 Quanren Pan
Quanren Pan Fengbiao Guo
Fengbiao Guo Yanyan Huang
Yanyan Huang Aifen Li
Aifen Li Shuxian Chen
Shuxian Chen Jiaxuan Chen
Jiaxuan Chen Hua-feng Liu
Hua-feng Liu Qingjun Pan
Qingjun Pan- Key Laboratory of Prevention and Management of Chronic Kidney Disease of Zhanjiang City, Institute of Nephrology, Affiliated Hospital of Guangdong Medical University, Zhanjiang, China
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease that was traditionally thought to be closely related to genetic and environmental risk factors. Although treatment options for SLE with hormones, immunosuppressants, and biologic drugs are now available, the rates of clinical response and functional remission of these drugs are still not satisfactory. Currently, emerging evidence suggests that gut microbiota dysbiosis may play crucial roles in the occurrence and development of SLE, and manipulation of targeting the gut microbiota holds great promises for the successful treatment of SLE. The possible mechanisms of gut microbiota dysbiosis in SLE have not yet been well identified to date, although they may include molecular mimicry, impaired intestinal barrier function and leaky gut, bacterial biofilms, intestinal specific pathogen infection, gender bias, intestinal epithelial cells autophagy, and extracellular vesicles and microRNAs. Potential therapies for modulating gut microbiota in SLE include oral antibiotic therapy, fecal microbiota transplantation, glucocorticoid therapy, regulation of intestinal epithelial cells autophagy, extracellular vesicle-derived miRNA therapy, mesenchymal stem cell therapy, and vaccination. This review summarizes novel insights into the mechanisms of microbiota dysbiosis in SLE and promising therapeutic strategies, which may help improve our understanding of the pathogenesis of SLE and provide novel therapies for SLE.
1 Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by the generation of autoantibodies and immune complexes, which can cause multiple organ damage to the skin, kidney, and central nervous system (1). The pathogenesis of SLE is very complex and is traditionally thought to be closely related to genetic and environmental risk factors (2). Infection is a significant cause of morbidity, disease exacerbation, and death in patients with SLE (3, 4). Recently, increasing evidence has shown that different degrees of intestinal-infection-related dysbacteriosis exist in patients with SLE and SLE mice, which are closely related to the development of SLE (5, 6). Previous studies have shown that the mechanisms associating gut microbiota dysbiosis and SLE pathogenesis include immune system imbalance, molecular mimicry, impaired intestinal barrier function, biofilms, and sex hormones. Under normal circumstances, the special barrier functions of the intestine include physical, biochemical, and immune barriers, which can separate the host from the environment. Intestinal epithelial cells are joined by tight junction proteins to form the intestinal physical barrier (7). In patients with SLE, impaired intestinal barrier function leads to increased intestinal permeability, allowing pathogens, toxins, and bacteria to leak out of the gut lumen and translocate to other organs, which is called a “leaky gut” (8). In addition, the antigens of the translocated bacteria are similar to some of the host’s structures, which cause cross-reactivity to produce autoantibodies and damage target organs in patients with SLE, a process called molecular mimicry (9, 10). Furthermore, curli amyloid in biofilms is associated with autoantibody production. Gut microbiota dysbiosis in SLE is sex-biased, which may be due to sex hormones (11).
The treatment of SLE mainly includes immune regulation and immunosuppression, with the aim of maintaining long-term remission or low disease activity, protecting organ function, and avoiding complications and adverse drug reactions (12, 13). Currently, although treatment options for SLE with hormones, immunosuppressants, and biologic drugs are now available, the rates of the clinical response and functional remission of these drugs are still not satisfactory, which may lead to serious side effects (14, 15). Therefore, there is an urgent need to develop treatment options that have good therapeutic effects in patients with few adverse effects. Emerging evidence has shown that intestinal dysbacteriosis may play an essential role in the pathogenesis of SLE and may be a novel therapeutic target for SLE. Previous studies have shown that interventions targeting the gut microbiota for SLE include dietary interventions, probiotics or prebiotics, antibiotic therapy, vaccination, and fecal microbiota transplantation (FMT). These treatments are currently only studied in lupus murine models, and further clinical trials are required to confirm their efficacy.
In this review, we summarize the gut microbiota dysbiosis in patients with SLE and mouse models, and first described the possible role of IECs autophagy, and extracellular vesicles (EVs) and miRNA, in the gut microbiota homeostasis of SLE, as shown in Figure 1. In addition, we propose several novel treatment strategies targeting gut microbiota, including regulation of IECs autophagy, EV-derived miRNA therapy, and mesenchymal stem cell therapy, which may have great value for SLE treatment in the future, as shown in Figure 2.
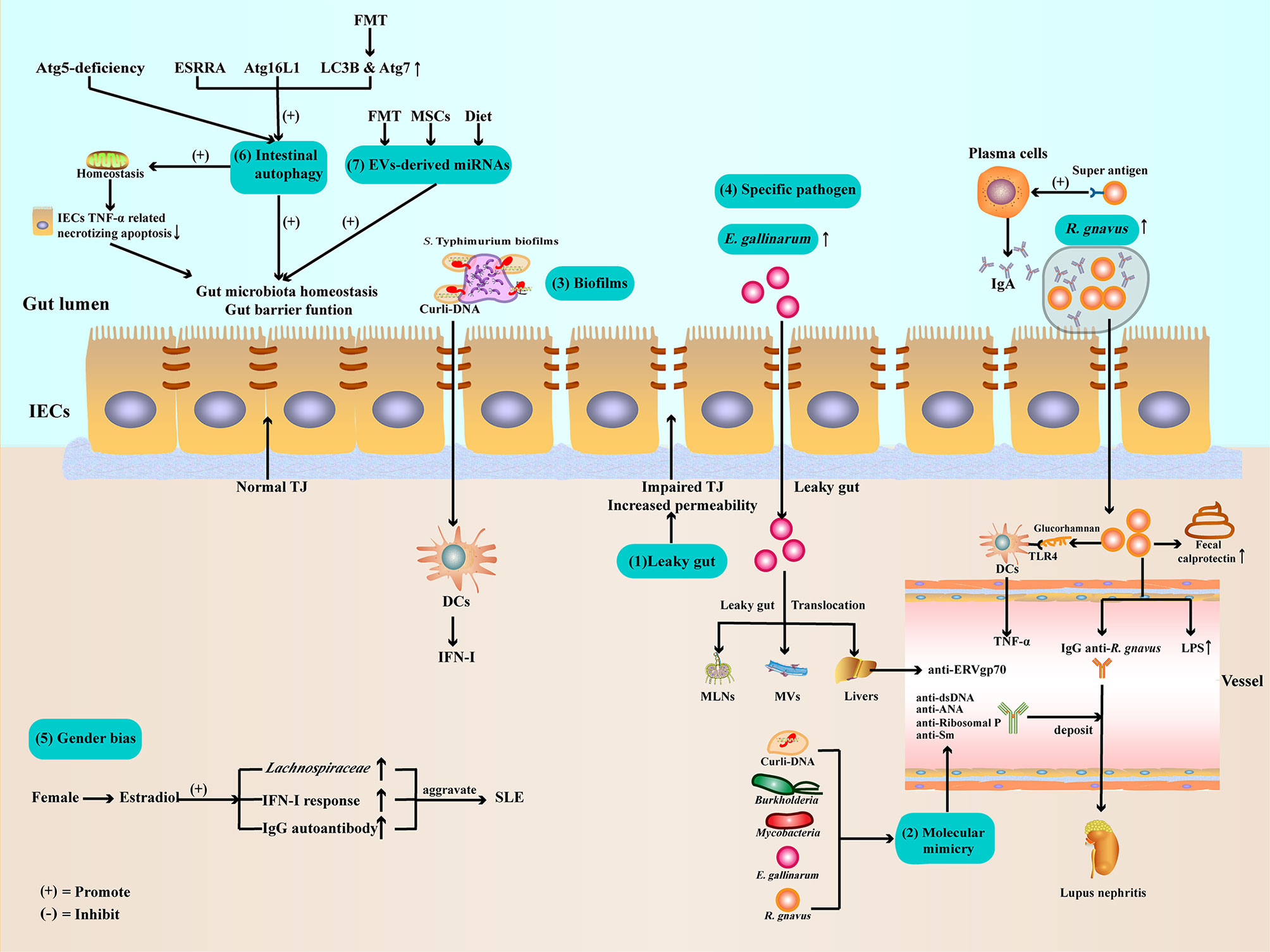
Figure 1 Potential mechanisms of gut microbiota dysbiosis in SLE (1). Gut barrier function impaired and leaky gut allow pathogen leak out of the gut lumen and translocate to other organs (2). Gut microbiota and Curli-DNA of biofilms produce autoantibodies through molecular mimicry, which deposit in kidneys, leading to lupus nephritis (3). Curli-DNA of biofilms activated DCs to secrete pathogenic IFN-I (4). E. gallinarum can disrupt intestinal barrier function and translocate to MLNs, MVs, and livers. At the same time, E. gallinarum promoted systemic autoimmunity by inducing ERV gp70 overexpression in the liver. R. gnavus express a B-cell superantigen to stimulate IgA antibodies production and encapsulate itself to facilitate intestinal colonization. Furthermore, R. gnavus can produce a glucorhamnan inflammatory polysaccharide that promotes DCs to secrete the inflammatory factor TNF-α via TLR4. In addition, R. gnavus can disrupt intestinal barrier function, resulting in increased calprotectin levels in stool samples and LPS levels in sera. Subsequently, the impaired intestinal barrier function exposes the intestinal commensal R. gnavus antigen, leading to mimicry of the molecule to produce anti-dsDNA autoantibodies, aggravating lupus nephritis (5). Estradiol promotes pathogen like Lachnospiraceae colonization, IFN-I response, and IgG autoantibody production (6). Regulate ESRRA, Atg16L1, LC3B, and Atg7 can activate IECs autophagy to maintenance gut microbiota homeostasis and intestinal barrier function (7). Evs-derived miRNAs from FMT, MSCs therapy, or dietary improve gut microbiota balance and enhance intestinal barrier function. ATG, autophagy-related protein; DCs, dendritic cells; E. gallinarum, Enterococcus gallinarum; ESRRA, estrogen related receptor alpha; FMT, Fecal microbiota transplantation; IECs, intestinal epithelial cells; IFN-I, type I interferon; LC3B, microtubule-associated protein 1 light chain 3B; LPSs, lipopolysaccharides; MLNs, mesenteric lymph nodes; MVs, Mesenteric veins; MSCs, mesenchymal stem cells; R. gnavus, Ruminococcus gnavus; TJ, tight junction; TLR4, toll-like receptor4; TNF-α, Tumor necrosis factor-α.
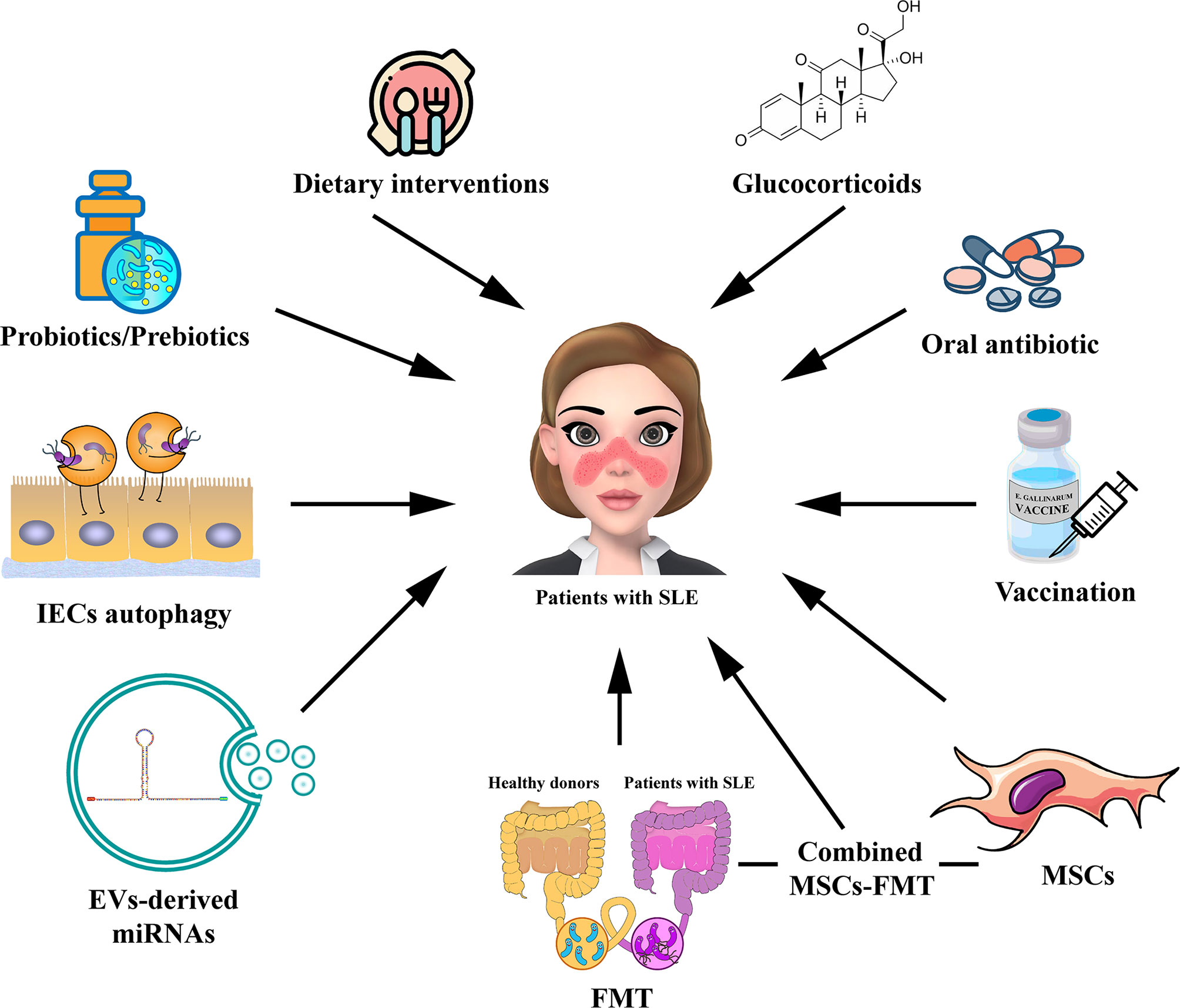
Figure 2 Potential strategies for targeting gut microbiota in the treatment of patients with SLE. The potential therapies for modulating gut microbiota for SLE, including probiotic or prebiotic therapy, dietary interventions, oral antibiotic therapy, GC therapy, vaccination, FMT, regulation of IECs autophagy, EV-derived miRNA therapy, and MSC therapy. The combined MSC-FMT transplantation approach may have a better therapeutic effect for SLE. EV, extracellular vesicle; GC, glucocorticoid; FMT, Fecal microbiota transplantation; MSCs, mesenchymal stem cells.
2 Gut Microbiota Dysbiosis in Patients With SLE
Recently, many studies have attempted to determine the correlation between gut microbiota dysbiosis and SLE pathogenesis, as shown in Table 1. A study showed that compared with healthy controls, patients with SLE suffered from intestinal dysbiosis and had a significantly lower ratio of Firmicutes/Bacteroidetes (F/B) (22). This result was confirmed by subsequent studies (18, 19, 23, 24). Importantly, Firmicutes are inversely correlated with the SLE disease activity index (SLEDAI score) (20), indicating that Firmicutes can delay lupus progression. It follows that the reduced F/B ratio is an important manifestation of gut microbiota dysbiosis in patients with SLE. A recent study analyzed stool samples from 117 untreated patients with SLE and reported that the gut microbiota of patients with SLE showed a pro-inflammatory and autoimmune profile compared to healthy controls (6). Furthermore, patients with SLE mostly show decreased richness and diversity of intestinal microbiota compared to healthy controls (6, 8, 25), and this was particularly severe in patients with high SLEDAI scores (8).
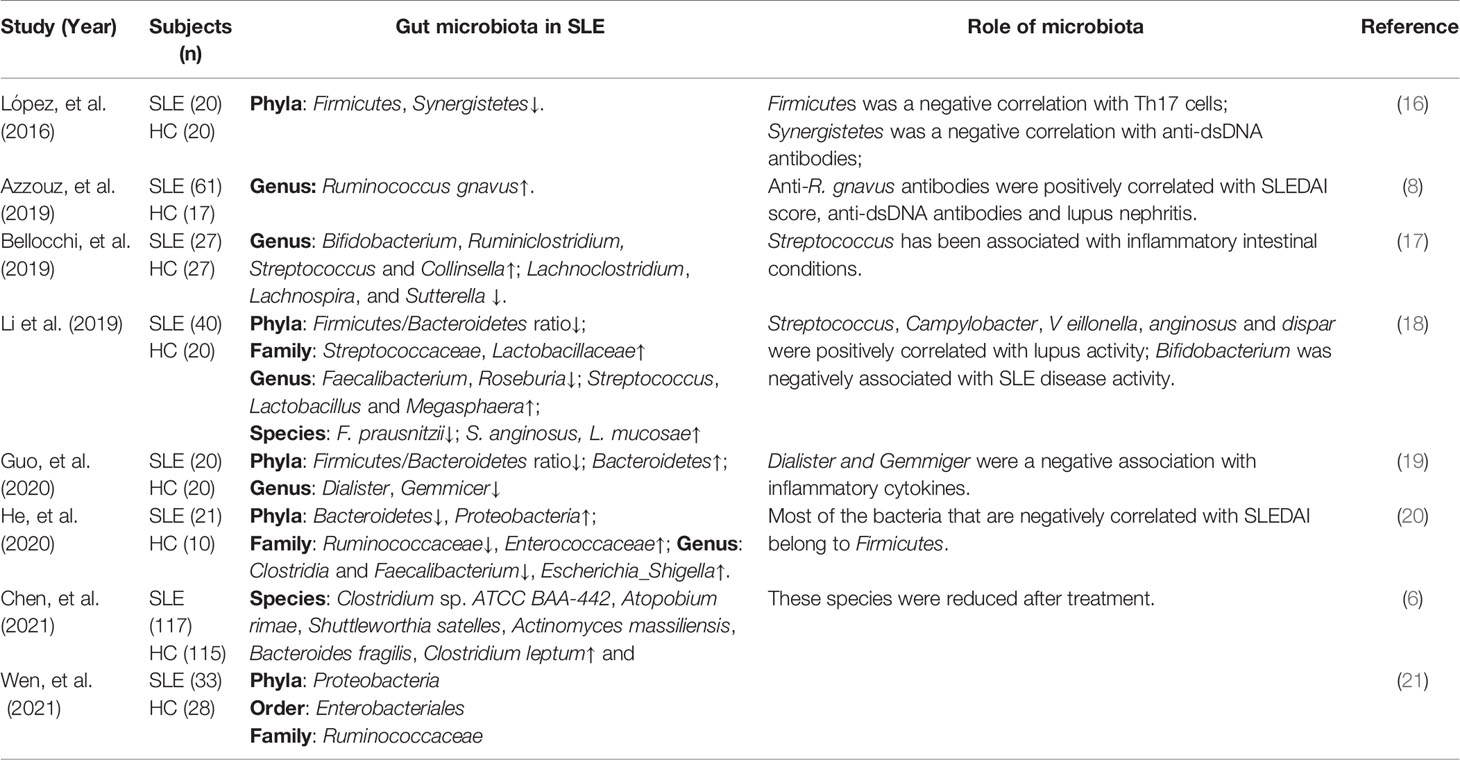
Table 1 Gut microbiota dysbiosis in patients with SLE.
Interestingly, the abundance of Ruminococcus gnavus (R. gnavus) was elevated 5-fold in the gut microbiota of 61 patients with SLE compared to that in healthy control and was strongly associated with SLE disease activity (8). And serum anti-R. gnavus antibodies were positively correlated with the SLEDAI score and anti-dsDNA levels (8).
Collectively, gut microbiota dysbiosis in patients with SLE typically displays a decreased F/B ratio, richness, and diversity. Meanwhile, impaired intestinal barrier function leads to microbiome translocation, which exacerbates disease progression in patients with lupus. Furthermore, proliferation of some specific microbiota such as R. gnavus may be significantly related to lupus progression.
3 Gut Microbiota Dysbiosis in Lupus Mouse Model
Recently, many studies have also revealed gut microbiota dysbiosis in lupus mouse models, as shown in Table 2. Zhang et al. (26) revealed that lactobacilli significantly reduced and Lachnospiraceae increased in the gut microbiota of MRL/lpr mice, which was more severe in female mice. Moreover, Lachnospiraceae is strongly associated with lupus progression in MRL/lpr mice. In contrast, the intestinal colonization of Lactobacillaceae was negatively correlated to the lupus activity in mice. Those results suggested that Lactobacillaceae may be a probiotic in the treatment of SLE. Another study reported similar results; antibiotic treatment can eliminate harmful microbiota Lachnospiraceae and enrich the probiotic Lactobacillus Spp., thereby attenuating lupus (27). On the other hand, many studies have found that leaky gut occurs in lupus-prone mice with impaired intestinal barrier function, resulting in increased microbial translocation, endotoxemia, and lupus progression, which can be reversed after treatment of lupus-prone mice (11, 28, 29, 34). These results suggested that impaired gut barrier function significantly influenced lupus progression.

Table 2 Gut microbiota dysbiosis in lupus mouse models.
In addition, Enterococcus gallinarum, a specific pathogenic bacterium in (NZW × BXSB) F1 lupus mice, was shown to induce intestinal barrier impairments and translocate to the liver to cause autoimmune hepatitis (28). More importantly, E. gallinarum was also found in the liver tissues of patients with SLE and autoimmune hepatitis, but not in healthy controls and non-autoimmune hepatitis patients.
Taken together, the above results indicate that intestinal microbiota dysbiosis in an SLE mouse model presents with decreased microbial diversity, increased colonization of harmful bacteria such as E. gallinarum, or decreased probiotics. At the same time, impaired intestinal barrier function plays a very important role, which can increase gut microbiota translocation and promote lupus progression.
4 Mechanisms of Gut Microbiota Dysbiosis in SLE
Currently, it is unclear whether gut microbiota dysbiosis is the cause or consequence of SLE. Genetic susceptibility is an important factor leading to gut microbiota dysbiosis and autoimmune disease progression in lupus-prone mice (35). However, the data from 1,046 healthy individuals suggested that environmental factors are more important than host genetics in shaping the human gut microbiota (36). In the past decades, the rising incidence of autoimmune diseases has been associated with environmental factors, including a high-salt diet (HSD) (37). Previous studies have shown that HSD could activate DCs and induce the production of pathogenic T Helper 17 (Th17) cells through the p38/MAPK-STAT1 signaling pathway, resulting in gut microbiota dysbiosis, hypertension and autoimmune progression (37–39). Moreover, gut microbiota dysbiosis may induced immune system imbalance and aggravates SLE (40). There are other potential mechanisms underlying for the role of gut microbiota dysbiosis in SLE, which we will elaborate on the following aspects.
4.1 Intestinal Barrier Function and Leaky Gut
At present, the pathogenesis of SLE is still not well known, but growing evidence suggests that the impaired intestinal barrier may be one of the essential factors (41). The intestinal mucosa needs the intestinal barrier function to defend against the invasion of foreign antigens, such as food antigens, bacteria, and toxins (7). As previously mentioned, a leaky gut was observed in patients with SLE and in mice. Calprotectin, a calcium-containing protein from neutrophils and macrophages, is a well-recognized biomarker of impaired intestinal barrier function (42). Calprotectin levels were significantly increased in stool samples from patients with SLE, indicating impaired intestinal barrier function (8, 28). At the same time, serum soluble CD14, α1-acid glycoprotein, and lipopolysaccharides (LPSs) levels were increased in patients with SLE, indicating the presence of intestinal bacterial translocation (8).
Interestingly, Thim-Uam et al. (43) used dextran sulfate solution to induce a leaky gut in FcgRIIb−/− lupus mice and pristane-induced lupus mice. They found that the leaky gut aggravated the progression and disease activity of these two murine models of lupus. Leaky gut increases the intestinal translocation of endotoxins or other organic molecules, thereby promoting apoptosis. Most notably, leaky gut promotes the production of anti-dsDNA autoantibodies and immune complex deposition, ultimately leading to lupus exacerbation. Recently, another study indicated that impaired intestinal barrier function is associated with intestinal oxidative stress in MRL/lpr lupus mice (44). This result further complements the mechanism involved in the development of the leaky gut in lupus mice. In addition, impaired gut barrier function and lupus were significantly ameliorated after treatment with antibiotics, probiotics, or dietary interventions in lupus mice (27, 34, 45).
Taken together, these results suggest that impaired intestinal barrier function is associated with SLE disease severity. Both patients with SLE and mice have varying degrees of impaired intestinal barrier function and a leaky gut. Mechanistically, impaired intestinal barrier function allows symbiotic bacteria or their contents to leak out of the intestinal lumen, which may be related to intestinal oxidative stress. Translocated gut bacteria or bacterial components can promote the production of autoantibodies through molecular mimicry. Finally, the deposition of immune complexes aggravates SLE progression.
4.2 Molecular Mimicry
Molecular mimicry is another critical condition that leads to the development of autoimmunity (6, 9, 46). Molecular mimicry means that certain structures of a microorganism are similar to the self-structures of the host, which causes an autoimmune response and tissue damage (47). Therefore, certain bacteria with epitope structures similar to self-antigens can stimulate patients with SLE to produce cross-reactive autoantibodies. Zhang et al. (9) found that Burkholderia bacterial partial purified antigen and transcriptional regulatory peptide RAGTDEGFG could bind to dsDNA antibodies in sera from patients with SLE (9). These results suggest that the production of anti-dsDNA antibodies in patients with SLE is associated with Burkholderia bacterial molecular mimicry. Interestingly, another study found that glycolipids of the mycobacterial cell wall can bind to anti-dsDNA autoantibodies derived from patients with SLE and mice (10). Thus, the production of autoantibodies can result from the molecular mimicry caused by different bacterial infections in SLE. Recently, it has been shown that peptides produced by Odoribacter splanchnicus and Akkermansia muciniphila bacteria are highly similar to Sm antigen and Fas antigen epitopes (6). More importantly, peptides from these bacteria can activate CD4+ T cells or B cells to produce autoantibodies (6). However, these results were limited in vitro experiments, in vivo experiments need to be designed to confirm these standpoints. In another study, molecular mimicry of commensal or environmental microbes was shown to promote autoantibody production in SLE, which was driven by T cells and HLA-DR restriction (48).
Molecular mimicry has also been associated to the pathogenesis of other autoimmune diseases, such as antiphospholipid syndrome (APS) (49). APS is an autoimmune disease characterized by anti-β2-glycoprotein I (β2GPI) autoantibodies production (50), which can be secondary to SLE (51). On the other hand, the intestinal commensal Roseburia intestinalis (R. int) mimotope cross-react with β2GPI-reactive memory CD4+ Th1 cells and produce anti-R. int autoantibodies in patients with APS (49). And oral gavage with R. int in BALB/c mice induced anti-human β2GPI autoantibodies and APS-associated autoimmune pathologies (49). Therefore, R. int promotes anti-β2GPI autoantibodies production and contributes to APS pathogenesis. In addition, aPL also targeted to β2GPI in SLE (52). Thus, the intestinal commensal R. int may be related to the pathogenesis of SLE, but further studies will be required.
In summary, bacterial molecular mimicry is an important factor in the pathogenesis of autoimmune diseases, including SLE and APS. Different bacteria can promote autoantibody production through molecular mimicry. T and B cells are involved in the bacterial molecular mimicry process; however, the precise mechanism remains unclear.
4.3 The Pathogenic Role of Bacterial Biofilms
Biofilms are considered to be a membrane in which the bacterial community produces an extracellular matrix and wraps itself (53), which can protect bacteria from the host immune response (54) and enable bacteria to develop drug resistance (55). The main structure of biofilms is amyloid protein rich in β-folding, which is associated with human autoimmune diseases (56–58). Curli fibrils in Salmonella enterica serovar Typhimurium (S. Typhimurium) amyloid could combined to the DNA in bacterial, and these complexes could promote biofilm formation, also contributing to SLE pathogenesis (57). In pre-lupus NZBxW/F1 mice, curli-DNA complexes activated the innate immune cells such as dendritic cells (DCs) to secrete pathogenic type I interferons (IFNs). NZBxW/F1 lupus-prone mice rapidly developed anti-dsDNA and ANA autoantibodies after intraperitoneal injection of curli-DNA complexes at six weeks of age, whereas injection of BSA did not show the same effect. Most importantly, normal control C57BL/6 mice also developed anti-dsDNA and ANA autoantibodies two weeks after intraperitoneal injection of curli-DNA complexes at six weeks of age. In addition, curli-DNA complexes promoted the proliferation of activated T cells, activated B cells, and inflammatory monocytes. Finally, infection with curli biofilm of S. Typhimurium promoted autoantibody production in lupus mice (57). These results suggest that curli-DNA complexes of bacterial biofilms not only promote the production of autoantibodies in lupus-prone mice, but also disrupt self-tolerance in non-autoimmune mice, causing lupus pathogenesis. This has also been shown in another study that curli-bacterial DNA complexes of urinary tract infections in patients with SLE cross-reacted with lupus autoantigens such as dsDNA (59). The above two studies suggest that bacterial proteins that interact with DNA may cause loss of immune tolerance to autoantigens and induce the production of autoantibodies, leading to SLE pathogenesis. Interestingly, a recent study by Fu et al. (58) suggested that DNABII proteins interacting with DNA in biofilms may not directly contribute to anti-dsDNA production but other mechanisms may be involved. Sera from patients with SLE specifically recognize the DNAB II protein-derived HU1 peptide in bacterial biofilms. Anti-HU1 aggravates the progression of lupus nephritis (LN) in patients with SLE and a pristane-induced lupus murine model. Although anti-HU1 antibodies can inhibit biofilm formation by Staphylococcus aureus, it is accompanied by cross-reactivity with the autoantigen P4HB on the glomerular cell membrane to induce LN (58).
In conclusion, certain components in bacterial biofilms such as curli and curli-DNA complexes can cross-react with autoantigens and induce the production of autoantibodies, resulting in SLE pathogenesis or disease aggravation.
4.4 Intestinal Specific Pathogens Infection
Intestinal infections with specific pathogens have been reported to be associated with the onset and progression of SLE. It is of great significance to study the mechanism of action of these specific pathogens in SLE.
4.4.1 Enterococcus gallinarum
Enterococcus gallinarum (E. gallinarum) is a human intestinal commensal bacterium that can invade the blood to induce sepsis when the immunity of the organism is low (60). Interestingly, Vieira et al. (28) observed that E. gallinarum plays an important role in the pathogenesis of SLE. Pathogenic E. gallinarum disrupted intestinal barrier function and promoted Th17 and Tfh cell proliferation in (NZW × BXSB) F1 lupus mice. Subsequently, the damaged intestinal barrier promoted translocation of E. gallinarum to mesenteric lymph nodes, mesenteric veins, and liver. At the same time, E. gallinarum promoted systemic autoimmunity by inducing ERV gp70 overexpression in the liver (28). Thus, E. gallinarum is a pathogenic bacterium that is closely related to the pathogenesis of SLE in (NZW × BXSB) F1 lupus mice. Surprisingly, E. gallinarum was detected in liver biopsies from patients with SLE and autoimmune hepatitis, but not in healthy controls and non-autoimmune hepatitis patients. This suggests that E. gallinarum of lupus mice were also present in patients with SLE; most importantly, after inoculation with the E. gallinarum vaccine, serum autoantibody levels were reduced, the survival time was prolonged, and bacterial translocation was inhibited in (NZW × BXSB) F1 mice (28). Therefore, pathogen-specific therapy can suppress host autoimmune processes without the use of immunosuppressants. More recently, another study showed that E. gallinarum is associated with autoimmune responses to autoantibodies such as anti-Ribosomal P, anti-dsDNA, and anti-Sm in patients with SLE (61). This study further confirmed that E. gallinarum is a specific pathogen of SLE-susceptible individuals. However, this study did not prove that whether E. gallinarum acts as the same role in other lupus mouse models and patients with SLE.
Taken together, the pathogenic bacteria E. gallinarum can be translocated into systemic organs by disrupting the intestinal barrier, which leads to SLE pathogenesis. Translocated E. gallinarum promotes Th17 and Tfh cell proliferation and autoantibody production. At the same time, E. gallinarum may also directly induce autoantigens, ERV proteins, and other substances to promote autoimmune processes.
4.4.2 Ruminococcus gnavus
As previously mentioned, Ruminococcus gnavus (R. gnavus) plays an important role in SLE (6, 8). Studies have shown that R. gnavus expresses a B-cell superantigen that stimulates the gut of mice to produce large amounts of plasma cells that secrete IgA antibodies (62) These IgA antibodies recognize and highly encapsulate R. gnavus, which may be associated with intestinal colonization of R. gnavus. Furthermore, R. gnavus can produce a glucorhamnan inflammatory polysaccharide that promotes dendritic cells to secrete the inflammatory factor Tumor necrosis factor-α (TNF-α) via toll-like receptor4 (TLR4) (63). A recent study has shown that some isolated strains of R. gnavus could produce capsular polysaccharides that promote the immune tolerance of R. gnavus. However, R. gnavus isolates without capsular polysaccharide produced a strong pro-inflammatory response and increased intestinal inflammatory indicators in sterile mice (64).
Interestingly, Azzouz et al. found that sIgA-coated R. gnavus increased in stool samples from patients with SLE, and the proliferation of R. gnavus was proportional to SLE disease activity (8). Thus, aberrant superantigen expression of R. gnavus may facilitate intestinal colonization of R. gnavus, thereby aggravating SLE progression. In addition, R. gnavus can disrupt intestinal barrier function, resulting in increased levels of calprotectin in stool samples and lipopolysaccharides (LPSs) in sera. Subsequently, the impaired intestinal barrier function exposes the intestinal commensal R. gnavus antigen, leading to mimicry of the molecule to produce anti-dsDNA autoantibodies, aggravating lupus (8).
In summary, R. gnavus may affect disease progression in SLE, but the causal relationship remains unresolved.
4.5 Gender Bias
Generally, SLE shows a strong female bias with a male-to-female ratio of 9:1 (65). In fact, there was also a gender bias in the intestinal microbiota in SLE. For example, over-colonization of Lachnospiraceae in the intestinal tract of female MRL/lpr lupus mice was associated with early onset or exacerbation of lupus, but not in male mice (26). Another study showed an increase in Lachnospiraceae and exacerbation of lupus in the gut microbiota of MRL/lpr mice after administering a phytoestrogen-supplemented diet (66). These results suggest that estrogen may account for gender bias in gut microbiota dysbiosis in SLE, but the underlying mechanism remains to be clarified. Moreover, estradiol exacerbates SLE disease severity by promoting type I interferon responses and IgG autoantibody production from B cells (67, 68). Abnormal modification of steroid receptors in T cells may alter the expression of estrogen receptor (ERα), thereby promoting the effect of estrogen (65). In contrast, testosterone is generally considered to be a beneficial sex hormone that inhibits B-cell activation and autoantibody production to alleviate LN (69). On the other hand, Mu et al. found that Lactobacillus treatment ameliorated lupus nephritis, increased IL-10, and decreased luteinizing hormone in female and emasculated male MRL/lpr mice, but not in intact male mice (11). These results suggest that Lactobacillus treatment ameliorates LN in MRL/lpr mice in a sex hormone-dependent manner. In addition, antibiotic treatment has been shown to inhibit SLE progression in lupus-prone (SWR × NZB) F1 female mice, but not in male mice. Orchiectomy alters the composition of the gut microbiota and promotes autoimmune progression in male mice (70).
In conclusion, estrogen can alter gut microbiota and promote type I interferon response and autoantibody production to aggravate SLE progression; conversely, androgen plays a protective role.
4.6 Intestinal Epithelial Cells Autophagy
At present, the relationship between autophagy and intestinal bacteria in SLE has not been reported. However, in another autoimmune disease, inflammatory bowel disease (IBD), autophagy is crucial for the homeostasis of intestinal bacteria and intestinal barrier function. On the one hand, autophagy may be beneficial to gut barrier function. The autophagic protein Atg16L1 prevents necrotizing apoptosis mediated by TNF-α in intestinal epithelial cells (IECs) by promoting mitochondrial homeostasis (71). Autophagy can also reduce epithelial permeability by inducing lysosomal degradation of the pore-forming tight junction protein claudin-2, thus enhancing intestinal barrier function (72). On the other hand, IECs autophagy plays a crucial role in regulating the diversity and composition of the gut microbiota. For example, the estrogen-associated receptor alpha (ESRRA) protects the host from mitochondrial dysfunction by activating autophagy and maintaining intestinal microbiota homeostasis, thereby attenuating intestinal inflammation (73). IECs-specific knockout of autophagy-associated gene 5 (Atg5) resulted in significant changes and decreased diversity of gut microbiota in mice. In Atg5-deficient mice, the abundance of inflammation-inhibiting Akkermansia muciniphila decreased, but the abundance of pro-inflammatory Candidatus Athromitus and potentially pathogenic Pasteurellaceae increased (74). In addition, fecal microbiota transplantation could increase the expression of LC3B and Atg7 to activate intestinal mucosal autophagy, thereby improving intestinal barrier function in piglets (75). These studies suggest that autophagy of host intestinal mucosal cells may affect the gut microbiota to ameliorate intestinal injury.
The previous discussion indicates that dysregulation of gut microbiota and impaired intestinal barrier function can lead to aggravated SLE progression. IECs autophagy contributes to the maintenance of gut microbiota homeostasis and intestinal barrier function. A review by Bhattacharya et al. (76) indicated that exploring the mechanism of the interaction between autophagy and gut microbiota is beneficial for the study of autoimmune diseases. Therefore, we hypothesized that IECs autophagy is closely related to the dysregulation of gut microbiota in SLE and affects the progression of SLE. However, further studies are required to confirm our findings.
4.7 Extracellular Vesicle and miRNA
Extracellular vesicles (EVs) are a group of membrane-enclosed nanoscale vesicles that carry various RNA, DNA, proteins, and lipids and transmit information between cells (77). Exosomes are EVs ranging in diameter from approximately 40 to 160 nm that carry miRNAs and other non-coding RNAs with the potential for diagnosis and treatment of diseases (78). miRNAs are single-stranded non-coding RNA molecules of approximately 22 nucleotides in length that play important roles in regulating gene expression and biological function (79). In recent years, studies have shown that EV-derived miRNA expression is related to gut microbiota and intestinal barrier function (80–82). Mice deficient in IECs miRNA showed intestinal dysbiosis and exacerbation of colitis, which ameliorated after transplantation with fecal EV-derived miRNA from wild-type mice (80). This study suggests that fecal EV-derived miRNAs can regulate the gut microbiota and ameliorate the progression of intestinal inflammation. Another study found that EV-derived miRNAs of dietary ginger can induce IL-22 production to improve intestinal barrier function and thus ameliorate intestinal inflammation (83). A recent study showed that exosome miR-181a derived from MSCs alleviated colitis by improving gut microbiota imbalance and intestinal barrier function and reducing pro-inflammatory factor secretion (82). Taken together, the above results suggest that some EV-derived miRNAs in the intestinal tract may inhibit the progression of SLE by improving gut microbiota homeostasis and intestinal barrier function. More studies are required to confirm this hypothesis.
5 Potential Therapy for SLE: Modulating Gut Microbiota
At present, the study of intestinal bacteria intervention in the treatment of SLE is still in its infancy, but it can learn from other dysbacteriosis-associated diseases and predict future regimens in the treatment of SLE. As described in a recent review (40), probiotics/prebiotic therapy are currently practical approaches for ameliorating intestinal dysbacteriosis to treat SLE. Probiotics and prebiotics can induce differentiation of Treg cells, improve Th17/Th1 imbalance, and reduce the production of autoantibodies, thereby reducing the severity of lupus (40). Nevertheless, the efficacy of probiotics/prebiotic in the treatment of SLE remains unclear and has not been confirmed by clinical trials. There are differences in phenotypic manifestations caused by gut microbiota in SLE. To illustrate, intestinal commensal E. gallinarum can translocation to the liver and cause autoimmune hepatitis in patients with SLE (28). R. gnavus could increase serum anti-dsDNA antibody and LPS levels (8). The curli-DNA complex of biofilms containing S. Typhimurium promoted lupus progression. These differences may influence the approaches to targeting gut microbiota for SLE. These differences may have an impact on choosing the most appropriate modulation method of gut microbiota. Next, we discuss several options for the intervention of gut microbiota in the treatment of SLE.
5.1 Dietary Intervention
Dietary intervention may regulate the imbalance of gut microbiota, and thus ameliorate SLE progression. The alteration of the pH value of drinking water could beneficially influence on gut microbiota composition and disease progression in SWR×NZB F1(SNF1) lupus mice (84). Dietary retinoic acids supplementation could upregulate lactobacilli and ameliorate lupus in MRL/lpr mice (26). However, the efficacy of dietary retinoic acids in SLE treatment remains controversial (45) and still needs further study.
Also, the high-salt diet plays an important role in the pathogenesis of gut microbiota dysbiosis in autoimmune diseases. Interestingly, a recent randomized controlled trial demonstrated that a low-salt diet increased circulating SCFAs and decreased blood pressures by affecting the gut microbiota in humans (85). Therefore, reducing dietary salt intake or targeting salt-sensitive associated protein may be a new therapeutic strategy for SLE treatment. But this strategy still needs to be confirmed by further studies.
In addition, celiac disease (CeD) is an autoimmune enteropathy that is proposed to be associated with SLE (86, 87). An analysis of 29,000 patients with biopsy-confirmed CeD found that patients with CeD had a three-fold increased risk of developing SLE compared with healthy controls (86). In contrast, a large case-control study involving 5018 patients with SLE reported a significantly higher prevalence of CeD in patients with SLE compared with matched controls (87). And gluten, the major protein of wheat grains, is one of the factors contributed to the coexistence of SLE and CeD (87). The gliadin polypeptide of gluten increased intestinal permeability and activated CD4+ T cells resulted in CeD (88, 89). Therefore, gluten may be one of the causes of impaired intestinal barrier function in patients with SLE. Currently, the gold standard treatment for CeD is a strict and life-long gluten-free diet (GFD) (90). However, the implementation of GFD is limited by high cost, decreased quality of life of patients and complex pathogenesis (90). GFD may contribute to improve gut barrier function but still requires additional study.
Altogether, dietary intervention may be an important and new therapy in SLE.
5.2 Oral Antibiotic Therapy
In recent years, many studies have attempted to use antibiotics to treat lupus mice. For example, treatment with broad-spectrum antibiotics or vancomycin after onset in lupus MRL/lpr mice removes harmful bacteria from the gut, enriches probiotics, and restores gut barrier function, thereby ameliorating lupus (27). Moreover, antibiotic treatment alleviates Treg/Th17 imbalance in lupus mice (27) and inhibits the high blood pressure caused by Th17 cell infiltration (32). Vieira et al. (28) found that vancomycin treatment of NZB/WF1 lupus mice cleared E. gallinarum, a specific pathogen in the intestine, improved intestinal barrier function, and delayed lupus progression. However, another study showed that treatment with antibiotics has no significant effect on both the gut microbiota and SLE progression in NZB/WF1 lupus mice, the mechanism of which is unclear (91). Similarly, Zhang et al. showed that antibiotic treatment exacerbated the disease in MRL/lpr mice, possibly due to a short course and insufficient dose of antibiotics before lupus onset (31). Alternatively, another study found that vancomycin treatment ameliorated lupus symptoms in common MRL/lpr mice, but aggravated lupus in pregnant and postpartum (PP) mice. Mechanistically, vancomycin treatment aggravates LN in PP mice by downregulating the expression of Treg cells through inhibition of IDO and upregulation of IFN-γ (29).
In conclusion, the antibiotic therapy regimen for SLE is controversial. In general, antibiotic treatment decreases pathogenic bacteria, enriches probiotics, and ameliorates intestinal leakage in lupus mice, thereby inhibiting lupus progression. However, antibiotics may also exacerbate lupus severity in premorbid and pregnant or lactating mice. Moreover, there are some limitations in the routine use of antibiotics to treat patients with SLE. Because antibiotic treatment may inhibit the therapeutic effect of prednisone on lupus in MRL/lpr mice (31), while prednisone is a common drug for patients with SLE in clinical practice. Furthermore, antibiotic abuse may lead to drug-resistant bacterial infection (92), which is an important cause of death in patients with SLE (93). Therefore, the use of antibiotics in the treatment of SLE needs to be further studied to specifically remove pathogenic bacteria without causing gut microbiota disorders as much as possible.
5.3 Fecal Microbiota Transplantation
Fecal microbiota transplantation (FMT) is defined as the transplantation of bacteria from the feces of healthy donors into the patient’s intestine to restore microecology homeostasis and thus treat diseases associated with gut microbiota imbalance (94). In 2013, FMT was included in the official therapeutic guidelines for Clostridium difficile infection (CDI) (95). In recent years, studies have shown that FMT is effective in the treatment of SLE mouse models (31, 33, 84). An acidic water diet can restore the balance of gut microbiota in lupus mice, and that this repaired gut microbiota can be used for FMT to treat control lupus mice [25703185]. In addition, short-term antibiotic treatment of early-stage MRL/lpr lupus mice promoted SLE progression, and the disease severity in these mice was reduced after FMT treatment in the following week. However, short-term premorbid antibiotic treatment or FMT could inhibit the therapeutic effect of prednisone on lupus in MRL/lpr mice aged 9 to 13 weeks (31). This study suggests that performing FMT early in the onset of lupus suppresses the progression of lupus, but, at the same time, affects the therapeutic effect of glucocorticoid therapy. If patients with SLE are routinely treated with glucocorticoids, treatment with FMT should be carefully considered. More recently, a study found that untreated lupus MRL/lpr mice transplanted with fecal microbiota from prednisone-treated mice experienced lupus attenuated without the side effects of prednisone-treated mice (33). These results suggest that FMT may be an effective SLE therapy to avoid adverse glucocorticoid reactions. The effect of the interaction between FMT and glucocorticoid therapy on the progression of SLE requires further study. FMT clinical trials have been studied for other autoimmune diseases, such as ulcer colitis and type 1 diabetes, and some efficacy has been achieved (96, 97). Therefore, clinical trials of FMT in patients with SLE are promising, but further studies are needed.
A recent article reported that a patient succumbed to infection due to drug-resistant E. coli bacteria in donor stool samples (98). Therefore, donor screening must be improved to prevent transmission of microorganisms leading to infectious events. In conclusion, the benefits and risks of FMT in the treatment of SLE need to be assessed, and how to apply it in clinical practice still needs further study.
5.4 Glucocorticoid Therapy
Glucocorticoids (GCs) are steroids that can bind and activate the cytosolic glucocorticoid receptors (GRs) to exert an anti-inflammatory effect (99). GCs have become one of the main traditional drugs for SLE due to their rapid and potent anti-inflammatory effects, low cost, and easy availability. Moreover, long-term high-dose GC treatment regimens are accompanied by an increase in side effects and infections (100). Enhancing the efficacy of GCs and reducing their side effects in patients with SLE is a challenge. In recent years, studies have shown that the efficacy of GCs in the treatment of SLE is related to changes in the gut microbiota. For example, NZB/W F1 mice treated with dexamethasone had increased diversity of intestinal bacteria and a significant reduction in a certain Lactobacillus species associated with lupus progression (101). In another study, prednisone treatment caused alterations in the gut microbiota, including a decrease in Mucispirillum and an increase in Anaerostipes, which were inversely associated with disease activity in SLE. Bromofuranone did not alleviate lupus but enhanced the efficacy of prednisone in the treatment of SLE (30). As previously mentioned, Wang et al. demonstrated that prednisone ameliorates gut microbiota dysbiosis in SLE mouse models, and FMT treatment of SLE may prevent glucocorticoid adverse reactions (33). Moreover, GCs treatment restored the gut Firmicutes/Bacteroidetes ratio and increased the abundance of probiotics such as Lactobacillus and Bifidobacterium in patients with SLE (19).
All those studies suggest that intestinal dysbacteriosis may be a target for GCs in the treatment of SLE, but the mechanism remains unclear. A comparative study indicated that an increased levels of Lactobacillus in patients with SLE under GCs treatment (19). Moreover, Lactobacillus contributes to the alleviation of lupus severity by upregulating Foxp3+ regulatory T (Treg) cells (102). Treg cells are indispensable GC target cells in vivo (103). And GCs directly act on GRs in Treg cells and regulate miR-342-3p dependent metabolic programming to exert therapeutic effects (103). Therefore, Lactobacillus may affect the therapeutic efficacy of GCs by promoting the proliferation of Treg cells.
Overall, intestinal dysbacteriosis is one of the targets of GCs in the treatment of SLE. Certain drugs such as bromofuranone are associated with enhancing the therapeutic effects of GCs, and FMT may be an effective treatment regimen to reduce the side effects of GCs. Certain intestinal bacteria such as Lactobacillus may affected the therapeutic effect of GCs by regulating Treg cells. However, the specific mechanism by which GCs modulate intestinal bacteria in the treatment of SLE needs to be elucidated in future studies.
5.5 Regulate IECs Autophagy and EV-Derived miRNA Therapy
Autophagy is crucial for maintaining the homeostasis of gut microbiota and intestinal barrier function (71, 72). Therefore, regulating IECs autophagy may help improve the gut microbiota balance for the treatment of SLE. First, some drugs can improve gut microbiota composition and intestinal barrier function by promoting IECs autophagy, thus reducing intestinal inflammation and inhibiting autoimmune (104–106). For example, rapamycin can inhibit the progression of multiple sclerosis by promoting IECs autophagy and restoring intestinal microbiota balance (107). Galangin increases the expression of autophagy-related proteins and promotes the formation of colonic autophagy, increases the richness of intestinal probiotics, and reduces intestinal inflammation (108). Second, FMT can increase the expression of intestinal mucosal autophagy-related proteins and reduce intestinal permeability in piglets (75).
In contrast, EV-derived miRNAs may treat SLE by modulating the gut microbiota. A recent review reported that food-derived miRNAs could regulate the composition of gut microbiota and enhance intestinal barrier function, which is beneficial to human health (109). For instance, dietary ginger-derived miRNAs can induce IL-22 production to improve intestinal barrier function and thus ameliorate intestinal inflammation (83). Furthermore, MSC-derived exosome miR-181a can alleviate colitis by improving gut microbiota imbalance and intestinal barrier function and reducing pro-inflammatory factor secretion (82). Mice deficient in IECs miRNA showed intestinal dysbiosis and exacerbation of colitis, which ameliorated after transplantation with fecal EV-derived miRNA from wild-type mice (80).
In summary, IECs autophagy and EV-derived miRNAs can restore gut microbiota balance and intestinal barrier function, thereby inhibiting autoimmune-related intestinal inflammation. Therefore, we believe that regulating autophagy and EV-derived miRNAs is a promising therapeutic option for SLE.
5.6 Mesenchymal Stem Cell Therapy
MSCs are stromal cells with self-renewal and multi-lineage differentiation potential that can be obtained from tissues such as bone marrow (110). With low immunogenicity and strong immunomodulatory effects (111), MSCs can be used to treat SLE (112, 113). Allogeneic MSC transplantation ameliorates clinical symptoms, decreases SLEDAI score, and ameliorates LN in patients with refractory SLE (113, 114). Moreover, studies have shown that MSCs can regulate gut microbiota, increase insulin-like growth factor-1 (IGF-1), promote intestinal healing, and ameliorate the mouse model of IBD (115, 116). In contrast, miRNA-181a of MSC-derived exosomes can attenuate intestinal inflammation in mouse models of colitis by improving the composition of gut microbiota and restoring barrier function. These studies suggest that MSCs or MSC-derived exosomes can improve gut microbiota dysbiosis and intestinal barrier function and ameliorate intestinal inflammation (82).
Recently, it has been shown that human umbilical mesenchymal stem cells (hUC-MSCs) treat rheumatoid arthritis (RA) by regulating the interaction between gut microbiota and host immunity through the aryl hydrocarbon receptor (AhR) (117). Thus, we hypothesized that MSCs can inhibit SLE progression by ameliorating gut microbiota dysbiosis and intestinal barrier function. However, the specific mechanism of action of MSCs in the treatment of SLE is unknown, and the therapeutic effects of clinical trials remain controversial. In a clinical trial, hUC-MSCs did not have a positive therapeutic effect in patients with severe LN compared with placebo control (118). Meanwhile, a review suggested that the immunomodulatory effects of MSCs depend on the inflammation status, and that MSCs can both suppress and promote immune responses (119). Moreover, gut microbiota dysbiosis may inhibit the therapeutic effect of MSCs. For example, chronic hypoxia has been found to lead to intestinal dysbiosis and promote senescence of bone marrow MSCs (120). Another study demonstrated that intestinal dysbacteriosis might inhibit the therapeutic effect of MSCs in diabetic mice, while modulation of intestinal bacteria may help to enhance the therapeutic effect of MSC transplantation (121). These results suggest that intestinal bacteria can affect the immunomodulatory effects of MSCs, which may be one of the reasons for the poor efficacy of some MSCs in the treatment of SLE. In addition, Ocansey’s review (122) suggested that there would be a higher clinical remission rate in patients with IBD treated with the combined MSC-FMT transplantation approach compared with MSC transplantation alone or FMT transplantation. Similarly, we believe that the combined MSC-FMT transplantation approach will have a better therapeutic effect in the treatment of SLE.
Taken together, the study of MSCs in the treatment of SLE has fallen into a bottleneck, and gut microbiota will be a very promising direction for future research.
5.7 Vaccination
To prevent infection, the EULAR guidelines recommend vaccinations such as pneumococcal vaccines (PCV13) for patients with SLE during inactive periods (123). Vaccination of MRL/lpr mice with PCV13 ameliorated lupus severity (124). As previously described, SLE mouse models may suffer from infection with specific intestinal pathogens, such as E. gallinarum and R. gnavus (8, 28). The development of vaccines against these specific pathogenic bacteria could contribute to the treatment of SLE. Accordingly, Vieira et al. (28) demonstrated that after inoculation of E. gallinarum vaccine in (NZW × BXSB) F1 lupus mice, intestinal barrier function was restored and SLE was alleviated. This study shows that specific targeted therapy for intestinal pathogens can inhibit host autoimmune progression independent of other drugs. Importantly, E. gallinarum was also detected in the gut and liver of patients with SLE (28). This illustrates that the E. gallinarum vaccine is very promising for the treatment of patients with SLE, but further studies are still needed.
At present, research on intestinal microbiota vaccines is still in the preliminary stage, but there is no doubt that targeted vaccine therapy of specific intestinal pathogens is a very promising treatment for SLE.
6 Future Perspectives
Gut microbiota dysbiosis is closely related to the occurrence and development of SLE. The interaction of factors such as impaired intestinal barrier function, molecular mimicry, biofilms, specific pathogens, and sex hormones can disrupt gut microbiota balance and aggravate SLE. In addition, we suggest that IECs autophagy and EV-derived miRNAs may also affect progression in SLE by regulating the gut microbiota.
Traditionally, it is difficult to treat SLE due to its heterogeneity and complex pathogenesis, while targeting intestinal bacteria may be a breakthrough. In previous studies, probiotics or prebiotic modulation of intestinal bacteria have shown some efficacy in the treatment of SLE, but it is still controversial and has not been confirmed in clinical trials. Dietary interventions such as oral retinoic acids, low-salt diets and gluten-free diets may be beneficial in the treatment of SLE, but further research is needed. Oral antibiotics have some efficacy but may lead to more severe intestinal dysbacteriosis or the development of drug-resistant bacteria. Vaccination against gut pathogenic bacteria suppresses lupus progression in (NZW × BXSB) F1 mice without antibiotic-related side effects, but it is not yet available to treat all lupus mouse and patients. Notably, FMT significantly ameliorates disease in lupus mice by restoring the intestinal bacterial balance and intestinal barrier function. Clinical trials of FMT in patients with SLE are promising; however, donor stool screening must be improved to prevent infectious events.
We also propose new insights into the regulation of gut microbiota in SLE, including GCs, autophagy, EV-derived miRNAs, and MSC therapy. First, GCs, which are commonly used in SLE, can ameliorate intestinal dysbacteriosis but have side effects. The regulation of gut microbiota may help enhance the efficacy of GCs in the treatment of SLE and prevent side effects. Second, regulating autophagy and EV-derived miRNAs may treat SLE by regulating the gut microbiota. Finally, promising results have been achieved for the use of MSCs in patients with refractory SLE in current clinical trials. However, disturbed gut microbiota may inhibit the therapeutic effects of MSCs. In contrast, MSCs can ameliorate intestinal dysbacteriosis, restore intestinal barrier function, and inhibit autoimmune progression. In addition, MSC-derived EVs could ameliorate RA in rats by modulating the gut microbiota. Therefore, the gut microbiota may be a target for MSCs in the treatment of SLE. Moreover, the combination of MSC-FMT transplantation has the potential to enhance the effect of MSCs in the treatment of SLE. Therefore, MSC regulation of gut microbiota for the treatment of SLE is a promising direction for future study.
Here, we summarize novel insights into the mechanisms of microbiota dysbiosis in SLE and provide promising therapeutic strategies, which may help improve our understanding of the pathogenesis of SLE and provide novel therapies for SLE.
Author Contributions
QRP and FG wrote the manuscript and designed the figures. YH, AL, SC, JC, and H-FL revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Natural Science Foundation of China (no.82070757), the Project of “Dengfeng Plan” and Department of established positions for the Zhujiang Scholar from Guangdong Medical University, and Guangdong Basic and Applied Basic Research Foundation (no.2019A1515012203), the Zhanjiang City Program for Tackling Key Problems in Science and Technology (no. 2019B01179, no. 2017A01010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic Lupus Erythematosus. Nat Rev Dis Primers (2016) 2:16039. doi: 10.1038/nrdp.2016.39
2. Tsokos GC. Autoimmunity and Organ Damage in Systemic Lupus Erythematosus. Nat Immunol (2020) 21:605–14. doi: 10.1038/s41590-020-0677-6
3. Gladman DD, Hussain F, Ibañez D, Urowitz MB. The Nature and Outcome of Infection in Systemic Lupus Erythematosus. Lupus (2002) 11:234–9. doi: 10.1191/0961203302lu170oa
4. Esposito S, Bosis S, Semino M, Rigante D. Infections and Systemic Lupus Erythematosus. Eur J Clin Microbiol Infect Dis (2014) 33:1467–75. doi: 10.1007/s10096-014-2098-7
5. Zegarra-Ruiz DF, El Beidaq A, Iñiguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell Host Microbe (2019) 25:113–27.e6. doi: 10.1016/j.chom.2018.11.009
6. Chen BD, Jia XM, Xu JY, Zhao LD, Ji JY, Wu BX, et al. An Autoimmunogenic and Proinflammatory Profile Defined by the Gut Microbiota of Patients With Untreated Systemic Lupus Erythematosus. Arthritis Rheumatol (2021) 73:232–43. doi: 10.1002/art.41511
7. Mu Q, Kirby J, Reilly CM, Luo XM. Leaky Gut As a Danger Signal for Autoimmune Diseases. Front Immunol (2017) 8:598. doi: 10.3389/fimmu.2017.00598
8. Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. Lupus Nephritis Is Linked to Disease-Activity Associated Expansions and Immunity to a Gut Commensal. Ann Rheumatic Dis (2019) 78:947–56. doi: 10.1136/annrheumdis-2018-214856
9. Zhang W, Reichlin M. A Possible Link Between Infection With Burkholderia Bacteria and Systemic Lupus Erythematosus Based on Epitope Mimicry. Clin Dev Immunol (2008) 2008:683489. doi: 10.1155/2008/683489
10. Shoenfeld Y, Vilner Y, Coates AR, Rauch J, Lavie G, Shaul D, et al. Monoclonal Anti-Tuberculosis Antibodies React With DNA, and Monoclonal Anti-DNA Autoantibodies React With Mycobacterium Tuberculosis. Clin Exp Immunol (1986) 66:255–61.
11. Mu Q, Zhang H, Liao X, Lin K, Liu H, Edwards MR, et al. Control of Lupus Nephritis by Changes of Gut Microbiota. Microbiome (2017) 5:73. doi: 10.1186/s40168-017-0300-8
12. Fanouriakis A, Tziolos N, Bertsias G, Boumpas DT. Update on the Diagnosis and Management of Systemic Lupus Erythematosus. Ann Rheum Dis (2021) 80:14–25. doi: 10.1136/annrheumdis-2020-218272
13. Pan Q, Xiao H, Shi L, He Y, Cai J, Wu J, et al. IgG4 Autoantibodies Attenuate Systemic Lupus Erythematosus Progression by Suppressing Complement Consumption and Inflammatory Cytokine Production. Front Immunol (2020) 11:1047. doi: 10.3389/fimmu.2020.01047
14. Kiriakidou M, Ching CL. Systemic Lupus Erythematosus. Ann Intern Med (2020) 172:Itc81–itc96. doi: 10.7326/aitc202006020
15. Pan Q, Gong L, Xiao H, Feng Y, Li L, Deng Z, et al. Basophil Activation-Dependent Autoantibody and Interleukin-17 Production Exacerbate Systemic Lupus Erythematosus. Front Immunol (2017) 8:348. doi: 10.3389/fimmu.2017.00348
16. López P, de Paz B, Rodríguez-Carrio J, Hevia A, Sánchez B, Margolles A, et al. Th17 Responses and Natural IgM Antibodies Are Related to Gut Microbiota Composition in Systemic Lupus Erythematosus Patients. Sci Rep (2016) 6:24072. doi: 10.1038/srep24072
17. Bellocchi C, Fernández-Ochoa Á, Montanelli G, Vigone B, Santaniello A, Quirantes-Piné R, et al. Identification of a Shared Microbiomic and Metabolomic Profile in Systemic Autoimmune Diseases. J Clin Med (2019) 8:1291. doi: 10.3390/jcm8091291
18. Li Y, Wang HF, Li X, Li HX, Zhang Q, Zhou HW, et al. Disordered Intestinal Microbes Are Associated With the Activity of Systemic Lupus Erythematosus. Clin Sci (Lond) (2019) 133:821–38. doi: 10.1042/CS20180841
19. Guo M, Wang H, Xu S, Zhuang Y, An J, Su C, et al. Alteration in Gut Microbiota is Associated With Dysregulation of Cytokines and Glucocorticoid Therapy in Systemic Lupus Erythematosus. Gut Microbes (2020) 11:1758–73. doi: 10.1080/19490976.2020.1768644
20. He J, Chan T, Hong X, Zheng F, Zhu C, Yin L, et al. Microbiome and Metabolome Analyses Reveal the Disruption of Lipid Metabolism in Systemic Lupus Erythematosus. Front Immunol (2020) 11:1703. doi: 10.3389/fimmu.2020.01703
21. Wen M, Liu T, Zhao M, Dang X, Feng S, Ding X, et al. Correlation Analysis Between Gut Microbiota and Metabolites in Children With Systemic Lupus Erythematosus. J Immunol Res (2021) 2021:5579608. doi: 10.1155/2021/5579608
22. Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. Intestinal Dysbiosis Associated With Systemic Lupus Erythematosus. mBio (2014) 5:e01548–14. doi: 10.1128/mBio.01548-14
23. He Z, Shao T, Li H, Xie Z, Wen C. Alterations of the Gut Microbiome in Chinese Patients With Systemic Lupus Erythematosus. Gut Pathog (2016) 8:64. doi: 10.1186/s13099-016-0146-9
24. Rodríguez-Carrio J, López P, Sánchez B, González S, Gueimonde M, Margolles A, et al. Intestinal Dysbiosis Is Associated With Altered Short-Chain Fatty Acids and Serum-Free Fatty Acids in Systemic Lupus Erythematosus. Front Immunol (2017) 8:23. doi: 10.3389/fimmu.2017.00023
25. van der Meulen TA, Harmsen HJM, Vila AV, Kurilshikov A, Liefers SC, Zhernakova A, et al. Shared Gut, But Distinct Oral Microbiota Composition in Primary Sjögren's Syndrome and Systemic Lupus Erythematosus. J Autoimmun (2019) 97:77–87. doi: 10.1016/j.jaut.2018.10.009
26. Zhang H, Liao X, Sparks JB, Luo XM. Dynamics of Gut Microbiota in Autoimmune Lupus. Appl Environ Microbiol (2014) 80:7551–60. doi: 10.1128/AEM.02676-14
27. Mu Q, Tavella VJ, Kirby JL, Cecere TE, Chung M, Lee J, et al. Antibiotics Ameliorate Lupus-Like Symptoms in Mice. Sci Rep (2017) 7:13675. doi: 10.1038/s41598-017-14223-0
28. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a Gut Pathobiont Drives Autoimmunity in Mice and Humans. Science (2018) 359:1156–61. doi: 10.1126/science.aar7201
29. Mu Q, Cabana-Puig X, Mao J, Swartwout B, Abdelhamid L, Cecere TE, et al. Pregnancy and Lactation Interfere With the Response of Autoimmunity to Modulation of Gut Microbiota. Microbiome (2019) 7:105. doi: 10.1186/s40168-019-0720-8
30. He Z, Kong X, Shao T, Zhang Y, Wen C. Alterations of the Gut Microbiota Associated With Promoting Efficacy of Prednisone by Bromofuranone in MRL/lpr Mice. Front Microbiol (2019) 10:978. doi: 10.3389/fmicb.2019.00978
31. Zhang Y, Liu Q, Yu Y, Wang M, Wen C, He Z. Early and Short-Term Interventions in the Gut Microbiota Affects Lupus Severity, Progression, and Treatment in MRL/lpr Mice. Front Microbiol (2020) 11:628. doi: 10.3389/fmicb.2020.00628
32. de la Visitación N, Robles-Vera I, Toral M, Gómez-Guzmán M, Sánchez M, Moleón J, et al. Gut Microbiota Contributes to the Development of Hypertension in a Genetic Mouse Model of Systemic Lupus Erythematosus. Br J Pharmacol (2021) 178:3708–29. doi: 10.1111/bph.15512
33. Wang M, Zhu Z, Lin X, Li H, Wen C, Bao J, et al. Gut Microbiota Mediated the Therapeutic Efficacies and the Side Effects of Prednisone in the Treatment of MRL/lpr Mice. Arthritis Res Ther (2021) 23:240. doi: 10.1186/s13075-021-02620-w
34. Toral M, Robles-Vera I, Romero M, de la Visitación N, Sánchez M, O'Valle F, et al. Lactobacillus Fermentum CECT5716: A Novel Alternative for the Prevention of Vascular Disorders in a Mouse Model of Systemic Lupus Erythematosus. FASEB J (2019) 33:10005–18. doi: 10.1096/fj.201900545RR
35. Choi SC, Brown J, Gong M, Ge Y, Zadeh M, Li W, et al. Gut Microbiota Dysbiosis and Altered Tryptophan Catabolism Contribute to Autoimmunity in Lupus-Susceptible Mice. Sci Transl Med (2020) 12:eaax2220. doi: 10.1126/scitranslmed.aax2220
36. Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, et al. Environment Dominates Over Host Genetics in Shaping Human Gut Microbiota. Nature (2018) 555:210–5. doi: 10.1038/nature25973
37. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium Chloride Drives Autoimmune Disease by the Induction of Pathogenic TH17 Cells. Nature (2013) 496:518–22. doi: 10.1038/nature11868
38. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-Responsive Gut Commensal Modulates T(H)17 Axis and Disease. Nature (2017) 551:585–9. doi: 10.1038/nature24628
39. Xiao ZX, Hu X, Zhang X, Chen Z, Wang J, Jin K, et al. High Salt Diet Accelerates the Progression of Murine Lupus Through Dendritic Cells. via p38 MAPK STAT1 Signaling pathways Signal Transduct Target Ther (2020) 5:34. doi: 10.1038/s41392-020-0139-5
40. Zhang L, Qing P, Yang H, Wu Y, Liu Y, Luo Y. Gut Microbiome and Metabolites in Systemic Lupus Erythematosus: Link, Mechanisms and Intervention. Front Immunol (2021) 12:686501. doi: 10.3389/fimmu.2021.686501
41. Fasano A. Leaky Gut and Autoimmune Diseases. Clin Rev Allergy Immunol (2012) 42:71–8. doi: 10.1007/s12016-011-8291-x
42. Gisbert JP, Bermejo F, Pérez-Calle JL, Taxonera C, Vera I, McNicholl AG, et al. Fecal Calprotectin and Lactoferrin for the Prediction of Inflammatory Bowel Disease Relapse. Inflammation Bowel Dis (2009) 15:1190–8. doi: 10.1002/ibd.20933
43. Thim-Uam A, Surawut S, Issara-Amphorn J, Jaroonwitchawan T, Hiengrach P, Chatthanathon P, et al. Leaky-Gut Enhanced Lupus Progression in the Fc Gamma Receptor-IIb Deficient and Pristane-Induced Mouse Models of Lupus. Sci Rep (2020) 10:777. doi: 10.1038/s41598-019-57275-0
44. Wang H, Wang G, Banerjee N, Liang Y, Du X, Boor PJ, et al. Aberrant Gut Microbiome Contributes to Intestinal Oxidative Stress, Barrier Dysfunction, Inflammation and Systemic Autoimmune Responses in MRL/lpr Mice. Front Immunol (2021) 12:651191. doi: 10.3389/fimmu.2021.651191
45. Abdelhamid L, Cabana-Puig X, Swartwout B, Lee J, Li S, Sun S, et al. Retinoic Acid Exerts Disease Stage-Dependent Effects on Pristane-Induced Lupus. Front Immunol (2020) 11:408. doi: 10.3389/fimmu.2020.00408
46. Deng GM, Tsokos GC. Cholera Toxin B Accelerates Disease Progression in Lupus-Prone Mice by Promoting Lipid Raft Aggregation. J Immunol (2008) 181:4019–26. doi: 10.4049/jimmunol.181.6.4019
47. Blank M, Barzilai O, Shoenfeld Y. Molecular Mimicry and Auto-Immunity. Clin Rev Allergy Immunol (2007) 32:111–8. doi: 10.1007/BF02686087
48. Zhao Z, Ren J, Dai C, Kannapell CC, Wang H, Gaskin F, et al. Nature of T Cell Epitopes in Lupus Antigens and HLA-DR Determines Autoantibody Initiation and Diversification. Ann Rheum Dis (2019) 78:380–90. doi: 10.1136/annrheumdis-2018-214125
49. Ruff WE, Dehner C, Kim WJ, Pagovich O, Aguiar CL, Yu AT, et al. Pathogenic Autoreactive T and B Cells Cross-React With Mimotopes Expressed by a Common Human Gut Commensal to Trigger Autoimmunity. Cell Host Microbe (2019) 26:100–13.e8. doi: 10.1016/j.chom.2019.05.003
50. Tsutsumi A, Matsuura E, Ichikawa K, Fujisaku A, Mukai M, Kobayashi S, et al. Antibodies to Beta 2-Glycoprotein I and Clinical Manifestations in Patients With Systemic Lupus Erythematosus. Arthritis Rheum (1996) 39:1466–74. doi: 10.1002/art.1780390905
51. Sciascia S, Cuadrado MJ, Khamashta M, Roccatello D. Renal Involvement in Antiphospholipid Syndrome. Nat Rev Nephrol (2014) 10:279–89. doi: 10.1038/nrneph.2014.38
52. Rauch J, Salem D, Subang R, Kuwana M, Levine JS. β2-Glycoprotein I-Reactive T Cells in Autoimmune Disease. Front Immunol (2018) 9:2836. doi: 10.3389/fimmu.2018.02836
53. Costerton JW. Introduction to Biofilm. Int J Antimicrob Agents (1999) 11:217–21. doi: 10.1016/s0924-8579(99)00018-7
54. Suh JD, Ramakrishnan V, Palmer JN. Biofilms. Otolaryngol Clin North Am (2010) 43:521–30. doi: 10.1016/j.otc.2010.02.010
55. Stewart PS, Costerton JW. Antibiotic Resistance of Bacteria in Biofilms. Lancet (2001) 358:135–8. doi: 10.1016/s0140-6736(01)05321-1
56. Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR. Diversity, Biogenesis and Function of Microbial Amyloids. Trends Microbiol (2012) 20:66–73. doi: 10.1016/j.tim.2011.11.005
57. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA Composites of Bacterial Biofilms Stimulate Autoimmunity. Immunity (2015) 42:1171–84. doi: 10.1016/j.immuni.2015.06.002
58. Fu W, Liu Y, Liu F, Liu C, Li J, Niu J, et al. A Novel Autoantibody Induced by Bacterial Biofilm Conserved Components Aggravates Lupus Nephritis. Front Immunol (2021) 12:656090. doi: 10.3389/fimmu.2021.656090
59. Pachucki RJ, Corradetti C, Kohler L, Ghadiali J, Gallo PM, Nicastro L, et al. Persistent Bacteriuria and Antibodies Recognizing Curli/eDNA Complexes From Escherichia Coli Are Linked to Flares in Systemic Lupus Erythematosus. Arthritis Rheumatol (2020) 72:1872–81. doi: 10.1002/art.41400
60. Choi SH, Lee SO, Kim TH, Chung JW, Choo EJ, Kwak YG, et al. Clinical Features and Outcomes of Bacteremia Caused by Enterococcus Casseliflavus and Enterococcus Gallinarum: Analysis of 56 Cases. Clin Infect Dis (2004) 38:53–61. doi: 10.1086/380452
61. Bagavant H, Araszkiewicz AM, Ingram JK, Cizio K, Merrill JT, Arriens C, et al. Immune Response to Enterococcus Gallinarum in Lupus Patients Is Associated With a Subset of Lupus-Associated Autoantibodies. Front Immunol (2021) 12:635072. doi: 10.3389/fimmu.2021.635072
62. Bunker JJ, Drees C, Watson AR, Plunkett CH, Nagler CR, Schneewind O, et al. B Cell Superantigens in the Human Intestinal Microbiota. Sci Transl Med (2019) 11:eaau9356. doi: 10.1126/scitranslmed.aau9356
63. Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus Gnavus, a Member of the Human Gut Microbiome Associated With Crohn's Disease, Produces an Inflammatory Polysaccharide. Proc Natl Acad Sci USA (2019) 116:12672–7. doi: 10.1073/pnas.1904099116
64. Henke MT, Brown EM, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Capsular Polysaccharide Correlates With Immune Response to the Human Gut Microbe Ruminococcus Gnavus. Proc Natl Acad Sci USA (2021) 118:e2007595118. doi: 10.1073/pnas.2007595118
65. Rider V, Abdou NI, Kimler BF, Lu N, Brown S, Fridley BL. Gender Bias in Human Systemic Lupus Erythematosus: A Problem of Steroid Receptor Action? Front Immunol (2018) 9:611. doi: 10.3389/fimmu.2018.00611
66. Edwards MR, Dai R, Heid B, Cecere TE, Khan D, Mu Q, et al. Commercial Rodent Diets Differentially Regulate Autoimmune Glomerulonephritis, Epigenetics and Microbiota in MRL/lpr Mice. Int Immunol (2017) 29:263–76. doi: 10.1093/intimm/dxx033
67. Hughes GC, Choubey D. Modulation of Autoimmune Rheumatic Diseases by Oestrogen and Progesterone. Nat Rev Rheumatol (2014) 10:740–51. doi: 10.1038/nrrheum.2014.144
68. Kanda N, Tsuchida T, Tamaki K. Estrogen Enhancement of Anti-Double-Stranded DNA Antibody and Immunoglobulin G Production in Peripheral Blood Mononuclear Cells From Patients With Systemic Lupus Erythematosus. Arthritis Rheum (1999) 42:328–37. doi: 10.1002/1529-0131(199902)42:2<328::Aid-anr16>3.0.Co;2-#
69. Kanda N, Tsuchida T, Tamaki K. Testosterone Suppresses Anti-DNA Antibody Production in Peripheral Blood Mononuclear Cells From Patients With Systemic Lupus Erythematosus. Arthritis Rheum (1997) 40:1703–11. doi: 10.1002/art.1780400921
70. Johnson BM, Gaudreau MC, Gudi R, Brown R, Gilkeson G, Vasu C. Gut Microbiota Differently Contributes to Intestinal Immune Phenotype and Systemic Autoimmune Progression in Female and Male Lupus-Prone Mice. J Autoimmun (2020) 108:102420. doi: 10.1016/j.jaut.2020.102420
71. Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, et al. Autophagy Protein ATG16L1 Prevents Necroptosis in the Intestinal Epithelium. J Exp Med (2017) 214:3687–705. doi: 10.1084/jem.20170558
72. Nighot PK, Hu CA, Ma TY. Autophagy Enhances Intestinal Epithelial Tight Junction Barrier Function by Targeting Claudin-2 Protein Degradation. J Biol Chem (2015) 290:7234–46. doi: 10.1074/jbc.M114.597492
73. Kim S, Lee JY, Shin SG, Kim JK, Silwal P, Kim YJ, et al. ESRRA (Estrogen Related Receptor Alpha) Is a Critical Regulator of Intestinal Homeostasis Through Activation of Autophagic Flux via Gut Microbiota. Autophagy (2020) 17:2856–75. doi: 10.1080/15548627.2020.1847460
74. Yang L, Liu C, Zhao W, He C, Ding J, Dai R, et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl Environ Microbiol (2018) 84:e00880–18. doi: 10.1128/aem.00880-18
75. Cheng S, Ma X, Geng S, Jiang X, Li Y, Hu L, et al. Fecal Microbiota Transplantation Beneficially Regulates Intestinal Mucosal Autophagy and Alleviates Gut Barrier Injury. mSystems (2018) 3:e00137–18. doi: 10.1128/mSystems.00137-18
76. Bhattacharya A, Eissa NT. Autophagy and Autoimmunity Crosstalks. Front Immunol (2013) 4:88. doi: 10.3389/fimmu.2013.00088
77. O'Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA Delivery by Extracellular Vesicles in Mammalian Cells and its Applications. Nat Rev Mol Cell Biol (2020) 21:585–606. doi: 10.1038/s41580-020-0251-y
78. Kalluri R, LeBleu VS. The Biology, Function, and Biomedical Applications of Exosomes. Science (2020) 367:eaau6977. doi: 10.1126/science.aau6977
79. Ambros V. microRNAs: Tiny Regulators With Great Potential. Cell (2001) 107:823–6. doi: 10.1016/s0092-8674(01)00616-x
80. Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, Bry L, et al. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe (2016) 19:32–43. doi: 10.1016/j.chom.2015.12.005
81. Muenchau S, Deutsch R, de Castro IJ, Hielscher T, Heber N, Niesler B, et al. Hypoxic Environment Promotes Barrier Formation in Human Intestinal Epithelial Cells Through Regulation of MicroRNA 320a Expression. Mol Cell Biol (2019) 39:e00553–18. doi: 10.1128/mcb.00553-18
82. Gu L, Ren F, Fang X, Yuan L, Liu G, Wang S. Exosomal MicroRNA-181a Derived From Mesenchymal Stem Cells Improves Gut Microbiota Composition, Barrier Function, and Inflammatory Status in an Experimental Colitis Model. Front Med (Lausanne) (2021) 8:660614. doi: 10.3389/fmed.2021.660614
83. Teng Y, Ren Y, Sayed M, Hu X, Lei C, Kumar A, et al. Plant-Derived Exosomal MicroRNAs Shape the Gut Microbiota. Cell Host Microbe (2018) 24:637–52.e8. doi: 10.1016/j.chom.2018.10.001
84. Johnson BM, Gaudreau MC, Al-Gadban MM, Gudi R, Vasu C. Impact of Dietary Deviation on Disease Progression and Gut Microbiome Composition in Lupus-Prone SNF1 Mice. Clin Exp Immunol (2015) 181:323–37. doi: 10.1111/cei.12609
85. Chen L, He FJ, Dong Y, Huang Y, Wang C, Harshfield GA, et al. Modest Sodium Reduction Increases Circulating Short-Chain Fatty Acids in Untreated Hypertensives: A Randomized, Double-Blind, Placebo-Controlled Trial. Hypertension (2020) 76:73–9. doi: 10.1161/hypertensionaha.120.14800
86. Ludvigsson JF, Rubio-Tapia A, Chowdhary V, Murray JA, Simard JF. Increased Risk of Systemic Lupus Erythematosus in 29,000 Patients With Biopsy-Verified Celiac Disease. J Rheumatol (2012) 39:1964–70. doi: 10.3899/jrheum.120493
87. Dahan S, Shor DB, Comaneshter D, Tekes-Manova D, Shovman O, Amital H, et al. All Disease Begins in the Gut: Celiac Disease Co-Existence With SLE. Autoimmun Rev (2016) 15:848–53. doi: 10.1016/j.autrev.2016.06.003
88. D'Avino P, Serena G, Kenyon V, Fasano A. An Updated Overview on Celiac Disease: From Immuno-Pathogenesis and Immuno-Genetics to Therapeutic Implications. Expert Rev Clin Immunol (2021) 17:269–84. doi: 10.1080/1744666x.2021.1880320
89. Christophersen A, Lund EG, Snir O, Solà E, Kanduri C, Dahal-Koirala S, et al. Distinct Phenotype of CD4(+) T Cells Driving Celiac Disease Identified in Multiple Autoimmune Conditions. Nat Med (2019) 25:734–7. doi: 10.1038/s41591-019-0403-9
90. Kivelä L, Caminero A, Leffler DA, Pinto-Sanchez MI, Tye-Din JA, Lindfors K. Current and Emerging Therapies for Coeliac Disease. Nat Rev Gastroenterol Hepatol (2021) 18:181–95. doi: 10.1038/s41575-020-00378-1
91. Schäfer AL, Eichhorst A, Hentze C, Kraemer AN, Amend A, Sprenger DTL, et al. Low Dietary Fiber Intake Links Development of Obesity and Lupus Pathogenesis. Front Immunol (2021) 12:696810. doi: 10.3389/fimmu.2021.696810
92. Guo Y, Song G, Sun M, Wang J, Wang Y. Prevalence and Therapies of Antibiotic-Resistance in Staphylococcus Aureus. Front Cell Infect Microbiol (2020) 10:107. doi: 10.3389/fcimb.2020.00107
93. Kedves M, Kósa F, Kunovszki P, Takács P, Szabó MZ, Karyekar C, et al. Large-Scale Mortality Gap Between SLE and Control Population is Associated With Increased Infection-Related Mortality in Lupus. Rheumatol (Oxford) (2020) 59:3443–51. doi: 10.1093/rheumatology/keaa188
94. Zeng W, Shen J, Bo T, Peng L, Xu H, Nasser MI, et al. Cutting Edge: Probiotics and Fecal Microbiota Transplantation in Immunomodulation. J Immunol Res (2019) 2019:1603758. doi: 10.1155/2019/1603758
95. Surawicz CM, Brandt LJ, Binion DG, Ananthakrishnan AN, Curry SR, Gilligan PH, et al. Guidelines for Diagnosis, Treatment, and Prevention of Clostridium Difficile Infections. Am J Gastroenterol (2013) 108:478–98; quiz 499. doi: 10.1038/ajg.2013.4
96. Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C, et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology (2015) 149:102–9.e6. doi: 10.1053/j.gastro.2015.04.001
97. de Groot P, Nikolic T, Pellegrini S, Sordi V, Imangaliyev S, Rampanelli E, et al. Faecal Microbiota Transplantation Halts Progression of Human New-Onset Type 1 Diabetes in a Randomised Controlled Trial. Gut (2021) 70:92–105. doi: 10.1136/gutjnl-2020-322630
98. DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, et al. Drug-Resistant E. Coli Bacteremia Transmitted by Fecal Microbiota Transplant. N Engl J Med (2019) 381:2043–50. doi: 10.1056/NEJMoa1910437
99. Cain DW, Cidlowski JA. Immune Regulation by Glucocorticoids. Nat Rev Immunol (2017) 17:233–47. doi: 10.1038/nri.2017.1
100. Porta S, Danza A, Arias Saavedra M, Carlomagno A, Goizueta MC, Vivero F, et al. Glucocorticoids in Systemic Lupus Erythematosus. Ten Questions and Some Issues. J Clin Med (2020) 9:2709. doi: 10.3390/jcm9092709
101. Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Appl Environ Microbiol (2018) 84:e02288–17. doi: 10.1128/aem.02288-17
102. Khorasani S, Mahmoudi M, Kalantari MR, Lavi Arab F, Esmaeili SA, Mardani F, et al. Amelioration of Regulatory T Cells by Lactobacillus Delbrueckii and Lactobacillus Rhamnosus in Pristane-Induced Lupus Mice Model. J Cell Physiol (2019) 234:9778–86. doi: 10.1002/jcp.27663
103. Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, et al. Anti-Inflammatory Roles of Glucocorticoids Are Mediated by Foxp3(+) Regulatory T Cells via a miR-342-Dependent Mechanism. Immunity (2020) 53:581–96.e5. doi: 10.1016/j.immuni.2020.07.002
104. Fan Q, Guan X, Hou Y, Liu Y, Wei W, Cai X, et al. Paeoniflorin Modulates Gut Microbial Production of Indole-3-Lactate and Epithelial Autophagy to Alleviate Colitis in Mice. Phytomedicine (2020) 79:153345. doi: 10.1016/j.phymed.2020.153345
105. Fan W, Zhang S, Wu Y, Lu T, Liu J, Cao X, et al. Genistein-Derived ROS-Responsive Nanoparticles Relieve Colitis by Regulating Mucosal Homeostasis. ACS Appl Mater Interfaces (2021) 13:40249–66. doi: 10.1021/acsami.1c09215
106. Zha Z, Lv Y, Tang H, Li T, Miao Y, Cheng J, et al. An Orally Administered Butyrate-Releasing Xylan Derivative Reduces Inflammation in Dextran Sulphate Sodium-Induced Murine Colitis. Int J Biol Macromol (2020) 156:1217–33. doi: 10.1016/j.ijbiomac.2019.11.159
107. Xu L, Zhang C, He D, Jiang N, Bai Y, Xin Y. Rapamycin and MCC950 Modified Gut Microbiota in Experimental Autoimmune Encephalomyelitis Mouse by Brain Gut Axis. Life Sci (2020) 253:117747. doi: 10.1016/j.lfs.2020.117747
108. Xuan H, Ou A, Hao S, Shi J, Jin X. Galangin Protects Against Symptoms of Dextran Sodium Sulfate-Induced Acute Colitis by Activating Autophagy and Modulating the Gut Microbiota. Nutrients (2020) 12:347. doi: 10.3390/nu12020347
109. Díez-Sainz E, Lorente-Cebrián S, Aranaz P, Riezu-Boj JI, Martínez JA, Milagro FI. Potential Mechanisms Linking Food-Derived MicroRNAs, Gut Microbiota and Intestinal Barrier Functions in the Context of Nutrition and Human Health. Front Nutr (2021) 8:586564. doi: 10.3389/fnut.2021.586564
110. Ding DC, Shyu WC, Lin SZ. Mesenchymal Stem Cells. Cell Transplant (2011) 20:5–14. doi: 10.3727/096368910x
111. Naji A, Eitoku M, Favier B, Deschaseaux F, Rouas-Freiss N, Suganuma N. Biological Functions of Mesenchymal Stem Cells and Clinical Implications. Cell Mol Life Sci (2019) 76:3323–48. doi: 10.1007/s00018-019-03125-1
112. Yuan L, Xiao ZT, Huang XZ, Wu MJ, Shi H, Liu AF. Human Embryonic Mesenchymal Stem Cells Alleviate Pathologic Changes of MRL/Lpr Mice by Regulating Th7 Cell Differentiation. Ren Fail (2016) 38:1432–40. doi: 10.3109/0886022x.2015.1136894
113. Wang D, Zhang H, Liang J, Wang H, Hua B, Feng X, et al. A Long-Term Follow-Up Study of Allogeneic Mesenchymal Stem/Stromal Cell Transplantation in Patients With Drug-Resistant Systemic Lupus Erythematosus. Stem Cell Rep (2018) 10:933–41. doi: 10.1016/j.stemcr.2018.01.029
114. Sun L, Wang D, Liang J, Zhang H, Feng X, Wang H, et al. Umbilical Cord Mesenchymal Stem Cell Transplantation in Severe and Refractory Systemic Lupus Erythematosus. Arthritis Rheum (2010) 62:2467–75. doi: 10.1002/art.27548
115. Soontararak S, Chow L, Johnson V, Coy J, Wheat W, Regan D, et al. Mesenchymal Stem Cells (MSC) Derived From Induced Pluripotent Stem Cells (iPSC) Equivalent to Adipose-Derived MSC in Promoting Intestinal Healing and Microbiome Normalization in Mouse Inflammatory Bowel Disease Model. Stem Cells Transl Med (2018) 7:456–67. doi: 10.1002/sctm.17-0305
116. Xu J, Wang X, Chen J, Chen S, Li Z, Liu H, et al. Embryonic Stem Cell-Derived Mesenchymal Stem Cells Promote Colon Epithelial Integrity and Regeneration by Elevating Circulating IGF-1 in Colitis Mice. Theranostics (2020) 10:12204–22. doi: 10.7150/thno.47683
117. Li X, Lu C, Fan D, Lu X, Xia Y, Zhao H, et al. Human Umbilical Mesenchymal Stem Cells Display Therapeutic Potential in Rheumatoid Arthritis by Regulating Interactions Between Immunity and Gut Microbiota. via Aryl Hydrocarbon Receptor Front Cell Dev Biol (2020) 8:131. doi: 10.3389/fcell.2020.00131
118. Deng D, Zhang P, Guo Y, Lim TO. A Randomised Double-Blind, Placebo-Controlled Trial of Allogeneic Umbilical Cord-Derived Mesenchymal Stem Cell for Lupus Nephritis. Ann Rheumatic Dis (2017) 76:1436–9. doi: 10.1136/annrheumdis-2017-211073
119. Wang Y, Chen X, Cao W, Shi Y. Plasticity of Mesenchymal Stem Cells in Immunomodulation: Pathological and Therapeutic Implications. Nat Immunol (2014) 15:1009–16. doi: 10.1038/ni.3002
120. Xing J, Ying Y, Mao C, Liu Y, Wang T, Zhao Q, et al. Hypoxia Induces Senescence of Bone Marrow Mesenchymal Stem Cells. via altered gut microbiota Nat Commun (2018) 9:2020. doi: 10.1038/s41467-018-04453-9
121. Lv W, Graves DT, He L, Shi Y, Deng X, Zhao Y, et al. Depletion of the Diabetic Gut Microbiota Resistance Enhances Stem Cells Therapy in Type 1 Diabetes Mellitus. Theranostics (2020) 10:6500–16. doi: 10.7150/thno.44113
122. Ocansey DKW, Wang L, Wang J, Yan Y, Qian H, Zhang X, et al. Mesenchymal Stem Cell-Gut Microbiota Interaction in the Repair of Inflammatory Bowel Disease: An Enhanced Therapeutic Effect. Clin Transl Med (2019) 8:31. doi: 10.1186/s40169-019-0251-8
123. Fanouriakis A, Kostopoulou M, Alunno A, Aringer M, Bajema I, Boletis JN, et al. 2019 Update of the EULAR Recommendations for the Management of Systemic Lupus Erythematosus. Ann Rheum Dis (2019) 78:736–45. doi: 10.1136/annrheumdis-2019-215089
Keywords: systemic lupus erythematosus, autoimmune disease, gut microbiota dysbiosis, extracellular vesicle, miRNA, mesenchymal stem cell therapy
Citation: Pan Q, Guo F, Huang Y, Li A, Chen S, Chen J, Liu H-f and Pan Q (2021) Gut Microbiota Dysbiosis in Systemic Lupus Erythematosus: Novel Insights into Mechanisms and Promising Therapeutic Strategies. Front. Immunol. 12:799788. doi: 10.3389/fimmu.2021.799788
Received: 22 October 2021; Accepted: 18 November 2021;
Published: 03 December 2021.
Edited by:
Michele Maria Luchetti Gentiloni, Marche Polytechnic University, ItalyReviewed by:
Carlo Perricone, University of Perugia, ItalyMatteo Piga, University of Cagliari, Italy
Simone Baldovino, University of Turin, Italy
Lingyun Sun, Nanjing University, China
Copyright © 2021 Pan, Guo, Huang, Li, Chen, Chen, Liu and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua-feng Liu, bGl1aGZAZ2RtdS5lZHUuY24=; Qingjun Pan, cHFqQGdkbXUuZWR1LmNu
†These authors have contributed equally to this work