
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 29 March 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.663883
 Milica Miladinovic1Boris Wittekindt1Sebastian Fischer2
Milica Miladinovic1Boris Wittekindt1Sebastian Fischer2 Elise Gradhand3Steffen Kunzmann4
Elise Gradhand3Steffen Kunzmann4 Stefanie Y. Zimmermann1
Stefanie Y. Zimmermann1 Shahrzad Bakhtiar1
Shahrzad Bakhtiar1 Thomas Klingebiel1Rolf Schlösser1
Thomas Klingebiel1Rolf Schlösser1 Thomas Lehrnbecher1*
Thomas Lehrnbecher1*Chronic granulomatous disease (CGD) is a primary immunodeficiency, which is diagnosed in most patients between one and three years of age. Here we report on a boy who presented at birth with extensive skin lesions and lymphadenopathy which were caused by CGD. An analysis of the literature revealed 24 patients with CGD who became symptomatic during the first six weeks of life. Although pulmonary complications and skin lesions due to infection were the leading symptoms, clinical features were extremely heterogenous. As follow-up was not well specified in most patients, the long-term prognosis of children with very early onset of CGD remains unknown.
Chronic granulomatous disease (CGD) is a rare primary immunodeficiency which occurs with a frequency of approximately 1:200.000 in the United States and Europe and is characterized by an increased susceptibility to bacterial and fungal infections (1–3). The disease is caused by a defect of the NADPH oxidase complex and most of the mutations are located in the genes gp91phox, p47phox, p22phox, p67phox or p40phox (4, 5). In affected families, patients may be diagnosed prior to any signs of the disease, even prenatally. However, the vast majority of patients with CGD is diagnosed between one and three years of age when they become clinically symptomatic with recurrent and severe infectious complications, mostly affecting the lung (79%), lymph nodes (53%), liver (27%) and skin (42%) (3). Typical pathogens include Staphylococcus aureus, Burkholderia cepacia, Serratia marcenscens, Nocardia spp, and Aspergillus spp, in particular A. fumigatus and A. nidulans (2–4, 6). Notably, CGD can also present in unusual forms such as gastrointestinal mucormycosis, cardiac empyema or phlebitis, which makes early diagnosis difficult, in particular in the neonatal period (3, 7). A milder phenotype of the disease has been associated with later diagnosis, but also with longer survival (2).
Here we describe the unusual case of a neonate with CGD who presented with extensive skin lesions and lymphadenopathy at birth, which prompted us to review and analyze the current literature for patients with an extremely early onset of CGD.
After uneventful pregnancy, a full-term neonate presented at birth with extensive papulo-pustular lesions on both hands and feet and scattered papules with central vesicles on the body (Figure 1). The boy was in good clinical condition, no other abnormality was seen. The father and the 6-year-old half-brother were healthy, whereas the mother was diagnosed at the age of 20 years with Crohn´s disease and was currently under therapy with the monoclonal antibody vedolizumab.
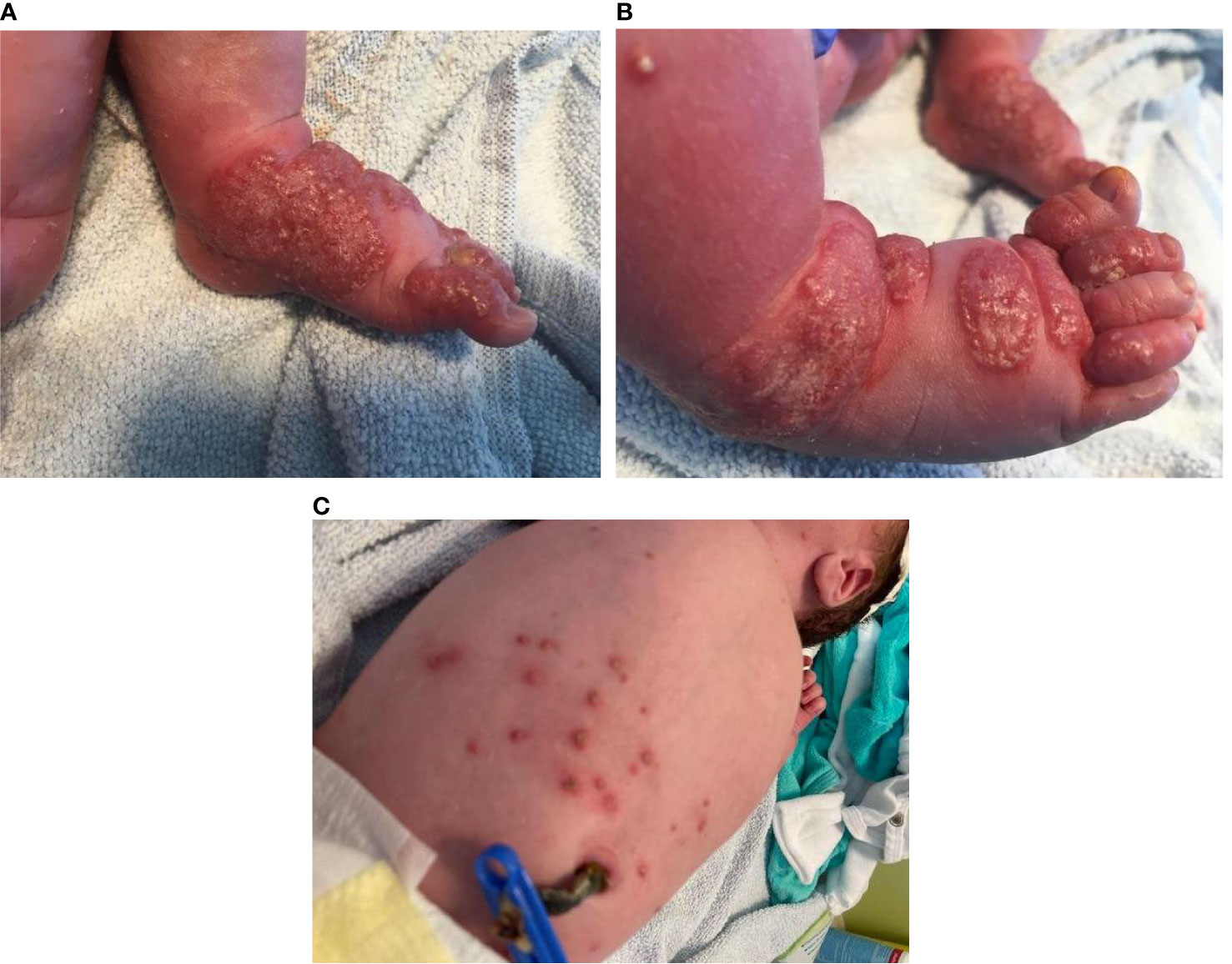
Figure 1 (A, B) Papulo-pustular lesions on an erythematous base on the feet. (C) Scattered papules with central vesicles on the body in a newborn with chronic granulomatous disease.
Laboratory tests revealed leukocytosis (25,560/µl, upper limit 16,200/µl), a high absolute number of eosinophils (up to 5340/µl, upper limit 950/µl) and an elevated C-reactive protein (initial evaluation 7.7 mg/dl, maximum 21.77 mg/dl; normal range <0.4mg/dl). Immunologic parameters including lymphocyte subsets and immunoglobulins were within normal range. Tumor markers including alpha-fetoprotein and ß-HCG were negative, and a chromosomal analysis did not reveal abnormalities. The blood level of vedolizumab six weeks after birth was with <4.0 µg/ml below the detection limit. Despite cultures of blood and skin lesions remained negative, antibiotic therapy was initiated, but showed no significant effect on leukocytes and C-reactive protein. Imaging studies revealed an enlarged thymus with multiple jagged-edged cysts (Figure 2), and axillary lymph nodes as well as those located along the lateral thoracic wall, parailiacal and inguinal were also increased in size with a maximum diameter of 1.8 cm. Magnetic resonance imaging showed bulky soft tissue masses surrounding the abdominal aorta and its branches from the coeliac trunk to the external iliac artery (Figure 2).
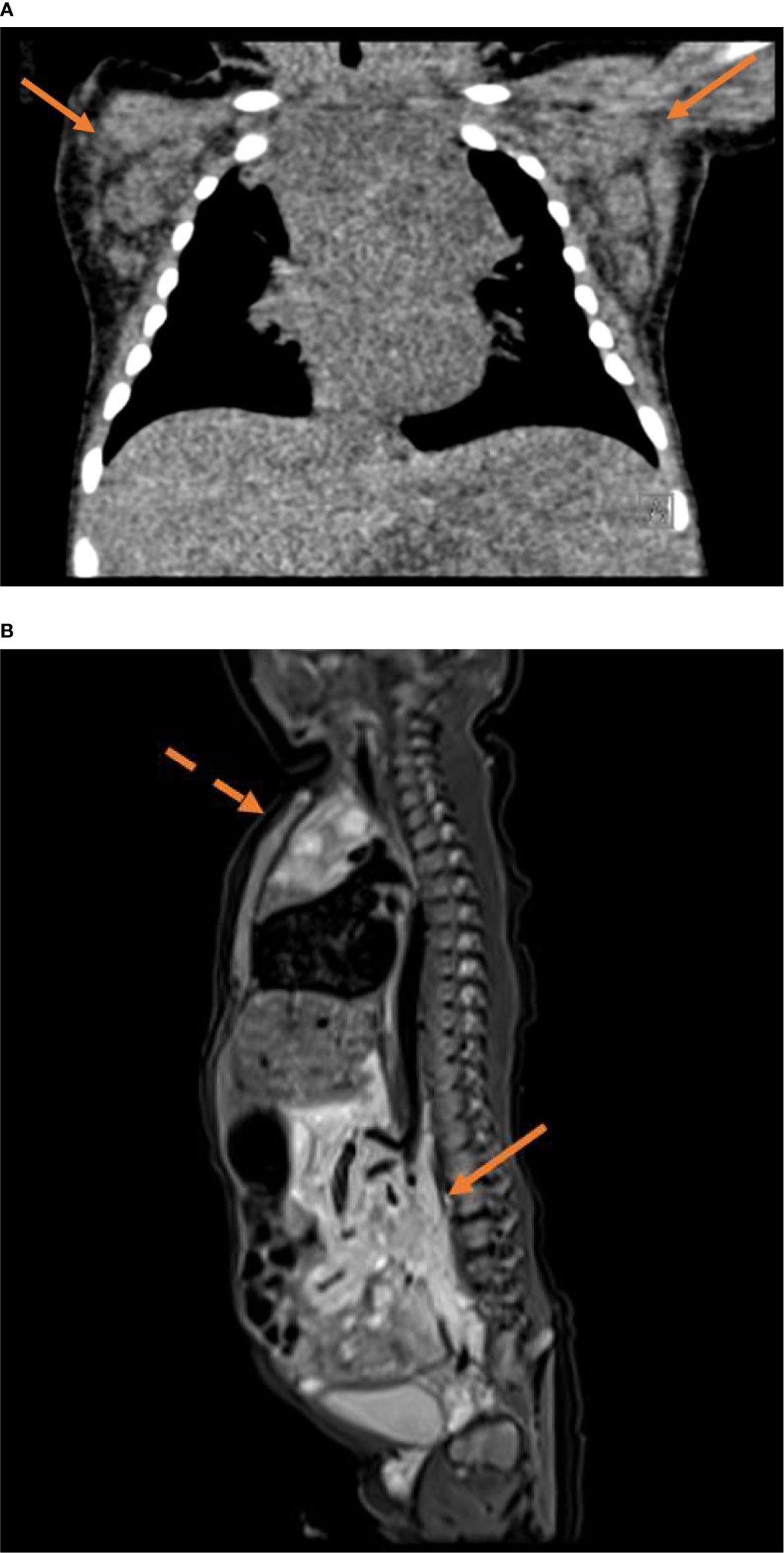
Figure 2 (A) Noncontrast CT (80kV, 48mAs, FOV 170x130mm) with coronal reconstruction using a soft tissue kernel shows a distinct bilateral axillary lymphadenopathy (arrows) and a prominent inhomogeneous thymus. (B) Sagittal T2 STIR sequence of a whole body MRI (TE 33ms, TR 3800ms, FOV 300x300mm, matrix 256x256px) shows bulky hyperintense soft tissue masses surrounding the aortocaval and mesenteric vasculature (solid arrow). The enlarged thymus features multiple jagged-edged cystic lesions (dotted arrow).
A biopsy of the skin and a lymph node revealed granulomatous inflammation with eosinophilic infiltrates. The granuloma showed a central collection of amorphous necrotic but not caseating material of fragmented fibres and prominent multi-nucleated giant cells. No atypical mycobacteria were detected, and CD1a and langerin expression were absent (Figure 3).
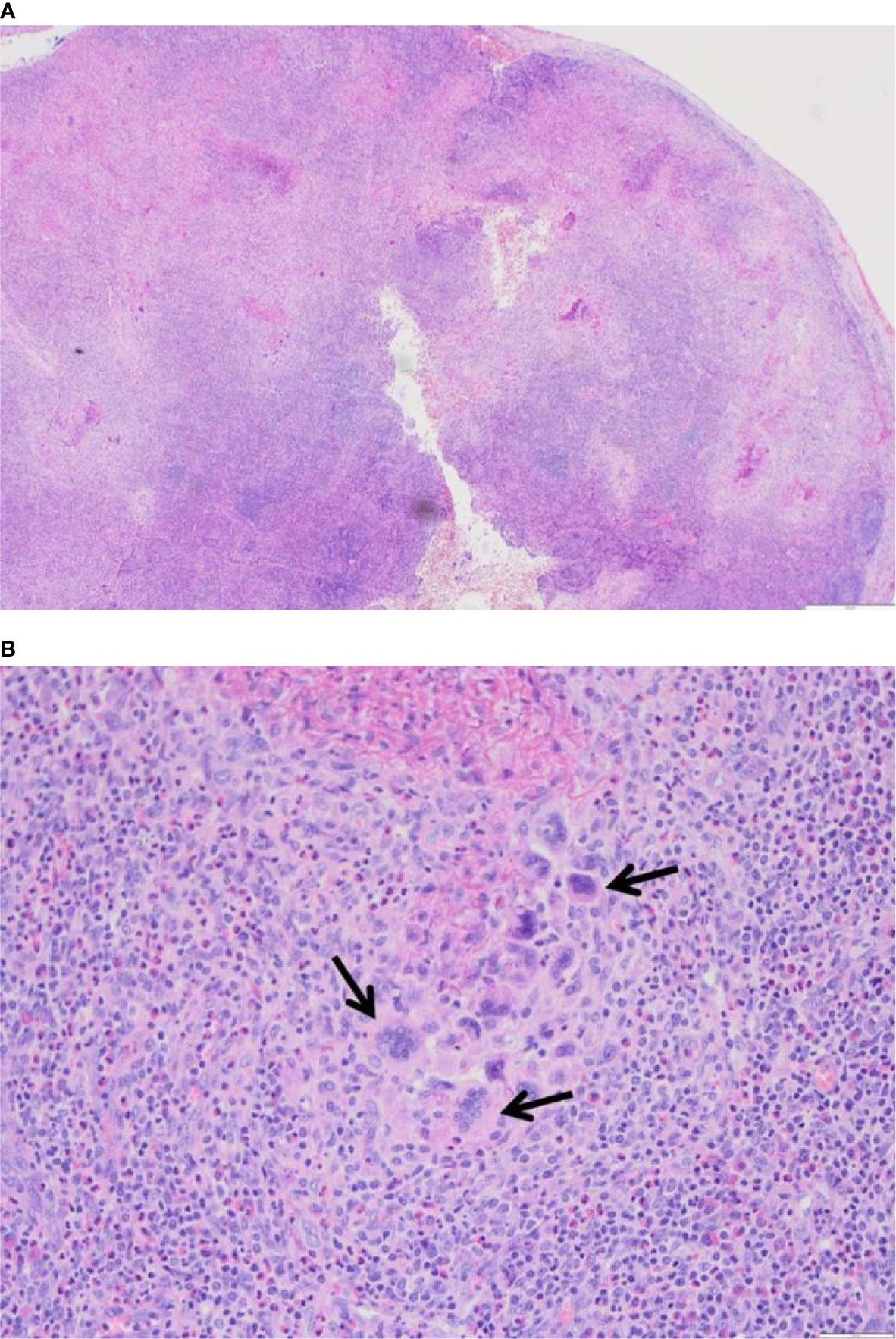
Figure 3 (A) H+E, 4x, Lymph node with severely disturbed architecture by a diffuse necrotizing and granulomatous inflammation. (B) H+E, 4x, Lymph node, close-up of the granulomatous inflammation with abundant multinucleated giant cells (see arrows) in a background of neutrophils and eosinophils.
Additional Immunologic investigations revealed a pathologic function of the NADPH-oxidase (DHR assay 1.40%, normal >98%), and a mutation within the hemizygous CYBB gene (c.742dupA, which results in a premature stop of translation) confirmed the diagnosis for X-linked CGD. This mutation was not found in the half-brother, genetic testing of the mother is planned. Several weeks later, the boy developed pulmonary granuloma due to probable invasive aspergillosis and/or auto-inflammation. Allogeneic hematopoietic stem cell transplantation was performed at the age of 4 months, without major complication during the first three weeks post-transplant.
References without language restriction were retrieved from MEDLINE (including MEDLINE In-Process) database up to February 28, 2021). The search included terms such as neonate, newborn, CGD, and chronic granulomatous disease. Retrieved publications were manually screened for additional references. Patients were included in the analysis if they developed symptoms compatible with CGD within the first six weeks of life, and CGD was diagnosed either by functional tests of neutrophils (including nitroblue tetrazolium test (NBT), dihydrorhodamine (DHR) assay or chemiluminescence test) and/or by genetic analysis. Patients without clinical symptoms detected by screening as family members of CGD-index candidates were not included in the analysis.
The search identified 24 patients, eight girls and sixteen boys (Table 1). Four of the patients were symptomatic already at birth. The diagnosis of CGD was made at an average age of 8 months (range, 1 month to 2 years 8 months), and a mutational analysis was reported in 18 out of the 24 patients. Eleven patients had a mutation in the gp91phox gene, whereas a mutation p67phox was found in three patients and in the p22phox and p47phox gene in two patients each (Table 1). The most common symptoms were respiratory problems (n=13; pulmonary nodules detected by imaging studies in 12 patients), skin lesions such as papules or abscess (11/24 patients) and fever (12/24 patients). Less often, lymphadenopathy (5/24 patients) or gastrointestinal symptoms (5/24 patients) were seen. Most patients had elevated inflammatory parameters (17/24 patients).
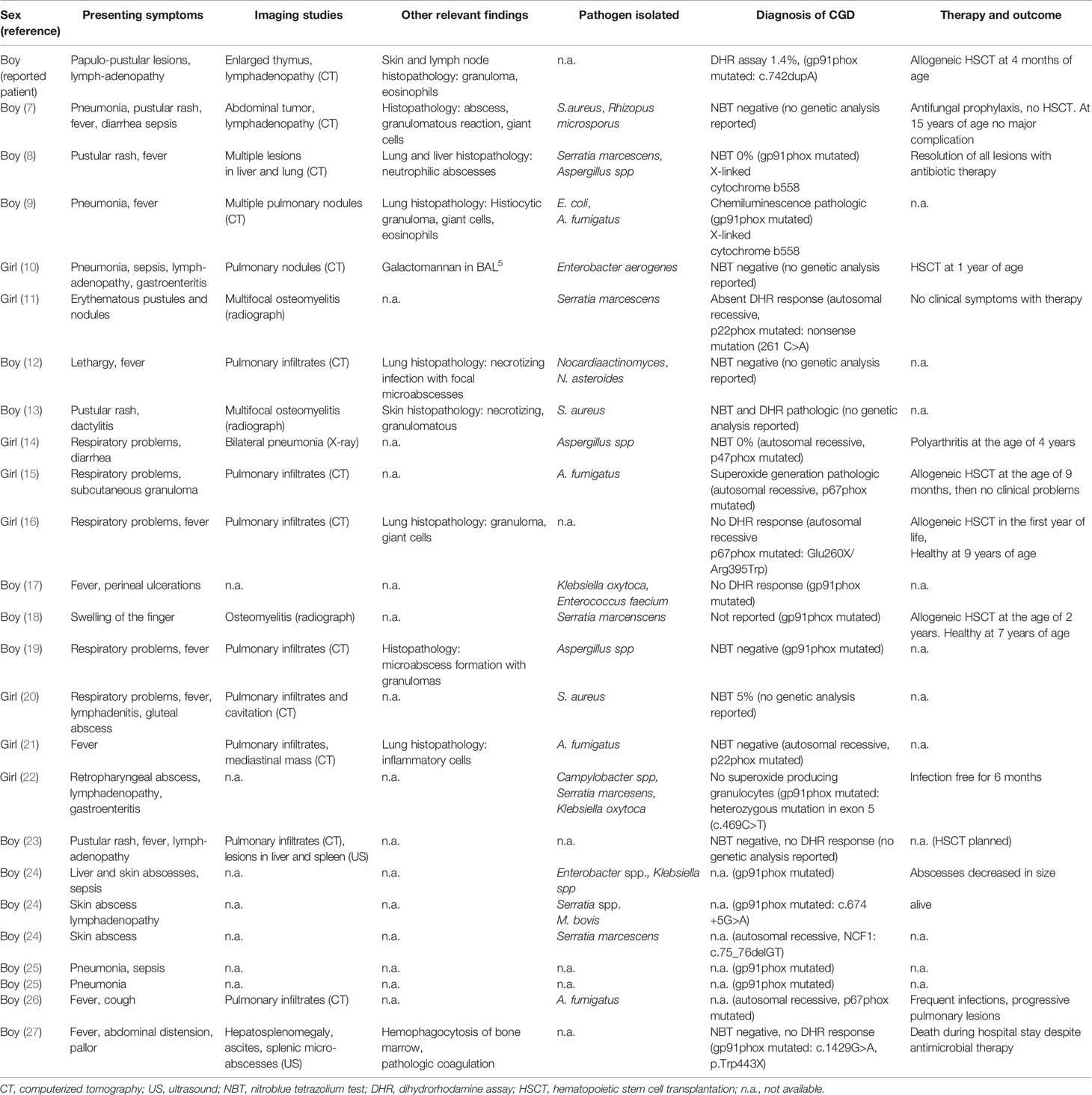
Table 1 Summary of patients reported in the literature with onset of chronic granulomatous disease (CGD) within the first six weeks of life.
In 19 out of the 24 patients, a pathogen was isolated (bacteria in 11 patients, a fungus in 5 patients, both bacterial and fungal pathogens in 3 patients) (Table 2). The most frequent pathogens were Aspergillus spp (7/24 patients). Bacterial infections were mainly caused by Gram- negative bacteria, mostly by Serratia spp. (+6/24 patients).
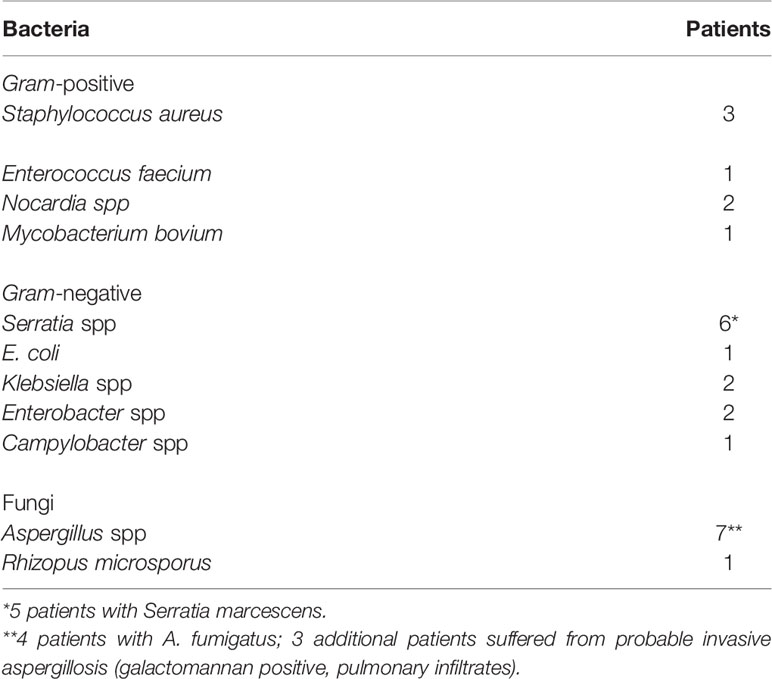
Table 2 Pathogens isolated in 19 patients with onset of chronic granulomatous disease within the first six weeks of life.
Chronic granulomatous disease is a rare primary immunodeficiency which is caused by a defect of the NADPH oxidase (1). Due to the impairment of the phagocytic function, patients have a high risk of bacterial and fungal infections (1). The majority of patients become symptomatic in childhood, but rarely within the first weeks of life (2, 3, 6). As we saw a boy with extended skin lesions caused by CGD already at birth, we thought to review the current literature for patients with extremely early onset of CGD. In total, we identified 24 patients who developed symptoms of CGD within the first six weeks of life. As in our analysis, corresponding studies in older children and adults report on a slight preponderance of boys (2, 3). Corroborating previous reports of older patients, the majority of symptomatic neonates suffered from the X-linked form of CGD, as did our patient (2, 3). Similarly, respiratory problems, skin lesions and fever were the most common initial symptoms of CGD in patients with very early onset of disease, which is comparable to older patients (2, 3). Abscesses are typical skin lesions adults with CGD, whereas in younger children, the lesions are extremely variable (2, 3). It has been reported that life-threatening hemophagocytic lymphohistiocytosis (HLH) presenting with a number of signs and symptoms including persistent fever, hepatosplenomegaly, lymphadenopathy, and low counts of red blood cells, white blood cells and platelets may be the first manifestation of CGD, which is not surprising as any infection can trigger secondary HLH (27–29). In our analysis, erythematous or vesiculo-pustular lesions were found in five patients, and six of them presented with skin abscesses. None of the neonates suffered from extended papulo-pustular and erythematous skin lesions comparable to our patient, which were recently described in two infants of 4 and 9 months of age, respectively (30). Interestingly, these patients had similar histopathologic findings and the same CYBB mutation as our patient (30). Eosinophilic inflammation and an elevated number of eosinophils, as observed in our patient, has been described in patients with X-linked CGD (31). This fact might be due to a compensatory mechanism for the neutrophil defect, as eosinophils may be able to produce gp91phox due to differential regulation of expression of this protein (32). In addition, eosinophilic major basic protein has been shown to activate neutrophils by increasing NADPH oxidase activity, and therefore, one can speculated whether the elevated number of eosinophils are a response to the deficient NADPH oxidase system (31).
In contrast to our newborn patient, a bacterial and/or fungal pathogen was isolated in most of the neonatal patients reported in the literature, with Staphylococcus aureus and Serratia spp as the predominant bacterial pathogens. This observation corroborates the findings in adults patients with CGD that Staphylococcus aureus is the most frequent pathogen causing abscesses and pulmonary infiltrates and that Serratia spp is frequently isolated in subcutaneous abscesses and osteomyelitis (3, 33). Almost half of the patients of our analysis suffered from probable or proven invasive fungal infection, mostly caused by Aspergillus spp. A large US registry including 386 children and adults with CGD reported that over time, invasive aspergillosis was diagnosed in 41% of the patients, and that Aspergillus spp was responsible in 35% of all infections with lethal outcome (3). Interestingly, in none of the patients in our analysis, A. nidulans was isolated, which is the second most encountered mold in CGD patients (34). Due to highly variable symptoms, early diagnosis of CGD in the very young is difficult, which explains the fact that in neonates with CGD, the diagnosis of CGD was made at an average age of 8 months. Newborn screening tests may help to diagnose and treat patients with CGD early, and recently, a robust novel method was reported which allowed the identification of neonates with various primary immunodeficiencies including X-linked CGD (35). Notably, in our patient, the mother suffered from Crohn’s disease, which has been described in female carriers (36), and therefore, genetic testing of the mother is being planned. The fact that the mother was treated with vedolizumab, a humanized monoclonal antibody to α4β7 integrin detected on a specific subpopulation of memory T-lymphocytes for down-regulation of inflammatory processes in the gastrointestinal tract did not explain the symptoms of our patient according to the information given in the literature (37–39). In addition, studies show that vedolizumab levels assessed in cord blood are lower than maternal levels and clear rapidly, with blood levels below the detection limit at 6 weeks after birth (40).
It is important to note that the histopathologic findings of cutaneous granulomas may be indicative of primary immunodeficiency (41), but also feature associations with Crohn´s disease, sarcoidosis, Langerhanscell histiocytosis or tuberculosis (42). Comparable histopathological findings were reported in five out of the 24 patients of our analysis, but in four of them an additional pathogen was detected. Eosinophils were not abundant.
Standard of care of patients with CGD consists of prophylaxis and treatment with antibacterial and antifungals agents (43, 44). However, it is important to note that in the neonatal age group, none of the commonly used broad-spectrum triazoles such as itraconazole or posaconazole is approved nor an adequate dosage has been established (45–47). Similarly, the benefit of interferon-γ, which significantly reduced the incidence of serious infections in a double-blind placebo-controlled study enrolling 128 patients with CGD (median age, 15 years), is not clear at all in the very young age group (48). To date, cure is only achieved by hematopoietic stem cell transplantation (HSCT), which results in a survival rate of more than 80% (43, 49). In our analysis, limited data regarding follow-up was provided for only ten patients. Unfortunately, the information is insufficient for a solid conclusion whether patients with an extremely early onset of CGD have a worse outcome compared to those with a later onset, and is clearly a limitation of this analysis.
Our data demonstrate that unspecific skin lesions and pulmonary symptoms during the first weeks of life may indicate very early onset of CGD. To date, it is unclear whether these patients have a worse prognosis than those which a later onset of the disease.
Chronic granulomatous disease is a life-threatening genetic immunodeficiency, which is diagnosed in the majority of patients between one and three years of age when they become clinically symptomatic. Our patient presented already at birth with unusual skin lesions and lymphadenopathy. A review of the literature revealed only 24 patients who presented with symptomatic CGD within the first weeks of life. Clinical features were extremely heterogenous. As follow-up data of these patients are limited, it remains unclear whether patients with an extremely early onset of CGD have a worse prognosis than those with a later onset of disease.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
MM and TL performed the literature research, analyzed data, and drafted the manuscript. BW, SK, SYZ, SB, TK, and RS analyzed clinical data. SF analyzed radiological data. EG analyzed pathological data. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Rider NL, Jameson MB, Creech CB. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. J Pediatr Infect Dis Soc (2018) 7:S2–5. doi: 10.1093/jpids/piy008
2. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PloS One (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
3. Winkelstein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Med (Baltimore) (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
4. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Med (Baltimore) (2000) 79:170–200. doi: 10.1097/00005792-200005000-00004
5. Matute JD, Arias AA, Dinauer MC, Patiño PJ. p40phox: the last NADPH oxidase subunit. Blood Cells Mol Dis (2005) 35:291–302. doi: 10.1016/j.bcmd.2005.06.010
6. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol (2008) 126:155–64. doi: 10.1016/j.clim.2007.09.008
7. Dekkers R, Verweij PE, Weemaes CM, Severijnen RS, van Krieken JH, Warris A. Gastrointestinal zygomycosis due to Rhizopus microsporus var. rhizopodiformis as a manifestation of chronic granulomatous disease. Med Mycol (2008) 46:491–4. doi: 10.1080/13693780801946577
8. Herman TE, Siegel MJ. Chronic granulomatous disease of childhood: neonatal serratia, hepatic abscesses, and pulmonary aspergillosis. J Perinatol (2002) 22:255–6. doi: 10.1038/sj.jp.7210708
9. Mouy R, Ropert JC, Donadieu J, Hubert P, de Blic J, Revillon Y, et al. Granulomatose septique chronique révélée par une aspergillose pulmonaire néonatale. Arch Pédiatrie (1995) 2:861–4. doi: 10.1016/0929-693X(96)81264-4
10. Lee E, Oh SH, Kwon JW, Kim BJ, Yu J, Park CJ, et al. A case report of chronic granulomatous disease presenting with aspergillus pneumonia in a 2-month old girl. Korean J Pediatr (2010) 53:722–6. doi: 10.3345/kjp.2010.53.6.722
11. McElroy JA, Monahan SH, Williams JV. A female neonate presenting with fever and rash. Clin Pediatr (Phila) (2011) 50:779–81. doi: 10.1177/0009922810379046
12. Johnston HC, Shigeoka AO, Hurley DC, Pysher TJ. Nocardia pneumonia in a neonate with chronic granulomatous disease. Pediatr Infect Dis J (1989) 8:526–8. doi: 10.1097/00006454-198908000-00011
13. Afrough R, Mohseni SS, Sagheb S. An Uncommon Feature of Chronic Granulomatous Disease in a Neonate. Case Rep Infect Dis (2016) 2016:5943783. doi: 10.1155/2016/5943783
14. Lee BW, Yap HK. Polyarthritis resembling juvenile rheumatoid arthritis in a girl with chronic granulomatous disease. Arthritis Rheum (1994) 37:773–6. doi: 10.1002/art.1780370524
15. Shigemura T, Nakazawa Y, Yoshikawa K, Hirabayashi K, Saito S, Kobayashi N, et al. Successful cord blood transplantation after repeated transfusions of unmobilized neutrophils in addition to antifungal treatment in an infant with chronic granulomatous disease complicated by invasive pulmonary aspergillosis. Transfusion (2014) 54:516–21. doi: 10.1111/trf.12325
16. Saito S, Oda A, Kasai M, Minami K, Nagumo H, Shiohara M, et al. A neonatal case of chronic granulomatous disease, initially presented with invasive pulmonary aspergillosis. J Infect Chemother (2014) 20:220–3. doi: 10.1016/j.jiac.2013.10.008
17. Prindaville B, Nopper AJ, Lawrence H, Horii KA. Chronic granulomatous disease presenting with ecthyma gangrenosum in a neonate. J Am Acad Dermatol (2014) 71:e44–5. doi: 10.1016/j.jaad.2013.12.038
18. Salfa I, Cantarutti N, Angelino G, Di Matteo G, Capo V, Farinelli G, et al. Serratia marcescens osteomyelitis in a newborn with chronic granulomatous disease. Pediatr Infect Dis J (2013) 32:926. doi: 10.1097/INF.0b013e31828f682a
19. Davoodi P, Wright SA, Brown EV, Perry JR. Rare diagnosis in a neonate who presents with fever. Clin Pediatr (Phila) (2015) 54:91–3. doi: 10.1177/0009922814541809
20. Narchi H, Gammoh S. Multiple nodular pneumonitis in a three-week-old female infant. Pediatr Infect Dis J (1999) 18:471:485–6. doi: 10.1097/00006454-199905000-00016
21. Chang JH, Boxer LA. Case 2: Infant with lung nodules. Paediatr Child Health (2007) 12:313–6. doi: 10.1093/pch/12.4.229
22. Alberdi T, Morrow MR, Leiding JW. Case Report of an Infant Female with X-Linked Chronic Granulomatous Disease Due to a De Novo Mutation in CYBB and Extremely Skewed X-Chromosome Inactivation (Lyonization). J Allergy Clin Immunol (2016) 137:AB221. doi: 10.1016/j.jaci.2015.12.854
23. Agarwal S. Chronic Granulomatous Disease. J Clin Diagn Res (2015) 9:SD01–2. doi: 10.7860/JCDR/2015/12139.5945
24. Baba LA, Ailal F, El Hafidi N, Hubeau M, Jabot-Hanin F, Benajiba N, et al. Chronic granulomatous disease in Morocco: genetic, immunological, and clinical features of 12 patients from 10 kindreds. J Clin Immunol (2014) 34:452–8. doi: 10.1007/s10875-014-9997-3
25. Hou L, Niu W-T, Ji H-Y, Hu X-F, Fang F, Ying Y-Q. Serum Biomarkers for Early Diagnosis of Chinese X-CGD Children: Case Reports and a Literature Review. Curr Med Sci (2019) 39:343–8. doi: 10.1007/s11596-019-2041-3
26. Guo C, Chen X, Wang J, Liu F, Liang Y, Yang J, et al. Clinical manifestations and genetic analysis of 4 children with chronic granulomatous disease. Med (Baltimore) (2020) 99:e20599. doi: 10.1097/MD.0000000000020599
27. Vignesh P, Loganathan SK, Sudhakar M, Chaudhary H, Rawat A, Sharma M, et al. Hemophagocytic Lymphohistiocytosis in Children with Chronic Granulomatous Disease-Single-Center Experience from North India. J Allergy Clin Immunol Pract (2021) 9:771–82.e3. doi: 10.1016/j.jaip.2020.11.041
28. Valentine G, Thomas TA, Nguyen T, Lai Y-C. Chronic granulomatous disease presenting as hemophagocytic lymphohistiocytosis: a case report. Pediatrics (2014) 134:e1727–30. doi: 10.1542/peds.2014-2175
29. Favara BE. Hemophagocytic lymphohistiocytosis: a hemophagocytic syndrome. Semin Diagn Pathol (1992) 9:63–74.
30. Rajani PS, Slack MA. Papulopustular Dermatitis in X-Linked Chronic Granulomatous Disease. Front Pediatr (2018) 6:429. doi: 10.3389/fped.2018.00429
31. Jaggi P, Freeman AF, Katz BZ. Chronic granulomatous disease presenting with eosinophilic inflammation. Pediatr Infect Dis J (2005) 24:1020–1. doi: 10.1097/01.inf.0000183775.69035.33
32. Weening RS, de Boer M, Kuijpers TW, Neefjes VM, Hack WW, Roos D. Point mutations in the promoter region of the CYBB gene leading to mild chronic granulomatous disease. Clin Exp Immunol (2000) 122:410–7. doi: 10.1046/j.1365-2249.2000.01405.x
33. Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, et al. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. J Pediatr (2000) 137:687–93. doi: 10.1067/mpd.2000.109112
34. Henriet SS, Verweij PE, Warris A. Aspergillus nidulans and chronic granulomatous disease: a unique host-pathogen interaction. J Infect Dis (2012) 206:1128–37. doi: 10.1093/infdis/jis473
35. Collins CJ, Yi F, Dayuha R, Whiteaker JR, Ochs HD, Freeman A, et al. Multiplexed Proteomic Analysis for Diagnosis and Screening of Five Primary Immunodeficiency Disorders From Dried Blood Spots. Front Immunol (2020) 11:464. doi: 10.3389/fimmu.2020.00464
36. Battersby AC, Braggins H, Pearce MS, Cale CM, Burns SO, Hackett S, et al. Inflammatory and autoimmune manifestations in X-linked carriers of chronic granulomatous disease in the United Kingdom. J Allergy Clin Immunol (2017) 140:628–630.e6. doi: 10.1016/j.jaci.2017.02.029
37. Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JW, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med (2005) 352:2499–507. doi: 10.1056/NEJMoa042982
38. Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel J-F, Sands BE, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med (2013) 369:711–21. doi: 10.1056/NEJMoa1215739
39. Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JW, et al. Treatment of active Crohn’s disease with MLN0002, a humanized antibody to the alpha4beta7 integrin. Clin Gastroenterol Hepatol (2008) 6:1370–7. doi: 10.1016/j.cgh.2008.06.007
40. Flanagan E, Gibson PR, Wright EK, Moore GT, Sparrow MP, Connell W, et al. Infliximab, adalimumab and vedolizumab concentrations across pregnancy and vedolizumab concentrations in infants following intrauterine exposure. Aliment Pharmacol Ther (2020) 52:1551–62. doi: 10.1111/apt.16102
41. Harp J, Coggshall K, Ruben BS, Ramírez-Valle F, He SY, Berger TG. Cutaneous granulomas in the setting of primary immunodeficiency: a report of four cases and review of the literature. Int J Dermatol (2015) 54:617–25. doi: 10.1111/ijd.12765
42. Leclerc-Mercier S, Moshous D, Neven B, Mahlaoui N, Martin L, Pellier I, et al. Cutaneous granulomas with primary immunodeficiency in children: a report of 17 new patients and a review of the literature. J Eur Acad Dermatol Venereol (2019) 33:1412–20. doi: 10.1111/jdv.15568
43. Arnold DE, Heimall JR. A Review of Chronic Granulomatous Disease. Adv Ther (2017) 34:2543–57. doi: 10.1007/s12325-017-0636-2
44. Seger RA. Modern management of chronic granulomatous disease. Br J Haematol (2008) 140:255–66. doi: 10.1111/j.1365-2141.2007.06880.x
45. Gallin JI, Alling DW, Malech HL, Wesley R, Koziol D, Marciano B, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med (2003) 348:2416–22. doi: 10.1056/NEJMoa021931
46. Beauté J, Obenga G, Le Mignot L, Mahlaoui N, Bougnoux M-E, Mouy R, et al. Epidemiology and outcome of invasive fungal diseases in patients with chronic granulomatous disease: a multicenter study in France. Pediatr Infect Dis J (2011) 30:57–62. doi: 10.1097/INF.0b013e3181f13b23
47. Welzen ME, Brüggemann RJ, van den Berg JM, Voogt HW, Gilissen JH, Pajkrt D, et al. A twice daily posaconazole dosing algorithm for children with chronic granulomatous disease. Pediatr Infect Dis J (2011) 30:794–7. doi: 10.1097/INF.0b013e3182195808
48. Gallin JI, Malech HL, Weening RS, Curnutte JT, Quie PG, Jaffe HS, et al. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N Engl J Med (1991) 324:509–16. doi: 10.1056/NEJM199102213240801
Keywords: chronic granulomatous disease, neonate, early onset, symptoms, outcome
Citation: Miladinovic M, Wittekindt B, Fischer S, Gradhand E, Kunzmann S, Zimmermann SY, Bakhtiar S, Klingebiel T, Schlösser R and Lehrnbecher T (2021) Case Report: Symptomatic Chronic Granulomatous Disease in the Newborn. Front. Immunol. 12:663883. doi: 10.3389/fimmu.2021.663883
Received: 03 February 2021; Accepted: 08 March 2021;
Published: 29 March 2021.
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
Marco Antonio Yamazaki-Nakashimada, National Institute of Pediatrics, MexicoCopyright © 2021 Miladinovic, Wittekindt, Fischer, Gradhand, Kunzmann, Zimmermann, Bakhtiar, Klingebiel, Schlösser and Lehrnbecher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Lehrnbecher, VGhvbWFzLkxlaHJuYmVjaGVyQGtndS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.