
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 31 March 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.654649
 Chendi Jing1,2†Chenghua Liu1†Yu Liu1†Ruli Feng3Run Cao4Zhangchun Guan1Bo Xuan1Yaping Gao1Qi Wang5Nana Yang6Yuanfang Ma4Lefu Lan6Jiannan Feng1Beifen Shen1Hui Wang5*Yanyan Yu2*Guang Yang1*
Chendi Jing1,2†Chenghua Liu1†Yu Liu1†Ruli Feng3Run Cao4Zhangchun Guan1Bo Xuan1Yaping Gao1Qi Wang5Nana Yang6Yuanfang Ma4Lefu Lan6Jiannan Feng1Beifen Shen1Hui Wang5*Yanyan Yu2*Guang Yang1*Extracellular traps released by neutrophils (NETs) are essential for the clearance of Pseudomonas aeruginosa. Alkaline protease (AprA) secreted by P. aeruginosa negatively correlates with clinical improvement. Moreover, anti-AprA in patients with cystic fibrosis (CF) can help identify patients with aggressive forms of chronic infection. However, the mechanism underlying the clinical outcomes remains unclear. We demonstrated that aprA deficiency in P. aeruginosa decreased the bacterial burden and reduced lung infection. AprA degraded NET components in vitro and in vivo but did not affect NET formation. Importantly, antibodies induced by AprA acted as an agonist and directly enhanced the degrading activities of AprA. Moreover, antisera from patients with P. aeruginosa infection exhibited antibody-dependent enhancement (ADE) similar to that of the antibodies we prepared. Our further investigations showed that the interaction between AprA and the specific antibodies might make the enzyme active sites better exposed, and subsequently enhance the recognition of substrates and accelerate the degradation. Our findings revealed that AprA secreted by P. aeruginosa may aggravate infection by destroying formed NETs, an effect that was further enhanced by its antibodies.
Pseudomonas aeruginosa is an opportunistic gram-negative bacterium that is ubiquitous in the environment. Normally, serious P. aeruginosa infections are prevalent in immunocompromised individuals, including patients with neutropenia, severe burns, cystic fibrosis (CF), and chronic obstructive pulmonary disease (COPD) (1, 2). As a consequence, P. aeruginosa is a leading cause of nosocomial infections and is one of the most common pathogens associated with ventilator-associated pneumonia, which contributes to a high morbidity and mortality in critically ill patients (3). In recent years, therapeutic options have become increasingly limited due to the emergence of multidrug-resistant P. aeruginosa strains, highlighting the fact that this bacterium has become a serious clinical problem (4).
P. aeruginosa secretes a number of extracellular proteases that allow it to escape the host immune attack, such as elastase and alkaline protease (AprA) (5, 6). AprA is a 50-kDa zinc metalloprotease secreted by the type I secretion system. In addition, P. aeruginosa encodes alkaline protease inhibitor (AprI), a specific inhibitor of AprA, that is predicted to be a periplasmic protein and protects self-proteins from degradation by AprA (7). Previous studies have demonstrated that AprA is detected in the majority of human patients with CF infected with P. aeruginosa and that AprA levels correlate inversely with clinical improvement (8, 9). However, the role of AprA in the pathogenesis of P. aeruginosa infection and its precise mechanism remain obscure.
Neutrophils are the most abundant innate immune cells in humans and play a critical role in controlling bacterial infections. In addition to phagocytosis and degranulation, neutrophils can also form neutrophil extracellular traps (NETs) to combat pathogens (10, 11). NETs are extracellular fibrous structures composed of decondensed chromatin, histones, and granular-derived antimicrobial peptides such as cathepsin G, myeloperoxidase (MPO), and neutrophil elastase (NE) (11, 12). It has been demonstrated that P. aeruginosa is able to induce a robust release of NETs from human neutrophils (13, 14). In addition, extensive neutrophilic infiltration and numerous NETs can be detected in most diseases associated with P. aeruginosa lung infection (15–17).
In the present study, we found that AprA degraded the components of NETs and thereby ensured effective immune evasion by P. aeruginosa. Moreover, we discovered that antibodies against AprA acted as an agonist and enhanced the degradation activity of AprA, contributing to the pathogenesis of P. aeruginosa infection.
Trial experiments done previously were used to determine sample size with adequate statistical power. Participants are randomly allocated to each independent variable group. The investigators were blinded to group allocation during data collection and analysis.
The Escherichia coli strains BL21 and DH5α were purchased from Novagen. PAO1 were kindly provided by professor Lefu Lan (Department of Molecular Pharmacology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences). E.coli cells and P. aeruginosa cells were grown in Luria-Bertani (LB) medium at 37°C for 12 h with shaking at 220 rpm.
To construct the aprA mutant strain (PAOaprA-), a SacB-based strategy (18) was employed. Briefly, the upstream fragment of the intended deletion was amplified from PAO1 genomic DNA using the specific primers (D-aprA-up: F 5’- CCCAAGCTTCGGAGAACACCTACAACCAG -3’, R 5’- CGGGATCCGCAGTTATCGGCCAGATCAG -3’), while the downstream fragment was amplified with specific primers (D-aprA-down: F 5’- CGGGATCCCCAGGACAAGATCGACCTGT -3’, R 5’- GGAATTCATGATCGGTCGCTTCGTGGT -3’). The two PCR products were digested and then cloned into vector pEX18Ap, yielding pEX18Ap::aprAUD. Subsequently, a ca. 1.8 kb gentamicin resistance cassette cut from pPS858 with BamHI was cloned into pEX18Ap::aprAUD, yielding pEX18Ap::aprAUGD. The resultant plasmids were electroporated into Wild Type PAO1 with selection for gentamicin resistance. Colonies were screened for resistance to gentamicin and sucrose and sensitivity to carbenicillin, which typically indicates a double-cross-over event and thus of gene replacement occurring. The deletion of aprA in PAO1 was further confirmed by PCR and gene sequencing.
To construct the aprA rescued strain-PAOaprA- (pAK1900-aprA), PCRs were performed to amplify aprA from PAO1 genomic using the specific primers (aprA: F 5’-AACAAGCTTGTGTCAGTTTGGACTTCGGC-3’, R 5’-TTTGGATCCAACCGCGCCCGTGTCAGCGCG-3’). The PCR products were cloned into the vector pAK1900, then the resultant plasmids were electroporated into PAOaprA- with selection for gentamicin resistance. Colonies with dual resistance to gentamicin and carbenicillin were selected on LB agar plates. The PAOaprA- (pAK1900-aprA) strain were further confirmed by PCR and gene sequencing.
The aprA, aprI and flagellin were amplified by PCR, using the genome of PAO1 with the specific primers (aprA: F 5’-CGCGGCAGCATATGGACGAATTGGTCAATGGC-3’, R 5’-CACCACCACTCGAGTCAGACGACGATGTCGG-3’; aprI: F 5’-CACCACCAGCTAGCAGTCTGATTCTTCTCAGCG-3’, R 5’-CACCACCACTCGAGCTAGGGCGTCGCGCGGCGCAGC-3’; flagellin: F 5’-CATGACCAGCTAGCATGGCCCTTACAGTCAACACGAAC-3’, R 5’-TGGTCATGCTCGAGTTAGCGCAGCAGGCTCAGGACCG-3’). The PCR products were cloned into the expression vector pET-28 (a), which were subsequently expressed in E.coli (BL21) and purified using Ni-NTA agarose (GE Healthcare, 17-5318-01) according to manufacturer’s instructions. The site-directed mutagenesis of AprA (ΔAprA) plasmid was constructed by the pET-28 (a)-AprA plasmid with specific primers (F1 5’-GTATGTCAGCGATATCTACGCGCTGGGCGCGTTCAGCGCCTTTTCCGC-3’, R1 5’-AAGGCGAACGCCGCACCGCCGACCGCCGCGCTGAAGTTGCCGAAGGTCA-3’; F2 5’-ATCAACGCGAGCTACAGCGCCAACGTCAATC-3’, R2 5’-TGTAGCTCGCGTTGATCAGGTACCAGGATTGC-3’; F3 5’-GGTGCGGCGTTCGCCTT-3’, R3 5’-GTAGATATCGCTGACATACTTCCAC-3’) via a Fast MultiSite Mutagenesis System (Transgen, Cat. No#FM201-01). The mutant was screened and further confirmed by DNA sequencing. The plasmid with desired mutants was then transformed into E.coli (BL21) for protein expression as described above. Protein concentrations were quantified by use of the bicinchoninic acid protein assay, and proteins were stored at -80°C until use.
HL-60 cells (5x106/ml) were collected and lysed in 1ml hypotonic lysis buffer for 30 min at 4°C. After centrifugation, the samples were resuspended in 400 μl 0.4N H2SO4 and incubated at 4°C for at least 30 min. Then the histones in the supernatant were precipitated with TCA and washed twice with ice-cold acetone. Protein concentrations were quantified as described above.
Ra-anti-AprA was induced after the administration of AprA in Female New Zealand White rabbits after three immunizations with 1mg of AprA mixed with complete Freund’s adjuvant (first immunization) and incomplete Freund’s adjuvant (second and third immunization). Then, the antibody titer and specificity of the sera were analyzed by ELISA and Western blotting. Ra-anti-AprA was purified using an immune-affinity column which was prepared by conjugating protein A according to the manufacturer’s instructions.
Hu-anti-AprA was isolated from a phage display antibody library using the standard procedure (19). In brief, AprA was coated on Nunc-immunotube (Thermo Scientific Nunc®, 444202) at 4°C overnight. Then phages (1012 plaque-forming units [PFU]/ml) were incubated with AprA for 1h, unbound phages were removed. The bound phages were eluted using 0.1M Gly-HCl (pH 2.2). E. coli TG1 cells were infected with the eluted phages. The amplified phages were then subjected to the next round of panning. After three rounds of panning, single colonies were randomly selected and further screened by ELISA. The sequences of the specific phage clones binding to AprA were analyzed.
Human neutrophils were isolated from peripheral blood of healthy volunteers using standard density gradient as described previously (12). Cells were resuspended in complete medium (RPMI 1640 with 2% heat-inactivated human serum), 2x105 neutrophils per well were seeded on the bottom of a cell culture dish and stimulated with PAO1, PAOaprA−, or PAOaprA- (pAK1900-aprA) (MOI = 5, 10, and 50) for 3 h or with PAO1, PAOaprA−, or PAOaprA- (pAK1900-aprA) (MOI = 50) for 30 min, 90 min, and 180 min.
ROS production was measured by dichlorofluorescin diacetate (DCFH-DA, Beyotime). Briefly, neutrophils were incubated with 10 μM DCFH-DA at 37°C for 30 min, cells were then washed and stimulated by different strains of P. aeruginosa (PAO1, PAOaprA-, PAOaprA- (pAK1900-aprA), MOI=50), PMA (100 nM) or were left unstimulated. Finally, samples were analyzed by flow cytometry (BD).
Human neutrophils (2x105 per well) were resuspended in RPMI containing 2% heat-inactivated human serum and seeded into 96 well plates. After stimulated with PMA (100 nM, Sigma) for 30 min at 37°C, the samples were treated with the phagocytosis inhibitor cytochalasin D (10 μg/ml) and different concentrations of AprA (0, 10 μg/ml) or AprI (0, 100 μg/ml). After incubation for 2 h at 37°C, PAO1 were added to the neutrophils at MOI of 2. Then the plates were centrifuged for 10 min at 800 g and followed by incubation at 37°C for 90 min. Finally, the sample suspensions were serially diluted and plated on LB plates to determine the surviving bacterial. The result was normalized to bacterial growth without neutrophils under the same conditions.
Specific pathogen free BALB/c mice (8-week-old females) were inoculated intranasally with different strains of P. aeruginosa (PAO1, PAOaprA-, PAOaprA- (pAK1900-aprA), 5x106 colony forming units (c.f.u.) per mouse) under anesthesia. The body weight was recorded at baseline, 6 h and 24 h post-challenge. Mice were sacrificed at different time points (at 6 h, 24 h and 48 h) post-challenge for further analyzes. Lungs were harvested and homogenized in phosphate-buffered saline plus 0.1% Triton X-100 under sterile conditions, serial dilutions of lysate were plated on LB plates to determine the bacterial load at 6h post-infection.
The experiment was performed as previously described with some modifications (20). Briefly, mice were injected intraperitoneally with neutrophil elastase inhibitor (50 μg per mouse, GW311616A, AxonMedChem, Groningen, Netherlands) or PBS 12 h before P. aeruginosa infection and every 12 h post-challenge until final analyzes. Mice were sacrificed for NETs detection and lung bacterial burden evaluation at 6 h or 24 h post-challenge.
To analyze the neutrophil recruitment in the airways, BALF was collected by lavaging the lung with 1 ml PBS in tubes containing EDTA. After centrifugation, acquired cells were resuspended in flow cytometry buffer, followed by incubation with fluorochrome-conjugated anti-mouse antibodies against CD11b (Biolegend), Gr-1 (Biolegend) for 30 min at RT. Data were collected via flow cytometry (BD) and analyzed using FlowJo software (TreeStar).
To analyze NETs formation in vitro, cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton X-100 prior to blocking with 5% Normal Donkey serum for 1 h at room temperature (RT). The samples were incubated with primary antibodies (goat anti-human/mouse myeloperoxidase/MPO [10 μg/ml Catalog # AF3667], rabbit anti-histone H3 (citrulline R2+ R8 + R17) [1:200 ab5103]) overnight at 4°C. For MPO and Cit-H3 staining, tetramethylrhodamine (TRITC)-conjugated goat anti-rabbit (1:100) and FITC-conjugated Donkey anti-goat antibody (1:100) were added for 1 h at RT, nuclei were stained with DAPI (1:5000) for 10 min at RT. The labeled cells were analyzed with a Carl Zeiss LSM 510 laser scanning confocal microscope.
Immunofluorescence staining of AprA expression and NETs formation in vivo was performed as previously described (21). Briefly, lungs were collected 6 h and 24 h after PA infection and fixed 48 h with 10% formalin. Lung tissues were embedded in paraffin and 5-μm-thick sections were cut for staining. After deparaffinization and rehydration, these sections were boiled for antigen retrieval, then permeabilized in 0.5% Triton X-100 prior to blocking with 4% Normal Donkey serum for 1 h at room temperature (RT). For detection of AprA, samples were stained with Ra-anti-AprA antibody, followed by a secondary tetramethylrhodamine (TRITC)-conjugated goat anti-rabbit antibody. For detection of NETs, lung sections were stained with antibodies described above. The samples were imaged and analyzed by a laser scanning confocal microscope. The immunofluorescence images were quantified using NIH ImageJ analysis software (ImageJ), according to previously established procedures (22).
After collection of human neutrophils and murine lung tissues (6 h and 24 h post-challenge), the samples were homogenized in RIPA lysis buffer containing protease inhibitor cocktails on ice. An equal amount of protein per sample was subjected to 12% SDS-PAGE and the proteins were blotted onto a Hybond-enhanced chemilumescent nitrocellulose membrane, which were then incubated with the primary antibodies (anti-human/mouse myeloperoxidase/MPO (AF3667, R&D), anti-histone H3 (citrulline R2 + R8 + R17) (ab5103, abcam), anti-histone H2A (ab177308, abcam), anti-histone H2B (ab52599, abcam), anti-histone H3 (ab1791, abcam), anti-histone H4 (ab10158, abcam), anti-His (HT501, Transgen Biotech), Ra-anti-AprA, Hu-anti-AprA), then subsequently with appropriate HRP-conjugated secondary antibodies (goat anti-rabbit IgG, goat anti-mouse IgG; rabbit anti-goat IgG,; goat anti-human IgG, Jackson ImmunoResearch Laboratories). Equal loading was confirmed by probing for β-actin (HC201, Transgen Biotech).
Lung tissues were routinely processed for paraffin embedding and were sectioned at 5-μm-thick for hematoxylin-eosin staining at 48 h post-infection. Histology was scored for intraluminal infiltrate, peribronchial infiltrate and alveolar involvement using a scoring system that described previously (23). At least ten representative, non-overlapping fields from a single lung section were independently and blindly evaluated by two pathologist.
AprA protein (10 μg/ml) was coated onto 96-well plates overnight. After washing and blocking, 100 μl of serially diluted Ra-anti-AprA, Hu-anti-AprA, and P1 through P5 were added to the wells respectively. Bound antibodies were detected with HRP-conjugated goat anti-rabbit IgG or HRP-conjugated goat anti-human IgG (Jackson ImmunoResearch Laboratories).
To study the antibodies role in vitro, antibodies (Ra-anti-AprA, Hu-anti-AprA, P1 through P5) were serial diluted and mixed with AprA or ΔAprA for 30 min at RT. Then the mixture were added to the samples (NETs, flagellin) and further incubated for 2 h at 37°C. After incubation, the samples were analyzed using a laser scanning confocal microscope or western blotting as described above.
To evaluate the antibodies role in vivo, mice were injected intraperitoneally with Hu-anti-AprA (100 μg per mouse) or control-IgG 24 h before PAO1 infection. At 6 h post-challenge, mice were sacrificed and the bacterial load in lungs was calculated as described above.
Five patients with P. aeruginosa infection were recruited from Peking University People’s Hospital. Clinical features were obtained from the medical record database of Peking University People’s Hospital. The characteristics of these patients were presented in Table S1, healthy blood donors were served as controls.
All experiments were repeated at least three times and the data were presented as the mean ± SD unless noted otherwise. Data were evaluated using a two-tailed Student’s t test or Mann-Whitney test. A P-value of less than 0.05 was considered to be statistically significant. GraphPad Prism software was used for statistical analyzes.
This study was approved by the ethics committee of the Peking University People’s Hospital (2019PHB077-01) and have been performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed consent was not needed due to that the medical records and patient information were anonymously reviewed and collected in this study. All animal experimental protocols of the study are in accordance with the national guidelines for the use of animals in scientific research “Regulations for the Administration of Affairs Concerning Experimental Animals” and were approved by the Animal Care and Use Committee of Beijing Institute of Pharmacology and Toxicology.
To identify the role of AprA in the pathogenesis of P. aeruginosa, we developed a murine pneumonia model of bacterial infection through the intranasal inoculation of 5 × 106 c.f.u. of different strains of P. aeruginosa, including a wild-type parental PAO1 and an aprA deletion mutant PAOaprA−. As expected, the number of bacteria in the lungs of mice in the PAOaprA− group was lower than that in the parent strain (PAO1) group at 6 h post-challenge (Figure 1A). Histopathological examination of the lungs also showed that the PAOaprA− group exhibited attenuated tissue damage compared to that of the PAO1 group at 48 h post-challenge (Figure 1B), this phenomenon was also confirmed using a quantitative scoring system (23) (Figure 1C). Moreover, the weight loss post-infection for the mice in the PAOaprA− group tended to be less than that of the PAO1 group, although it did not reach a statistically significant difference (Figure S1).
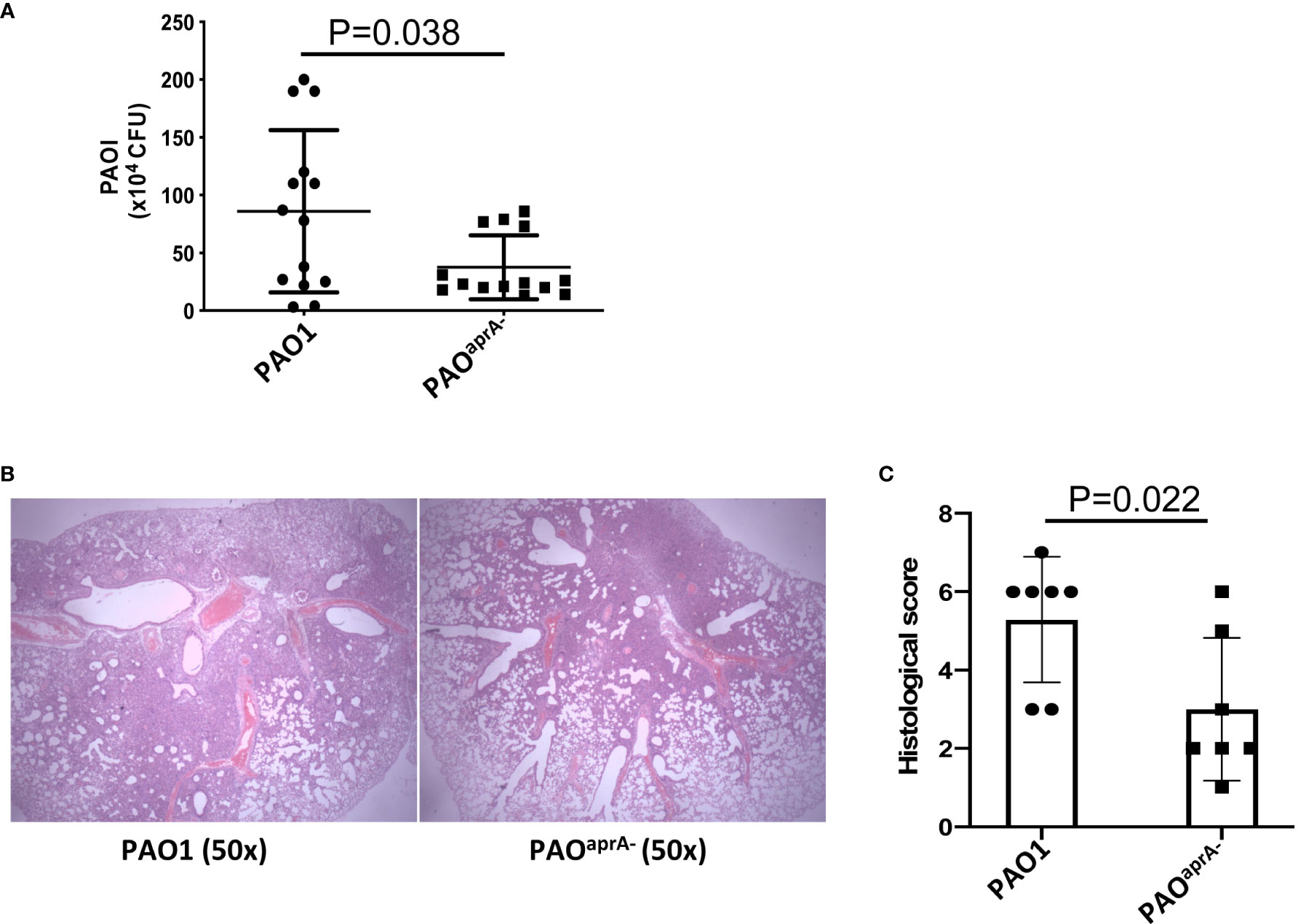
Figure 1 AprA is involved in the lung infection caused by P. aeruginosa. (A) Bacterial burden in the lung tissues of the indicated groups of mice. BALB/c mice (n = 14 per group) were inoculated intranasally with PAO1 or PAOaprA- (5×106 c.f.u. per mouse), respectively. The number of bacteria in lung tissues was evaluated at 6 h post-challenge. (B) Histological examination of lung tissues. Lung specimens were collected 48 h post-challenge and stained with hematoxylin-eosin. (C) Histopathology scores of lung tissues (n=7 per group). All data are representative of three independent experiments. Data in (A, C) are represented as mean ± SD. Significant differences between groups in (A, C) were evaluated using Mann-Whitney test.
To further explore the mechanism of AprA enhancement of P. aeruginosa pathogenesis, we generated an aprA-rescue strain of PAOaprA− using the plasmid pAK1900-aprA. Compared with that in PAO1, the expression level of AprA was absent in PAOaprA−, with expression level being recovered in PAOaprA- (pAK1900-aprA) (Figure S2A, B). Moreover, we also demonstrated that compared with PAO1 and PAOaprA- (pAK1900-aprA), the enzymatic activity of AprA was significantly reduced in the supernatant of PAOaprA- (Figure S2C).
Given that P. aeruginosa is able to induce robust infiltration of neutrophils during lung infections and neutrophils are considered to play an essential role in the clearance of P. aeruginosa (24, 25), we determined whether neutrophil levels were altered for the different P. aeruginosa strains in the murine pneumonia model. Female BALB/c mice (n = 10 per group) were challenged with a sub-lethal dose of PAO1, PAOaprA−, or PAOaprA- (pAK1900-aprA) (5 × 106 c.f.u. per mouse). Bronchoalveolar lavage fluid (BALF) was collected from the mice 6 h post-challenge and neutrophil numbers were evaluated by flow cytometry. There was no significant difference among the three groups of mice (Figure S2D).
As reported in previous studies, neutrophils are also able to release NETs to trap and kill bacteria (10, 11). We suspected that AprA secreted by P. aeruginosa may influence the level of NETs. First, we investigated whether AprA was detectable in vivo after mice were intratracheally challenged with P. aeruginosa. Lung tissue specimens were stained using specific antibody against AprA. As expected, we found that AprA was present in the lung tissues of mice challenged with PAO1 and PAOaprA- (pAK1900-aprA), but not in mice challenged with PAOaprA− (Figures 2A, B). We then determined the level of NETs in the lung tissues by staining for citrullinated histone H3 (Cit-H3), which is considered a hallmark of NETs (26, 27). Laser scanning confocal microscopy revealed that Cit-H3 levels were increased in the lung tissues of PAOaprA−-infected mice compared to those in PAO1-infected and PAOaprA- (pAK1900-aprA)-infected counterparts (Figures 2C, D). NET levels were also measured by western blotting using an anti-Cit-H3 antibody. Consistent with the confocal microscopy results, it was demonstrated that NETs were more abundantly present in lung tissues of PAOaprA–infected mice than in lung tissues of the other two groups (Figure 2E).
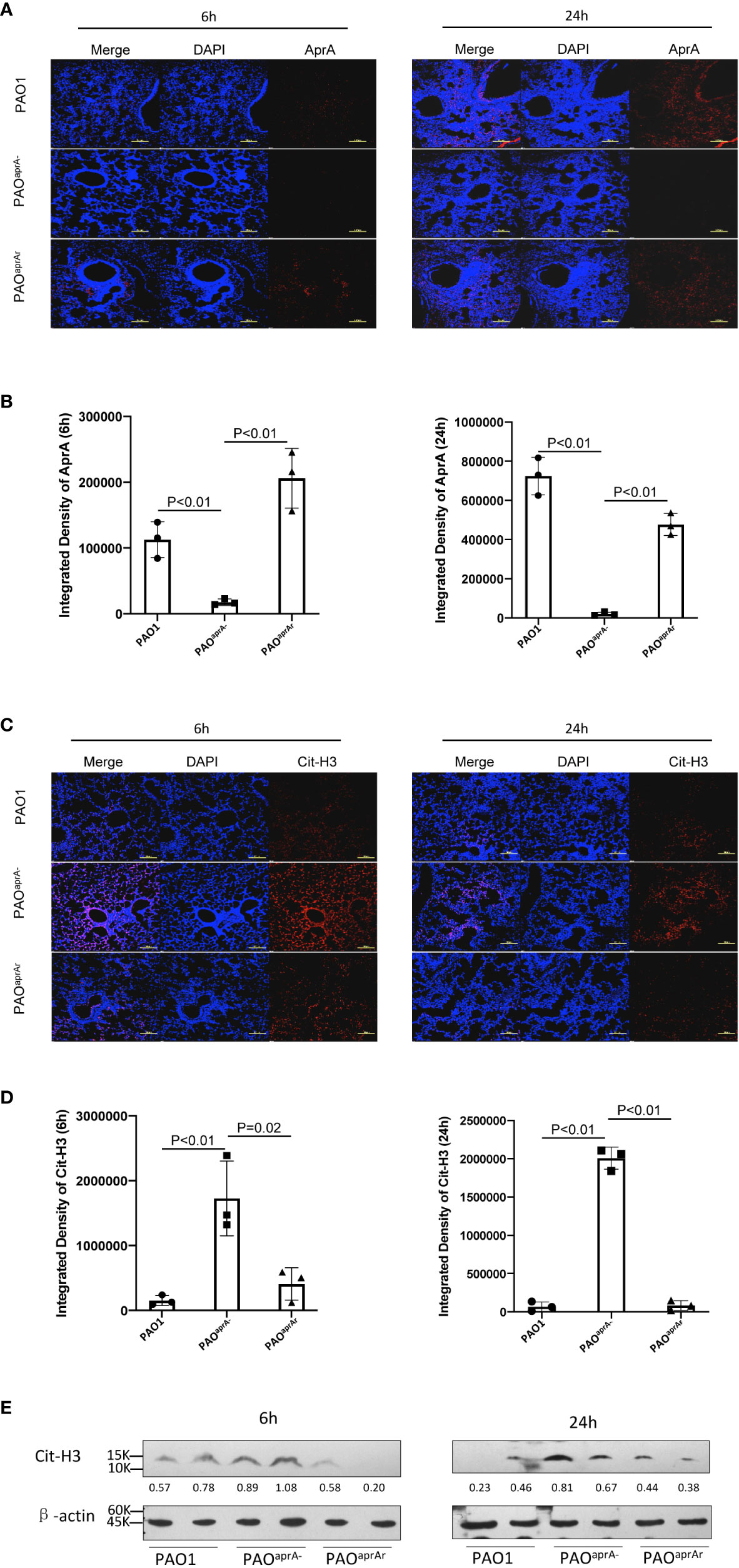
Figure 2 Deficiency of aprA increased NET levels in vivo. (A–D) Confocal scanning laser microscopic analysis of AprA and Cit-H3 in lung tissues of the indicated groups of mice (n = 10 per group). Lung tissue specimens were fixed at 6 h and 24 h post-challenge and stained using rabbit anti-AprA antibody (A) or rabbit anti-histone H3 (citrulline R2 + R8 + R17) antibody (C), followed by a secondary tetramethylrhodamine (TRITC)-conjugated goat anti-rabbit antibody (red). Confocal laser microscopy, ×20. Scale bars, 100 μm. (B, D) Quantitation of immunofluorescence images shown in (A, C). (E) The Cit-H3 levels in lung tissues of the indicated groups of mice were observed by western blotting. Lung tissues were homogenized at 6 h and 24 h post-challenge. The lysates were analyzed by western blotting using anti-β-actin and anti-histone H3 (citrulline R2 + R8 + R17) antibodies. All data are representative of three independent experiments. Data in (B, D) are represented as mean ± SD. Significant differences between groups were evaluated using two-tailed Student’s t tests.
To further investigate the mechanism of decreased NET levels in vivo, we first determined whether aprA deficiency affected NET formation. Previous studies have shown that reactive oxygen species (ROS) were released during P. aeruginosa-induced or phorbol myristate acetate (PMA)-induced NET formation (12, 28). Human neutrophils were isolated and incubated with P. aeruginosa (PAO1, PAOaprA−, or PAOaprA- (pAK1900-aprA)) at a multiplicity of infection (MOI) of 50. ROS production was measured using flow cytometry, which showed that P. aeruginosa-triggered neutrophils released large amounts of ROS, but no significant difference was observed among the groups (Figures 3A, B). In addition, NET formation induced by the different P. aeruginosa strains was also detected by immunofluorescence. We found that PAO1, PAOaprA−, and PAOaprA- (pAK1900-aprA) each stimulated NET formation in dose-dependent and time-dependent manners with no significant difference being observed among the strains (Figures 3C, D, Figure S3A, B). These results suggested that aprA deficiency had no significant effect on NET formation.
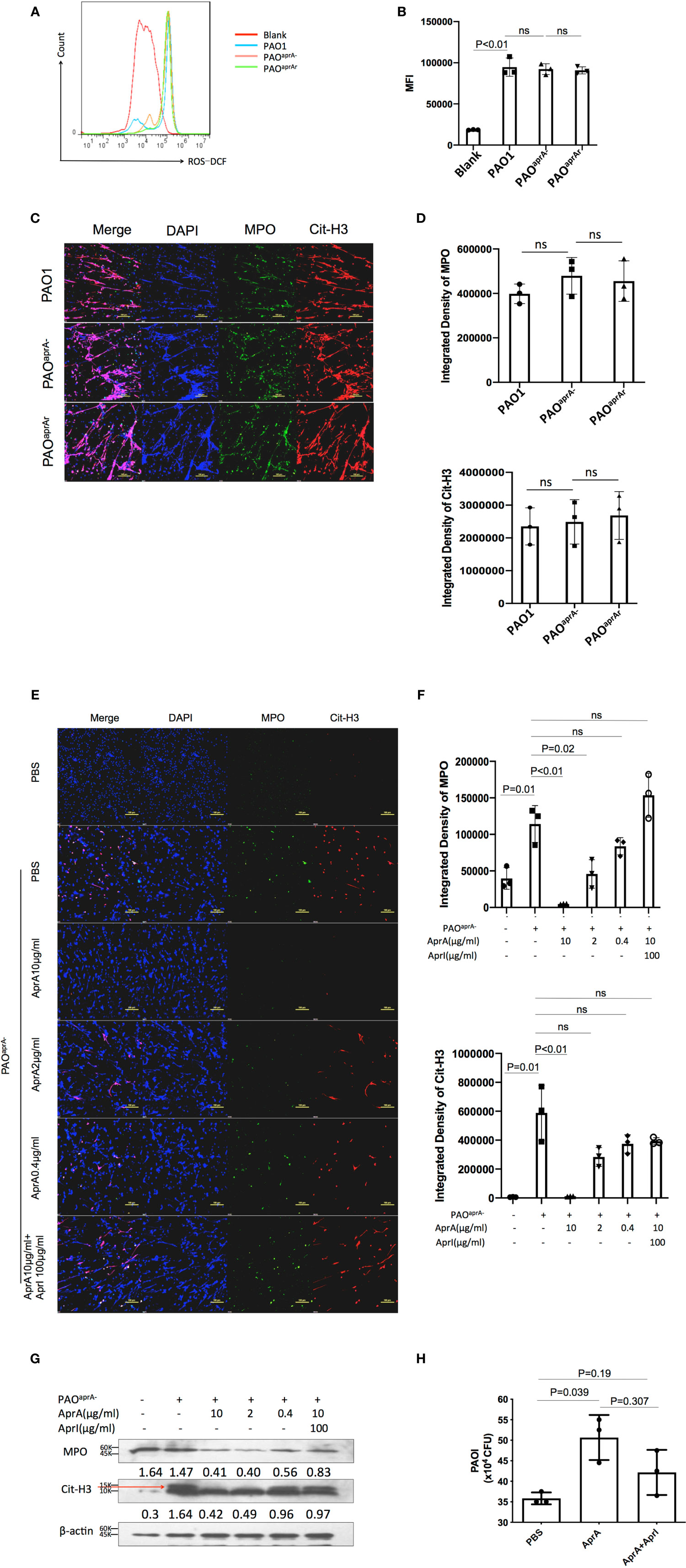
Figure 3 AprA degraded NET components but did not inhibit NET formation. Representative (A) or averages (B) of ROS production measured by flow cytometry (n=3). Human neutrophils were isolated and incubated with P. aeruginosa strains PAO1, PAOaprA−, or PAOaprA- (pAK1900-aprA) (MOI=50). ROS production was detected using dichlorofluorescin dacetate (DCFH-DA). (C, D) Confocal scanning laser microscopic analysis of NET formation. (C) Neutrophils were isolated and stimulated with PAO1, PAOaprA−, or PAOaprA- (pAK1900-aprA) (MOI = 50) for 3 h. Confocal laser microscopy, ×20. Scale bars, 100 μm. (D) Quantitation of immunofluorescence images shown in (C). (E–G) Neutrophils were stimulated with PAOaprA− (MOI = 50) for 3 h and then incubated with various concentrations of AprA and AprI for 2 h. NET components were then quantified by immunostaining (E) and western blotting (G). Confocal laser microscopy, ×20. Scale bars, 100 μm. (F) Quantitation of immunofluorescence images shown in (E). (H) The effect of AprA on NET-mediated bacterial killing. The surviving bacteria were counted, and the results normalized to the bacterial growth under the same conditions without neutrophils. All data are representative of three independent experiments. Data in (B, D, F, H) are represented as mean ± SD. Significant differences between groups were evaluated using two-tailed Student’s t tests ns, not significant.
Considering that aprA deficiency induced elevated levels of Cit-H3, and AprA is a type of metalloprotease, we investigated whether AprA directly degraded Cit-H3 via its proteolytic activity. We first expressed and purified AprA, AprI, and flagellin in Escherichia coli. The activity of AprA was determined by incubation with flagellin in the presence and absence of AprI. Consistent with a previous study (29), AprA was found to efficiently degrade flagellin, which was blocked by AprI (Figure S3C). We then determined whether the purified AprA affected the formation of NETs. Human neutrophils were stimulated with PMA in the presence and absence of AprA. As expected, ROS production was not affected by AprA during PMA-induced NET formation (Figure S3D).
In further investigations, human neutrophils were incubated with PAOaprA− or PMA for 3 h. Different concentrations of AprA were then added in the presence and absence of AprI. Components of NETs were stained using specific antibodies and detected by immunofluorescent labeling. The results of laser scanning confocal microscopy revealed that Cit-H3 was dramatically degraded by AprA in a concentration-dependent manner and this proteolytic activity of AprA was neutralized by AprI. Moreover, we found that MPO, another major component of NETs, was also degraded by AprA (Figures 3E, F, Figure S3E). These findings were confirmed by western blotting (Figure 3G). It has been demonstrated previously that histone H3 is citrullinated by peptidylarginine deiminase 4 (PAD4) during the formation of NETs (26, 27). Given our observed degradation of Cit-H3, we investigated whether AprA degraded only the modified form of histone H3. Histones from HL-60 cells were extracted and incubated with AprA. It was determined that AprA also efficiently degraded non-citrullinated histone H3. Moreover, we observed that other histones (H2A, H2B, and H4) were also degraded by AprA in a time-dependent manner (Figure S3F–H).
As histones and MPO have been previously shown to play an important role in NET antimicrobial activity (11), we examined whether the addition of AprA affected NET-mediated bacterial killing. We determined that the presence of AprA attenuated the bactericidal ability of NETs, which tended to be neutralized by AprI, although it did not reach a statistically significant difference (Figure 3H).
As demonstrated above, AprA effectively degraded NET components and attenuated NET-mediated bacterial killing. To assess the contribution of these findings in the pathogenesis of P. aeruginosa, we used neutrophil elastase inhibitor (NEi) to inhibit NET formation in the murine pneumonia model as described previously (20). We determined that NEi-treated mice exhibited a reduction in NET components compared to that of PBS-treated counterparts (Figures 4A, B).
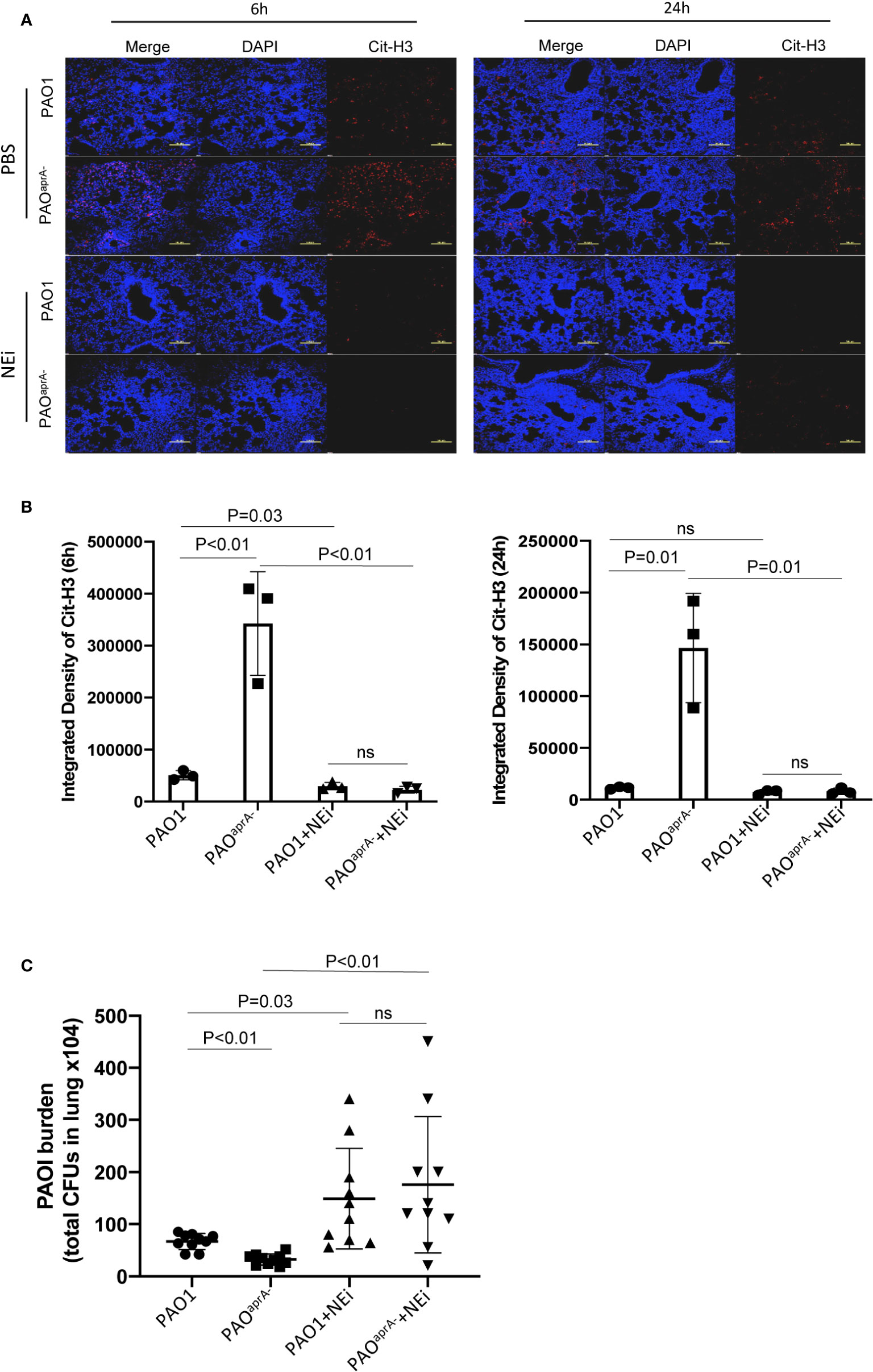
Figure 4 NETs play an important role in murine lung infection caused by P. aeruginosa. (A) Confocal scanning laser microscopic analysis of Cit-H3 in lung tissues of the indicated groups of mice. Mice (n = 16 per group) were injected with NEi [50 μg per mouse) or PBS 12 h before P. aeruginosa infection and every 12 h post-challenge. Lung tissue sections were obtained at 6 h and 24 h post-challenge. Confocal laser microscopy, ×20. Scale bars, 100 μm. (B) Quantitation of immunofluorescence images shown in (A). (C) Bacterial burden in lung tissues of the indicated groups of mice. The number of bacteria in lung tissues was evaluated at 6 h post-challenge. All data are representative of three independent experiments. Data in (B) are represented as mean ± SD. Significant differences between groups were evaluated using two-tailed Student’s t tests. ns, not significant.
The bacterial burden in lung tissues was also evaluated. Notably, the administration of NEi increased the number of bacteria in the lung tissues. Although the bacterial burden was higher in the PAO1 group than in the PAOaprA− group of PBS-treated mice, no statistically significant difference was observed between the two P. aeruginosa strains in NEi-treated mice (Figure 4C).
To further validate the direct effect of AprA in our mouse pneumonia model, mice were treated with AprA or PBS before infected with PAOaprA-. Consistent with our previous results, AprA could efficiently degraded NETs components, and accompanied with higher levels of bacterial burden. However, no significant difference was observed between the two groups with administration of NEi (Figure S4A and B ).
Since anti-AprA can be detected in patients with P. aeruginosa infection (30), we investigated whether anti-AprA could neutralize the degradation activity of AprA. First, we prepared rabbit polyclonal antibodies (Ra-anti-AprA) by immunization of the rabbit with AprA. The interaction between AprA and Ra-anti-AprA was evaluated by enzyme-linked immunosorbent assay (ELISA) and western blotting (Figure S5A, C). We then evaluated the effect of Ra-anti-AprA on the enzymatic activities of AprA. Surprisingly, we determined that Ra-anti-AprA did not inhibit, but rather enhanced, the degradation of flagellin by AprA (Figure 5A, Figure S5E). We then determined the effect of Ra-anti-AprA on the AprA-mediated degradation of NET components. After NET formation, Ra-anti-AprA increased the degradation of Cit-H3 and MPO in a dose-dependent manner in the presence of AprA (Figures 5B, C, F).
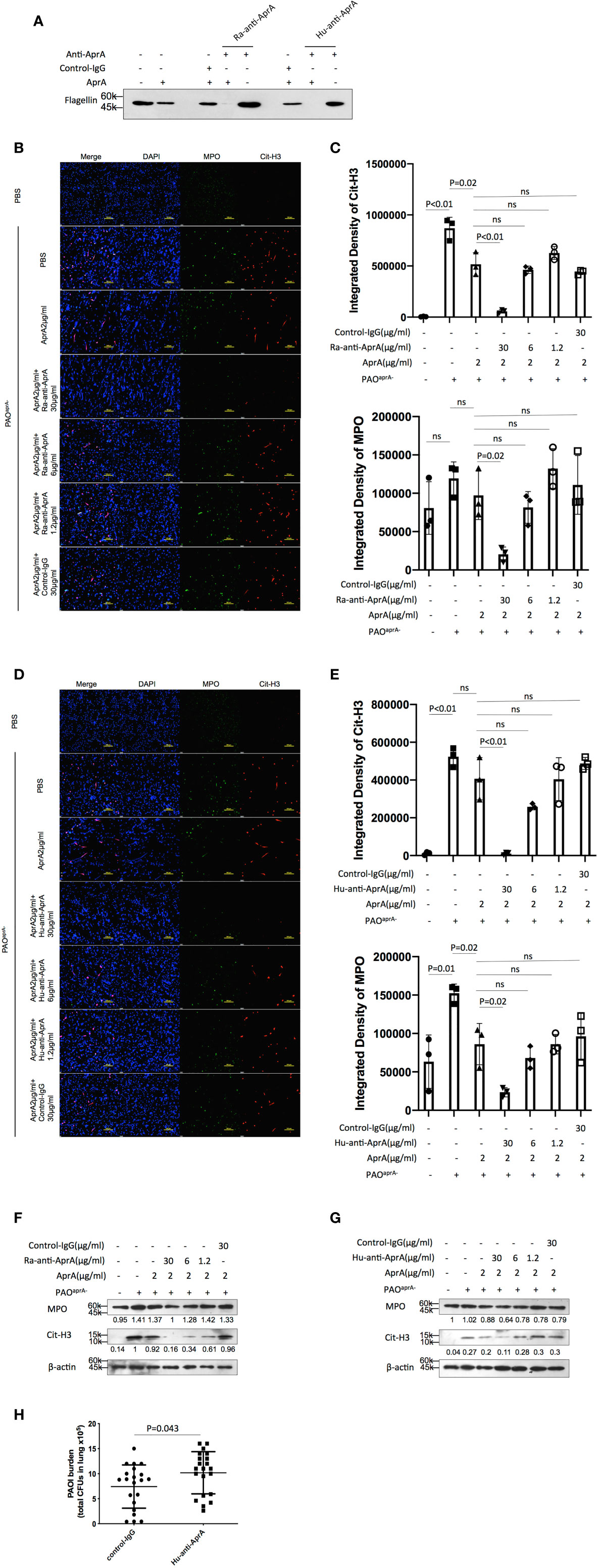
Figure 5 Antibody-dependent enhancement (ADE) of P. aeruginosa infection. (A) The effect of Ra-anti-AprA and Hu-anti-AprA on the degradation of flagellin mediated by AprA. The samples were analyzed by western blotting using anti-His antibody. (B–G) The effect of Ra-anti-AprA and Hu-anti-AprA on the degradation of NET components mediated by AprA. The samples were analyzed using a laser scanning confocal microscope (B, D) or western blotting (F, G). Confocal laser microscopy, ×20. Scale bars, 100 μm. (C, E) Quantitation of immunofluorescence images shown in (B, D). (H) Bacterial burden in lung tissues of the indicated groups of mice. Mice (n = 21 per group) were injected intraperitoneally with Hu-anti-AprA (100 μg per mouse) or control-IgG 24 h before PAO1 infection. The bacterial load in the lungs was calculated at 6 h post-challenge. All data are representative of three independent experiments. Data in (C, E, H) are represented as mean ± SD. Significant differences between groups were evaluated using two-tailed Student’s t tests. ns, not significant.
We then screened for the specific full human monoclonal antibody against AprA from human antigen binding fragment (Fab) libraries using phage display. After three rounds of screening, a specific antibody was generated and named Hu-anti-AprA. The interaction between AprA and Hu-anti-AprA was evaluated by ELISA and western blotting (Figure S5B, D). We then determined whether Hu-anti-AprA was able to neutralize the enzymatic activities of AprA. After incubation with Hu-anti-AprA, AprA-mediated degradation of flagellin and NET components was increased (Figures 5A, D, E, G and Figure S5F), suggesting that Hu-anti-AprA was also an agonist of AprA, similar to what was observed for Ra-anti-AprA.
To further assess the effect of Hu-anti-AprA in the murine pneumonia model using P. aeruginosa, mice were injected with Hu-anti-AprA or control-IgG 12 h prior to intranasal administration of PAO1. As expected, mice pretreated with Hu-anti-AprA exhibited increased numbers of bacteria in their lung tissues compared with those of the control mice (Figure 5H).
In subsequent studies, antibodies were purified from the individual sera of five patients with P. aeruginosa infection and named P1 through P5, respectively. The level of anti-AprA was determined by ELISA (Figure 6A). We then tested the effect of P1 through P5 on the enzymatic activities of AprA. We determined that P5 significantly enhanced the AprA-mediated degradation of flagellin, which correlated with its higher anti-AprA titer (Figure 6B, Figure S5G). Similar results were also observed for the AprA-mediated degradation of NET components (Figures 6C, D).
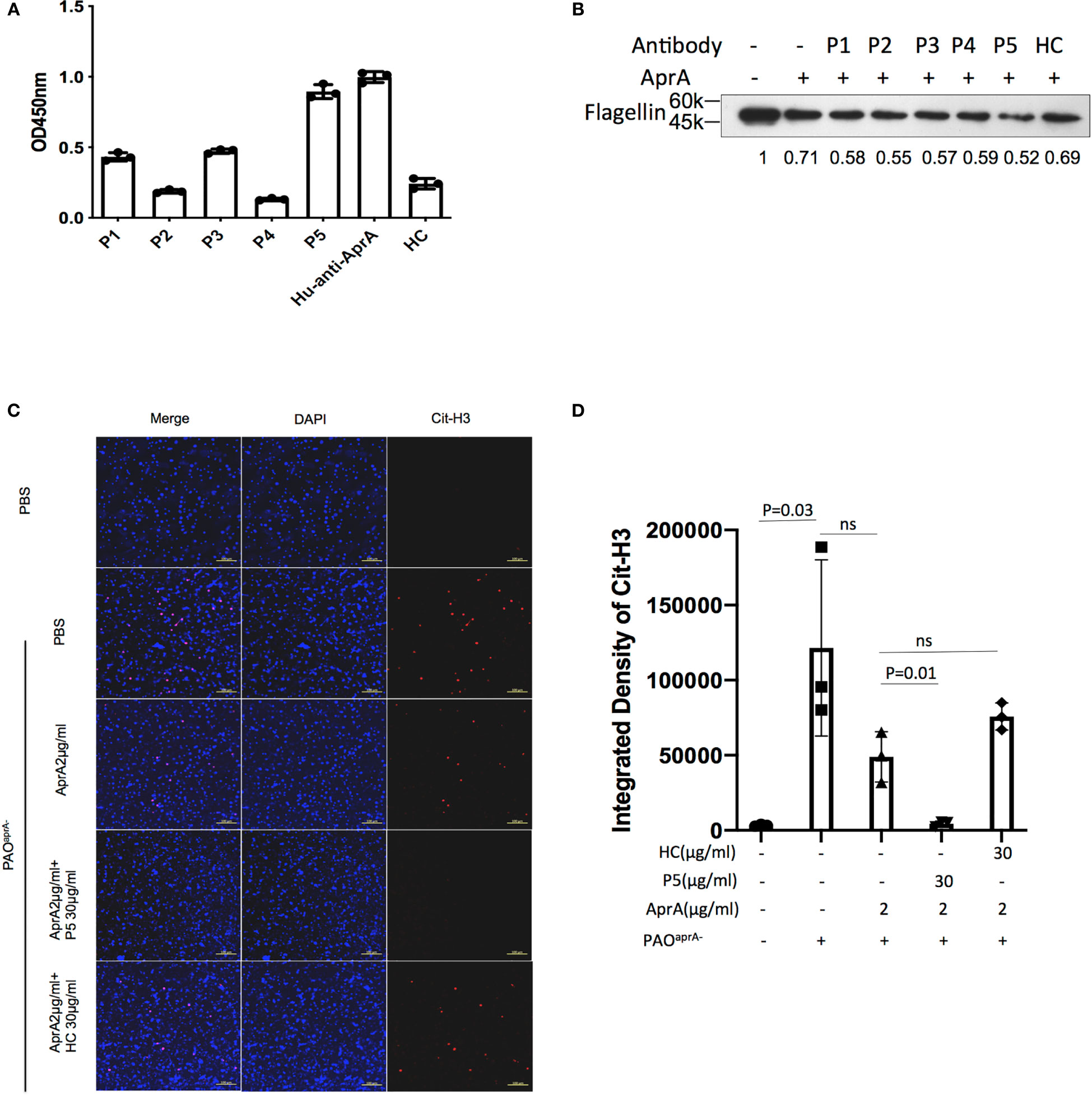
Figure 6 The effect of antibodies from patients with P. aeruginosa infection on AprA activities. (A) The levels of anti-AprA in P1 through P5 were detected by ELISA. AprA protein (10 μg/ml) was coated onto 96-well plates. P1 through P5 (10 μg/ml) were added to each well and then visualized using horseradish peroxidase (HRP)-conjugated goat anti-human IgG. (B) The effect of P1 through P5 on the degradation of flagellin mediated by AprA. The samples were analyzed by western blotting using anti-His antibody. (C) The effect of P5 on the degradation of NET components mediated by AprA. The samples were analyzed using a laser scanning confocal microscope. Confocal laser microscopy, ×20. Scale bars, 100 μm. (D) Quantitation of immunofluorescence images shown in (C). All data are representative of three independent experiments. Data in (A, D) are represented as mean ± SD. Significant differences between groups were evaluated using two-tailed Student’s t tests. HC, healthy control. ns, not significant.
Based on the results given above, we speculated that ADE effect of anti-AprA was dependent on AprA enzyme activity. To test this hypothesis, we used the computer-guided homology modeling method and predicted the potential enzyme active sites (Ser90, Lys93, Ser138, Ser139 and Ser165) of AprA. Then we constructed and purified the mutant AprA protein (ΔAprA) with substitution of all the five amino acids with Ala. It was shown that the degradation of flagellin by ΔAprA was decreased compared with AprA (Figure S5H). In the further investigation, we detected whether Hu-anti-AprA enhanced the activities of ΔAprA. The results showed that Hu-anti-AprA recognized the ΔAprA as well as AprA (Figure S5I). However, compared with AprA, the enhancing activity of Hu-anti-AprA was disappeared when incubated with ΔAprA (Figure 7A).

Figure 7 The potential mechanism of ADE effect of antibodies against AprA. (A) The effect of Hu-anti-AprA on the degradation of flagellin mediated by AprA or ΔAprA. The samples were analyzed by western blotting using anti-His antibody. (B, C) The distance of the flagellin binding pocket in AprA (B) or the complex of Hu-anti-AprA-AprA (C). Data in (A) is representative of three independent experiments.
To investigate how Hu-anti-AprA increased enzymatic activities of AprA, we constructed the complex structure of the Hu-anti-AprA-AprA. With the assistance of computer-guided homology modeling, the 3-D structures of the variable region of Hu-anti-AprA heavy and light chain (i.e. VH and VL domain) were modeled and optimized. After the 3-D structure model of the agonist antibody Fv fragment was constructed, the spatial complex structure of AprA and its agonist antibody Fv was obtained theoretically. The potential key domain and important residues identified by Hu-anti-AprA were predicted using the theoretical model (Figure S6A) and the key amino acids in antibody CDR (Arg11, Asp13, and Glu31) domain and AprA (Asn230, Thr231, Asn20, Ser12, Gln10, and Thr16) were demonstrated (Figure S6B).
Furthermore, we theoretically analyzed the conformational changes that occurred while the antibody binds to AprA. Based on the distance geometry method, the distance of the flagellin binding pocket in AprA (i.e., the distance between the main chain carbon atoms of Ser90 and Ser165) was calculated. When Hu-anti-AprA bound to AprA, the distance of the substrate-binding pocket changed from 18.29Å to 20.65Å (Figures 7B, C). These results suggested that the binding activity between AprA and flagellin could be strengthened when Hu-anti-AprA, the agonistic antibody, binding AprA. Furthermore, the solvent-accessible area of the binding pocket of AprA identified by its substrate was increased to about 82.5 Å2 when AprA bound to its agonist antibody.
Recently, NETs have been identified in several lung diseases associated with P. aeruginosa, suggesting that NETs can be induced in vivo after P. aeruginosa infection and may therefore contribute to host immune defenses (15–17, 31, 32). Previous studies reported that some bacteria have evolved strategies to escape being trapped and killed by NETs. For instance, by producing a nuclease, Staphylococcus aureus is able to degrade the DNA backbone and reduce its entrapment by NETs (33). In addition, Group A Streptococcus (GAS) is able to suppress NET formation by expressing streptococcal collagen-like protein 1 (Scl-1) (34). Other researchers have shown that P. aeruginosa isolated from patients with CF also has the ability to escape NET-mediated killing, but the precise mechanisms have not been elucidated (17). In the current study, we identified a potential mechanism for P. aeruginosa to escape NET-related immune clearance. Using a mouse pneumonia model, we determined that AprA levels were inversely correlated with NET components. Moreover, AprA was found to efficiently degrade Cit-H3 and MPO, which are known to coat DNA fibers to kill the entrapped bacteria, thereby suppressing the bactericidal ability of NETs in vitro. We also provided direct evidence that AprA-mediated degradation of NET components plays a critical role in lung infections caused by P. aeruginosa, which may partly explain the observation that AprA levels are negatively correlated with the prognosis of patients with P. aeruginosa infection.
Heterologous antigens can induce the activation of adaptive immunity to generate specific antibodies that typically neutralize the functions of the particular antigens. However, previous works reported that the detection of anti-AprA in patients with CF may help identify patients with aggressive forms of chronic infection (30, 35), which suggests that AprA-induced antibodies may be detrimental to the process of P. aeruginosa infection. Here, we demonstrated that anti-AprA in the serum of a rabbit immunized with AprA or in the serum of patients with P. aeruginosa infection increased the ability of AprA to degrade NET components. These results indicated that antibodies induced by AprA may enhance, not block, the activities of this enzyme. Moreover, we determined that AprA-specific antibody aggravated the infection caused by P. aeruginosa in the murine pneumonia model.
ADE of infection has been identified for numerous viruses, including Dengue virus (DENV), Ebola virus, human immunodeficiency virus, Zika virus, and measles virus (36–39). It has been determined in primary human macrophages that antibody-mediated cell entry of DENV enhances the fusion potential of the virus (40). However, the ADE of viral infection is obviously different from our current observations for P. aeruginosa infection in that the AprA-specific antibody in our study directly enhanced the activities of the antigen (AprA). Our results demonstrated that the ADE of anti-AprA was dependent on AprA enzyme activity. We speculated that the combination of AprA and anti-AprA could change the conformation of AprA, and make the enzyme active sites better exposed. More details of the enhancement of AprA activities mediated by Hu-anti-AprA will be investigated in the future.
In addition to flagellin, Cit-H3, MPO, and histones, previous studies have demonstrated that several proteins of the host immune system, such as complement C1q, C2, and C3, and cytokines like IFN-γ and TNF-α, are also substrates of AprA (29, 41, 42). Antibodies against AprA may also accelerate the degradation of these substrates and contribute further to the pathogenesis of P. aeruginosa. This will be investigated in future studies. Moreover, whether antibodies against other P. aeruginosa enzymes have similar ADE effects also needs to be addressed in the future.
In conclusion, our findings provided insight into two aspects of the effect of AprA on the pathogenesis of P. aeruginosa (Figure 8). First, AprA degraded NET components to help mediate immune evasion by P. aeruginosa. Second, AprA induced the production of antibodies that acted as agonists to enhance the activities of AprA and further exacerbate the P. aeruginosa infection.
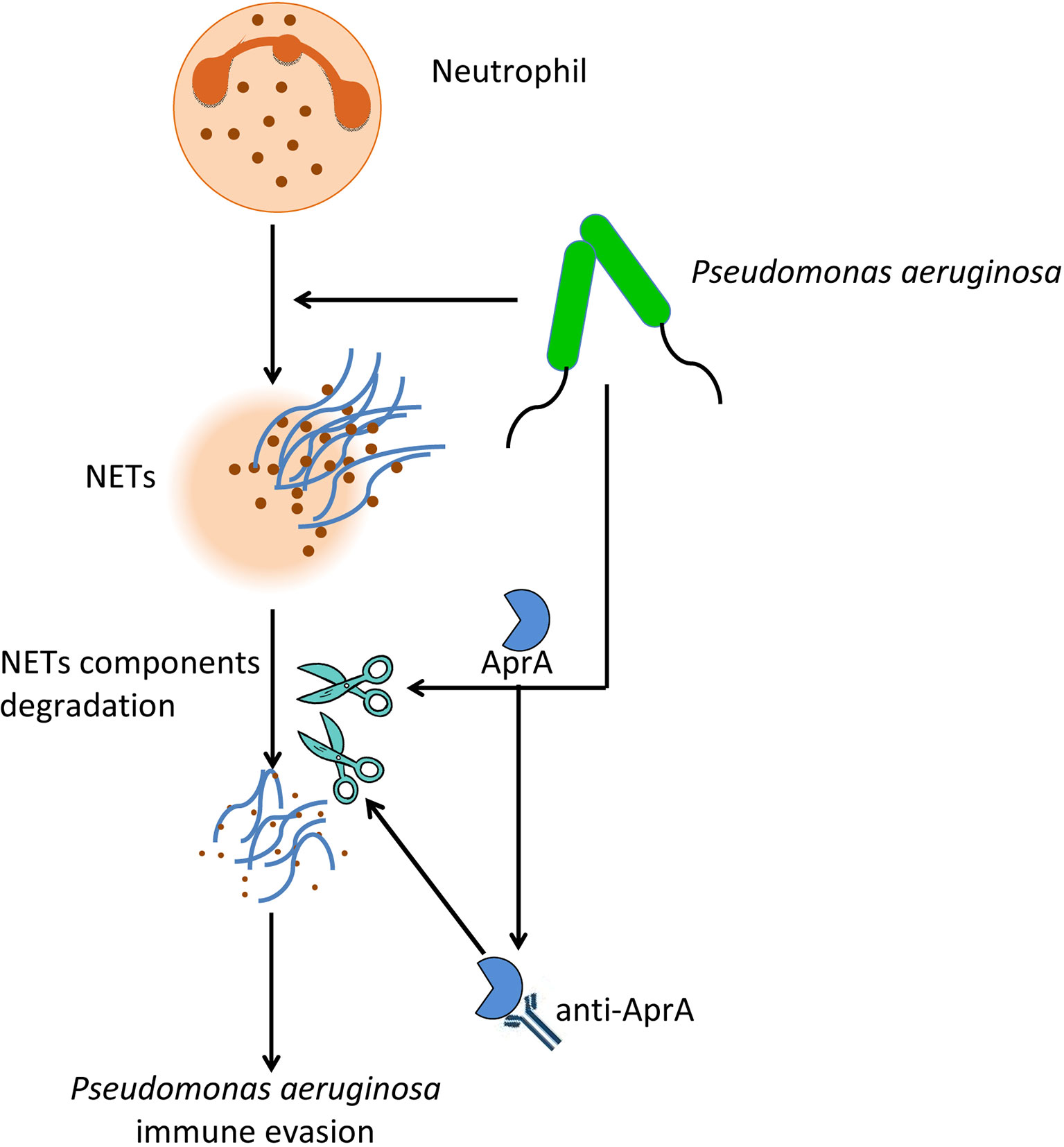
Figure 8 The effect of AprA on the pathogenesis of P. aeruginosa. P. aeruginosa can evade NETs killing by secreting AprA, which not only degrades NETs components, but also induce the production of specific antibodies that further enhance its effect.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by The ethics committee of the Peking University People’s Hospital. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
CJ acquired, analyzed, and interpreted data. YL and CL acquired and analyzed data. RF, RC, ZG, BX, YG, QW, and NY provided administrative, technical, and material support. YM, LL, JF, and BS supervised the study. YY and HW supervised the study and drafted the manuscript. GY conceived and designed the study, obtained funding and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from National Natural Science Foundation of China (http://www.nsfc.gov.cn) [81871618] and [31870127], and Shandong Provincial Major Scientific and Technological Innovation Project (MSTIP)[2019JZZY011012]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.654649/full#supplementary-material
AprA, alkaline protease; AprI, alkaline protease inhibitor; ADE, antibody-dependent enhancement; BALF, bronchoalveolar lavage fluid; CF, cystic fibrosis; COPD, chronic obstructive pulmonary disease; Cit-H3, citrullinated histone H3; MPO, myeloperoxidase; MOI, multiplicity of infection; NETs, neutrophil extracellular traps; NE, neutrophil elastase; NEi, neutrophil elastase inhibitor; PAD4, peptidylarginine deiminase 4; ROS, reactive oxygen species.
1. Gellatly Shaan L, Hancock Robert EW. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis (2013) 67:159–73. doi: 10.1111/2049-632X.12033
2. Mayer-Hamblett N, Rosenfeld M, Gibson RL, Ramsey BW, Kulasekara HD, Retsch-Bogart GZ, et al. Pseudomonas aeruginosa in vitro phenotypes distinguish cystic fibrosis infection stages and outcomes. Am J Respir Crit Care Med (2014) 190:289–97. doi: 10.1164/rccm.201404-0681OC
3. Parker CM, Kutsogiannis J, Muscedere J, Cook D, Dodek P, Day AG, et al. Ventilator-associated pneumonia caused by multidrug-resistant organisms or Pseudomonas aeruginosa: prevalence, incidence, risk factors, and outcomes. J Crit Care (2008) 23:18–26. doi: 10.1016/j.jcrc.2008.02.001
4. Zilberberg MD, Shorr AF. Prevalence of multidrug-resistant Pseudomonas aeruginosa and carbapenem-resistant Enterobacteriaceae among specimens from hospitalized patients with pneumonia and bloodstream infections in the United States from 2000 to 2009. J Hosp Med (2013) 8:559–63. doi: 10.1086/432803
5. Livermore DM. Multiple mechanisms of antimicrobial resistance in Pseudomonas aeruginosa: our worst nightmare? Clin Infect Dis (2002) 34:634–40. doi: 10.1002/jhm.2080
6. Matsumoto K. Role of bacterial proteases in pseudomonal and serratial keratitis. Biol Chem (2004) 385:1007–16. doi: 10.1086/338782
7. Bardoel BW, van Kessel KP, van Strijp JA, Milder FJ. Inhibition of Pseudomonas aeruginosa virulence: characterization of the AprA-AprI interface and species selectivity. J Mol Biol (2012) 415:573–83. doi: 10.1016/j.jmb.2011.11.039
8. Jaffar-Bandjee MC, Lazdunski A, Bally M, Carrère J, Chazalette JP, Galabert C. Production of elastase, exotoxin A, and alkaline protease in sputa during pulmonary exacerbation of cystic fibrosis in patients chronically infected by Pseudomonas aeruginosa. J Clin Microbiol (1995) 33:924–9. doi: 10.1016/j.jmb.2011.11.039
9. Döring G, Obernesser HJ, Botzenhart K, Flehmig B, Høiby N, Hofmann A. Proteases of Pseudomonas aeruginosa in patients with cystic fibrosis. J Infect Dis (1983) 147:744–50. doi: 10.1128/JCM.33.4.924-929.1995
10. Segal AW. How neutrophils kill microbes. Annu Rev Immunol (2005) 23:197–223. doi: 10.1093/infdis/147.4.744
11. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
12. Winterbourn CC, Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal (2013) 18:642–60. doi: 10.1089/ars.2012.4827
13. Yoo DG, Winn M, Pang L, Moskowitz SM, Malech HL, Leto TL, et al. Release of cystic fibrosis airway inflammatory markers from Pseudomonas aeruginosa-stimulated human neutrophils involves NADPH oxidase-dependent extracellular DNA trap formation. J Immunol (2014) 192:4728–38. doi: 10.4049/jimmunol.1301589
14. Yoo DG, Floyd M, Winn M, Moskowitz SM, Rada B. NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol Lett (2014) 160:186–94. doi: 10.1016/j.imlet.2014.03.003
15. Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner WD, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res (2015) 16:59. doi: 10.1186/s12931-015-0221-7
16. Marcos V, Zhou-Suckow Z, Önder Yildirim A, Bohla A, Hector A, Vitkov L, et al. Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediators Inflammation (2015) 2015:408935. doi: 10.1155/2015/408935
17. Sousa AM, Pereira MO. Pseudomonas aeruginosa Diversification during Infection Development in Cystic Fibrosis Lungs-A Review. Pathogens (2014) 3:680–703. doi: 10.3390/pathogens3030680
18. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene (1998) 212(1):77–86. doi: 10.1016/s0378-1119(98)00130-9
20. Toussaint M, Jackson DJ, Swieboda D, Guedán A, Tsourouktsoglou TD, Ching YM, et al. Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat Med (2017) 23:681–91. doi: 10.1038/nm.4332
21. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207:1853–62. doi: 10.1084/jem.20100239
22. Jensen EC. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec (Hoboken) (2013) 296(3):378–81. doi: 10.1002/ar.22641
23. Dubin PJ, Kolls JK. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol (2007) 292:L519–28. doi: 10.1152/ajplung.00312.2006
24. Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun (2009) 77:5300–10. doi: 10.1128/IAI.00501-09
25. Lovewell RR, Patankar YR, Berwin B. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol (2014) 306:L591–603. doi: 10.1152/ajplung.00335.2013
26. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184:205–13. doi: 10.1083/jcb.200806072
27. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol (2012) 3:307. doi: 10.3389/fimmu.2012.00307
28. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
29. Bardoel BW, van der Ent S, Pel MJ, Tommassen J, Pieterse CM, van Kessel KP, et al. Pseudomonas evades immune recognition of flagellin in both mammals and plants. PloS Pathog (2011) 7:e1002206. doi: 10.1371/journal.ppat.1002206
30. Gaweł J, Pogorzelski A, Działek-Smętek E, Sochań B, Ligarska R, Lącka M, et al. Distribution of antibodies to selected antigens of Pseudomonas aeruginosa in children and young adults with cystic fibrosis. Pneumonol Alergol Pol (2014) 82:336–41. doi: 10.5603/PiAP.2014.0042
31. Dwyer M, Shan Q, D’Ortona S, Maurer R, Mitchell R, Olesen H, et al. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. Version 2. J Innate Immun (2014) 6:765–79. doi: 10.1159/000363242
32. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
33. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun (2010) 2:576–86. doi: 10.1159/000319909
34. Döhrmann S, Anik S, Olson J, Anderson EL, Etesami N, No H, et al. Role for streptococcal collagen-like protein 1 in M1T1 group A Streptococcus resistance to neutrophil extracellular traps. Infect Immun (2014) 82:4011–20. doi: 10.1128/IAI.01921-14
35. Høiby N. Antibodies against Pseudomonas aeruginosa in patients with bronchiectasis: helpful or harmful? Thorax (2001) 56:667–8. doi: 10.1136/thorax.56.9.667
36. Sasaki T, Setthapramote C, Kurosu T, Nishimura M, Asai A, Omokoko MD, et al. Dengue virus neutralization and antibody-dependent enhancement activities of human monoclonal antibodies derived from dengue patients at acute phase of secondary infection. Antiviral Res (2013) 98:423–31. doi: 10.1016/j.antiviral.2013.03.018
37. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-dependent enhancement of Ebola virus infection. J Virol (2003) 77:7539–44. doi: 10.1128/jvi.77.13.7539-7544.2003
38. Tóth FD, Mosborg-Petersen P, Kiss J, Aboagye-Mathiesen G, Zdravkovic M, Hager H, et al. Antibody-dependent enhancement of HIV-1 infection in human term syncytiotrophoblast cells cultured in vitro. Clin Exp Immunol (1994) 96:389–94. doi: 10.1111/j.1365-2249.1994.tb06040.x
39. Iankov ID, Pandey M, Harvey M, Griesmann GE, Federspiel MJ, Russell SJ. Immunoglobulin g antibody-mediated enhancement of measles virus infection can bypass the protective antiviral immune response. J Virol (2006) 80:8530–40. doi: 10.1128/JVI.00593-06
40. Halstead SB. Neutralization and antibody-dependent enhancement of dengue viruses. Adv Virus Res (2003) 60:421–67. doi: 10.1016/s0065-3527(03)60011-4
41. Hong YQ, Ghebrehiwet B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clin Immunol Immunopathol (1992) 62:133–8. doi: 10.1016/0090-1229(92)90065-v
Keywords: Pseudomonas aeruginosa, alkaline protease, alkaline protease inhibitor, neutrophil extracellular traps, antibody-dependent enhancement
Citation: Jing C, Liu C, Liu Y, Feng R, Cao R, Guan Z, Xuan B, Gao Y, Wang Q, Yang N, Ma Y, Lan L, Feng J, Shen B, Wang H, Yu Y and Yang G (2021) Antibodies Against Pseudomonas aeruginosa Alkaline Protease Directly Enhance Disruption of Neutrophil Extracellular Traps Mediated by This Enzyme. Front. Immunol. 12:654649. doi: 10.3389/fimmu.2021.654649
Received: 17 January 2021; Accepted: 17 March 2021;
Published: 31 March 2021.
Edited by:
Dane Parker, The State University of New Jersey, United StatesReviewed by:
Alexander Dalpke, Technische Universität Dresden, GermanyCopyright © 2021 Jing, Liu, Liu, Feng, Cao, Guan, Xuan, Gao, Wang, Yang, Ma, Lan, Feng, Shen, Wang, Yu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guang Yang, yangg62033@outlook.com; Hui Wang, wanghui@pkuph.edu.cn; Yanyan Yu, yyy@bjmu.edu.cn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.