 Prerna Malaney1†
Prerna Malaney1† María Velasco-Estevez2†
María Velasco-Estevez2† Pedro Aguilar-Garrido2†
Pedro Aguilar-Garrido2† Marisa J. L. Aitken1Lauren E. Chan1
Marisa J. L. Aitken1Lauren E. Chan1 Xiaorui Zhang1
Xiaorui Zhang1 Sean M. Post1
Sean M. Post1 Miguel Gallardo2*
Miguel Gallardo2*- 1Department of Leukemia, MD Anderson Cancer Center, Houston, TX, United States
- 2H12O–CNIO Haematological Malignancies Clinical Research Unit, CNIO, Madrid, Spain
B-cell lymphomas are one of the most biologically and molecularly heterogeneous group of malignancies. The inherent complexity of this cancer subtype necessitates the development of appropriate animal model systems to characterize the disease with the ultimate objective of identifying effective therapies. In this article, we discuss a new driver of B-cell lymphomas – hnRNP K (heterogenous nuclear ribonucleoprotein K)—an RNA-binding protein. We introduce the Eµ-Hnrnpk mouse model, a murine model characterized by hnRNP K overexpression in B cells, which develops B-cell lymphomas with high penetrance. Molecular analysis of the disease developed in this model reveals an upregulation of the c-Myc oncogene via post-transcriptional and translational mechanisms underscoring the impact of non-genomic MYC activation in B-cell lymphomas. Finally, the transplantability of the disease developed in Eµ-Hnrnpk mice makes it a valuable pre-clinical platform for the assessment of novel therapeutics.
Introduction to B-Cell Malignancies
B-cell neoplasms comprise a heterogeneous cohort of hematological malignancies originating from B cells. They are classified, according to WHO criteria, into precursor B-cell neoplasms (B-lymphoblastic leukemia) and mature B-cell neoplasms (1). Mature B-cell neoplasms encompass a plethora of malignancies, such as leukemias (chronic lymphocytic leukemia, small lymphocytic lymphoma), plasma cell malignancies (e.g. myeloma), and lymphomas. Specifically, lymphomas are classified as Hodgkin’s and non-Hodgkin’s lymphomas, which account for 10% and 90% of cases, respectively (2). Nearly half of all non-Hodgkin’s lymphomas are diffuse large B-cell lymphomas (DLBCL), making them the most commonly diagnosed lymphoma sub-type. Other non-Hodgkin’s lymphomas include follicular lymphomas, marginal zone lymphoma, mantle cell lymphoma and Burkitt’s lymphoma (3).
B cells are part of the adaptive immune system and play a vital role in the production of antigen-specific immunoglobulins (antibodies) in response to invasive pathogens. In order to do so, B cells must maintain an extensive array of antigen receptors. B cells must also demonstrate high variability in antigen recognition sites in order to protect organisms against a plethora of pathogens. To achieve this, B cells employ a complex mechanism called VDJ recombination, which enables the generation of an almost unlimited repertoire of antigenic B-cell receptors. The VDJ recombination process involves the formation and repair of several highly regulated DNA breaks. Aberrations in this intricate process along with spontaneous mutations and breaks in the DNA lead to alterations that in turn promote the malignant transformation of B cells (4, 5).
Aberrant VDJ recombination often results in chromosomal translocations. A clear example of this is Burkitt’s lymphoma wherein a chromosomal translocation results in constitutive activation of the MYC oncogene (6–8). Other examples include the t(14;18) translocation in follicular lymphoma, resulting in constitutive expression of BCL2 (4) and the t(11;14) translocation involving the cyclin D1 gene in mantle-cell lymphoma (9, 10). In addition to translocations, somatic mutations in various genes have also been implicated in the pathogenesis of other non-Hodgkin’s lymphomas (11). Beyond mutations, overexpression of oncogenes and loss of tumor suppressor genes have also been identified as driver events for lymphomagenesis (12–14). Well-defined genetic basis of disease has helped establish several genetically defined transgenic models of B-cell malignancies, as discussed below.
Microscopic analysis of lymphomas show that tumors contain not only cancerous cells, but also host immune cells, stromal cells, blood vessels and the extracellular matrix, comprising the tumor microenvironment (15). Tumor cells create this microenvironment by first homing to sites that promote or help their growth, and later recruiting support cells and/or causing cells in their microenvironment to differentiate to their benefit (3). In recent years, it has become clear that the tumor microenvironment is a key player in the onset and progression of cancer, as well as therapy resistance, underscoring the need for better understanding in this area. Investigating B-cell lymphomas using in vitro systems is an important initial approach, but its predictive value is limited as it leaves out other factors such as the host immune system and the tumor microenvironment. Therefore, the use of more complex tools such as genetically engineered mouse models are needed to more fully study B-cell malignancies.
Need for the Development of New Murine Models for B-Cell Malignancies
Murine models of hematological malignancies can be classified as genetically modified models or patient-derived xenograft (PDX) models. The former can be used to investigate the onset, progression and therapeutic approaches for leukemia and lymphomas, while the latter is a valuable platform for drug testing and pre-clinical research.
PDX models are ideal for preclinical studies, wherein a heterogeneous population of tumor cells extracted from a patient is implanted into an immunocompromised mouse, thereby facilitating studies that assess the response or resistance of the disease to existing and novel therapeutic agents (16). Despite the caveat of lacking the tumor-immune cell component, B-cell lymphoma PDX mouse models can show biological, histopathological and clinical features of the original patient tumor, making it an ideal platform for preclinical studies (17). The use of humanized mice for transplantation is gaining popularity and allows for the assessment of immunotherapies as well as the study of the interactions between the tumor and the immune system (18). However, PDX models cannot be used to investigate the onset and progression of tumors. They are also not amenable for the identification of novel oncogenes or tumor suppressor genes, making transgenic and knockout mouse models the preferred platform for this aim. While PDX models enable the assessment of developed disease, transgenic animal models can be used to investigate the origin and onset of disease. Moreover, PDX models carry the diversity of the patients they are derived from and are therefore diverse in their pathology and drug responses, complicating interpretation of data. Transgenic models, however, have a common background allowing for a replicable and controlled phenotype for research. Taken together, the research hypotheses and objectives must be taken into consideration while selecting an appropriate animal model.
There are several well-defined genetic alterations observed in human patients that give rise to B-cell lymphomas due to uncontrolled B-cell proliferation and/or maturation. Some of these genetic alterations have already been recreated in transgenic mice to study the spontaneous onset and progression of lymphomas and leukemia-like phenotypes. Notable models include a BCL6 knock-in which results in a DLBCL-like phenotype (19), VavP-Bcl2 mice, mimicking the t(14; 18) chromosomal translocation, which develop follicular lymphomas, a targeted deletion of Trp53 – CD21-Tp53lox – that develops non-germinal center B-cell lymphomas, and Eµ-Tcl1 mice which develop aggressive chronic lymphocytic leukemia (20–22).
However, the most recurrent and extensively investigated genetic alterations are translocations of the MYC oncogene. In the 1980s, MYC was identified as the first proto-oncogene in B-cell lymphomas. Transgenic mouse models with translocations in the Myc gene were first introduced in 1985 (23). Of these models, the most extensively used is the Eµ-Myc model. This mouse obtained by the translocation of Myc to the Ig Heavy chain (IgH) locus, causing overexpression of c-Myc and abnormal B-cell proliferation (24). Similar to humans, Eµ-Myc mice develop B-cell lymphoma-like malignancies with a >90% penetrance and variable onset. These Eµ-Myc mice exhibit either a DLBCL phenotype or a Burkitt’s lymphoma phenotype depending on the time of development (25, 26). Another model of c-Myc driven Burkitt’s lymphoma is one driven solely by the 3’ regulatory region of the IgH locus rather than the Eµ enhancer. This model results in the development of B-cell malignancies with a mature B-cell phenotype in contrast to the Eµ model wherein a pro-B phenotype is predominant (27, 28). However, multiple mouse models with other Myc translocations develop different B-cell malignancies, such as Vκ-Myc mice for myeloma (29), λ-Myc for Burkitt’s lymphoma (30), iMycEμ for endemic Burkitt’s lymphoma (31), and iMycCμ mice for sporadic and immunodeficiency-associated Burkitt’s lymphoma (32), demonstrating the critical role of c-Myc in B cell biology.
New Driver of B-Cell Lymphomas: hnRNP K
Altered genes have historically been designated as either tumor suppressors or oncogenes. However, recent studies have shown that some genes can have dual oncogenic and tumor-suppressive functions in different contexts (33). One such example of a dual regulator is heterogeneous nuclear ribonucleoprotein K (HNRNPK) (34).
hnRNP K is an ss-DNA and RNA-binding protein that regulates a myriad of cellular processes via transcriptional, posttranscriptional and translational mechanisms. It contains three K homology (KH) domains – responsible for nucleic acid binding – one K-protein-interactive domain (KI), and one nuclear-cytoplasmic shuttling domain (KNS) (35, 36). Due to hnRNP K’s pleiotropic nature, both its over- and under-expression can be pathogenic (37–41), likely by deregulating the transcription and/or translation of multiple cellular oncogenes or tumor suppressors. For instance, there is a clinical correlation between high levels of hnRNP K and the onset and treatment resistance of various tumors types in patients, such as lung (42), breast (43), rectal adenocarcinoma (44), and melanoma (45). In the context of hematological malignancies, ~2% of acute myeloid leukemia patients have a 9q21.32 deletion that encompasses the HNRNPK gene, resulting in loss of one copy of HNRNPK. Consequently, haploinsufficiency of HNRNPK was shown to be pathogenic in mice, as an Hnrnpk+/- mouse model showed a myeloproliferative phenotype and reduced survival. In addition, Hnrnpk haploinsufficiency also triggered B-cell lymphoma phenotypes in 30% of the animals. This role of hnRNP K as a tumor suppressor is partially due to its regulation of the p53/p21 pathway (40, 46). However, it is of note that hnRNP K acts not only as a tumor suppressor, but also an oncogene. It has been shown that hnRNP K promotes the expression of classical oncogenes such c-Myc, resulting in the development of B-cell malignancies (39, 45), as well as c-Src (47) and eIF4E (38). As an ss-DNA and RNA-binding protein, hnRNP K can regulate both the transcription and translation of certain genes, as it does in the case of MYC. It has been observed that hnRNP K binds to the CT-rich regions of the MYC promoter, enhancing transcription of this gene (48–50). Additionally, hnRNP K positively regulates translation of MYC mRNA by binding to the IRES (Internal Ribosome Entry Site) sequence, aiding its entry to the ribosome through a cap-independent mechanism (37, 51). Control of MYC translation via this mechanism through hnRNP K is significant in the context of tumorigenesis, as it has been observed that even a single CT mutation in the IRES sequence – present in 42% of myeloma multiple patients – could enhance the binding of hnRNP K to this sequence and promote an increased aberrant expression of c-Myc (52). In fact, hnRNP K has been described as an (IRES)-trans-acting factors (ITAF) and can potentially regulating cap-independent translation of several oncogenes resulting in the systemic activation of an oncogenic program (53, 54).
Given the role of hnRNP K in regulating the expression levels of oncogenic molecules, elevated hnRNP K levels are observed in several solid and hematological malignancies (42, 44, 45). Specific to B-cell lymphomas, as we have previously described in Gallardo et al., hnRNP K is overexpressed in human patients with diffuse large B-cell lymphoma and is associated with poor clinical outcomes and non-responsiveness to chemotherapy (39). Overexpression of hnRNP K is also observed in Burkitt’s lymphoma where sumoylated hnRNP K is elevated, which regulates the expression of c-Myc at the translational level (55). Taken together, a dual role of hnRNP K has been observed in the formation of hematological malignancies, wherein an under-expression of the protein leads to a deficit in its role inducing the expression of tumor suppressors p53/p21, thus leading to a myeloproliferative phenotype and lymphoma in rodents (40), while on the other hand, an increment in the levels of hnRNP K has been related to higher levels of c-Myc and the formation of lymphomas (39, 55).
Critically, with respect to B-cell malignancies, we observe an oncogenic function for hnRNP K. Our data showed that hnRNP K is overexpressed in DLBCL patients and higher expression of hnRNP K correlates with poor clinical outcomes and lack of response to chemotherapy in these patients. Interestingly, we observed that hnRNP K is overexpressed in patients who do not harbor any MYC genomic alterations. The regulation of MYC by hnRNP K occurs at the post-transcriptional and translational level, indicating that hnRNP K over-expression represents a key non-genomic mechanism of MYC regulation in B-cell lymphomas (37).
The role of hnRNP K is especially critical given the increasing clinical relevance of RNA-binding proteins (RBPs) and splicing factors in hematological malignancies. Mutations and aberrant expression of RBPs is increasingly being observed in leukemia, myelodysplastic disease, and lymphomas (56–60). Moreover, taking into consideration the critical role of c-Myc not only in B-cell biology and B-cell malignancies, but also in other heme malignancies, hnRNP K-mediated regulation of c-Myc warrants extensive study. The B-cell specific murine model of hnRNP K, Eµ-Hnrnpk, described below, is therefore crucial for unraveling the complex and nuanced role of this protein in B-cell malignancies.
Eµ-Hnrnpk Murine Model as a Platform to Study B-Cell Malignancies
We previously generated the Eµ-Hnrnpk mouse model by placing the Hnrnpk cDNA downstream of the extensively characterized immunoglobulin heavy-chain (Igh) enhancer Eµ. The resulting mice specifically overexpress hnRNP K in B cells, have reduced survival, and develop B-cell malignancies (39). The Eµ-Hnrnpk model has several utilities and applications. First, the model helps establish the RNA-binding protein, hnRNP K, as a bona fide oncogene. To the best of our knowledge, this is the first published transgenic mouse model that demonstrates the oncogenicity of hnRNP K over-expression. Second, the model provides an in-vivo system to assay the role of hnRNP K in B-cell biology and malignancy. Third, the mice provide a pre-clinical platform to test existing and experimental therapeutics.
Phenotype of the Eµ-Hnrnpk Mouse Model
The Eµ-Hnrnpk mice develop B-cell malignancies with a high latency. Roughly 70% of mice survived the first year but only 20% survived until the end of the second year. This accelerated mortality between years one and two may be due to yet-unknown secondary genomic aberrations or epigenetic changes warranting further in-depth analyses of this disease model. In contrast with the slow generation of the phenotype, the disease penetrance is incredibly high – almost 100% of the mice developed some type of B-cell malignancy.
Gross analyses revealed that Eµ-Hnrnpk mice have marked hepatosplenomegaly, a 12-fold increase in spleen weight, and a 3.5-fold increase in the hepatic weight. The splenomegaly is accompanied by loss of splenic architecture, expansion of B-cell lineages as evidenced by increased PAX5 staining, and the presence of highly proliferative Ki67+ cells. In addition to their proliferative nature, the malignant cells have invasive properties as well. Livers of tumor-burdened mice showed extensive B-cell (B220+) infiltration and minimal T-cell (CD3+) infiltration.
The malignancy of Eµ-Hnrnpk cells was confirmed through transplantation assays. Immunodeficient mice injected with cells from Eµ-Hnrnpk mice bearing disease recapitulate the same phenotypes (reduced survival, B cell proliferation), confirming the cell-autonomous nature of Eµ-Hnrnpk cells.
The preponderance of large malignant cells observed in hematopoietic tissues of the Eµ-Hnrnpk mice and clinical data from DLBCL patients pinpoint the role of hnRNP K overexpression in this disease. However, a small proportion (2-5%) of the Eµ-Hnrnpk mice developed other types of B-cell malignancies, not described herein, consistent with plasma cell-like malignancy. This observation falls in line with some late-arising tumors seen in Eµ-Myc mice that tend to develop lymphomas derived from plasma cells or with plasma cell differentiation (25).
Molecular Mechanism of hnRNP K-Mediated Lymphomagenesis
Due to the plurality of hnRNP K’s cellular roles, the molecular mechanism of hnRNP K-mediated lymphomagenesis is likely to be multifaceted and complex. However, hnRNP K has been previously demonstrated to bind to C-rich regions in DNA and RNA (38, 61). Data obtained from RIP-Seq (RNA immunoprecipitation followed by sequencing) and its formaldehyde-fixed molecular cousin (fRIP-seq) (62) experiments, previously published in Gallardo et al., revealed that one of the top targets of hnRNP K is the MYC transcript, and that this interaction enhances its stability and translation (39). We confirmed a direct interaction between hnRNP K protein and the MYC transcript using fluorescence anisotropy assays. Actinomycin-chase experiments revealed an increased stability of the MYC transcript associated with hnRNP K overexpression. Finally, polysome profiling assays revealed the essential role of hnRNP K in regulating MYC translation and that knockdown of hnRNP K adversely affected the loading of the MYC transcript onto monosomes. Taken together, our biochemical and molecular assays establish a physical and functional link between hnRNP K and MYC (Figure 1). This finding is borne out in the in vivo model: lymphomas derived from Eµ-Hnrnpk mice show elevated c-Myc levels. These findings are particularly relevant when coupled with the observation that hnRNP K expression levels are elevated in DLBCL patients without MYC genomic alterations, suggesting that hnRNP K can drive c-Myc signaling in the absence of MYC mutations. Therefore, hnRNP K overexpression represents an alternate mechanism of c-Myc pathway activation in B-cell malignancies. Interestingly, Smurf2 ablation in mice also results in the development of B-cell tumors due to transcriptional upregulation of the c-Myc protein representing yet another non-genomic mechanism of aberrant c-Myc expression in tumors (63). Considering the fact that hnRNP K binds to a multitude of transcripts, it would be reasonable to assume that it causes lymphomagenesis via several mechanisms and not just via regulation of c-Myc. An overlap of RIP-Seq data from two hematological cell lines (K562 and OCI-AML3) with a set of genes implicated in lymphomas reveal several interesting candidates for further study (Figure 2) (19, 64).
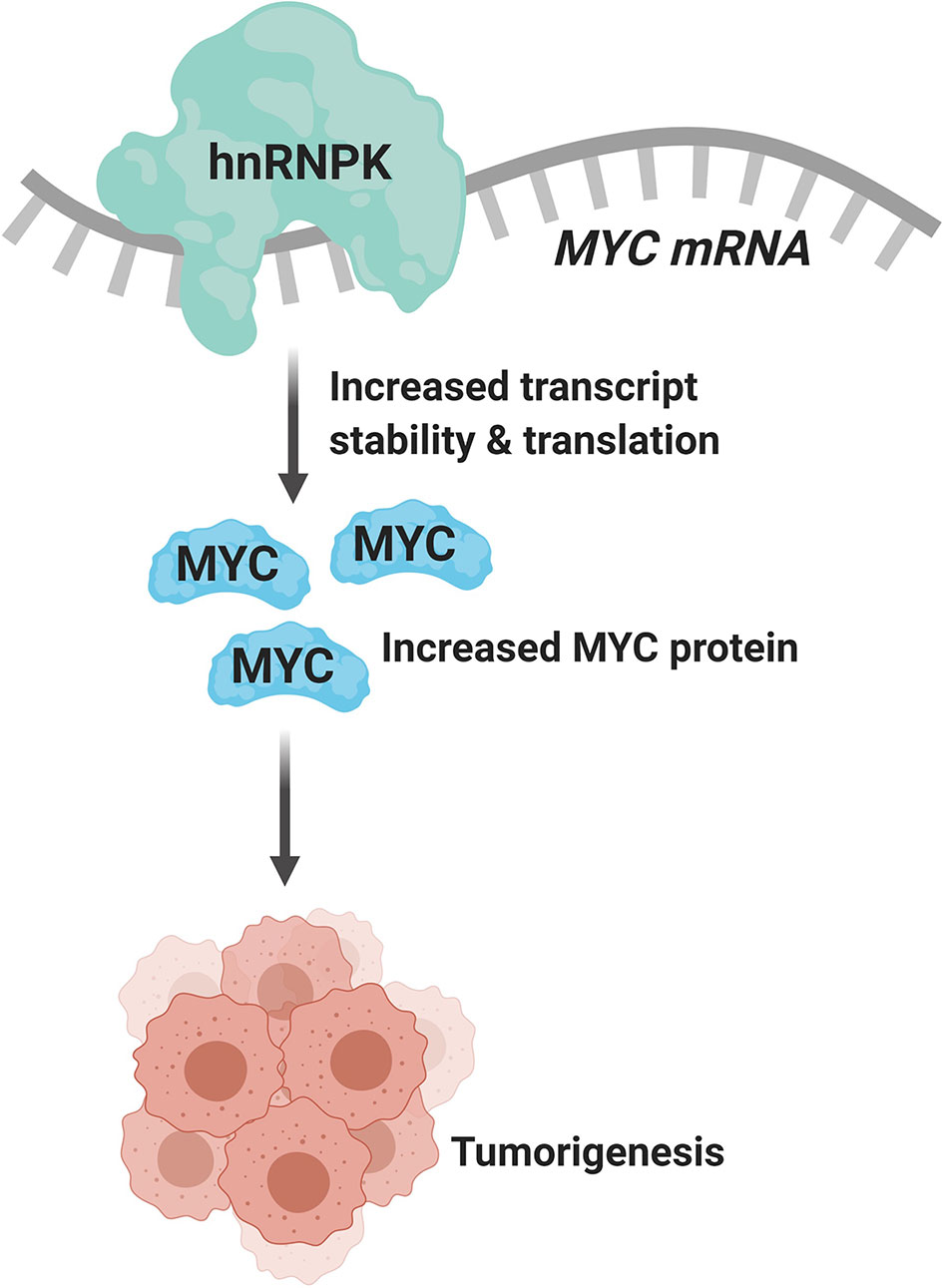
Figure 1 hnRNP K binds to and regulates the stability and translation of the MYC transcript (created with BioRender.com).
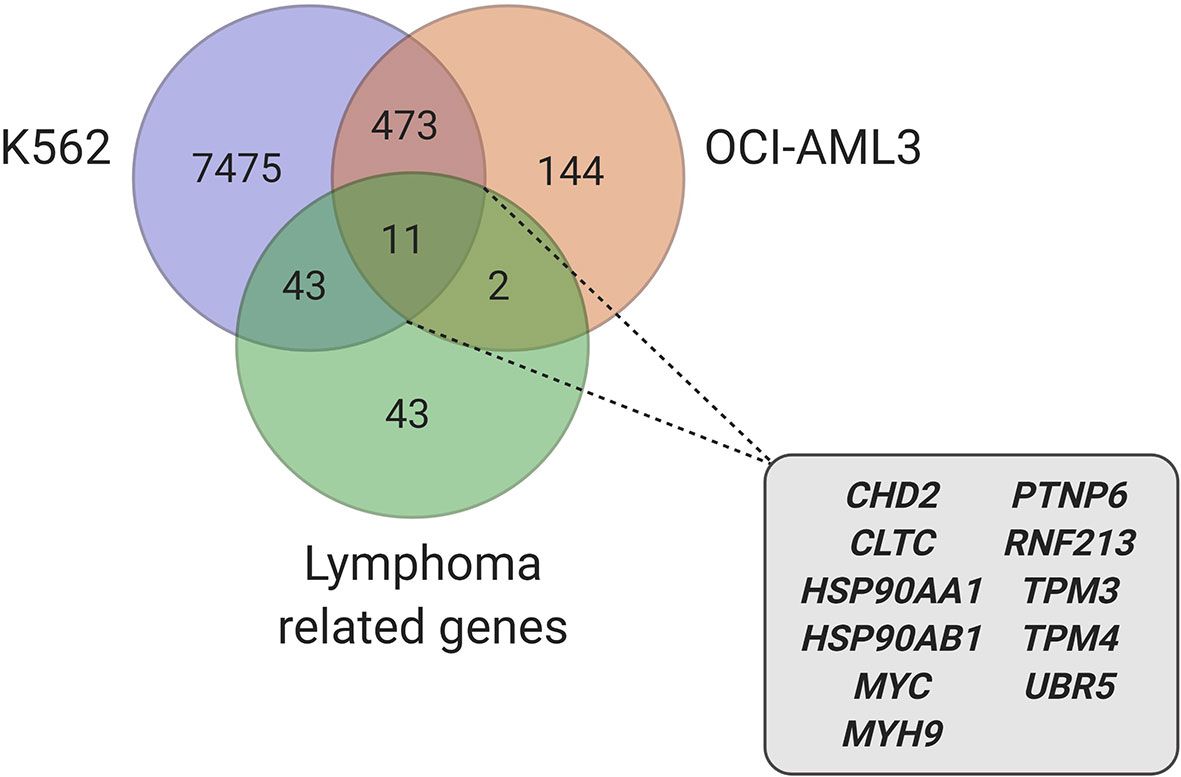
Figure 2 Candidate genes, involved in lymphoma, identified in hnRNP K RIP-Seq datasets (39, 64) (created with BioRender.com).
Beyond its RNA-binding functions, hnRNP K also interacts with a host of proteins via its KI domain. Therefore, it is entirely plausible that the protein interactome of hnRNP K contributes to its oncogenicity. Immunoprecipitation of hnRNP K followed by mass spectrometry, as described in Gallardo et al., revealed that hnRNP K associates with a host of ribosomal and RNA processing proteins (39). Given the preponderance of ribosomal subunits in the hnRNP K interactome, the impact that hnRNP K has on global translational profiles and its relationship to p53 allude to the possibility that aberrations in hnRNP K are likely to contribute to a nucleolar stress response (NSR). In fact, IP/MS experiments reveal a physical interaction between hnRNP K and the NSR sensor Nucleolin (NCL). The impact of hnRNP K on ribosome biogenesis and nucleolar stress pathways and their relevance to disease is currently under investigation in our laboratories.
In conclusion, the Eµ-Hnrnpk mice demonstrate a level of biological heterogeneity that is also seen in human disease. Consequently, the model represents a valuable tool for the assessment of existing and novel drugs and therapies. The transplantability of malignant cells from this model and the aggressive nature of the transplanted disease allow for a rapid evaluation of therapeutic modalities. Although current transplantation studies with the Eµ-Hnrnpk are limited to immunodeficient mice, future research with syngeneic models may be of value, particularly to test immunotherapeutic agents.
Discussion
The generation, maturation, and genetic reprogramming of B-cell lymphocytes represent some of the most complex biological processes outside of development. The maturation process occurs in a bevy of host hematologic tissues. Germinal centers (GCs) – specific structures in secondary lymphoid organs, such as lymph nodes and the spleen – are where the generation and selection of B cells occurs. GCs are typically divided into the dark and the light zone. In the dark zone, B cells undergo rapid and mutative cell division called immunoglobulin somatic hypermutation (SHM), while in the light zone, B cells are selected based on antigen affinity. The whole process of GC initiation, dark zone formation, and the passage of B cells between the dark and light zones to final differentiation is a concatenated process involving the participation of a plethora of proteins such as BCL6, BCL2, TCL1, PAX5, IRF4, NFKB, MLL2 and c-Myc (65). All of these molecules are differentially expressed based on areas within the GC and/or stage of B-cell differentiation and are intricately balanced via multiple feedback loops. The inherent process of B-cell maturation and function relies on the inaccuracy of targeted DNA breaks and subsequent repair during VDJ recombination, SHM, and class-switch recombination (CSR). The rapid mutative divisions in B cells therefore put their genomic integrity in jeopardy. When an error occurs, the two most common genomic sequelae are chromosomal translocations and aberrant SHMs (ASHMs) (66). These alterations are due to mistakes in recombination activating gene (RAG) mediated VDJ recombination process, mistakes in the activation-induced cytidine deaminase (AID) dependent-CSR process, or translocations involving IGH (66). The result of these aberrancies is commonly the juxtaposition of strong promoters/enhancers (e.g. IGH) in front of oncogenes related with B-cell biology. The dysregulation of these oncogenes drives B-cell hematological malignancies such as follicular lymphoma (BCL2 translocation, MLL2 inactivation) (11, 67) DLBCL (e.g. BCL6, BCL2 and MYC translocation) (8, 68, 69) or Burkitt’s lymphoma (MYC translocation) (70, 71).
In the last 40 years, researchers have tried to reproduce the same genetic alterations and translocations found in human B-cell malignancies in animal models in order to develop tools to study the pathophysiology of the disease and develop pre-clinical therapeutic platforms. However, there are some challenges involved in the creation of these models. The vast majority of models of B-cell disease – such as Eµ-Myc, VavP-Bcl2 or Iµ-Bcl6 – are not “clean” models, i.e. they do not perfectly recapitulate human disease or often result in the development of multiple phenotypes due to two main reasons. First, “timing” and “location” in B-cell development plays a pivotal role in its biology, and heavily impact the development of B-cell malignancies. Thus, expression of the oncogenes in a more mature or immature B-cell subtype could result in completely different phenotypes than those observed in human disease. Consequently, the use of universal B-cell promoters that are common to multiple B-cell subtypes will often result in mixed phenotypes (25). Second, most drivers of B-cell malignancies are master regulators of a slew of different biological processes. Therefore, dysregulation of these molecules inhibits DNA damage response, increases genomic instability, or promotes other biological phenomena that enhance the accumulation of genomic aberrations in lymphoma cells. These genetic alterations will likely be different in each individual mouse, and its relationship with the driver alteration could explain the diversity of phenotypes. As an example, Lefebure et al. studied the genetic landscape of Eµ-Myc mice and observed multiple single nucleotide variants, deletions, or indels in Bcor, Trp53, Cdkn2a, Kras or Nras, suggesting that genomic instability driven by the genetic modification i.e., Myc, and the collaboration of other oncogenes and tumor suppressors contribute to the final malignant phenotype (72). The Eµ-Hnrnpk mouse model is not an exception to these problems. The limitations of the model are in fact consistent with other B-cell malignancy models. hnRNP K is also a master regulator of multiple biological process, as it regulates transcription, translation, DNA damage response, and splicing (36), which may contribute to the diversity of phenotypes observed in the Eµ-Hnrnpk model. Moreover, hnRNP K, as an RNA-binding protein, governs the expression of other master regulators beyond c-Myc (39). There are also multiple lines of evidence that suggest hnRNP K can function as a master cancer switch, driving not only hematological neoplasms, but also solid tumors (42–45).
One of the most common lymphomagenesis mouse models used is the Eµ-Myc mouse model. The extensive use of the Eµ-Myc model is not only due to historical reasons (model was developed in 1985), but also due to the biological mechanisms of lymphomagenesis. c-Myc is the most widely studied master regulator of B-cell malignancies and is altered in follicular lymphoma, DLBCL, Burkitt’s lymphoma, and myeloma. However, dysregulation of the c-Myc pathway is observed more frequently than can be explained via MYC genomic aberrations. Thus, it is critical to identify c-Myc regulators that contribute to its non-genomic regulation. To this end, hnRNP K as an RNA-binding protein emerged as an idyllic candidate, and the Eµ-Hnrnpk mouse model allows exploration of B-cell malignancies in the absence of MYC alterations. Moreover, we recently found that hnRNP K is overexpressed in DLBCL patients, suggesting its direct role in B-cell malignancies and thereby making the Eµ-Hnrnpk model relevant beyond its function as a genetic and mechanistic tool. However, hnRNP K is a relatively under-studied molecule in cancer, particularly in B-cell neoplasms. Our studies with Hnrnpk haploinsufficient mice demonstrated that hnRNP K plays a vital role in lymphomagenesis beyond its role as a c-Myc regulator (40). Rigorous experimentation is required to better determine the role of hnRNP K in lymphocyte biology and its implications in development of DLBCL and other B-cell malignancies. Further work also needs to be done to identify the mechanisms underlying hnRNP K overexpression in human disease.
Perhaps the biggest drawback of most common B-cell malignancy mouse models is the diversity in phenotype development that makes them divergent from human disease. As previously discussed, the nature of the oncogenic drivers and their relationship with other cancer regulators combined with the spatio-temporal expression of the genetic modifications contributes to these divergences. Thus, more selective and specific mouse models, using promoters of one-stage/one-grade of B-cell maturation could help to surpass these limitations. Moreover, the generation of more complex mouse models that could recapitulate human diseases more precisely, with a combination of different genetic alterations, could guarantee more consistent models. In fact, complex models, such as Eμ-Myc/BCRHEL/sHEL to mimic Burkitt’s lymphoma, or IL-14α TG × c-Myc TG (DTG) mice for blastoid-variant mantle-cell lymphoma (MCL-BV) are good examples (73, 74).
The ability of a murine model to accurately mimic human disease is more relevant when animal models are used as therapeutic platforms. The selection of a model that does not accurately recapitulate human disease could contribute to the low correlation between results observed in pre-clinical and clinical trials. Indeed, only 8% of pre-clinical animal model research successfully advances to clinical trials, and of the experimental agents that are successful in preclinical models, 85% of them fail in early stages of human trials (75). Hence, a battery of B-cell malignancies drivers and promoters is needed to more precisely mimic the spatio-temporal compartmentalization of alterations to ensure better clinical correlations. To this end, the Eµ-Hnrnpk model adds to the catalog of mouse models available to study B-cell malignancies and establish therapeutic platforms, alone or in combination with other drivers.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee at MD Anderson under protocol 0000787-RN02.
Author Contributions
PM, MV-E, and PA-G: conceptualized, organized, and wrote the manuscript and generated figures. MJLA, LEC, and XZ: critically revised the manuscript. SMP and MG supervised and critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study has been supported a National Cancer Institute/National Institutes of Health Award (R01CA207204) and Leukemia and Lymphoma Society (6577–19) to SMP. PM is a Jane Coffin Childs Memorial Fund for Medical Research Fellow and MA is a recipient of the Dr. John J. Kopchick Fellowship. MG is supported by financial support from the Cris contra el Cancer association and Instituto de Salud Carlos III (Ministry of Economy, Industry and Competitiveness) and cofunded by the European Regional Development Fund with Miguel Servet (CP19/00140) and PI (PI18/00295).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Carmen Delgado for her support in cytopathologic analysis.
Abbreviations
DLBCL, diffuse large B-cell lymphoma; hnRNP K, heterogeneous nuclear ribonucleoprotein K; VDJ, variability, diversity, and joining; PDX, patient-derived xenograft; IgH, immunoglobulin heavy; RBP, RNA-binding protein; GC, germinal center; SHM, somatic hypermutation; CSR, class-switch recombination.
References
1. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
3. Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer (2014) 14(8):517–34. doi: 10.1038/nrc3774
4. Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science (New York NY) (1985) 228(4706):1440–3. doi: 10.1126/science.3874430
5. Tsujimoto Y, Jaffe E, Cossman J, Gorham J, Nowell PC, Croce CM. Clustering of breakpoints on chromosome 11 in human B-cell neoplasms with the t(11 ; 14) chromosome translocation. Nature (1985) 315(6017):340–3. doi: 10.1038/315340a0
6. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci (1982) 79(24):7824–7. doi: 10.1073/pnas.79.24.7824
7. Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A (1982) 79(24):7837–41. doi: 10.1073/pnas.79.24.7837
8. Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med (2006) 354(23):2419–30. doi: 10.1056/NEJMoa055351
9. Leroux D, Le Marc’Hadour F, Gressin R, Jacob MC, Keddari E, Monteil M, et al. Non-Hodgkin’s lymphomas with t(11;14)(q13;q32): a subset of mantle zone/intermediate lymphocytic lymphoma? Br J Haematol (1991) 77(3):346–53. doi: 10.1111/j.1365-2141.1991.tb08582.x
10. Rimokh R, Berger F, Cornillet P, Wahbi K, Rouault J-P, Ffrench M, et al. Break in the BCL1 locus is closely associated with intermediate lymphocytic lymphoma subtype. Genes Chromosomes Cancer (1990) 2(3):223–6. doi: 10.1002/gcc.2870020310
11. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature (2011) 476(7360):298–303. doi: 10.1038/nature10351
12. Watanabe T, Hotta T, Ichikawa A, Kinoshita T, Nagai H, Uchida T, et al. The MDM2 oncogene overexpression in chronic lymphocytic leukemia and low-grade lymphoma of B-cell origin. Blood (1994) 84(9):3158–65. doi: 10.1182/blood.V84.9.3158.bloodjournal8493158
13. Hachem A, Gartenhaus RB. Oncogenes as molecular targets in lymphoma. Blood (2005) 106(6):1911–23. doi: 10.1182/blood-2004-12-4621
14. Geradts J, Andriko JW, Abbondanzo SL. Loss of Tumor Suppressor Gene Expression in High-Grade But Not Low-Grade Non-Hodgkin’s Lymphomas. Am J Clin Pathol (1998) 109(6):669–74. doi: 10.1093/ajcp/109.6.669
15. Cioroianu AI, Stinga PI, Sticlaru L, Cioplea MD, Nichita L, Popp C, et al. Tumor Microenvironment in Diffuse Large B-Cell Lymphoma: Role and Prognosis. Anal Cell Pathol (2019) 2019:8586354. doi: 10.1155/2019/8586354
16. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov (2014) 4(9):998–1013. doi: 10.1158/2159-8290.Cd-14-0001
17. Zhang L, Nomie K, Zhang H, Bell T, Pham L, Kadri S, et al. B-Cell Lymphoma Patient-Derived Xenograft Models Enable Drug Discovery and Are a Platform for Personalized Therapy. Clin Cancer Res (2017) 23(15):4212–23. doi: 10.1158/1078-0432.Ccr-16-2703
18. Morton JJ, Bird G, Refaeli Y, Jimeno A. Humanized Mouse Xenograft Models: Narrowing the Tumor-Microenvironment Gap. Cancer Res (2016) 76(21):6153–8. doi: 10.1158/0008-5472.CAN-16-1260
19. Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell (2005) 7(5):445–55. doi: 10.1016/j.ccr.2005.03.037
20. Gostissa M, Bianco JM, Malkin DJ, Kutok JL, Rodig SJ, Morse HC,3, et al. Conditional inactivation of p53 in mature B cells promotes generation of nongerminal center-derived B-cell lymphomas. Proc Natl Acad Sci U S A (2013) 110(8):2934–9. doi: 10.1073/pnas.1222570110
21. Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood (2004) 103(6):2276–83. doi: 10.1182/blood-2003-07-2469
22. Lee HJ, Gallardo M, Ma H, Zhang X, Larsson CA, Mejia A, et al. p53-independent ibrutinib responses in an Eμ-TCL1 mouse model demonstrates efficacy in high-risk CLL. Blood Cancer J (2016) 6(6):e434. doi: 10.1038/bcj.2016.41
23. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature (1985) 318(6046):533–8. doi: 10.1038/318533a0
24. Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med (1988) 167(2):353–71. doi: 10.1084/jem.167.2.353
25. Mori S, Rempel RE, Chang JT, Yao G, Lagoo AS, Potti A, et al. Utilization of pathway signatures to reveal distinct types of B lymphoma in the Emicro-myc model and human diffuse large B-cell lymphoma. Cancer Res (2008) 68(20):8525–34. doi: 10.1158/0008-5472.Can-08-1329
26. Donnou S, Galand C, Touitou V, Sautès-Fridman C, Fabry Z, Fisson S. Murine Models of B-Cell Lymphomas: Promising Tools for Designing Cancer Therapies. Adv Hematol (2012) 2012:701704. doi: 10.1155/2012/701704
27. Ghazzaui N, Issaoui H, Ferrad M, Carrion C, Cook-Moreau J, Denizot Y, et al. Eμ and 3′RR transcriptional enhancers of the IgH locus cooperate to promote c-myc–induced mature B-cell lymphomas. Blood Adv (2020) 4(1):28–39. doi: 10.1182/bloodadvances.2019000845
28. Ghazzaui N, Saintamand A, Issaoui H, Vincent-Fabert C, Denizot Y. The IgH 3’ regulatory region and c-myc-induced B-cell lymphomagenesis. Oncotarget (2017) 8(4):7059–67. doi: 10.18632/oncotarget.12535
29. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell (2008) 13(2):167–80. doi: 10.1016/j.ccr.2008.01.007
30. Kovalchuk AL, Qi CF, Torrey TA, Taddesse-Heath L, Feigenbaum L, Park SS, et al. Burkitt lymphoma in the mouse. J Exp Med (2000) 192(8):1183–90. doi: 10.1084/jem.192.8.1183
31. Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, et al. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res (2005) 65(4):1306–15. doi: 10.1158/0008-5472.Can-04-0268
32. Cheung WC, Kim JS, Linden M, Peng L, Van Ness B, Polakiewicz RD, et al. Novel targeted deregulation of c-Myc cooperates with Bcl-X(L) to cause plasma cell neoplasms in mice. J Clin Invest (2004) 113(12):1763–73. doi: 10.1172/jci20369
33. Manfredi JJ. The Mdm2–p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev (2010) 24(15):1580–9. doi: 10.1101/gad.1941710
34. Gallardo M, Hornbaker MJ, Zhang X, Hu P, Bueso-Ramos C, Post SM. Aberrant hnRNP K expression: All roads lead to cancer. Cell Cycle (Georgetown Tex) (2016) 15(12):1552–7. doi: 10.1080/15384101.2016.1164372
35. Barboro P, Ferrari N, Balbi C. Emerging roles of heterogeneous nuclear ribonucleoprotein K (hnRNP K) in cancer progression. Cancer Lett (2014) 352(2):152–9. doi: 10.1016/j.canlet.2014.06.019
36. Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. BioEssays (2004) 26(6):629–38. doi: 10.1002/bies.20048
37. Notari M, Neviani P, Santhanam R, Blaser BW, Chang JS, Galietta A, et al. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood (2006) 107(6):2507–16. doi: 10.1182/blood-2005-09-3732
38. Lynch M, Chen L, Ravitz MJ, Mehtani S, Korenblat K, Pazin MJ, et al. hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol Cell Biol (2005) 25(15):6436–53. doi: 10.1128/mcb.25.15.6436-6453.2005
39. Gallardo M, Malaney P, Aitken MJL, Zhang X, Link TM, Shah V, et al. Uncovering the Role of RNA-Binding Protein hnRNP K in B-Cell Lymphomas. J Natl Cancer Inst (2020) 112(1):95–106. doi: 10.1093/jnci/djz078
40. Gallardo M, Lee HJ, Zhang X, Bueso-Ramos C, Pageon LR, McArthur M, et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell (2015) 28(4):486–99. doi: 10.1016/j.ccell.2015.09.001
41. Enge M, Bao W, Hedström E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell (2009) 15(3):171–83. doi: 10.1016/j.ccr.2009.01.019
42. Tang F, Li W, Chen Y, Wang D, Han J, Liu D. Downregulation of hnRNP K by RNAi inhibits growth of human lung carcinoma cells. Oncol Lett (2014) 7(4):1073–7. doi: 10.3892/ol.2014.1832
43. Mandal M, Vadlamudi R, Nguyen D, Wang R-A, Costa L, Bagheri-Yarmand R, et al. Growth Factors Regulate Heterogeneous Nuclear Ribonucleoprotein K Expression and Function. J Biol Chem (2001) 276(13):9699–704. doi: 10.1074/jbc.M008514200
44. Daskalaki W, Wardelmann E, Port M, Stock K, Steinestel J, Huss S, et al. Expression levels of hnRNP K and p21WAF1/CIP1 are associated with resistance to radiochemotherapy independent of p53 pathway activation in rectal adenocarcinoma. Int J Mol Med (2018) 42(6):3269–77. doi: 10.3892/ijmm.2018.3898
45. Wen F, Shen A, Shanas R, Bhattacharyya A, Lian F, Hostetter G, et al. Higher expression of the heterogeneous nuclear ribonucleoprotein k in melanoma. Ann Surg Oncol (2010) 17(10):2619–27. doi: 10.1245/s10434-010-1121-1
46. Moumen A, Masterson P, O’Connor MJ, Jackson SP. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell (2005) 123(6):1065–78. doi: 10.1016/j.cell.2005.09.032
47. Ritchie SA, Pasha MK, Batten DJ, Sharma RK, Olson DJ, Ross AR, et al. Identification of the SRC pyrimidine-binding protein (SPy) as hnRNP K: implications in the regulation of SRC1A transcription. Nucleic Acids Res (2003) 31(5):1502–13. doi: 10.1093/nar/gkg246
48. Lee MH, Mori S, Raychaudhuri P. trans-Activation by the hnRNP K protein involves an increase in RNA synthesis from the reporter genes. J Biol Chem (1996) 271(7):3420–7. doi: 10.1074/jbc.271.7.3420
49. Michelotti EF, Michelotti GA, Aronsohn AI, Levens D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol Cell Biol (1996) 16(5):2350–60. doi: 10.1128/mcb.16.5.2350
50. Michelotti EF, Tomonaga T, Krutzsch H, Levens D. Cellular nucleic acid binding protein regulates the CT element of the human c-myc protooncogene. J Biol Chem (1995) 270(16):9494–9. doi: 10.1074/jbc.270.16.9494
51. Evans JR, Mitchell SA, Spriggs KA, Ostrowski J, Bomsztyk K, Ostarek D, et al. Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene (2003) 22(39):8012–20. doi: 10.1038/sj.onc.1206645
52. Chappell SA, LeQuesne JP, Paulin FE, deSchoolmeester ML, Stoneley M, Soutar RL, et al. A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: a novel mechanism of oncogene de-regulation. Oncogene (2000) 19(38):4437–40. doi: 10.1038/sj.onc.1203791
53. Horvilleur E, Wilson LA, Bastide A, Piñeiro D, Pöyry TAA, Willis AE. Cap-Independent Translation in Hematological Malignancies. Front Oncol (2015) 5:293. doi: 10.3389/fonc.2015.00293
54. Spriggs KA, Bushell M, Mitchell SA, Willis AE. Internal ribosome entry segment-mediated translation during apoptosis: the role of IRES-trans-acting factors. Cell Death Differ (2005) 12(6):585–91. doi: 10.1038/sj.cdd.4401642
55. Suk FM, Lin SY, Lin RJ, Hsine YH, Liao YJ, Fang SU, et al. Bortezomib inhibits Burkitt’s lymphoma cell proliferation by downregulating sumoylated hnRNP K and c-Myc expression. Oncotarget (2015) 6(28):25988–6001. doi: 10.18632/oncotarget.4620
56. Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res (2015) 25(1):14–26. doi: 10.1101/gr.181016.114
57. Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, et al. Disease-Causing Mutations in SF3B1 Alter Splicing by Disrupting Interaction with SUGP1. Mol Cell (2019) 76(1):82–95.e7. doi: 10.1016/j.molcel.2019.07.017
58. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell (2015) 27(5):617–30. doi: 10.1016/j.ccell.2015.04.006
59. Kharas MG, Lengner CJ, Al-Shahrour F, Bullinger L, Ball B, Zaidi S, et al. Musashi-2 regulates normal hematopoiesis and promotes aggressive myeloid leukemia. Nat Med (2010) 16(8):903–8. doi: 10.1038/nm.2187
60. Palanichamy JK, Tran TM, Howard JM, Contreras JR, Fernando TR, Sterne-Weiler T, et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J Clin Invest (2016) 126(4):1495–511. doi: 10.1172/JCI80046
61. Paziewska A, Wyrwicz LS, Bujnicki JM, Bomsztyk K, Ostrowski J. Cooperative binding of the hnRNP K three KH domains to mRNA targets. FEBS Lett (2004) 577(1):134–40. doi: 10.1016/j.febslet.2004.08.086
62. G Hendrickson D, Kelley DR, Tenen D, Bernstein B, Rinn JL. Widespread RNA binding by chromatin-associated proteins. Genome Biol (2016) 17(1):28. doi: 10.1186/s13059-016-0878-3
63. Ramkumar C, Cui H, Kong Y, Jones SN, Gerstein RM, Zhang H. Smurf2 suppresses B-cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat Commun (2013) 4:2598–. doi: 10.1038/ncomms3598
64. Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, et al. A large-scale binding and functional map of human RNA-binding proteins. Nature (2020) 583(7818):711–9. doi: 10.1038/s41586-020-2077-3
65. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat Rev Immunol (2011) 12(1):24–34. doi: 10.1038/nri3128
66. Shaffer AL, Rosenwald A, Staudt LM. Lymphoid malignancies: the dark side of B-cell differentiation. Nat Rev Immunol (2002) 2(12):920–32. doi: 10.1038/nri953
67. Yunis JJ, Oken MM, Kaplan ME, Ensrud KM, Howe RR, Theologides A. Distinctive chromosomal abnormalities in histologic subtypes of non-Hodgkin’s lymphoma. N Engl J Med (1982) 307(20):1231–6. doi: 10.1056/nejm198211113072002
68. Huang JZ, Sanger WG, Greiner TC, Staudt LM, Weisenburger DD, Pickering DL, et al. The t(14;18) defines a unique subset of diffuse large B-cell lymphoma with a germinal center B-cell gene expression profile. Blood (2002) 99(7):2285–90. doi: 10.1182/blood.v99.7.2285
69. Iqbal J, Greiner TC, Patel K, Dave BJ, Smith L, Ji J, et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia (2007) 21(11):2332–43. doi: 10.1038/sj.leu.2404856
70. Zech L, Haglund U, Nilsson K, Klein G. Characteristic chromosomal abnormalities in biopsies and lymphoid-cell lines from patients with Burkitt and non-Burkitt lymphomas. Int J Cancer (1976) 17(1):47–56. doi: 10.1002/ijc.2910170108
71. Manolov G, Manolova Y. Marker Band in One Chromosome 14 from Burkitt Lymphomas. Nature (1972) 237(5349):33–4. doi: 10.1038/237033a0
72. Lefebure M, Tothill RW, Kruse E, Hawkins ED, Shortt J, Matthews GM, et al. Genomic characterisation of Eμ-Myc mouse lymphomas identifies Bcor as a Myc co-operative tumour-suppressor gene. Nat Commun (2017) 8(1):14581. doi: 10.1038/ncomms14581
73. Ford RJ, Shen L, Lin-Lee YC, Pham LV, Multani A, Zhou HJ, et al. Development of a murine model for blastoid variant mantle-cell lymphoma. Blood (2007) 109(11):4899–906. doi: 10.1182/blood-2006-08-038497
74. Refaeli Y, Young RM, Turner BC, Duda J, Field KA, Bishop JM. The B cell antigen receptor and overexpression of MYC can cooperate in the genesis of B cell lymphomas. PloS Biol (2008) 6(6):e152. doi: 10.1371/journal.pbio.0060152
Keywords: B-cell malignancies, lymphoma, diffuse large B cell lymphoma, mouse model, hnRNP K, Eµ-Hnrnpk, RNA-binding protein, MYC
Citation: Malaney P, Velasco-Estevez M, Aguilar-Garrido P, Aitken MJL, Chan LE, Zhang X, Post SM and Gallardo M (2021) The Eµ-hnRNP K Murine Model of Lymphoma: Novel Insights into the Role of hnRNP K in B-Cell Malignancies. Front. Immunol. 12:634584. doi: 10.3389/fimmu.2021.634584
Received: 28 November 2020; Accepted: 23 March 2021;
Published: 12 April 2021.
Edited by:
Christelle Vincent-Fabert, UMR7276 Contrôle des réponses immunes B et des lymphoproliférations (CRIBL), FranceReviewed by:
Michel Cogne, University of Limoges, FranceRachel Maurie Gerstein, University of Massachusetts Medical School, United States
Copyright © 2021 Malaney, Velasco-Estevez, Aguilar-Garrido, Aitken, Chan, Zhang, Post and Gallardo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Gallardo, bWdhbGxhcmRvZEBjbmlvLmVz, bWlndWVsZ2FsbGFyZG9kZWxnYWRvQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work