 Jun Zhao
Jun Zhao Chao Qin
Chao Qin Yongzhen Liu
Yongzhen Liu Youliang Rao
Youliang Rao Pinghui Feng
Pinghui Feng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 29 January 2021
Sec. Viral Immunology
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.613799
This article is part of the Research Topic Sensing DNA in Antiviral Innate Immunity View all 14 articles
Herpes simplex viruses (HSVs) are experts in establishing persistent infection in immune-competent humans, in part by successfully evading immune activation through diverse strategies. Upon HSV infection, host deploys pattern recognition receptors (PRRs) to recognize various HSV-associated molecular patterns and mount antiviral innate immune responses. In this review, we describe recent advances in understanding the contributions of cytosolic PRRs to detect HSV and the direct manipulations on these receptors by HSV-encoded viral proteins as countermeasures. The continuous update and summarization of these mechanisms will deepen our understanding on HSV-host interactions in innate immunity for the development of novel antiviral therapies, vaccines and oncolytic viruses.
Herpesviridae is a family of large DNA viruses that establish persistent infection within their immune-competent host. Members of the family are further grouped into three subfamilies, i.e., alpha-, beta-, and gamma-herpesviruses based on their genome organization, biological characteristics, and cell tropism (1, 2). Herpes simplex virus type 1 (HSV-1 or human herpesvirus 1, HHV-1) and type 2 (HSV-2 or human herpesvirus 2, HHV-2) belong to the alpha-herpesvirus subfamily and the genera simplex virus. They are neurotropic viruses that establish latent infection in the trigeminal ganglia (TG) and dorsal root ganglia (DRG) for the entire life of the host (3). Seropositive for HSV are high, averaging nearly 70% in the general population and approaching 100% in senior citizens of 65-year or older (4, 5). Clinical manifestations of HSV-1 infections include various mucocutaneous diseases, such as herpes labialis, genital herpes, herpetic whitlow, and keratitis (6). It can cause encephalitis that is often life-threatening, in a small portion of the infected individuals who are immune-compromised (6). HSV-2 infection frequently causes genital sores (7).
HSV-1 and HSV-2 are structurally closely related. Herpes simplex virions are spherical particles with a diameter of 186 nm, with glycoprotein protrusions on the surface, making the full diameter approximately 225 nm (8). Both viruses contain a linear double-stranded DNA (dsDNA) genome that is ~150 kilobase (kb) in size and encodes more than 70 open reading frames (ORFs). The viral genomes are caged by a 125 nm icosahedral capsid, which is surrounded by an amorphous layer called tegument (9). Packaged within the tegument compartment, a large number of viral structural proteins are released into the infected cell to establish an environment that is conducive for viral replication. The tegument is enveloped by a lipid bilayer within which multiple viral glycoproteins are embedded. These surface glycoproteins mediate the entry and fusion of the virus with the target cell (10).
HSV-1 and HSV-2 share almost identical replication cycles. Viral entry into host cells is mediated by the interactions between cellular receptors and viral glycoproteins anchored within the virion envelope. The initial binding occurs through the binding of envelope glycoprotein C (gC) and/or gB to heparan sulfate proteoglycan, which is immediately followed by gD association with one of the three known receptors to initiate virus entry (11). The receptors involved are cell-type dependent. While nectin-1 is the main receptor of epithelial cells, neuronal cells and fibroblasts (12), HVEM is the main receptor of T cells and cornea epithelial cells, for HSV infection (13, 14). Upon fusion of the virion envelope with the host cell membrane, tegument proteins are released into the cytoplasm of the infected cells to facilitate capsid trafficking and evade host antiviral immunity. The de-enveloped nucleocapsid is transported along microtubules to the nuclear pore, where the viral genome is injected into the nucleus. At this point, HSVs adopt two modes of infection. In neuronal cells located at the peripheral ganglia region and lab-isolated primary neurons, the viral genome stays as a circularized episome with no active gene transcription except for the latent-associated transcripts (LATs) (15). LATs do not encode proteins, but two major RNA species and several small non-coding RNAs that regulate cell survival and viral lytic gene expression (16). Therefore, this stage is termed as viral latency with no clear clinical manifestation. However, the virus can be periodically reactivated and enters the lytic cycle, largely due to stress responses and other stimuli not fully understood. During the lytic cycle, the viral genome serves as the template for transcription, leading to the sequential production of viral messenger RNAs and polypeptides of the immediate early (IE), early (E), and late (L) phases (17). Tegument protein VP16 and cellular factors promote transcription of IE genes [e.g., infected cell polypeptide 0 (ICP0), ICP4, ICP22, ICP27 and ICP47]. IE proteins then promote transcription and translation of E genes, which produce the necessary components for viral DNA replication. Replicated viral genomes collaborate with transcription factors to promote the expression of L proteins that are structural components of HSV virions (such as glycoproteins and capsid proteins VP5, VP21, VP23, VP24 and VP26), thereby maximizing viral protein production in preparation for viral assembly and egress. Nucleocapsids assemble in the nucleus, undergo envelopment and de-envelopment at the nuclear membrane, and re-envelopment in the TGN to acquire their tegument and glycoprotein-embedded membrane, en route to the maturation and release of amplified virion progeny (18). Importantly, viral latency, reactivation and lytic replication collectively contribute to the life-long ‘persistent infection’ of HSV in an immune-competent host, leading to the recurrent pathogenesis associated with the virus.
Upon infection, host cells sense invading viruses via cellular pattern-recognition receptors (PRRs) to initiate the antiviral innate immune defense. Structurally, PRRs can be generally classified into several major families, including Toll‐like receptors (TLRs), RIG‐I like receptor (RLRs), NOD‐like receptors (NLRs), C‐type lectin receptors (CLRs), AIM2‐like receptors (ALRs), and cyclic GMP‐AMP synthase (cGAS). These PRRs can recognize various pathogen-associated molecular patterns (PAMPs) from bacteria, viruses, fungi and protozoa. Microbial PAMPs can be lipoproteins, carbohydrates, lipopolysaccharides and nucleic acids. PRRs also recognize endogenous damage- or danger-associated molecular patterns (DAMP) from the host, which are related to immune homeostasis and autoimmune diseases. Among PAMPs, the nucleic acid RNA and DNA have attracted much attention. PRRs recognizing the nucleic acids include: DNA sensors such as endosomal Toll-Like Receptor 9 (TLR9), cytosolic Absent In Melanoma 2 (AIM2), Interferon Gamma Inducible Protein 16 (IFI16), DNA-dependent Activator of Interferon-regulatory factors (DAI) and cyclic GMP-AMP synthase (cGAS); RNA sensors TLR3, TLR7, TLR8, and cytosolic Retinoic acid-Inducible Gene I (RIG‐I), Melanoma Differentiation-Associated protein 5 (MDA5), NLR Family Pyrin Domain Containing 3 (NLRP3), and Nucleotide-binding Oligomerization Domain-containing protein 2 (NOD2) (19). TLRs are transmembrane receptors, while cytosolic or nuclear receptors are soluble within their corresponding compartments. After sensing PAMPs or DAMPs, PRRs activate their adaptors and downstream Interferon Regulatory Factors (IRFs) and Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB), leading to the transcription and translation of cytokines, chemokines, MHC, and co-stimulatory molecules. In addition, PRRs can trigger signal transduction and induce cellular processes that do not rely on transcription, such as phagocytosis, autophagy, cell death, and inflammasome activation. These processes work in concert with innate immune response to mesh a network of antiviral host defense (19). In this review, we will summarize the recent findings on the contribution of cytosolic PRRs to sense HSV in host defense, and the counteractive measures deployed by HSV to deflect these PRRs to establish persistent infection.
RLRs, including RIG-I (20), MDA5 (21, 22), and probable ATP-dependent RNA helicase DHX58 (LGP2), are cytoplasmic PRRs that recognize virus-derived or viral infection-associated cellular double-stranded RNA (dsRNA). RIG-I recognizes short, blunt-ended dsRNA carrying terminal 5’-triphosphate or 5’-diphosphate moieties (23), while MDA5 prefers longer dsRNA independent of its terminal phosphate groups.
Upon engaging viral dsRNA, RIG-I and MDA-5 hydrolyze ATP to induce their oligomerization on the dsRNA, thereby exposing their N-terminal caspase activation and recruitment domains (CARDs) to relay immune activation via seeding the oligomerization of the adaptor protein MAVS (also known as IPS-1, CARDIF, and VISA) (24–28). LGP2 lacks the CARD domain and is reported to inhibit RIG-I-mediated antiviral responses. Once activated, MAVS forms prion-like oligomers on the outer membrane of mitochondria (29), which further recruits the tank-binding kinase-1(TKB1) and IκB kinase (IKK) complex to activate IRF and NF-κB transcription factors, respectively. Therefore, RIG-I and MDA5 exhibit antiviral activities to a broad spectrum of RNA viruses, including influenza A virus, hepatitis C virus, dengue virus, encephalomyocarditis virus, coronavirus, etc (30). Post-translational modifications, such as phosphorylation and ubiquitination, are discovered to tightly regulate the activation of RIG-I (31–35).
Unlike RNA viruses, genomes of DNA viruses such as herpes simplex viruses (HSV-1 and -2) do not carry the structural features required for binding to RLRs. Remarkably, RLRs demonstrate antiviral activities against HSVs. During HSV-1 latency, two small non-coding RNAs (sncRNAs) coded by the LAT, sncRNA1 and sncRNA2, were shown to interact with and activate RIG-I in neuronal cells, resulting in type I interferon induction and NF-κB activation that promote viral latency and neuronal survival (36). Upon viral entry, early studies have shown that RIG-I and MDA5 non-redundantly activate type I IFN genes upon cytosolic DNA stimulation (37). In support of this, DNA-dependent RNA polymerase III (Pol III) is reported to convert cytosolic DNA to 5’-ppp RNA that activates RIG-I (38). Regarding the source of cytosolic DNA, in macrophages, HSV-1 capsid is found to be degraded by the ubiquitin-mediated proteasome system, thereby releasing viral DNA into the cytosol (39). As such, RIG-I and TLR9 is reported can cooperate to enable the production of type I IFN in HSV-2–infected mouse macrophages (40). However, MDA5 mediates a Pol III-independent pathway to sense HSVs in primary human macrophages (41). The identity of viral RNA or other ligands activating MDA5 remains unknown. In nonimmune cells infected with HSV, studies have detected dsRNA localized in the cytosol, which activates the RIG-I-mediated IFN induction (42). It is proposed that dsRNA molecules originated from the complementary transcription of HSV activate RIG-I. Interestingly, transcripts derived from a cellular 5S ribosomal RNA pseudogene are found to be unmasked by HSV-1 to induce RIG-I activation (43). These findings collectively support the role of RIG-I and MDA5 to sense herpes simplex viruses and induce IFN response.
To counteract RIG-I- and MDA-mediated type I IFN responses, HSV has evolved strategies to directly target these receptors. HSV-2 virion host shutoff (Vhs) protein selectively suppresses the expression of TLR2, TLR3, RIG-I and MDA-5 in human vaginal epithelial cells (44). Given that Vhs is not a sequence-specific endonuclease, it remains unknown how Vhs selectively targets these mRNAs of innate immune function for destruction. It was shown that Vhs targets mRNA for degradation, via associating with translation initiation factors (45, 46). Thus, infection-induced translational activation of mRNAs of immune function may be preferentially degraded by Vhs. US11, a dsRNA-binding protein packaged in the virion, binds to RIG-I and MDA5 in a manner independent of its RNA-binding domain and inhibits their interactions with MAVS (47). Released from the tegument upon infection, UL37 displays an intrinsic enzyme activity to deamidate RIG-I during HSV-1 infection (42). Deamidation of two asparagine residues in the helicase domain of RIG-I abrogates its binding to dsRNA and subsequent RNA-stimulated helicase activity. As such, recombinant HSV-1 containing a point mutation that abolishes UL37 deamidase activity triggers more robust RIG-I activation and potent IFN responses than wild-type HSV-1. This recombinant HSV-1 is highly attenuated in vitro and in mice.
Stimulator of interferon genes (STING), also known as Met-Pro-Tyr-Ser (MPYS), mediator of IRF3 activation (MITA) (48), Endoplasmic Reticulum IFN stimulator (ERIS) (49), transmembrane protein 173 (TMEM173), is an endoplasmic reticulum adaptor that mediates innate immune activation in response to cyclic dinucleotides (CDNs) (48, 50). These CDNs include cyclic-di-AMP, cyclic-di-GMP, and cyclic-GMP-AMP. Upon activation, STING oligomerizes and translocates to the trans-Golgi network (TGN) where STING recruits TBK1 and IKK kinase complex to activate IRF and NF-κB, leading to the production of type I interferons and inflammatory cytokines. Notably, K27- and K63-linked polyubiquitin chains of STING are essential for the activation of the transcription activity of IRF3 (51).
In response to HSV-1 infection, STING is required for IFN production in multiple cell lines, including murine embryonic fibroblasts, macrophages and dendritic cells (52). Moreover, STING protects mice from HSV-1 lethal infection via intravenous and intracerebral routes, while mucosal infection of HSV-1 in STING−/− mice results in the increased corneal and trigeminal ganglia viral titers, demonstrating the importance of STING in host defense against HSV-1 in vivo (53). As one of the countermeasures, HSV-1 deploys UL36 (also known as VP1–2) to deubiquitinate STING, thus impeding the activation of TBK1 and IRF3. UL36 is the largest protein encoded within HSV and likely provides a scaffold for tegument protein incorporation (54). In fact, HSV-1 ΔDUB mutant induces more robust IFN induction in microglia and shows reduced replication in the brain compared with wild-type HSV-1 (55). Besides UL36, γ134.5 (ICP34.5) interacts with STING and disrupts its translocation from endoplasmic reticulum to Golgi apparatus, a step that is essential for STING to transduce innate immune signals (56). Lastly, ICP27, expressed during HSV-1 de novo infection in macrophages, interacts with the activated TBK1-STING signalosome to inhibit IRF3 activation (57), thereby evading immune response downstream of STING.
Paradoxically, in several cell lines, including HEp-2 and HeLa, STING is found to be stabilized by HSV-1 viral proteins, and depletion of STING impedes HSV-1 productive infection (58). The mechanism by which STING enhances HSV-1 replication in these cell lines remains unclear. Nevertheless, these findings suggest the opposing function of STING in host defense is cell type-dependent. One possibility is that the STING-dependent immune defense pathway is rewired by the tumor cell to promote proliferation or growth, which is usurped by HSV-1.
Cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS), is a sensor that binds to virus or cell-associated DNA in a sequence-independent manner (59). cGAS is previously demonstrated to mainly reside in the cytoplasm to detect cytoplasmic DNA as it represents a danger signal. Recent finding also suggests that cGAS enters the nucleus to inhibit DNA double-stranded breaks and promotes tumorigenesis (60). The binding of cGAS to DNA induces its oligomerization and concomitant conformational changes, enabling its enzymatic domain to catalyze the synthesis of a second messenger, cyclic GMP-AMP (cGAMP), from cellular GTP and ATP. cGAMP serves as a ligand to activate STING and the downstream IRF and NF-κB branched pathways (61). Therefore, the cGAS-STING pathway plays pivotal roles in inducing type I IFNs and cytokines to mount innate immune responses against bacterial, DNA viruses, cellular genome instability and other related danger signals.
Soon after its discovery, the contribution of cGAS to antagonize HSV-1 was demonstrated by that cGAS−/− mice were more susceptible to HSV-1 challenge than wild-type mice (62). cGAS deficiency also led to impaired IFN expression in microglia, thus resulting in the susceptibility of the mice to herpes simplex encephalitis (HSE) upon ocular infection (63). As cGAS senses HSV-1 DNA to trigger innate immune responses, it is not surprising that HSV-1 evolved diverse strategies to antagonize this pattern recognition receptor and its downstream signaling. HSV-1 tegument protein UL41, an mRNA-specific endonuclease, downregulates the mRNA and protein level of cGAS to abrogate cGAS- and STING-mediated signaling (64). In addition, another tegument protein VP22 is found to interact with cGAS and directly inhibit its enzymatic activity (65). β-catenin is found to be required for the optimal induction of IFN induced by cGAS. As such, HSV-1 US3 phosphorylates β-catenin at Thr556 and blocks its nuclear translocation to dampen cGAS-dependent host antiviral responses (66). We identify that HSV-1 tegument deamidase UL37 targets cGAS, in addition to RIG-I, for deamidation (67). Deamidation of N210, which is in close proximity to the catalytic triad of cGAS, abolishes its catalytic activity to synthesize cGAMP, thereby shutting down cGAMP production and downstream signaling. Interestingly, deamidation does not impair DNA-binding and oligomerization of cGAS, implying the dominant negative effect of deamidated cGAS on the cGAS-IFN pathway. Importantly, non-human primates are resistant to HSV-1 infection and their cGAS proteins contain histine or arginine at the equivalent location of residue 210, which makes cGAS resistant to HSV-1–induced deamidation (67). These findings suggest that cGAS deamidation contributes to the host susceptibility of HSV-1. Altogether, our studies highlight the utmost immune evasion functions of UL37 by targeting multiple sensors for deamidation.
IFI16 belongs to the IFN-inducible PYHIN-200 gene family. Members in this family carry the signature HIN domain (IFI16 has two) that binds to dsDNA or ssDNA in a sequence-independent manner. In addition to a DNA-binding domain, IFI16 contains a PYRIN domain (PYD) that mediates protein-protein interactions. Binding to DNA can trigger two distinct signaling pathways, i.e., IFN signaling and inflammasome signaling, depending on the nature of the stimulating signal (68). During viral infection, IFI16 is proposed to bind viral DNA and trigger the activation of STING and induction of IFN, although the detailed mechanism remains unknown (68).
Depletion of IFI16 in the cornea by in vivo siRNA transfection results in the decrease of IRF3 phosphorylation and correspondingly increase of HSV-1 viral replication, while MyD88−/− and Trif−/− double knockout mice demonstrate similar IFN production compared to WT controls. This result suggests that IFI16, rather than TLRs, mediates the innate immune response in corneal epithelium against HSV-1 (69). Unlike the cornea, IFI16 is largely dispensable for host defense against HSV-2 in the urogenital system, while TLR2, TLR9, and DAI are essential for IFN and cytokine production (70). In primary human foreskin fibroblasts (HFFs), nuclear resident IFI16 senses the HSV-1 DNA to induce IFN production in a STING-dependent manner, while cGAS promotes IFI16-mediated IFN induction via stabilizing IFI16 protein (71). However, how nuclear IFI16 triggers STING activation remains to be addressed. Another study reports a different mechanism of IFI16 to restrict HSV-1 replication in multiple cell lines, where IFI16 selectively binds to HSV-1 transcription start sties to block viral gene transcription via inducing repressive histone modifications (72). These studies demonstrate multiple functions of IFI16 to restrict HSV-1 replication. The controversy on IFI16 and cGAS as the HSV-1 sensor could be explained by the differential compartmentalization of the two sensors. For example, cGAS plays major roles in sensing HSV-1 DNA in macrophages, where viral DNA is exposed in the cytosol due to capsid degradation (39). In contrast, IFI16 may detect and mount innate immune response in cells where DNA is delivered into the nucleus. However, a number of recent studies reported that part of the cGAS resides in the nucleus (60, 73), adding to the complexity of the nuclear DNA-sensing mechanism against HSV-1. Following the sensing of nuclear DNA, an immediate question is how nuclear signal of activated IFI16 is relayed to the cytoplasmic STING and downstream signaling events. These questions call for further investigation.
To counteract IFI16, HSV-1 encodes ICP0, a E3 ligase, to induce IFI16 degradation in a proteasome-dependent manner (74). Interesting, ICP0 reduces IFI16 protein in HFFs and oral keratinocytes (NOKs), whereas HSV-1–induced loss in IFI16 protein is dependent on Vhs-mediated mRNA decay (75). These findings highlight distinct mechanisms by which HSV-1 antagonizes the expression of IFI16 in a cell type-specific fashion.
Transfection of bacterial, viral and cellular DNA into macrophages leads to the formation of inflammasomes (76, 77). The inflammasome is a protein complex formed in response to the activation of several sensors, including the NLR (NOD-like receptor) or PYHIN (containing pyran and HIN domains) proteins upon recognizing varieties of viral PAMPs (78). Genetic manipulation via RNA interference in cultured cells and knockout in mice demonstrates that AIM2 is a cytosolic DNA sensor (79–84). AIM2 consists of a C-terminal HIN-200 domain and an N-terminal pyrin domain, which form an intramolecular loop to establish a self-repressing state (85). Upon stimulation, the HIN-200 domain binds directly to the sugar-phosphate backbone of dsDNA, releasing the pyrin domain which forms homotypic interaction with the pyrin domain of apoptosis-associated speck-like protein containing a carboxy-terminal CARD (ASC) (86). The CARD of ASC then interacts with the CARD of pro-caspase-1 to activate caspase-1 and form the AIM2 inflammasome. Finally, the activated caspase-1 cleaves the pro-IL-1β and pro-IL-18 and induces the release of the mature IL-1β and IL-18 from the cell (85, 87). Importantly, the expression of the sensors and the cytokine precursors requires a priming step that is stimulated by pro-inflammatory signals such as LPS. Besides cytokine releasing, activated AIM2 inflammasome also induces an inflammatory cell death to protect infected host from invading pathogens, including intracellular bacteria (88, 89), vaccinia virus (79, 89), and murine cytomegalovirus (a beta-herpesvirus) (89). In the absence of microbial infection, AIM2 also plays an important role in sensing damage-associated molecular patterns (DAMPs) released by distressed or damaged cells (90). The cellular DNA, as one of the DAMPs produced by nuclear DNA damage or immunogenic cell death, activates AIM2 and initiates inflammasome assembly to promote the secretion of IL-1β and IL-18 (91–93). It was reported that inhibition of potassium efflux inhibited the secretion of IL-1β mediated by AIM2 (94), suggesting that like NLRP3, AIM2 inflammasome activation may depend on distinct ion fluxes and concentrations (95).
Because viral DNA can be released into the cytoplasm during HSV-1 infection in macrophages, it should engage cytoplasmic DNA sensors such as AIM2 (39, 96). However, HSV-1 infection of macrophages induces inflammasome activation independent of AIM2, in stark contrast to murine cytomegalovirus that efficiently induces AIM2-dependent inflammasome activation (89). Based on this observation, it is hypothesized that HSV-1 may have evolved a mechanism(s) to evade AIM2-dependent inflammasome activation. Indeed, HSV-1 tegument protein VP22 was reported to inhibit AIM2-dependent inflammasome activation and IL-1β secretion in infected macrophages (97). VP22 interacts with AIM2 and prevents its oligomerization, an essential step in AIM2 inflammasome activation. Consequently, recombinant VP22-deficient HSV-1 (HSV-1ΔVP22) potently induces AIM2 inflammasome activation and subsequent secretion of IL-1β and IL-18. Similarly, HSV-2 and PRV VP22 homologues also demonstrate inhibitory effect on AIM2-dependent inflammasome activation (97). Interestingly, KSHV tegument protein ORF63 interacts with an inflammasome sensor NLRP1 and prevents its oligomerization to block inflammasome activation in ways similar to VP22 (98). Collectively, these findings reveal a mechanism that the inhibition of AIM2-dependent inflammasome activation appears to be shared by diverse herpesviruses.
DNA-dependent activator of IRFs (DAI, also known as ZBP-1) is the first putative cytosolic DNA receptor identified (99). DAI recruits TBK1 and IRF3, and induces type I IFN production after binding to dsDNA. HSV-1 induces DAI activation in the murine fibroblast cell line L929 (99). Structurally, DAI contains tandem amino-terminal Z-DNA-binding domains, Zα1 and Zα2 (also called Zβ), which binds double-stranded Z-form DNA (99, 100). In addition to Z-DNA-binding domains, DAI also contains RIP homotypic interaction motifs (RHIMs) that trigger necroptosis and activate NF-κB pathway by interacting with the receptor-interacting kinase-3 (RIPK3) (101, 102). RIPK3 and its downstream substrate Mixed Lineage Kinase domain-Like protein (MLKL) contributes to the programmed necrotic cell death, which curtails viral replication and restricts dissemination of virions (103, 104). In this pathway, DAI acts as a nucleic acid sensor to detect viral RNA transcripts rather than the cytoplasmic viral DNA during the infection of influenza (105–108), vaccinia (109), MCMV (110, 111), and HSV1 (112, 113), and triggers necroptosis. On the other hand, these viruses manage to inhibit necroptosis by encoding gene products to target DAI-mediated signaling (109, 114, 115). MCMV M45 inhibits virus-induced necroptosis by blocking DAI-dependent oligomerization and activation of RIPK3 (115), while HSV-1 deploys ICP6 (UL39) to prevent the formation of DAI-RIPK3-MLKL complex induced by virus infection (116). Therefore, MCMV and HSV1 deploy similar strategies to block DAI-mediated necroptosis and maintain the viability of the infected cells.
DsRNA-dependent protein kinase R (PKR), an interferon-stimulated serine/threonine kinase, is a potent antiviral protein whose activity depends on dsRNA binding (117, 118). PKR consists of two dsRNA-binding domains (dsRBDs) and a kinase domain (119). Encountering dsRNA, DsRBDs bind to the backbone of a RNA in a sequence-independent manner, thus triggering a conformational change and subsequent oligomerization of PKR (120). PKR undergoes cis-phosphorylation within the activation loop by its kinase domain (121). Once activated, PKR phosphorylates the translation initiation factor eIF2α, leading to the suppression of eIF2 in cap-dependent translation and a global shutdown of translation (122). As such, PKR exerts its antiviral activity on a broad spectrum of DNA and RNA viruses by blocking the translation of cellular and viral mRNAs. Besides inhibiting protein synthesis, PKR was reported to promote the RLR-mediated type I interferon signaling via phosphorylation of IκB (123) and stabilization of mRNAs of type I interferon genes (124). Nevertheless, the molecular mechanism underpinning the PKR-dependent amplification of interferon signaling is not fully understood.
In HSV-1–infected cells, PKR is shown to be activated, which is required for the activation of NF-κB (125). It remains unclear whether dsRNAs activating PKR originate from HSV-1–encoded symmetrical transcripts or the HSV-1–infected host genome. Interestingly, HSV-1 may activate PKR via a cellular protein activator known as PACT (126). To escape PKR-mediated antiviral responses, HSV-1 deploys γ134.5 (ICP34.5) to recruit cellular protein phosphatase 1α (PP1) that counteracts PKR-mediated eIF2α phosphorylation and restores translation (127–129). Moreover, ICP34.5 antagonizes Beclin 1-mediated autophagy, an antiviral process that is dependent on PKR (130). US11, a tegument protein, directly binds PKR to inhibit its conformational change and activation by PACT (126). It was later demonstrated that virion-associated US11, rather than its expression during replication, mediates the inhibition of PKR autophosphorylation (131). Additionally, Vhs degrades RNAs to block PKR activation during early stages of HSV-1 infection (132). These diverse viral strategies to antagonize PKR further emphasize the importance of PKR as a potent anti-HSV molecule.
In the current review, we summarized the recent findings on the contribution of cytosolic PRRs in sensing HSVs and the direct countermeasures evolved by these viruses (Figure 1). During HSV-1 infection, diverse molecular patterns throughout the virus life cycle, including viral DNA genome, transcription-derived RNA species, unmasked cellular RNA, etc., are dynamically sensed by the PRRs to trigger innate immune signaling. On the other side of the coin, HSV develops various countermeasures, ranging from transcription shutoff, protein degradation, interaction competition to enzymatic activity disruption, to escape PRR detection. These lessons learnt from our characterization of HSV-PRR interactions deepen the understanding of the nature and regulations of PRR-mediated innate immune signaling, and may lead to the discovery of novel antiviral modalities. Importantly, strategies interfering with these manipulations can be potentially developed into novel antiviral therapies, while immune modulatory-deficient HSV mutants are good candidates for vaccine and oncolytic virus strains, further highlighting the translational value of the basic research.
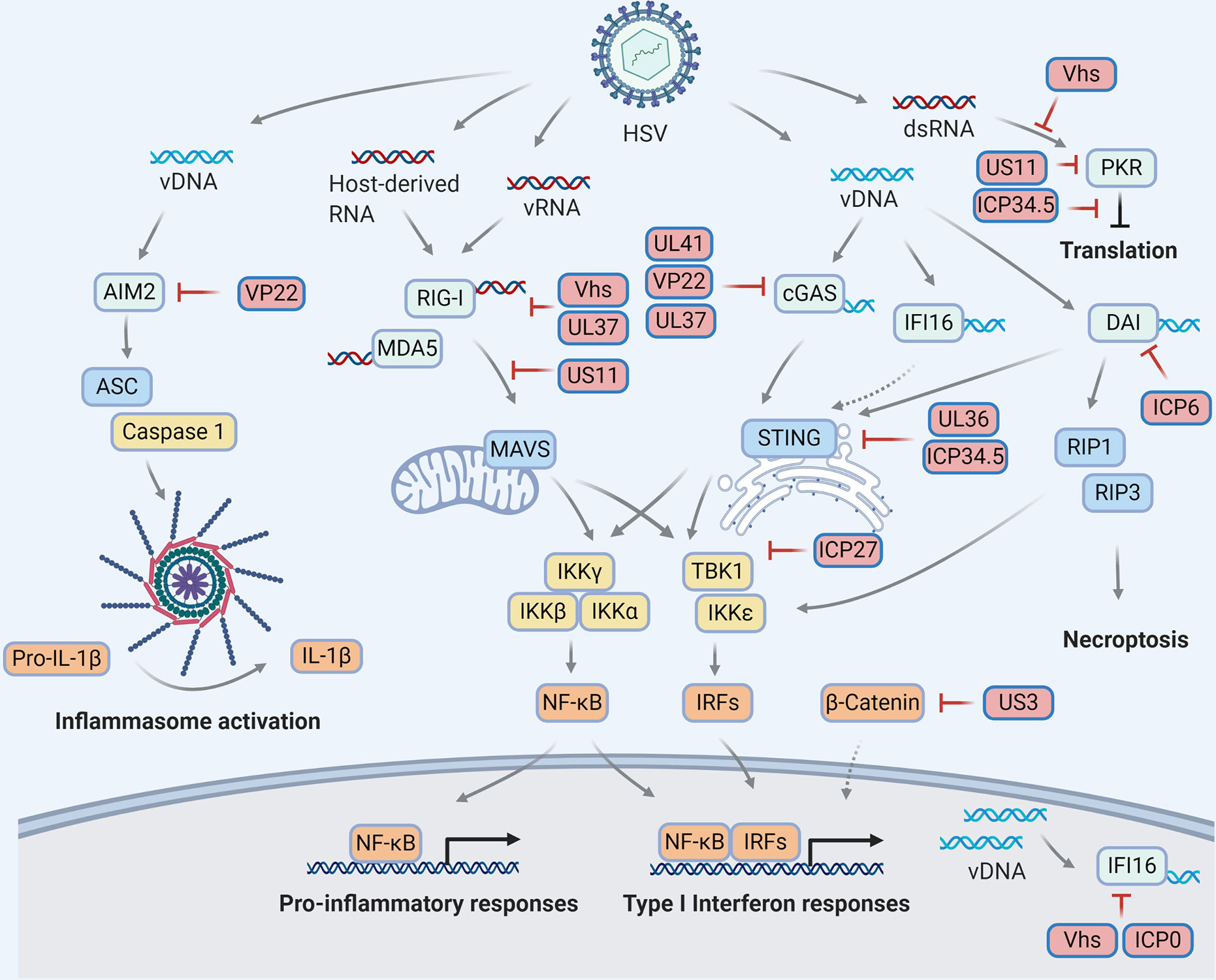
Figure 1 HSV manipulations on the cytosolic pattern recognition receptors. Viral infection derives molecular patterns (DNAs and RNAs) which activate pattern recognition receptors (light blue) to transduce innate immune signaling through distinct adaptor proteins (blue) and ultimately trigger antiviral responses, including but not limited to cytokine production, inflammasome activation, translational inhibition and necroptosis. To escape innate immune surveillance, HSV encode viral proteins (red) to manipulate multiple steps of each signaling pathway via diverse mechanisms, resulting in a complex HSV-host interaction network on innate immunity.
One of the knowledge gaps to fill is on the functional redundancy of the PRRs in sensing HSV, as controversy remains on defining the ‘true’ sensor for HSV. While a simple explanation is that such redundancy may have been evolved by the host as backup protections during the arms races with the virus, emerging studies have implicated these PRRs have unique roles in mounting immune responses and antagonizing HSV-1 infection in a temporal and cell/tissue-specific manner. Notably, part of the previous studies relies heavily on a single model cell line, sometimes cancer cell lines, to characterize HSV-PRR interactions, which limits the scope of the findings as some PRRs or signaling pathways may be missing. Thus, more investigations are needed to systematically address the contributions of PRRs, including more in vivo studies of HSV infection using tissue-specific knockout mouse models.
Interestingly, ‘functional redundancy’ applies to the virus too, because HSVs deploy multiple proteins to target the same sensor, e.g. RIG-I and cGAS, though via distinct molecular mechanism. One possibility is that these viral proteins sequentially work on the sensor throughout the HSV life cycle to maintain constant immune evasion. Alternatively, these viral proteins are cooperating to synergistically antagonize PRR functions or operate in a tissue-specific manner. It will require more work to define the ‘major players’ in these viral proteins that potently antagonize innate immune responses, as efforts in manipulating such proteins confer the greatest susceptibility of the virus to immune response and thus could serve as the best antiviral strategy.
JZ and PF conceived the paper. JZ, CQ, YL, YR, and PF wrote the paper. All authors contributed to the article and approved the submitted version.
Work in the Feng laboratory is supported by grants from National Institute of Health (DE027556, DE026003 and CA221521 to PF and DE028973 to JZ) and startup funds from the Herman Ostrow School of Dentistry of University of Southern California.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Sehrawat S, Kumar D, Rouse BT. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front Cell Infect Microbiol (2018) 8:177. doi: 10.3389/fcimb.2018.00177
2. Sharma V, Mobeen F, Prakash T. Comparative Genomics of Herpesviridae Family to Look for Potential Signatures of Human Infecting Strains. Int J Genomics (2016) 2016:9543274. doi: 10.1155/2016/9543274
3. Steiner I, Benninger F. Update on herpes virus infections of the nervous system. Curr Neurol Neurosci Rep (2013) 13:414. doi: 10.1007/s11910-013-0414-8
4. Looker KJ, Magaret AS, May MT, Turner KM, Vickerman P, Gottlieb SL, et al. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS One (2015) 10:e0140765. doi: 10.1371/journal.pone.0140765
5. Looker KJ, Magaret AS, Turner KM, Vickerman P, Gottlieb SL, Newman LM. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS One (2015) 10:e114989. doi: 10.1371/journal.pone.0114989
6. Danastas K, Miranda-Saksena M, Cunningham AL. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int J Mol Sci (2020) 21(14):5150. doi: 10.3390/ijms21145150
7. Tognarelli EI, Palomino TF, Corrales N, Bueno SM, Kalergis AM, Gonzalez PA. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front Cell Infect Microbiol (2019) 9:127. doi: 10.3389/fcimb.2019.00127
8. Grünewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, et al. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science (2003) 302:1396–8. doi: 10.1126/science.1090284
9. Wu W, Newcomb WW, Cheng N, Aksyuk A, Winkler DC, Steven AC. Internal Proteins of the Procapsid and Mature Capsids of Herpes Simplex Virus 1 Mapped by Bubblegram Imaging. J Virol (2016) 90:5176–86. doi: 10.1128/JVI.03224-15
10. Kukhanova MK, Korovina AN, Kochetkov SN. Human herpes simplex virus: life cycle and development of inhibitors. Biochem (Mosc) (2014) 79:1635–52. doi: 10.1134/S0006297914130124
11. Wirtz L, Möckel M, Knebel-Mörsdorf D. Invasion of Herpes Simplex Virus 1 into Murine Dermis: Role of Nectin-1 and Herpesvirus Entry Mediator as Cellular Receptors during Aging. J Virol (2020) 94(5):e02046–19. doi: 10.1128/JVI.02046-19
12. Petermann P, Thier K, Rahn E, Rixon FJ, Bloch W, Ozcelik S, et al. Entry mechanisms of herpes simplex virus 1 into murine epidermis: involvement of nectin-1 and herpesvirus entry mediator as cellular receptors. J Virol (2015) 89:262–74. doi: 10.1128/JVI.02917-14
13. Wang K, Tomaras GD, Jegaskanda S, Moody MA, Liao HX, Goodman KN, et al. Monoclonal Antibodies, Derived from Humans Vaccinated with the RV144 HIV Vaccine Containing the HVEM Binding Domain of Herpes Simplex Virus (HSV) Glycoprotein D, Neutralize HSV Infection, Mediate Antibody-Dependent Cellular Cytotoxicity, and Protect Mice from Ocular Challenge with HSV-1. J Virol (2017) 91(19):e00411–17. doi: 10.1128/JVI.00411-17
14. Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell (1996) 87:427–36. doi: 10.1016/S0092-8674(00)81363-X
15. Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science (1987) 235:1056–9. doi: 10.1126/science.2434993
16. Nicoll MP, Proenca JT, Efstathiou S. The molecular basis of herpes simplex virus latency. FEMS Microbiol Rev (2012) 36:684–705. doi: 10.1111/j.1574-6976.2011.00320.x
17. Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, et al. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog (2010) 6:e1000991. doi: 10.1371/journal.ppat.1000991
18. Alandijany T. Host Intrinsic and Innate Intracellular Immunity During Herpes Simplex Virus Type 1 (HSV-1) Infection. Front Microbiol (2019) 10:2611. doi: 10.3389/fmicb.2019.02611
19. Chen N, Xia P, Li S, Zhang T, Wang TT, Zhu J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life (2017) 69:297–304. doi: 10.1002/iub.1625
20. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5:730–7. doi: 10.1038/ni1087
21. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA (2002) 99:637–42. doi: 10.1073/pnas.022637199
22. Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA (2004) 101:17264–9. doi: 10.1073/pnas.0407639101
23. Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, et al. Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity (2009) 31:25–34. doi: 10.1016/j.immuni.2009.05.008
24. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi: 10.1016/j.cell.2005.08.012
25. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell (2005) 19:727–40. doi: 10.1016/j.molcel.2005.08.014
26. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol (2005) 6:981–8. doi: 10.1038/ni1243
27. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature (2005) 437:1167–72. doi: 10.1038/nature04193
28. Peisley A, Wu B, Xu H, Chen ZJ, Hur S. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature (2014) 509:110–4. doi: 10.1038/nature13140
29. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell (2011) 146:448–61. doi: 10.1016/j.cell.2011.06.041
30. Loo YM, Gale M Jr. Immune signaling by RIG-I-like receptors. Immunity (2011) 34:680–92. doi: 10.1016/j.immuni.2011.05.003
31. Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature (2007) 446:916–20. doi: 10.1038/nature05732
32. Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem (2009) 284:807–17. doi: 10.1074/jbc.M804259200
33. Gack MU, Nistal-Villan E, Inn KS, Garcia-Sastre A, Jung JU. Phosphorylation-mediated negative regulation of RIG-I antiviral activity. J Virol (2010) 84:3220–9. doi: 10.1128/JVI.02241-09
34. Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, et al. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity (2013) 38:437–49. doi: 10.1016/j.immuni.2012.11.018
35. Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell (2010) 141:315–30. doi: 10.1016/j.cell.2010.03.029
36. da Silva LF, Jones C. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res (2013) 175:101–9. doi: 10.1016/j.virusres.2013.04.005
37. Choi MK, Wang Z, Ban T, Yanai H, Lu Y, Koshiba R, et al. A selective contribution of the RIG-I-like receptor pathway to type I interferon responses activated by cytosolic DNA. Proc Natl Acad Sci USA (2009) 106:17870–5. doi: 10.1073/pnas.0909545106
38. Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell (2009) 138:576–91. doi: 10.1016/j.cell.2009.06.015
39. Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J Immunol (2013) 190:2311–9. doi: 10.4049/jimmunol.1202749
40. Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, et al. Herpes simplex virus infection is sensed by both Toll-like receptors and retinoic acid-inducible gene- like receptors, which synergize to induce type I interferon production. J Gen Virol (2009) 90:74–8. doi: 10.1099/vir.0.005389-0
41. Melchjorsen J, Rintahaka J, Soby S, Horan KA, Poltajainen A, Ostergaard L, et al. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J Virol (2010) 84:11350–8. doi: 10.1128/JVI.01106-10
42. Zhao J, Zeng Y, Xu S, Chen J, Shen G, Yu C, et al. A Viral Deamidase Targets the Helicase Domain of RIG-I to Block RNA-Induced Activation. Cell Host Microbe (2016) 20:770–84. doi: 10.1016/j.chom.2016.10.011
43. Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol (2018) 19:53–62. doi: 10.1038/s41590-017-0005-y
44. Yao XD, Rosenthal KL. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J Gen Virol (2011) 92:1981–93. doi: 10.1099/vir.0.030296-0
45. Feng P, Everly DN Jr., Read GS. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J Virol (2001) 75:10272–80. doi: 10.1128/JVI.75.21.10272-10280.2001
46. Feng P, Everly DN Jr., Read GS. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J Virol (2005) 79:9651–64. doi: 10.1128/JVI.79.15.9651-9664.2005
47. Xing J, Wang S, Lin R, Mossman KL, Zheng C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol (2012) 86:3528–40. doi: 10.1128/JVI.06713-11
48. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity (2008) 29:538–50. doi: 10.1016/j.immuni.2008.09.003
49. Sun W, Li Y, Chen L, Chen H, You F, Zhou X, et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA (2009) 106:8653–8. doi: 10.1073/pnas.0900850106
50. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
51. Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity (2010) 33:765–76. doi: 10.1016/j.immuni.2010.10.013
52. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
53. Parker ZM, Murphy AA, Leib DA. Role of the DNA Sensor STING in Protection from Lethal Infection following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. J Virol (2015) 89:11080–91. doi: 10.1128/JVI.00954-15
54. Cardone G, Newcomb WW, Cheng N, Wingfield PT, Trus BL, Brown JC, et al. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J Virol (2012) 86:4058–64. doi: 10.1128/JVI.00012-12
55. Bodda C, Reinert LS, Fruhwurth S, Richardo T, Sun C, Zhang BC, et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J Exp Med (2020) 217(7):e20191422. doi: 10.1084/jem.20191422
56. Pan S, Liu X, Ma Y, Cao Y, He B. Herpes Simplex Virus 1 gamma134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J Virol (2018) 92(20):e01015–18. doi: 10.1128/JVI.01015-18
57. Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J (2016) 35:1385–99. doi: 10.15252/embj.201593458
58. Kalamvoki M, Roizman B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci USA (2014) 111:E611–7. doi: 10.1073/pnas.1323414111
59. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (2013) 339:786–91. doi: 10.1126/science.1232458
60. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature (2018) 563:131–6. doi: 10.1038/s41586-018-0629-6
61. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi: 10.1126/science.1229963
62. Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science (2013) 341:1390–4. doi: 10.1126/science.1244040
63. Reinert LS, Lopusna K, Winther H, Sun C, Thomsen MK, Nandakumar R, et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun (2016) 7:13348. doi: 10.1038/ncomms13348
64. Su C, Zheng C. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J Virol (2017) 91(6):e02414–16. doi: 10.1128/JVI.02414-16
65. Huang J, You H, Su C, Li Y, Chen S, Zheng C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J Virol (2018) 92(15):e00841–18. doi: 10.1128/JVI.00841-18
66. You H, Lin Y, Lin F, Yang M, Li J, Zhang R, et al. beta-Catenin Is Required for the cGAS/STING Signaling Pathway but Antagonized by the Herpes Simplex Virus 1 US3 Protein. J Virol (2020) 94(5):e01847–19. doi: 10.1128/JVI.01847-19
67. Zhang J, Zhao J, Xu S, Li J, He S, Zeng Y, et al. Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell Host Microbe (2018) 24:234–48.e5. doi: 10.1016/j.chom.2018.07.004
68. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi: 10.1038/ni.1932
69. Conrady CD, Zheng M, Fitzgerald KA, Liu C, Carr DJ. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol (2012) 5:173–83. doi: 10.1038/mi.2011.63
70. Triantafilou K, Eryilmazlar D, Triantafilou M. Herpes simplex virus 2-induced activation in vaginal cells involves Toll-like receptors 2 and 9 and DNA sensors DAI and IFI16. Am J Obstet Gynecol (2014) 210:122.e1–122 e10. doi: 10.1016/j.ajog.2013.09.034
71. Orzalli MH, Broekema NM, Diner BA, Hancks DC, Elde NC, Cristea IM, et al. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc Natl Acad Sci USA (2015) 112:E1773–81. doi: 10.1073/pnas.1424637112
72. Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PloS Pathog (2014) 10:e1004503. doi: 10.1371/journal.ppat.1004503
73. Volkman HE, Cambier S, Gray EE, Stetson DB. Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife (2019) 8:e47491. doi: 10.7554/eLife.47491
74. Orzalli MH, DeLuca NA, Knipe DM. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci USA (2012) 109:E3008–17. doi: 10.1073/pnas.1211302109
75. Orzalli MH, Broekema NM, Knipe DM. Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J Virol (2016) 90:8351–9. doi: 10.1128/JVI.00939-16
76. Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature (2008) 452:103–7. doi: 10.1038/nature06664
77. Stacey KJ, Ross IL, Hume DA. Electroporation and DNA-dependent cell death in murine macrophages. Immunol Cell Biol (1993) 71(Pt 2):75–85. doi: 10.1038/icb.1993.8
78. Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell (2016) 165:792–800. doi: 10.1016/j.cell.2016.03.046
79. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature (2009) 458:514–8. doi: 10.1038/nature07725
80. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458:509–13. doi: 10.1038/nature07710
81. Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol (2009) 10:266–72. doi: 10.1038/ni.1702
82. Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science (2009) 323:1057–60. doi: 10.1126/science.1169841
83. Jin T, Perry A, Jiang J, Smith P, Curry JA, Unterholzner L, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity (2012) 36:561–71. doi: 10.1016/j.immuni.2012.02.014
84. Jin T, Perry A, Smith P, Jiang J, Xiao TS. Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J Biol Chem (2013) 288:13225–35. doi: 10.1074/jbc.M113.468033
85. Man SM, Karki R, Kanneganti TD. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol (2016) 46:269–80. doi: 10.1002/eji.201545839
86. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev (2015) 265:6–21. doi: 10.1111/imr.12296
87. Brunette RL, Young JM, Whitley DG, Brodsky IE, Malik HS, Stetson DB. Extensive evolutionary and functional diversity among mammalian AIM2-like receptors. J Exp Med (2012) 209:1969–83. doi: 10.1084/jem.20121960
88. Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep (2014) 6:196–210. doi: 10.1016/j.celrep.2013.12.014
89. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol (2010) 11:395–402. doi: 10.1038/ni.1864
90. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
91. Komada T, Chung H, Lau A, Platnich JM, Beck PL, Benediktsson H, et al. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J Am Soc Nephrol (2018) 29:1165–81. doi: 10.1681/ASN.2017080863
92. Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science (2016) 354:765–8. doi: 10.1126/science.aaf7532
93. Lian Q, Xu J, Yan S, Huang M, Ding H, Sun X, et al. Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res (2017) 27:784–800. doi: 10.1038/cr.2017.54
94. Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol (2010) 11:385–93. doi: 10.1038/ni.1859
95. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
96. Morrison EE, Wang YF, Meredith DM. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J Virol (1998) 72:7108–14. doi: 10.1128/JVI.72.9.7108-7114.1998
97. Maruzuru Y, Ichinohe T, Sato R, Miyake K, Okano T, Suzuki T, et al. Herpes Simplex Virus 1 VP22 Inhibits AIM2-Dependent Inflammasome Activation to Enable Efficient Viral Replication. Cell Host Microbe (2018) 23:254–65.e7. doi: 10.1016/j.chom.2017.12.014
98. Gregory SM, Davis BK, West JA, Taxman DJ, Matsuzawa S, Reed JC, et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science (2011) 331:330–4. doi: 10.1126/science.1199478
99. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature (2007) 448:501–5. doi: 10.1038/nature06013
100. Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, et al. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA (2008) 105:5477–82. doi: 10.1073/pnas.0801295105
101. Kuriakose T, Kanneganti TD. ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol (2018) 39:123–34. doi: 10.1016/j.it.2017.11.002
102. Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep (2009) 10:916–22. doi: 10.1038/embor.2009.109
103. Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol (2008) 181:6427–34. doi: 10.4049/jimmunol.181.9.6427
104. Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature (2008) 451:725–9. doi: 10.1038/nature06537
105. Kuriakose T, Zheng M, Neale G, Kanneganti TD. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J Immunol (2018) 200:1489–95. doi: 10.4049/jimmunol.1701538
106. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe (2016) 20:674–81. doi: 10.1016/j.chom.2016.09.014
107. Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol (2016) 1(2):aag2045. doi: 10.1126/sciimmunol.aag2045
108. Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, Mishra A, et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J Exp Med (2017) 214:2217–29. doi: 10.1084/jem.20170550
109. Koehler H, Cotsmire S, Langland J, Kibler KV, Kalman D, Upton JW, et al. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc Natl Acad Sci USA (2017) 114:11506–11. doi: 10.1073/pnas.1700999114
110. Sridharan H, Ragan KB, Guo H, Gilley RP, Landsteiner VJ, Kaiser WJ, et al. Murine cytomegalovirus IE3-dependent transcription is required for DAI/ZBP1-mediated necroptosis. EMBO Rep (2017) 18:1429–41. doi: 10.15252/embr.201743947
111. Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J (2017) 36:2529–43. doi: 10.15252/embj.201796476
112. Guo H, Gilley RP, Fisher A, Lane R, Landsteiner VJ, Ragan KB, et al. Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis (2018) 9:816. doi: 10.1038/s41419-018-0868-3
113. Pham TH, Kwon KM, Kim YE, Kim KK, Ahn JH. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J Virol (2013) 87:3076–86. doi: 10.1128/JVI.02860-12
114. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe (2012) 11:290–7. doi: 10.1016/j.chom.2012.01.016
115. Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe (2010) 7:302–13. doi: 10.1016/j.chom.2010.03.006
116. Wang X, Li Y, Liu S, Yu X, Li L, Shi C, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA (2014) 111:15438–43. doi: 10.1073/pnas.1412767111
117. Roberts WK, Hovanessian A, Brown RE, Clemens MJ, Kerr IM. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature (1976) 264:477–80. doi: 10.1038/264477a0
118. Sen GC, Taira H, Lengyel P. Interferon, double-stranded RNA, and protein phosphorylation. Characteristics of a double-stranded RNA-activated protein kinase system partially purified from interferon treated Ehrlich ascites tumor cells. J Biol Chem (1978) 253:5915–21.
119. Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J (1998) 17:5458–65. doi: 10.1093/emboj/17.18.5458
120. Dey M, Cao C, Dar AC, Tamura T, Ozato K, Sicheri F, et al. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell (2005) 122:901–13. doi: 10.1016/j.cell.2005.06.041
121. Dey M, Mann BR, Anshu A, Mannan MA. Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J Biol Chem (2014) 289:5747–57. doi: 10.1074/jbc.M113.527796
122. Hovanessian AG. The double stranded RNA-activated protein kinase induced by interferon: dsRNA-PK. J Interferon Res (1989) 9:641–7. doi: 10.1089/jir.1989.9.641
123. Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol (2000) 20:1278–90. doi: 10.1128/MCB.20.4.1278-1290.2000
124. Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe (2010) 7:354–61. doi: 10.1016/j.chom.2010.04.007
125. Taddeo B, Luo TR, Zhang W, Roizman B. Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc Natl Acad Sci USA (2003) 100:12408–13. doi: 10.1073/pnas.2034952100
126. Peters GA, Khoo D, Mohr I, Sen GC. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J Virol (2002) 76:11054–64. doi: 10.1128/JVI.76.21.11054-11064.2002
127. Mohr I, Gluzman Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J (1996) 15:4759–66. doi: 10.1002/j.1460-2075.1996.tb00853.x
128. Chou J, Roizman B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc Natl Acad Sci USA (1994) 91:5247–51. doi: 10.1073/pnas.91.12.5247
129. He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc Natl Acad Sci USA (1997) 94:843–8. doi: 10.1073/pnas.94.3.843
130. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe (2007) 1:23–35. doi: 10.1016/j.chom.2006.12.001
131. Ishioka K, Ikuta K, Sato Y, Kaneko H, Sorimachi K, Fukushima E, et al. Herpes simplex virus type 1 virion-derived US11 inhibits type 1 interferon-induced protein kinase R phosphorylation. Microbiol Immunol (2013) 57:426–36. doi: 10.1111/1348-0421.12048
Keywords: herpes simplex virus, pattern recognition receptors, RIG-I/MDA5, CGAS, IFI16, AIM2, DAI, PKR
Citation: Zhao J, Qin C, Liu Y, Rao Y and Feng P (2021) Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 11:613799. doi: 10.3389/fimmu.2020.613799
Received: 03 October 2020; Accepted: 14 December 2020;
Published: 29 January 2021.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Angello Retamal-Diaz, Universidad de Antofagasta, ChileCopyright © 2021 Zhao, Qin, Liu, Rao and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Zhao, anVuekB1c2MuZWR1; Pinghui Feng, cGluZ2h1aWZAdXNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.