 Yifan Zhou1
Yifan Zhou1 Wei Qiu
Wei Qiu- 1Department of Neurology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 2Department of Internal Medicine, The Ohio State University Wexner Medical Center, Columbus, OH, United States
The nuclear factor κB (NF-κB) signaling cascade has been implicating in a broad range of biological processes, including inflammation, cell proliferation, differentiation, and apoptosis. The past three decades have witnessed a great progress in understanding the impact of aberrant NF-κB regulation on human autoimmune and inflammatory disorders. In this review, we discuss how aberrant NF-κB activation contributes to multiple sclerosis, a typical inflammatory demyelinating disease of the central nervous system, and its involvement in developing potential therapeutic targets.
Introduction
Nuclear factor κB (NF-κB) comprises a family of transcription factors that coordinate hundreds of genes expression by forming homodimers or heterodimers (http://www.bu.edu/NF-κB/gene-resources/targetgenes/). In mammals, there are five members of NF-κB family: RelA (p65), RelB, c-Rel, p105 (NF-κB1), and p100 (NF-κB2). In most resting cells, NF-κB is sequestered in the cytoplasm through interacting with any of a family of inhibitors of κB (IκB) proteins, such as IκBα, IκBβ, and p100. Upon stimulated signals, IκB kinase (IKK) rapidly phosphorylates IκB and facilitates its ubiquitination and proteasomal degradation, which ultimately enables the entrance of NF-κB into the nucleus and elicits its transcriptional activity (1).
The general NF-κB signaling cascade can be categorized as canonical (classical) and non-canonical (alternative) pathways (Figure 1). The canonical NK-κB signaling pathway can be induced by extensive numbers of stimuli including Toll-like receptor ligands, proinflammatory cytokines [e.g., tumor necrosis factor α (TNF-α)], and antigens, leading to the activation of IKK complex, which comprises IKKα (IKK1), IKKβ (IKK2), and NF-κB essential modulator (NEMO, also known as IKKγ). The IKK complex then phosphorylates IκB proteins, allowing the cytoplasmic NF-κB dimers (usually p50–p65) to be released from the degraded IκB. The activation of canonical pathway has been shown to mediate a wide range of biological functions within minutes (2, 3). In contrast, the non-canonical NK-κB signaling pathway responds to only a specific set of stimuli such as B cell-activation factor (BAFF), lymphotoxin β, CD40 ligand (CD40L), and receptor activator of nuclear factor kappa-B ligand (4–6). Once stimulated, NF-κB–inducing kinase specifically activates IKKα, which in turn processes the p52 precursor, p100, into p52, and mature p52 subsequently translocates to the nucleus via its dimerization with RelB. Unlike the canonical pathway, this process can be slow and is involved in a limited number of cellular responses (7).
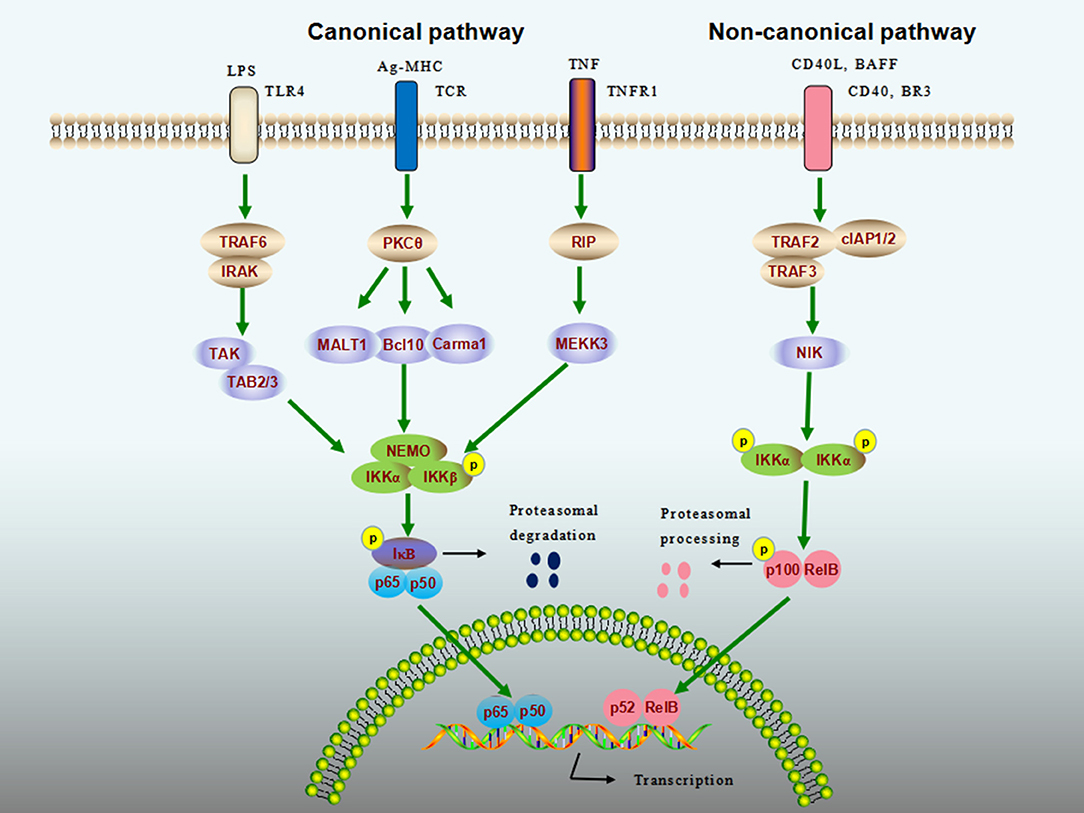
Figure 1. NF-κB signaling cascade. Nuclear factor-κB (NF-κB) activity is stimulated by canonical (classical) and noncanonical (alternative) pathways. The canonical pathway can be activated by extensive numbers of stimuli, such as lipopolysaccharide (LPS), antigens, and tumor-necrosis factor (TNF). The inhibitor of NF-κB (IκB) kinase (IKK) complex that comprises IKKα (IKK1), IKKβ (IKK2), and NF-κB essential modulator (NEMO, also known as IKKγ) is a point of convergence for the canonical pathway, which phosphorylates IκB proteins, allowing the cytoplasmic NF-κB to be released and to enter into the nucleus to elicit transcriptional activity. The noncanonical pathway responds to a different set of ligands, including CD40 ligand (CD40L) and B cell-activation factor (BAFF). Upon binding of these ligands to their cognate receptors, NF-κB-inducing kinase (NIK) specifically phosphorylates IKKα, which processes the p100 into mature p52. The p52 then translocates to the nucleus via its dimerization with RelB to activate noncanonical NF-κB target genes.
Given that NF-κB affects almost the entire arsenal of immune guardians and immune cells (1), special concern has gradually been focused on the pivotal role of NF-κB dysregulation in many autoimmune inflammatory diseases including multiple sclerosis (MS), systemic lupus erythematosus, and type 1 diabetes.
MS and Its Animal Models
Multiple sclerosis is a multifactorial inflammatory demyelinating disease of the central nervous system (CNS) marked by repeated demyelination and disabling outcomes (5, 8). Although the exact etiology of MS remains unclear, the interaction between predisposing genes and environment triggers MS at a preclinical phase, primarily through affecting the immune system (8). As aberrant peripheral immune cells invade the CNS through disrupted blood–brain barrier (BBB) and induce further inflammation, oligodendrocytes, and neurons are preferentially injured, thereby leading to demyelination and neurodegeneration. Analysis of active human MS lesions demonstrates a complicated immune repertoire that includes but is not limited to lymphocytes, antibodies, cytokines/chemokines, macrophages, microglia, and complement (Figure 2) (9, 10).
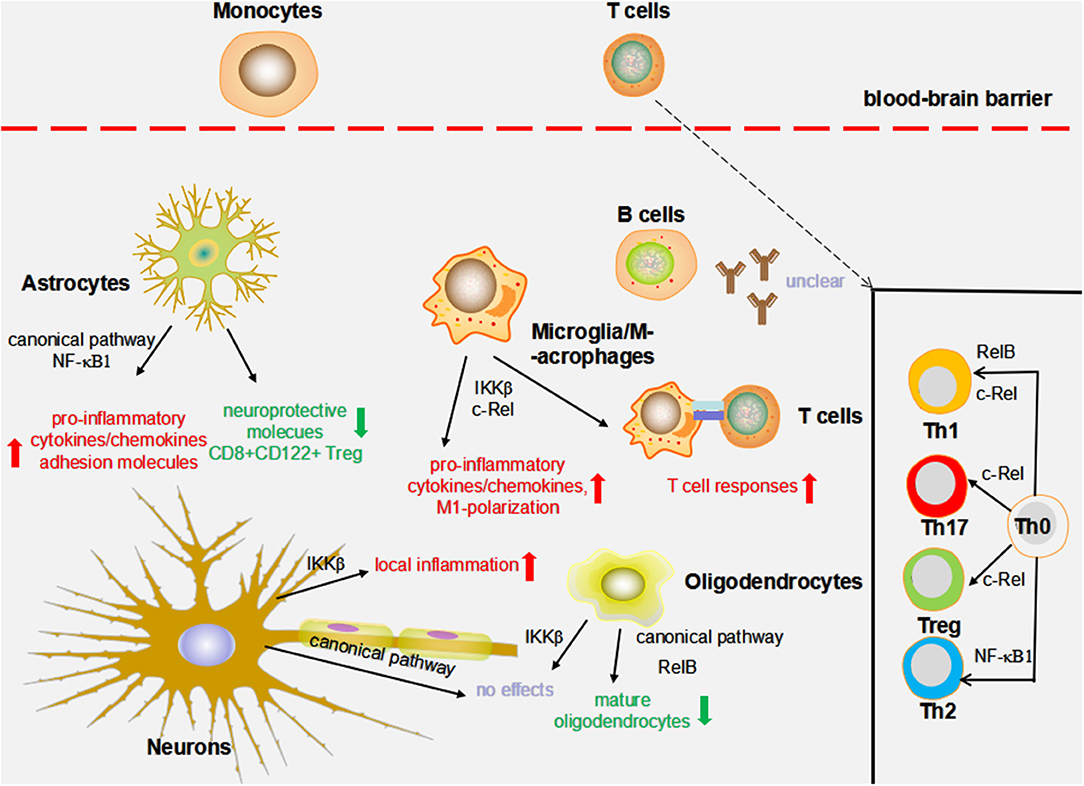
Figure 2. The impact of NF-κB on MS animal models. The effects of nuclear factor-κB (NF-κB) on experimental autoimmune encephalomyelitis (EAE) and cuprizone models are summarized as follows. c-Rel and IKKβ in macrophages/microglia might influence the production of pro-inflammatory cytokines/chemokines, M1 macrophage/microglia phenotype polarization, and T cell immune responses. The deficiency of IKKβ in oligodendrocytes does not alter myelin formation, demyelination, and remyelination; however, blocking RelB and the canonical pathway results in a decreased number of mature oligodendrocytes. NF-κB1 and the canonical pathway are required to augment local inflammation through driving the production of pro-inflammatory mediators and suppressing the levels of neuroprotective molecule adhesion molecules and CD8+ CD122+ regulatory T cells (Tregs). Neuronal IKKβ has been suggested to suppress CNS inflammation. By contrast, conditional deletion of the neuronal NF-κB pathway by the transgenic expression of an IκBα super-repressor did not influence the EAE course. c-Rel is essential in Treg, T helper 1 (Th1), and Th17 differentiation. In addition to c-Rel, Th1 differentiation is also regulated by RelB, whereas NF-κB1 is essential in mediating Th2 responses.
Among this intrinsic network, autoreactive T cells against myelin antigens are believed to initiate and augment disease once they migrate into the CNS, where they are reactivated (11). This idea has been reinforced by several lines of indirect evidence: first, myelin-reactive T cells were isolated from both the blood and cerebrospinal fluid of MS patients (12, 13); second, an exacerbated outcome was reported in MS patients treated with a myelin basic protein–derived altered peptide ligand (14); finally, some MS risk variants (e.g., HLA-DRB1*1501) were associated with antigen presenting process (15). Despite the fact that myelin-reactive T cells are present in healthy individuals and absent in some MS patients (16, 17), studies have identified several features, such as cytokine secreting profile, activation requirement, and the level of interleukin 2 (IL-2) receptor, which can help distinguish between MS and healthy subjects (18, 19). Further investigations are clearly needed to determine whether self-reactive T cells play a pathogenic role in MS pathogenesis. In this article, we discuss below the prevailing concept that T helper 1 (TH1), TH17, and CD8+ T cells are the major drivers of MS, whereas regulatory T cells (Tregs), perhaps plus TH2, confer protective properties.
Most of our understanding on how the immune system regulates MS has originated from experiments performed in experimental autoimmune encephalomyelitis (EAE), a widely used animal model induced by immunizing rodents with myelin proteins/peptides or passively transferring the myelin-reactive T cells to elicit a T cell–mediated autoimmunity (20–22). Experimental autoimmune encephalomyelitis, despite its limitations, mimics many clinical and immunopathological features of human MS. Other models for MS include viral-induced (e.g., Theiler's virus) and toxic-induced (e.g., cuprizone and lysolecithin) demyelinating disorders. Current work exploring the role of NF-κB in MS is based on EAE and the cuprizone model, which is applied to study demyelination and remyelination mechanisms independent of antigen-specific T cells and share some resemblance to those of pattern III MS lesions (23). Demyelination of cuprizone model is characterized by pronounced accumulation of microglia and astrocytes, whereas the contributions of blood-derived immune cells and BBB are minimal (24, 25). Therefore, the role of NF-κB cascade in MS will be discussed, in both humans and animal model (EAE and cuprizone model) with much attention paid to the T cell functioning.
The Evidence of NF-κB Involvement in Human MS
With the advent of genome-wide association studies and other genetic technologies, a large set of MS susceptibility variants has recently been reported, some of which fall in or near genes that regulate the NF-κB pathway, such as NFKBIZ and RELA (25–31). Subsequent studies have noted increased levels of NF-κB in total peripheral blood mononuclear cells (PBMCs), CD3+/CD4+ T cells, and monocytes from patients with MS (32, 33). In addition, CD4+ T cells from donors carrying rs228614-G, an MS risk variant proximal to NF-κB1, exhibited increased IκBα degradation and NF-κB p65 nuclear translocation after TNF-α stimulation (34). The altered NF-κB responses were due to the enhanced expression of NF-κB itself, with the rs228614-G variant inducing a 20-fold increase in NF-κB p50 and decrease in several negative regulators of NF-κB (34). Some studies have shown a link between increased NF-κB–related genes in T cells and MS relapse (35, 36). More recently, another MS risk allele, rs7665090-G, was shown to upregulate NF-κB signaling and target genes in astrocytes that increased lymphocytic infiltration and MS lesion size. These findings help to explain how NF-κB may contribute to MS progression in various respects (30). Combined with pathological studies that detected activated NF-κB subunits in macrophages, microglia, oligodendrocytes, astrocytes, and perivascular lymphocytes near or in active MS plaques (37, 38), studies to explore the impact of dysfunctional NF-κB on different cell types on MS would be of interest (Table 1).
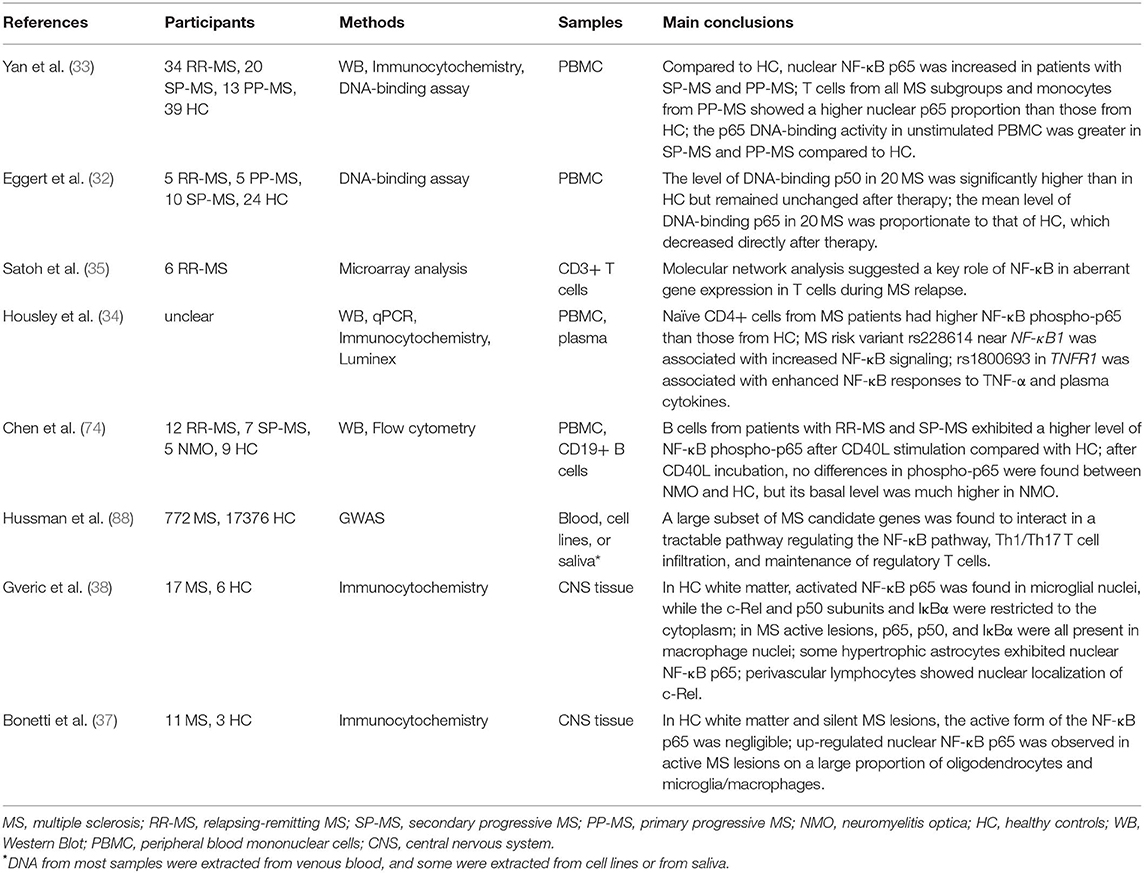
Table 1. Summary of NF-κB expression in different types of multiple sclerosis.
The Impact of NF-κB on Different Cell Types
Macrophages/Microglia
Notably, brain resident microglia, which are developmentally and functionally distinct from blood-borne myeloid cells, are now known to originate from embryonic yolk sac precursors. Because of the phenotypic similarities between microglia, CNS-resident macrophages, and circulating monocyte-derived macrophages, surface markers such as CD11b and F4/80 used by most studies actually stain all these populations (39). Thus, unless otherwise specified, the effects of macrophages/microglia in MS are discussed together.
Prominent macrophage and microglial activation at the site of actively demyelinating plaques is believed to play a central role in MS development (23). Consistent with this, increased nuclear expression of RelA, c-Rel, and NF-κB p50 in macrophages, as well as RelA in microglia, was found to correlate with MS lesion activity (38). Moreover, recent studies have suggested that M1 phenotype macrophages/microglia are crucial in driving EAE progression by secreting large amounts of proinflammatory cytokines and chemokines and reactivating encephalitogenic T cells as antigen-presenting cells (APCs), whereas M2 phenotype macrophages/microglia protect against disease and have a potent ability to release anti-inflammatory molecules and growth factors (40). Researchers found that mice in which Ikkβ was conditionally knocked out in myeloid cells, which targeted the majority of macrophages and microglia, exhibited ameliorated EAE progression accompanied by reduced levels of macrophage/microglia infiltration, M1 polarization, and CD4+ T cell responses (41). Similarly, the silencing of c-Rel in macrophages alleviated EAE symptoms through suppressing proinflammatory cytokines and T cell accumulation (42). Depletion of microglial transforming growth factor β-activated kinase 1 (TAK1), a molecule that modulates NF-κB, was shown to inhibit the NF-κB canonical pathway and attenuate EAE pathology (43). By contrast, knocking out NF-κB regulatory protein A20 in microglia was associated with massive microglia activation, neuroinflammation, and increased EAE pathology (44). And knocking out IκBα in mouse myeloid cells led to similar findings. These mutant mice displayed constitutively active NF-κB, which led to a prominent increase in macrophages/microglia, T cells, and key proinflammatory mediators (45).
Notably, because the mechanism of NF-κB regulation is not entirely clear, changing certain molecules may result in opposing outcomes. For instance, in contrast to the findings in IKKβ-deficient macrophages, blocking IKKα was shown to elicit aberrant NF-κB activation that made macrophages hyperactive to various stimuli (46).
Oligodendrocytes
By immunohistochemistry, strong immunoreactivity for NF-κB p65 was observed in approximately half of the oligodendrocytes in active, but not chronic silent, MS lesions (38). To assess the impact of NF-κB on normal oligodendrocyte maturation, demyelination, and remyelination in an MS background, Raasch et al. depleted IKKβ in CNS cells (IKKβCNS-KO) and oligodendrocytes alone (IKKβOligo−KO) (47). As a result, structurally intact myelin sheaths with similar numbers of oligodendrocyte progenitor cells and mature oligodendrocytes were observed in IKKβCNS-KO and IKKβOligo−KO mice compared with their wild-type littermates (47). Further studies revealed comparable degrees of demyelination and remyelination between IKKβOligo−KO and control in both EAE and cuprizone models, suggesting that NF-κB in oligodendrocytes is dispensable for myelin loss in patients with MS. However, because IKKβ modulates signaling pathways other than that of NF-κB, such as the extracellular signal–regulated kinase (ERK)-1/2 pathway (48, 49), NF-κB–independent effects on oligodendrocytes cannot be completely ruled out. Similar to mice in which IKKβ had been deleted, mice in which c-Rel, RelB, NF-κB p52, NEMO, or IKKα had been deleted were shown in previous reports to display proper brain myelination under normal conditions (50–52). Notably, NF-κB activity is crucial in orchestrating Schwann cell differentiation and myelination in the peripheral nervous system (PNS), which is in sharp contrast to its role in the CNS; NF-κB activity is crucial in orchestrating Schwann cell differentiation and myelination in the PNS (53).
Furthermore, opinions on the role of NF-κB differ. In a recent study, researchers generated a mouse model that specifically expressed IκBαΔN, a super-suppressor of NF-κB, in oligodendrocytes and documented identical demyelination and remyelination in cuprizone model between IκBαΔN+ and IκBαΔN− mice. However, IκBαΔN mice exhibited markedly impaired oligodendrocyte regeneration and remyelination compared to control mice in the presence of interferon γ (IFN-γ) (54). Consistently, IκBαΔN+ mice developed much more severe EAE because oligodendrocytes from these mice were more vulnerable to inflammation than those from control mice (54). These data indicate that NF-κB activation may, at least in some circumstances, promote oligodendrocyte survival during inflammation. In contrast, mice in which RelB was absent specifically in oligodendrocytes (RelBΔOligo) showed a decrease in the loss of mature oligodendrocytes, which in turn prevented demyelination upon EAE challenge (55). This protective phenotype was proposed to be the consequence of increased NF-κB p65 activity that protected oligodendrocytes against inflammation. Interestingly, the altered course of EAE in IκBαΔN and RelBΔOligo mice was more dependent on controlling oligodendrocytes themselves, while the change in inflammation was not significant. Additionally, there was evidence that patients with additional copies of IKBKG, the gene encoding NEMO, experience defective CNS myelination due to NF-κB inactivation (52). To date, the effects of NF-κB on oligodendrocytes in normal myelin formation and MS remain ambiguous, and as oligodendrocytes are the main target of MS, further efforts are required to provide clues for future therapeutic approaches.
Astrocytes
In addition to their role in forming the BBB and supporting neurons, astrocytes are crucial in regulating CNS inflammation (56). Previous reports found nuclear NF-κB p65 in hypertrophic astrocytes in the parenchyma of active MS lesions (38). Recently, Ponath et al. demonstrated that the MS risk variant rs7665090G, which is located near NFKB1, is associated with upregulated NF-κB and target gene expression (e.g., IFN-γ and TNF-α) in human astrocytes (30). Further characterization revealed stronger immunofluorescent staining for activated NF-κB, chemokines (e.g., CXCL10 and CCL5), and C3d located within the hypertrophic astrocytes and greater perivascular T lymphocyte infiltration in MS lesions with the rs7665090 risk than those with a protective genotype (30). In addition, MS-approved agent fingolimod (FTY720) has exhibited strong anti-inflammatory properties through inhibiting NF-κB activity in astrocytes (57, 58). Therefore, by modulating astroglial NF-κB and thereby relieving the inflamed CNS microenvironment, it is possible to reduce tissue injury and promote later recovery. Consistently, glial fibrillary acidic protein (GFAP)–IκBα-dominant-negative (dn) mice, in which NF-κB was inactivated specifically in astrocytes, manifested alleviated symptoms and steady functional improvement following EAE induction compared to those in wild-type control mice (59). Central nervous system of these mutant animals were characterized by the reduced expression of several proinflammatory cytokines/chemokines and increased levels of CD8+ CD122+ Tregs, neuroprotective molecules, and unexpectedly, CD45+ leukocytes and IL-6 (59). Another study provided similar results based on cuprizone model that the inactivation of astroglial NF-κB dramatically prevented axonal loss through inhibiting proinflammatory cytokines and gliosis in GFAP-IκBα-dn mice (47). Blocking astroglial RelB, however, had a very limited impact on the course of EAE and mainly delayed disease onset with a mild or no effect on CNS inflammation (47). Finally, several adhesion molecules required for BBB integrity, including intercellular cell adhesion molecule 1 and vascular cell adhesion molecule 1, were found to be reduced in GFAP-IκBα-dn EAE mice, further supporting the therapeutic value of interfering with NF-κB in astrocytes.
Although current studies consistently recognize that the activation of astroglial NF-κB exerts detrimental effects on EAE and MS, blocking NF-κB activation may also bring unfavorable outcomes. Many neurotrophic factors released by astrocytes, including nerve growth factor, glial cell line–derived neurotrophic factor, and brain-derived neurotrophic factor, are dependent on the NF-κB pathway (60, 61). Furthermore, IL-6 and leukocytes of GFAP-IκBα-dn EAE mice were significantly upregulated in the CNS (59). Later research conducted by the same group showed, however, a robust reduction in most immune cell populations in the CNS of GFAP-IκBα-dn mice at chronic EAE phase compared with controls (62).
Neurons
Although neurons generally do not express major histocompatibility complex class I and II molecules and therefore fail to participate in antigen presentation, a growing amount of evidence now suggests that neurons may suppress microglial/microphage activation through the CD220-CD220R interaction, act with T cells to control their survival, and induce the conversion of encephalitogenic T cells to Tregs (63, 64).
Constitutively high basal levels of NF-κB in neurons are essential for regulating cell morphology and plasticity and involved in behavior, learning, and memory (65). Neuronal IKKβ-deficient mice developed a severe, non-resolving form of EAE accompanied by the enhanced accumulation of TH1 and NK1.1+ cells and proinflammatory cytokines in the CNS (66). In contrast, conditional deletion of the neuronal NF-κB pathway by the transgenic expression of an IκBα super-repressor did not alter the course of EAE (67). One possible explanation for this finding is that IKK ablation, as mentioned above, simultaneously influences other signaling pathways in addition to NF-κB signaling (48). Therefore, other molecules triggered by IKKβ, such as tumor progression locus 2, an important element involved in ERK-1/2 pathways (63), might confer neuroprotection owing to the ability of ERK-1/2 to promote remyelination and decelerate EAE (68, 69). Additionally, the super-repressor was grossly absent in the hindbrain, brainstem, and spinal cord (67), which to some extent restricted the magnitude of neuronal NF-κB deficiency.
B Cells
Multiple sclerosis has historically been considered a T cell–mediated autoimmune demyelinating disorder. However, an increasing amount of data from neuropathological studies and anti-CD20–directed B cell therapy have made it clear that B lymphocytes also play a critical role in driving inflammation and MS progression (10). B cells have a strong capacity to secrete cytokines and antibodies and reactivate T cells, thereby enhancing immune responses. Further observations indicated that B cells, like T cells, have both pathogenic (effector B cells) and protective (regulatory B cells) effects in the setting of inflammation (70–73).
At present, no in vivo studies in an NF-κB–deficient MS model have been performed. It has been reported that a proportion of NF-κB p65 translocated to the nucleus was similar between progressive MS, relapsing MS, and healthy controls (33). By contrast, a recent study revealed that B cells from MS patients exhibited an increased level of activated NF-κB p65 following CD40 stimulation compared with healthy controls (74). The interaction between CD40 and CD40L is a pivotal step in mediating B cell activities (e.g., survival, proliferation, and differentiation) (75). Given the close relationship between aberrant CD40 and autoimmunity (76–78), downregulating the B cell NF-κB pathway, which is a major signaling cascade, by CD40 stimulation may reverse the hyperresponsiveness of B cells induced by CD40 in MS patients. Furthermore, blocking BAFF, a member of the TNF ligand superfamily that specifically regulates B cell functions, was found to promote T cell apoptosis in EAE mice through reducing osteopontin release in an NF-κB–dependent manner (79). The causal MS variant SP140 (rs28445040-T), which induces decreased SP140 expression, was recently suggested to exerts its function in B cells through upregulating NF-κB activity (80). Finally, the therapeutic mechanism of dimethyl fumarate, which is approved for MS management, has been shown to correlate with a dramatic reduction in proinflammatory B cell subsets in vitro partially due to the inhibition of NF-κB activation (81).
In addition, some indirect evidence also points to the benefit of downregulating B cell NF-κB to control excess inflammation. Studies in NF-κB knockout mice have identified distinct functions of different NF-κB proteins. B cells lacking RelB exhibited a proliferation deficit but normal maturation, Ig secretion, and Ig class switching (82). The c-Rel deficiency in B cells was associated with germinal center (GC) collapse and impaired cell growth, whereas RelA deficiency was associated with the weakened development of plasma cells (83). In addition, NF-κB2 deletion mice presented with deficits in antibody secretion, GC reactions, and splenic microarchitecture (84), and NF-κB1 blockade resulted in diminished T cell–dependent antibody responses (85). B cell NF-κB is also essential in preventing autoimmunity, as suggested by de Valle et al., who observed a multiorgan autoimmune disease in NF-κB1 knockout mice that was largely attributable to the dysregulated activity of B cells, which released aberrant levels of IL-6 (86). Overall, because it is difficult to predict whether these immune dysfunctions induced by B cell NF-κB blockade occur in EAE or MS and affect their pathophysiology, further investigations are needed to explore this unidentified issue.
T Cells
CD3+ T lymphocytes are predominant in the demyelinating tissues of patients with MS. However, as CD4+ T cells are the major mediators of CNS injury during the course of EAE, in studies of human MS, in which conspicuous CD8+ T cells are infiltrated throughout lesions at all stages, whereas CD4+ T cells are sparse or even absent, an added challenge for determining the precise role of CD4+ T cells is ascertaining whether these effectors are more critical in disease initiation than in established MS (11). And so far, no approved clinical trial that selectively eliminates CD4+ T cells has provided definitive evidence of clinical efficacy (87). The significance of T cells and key differences in the inflammatory response between MS patients and EAE animals has recently been reviewed in detail, and caution must therefore be taken when extrapolating animal findings to humans.
There are data, but no direct proof as of yet, associating T cell NF-κB signaling with the risk and maintenance of MS. First, the level of lymphocytic DNA-binding NF-κB p50 was found to be higher in MS patients than in their healthy counterparts, and NF-κB p65, despite its normal level of expression, decreased significantly during therapy (32). In another study, naive CD4+ T cells from MS patients were reported to exhibit enhanced activation of NF-κB p65 (34). Moreover, the team identified that the presence of MS risk variant rs228614 proximal to NF-κB1 resulted in increased degradation of IκBα and NF-κB p65 phosphorylation in both TNF-α-stimulated or PMA (phorbol 12-myristate 13-acetate)–stimulated CD4+ T cells (34). Similarly, Yan et al. observed that the amount of nuclear NF-κB p65 in CD3+ T cells of all MS subgroups was significantly higher compared with healthy controls (33). Second, a large number of MS candidate genes were found to interact in a tractable pathway regulating TH1/TH17 inflammation, Treg tolerance, and NF-κB induction (88). The imbalance between Treg and TH1/TH17 cells critically involves in the pathogenesis of EAE and other autoimmune and inflammatory diseases (89, 90). Finally, the data from a network analysis of the CD3+ T cell transcriptome implicate aberrant regulation of gene expression by NF-κB as a biomarker of acute MS relapse (35).
In T cells, the mediation of NF-κB subunits by the downstream of T cell receptor mainly requires p50-p50, p50-p65, or p50-c-Rel dimers (91). The importance of NF-κB1 (p50/p105) functioning can be seen in mouse models; NF-κB1–deficient mice develop normally and acquire a structurally normal immune system but are resistant to EAE. Further investigation suggested that this protection was due to the hindered activation and differentiation of MOG-specific TH1 and TH2 cells in these mutant EAE mice (92). Notably, as NF-κB p105 also belongs to the IκB protein family, previous studies have noted a remarkable increase in the activation of CD4+ T cells and a higher frequency of memory/effector T cells in mice specifically deficient for p105 compared to wild-type mice (93). Nuclear factor κB1, however, may also have beneficial effects because NF-κB1–deficient EAE mice showed more infiltrated inflammatory cells in the CNS than control mice. Consistently, p50-deficient mice exhibit augmented microglial proinflammatory responses after peripheral injection with lipopolysaccharide (94).
Although c-Rel knockout mice do not suffer from development defects, studies have clarified that T cells from these mice are impaired in their ability to activate and generate cytokines such as IL-2, IL-3, and granulocyte–macrophage colony-stimulating factor and differentiate into effector populations (95–97). Similar to mice lacking NF-κB1, mice lacking c-Rel were shown to be protected from EAE (98). In contrast to NF-κB1–deficient mice, in which TH2 cell differentiation was preferentially compromised (99), splenocytes derived from c-Rel–deficient mice produced undetectable IFN-γ and increased levels of IL-4 (98), denoting their non-overlapping capacities for different NF-κB molecules. Later in vitro studies led to the conclusion that c-Rel–deficient CD4+ T cells are intrinsically unable to generate IFN-γ under both TH0- and TH1-polarizing conditions (98). Moreover, c-Rel–deficient APCs displayed a substantially reduced level of IL-12 p40, an essential cytokine for TH1 cell differentiation, which further aggravated TH1 cell deficiency (98). In addition to promoting TH1 cells, c-Rel is involved in the development of TH17 cells through directly controlling expression of the Rorc gene, which encodes the TH17 cell–specific transcription factor retinoic acid–related orphan receptor γt, and indirectly facilitating APCs to generate IL-23, a critical molecule known to enhance IL-17 expression by CD4+ T cells (97, 100, 101). Consistent with these findings, the inability to provoke optimal TH1 and TH17 cell immune responses occurred in parallel with an ameliorated phenotype in c-Rel knockout mice after EAE induction (98, 100). Moreover, c-Rel may additionally influence cytotoxic T lymphocytes because cell survival was dramatically impaired in c-Rel-deficient CD8+ T cells that could be reversed with IL-2 addition (102); however, the capacity of c-Rel-deficient CD8+ T cells to clear viral infection was not affected (103). Although c-Rel deficiency confers resistance to several T cell–dependent autoimmune disorders such as EAE and collagen-induced arthritis (100, 104), novel data have demonstrated the anti-inflammatory effect of c-Rel in promoting the Treg cell lineage, as revealed in c-Rel–deficient mice, in which thymic and peripheral CD4+ Foxp3+ T cells were vastly reduced in number compared to wild-type counterparts (105, 106). Defects in Treg cells are now thought to be partially due to the direct regulation of Foxp3 enhanceosomes by c-Rel (107). Furthermore, as the addition of exogenous IL-2 was sufficient to rescue Foxp3 deficiency, decreased IL-2 generation in c-Rel deletion mice may amplify the lack of Treg cell expansion (106). Notably, despite their decreased frequency, c-Rel-deficient Tregs were indicated by in vitro and in vivo findings as capable of suppressing effector T cells at normal ranges (105).
There are no data exploring the changes in EAE under NF-κB p65-deficient conditions because of embryonic lethality and liver degeneration (108). Lymphocytes derived from SCID (severe combined immunodeficient) mice transplanted with p65-deficient fetal liver cells displayed normal development and IL-2 expression but were defective in their proliferative response to various mitogens (108). Moreover, p65 deficiency in T cells largely blocked TH17 cell differentiation in a manner similar to that of c-Rel deficiency, which was caused by reduced Rorg activity in the TH17 cell lineage (101). In contrast, studies utilizing T cell–specific p65 mutant mice have indicated that p65 is dispensable in TH17 cell differentiation but required for another important source of IL-17, γδ T cells (109). On the other hand, p65 might prevent EAE with its potent capacity to modulate Treg cell homeostasis. In recent years, mounting evidence has identified p65 as an essential component in mature Treg cell identity formation, tolerogenic function, and egress from the thymus (110–112). Finally, in addition to c-Rel, TH1 differentiation is also regulated by RelB. The absence of RelB in T cells led to a dramatic decrease in TH1 differentiation and IFN-γ production, but the conventional TH17 polarization was normal.
Therapeutic Potential For MS
The therapeutic efficacies of many approved treatments for MS are now thought to be attributed, at least in part, to blockade of NF-κB pathway of the peripheral nervous system and CNS immune response. Dimethyl fumarate, for instance, was shown to effectively inhibit the generation of IL-6, TNF, nitric oxide (NO), and NF-κB activation in stimulated microglia, and its active metabolite, monomethyl fumarate, was found to suppress myeloid dendritic cell (DC) maturation partially via NF-κB signaling, hence reducing proinflammatory activities in cocultured T cells (113–115). The effects of FTY720 and phosphorylated FTY720 were observed to decrease NF-κB activity in cultured astrocytes (57, 58). And the most widely used glucocorticoids were found to downregulate NF-κB through both directly inhibiting p65-dependent gene activation and indirectly enhancing IκBα synthesis (116, 117). Responsiveness to laquinimod, a novel immunomodulatory compound for relapsing-remitting MS, was linked to its ability to impair DC maturation and function through NF-κB interference (118). Moreover, laquinimod was reported to ameliorate CNS inflammation and myelin loss in a cuprizone model by attenuating astrocytic NF-κB activation (119).
Although no NF-κB inhibitors have been approved to the clinical treatment for MS, the beneficial effects of NF-κB interference by a considerable number of natural components (e.g., piperlongumine and denanthin) have been suggested in basic animal studies (120, 121). Moreover, the selective NF-κB inhibitor pyrrolidine dithiocarbamate markedly alleviated the incidence and severity of EAE in rats (122). The IKK1/2 inhibitor reduced plasma IL-17 and IFN-γ levels and reduced EAE symptoms (123). Administration of PS-1145, a compound that inhibits IKK2- and NEMO-dependent canonical NF-κB signaling but maintains the alternative NF-κB signaling pathway, effectively improved EAE, which was characterized by decreased lymphocytic proliferation and cytokine (IL-2 and IL-17) production (124). Similarly, peptides corresponding to the NEMO-binding domain displayed a potent propensity to suppress encephalitogenic T cell generation and activation and TH1 cell responses, thus significantly protecting against EAE (125). Finally, a novel peptide from glucocorticoid-induced leucine zipper (GILZ), a molecule that binds and inhibits NF-κB p65, was shown to increase the level of IL-10 and reduce IFN-γ, IL-12, and IL-17 levels in GILZ-treated EAE mice (126).
As discussed above, excessive and persistent immune reactions primarily contribute to MS tissue injury. This context gives rise to uncontrolled NF-κB activity, which further drives ongoing inflammation in a self-amplifying cycle. Several lines of evidence have highlighted the beneficial effects of NF-κB pathway inhibition based on clinical and basic data. However, because basal NF-κB is crucial to normal cellular physiology and pathogen clearance, the non-selective blockade of NF-κB may therefore lead to many unwanted side effects, as we recently reported that the blockade of TNFR1 or TNFRII had a completely difference consequence on TH17 and Treg cells (127). Furthermore, as NF-κB exerts diverse effects depending on the isoform member, type of activated cells, and strength of the triggering event, it would be difficult to predict the therapeutic outcome. Another hurdle lies in the differences between the EAE model and human MS, as well as the heterogeneous pathogenesis in patients with relapsing and progressive MS (11). As such, great effort has been made to increase the safety of NF-κB interference, including selectively diminishing NF-κB activity by targeting the IKK complex, IκB proteins, and the ubiquitin–proteasome system (124, 125). Organ-specific NF-κB interference has raised much attention owing to its potential effect to avoid systemic side events. Local administration of NF-κB decoy oligodeoxynucleotides (ODNs) encapsulated in a viral vesicle was shown to treat various models of inflammatory colitis without impairing extraintestinal NF-κB activation (128). Further study on the delivery of a naked NF-κB decoy ODN to inflamed tissue also indicated success in improving murine bowel disease and restoring colon homeostasis (129).
Conclusion
Multiple sclerosis is an autoimmune inflammatory disease driven by the complex interaction between environment and predisposing genes. Compelling data support the critical role of aberrant NF-κB activation, which triggers proinflammatory activities via multiple aspects, in the pathogenesis of MS and EAE. As NF-κB has both beneficial and detrimental effects, promising agents have been explored to retain essential NF-κB activity. In this regard, a better understanding of the molecular events that determine the point at which NF-κB responses switch from being protective to mediating damaging effects is needed for the therapeutic modulation of neuroinflammation and neurodegeneration.
Author Contributions
YZ completed the first draft. WQ and SZ reviewed and improved this manuscript. CC, WL, and XM polished the article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Zhang Q, Lenardo M J, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
2. Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. (2011) 21:71–85. doi: 10.1038/cr.2010.177
3. Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. (2002) 2:725–34. doi: 10.1038/nri910
4. Kong N, Lan Q, Su W, Chen M, Wang J, Yang Z, et al. Induced T regulatory cells suppress osteoclastogenesis and bone erosion in collagen-induced arthritis better than natural T regulatory cells. Ann Rheum Dis. (2012) 71:1567–72. doi: 10.1136/annrheumdis-2011-201052
5. Su W, Fan H, Chen M, Wang J, Brand D, He X, et al. Induced CD4+ forkhead box protein-positive T cells inhibit mast cell function and established contact hypersensitivity through TGF-beta1. J Allergy Clin Immunol. (2012) 130:444–52. doi: 10.1016/j.jaci.2012.05.011
6. Luo Y, Wu W, Gu J, Zhang X, Dang J, Wang J, et al. Human gingival tissue-derived MSC suppress osteoclastogenesis and bone erosion via CD39-adenosine signal pathway in autoimmune arthritis. EBioMedicine. (2019) 43:620–31. doi: 10.1016/j.ebiom.2019.04.058
7. Cildir G, Low KC, Tergaonkar V. Noncanonical NF-κB signaling in health and disease. Trends Mol Med. (2016) 22:414–29. doi: 10.1016/j.molmed.2016.03.002
8. Gong J, Qiu W, Zeng Q, Liu X, Sun X, Li H, et al. Lack of short-chain fatty acids and overgrowth of opportunistic pathogens define dysbiosis of neuromyelitis optica spectrum disorders: a Chinese pilot study. Mult Scler. (2019) 25:1316–25. doi: 10.1177/1352458518790396
9. Magliozzi R, Howell OW, Nicholas R, Cruciani C, Castellaro M, Romualdi C, et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann Neurol. (2018) 83:739–55. doi: 10.1002/ana.25197
10. Machado-Santos J, Saji E, Tröscher AR, Paunovic M, Liblau R, Gabriely G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain. (2018) 141:2066–82. doi: 10.1093/brain/awy151
11. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron. (2018) 97:742–68. doi: 10.1016/j.neuron.2018.01.021
12. Söderström M, Link H, Sun JB, Fredrikson S, Wang ZY, Huang WX. Autoimmune T cell repertoire in optic neuritis and multiple sclerosis: T cells recognising multiple myelin proteins are accumulated in cerebrospinal fluid. J Neurol Neurosurg Psychiatry. (1994) 57:544–51. doi: 10.1136/jnnp.57.5.544
13. Cao Y, Goods BA, Raddassi K, Nepom GT, Kwok WW, Love JC, et al. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci Transl Med. (2015) 7:274r−87r. doi: 10.1126/scitranslmed.aaa8038
14. Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83-99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. (2000) 6:1167–75. doi: 10.1038/80516
15. Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol. (2014) 13:700–9. doi: 10.1016/S1474-4422(14)70041-9
16. Diaz-Villoslada P, Shih A, Shao L, Genain CP, Hauser SL. Autoreactivity to myelin antigens: myelin/oligodendrocyte glycoprotein is a prevalent autoantigen. J Neuroimmunol. (1999) 99:36–43. doi: 10.1016/S0165-5728(99)00099-5
17. Sun JB, Olsson T, Wang WZ, Xiao BG, Kostulas V, Fredrikson S, et al. Autoreactive T and B cells responding to myelin proteolipid protein in multiple sclerosis and controls. Euro J Immunol. (1991) 21:1461–8. doi: 10.1002/eji.1830210620
18. Lovett-Racke AE, Trotter JL, Lauber J, Perrin PJ, June CH, Racke MK. Decreased dependence of myelin basic protein-reactive T cells on CD28-mediated costimulation in multiple sclerosis patients. A marker of activated/memory T cells. J Clin Invest. (1998) 101:725–30. doi: 10.1172/JCI1528
19. Hellings N, Barée M, Verhoeven C, D'hooghe MB, Medaer R, Bernard CC, et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. (2001) 63:290–302. doi: 10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4
20. Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc Natl Acad Sci USA. (2015) 112:E3246–54. doi: 10.1073/pnas.1421463112
21. Zhou X, Xia Z, Lan Q, Wang J, Su W, Han YP, et al. BAFF promotes Th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS ONE. (2011) 6:e23629. doi: 10.1371/journal.pone.0023629
22. Pan HF, Leng RX, Li XP, Zheng SG, Ye DQ. Targeting T-helper 9 cells and interleukin-9 in autoimmune diseases. Cytokine Growth Factor Rev. (2013) 24:515–22. doi: 10.1016/j.cytogfr.2013.09.001
23. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. (2000) 47:707–17. doi: 10.1002/1531-8249(200006)47:6<707::AID-ANA3>3.0.CO;2-Q
24. Bakker DA, Ludwin SK. Blood-brain barrier permeability during Cuprizone-induced demyelination. Implications for the pathogenesis of immune-mediated demyelinating diseases. J Neurol Sci. (1987) 78:125–37. doi: 10.1016/0022-510X(87)90055-4
25. Gudi V, Gingele S, Skripuletz T, Stangel M. Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front Cell Neurosci. (2014) 8:73. doi: 10.3389/fncel.2014.00073
26. Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. (2013) 45:1353–60. doi: 10.1038/ng.2770
27. De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. (2009) 41:776–82. doi: 10.1016/j.clim.2009.03.294
28. Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
29. Miterski B, Böhringer S, Klein W, Sindern E, Haupts M, Schimrigk S, et al. Inhibitors in the NFkappaB cascade comprise prime candidate genes predisposing to multiple sclerosis, especially in selected combinations. Genes Immun. (2002) 3:211–9. doi: 10.1038/sj.gene.6363846
30. Ponath G, Lincoln MR, Levine-Ritterman M, Park C, Dahlawi S, Mubarak M, et al. Enhanced astrocyte responses are driven by a genetic risk allele associated with multiple sclerosis. Nat Commun. (2018) 9:5337. doi: 10.1038/s41467-018-07785-8
31. Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. (2016) 7:12015. doi: 10.1038/ncomms12015
32. Eggert M, Goertsches R, Seeck U, Dilk S, Neeck G, Zettl UK. Changes in the activation level of NF-kappa B in lymphocytes of MS patients during glucocorticoid pulse therapy. J Neurol Sci. (2008) 264:145–50. doi: 10.1016/j.jns.2007.08.026
33. Yan J, Winterford CM, Catts VS, Pat BK, Pender MP, McCombe PA, et al. Increased constitutive activation of NF-kappaB p65 (RelA) in peripheral blood cells of patients with progressive multiple sclerosis. J Neuroimmunol. (2018) 320:111–6. doi: 10.1016/j.jneuroim.2018.04.002
34. Housley WJ, Fernandez SD, Vera K, Murikinati SR, Grutzendler J, Cuerdon N, et al. Genetic variants associated with autoimmunity drive NFkappaB signaling and responses to inflammatory stimuli. Sci Transl Med. (2015) 7:291r−3r. doi: 10.1126/scitranslmed.aaa9223
35. Satoh J, Misawa T, Tabunoki H, Yamamura T. Molecular network analysis of T-cell transcriptome suggests aberrant regulation of gene expression by NF-kappaB as a biomarker for relapse of multiple sclerosis. Dis Markers. (2008) 25:27–35. doi: 10.1155/2008/824640
36. Lindsey JW, Agarwal SK, Tan FK. Gene expression changes in multiple sclerosis relapse suggest activation of T and non-T cells. Mol Med. (2011) 17:95–102. doi: 10.2119/molmed.2010.00071
37. Bonetti B, Stegagno C, Cannella B, Rizzuto N, Moretto G, Raine CS. Activation of NF-kappaB and c-jun transcription factors in multiple sclerosis lesions. Implications for oligodendrocyte pathology. Am J Pathol. (1999) 155:1433–8. doi: 10.1016/S0002-9440(10)65456-9
38. Gveric D, Kaltschmidt C, Cuzner ML, Newcombe J. Transcription factor NF-kappaB and inhibitor I kappaBalpha are localized in macrophages in active multiple sclerosis lesions. J Neuropathol Exp Neurol. (1998) 57:168–78. doi: 10.1097/00005072-199802000-00008
39. Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. (2015) 7:a20537. doi: 10.1101/cshperspect.a020537
40. Liu B, Gu Y, Pei S, Peng Y, Chen J, Pham LV, et al. Interleukin-1 receptor associated kinase (IRAK)-M -mediated type 2 microglia polarization ameliorates the severity of experimental autoimmune encephalomyelitis (EAE). J Autoimmun. (2019) 102:77–88. doi: 10.1016/j.jaut.2019.04.020
41. Lee MJ, Bing SJ, Choi J, Jang M, Lee G, Lee H, et al. IKKbeta-mediated inflammatory myeloid cell activation exacerbates experimental autoimmune encephalomyelitis by potentiating Th1/Th17 cell activation and compromising blood brain barrier. Mol Neurodegener. (2016) 11:54. doi: 10.1186/s13024-016-0116-1
42. Zhang H, Bi J, Yi H, Fan T, Ruan Q, Cai L, et al. Silencing c-Rel in macrophages dampens Th1 and Th17 immune responses and alleviates experimental autoimmune encephalomyelitis in mice. Immunol Cell Biol. (2017) 95:593–600. doi: 10.1038/icb.2017.11
43. Goldmann T, Wieghofer P, Müller PF, Wolf Y, Varol D, Yona S, et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci. (2013) 16:1618–26. doi: 10.1038/nn.3531
44. Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, et al. A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun. (2018) 9:2036. doi: 10.1038/s41467-018-04376-5
45. Ellrichmann G, Thöne J, Lee DH, Rupec RA, Gold R, Linker RA. Constitutive activity of NF-kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J Neuroinflammation. (2012) 9:15. doi: 10.1186/1742-2094-9-15
46. Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. (2005) 434:1138–43. doi: 10.1038/nature03491
47. Raasch J, Zeller N, van Loo G, Merkler D, Mildner A, Erny D, et al. IkappaB kinase 2 determines oligodendrocyte loss by non-cell-autonomous activation of NF-kappaB in the central nervous system. Brain. (2011) 134(Pt 4):1184–98. doi: 10.1093/brain/awq359
48. Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. (2009) 19:404–13. doi: 10.1016/j.tcb.2009.05.006
49. Lee S, Andrieu C, Saltel F, Destaing O, Auclair J, Pouchkine V, et al. IkappaB kinase beta phosphorylates Dok1 serines in response to TNF, IL-1, or gamma radiation. Proc Natl Acad Sci USA. (2004) 101:17416–21. doi: 10.1073/pnas.0408061101
50. van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, et al. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol. (2006) 7:954–61. doi: 10.1038/ni1372
51. Lu ZY, Yu SP, Wei JF, Wei L. Age-related neural degeneration in nuclear-factor kappaB p50 knockout mice. Neuroscience. (2006) 139:965–78. doi: 10.1016/j.neuroscience.2005.12.062
52. Kretz A, Herrmann KH, Fischer S, Engelmann C, Witte OW, Reichenbach JR, et al. Dysfunctional NF-kappaB and brain myelin formation. Eur J Hum Genet. (2014) 22:724–5. doi: 10.1038/ejhg.2013.240
53. Nickols JC, Valentine W, Kanwal S, Carter BD. Activation of the transcription factor NF-kappaB in Schwann cells is required for peripheral myelin formation. Nat Neurosci. (2003) 6:161–7. doi: 10.1038/nn995
54. Stone S, Jamison S, Yue Y, Durose W, Schmidt-Ullrich R, Lin W. NF-kappaB activation protects oligodendrocytes against inflammation. J Neurosci. (2017) 37:9332–44. doi: 10.1523/JNEUROSCI.1608-17.2017
55. Gupta AS, Biswas DD, Brown SN, Mockenhaupt K, Marone M, Hoskins A, et al. A detrimental role of RelB in mature oligodendrocytes during experimental acute encephalomyelitis. J Neuroinflammation. (2019) 16:161. doi: 10.1186/s12974-019-1548-7
56. Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. (2014) 94:1077–98. doi: 10.1152/physrev.00041.2013
57. Trkov Bobnar S, Stenovec M, Miš K, Pirkmajer S, Zorec R. Fingolimod suppresses the proinflammatory status of interferon-gamma-activated cultured rat astrocytes. Mol Neurobiol. (2019) 56:5971–86. doi: 10.1007/s12035-019-1481-x
58. Dong YF, Guo RB, Ji J, Cao LL, Zhang L, Chen ZZ, et al. S1PR3 is essential for phosphorylated fingolimod to protect astrocytes against oxygen-glucose deprivation-induced neuroinflammation via inhibiting TLR2/4-NFkappaB signalling. J Cell Mol Med. (2018) 22:3159–66. doi: 10.1111/jcmm.13596
59. Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, et al. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. (2009) 182:2628–40. doi: 10.4049/jimmunol.0802954
60. He Y, Jackman NA, Thorn TL, Vought VE, Hewett SJ. Interleukin-1beta protects astrocytes against oxidant-induced injury via an NF-kappaB-dependent upregulation of glutathione synthesis. Glia. (2015) 63:1568–80. doi: 10.1002/glia.22828
61. Chu LF, Wang WT, Ghanta VK, Lin CH, Chiang YY, Hsueh CM. Ischemic brain cell-derived conditioned medium protects astrocytes against ischemia through GDNF/ERK/NF-kB signaling pathway. Brain Res. (2008) 1239:24–35. doi: 10.1016/j.brainres.2008.08.087
62. Brambilla R, Morton PD, Ashbaugh JJ, Karmally S, Lambertsen KL, Bethea JR. Astrocytes play a key role in EAE pathophysiology by orchestrating in the CNS the inflammatory response of resident and peripheral immune cells and by suppressing remyelination. Glia. (2014) 62:452–67. doi: 10.1002/glia.22616
63. Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. (2006) 12:518–25. doi: 10.1038/nm1402
64. Xie X, Luo X, Liu N, Li X, Lou F, Zheng Y, et al. Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J Neurochem. (2017) 141:222–35. doi: 10.1111/jnc.13972
65. Kaltschmidt B, Kaltschmidt C. NF-κB in the nervous system. Cold Spring Harb Perspect Biol. (2009) 1:a1271. doi: 10.1101/cshperspect.a001271
66. Emmanouil M, Taoufik E, Tseveleki V, Vamvakas SS, Tselios T, Karin M, et al. Neuronal I kappa B kinase beta protects mice from autoimmune encephalomyelitis by mediating neuroprotective and immunosuppressive effects in the central nervous system. J Immunol. (2009) 183:7877–89. doi: 10.4049/jimmunol.0900834
67. Lee DH, Kubera K, Rosenthal B, Kaltschmidt B, Kaltschmidt C, Gold R, et al. Neuronal NF-κB ablation does not influence neuro-axonal degeneration in experimental autoimmune demyelination. J Neuroimmunol. (2012) 246:38–42. doi: 10.1016/j.jneuroim.2012.03.005
68. Sun J, Fang Y, Chen T, Guo J, Yan J, Song S, et al. WIN55, 212-2 promotes differentiation of oligodendrocyte precursor cells and improve remyelination through regulation of the phosphorylation level of the ERK 1/2 via cannabinoid receptor 1 after stroke-induced demyelination. Brain Res. (2013) 1491:225–35. doi: 10.1016/j.brainres.2012.11.006
69. Agrawal A, Dillon S, Denning TL, Pulendran B. ERK1-/- mice exhibit Th1 cell polarization and increased susceptibility to experimental autoimmune encephalomyelitis. J Immunol. (2006) 176:5788–96. doi: 10.4049/jimmunol.176.10.5788
70. Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol. (2018) 19:696–707. doi: 10.1038/s41590-018-0135-x
71. Xu A, Liu Y, Chen W, Wang J, Xue Y, Huang F, et al. TGF-beta-induced regulatory T cells directly suppress B cell responses through a noncytotoxic mechanism. J Immunol. (2016) 196:3631–41. doi: 10.4049/jimmunol.1501740
72. Zhong H, Liu Y, Xu Z, Liang P, Yang H, Zhang X, et al. TGF-beta-induced CD8(+)CD103(+) regulatory T cells show potent therapeutic effect on chronic graft-versus-host disease lupus by suppressing B cells. Front Immunol. (2018) 9:35. doi: 10.3389/fimmu.2018.00035
73. Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature. (2012) 491:264–8. doi: 10.1038/nature11501
74. Chen D, Ireland SJ, Remington G, Alvarez E, Racke MK, Greenberg B, et al. CD40-mediated NF-kappaB Activation in B Cells Is Increased in Multiple Sclerosis and Modulated by Therapeutics. J Immunol. (2016) 197:4257–65. doi: 10.4049/jimmunol.1600782
75. Luo W, Weisel F, Shlomchik MJ. B cell receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity. (2018) 48:313–26. doi: 10.1016/j.immuni.2018.01.008
76. Aarts SABM, Seijkens TTP, van Dorst KJF, Dijkstra CD, Kooij G, Lutgens E. The CD40-CD40L Dyad in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol. (2017) 8:1791. doi: 10.3389/fimmu.2017.01791
77. Lee HJ, Lombardi A, Stefan M, Li CW, Inabnet WB, Owen RP, et al. CD40 Signaling in graves disease is mediated through canonical and noncanonical thyroidal nuclear factor kappaB activation. Endocrinology. (2017) 158:410–8. doi: 10.1210/en.2016-1609
78. Boon L, Brok HP, Bauer J, Ortiz-Buijsse A, Schellekens MM, Ramdien-Murli S, et al. Prevention of experimental autoimmune encephalomyelitis in the common marmoset (Callithrix jacchus) using a chimeric antagonist monoclonal antibody against human CD40 is associated with altered B cell responses. J Immunol. (2001) 167:2942–9. doi: 10.4049/jimmunol.167.5.2942
79. Ma N, He Y, Xiao H, Han G, Chen G, Wang Y, et al. BAFF maintains T-cell survival by inducing OPN expression in B cells. Mol Immunol. (2014) 57:129–37. doi: 10.1016/j.molimm.2013.08.014
80. Karaky M, Fedetz M, Potenciano V, Andrés-León E, Codina AE, Barrionuevo C, et al. SP140 regulates the expression of immune-related genes associated with multiple sclerosis and other autoimmune diseases by NF-kappaB inhibition. Hum Mol Genet. (2018) 27:4012–23. doi: 10.1093/hmg/ddy284
81. Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol. (2017) 198:691–8. doi: 10.4049/jimmunol.1601649
82. Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, et al. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med. (1996) 184:1537–41. doi: 10.1084/jem.184.4.1537
83. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal center B cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med. (2014) 211:2103–18. doi: 10.1084/jem.20132613
84. Franzoso G, Carlson L, Poljak L, Shores EW, Epstein S, Leonardi A, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. (1998) 187:147–59. doi: 10.1084/jem.187.2.147
85. Jacque E, Schweighoffer E, Visekruna A, Papoutsopoulou S, Janzen J, Zillwood R, et al. IKK-induced NF-kappaB1 p105 proteolysis is critical for B cell antibody responses to T cell-dependent antigen. J Exp Med. (2014) 211:2085–101. doi: 10.1084/jem.20132019
86. de Valle E, Grigoriadis G, O'Reilly LA, Willis SN, Maxwell MJ, Corcoran LM, et al. NFkappaB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J Exp Med. (2016) 213:621–41. doi: 10.1084/jem.20151182
87. van Oosten BW, Lai M, Hodgkinson S, Barkhof F, Miller DH, Moseley IF, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology. (1997) 49:351–7. doi: 10.1212/WNL.49.2.351
88. Hussman JP, Beecham AH, Schmidt M, Martin ER, McCauley JL, Vance JM, et al. GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. (2016) 17:305–12. doi: 10.1038/gene.2016.23
89. Chen W, Wang J, Xu Z, Huang F, Qian W, Ma J, et al. Apremilast ameliorates experimental arthritis via suppression of Th1 and Th17 cells and enhancement of CD4(+)Foxp3(+) regulatory T cells differentiation. Front Immunol. (2018) 9:1662. doi: 10.3389/fimmu.2018.01662
90. Luo Y, Xue Y, Wang J, Dang J, Fang Q, Huang G, et al. Negligible effect of sodium chloride on the development and function of TGF-beta-induced CD4(+) Foxp3(+) regulatory T cells. Cell Rep. (2019) 26:1869–79. doi: 10.1016/j.celrep.2019.01.066
91. Zheng Y, Vig M, Lyons J, Van Parijs L, Beg AA. Combined deficiency of p50 and cRel in CD4+ T cells reveals an essential requirement for nuclear factor kappaB in regulating mature T cell survival and in vivo function. J Exp Med. (2003) 197:861–74. doi: 10.1084/jem.20021610
92. Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y. Experimental autoimmune encephalomyelitis in NF-kappa B-deficient mice:roles of NF-kappa B in the activation and differentiation of autoreactive T cells. J Immunol. (1999) 163:2937–43.
93. Chang M, Lee AJ, Fitzpatrick L, Zhang M, Sun SC. NF-kappa B1 p105 regulates T cell homeostasis and prevents chronic inflammation. J Immunol. (2009) 182:3131–8. doi: 10.4049/jimmunol.0803637
94. Taetzsch T, Benusa S, Levesque S, Mumaw CL, Block ML. Loss of NF-kappaB p50 function synergistically augments microglial priming in the middle-aged brain. J Neuroinflammation. (2019) 16:60. doi: 10.1186/s12974-019-1446-z
95. Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol. (1999) 11:361–71. doi: 10.1093/intimm/11.3.361
96. Gerondakis S, Strasser A, Metcalf D, Grigoriadis G, Scheerlinck JY, Grumont RJ. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc Natl Acad Sci USA. (1996) 93:3405–9. doi: 10.1073/pnas.93.8.3405
97. Visekruna A, Volkov A, Steinhoff U. A key role for NF-κB transcription factor c-Rel in T-lymphocyte-differentiation and effector functions. Clin Dev Immunol. (2012) 2012:239368. doi: 10.1155/2012/239368
98. Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi SE, Liou HC, et al. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest. (2002) 110:843–50. doi: 10.1172/JCI0215254
99. Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. (2001) 2:45–50. doi: 10.1038/83158
100. Chen G, Hardy K, Pagler E, Ma L, Lee S, Gerondakis S, et al. The NF-κB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J Immunol. (2011) 187:4483–91. doi: 10.4049/jimmunol.1101757
101. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 immune response is controlled by the Rel-RORgamma-RORgamma T transcriptional axis. J Exp Med. (2011) 208:2321–33. doi: 10.1084/jem.20110462
102. Saibil SD, Jones RG, Deenick EK, Liadis N, Elford AR, Vainberg MG, et al. CD4+ and CD8+ T cell survival is regulated differentially by protein kinase Ctheta, c-Rel, and protein kinase B. J Immunol. (2007) 178:2932–9. doi: 10.4049/jimmunol.178.5.2932
103. Harling-Mcnabb L, Deliyannis G, Jackson DC, Gerondakis S, Grigoriadis G, Brown LE. Mice lacking the transcription factor subunit Rel can clear an influenza infection and have functional anti-viral cytotoxic T cells but do not develop an optimal antibody response. Int Immunol. (1999) 11:1431–9. doi: 10.1093/intimm/11.9.1431
104. Campbell IK, Gerondakis S, O'Donnell K, Wicks IP. Distinct roles for the NF-kappaB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J Clin Invest. (2000) 105:1799–806. doi: 10.1172/JCI8298
105. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. (2009) 206:3001–14. doi: 10.1084/jem.20091411
106. Visekruna A, Huber M, Hellhund A, Bothur E, Reinhard K, Bollig N, et al. c-Rel is crucial for the induction of Foxp3(+) regulatory CD4(+) T cells but not T(H)17 cells. Eur J Immunol. (2010) 40:671–6. doi: 10.1002/eji.200940260
107. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. (2009) 31:932–40. doi: 10.1016/j.immuni.2009.10.006
108. Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. (1995) 376:167–70. doi: 10.1038/376167a0
109. Powolny-Budnicka I, Riemann M, Tänzer S, Schmid RM, Hehlgans T, Weih F. RelA and RelB transcription factors in distinct thymocyte populations control lymphotoxin-dependent interleukin-17 production in gammadelta T cells. Immunity. (2011) 34:364–74. doi: 10.1016/j.immuni.2011.02.019
110. Fukazawa T, Hiraiwa N, Umemura T, Mise-Omata S, Obata Y, Doi T. Egress of mature murine regulatory T cells from the thymus requires RelA. J Immunol. (2015) 194:3020–8. doi: 10.4049/jimmunol.1302756
111. Messina N, Fulford T, O'Reilly L, Loh WX, Motyer JM, Ellis D, et al. The NF-kappaB transcription factor RelA is required for the tolerogenic function of Foxp3(+) regulatory T cells. J Autoimmun. (2016) 70:52–62. doi: 10.1016/j.jaut.2016.03.017
112. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-kappaB transcription-factor-dependent lineage-specific transcriptional program promotes regulatory T cell identity and function. Immunity. (2017) 47:450–65. doi: 10.1016/j.immuni.2017.08.010
113. Miljković D, BlaŽevski J, Petković F, Djedović N, Momčilović M, Stanisavljević S, et al. A comparative analysis of multiple sclerosis-relevant anti-inflammatory properties of ethyl pyruvate and dimethyl fumarate. J Immunol. (2015) 194:2493–503. doi: 10.4049/jimmunol.1402302
114. Mazzola MA, Raheja R, Regev K, Beynon V, von Glehn F, Paul A, et al. Monomethyl fumarate treatment impairs maturation of human myeloid dendritic cells and their ability to activate T cells. Mult Scler. (2019) 25:63–71. doi: 10.1177/1352458517740213
115. Peng H, Guerau-de-Arellano M, Mehta VB, Yang Y, Huss DJ, Papenfuss TL, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor kappaB (NF-kappaB) and extracellular signal-regulated kinase 1 and 2 (ERK1/2) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem. (2012) 287:28017–26. doi: 10.1074/jbc.M112.383380
116. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. (1995) 270:286–90. doi: 10.1126/science.270.5234.286
117. De Bosscher K, Schmitz ML, Vanden Berghe W, Plaisance S, Fiers W, Haegeman G. Glucocorticoid-mediated repression of nuclear factor-kappaB-dependent transcription involves direct interference with transactivation. Proc Natl Acad Sci USA. (1997) 94:13504–9. doi: 10.1073/pnas.94.25.13504
118. Jolivel V, Luessi F, Masri J, Kraus SH, Hubo M, Poisa-Beiro L, et al. Modulation of dendritic cell properties by laquinimod as a mechanism for modulating multiple sclerosis. Brain. (2013) 136(Pt 4):1048–66. doi: 10.1093/brain/awt023
119. Brück W, Pförtner R, Pham T, Zhang J, Hayardeny L, Piryatinsky V, et al. Reduced astrocytic NF-kappaB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. (2012) 124:411–24. doi: 10.1007/s00401-012-1009-1
120. Yin QQ, Liu CX, Wu YL, Wu SF, Wang Y, Zhang X, et al. Preventive and therapeutic effects of adenanthin on experimental autoimmune encephalomyelitis by inhibiting NF-kappaB signaling. J Immunol. (2013) 191:2115–25. doi: 10.4049/jimmunol.1203546
121. Zhang J, Zeng YQ, Zhang J, Pan XD, Kang DY, Huang TW, et al. Tripchlorolide ameliorates experimental autoimmune encephalomyelitis by down-regulating ERK1/2-NF-kappaB and JAK/STAT signaling pathways. J Neurochem. (2015) 133:104–12. doi: 10.1111/jnc.13058
122. Pahan K, Schmid M. Activation of nuclear factor-kB in the spinal cord of experimental allergic encephalomyelitis. Neurosci Lett. (2000) 287:17–20. doi: 10.1016/S0304-3940(00)01167-8
123. Lunin SM, Khrenov MO, Glushkova OV, Parfenyuk SB, Novoselova TV, Novoselova EG. Immune response in the relapsing-remitting experimental autoimmune encephalomyelitis in mice: The role of the NF-kappaB signaling pathway. Cell Immunol. (2019) 336:20–7. doi: 10.1016/j.cellimm.2018.12.003
124. Greve B, Weissert R, Hamdi N, Bettelli E, Sobel RA, Coyle A, et al. I kappa B kinase 2/beta deficiency controls expansion of autoreactive T cells and suppresses experimental autoimmune encephalomyelitis. J Immunol. (2007) 179:179–85. doi: 10.4049/jimmunol.179.1.179
125. Dasgupta S, Jana M, Zhou Y, Fung YK, Ghosh S, Pahan K. Antineuroinflammatory effect of NF-kappaB essential modifier-binding domain peptides in the adoptive transfer model of experimental allergic encephalomyelitis. J Immunol. (2004) 173:1344–54. doi: 10.4049/jimmunol.173.2.1344
126. Srinivasan M, Janardhanam S. Novel p65 binding glucocorticoid-induced leucine zipper peptide suppresses experimental autoimmune encephalomyelitis. J Biol Chem. (2011) 286:44799–810. doi: 10.1074/jbc.M111.279257
127. Yang S, Xie C, Chen Y, Wang J, Chen X, Lu Z, et al. Differential roles of TNFalpha-TNFR1 and TNFalpha-TNFR2 in the differentiation and function of CD4(+)Foxp3(+) induced Treg cells in vitro and in vivo periphery in autoimmune diseases. Cell Death Dis. (2019) 10:27. doi: 10.1038/s41419-018-1266-6
128. Fichtner-Feigl S, Fuss IJ, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J Clin Invest. (2005) 115:3057–71. doi: 10.1172/JCI24792
Keywords: NF-κB, multiple sclerosis, demyelination, inflammation, T cells
Citation: Zhou Y, Cui C, Ma X, Luo W, Zheng SG and Qiu W (2020) Nuclear Factor κB (NF-κB)–Mediated Inflammation in Multiple Sclerosis. Front. Immunol. 11:391. doi: 10.3389/fimmu.2020.00391
Received: 28 December 2019; Accepted: 19 February 2020;
Published: 24 March 2020.
Edited by:
Jorge Matias-Guiu, Complutense University of Madrid, SpainReviewed by:
Ulises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, SpainJorge Tolivia, University of Oviedo, Spain
Copyright © 2020 Zhou, Cui, Ma, Luo, Zheng and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song Guo Zheng, c29uZ2d1by56aGVuZ0Bvc3VtYy5lZHU=; Wei Qiu, cWl1d2VpMTIwQHZpcC4xNjMuY29t