 Wenpeng Song
Wenpeng Song Jing Ye
Jing Ye Nanfang Pan
Nanfang Pan Chunyu Tan
Chunyu Tan Martin Herrmann
Martin Herrmann- 1Department of Rheumatology, West China Hospital of Sichuan University, Chengdu, China
- 2Department of Internal Medicine 3, Universitätsklinik Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
In recent years, neutrophil extracellular traps at the forefront of neutrophil biology have proven to help capture and kill pathogens involved in the inflammatory process. There is growing evidence that persistent neutrophil extracellular traps drive the pathogenesis of autoimmune diseases. In this paper, we summarize the potential of neutrophil extracellular traps to drive the pathogenesis of rheumatoid arthritis and experimental animal models. We also describe the diagnosis and treatment of rheumatoid arthritis in association with neutrophil extracellular traps.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease with high disability and increased mortality. It is characterized by progressive joint damage and synovial membrane hypertrophy. There are stark differences in the prevalence among different ethnicities and populations. RA is a heavy burden for the patients, their families, and society. To date, studies have shown that RA is a multifactorial disease involving age, sex, environmental, epigenetic, and genetic factors. However, the pathogenesis of RA is not fully understood (1). Many studies have shown that both innate immune response and adaptive immune response contribute to the etiopathogenesis of RA (2). It’s considered that the formation of autoantibodies to citrullinated antigens (ACPA) is a critical pathogenic event involved in the development of RA. Neutrophils isolated from patients suffering from autoimmune diseases present enhanced formation of neutrophil extracellular traps (NETs). The role of neutrophils in autoimmune disease is still elusive (3). The release of cytotoxic products [e.g., degradation enzymes and reactive oxygen species (ROS)] from activated neutrophils into the synovial fluid and pannus in RA has been known for a long time and is considered important for RA (4, 5). In recent years, it was discovered that neutrophils participate in the inflammatory progression of RA through multiple regulatory immune mechanisms, including directly secreting cytokines and chemokines, and releasing neutrophil granules that activate or inactivate cytokines and chemokines. Enzymes upregulate the expression of MHC II and promote cell-cell interactions.
A novel role of neutrophils, the release of NETs, has attracted increasing attention. Upon pharmacological activation with phorbol myristate acetate (PMA) (6), interleukin 8 (IL-8) (7), or lipopolysaccharide (LPS) (8), neutrophils release granule proteins and chromatin to form NETs. The release of NETs (9) constitutes a novel programmed cell death that differs from apoptosis (10). LPS-induced NET formation increases with adhesion and substrate elasticity, while PMA-induced NET formation is independent of adhesion (6). NETs are composed of chromatin and granular proteins, which trap and kill bacteria (11, 12). Most DNA is derived from nuclei however, mitochondrial DNA is also included. The proteins consist of neutrophil elastase (NE), myeloperoxidase (MPO), histones, defensins, calprotectin (13), matrix metalloproteinase-9, and others (14). During NET formation, NE, MPO, and peptidyl arginine deiminase (PAD4) promote nuclear decondensation and histone citrullination, respectively (15). NETs can be quantified and analyzed by DNA area and NETosis analysis (DANA). Higher frequencies of NETs are detected in subjects with RA (16).
NET formation conventionally occurs via the NADPH oxidase (NOX) and ROS-dependent suicidal pathway in which neutrophils rupture and release NETs. Suicidal NETosis is triggered by the engagement of specific receptors or other biomolecules, such as IgG-Fc receptors, Toll-like receptors (TLRs), complement molecules, and cytokines on neutrophils (11, 17–19). The formation of suicidal NETosis is a gradual process that is commonly initiated by the generation of ROS. Then NE and MPO are transported into the nucleus where histones are modified. Finally, nuclear and cellular membranes break, and NETs are released (9, 10, 14). During this process, substantial morphological changes occur. Neutrophils flatten and form membrane protrusions after stimulation (11). Nuclear lobules disappear and chromatin decondense. The inner and outer nuclear membranes detach. The nuclear membrane disintegrates into vesicles, and nuclear material mixes with the cytoplasm to form a homogenous mass. Finally, the neutrophils round up and rupture to release the NETs (10). ROS are pivotal for suicidal NETosis formation (20). ROS are mainly generated by NOX during the “respiratory burst” of neutrophils (21). Patients with chronic granulomatous disease harbor mutations of NOX genes and show reduced NET formation (10, 22). Finally, the NOX complex converts molecular oxygen to hydrogen peroxide, which is a substrate of MPO and is sufficient to induce NET formation (10). The reaction of hydrogen peroxide and MPO can form hypochlorous acid. The latter induces the generation of chlorinated polyamines that may cross-link NET proteins, which maintains the ordered structure of NETs and increases the capacity to trap bacteria (23). MPO partly binds to NE to form the azurosome complex that spans granular membranes without dissolution of the granular membranes (24). NE is a critical enzyme involved in many pathways of NET formation. Methicillin-resistant Staphylococcus aureus (MRSA) infected mice with NE deficiency fail to form NETs (25). However, other studies have demonstrated that NE is not required for NET formation induced by noninfectious stimuli; meanwhile, NE deficiency has little effect on histone citrullination (26). NE combines with F-actin filaments to enter the nucleus before MPO. The proteolytic activity of NE is determined by MPO. Patients with mutant MPO also show reduced NET formation (22). As NE translocates from cytoplasm to nucleus, it cleaves histones and participates in chromatin decondensation (27).
In addition to chromatin decondensation performed by NE, another important chromatin modification is histone citrullination driven by PAD4. During NET formation, calcium influx of neutrophils activates a high amount of PAD4, which catalyze histone to citrullinated protein. This enzyme citrullinates arginine residues in the core histones H2A, H3, H4, thus reducing their positive charge, which weakens the interactions between histones and DNA and further promotes chromatin decondensation (28). Five calcium molecules are bound to every PAD4 molecule (29). Interestingly, citrullination driven by PAD4 is inhibited accompanied by inhibition of NOX (28), which may be due to the NOX-induced increase in cytoplasmic calcium levels that activate PAD4 (30). Citrullination driven by PAD4 is induced by LPS and PMA (31). PADI4-deficient mice failed to form NETs after treatment with certain stimuli (32–34).
However, Clark et al. first reported vital NETosis in which neutrophils remained impermeable for SYTOX Green after releasing NETs, which suggested that the neutrophils maintained an intact plasma membrane during NET formation (7). Subsequent researchers found that NETs were induced by blebbing of the nuclear envelope and vesicular exportation in S. aureus infection (35). Vital NETosis is activated by pathogen-associated molecular patterns (PAMPs) or endogenous damage-associated molecular patterns (DAMPs). Stimuli recognized by TLR4, such as LPS, may initiate vital NETosis. The nucleus loses its multilobular shape and becomes rounded. Then, the nuclear double membranes vanish, and vesicles composed of DNA filaments bud. These vesicles approach toward the plasma membrane. DNA is released through a small area on the cell surface. Suicidal NETosis and vital NETosis can be distinguished by cleaved N-terminal histone tails (36).
Roles of NETs in RA
Pathogenesis of RA Related to NETs Formation
Various elements in the peripheral blood of patients with RA, such as autoantibodies or immunostimulatory molecules, reportedly stimulate NET formation (Table 1). Excessive NET formation leads to the production of deaminated antigens such as citH2A, citH2B, and citH4 histones (Figure 1). Furthermore, NET-borne citrullinated vimentin is a pivotal autoantigen that stimulates the secretion of proinflammatory cytokines (e.g., TNF-α and IL-1) and the expression of PADI4 and receptor activator of nuclear factor kappa B ligand (RANKL) in fibroblast-like synoviocytes (FLSs) (44). FLSs, key effector cells of inflammation in RA, produce multiple cytokines that cause damage in the joints (45). In a joint with RA, presented citrullinated antigens induce antigen-driven autoimmune responses and lead to the generation of anti-NET autoantibodies. Thus, chronic inflammation and autoimmunity exist for a long time. Anti-NET RA recombinant monoclonal antibodies (rmAbs) derived from CD19+ synovial B cells of patients with RA constantly bind NETs.

Table 1 Stimuli inducing neutrophil extracellular traps (NETs) formation.
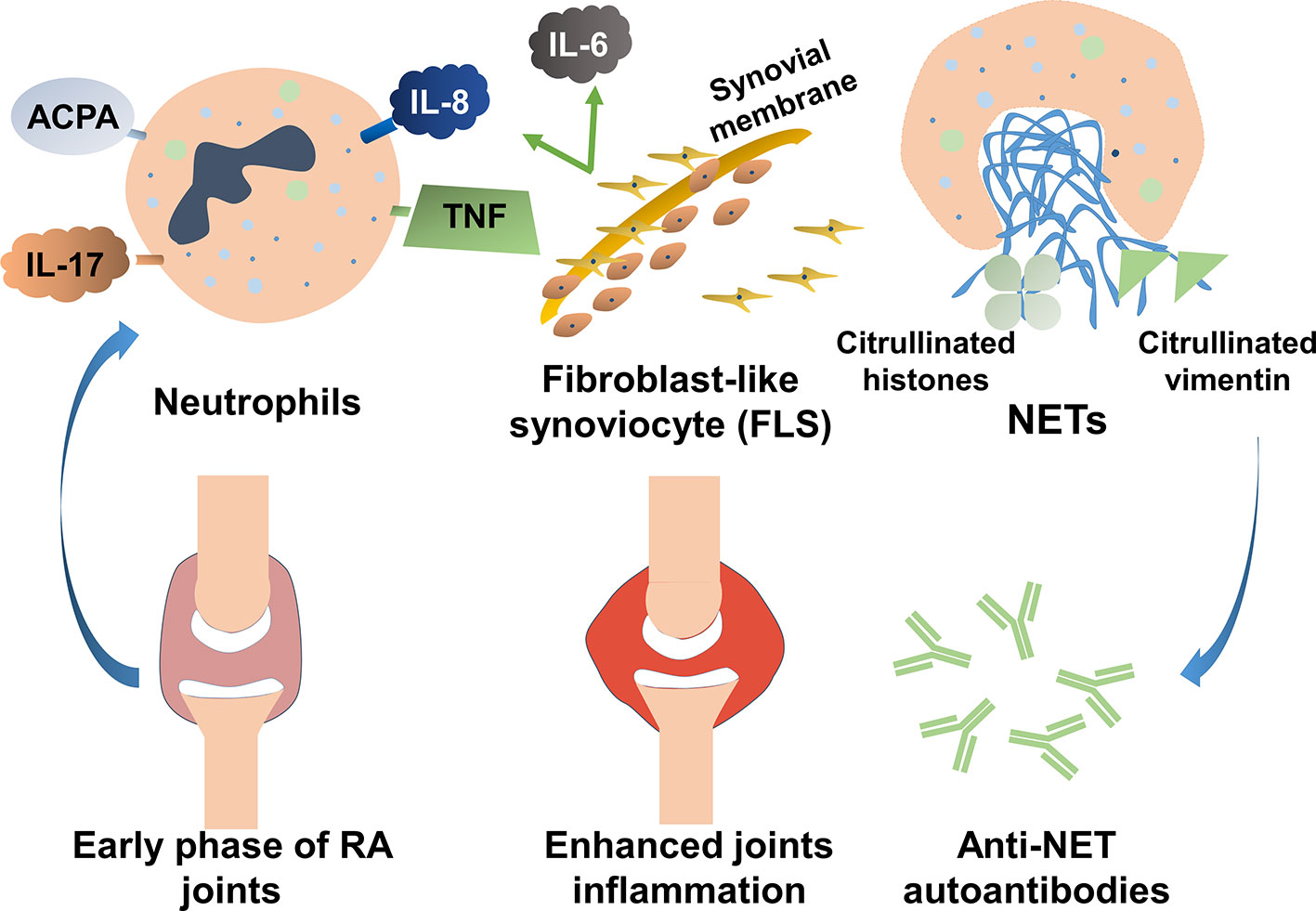
Figure 1 The role of neutrophil extracellular traps (NETs) in the pathogenesis of rheumatoid arthritis (RA). Various elements in the peripheral blood of patients with RA can stimulate NET formation. Excessive NET formation leads to the production of deaminated antigens such as citH2A, citH2B, and citH4 histones. In a joint with RA, presented citrullinated antigens induce antigen-driven autoimmune responses and lead to the generation of anti-NET autoantibodies. Thus, persistent inflammation of the synovial membranes occurs.
The immunoreactivity of NET-Ags depends on somatic hypermutation (SHM) within the Ig variable H (VH) and variable L (VL) chains of synovial B cells. Moreover, Fab-N-linked-glycosylation determines the reactivity of the autoantibodies (46). Rheumatoid factor (RF), anti-citrullinated protein antibodies (ACPAs), and other autoantibodies in peripheral blood or synovial fluid robustly support NET formation in RA (47). IgG or IgM collected from peripheral blood or synovial fluid of patients with RA induce more NET formation than antibodies from healthy controls (38). Recently, NET-derived elastase results in cartilage matrix disruption and induction of membrane-bound peptidylarginine deiminase-2 released by FLSs. Cartilage fragments are subsequently citrullinated and presented to antigen-specific CD4+ T cells (48). In the NOX/ROS pathway, PAD4-induced histone citrullination promotes chromatin decondensation and NET formation (49). The chromatin-associated protein DEK regulates the structure of extracellular chromatin (50, 51). In models of RA (51), NET formation and protein citrullination are shown to be prevented by depletion of DEK or administration of DEK-targeted aptamers. Both strategies alleviate the symptoms of RA.
In patients with RA, IL-8, IL-17A, and TNF-α reportedly induced NET formation (38). Upon exposure to IL-17A, neutrophils in RAlead to NET formation when the cells are primed with TNF-α. Correlations with NET formation have also been detected for a higher serum level of C-reactive protein (CRP) and a higher erythrocyte sedimentation rate. Furthermore, histone citrullination alone with NET formation can be triggered by the treatment of neutrophils with supernatants harvested from IL-15-stimulated CD69(+)CD8(+) T cells, leading to the extracellular release of citrullinated proteins (52). Conversely, immune complexes induce “incomplete” NET formation (53, 54). Ribonucleoprotein-containing immune complexes induce NET formation depending on mitochondrial ROS rather than NOX (55, 56), which correlates with hypercitrullinated proteins (57) and production of IFN by plasmacytoid dendritic cells (58).
Signal Transduction Pathways Correlating With NETs
Several underlying signal transduction pathways may promote NET formation in RA and the molecular mechanisms may be pleomorphic. In RA, high concentrations of NE, MPO, PAD4/DNA-complex, and ROS production correspond to the elevated formation of NETs (31, 59).
In neutrophils, Rac is a subunit of the NOX complex (60). Guanosine exchange factor (GEF) activator, Vav, and the p21-activated kinases (Paks) are involved in Rac signaling pathway (61). NOX is indispensable for oxidative burst-dependent NET formation (60–62). Inhibition of NOX reduces NET formation and induces non-canonical NETs (10). ROS are related to the lytic NET formation (63) and stimulate the activation of NE. NE and MPO are also released from azurophilic granules into the nucleoplasm (25). In the nucleus, NE proteolytically cleaves histones and thus interferes with the dense package of chromatin (27). Many physiological and artificial stimuli can activate the MPO-NE pathway (22).
In the peripheral blood of patients with RA, ACPAs stimulate neutrophils to release PAD enzymes (64). Porphyromonas gingivalis and smoking are known risk factors for RA, and overexpression of endogenous or bacterial PAD enzymes drives citrullination (65, 66). PAD4 depends on Ca2+ (67) and is activated via the ROS pathways to convert internal arginine to citrullines (8, 9). Upon PAD4 activation, locally released citrullinated histones enhance the generation of highly mutated clonal B cells resulting in the generation of high-affinity ACPAs (68). At a high titer, fibrinogen citrullinated by PAD 4 acts as the preferred targets for ACPAs (69). Additionally, human leukocyte antigen (HLA)-DR bound PAD4 is recognized by T cells and further contributes to the production of antibodies responded to citrullinated proteins, such as ACPAs and anti-PAD antibodies (70). Anti-PAD4 antibodies have been reported to be closely related to ACPAs (71–73). Kolfenbach et al. evaluated the prediagnosis serum samples of 83 patients with RA and found that 15 RA samples had anti-PAD4 antibodies with a high specificity of 98.8% (71). Interestingly, Erika Darrah et al. first detected PAD4-specific CD4+ T cells in peripheral blood mononuclear cells (PBMCs) of RA patients and found that protease granzyme B (GrB) induced structure changes of PAD4 and promoted the presentation of CD4+ T cell epitopes (74). Overall, further studies are need to demonstrate correlation between PAD4 and RA citrullinome.
Deficiency or inhibition of PADI4 reduces the formation and the size of NETs and alleviates arthritis symptoms in many models, except the murine K/BxN model (75). These findings suggest that PADI4 acts downstream of ROS in NET formation and generates autoantigens that amplify the inflammatory response that precipitates in the pathogenesis of RA (32, 76, 77). PADI4 thus participates in the initiation rather than the effector phase of RA. PAD4, which is associated with histone deamination, can catalyze hypercitrullination by immune-mediated membranolytic pathways (57, 78). Interestingly, in a TNF-induced model of citrullination and arthritis, protein citrullination is executed by PAD2 instead of PAD4. PAD2 is not associated with NET formation (79). Relatively high activity of putatively neutrophil-derived PAD4 has been reported in RA synovial fluid (80). The pathogenesis of RA is also related to T cells specific for citrullinated epitope (81). In brief, the NOX pathways and PAD4 activity can be regarded as critical elements that regulate NET formation and generation of citrullinated autoantigens in RA (82).
NETs Promote Autoantibody Production, Cytokine Expression, and Cell Activation
Citrullinated components of NETs often serve as self-antigens recognized in the serum of patients with RA (80). Aberrant NETs may promote the externalization of citrullinated autoantigens and immunostimulatory molecules, which enhances the expression of epitopes related to the pathogenesis of RA (38). In RA or osteoarthritis (OA), the levels of IL-6 and IL-8 are upregulated in the presence of NETs, resulting in the activation of FLSs (38, 83). LL-37/DNA complexes induce NETs that activate plasmacytoid dendritic cells via TLR7 and TLR9 to produce type I IFN (84). Moreover, NETs are abnormally accumulated in some patients with SLE due to the DNase I inhibitory factors (85, 86), leading to IFN-a release. IFN-a not only enhances NETosis but also induces activation of autoreactive T- and B cells to synthesize autoAbs, such as anti-dsDNA, anti-HNP, and anti-LL37 autoAbs. Moreover, NETs can trigger the production of IL-1β and IL-18, and further stimulate NETosis. These vicious cycles contribute to the imbalance immune homeostasis of SLE. Similarly, NETs are involved in multiple sclerosis (MS). NETosis secretion of antimicrobial proteins induces elevated T-cell activation resulting in tissue damage in MS (87).
NETs triggered by microscopic cholesterol crystals also take part in atherosclerosis (88). NETs induce the activation of leukocytes, platelets, and endothelial cells and further lead to endothelial dysfunction (89). Moreover, NETs promote the production of IL-6 and pro-IL-1β in macrophages (49). Accordingly, these increased cytokines accelerate T helper 17 (TH17) cells differentiation and subsequently induce immune cell recruitment in atherosclerotic lesions. Neutrophils infiltration of culprit lesions results in plaque rupture and erosion via NETs (90, 91). Very recently, significantly higher plasma levels of NETs are observed in the carotid lesion site (CLS) of stroke patients. NETs decorated with phosphatidylserine (PS) are detected in thrombi. NET formation requires the synergy of CLS plasma and activated platelets (PLTs). PS-bearing NETs can induce the formation of thrombin and fibrin as well as the conversion of endothelial cells to a procoagulant phenotype (92). These findings indicate that NETs are indispensable in the pathogenesis of many diseases, such as RA, SLE, MS, atherosclerosis, and stroke via multiple molecular mechanisms.
NET and Citrullinated Autoantigens Form a Vicious Cycle in RA
In RA, neutrophils infiltrate synovial tissue, rheumatoid nodules, and the skin (38), when neutrophils form NETs, deaminate proteins, and initiate ACPA production (57). Furthermore, circulating low-density granulocytes (LDGs) in patients with RA tend to increasingly form NETs (19). FLSs activated by NETs express IL-17A, TNF-α, and IL-8 and infiltrate the cartilage, where they enhanced proinflammatory responses (10). The enhanced release of inflammatory cytokines from FLSs driven by NETs causes joint damage and further worsens the condition (31). The secretory leukocyte protease inhibitors can prevent proteolytic maturation of cytokines related to NET formation. Skin lesions may be associated with the insufficient activity of the secretory leukocyte protease inhibitors (93). Importantly, these cytokines trigger the vicious cycle of NET formation and autoantibody biogenesis (38). IL-8 and IL-17 recruit neutrophils and promote the exposure of autoantigens (12, 94). Therefore, NET formation plays a critical role in the pathogenesis of RA. A comprehensive understanding of the mechanisms involved in NET formation may help us develop new therapies via targeting NETs to treat NET-related diseases.
NETs are Associated with RA in Mouse Models
Neutrophils Drive the Inflammation of Murine Arthritis
RA is an autoimmune disease characterized by progressive destruction of joints. The pathogenesis of RA is still elusive. Researchers usually establish murine models to analyze the pathogenetic sequelae of RA (95). Daisuke and colleagues established an experimental model of male BALB/cAnNCrj (BALB/c) mice injected with an anti-type II collagen antibody and LPS (95). Histological analysis showed that neutrophils were the vast majority of infiltrating cells in the joint space. To determine the effect of neutrophils on arthritis, monoclonal antibodies (mAbs) against Gr-1 (the RB6-8C5 mAb) were intravenously injected into arthritic mice to deplete circulating neutrophils. These experiments suggested that neutrophils are indispensable for the development of arthritis. It is commonly believed that neutrophils play a key role in inflammatory diseases due to their secretion of cytotoxic products (4). However, neutrophils are now considered to be not only effectors of the innate immune systems but also key players in the regulatory circuits of the immune system (96). FcγRs activate neutrophils and trigger a series of signaling events, including ROS generation, protease release as well as the production of chemokines and cytokines. These mediators recruit additional neutrophils and regulate the functions of other immune cells. Hence, they participate in the regulatory network and interplay of innate and adaptive immunity (97). Neutrophils isolated from patients with RA functionally differ from those from healthy controls. Blood- and synovial fluid-derived neutrophils from patients with RA trigger ROS production and display enhanced NET formation (38).
The Role of NETs in the Etiopathogenesis of RA
NETs are considered to contribute to the pathogenesis of RA (9). Degradation or citrullination of histones driven by PAD4 promotes chromatin decondensation and NET release (38). Furthermore, PAD4 exacerbates inflammatory arthritis and is crucial in some pathways of NET formation (79). Compared to wild-type mice, PADI4 (encoding PAD4)-deficient mice induced by glucose-6-phosphate isomerase showed less severe inflammatory arthritis and reduced autoantibody titers (75). Similarly, in murine collagen-induced arthritis (CIA), inhibition of PADI4 reduced the formation of NETs and arthritis relief (98). However, PAD4 was dispensable in spontaneous arthritis in the K/BxN mouse model (99). NET formation and arthritis in the murine TNFα-induced inflammatory arthritis were investigated to identify the roles of PAD2 and PAD4 for citrullination. PAD2 mediated TNFα-induced citrullination and arthritis but was dispensable for NET formation (99). PAD4, which is involved in NET formation, was dispensable for citrullination. These studies indicate that various pathogenic pathways may cause murine arthritis. These can be dependent or independent of NETs.
Further evidence supported the roles of NETs in the pathogenesis of RA that blocked NET formation and protein citrullination was caused by treatment with DEK-targeted aptamers, as DEK is essential for certain pathways of NET formation (38, 100). Autoantibodies that recognize DEK have been detected in the sera of patients with autoimmune diseases, such as systemic lupus erythematosus (SLE) and adolescent idiopathic arthritis (JIA) (100). DEK acts as a chemoattractant, triggers inflammatory responses, and plays an important role in a murine model of arthritis. Aptamers targeting DEK could reduce NETs formation, slow the progression of joint inflammation, and ameliorate the disease symptoms in arthritic mice (51).
Another established murine model of RA is the K/BxN mice. The pathology is similar to that of human RA. The K/BxN mice are generated by crossing KRN-C57BL/6 mice, which carry a transgenic T cell receptor, with autoimmunity-prone non-obese diabetes (NOD) mice (101). K/BxN mice develop IgG autoantibodies against glucose-6-phosphate isomerase, which precipitate joint damage.
Mice lacking functional NOX have the further aggravation of arthritic symptoms. ROS suppression occurs in patients with chronic granulomas disease (CGD) due to impaired function of NOX (35). This implies that ROS in NETs are derived from additional mechanisms beyond the NOX pathway (86). NET formation triggered by nicotine (102) was found to be dependent on mitochondrial ROS rather than depend on NOX (20). However, Cl-amidine, a PAD inhibitor, did not inhibit the formation of mitochondrial ROS but inhibit NETs in the New Zealand mixed 2328 (NZM) murine mice (34).
Diagnosing RA with NETs
Currently, the laboratory diagnosis of RA relies on the detection of RF and ACPAs (59, 103). These autoantibodies can be found in most RA patients, and the titer of ACPAs correlates with the severity of RA (4). Although many autoantibodies markers have been applied for patients’ diagnosis with RA, ACPAs are the most disease-specific markers with the highest specificity and sensitivity (103). Khandpur et al (38). analyzed 55 Patients with RA and 36 healthy volunteers or patients with OA. The results showed that NET formation was associated with the levels of ACPAs and indicated NETs were a potential target for ACPAs. ACPAs include antibodies targeting keratin (AKA), perinuclear factor (APF), profillagrin/fillagrin (AFA), Sa, and artificial cyclic citrulline peptide (CCP). The diagnostic specificity of four kinds of ACPAs (APF, AKA, AFA, and CCP (II)) for RA was more than 90%, which is significantly higher than that of RF (77.7%), suggesting that ACPAs can be employed as effective diagnostic antibodies for RA. However, the sensitivity of ACPAs for RA differs due to differences in antigens preparation and detection methods (104–106).
Recently, some studies have focused on the detection of potential signaling pathways that lead to the increase of NET formation in RA. This is to determine whether the products of NET formation are useful for diagnosis. NETs as target biomarkers have been reported in many autoimmune diseases. Levels of human neutrophil peptide 1–3 (HNP 1–3), a part of NETs, were found to be significantly higher in patients with lupus nephritis (LN) than in healthy controls. They are an independent indicator of LN [P = 0.006, odds ratio (OR) = 7.5, 95% confidence interval (CI), 1.782–31.842]. Moreover, the NET-inducing capacity might be a novel biomarker of ANCA-associated vasculitis (AAV). The levels of NET degradation products, such as circulating free DNA (cfDNA), free nucleosomes, NE-DNA, and MPO-DNA complexes, are reportedly increased in patients with RA (3). NET-derived products were previously analyzed in vitro by microscopy and enzyme-linked immunosorbent assay (ELISA). Receiver operating characteristic (ROC) curves showed spontaneously increased formation of NETs and production of ROS in patients with RA (59). NET-derived cell-free nucleosomes in RA serum showed diagnostic value with an area under the ROC > 97% with 91% sensitivity and 92% specificity (59). No significant differences were detected between ACPA-positive and ACPA-negative patients (59). Recently, the level of anti-NET antibodies (ANETA) in RA serum was reported to be significantly higher in rheumatoid factor-positive than that in seronegative patients (107). The collective evidence suggests that the quantitative detection of the NET-derived products may be a useful complementary tool to identify individuals at risk and to monitor patients with RA.
Therapeutic Targeting of NET Formation to Treat RA
Clearance of NETs
DNAse-1 dismantles NETs in vitro. In vivo, DNase-1 does not interfere with NET production, but fragments the DNA and destroys the backbone of the NETs (9). DNase-1 reportedly promotes the escape of group A Streptococcus (GAS) from being killed by NETs. Enhanced neutrophil depletion of GAS and reduced virulence occur in the presence of the DNase I inhibitor G-actin (108). Recently, several studies have reported that bacterial DNases degrade NETs, allowing the bacteria to escape killing in NETs (109–111). However, Bryan et al. injected S. aureus intraperitoneally into mice and monitored the infections with minimally invasive nonsurgical luminescent imaging, showing that DNase reduced bacterial growth (112). Kolaczkowska and colleagues also demonstrated that DNase effectively eliminated NET-borne DNA and inhibited the proteolytic activity of NE (25). Nevertheless, NETs still show some antimicrobial activities, as most of the histones remained. The circulating zymogen form of factor VII activating protease (FSAP) can be activated by histones and the nuclear lobules of NETs. NETs bound to FSAP fail to activate pro-FASP. However, histones release after the degradation of NETs by DNase dramatically stimulating pro-FASP activity (113). Pathogenic micro-organisms produce DNases that inhibit the generation of ROS in the later stage and lead to escape the killing in NETs (114). In addition, NETs are cleared via phagocytosis of macrophages, which increases the release of cfDNA (115). Whether the pathogenesis of RA involves macrophage dysfunction needs further examination. The cfDNA levels in synovial fluids were correlated with neutrophil counts but not with macrophage counts (80). The increased NETs levels in RA synovial fluids may be caused by either impaired activity of DNase-1 or by inhibitors of DNase-1. Serum DNase-1 activity is negatively correlated with inflammatory markers and neutrophil counts, suggesting that insufficient DNase-1 activity is an important factor in the regulation of NET persistence. The elevated cfDNA levels in the synovial fluid may be an important source of “altered self.” Only very few eosinophils and mast cells exist in the synovial fluid of patients with RA, suggesting that cfDNA are mainly derived from NETs. An advantage of DNase-1 is low toxicity, which has been verified in murine models of breast cancer (116), lupus (117), or lung damage (118). Exogenous administration of DNase I may be used to dismantle NETs and can, therefore, be considered for the treatment of RA.
Inhibition of NET Formation
Additionally, drugs that reduce the formation of NETs may also be used to treat and relieve RA. Delivery of the NE inhibitor sivelestat via a nanoparticle system to LPS-induced endotoxin shock mouse model inhibits NET formation, reduces circulating NE, and prevents mice from endotoxic shock (119). CI-amidine can reduce protein citrullination in the pGIA mouse model (120). Rituximab and belimumab reduce NET formation by blocking the immune-complexes formation (121). Emodin accelerates apoptosis and suppresses autophagy and NET formation by reducing IL-6, IFN-γ, and TNF-α in the murine adjuvant-induced arthritis (AA) (122). Polydatin (PD) reduces NET formation of bone marrow-derived neutrophils and in patients with RA. Similarly, in CIA mice, the deposition of NETs in the ankle joints is decreased by PD-treatment (123). Ascomycin and cyclosporine A can decrease IL-8 induced NET formation by inhibiting the calcineurin pathway (124). Triptolide (TP) exhibits potential as an RA therapeutic by lowering neutrophil recruitment and downregulating the expression of TNF-α and IL-6. TP is also able to suppress NET formation and autophagy of neutrophils (125). Moreover, celastrol can inhibit NOX-dependent NET formation (126). Tocilizumab likewise shows the potential to reduce autoantibody levels and, consequently, immune complex formation in patients with RA (127). Nevertheless, there are differences in the immune system and physiological function between murine models and humans. Given the limited scope of current clinical standard therapies, more clinical research is required to establish NET reducing therapies.
Perspectives and Conclusion
The accumulated data on the role of NETs in RA has brought NETs into focus as novel therapeutic targets for RA. The future will tell whether a blockage in NET formation or increased NET catabolism will win the race.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was partially supported by the German Research Foundation (DFG) (TRR241: B04; CRC1181: C03, Z02; FOR 2886 projects B03. This work was funded by the National Natural Science Foundation of China [grant number 81501412].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
2. Klareskog L, Lundberg K, Malmström V. Autoimmunity in rheumatoid arthritis: citrulline immunity and beyond. Adv Immunol (2013) 118:129–58. doi: 10.1016/b978-0-12-407708-9.00003-0
3. Pérez-Sánchez C, Ruiz-Limón P, Aguirre MA, Jiménez-Gómez Y, Arias-de la Rosa I, Ábalos-Aguilera MC, et al. Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in Rheumatoid Arthritis patients. J Autoimmun (2017) 82:31–40. doi: 10.1016/j.jaut.2017.04.007
4. Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol (2014) 10:593–601. doi: 10.1038/nrrheum.2014.80
5. Hallett MB, Lloyds D. Neutrophil priming: the cellular signals that say ‘amber’ but not ‘green’. Immunol Today (1995) 16:264–8. doi: 10.1016/0167-5699(95)80178-2
6. Erpenbeck L, Gruhn AL, Kudryasheva G, Günay G, Meyer D, Busse J, et al. Effect of Adhesion and Substrate Elasticity on Neutrophil Extracellular Trap Formation. Front Immunol (2019) 10:2320. doi: 10.3389/fimmu.2019.02320
7. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med (2007) 13:463–9. doi: 10.1038/nm1565
8. Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun (2009) 1:194–201. doi: 10.1159/000206974
9. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med (2017) 23:279–87. doi: 10.1038/nm.4294
10. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol (2007) 176:231–41. doi: 10.1083/jcb.200606027
11. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303:1532–5. doi: 10.1126/science.1092385
12. Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol (2005) 66:1146–54. doi: 10.1016/j.humimm.2005.11.003
13. Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PloS Pathog (2009) 5:e1000639. doi: 10.1371/journal.ppat.1000639
14. Pinegin B, Vorobjeva N, Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun Rev (2015) 14:633–40. doi: 10.1016/j.autrev.2015.03.002
15. Neubert E, Bach KM, Busse J, Bogeski I, Schön MP, Kruss S, et al. Blue and Long-Wave Ultraviolet Light Induce in vitro Neutrophil Extracellular Trap (NET) Formation. Front Immunol (2019) 10:2428:2428. doi: 10.3389/fimmu.2019.02428
16. Rebernick R, Fahmy L, Glover C, Bawadekar M, Shim D, Holmes CL, et al. DNA Area and NETosis Analysis (DANA): a High-Throughput Method to Quantify Neutrophil Extracellular Traps in Fluorescent Microscope Images. Biol Proced Online (2018) 20:7. doi: 10.1186/s12575-018-0072-y
17. Munks MW, McKee AS, Macleod MK, Powell RL, Degen JL, Reisdorph NA, et al. Aluminum adjuvants elicit fibrin-dependent extracellular traps in vivo. Blood (2010) 116:5191–9. doi: 10.1182/blood-2010-03-275529
18. Brinkmann V, Zychlinsky A. Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol (2007) 5:577–82. doi: 10.1038/nrmicro1710
19. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
20. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
21. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
22. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood (2011) 117:953–9. doi: 10.1182/blood-2010-06-290171
23. Csomós K, Kristóf E, Jakob B, Csomós I, Kovács G, Rotem O, et al. Protein cross-linking by chlorinated polyamines and transglutamylation stabilizes neutrophil extracellular traps. Cell Death Dis (2016) 7:e2332. doi: 10.1038/cddis.2016.200
24. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep (2014) 8:883–96. doi: 10.1016/j.celrep.2014.06.044
25. Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun (2015) 6:6673. doi: 10.1038/ncomms7673
26. Martinod K, Witsch T, Farley K, Gallant M, Remold-O'Donnell E, Wagner DD. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thromb Haemost (2016) 14:551–8. doi: 10.1111/jth.13239
27. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol (2010) 191:677–91. doi: 10.1083/jcb.201006052
28. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol (2009) 184:205–13. doi: 10.1083/jcb.200806072
29. Rohrbach AS, Slade DJ, Thompson PR, Mowen KA. Activation of PAD4 in NET formation. Front Immunol (2012) 3:360:360. doi: 10.3389/fimmu.2012.00360
30. Zimmerman MC, Takapoo M, Jagadeesha DK, Stanic B, Banfi B, Bhalla RC, et al. Activation of NADPH oxidase 1 increases intracellular calcium and migration of smooth muscle cells. Hypertension (2011) 58:446–53. doi: 10.1161/hypertensionaha.111.177006
31. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev (2017) 16:1160–73. doi: 10.1016/j.autrev.2017.09.012
32. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med (2010) 207:1853–62. doi: 10.1084/jem.20100239
33. Hemmers S, Teijaro JR, Arandjelovic S, Mowen KA. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PloS One (2011) 6:e22043. doi: 10.1371/journal.pone.0022043
34. Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest (2013) 123:2981–93. doi: 10.1172/jci67390
35. Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood (2009) 114:2619–22. doi: 10.1182/blood-2009-05-221606
36. Pieterse E, Rother N, Yanginlar C, Gerretsen J, Boeltz S, Munoz LE, et al. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann Rheum Dis (2018) 77:1790–8. doi: 10.1136/annrheumdis-2018-213223
37. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Differential Use of Human Neutrophil Fcγ Receptors for Inducing Neutrophil Extracellular Trap Formation. J Immunol Res (2016) 2016:2908034. doi: 10.1155/2016/2908034
38. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med (2013) 5:178ra140. doi: 10.1126/scitranslmed.3005580
39. Wong K-W, Jacobs WR Jr. Mycobacteriumtuberculosis Exploits Human Interferon γ to Stimulate Macrophage Extracellular Trap Formation and Necrosis. J Infect Dis (2013) 208:109–19. doi: 10.1093/infdis/jit097
40. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med (2009) 15:623–5. doi: 10.1038/nm.1959
41. Guimarães-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceição-Silva F, et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci U S A (2009) 106:6748–53. doi: 10.1073/pnas.0900226106
42. Oehmcke S, Mörgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun (2009) 1:225–30. doi: 10.1159/000203700
43. Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, et al. Neutrophil extracellular traps (NET) induced by different stimuli: A comparative proteomic analysis. PloS One (2019) 14:e0218946. doi: 10.1371/journal.pone.0218946
44. Fan LY, He DY, Wang Q, Zong M, Zhang H, Yang L, et al. Citrullinated vimentin stimulates proliferation, pro-inflammatory cytokine secretion, and PADI4 and RANKL expression of fibroblast-like synoviocytes in rheumatoid arthritis. Scand J Rheumatol (2012) 41:354–8. doi: 10.3109/03009742.2012.670263
45. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev (2010) 233:233–55. doi: 10.1111/j.0105-2896.2009.00859.x
46. Corsiero E, Carlotti E, Jagemann L, Perretti M, Pitzalis C, Bombardieri M. H and L Chain Affinity Maturation and/or Fab N-Glycosylation Influence Immunoreactivity toward Neutrophil Extracellular Trap Antigens in Rheumatoid Arthritis Synovial B Cell Clones. J Immunol (2020) 204:2374–9. doi: 10.4049/jimmunol.1901457
47. Lundberg K, Nijenhuis S, Vossenaar ER, Palmblad K, van Venrooij WJ, Klareskog L, et al. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res Ther (2005) 7:R458–467. doi: 10.1186/ar1697
48. Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight (2020) 5. doi: 10.1172/jci.insight.139388
49. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (2015) 349:316–20. doi: 10.1126/science.aaa8064
50. Mor-Vaknin N, Kappes F, Dick AE, Legendre M, Damoc C, Teitz-Tennenbaum S, et al. DEK in the synovium of patients with juvenile idiopathic arthritis: characterization of DEK antibodies and posttranslational modification of the DEK autoantigen. Arthritis Rheumatol (2011) 63:556–67. doi: 10.1002/art.30138
51. Mor-Vaknin N, Saha A, Legendre M, Carmona-Rivera C, Amin MA, Rabquer BJ, et al. DEK-targeting DNA aptamers as therapeutics for inflammatory arthritis. Nat Commun (2017) 8:14252. doi: 10.1038/ncomms14252
52. Jung J, Lee JS, Kim YG, Lee CK, Yoo B, Shin EC, et al. Synovial fluid CD69(+)CD8(+) T cells with tissue-resident phenotype mediate perforin-dependent citrullination in rheumatoid arthritis. Clin Transl Immunol (2020) 9:e1140. doi: 10.1002/cti2.1140
53. Behnen M, Leschczyk C, Möller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J Immunol (2014) 193:1954–65. doi: 10.4049/jimmunol.1400478
54. Kraaij T, Tengström FC, Kamerling SW, Pusey CD, Scherer HU, Toes RE, et al. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmun Rev (2016) 15:577–84. doi: 10.1016/j.autrev.2016.02.018
55. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol (2018) 18:134–47. doi: 10.1038/nri.2017.105
56. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
57. Romero V, Fert-Bober J, Nigrovic PA, Darrah E, Haque UJ, Lee DM, et al. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci Transl Med (2013) 5:209ra150. doi: 10.1126/scitranslmed.3006869
58. Gehrke N, Mertens C, Zillinger T, Wenzel J, Bald T, Zahn S, et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity (2013) 39:482–95. doi: 10.1016/j.immuni.2013.08.004
59. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther (2014) 16:R122. doi: 10.1186/ar4579
60. Röhm M, Grimm MJ, D’Auria AC, Almyroudis NG, Segal BH, Urban CF. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect Immun (2014) 82:1766–77. doi: 10.1128/iai.00096-14
61. Gavillet M, Martinod K, Renella R, Wagner DD, Williams DA. A key role for Rac and Pak signaling in neutrophil extracellular traps (NETs) formation defines a new potential therapeutic target. Am J Hematol (2018) 93:269–76. doi: 10.1002/ajh.24970
62. Stojkov D, Amini P, Oberson K, Sokollik C, Duppenthaler A, Simon HU, et al. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J Cell Biol (2017) 216:4073–90. doi: 10.1083/jcb.201611168
63. Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
64. Jones JE, Causey CP, Knuckley B, Slack-Noyes JL, Thompson PR. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr Opin Drug Discovery Devel (2009) 12:616–27.
65. Delbosc S, Alsac JM, Journe C, Louedec L, Castier Y, Bonnaure-Mallet M, et al. Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation. Proof of concept in rats. PloS One (2011) 6:e18679. doi: 10.1371/journal.pone.0018679
66. Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis (2008) 67:1488–92. doi: 10.1136/ard.2007.075192
67. Martinod K, Fuchs TA, Zitomersky NL, Wong SL, Demers M, Gallant M, et al. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood (2015) 125:1948–56. doi: 10.1182/blood-2014-07-587709
68. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
69. Blachère NE, Parveen S, Frank MO, Dill BD, Molina H, Orange DE. High-Titer Rheumatoid Arthritis Antibodies Preferentially Bind Fibrinogen Citrullinated by Peptidylarginine Deiminase 4. Arthritis Rheumatol (2017) 69:986–95. doi: 10.1002/art.40035
70. Arnoux F, Mariot C, Peen E, Lambert NC, Balandraud N, Roudier J, et al. Peptidyl arginine deiminase immunization induces anticitrullinated protein antibodies in mice with particular MHC types. Proc Natl Acad Sci U S A (2017) 114:E10169–e10177. doi: 10.1073/pnas.1713112114
71. Kolfenbach JR, Deane KD, Derber LA, O'Donnell CI, Gilliland WR, Edison JD, et al. Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheumatol (2010) 62:2633–9. doi: 10.1002/art.27570
72. Darrah E, Yu F, Cappelli LC, Rosen A, O'Dell JR, Mikuls TR. Association of Baseline Peptidylarginine Deiminase 4 Autoantibodies With Favorable Response to Treatment Escalation in Rheumatoid Arthritis. Arthritis Rheumatol (2019) 71:696–702. doi: 10.1002/art.40791
73. Martinez-Prat L, Lucia D, Ibarra C, Mahler M, Dervieux T. Antibodies targeting protein-arginine deiminase 4 (PAD4) demonstrate diagnostic value in rheumatoid arthritis. Ann Rheum Dis (2019) 78:434–6. doi: 10.1136/annrheumdis-2018-213818
74. Darrah E, Kim A, Zhang X, Boronina T, Cole RN, Fava A, et al. Proteolysis by Granzyme B Enhances Presentation of Autoantigenic Peptidylarginine Deiminase 4 Epitopes in Rheumatoid Arthritis. J Proteome Res (2017) 16:355–65. doi: 10.1021/acs.jproteome.6b00617
75. Seri Y, Shoda H, Suzuki A, Matsumoto I, Sumida T, Fujio K, et al. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci Rep (2015) 5:13041. doi: 10.1038/srep13041
76. Valesini G, Gerardi MC, Iannuccelli C, Pacucci VA, Pendolino M, Shoenfeld Y. Citrullination and autoimmunity. Autoimmun Rev (2015) 14:490–7. doi: 10.1016/j.autrev.2015.01.013
77. Neeli I, Radic M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol (2013) 4:38:38. doi: 10.3389/fimmu.2013.00038
78. Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet (2003) 34:395–402. doi: 10.1038/ng1206
79. Bawadekar M, Shim D, Johnson CJ, Warner TF, Rebernick R, Damgaard D, et al. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J Autoimmun (2017) 80:39–47. doi: 10.1016/j.jaut.2017.01.006
80. Spengler J, Lugonja B, Ytterberg AJ, Zubarev RA, Creese AJ, Pearson MJ, et al. Release of Active Peptidyl Arginine Deiminases by Neutrophils Can Explain Production of Extracellular Citrullinated Autoantigens in Rheumatoid Arthritis Synovial Fluid. Arthritis Rheumatol (2015) 67:3135–45. doi: 10.1002/art.39313
81. Pruijn GJ. Citrullination and carbamylation in the pathophysiology of rheumatoid arthritis. Front Immunol (2015) 6:192:192. doi: 10.3389/fimmu.2015.00192
82. Abbas AK, Le K, Pimmett VL, Bell DA, Cairns E, Dekoter RP. Negative regulation of the peptidylarginine deiminase type IV promoter by NF-κB in human myeloid cells. Gene (2014) 533:123–31. doi: 10.1016/j.gene.2013.09.108
83. Katz Y, Nadiv O, Beer Y. Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1,6, and 8 in skin and synovial fibroblasts: a possible role as a “fine-tuning cytokine” in inflammation processes. Arthritis Rheumatol (2001) 44:2176–84. doi: 10.1002/1529-0131(200109)44:9<2176::aid-art371>3.0.co;2-4
84. Guiducci C, Tripodo C, Gong M, Sangaletti S, Colombo MP, Coffman RL, et al. Autoimmune skin inflammation is dependent on plasmacytoid dendritic cell activation by nucleic acids via TLR7 and TLR9. J Exp Med (2010) 207:2931–42. doi: 10.1084/jem.20101048
85. Dwivedi N, Radic M. Burning controversies in NETs and autoimmunity: The mysteries of cell death and autoimmune disease. Autoimmunity (2018) 51:267–80. doi: 10.1080/08916934.2018.1523395
86. Bruschi M, Bonanni A, Petretto A, Vaglio A, Pratesi F, Santucci L, et al. Neutrophil Extracellular Traps Profiles in Patients with Incident Systemic Lupus Erythematosus and Lupus Nephritis. J Rheumatol (2020) 47:377–86. doi: 10.3899/jrheum.181232
87. Tani K, Murphy WJ, Chertov O, Oppenheim JJ, Wang JM. The neutrophil granule protein cathepsin G activates murine T lymphocytes and upregulates antigen-specific IG production in mice. Biochem Biophys Res Commun (2001) 282:971–6. doi: 10.1006/bbrc.2001.4676
88. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
89. Döring Y, Soehnlein O, Weber C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res (2017) 120:736–43. doi: 10.1161/circresaha.116.309692
90. Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation (2002) 106:2894–900. doi: 10.1161/01.cir.0000042674.89762.20
91. Megens RT, Vijayan S, Lievens D, Döring Y, van Zandvoort MA, Grommes J, et al. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost (2012) 107:597–8. doi: 10.1160/th11-09-0650
92. Zhou P, Li T, Jin J, Liu Y, Li B, Sun Q, et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine (2020) 53:102671. doi: 10.1016/j.ebiom.2020.102671
93. Zabieglo K, Majewski P, Majchrzak-Gorecka M, Wlodarczyk A, Grygier B, Zegar A, et al. The inhibitory effect of secretory leukocyte protease inhibitor (SLPI) on formation of neutrophil extracellular traps. J Leukoc Biol (2015) 98:99–106. doi: 10.1189/jlb.4AB1114-543R
94. Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol (2012) 181:8–18. doi: 10.1016/j.ajpath.2012.03.044
95. Tanaka D, Kagari T, Doi H, Shimozato T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology (2006) 119:195–202. doi: 10.1111/j.1365-2567.2006.02424.x
96. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi: 10.1038/nri3024
97. Corsiero E, Pratesi F, Prediletto E, Bombardieri M, Migliorini P. NETosis as Source of Autoantigens in Rheumatoid Arthritis. Front Immunol (2016) 7:485:485. doi: 10.3389/fimmu.2016.00485
98. Papadaki G, Kambas K, Choulaki C, Vlachou K, Drakos E, Bertsias G, et al. Neutrophil extracellular traps exacerbate Th1-mediated autoimmune responses in rheumatoid arthritis by promoting DC maturation. Eur J Immunol (2016) 46:2542–54. doi: 10.1002/eji.201646542
99. Rohrbach AS, Hemmers S, Arandjelovic S, Corr M, Mowen KA. PAD4 is not essential for disease in the K/BxN murine autoantibody-mediated model of arthritis. Arthritis Res Ther (2012) 14:R104. doi: 10.1186/ar3829
100. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
101. Ditzel HJ. The K/BxN mouse: a model of human inflammatory arthritis. Trends Mol Med (2004) 10:40–5. doi: 10.1016/j.molmed.2003.11.004
102. Hosseinzadeh A, Thompson PR, Segal BH, Urban CF. Nicotine induces neutrophil extracellular traps. J Leukoc Biol (2016) 100:1105–12. doi: 10.1189/jlb.3AB0815-379RR
103. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol (2010) 62:2569–81. doi: 10.1002/art.27584
104. Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA. Nat Rev Rheumatol (2012) 8:144–52. doi: 10.1038/nrrheum.2011.204
105. Nielen MM, van der Horst AR, van Schaardenburg D, van der Horst-Bruinsma IE, van de Stadt RJ, Aarden L, et al. Antibodies to citrullinated human fibrinogen (ACF) have diagnostic and prognostic value in early arthritis. Ann Rheum Dis (2005) 64:1199–204. doi: 10.1136/ard.2004.029389
106. Dejaco C, Klotz W, Larcher H, Duftner C, Schirmer M, Herold M. Diagnostic value of antibodies against a modified citrullinated vimentin in rheumatoid arthritis. Arthritis Res Ther (2006) 8:R119. doi: 10.1186/ar2008
107. de Bont CM, Stokman MEM, Faas P, Thurlings RM, Boelens WC, Wright HL, et al. Autoantibodies to neutrophil extracellular traps represent a potential serological biomarker in rheumatoid arthritis. J Autoimmun (2020) 113:102484. doi: 10.1016/j.jaut.2020.102484
108. Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M, et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol (2006) 16:396–400. doi: 10.1016/j.cub.2005.12.039
109. Halverson TW, Wilton M, Poon KK, Petri B, Lewenza S. DNA is an antimicrobial component of neutrophil extracellular traps. PloS Pathog (2015) 11:e1004593. doi: 10.1371/journal.ppat.1004593
110. Tran TM, MacIntyre A, Hawes M, Allen C. Escaping Underground Nets: Extracellular DNases Degrade Plant Extracellular Traps and Contribute to Virulence of the Plant Pathogenic Bacterium Ralstonia solanacearum. PloS Pathog (2016) 12:e1005686. doi: 10.1371/journal.ppat.1005686
111. de Buhr N, Neumann A, Jerjomiceva N, von Köckritz-Blickwede M, Baums CG. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology (2014) 160:385–95. doi: 10.1099/mic.0.072199-0
112. Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med (2012) 18:1386–93. doi: 10.1038/nm.2847
113. Grasso S, Neumann A, Lang IM, Etscheid M, von Köckritz-Blickwede M, Kanse SM. Interaction of factor VII activating protease (FSAP) with neutrophil extracellular traps (NETs). Thromb Res (2018) 161:36–42. doi: 10.1016/j.thromres.2017.11.012
114. Munafo DB, Johnson JL, Brzezinska AA, Ellis BA, Wood MR, Catz SD. DNase I inhibits a late phase of reactive oxygen species production in neutrophils. J Innate Immun (2009) 1:527–42. doi: 10.1159/000235860
115. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, et al. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun (2016) 67:19–28. doi: 10.1016/j.jaut.2015.08.018
116. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest (2013) 123:3446–58. doi: 10.1172/jci67484
117. Macanovic M, Sinicropi D, Shak S, Baughman S, Thiru S, Lachmann PJ. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin Exp Immunol (1996) 106:243–52. doi: 10.1046/j.1365-2249.1996.d01-839.x
118. Sayah DM, Mallavia B, Liu F, Ortiz-Muñoz G, Caudrillier A, DerHovanessian A, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med (2015) 191:455–63. doi: 10.1164/rccm.201406-1086OC
119. Okeke EB, Louttit C, Fry C, Najafabadi AH, Han K, Nemzek J, et al. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials (2020) 238:119836. doi: 10.1016/j.biomaterials.2020.119836
120. Kawaguchi H, Matsumoto I, Osada A, Kurata I, Ebe H, Tanaka Y, et al. Peptidyl arginine deiminase inhibition suppresses arthritis via decreased protein citrullination in joints and serum with the downregulation of interleukin-6. Mod Rheumatol (2019) 29:964–9. doi: 10.1080/14397595.2018.1532545
121. Kraaij T, Kamerling SWA, de Rooij ENM, van Daele PLA, Bredewold OW, Bakker JA, et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J Autoimmun (2018) 91:45–54. doi: 10.1016/j.jaut.2018.03.003
122. Zhu M, Yuan K, Lu Q, Zhu Q, Zhang S, Li X, et al. Emodin ameliorates rheumatoid arthritis by promoting neutrophil apoptosis and inhibiting neutrophil extracellular trap formation. Mol Immunol (2019) 112:188–97. doi: 10.1016/j.molimm.2019.05.010
123. Yang F, Luo X, Luo G, Zhai Z, Zhuang J, He J, et al. Inhibition of NET formation by polydatin protects against collagen-induced arthritis. Int Immunopharmacol (2019) 77:105919. doi: 10.1016/j.intimp.2019.105919
124. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PloS One (2014) 9:e97088. doi: 10.1371/journal.pone.0097088
125. Huang G, Yuan K, Zhu Q, Zhang S, Lu Q, Zhu M, et al. Triptolide inhibits the inflammatory activities of neutrophils to ameliorate chronic arthritis. Mol Immunol (2018) 101:210–20. doi: 10.1016/j.molimm.2018.06.012
126. Yu Y, Koehn CD, Yue Y, Li S, Thiele GM, Hearth-Holmes MP, et al. Celastrol inhibits inflammatory stimuli-induced neutrophil extracellular trap formation. Curr Mol Med (2015) 15:401–10. doi: 10.2174/1566524015666150505160743
127. Ruiz-Limón P, Ortega R, Arias de la Rosa I, Abalos-Aguilera MDC, Perez-Sanchez C, Jimenez-Gomez Y, et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Transl Res (2017) 183:87–103. doi: 10.1016/j.trsl.2016.12.003
Keywords: neutrophil extracellular traps, citrullinated autoantigens, anti-citrullinated protein antibodies, autoimmunity, chronic inflammation, protein-arginine deiminase type 4, rheumatoid arthritis
Citation: Song W, Ye J, Pan N, Tan C and Herrmann M (2021) Neutrophil Extracellular Traps Tied to Rheumatoid Arthritis: Points to Ponder. Front. Immunol. 11:578129. doi: 10.3389/fimmu.2020.578129
Received: 30 June 2020; Accepted: 14 December 2020;
Published: 29 January 2021.
Edited by:
Zhifeng Gu, Affiliated Hospital of Nantong University, ChinaReviewed by:
Shemin Lu, Xi’an Jiaotong University, ChinaRichard Williams, University of Oxford, United Kingdom
Copyright © 2021 Song, Ye, Pan, Tan and Herrmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunyu Tan, YW5uYXF1aW50ZXNzZW5jZUAxNjMuY29t