheng YC. Antisense Oligonucleotides as Therapeutic Agents–is the Bullet Really Magical? Sci (New York N.Y.) (1993) 261:1004–12. doi: 10.1126/science.8351515
63. Nagel KM, Holstad SG, Isenberg KE. Oligonucleotide Pharmacotherapy: An Antigene Strategy. Pharmacotherapy (1993) 13:177–88.
64. Horwitz DA, Bickerton S, Koss M, Fahmy TM, La Cava A. Suppression of Murine Lupus by CD4+ and CD8+ Treg Cells Induced by T Cell-Targeted Nanoparticles Loaded With Interleukin-2 and Transforming Growth Factor β. Arthritis Rheumatol (2019) 71:632–40. doi: 10.1002/art.40773
65. Horwitz DA, Fahmy TM, Piccirillo CA, La Cava A. Rebalancing Immune Homeostasis to Treat Autoimmune Diseases. Trends Immunol (2019) 40:888–908. doi: 10.1016/j.it.2019.08.003
66. Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung S, et al. Suppression of Rheumatoid Arthritis B Cells by XmAb5871, an Anti-CD19 Antibody That Coengages B Cell Antigen Receptor Complex and Fcγ Receptor IIb Inhibitory Receptor. Arthritis Rheumatol (2014) 66:1153–64. doi: 10.1002/art.38334
67. Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, et al. Inhibition of B Cell Receptor-Mediated Activation of Primary Human B Cells by Coengagement of CD19 and FcgammaRIIb With Fc-Engineered Antibodies. Mol Immunol (2008) 45:3926–33. doi: 10.1016/j.molimm.2008.06.027
68. Yu F, Haas M, Glassock R, Zhao MH. Redefining Lupus Nephritis: Clinical Implications of Pathophysiologic Subtypes. Nat Rev Nephrol (2017) 13:483–95. doi: 10.1038/nrneph.2017.85
70. Anders HJ, Saxena R, Zhao MH, Parodis I, Salmon JE, Mohan C. Lupus Nephritis. Nat Rev Dis Primers (2020) 6:7. doi: 10.1038/s41572-019-0141-9
71. Borchers AT, Leibushor N, Naguwa SM, Cheema GS, Shoenfeld Y, Gershwin ME. Lupus Nephritis: A Critical Review. Autoimmun Rev (2012) 12:174–94. doi: 10.1016/j.autrev.2012.08.018
72. Morillon YM 2nd, Sabzevari A, Schlom J, Greiner JW. The Development of Next-Generation PBMC Humanized Mice for Preclinical Investigation of Cancer Immunotherapeutic Agents. Anticancer Res (2020) 40:5329–41. doi: 10.21873/anticanres.14540
73. Schmidt MR, Appel MC, Giassi LJ, Greiner DL, Shultz LD, Woodland RT. Human BLyS Facilitates Engraftment of Human PBL Derived B Cells in Immunodeficient Mice. PloS One (2008) 3:e3192. doi: 10.1371/journal.pone.0003192
74. O’Connell RM, Balazs AB, Rao DS, Kivork C, Yang L, Baltimore D. Lentiviral Vector Delivery of Human Interleukin-7 (hIL-7) to Human Immune System (HIS) Mice Expands T Lymphocyte Populations. PloS One (2010) 5:e12009. doi: 10.1371/journal.pone.0012009
75. Chen Q, Khoury M, Chen J. Expression of Human Cytokines Dramatically Improves Reconstitution of Specific Human-Blood Lineage Cells in Humanized Mice. Proc Natl Acad Sci USA (2009) 106:21783–8. doi: 10.1073/pnas.0912274106
76. Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, et al. Development of Mature and Functional Human Myeloid Subsets in Hematopoietic Stem Cell-Engrafted NOD/SCID/Il2rγKO Mice. J Immunol (Baltimore Md: 1950) (2012) 188:6145–55. doi: 10.4049/jimmunol.1103660
77. Cashman JD, Eaves CJ. Human Growth Factor-Enhanced Regeneration of Transplantable Human Hematopoietic Stem Cells in Nonobese Diabetic/Severe Combined Immunodeficient Mice. Blood (1999) 93:481–7. doi: 10.1182/blood.V93.2.481
78. Ding Y, Wilkinson A, Idris A, Fancke B, O’Keeffe M, Khalil D, et al. FLT3-Ligand Treatment of Humanized Mice Results in the Generation of Large Numbers of CD141+ and CD1c+ Dendritic Cells In Vivo. J Immunol (Baltimore Md: 1950) (2014) 192:1982–9. doi: 10.4049/jimmunol.1302391
79. Billerbeck E, Barry WT, Mu K, Dorner M, Rice CM, Ploss A. Development of Human CD4+FoxP3+ Regulatory T Cells in Human Stem Cell Factor-, Granulocyte-Macrophage Colony-Stimulating Factor-, and Interleukin-3-Expressing NOD-SCID Il2rγ(Null) Humanized Mice. Blood (2011) 117:3076–86. doi: 10.1182/blood-2010-08-301507
80. Tournoy KG, Depraetere S, Pauwels RA, Leroux-Roels GG. Mouse Strain and Conditioning Regimen Determine Survival and Function of Human Leucocytes in Immunodeficient Mice. Clin Exp Immunol (2000) 119:231–9. doi: 10.1046/j.1365-2249.2000.01099.x
81. Santini SM, Rizza P, Logozzi MA, Sestili P, Gherardi G, Lande R, et al. The SCID Mouse Reaction to Human Peripheral Blood Mononuclear Leukocyte Engraftment. Neutrophil Recruitment Induced Expression of a Wide Spectrum of Murine Cytokines and Mouse Leukopoiesis, Including Thymic Differentiation. Transplantation (1995) 60:1306–14. doi: 10.1097/00007890-199512000-00020
Keywords: systemic lupus erythematosus, immunodeficient mouse, humanized SLE mouse, autoantibodies, pro-inflammatory cytokines, lupus nephritis
Citation: Chen J, Liao S, Zhou H, Yang L, Guo F, Chen S, Li A, Pan Q, Yang C, Liu H-f and Pan Q (2022) Humanized Mouse Models of Systemic Lupus Erythematosus: Opportunities and Challenges. Front. Immunol. 12:816956. doi: 10.3389/fimmu.2021.816956
Received: 17 November 2021; Accepted: 30 December 2021;
Published: 18 January 2022.
Edited by:
Kunihiro Ichinose, Nagasaki University, JapanReviewed by:
Laurence Morel, University of Florida, United StatesAmir Sharabi, Beth Israel Deaconess Medical Center and Harvard Medical School, United States
Copyright © 2022 Chen, Liao, Zhou, Yang, Guo, Chen, Li, Pan, Yang, Liu and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua-feng Liu, bGl1aGZAZ2RtdS5lZHUuY24=; Qingjun Pan, cHFqQGdkbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship