
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 05 January 2018
Sec. Inflammation
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01932
 Stefano Alivernini1
Stefano Alivernini1 Elisa Gremese1Charles McSharry2Barbara Tolusso1Gianfranco Ferraccioli1Iain B. McInnes2,3
Elisa Gremese1Charles McSharry2Barbara Tolusso1Gianfranco Ferraccioli1Iain B. McInnes2,3 Mariola Kurowska-Stolarska2,3*
Mariola Kurowska-Stolarska2,3*
MicroRNAs (miRNAs) are small non-coding RNAs that fine-tune the cell response to a changing environment by modulating the cell transcriptome. miR-155 is a multifunctional miRNA enriched in cells of the immune system and is indispensable for the immune response. However, when deregulated, miR-155 contributes to the development of chronic inflammation, autoimmunity, cancer, and fibrosis. Herein, we review the evidence for the pathogenic role of miR-155 in driving aberrant activation of the immune system in rheumatoid arthritis, and its potential as a disease biomarker and therapeutic target.
Epigenetic modification represents an important mechanism of regulating gene expression that enables cells to respond appropriately to a changing environment. The mechanisms of epigenetic modification include, e.g., acetylation of histones, methylation of gene promoters, and silencing of mRNA transcripts by microRNAs (miRNA) (1). miRNAs are a family of short, non-coding RNAs, acting at the posttranscriptional level (2, 3), that fine-tune mRNA transcription (4). More than 2,000 miRNAs have been identified in humans, and computational predictions show that these regulate the expression of approximately 60% of all human protein-coding genes (5). miRNAs are pleotropic; a single miRNA can regulate a cellular response by targeting multiple components of a biological pathway (2). More than 100 different miRNAs are expressed by cells of the immune system together influencing the pathways that control the development and function of cells of innate and adaptive immune responses (2, 6, 7). Identifying disease-specific miRNAs improved the understanding of molecular pathways involved in diseases, and provided an evidence-base for new therapeutic strategies, e.g., in cancer (8) and tendinopathy (9).
Rheumatoid arthritis (RA) is an autoimmune disease that affects approximately 1% of the global population and leads to progressive loss of joint function (10). Currently, more than half of RA patients do not achieve sustained drug-induced disease remission (11), which constitutes an important clinical unmet need. A better understanding of the disease process is required to improve the treatment options for patients resistant to current therapeutics and to provide an evidence base for personalized medicine.
A decade of research has shown that aberrant expression of miRNAs underlies the immune response and stromal cell activation in RA (2, 12). miR-155 is a master-regulator of the immune response and the aim of the present review is to discuss its role in the immuno-pathogenesis of RA and its potential as a disease activity biomarker and therapeutic target.
MicroRNAs are ~23 nt small non-coding RNAs that regulate mRNA expression at the posttranscriptional level by directing mRNA degradation or translational repression. They fine-tune expression of their target genes (by approximately 1.2-fold to fourfold) and correspondingly affect biological pathway function (2). miRNAs bind complementary seed-region sequences in the 3′ untranslated regions (UTRs) of specific target mRNAs leading to the repression of protein production. Each miRNA has the potential to repress many target mRNAs, often in the same molecular pathway, highlighting the sophistication of this epigenetic regulation.
MicroRNAs emerge from long primary transcripts (pri-miRNAs) transcribed from independent miRNA coding genes or from introns of protein-coding mRNAs (13, 14). After transcription, pri-miRNAs are capped, poly-adenylated, and then cleaved into ~70 nt hairpin structures (pre-miRNAs) by a nuclear microprocessor complex composed of RNase III-type endonuclease Drosha and the DiGeorge critical region 8 protein. DGCR8 is essential for Drosha activity and is capable of binding single-stranded fragments of the pri-miRNA that are required for proper processing. Pre-miRNAs are then exported from the nucleus to the cytoplasm through Exportin-5 in a GTP-dependent mechanism. In the cytoplasm, pre-miRNAs are then cleaved by Dicer; a RNase III-type enzyme that acts with its cofactor TAR RNA-binding protein 2 resulting in RNA duplexes of ~22–23 nt in length. Subsequently, an argonaute (Ago) protein and a 182-kDa glycine–tryptophan repeat-containing protein bind to these RNA duplexes forming the core of a multi-subunit complex called the miRNA-mediated silencing complex (miRISC). Once assembled into the miRISC, through base-pairing interactions between nucleotides 2 and 8 of the miRNA (the seed region) and complementary nucleotides predominantly in the 3′-UTR of mRNAs, they act as repressor of mRNA translation (15–18).
The miR-155 gene was initially described as the B-cell integration cluster gene (bic) in chickens, which induced leucocytosis when activated by a viral promoter insertion that increased bic transcription (19). Following this finding, a homologous gene to bic was identified in humans and mice (20). Human and murine bic cDNA have >70% identity (20). Bic is strongly expressed in the thymus and spleen, and can be detected in other tissues, including liver, lung, and kidney (20–22). At the cellular level, analysis of small RNA clone libraries by Landgraf and colleagues demonstrated that miR-155 is expressed in hematopoietic stem-progenitor cells and mature hematopoietic cells, including monocytes, granulocytes, B-cells, and T-cells (23). Subsequent experiments showed that miR-155 plays an essential role in controlling both myelopoiesis and erythropoiesis from CD34+ hematopoietic stem-progenitor cells (24, 25).
The miRNA duplex contains two strands identified with either the suffix -5p (from the 5′ arm of pre-miR; i.e., miR-155-5p) or -3p (from the 3′ arm of pre-miR; i.e., miR-155-3p) (26). One of the strands of the duplex is normally discarded (the passenger strand; annotated *) while the retained strand guides eventual mRNA target selection (the guide strand). Thermodynamic properties of the duplex appear to determine strand selection; the strand with the weakest binding at the 5′-end of the duplex is more likely to become the guide strand. Other key characteristics of miRNA guide strands are a U-bias at the 5′-end and an excess of purines (A/G rich), whereas the passenger strands have a C-bias at the 5′-end and an excess of pyrimidines (U/C rich) (27). However, the preferred guide strand can be changed by a single point mutation in the duplex (28), posttranscriptional modification of the duplex (29), and the type of proteins associated with Ago2 in the RISC complex (e.g., trans-activation response RNA-binding protein versus protein activator of dsRNA-dependent protein kinase) (30). Thus, there is increasing evidence demonstrating that both arms of the pre-miRNA hairpin can give rise to guide miRNAs (31, 32) that are biologically functional. In general, miR-155-3p is considered to be the passenger strand (*), and its expression levels are typically 20-fold to 200-fold lower than miR-155-5p. However, despite this difference in expression level, miR-155-3p (*) can be functional, e.g., following TLR7 ligand stimulation of plasmocytoid dendritic cells (pDCs), miR-155-3p is rapidly upregulated while miR-155-5p is induced at a later stage (33, 34). miR-155-3p acts at an early stage by targeting interleukin-1 receptor-associated kinase 3 (IRAK3/IRAKM) mRNA which is a negative regulator of toll-like receptor signaling, facilitating TLR7-induced IFNα/β production, and the later induction of miR-155-5p (miR-155) terminates this production by targeting TGF-β activated kinase 1/MAP3K7 binding protein 2 (TAB 2) mRNA, a key signaling molecule of TLRs (33, 34). Thus, both strands of the miR-155 duplex are required for an efficiently co-ordinated pDCs response. Most studies have investigated the biology of the miR-155-5p strand and further studies are recommended to investigate the evidence for a role of miR-155*(3p) in the regulation of the immune system and disease.
miR-155 expression is rapidly increased in response to infection or injury. Inducing factors include pathogen-associated molecular patterns and damage-associated molecular patterns (PAMPs/DAMPs (35)), alarmins (e.g., IL-1α) (36), and inflammatory stimuli, e.g., TNF, IL-1β, interferons (35), and hypoxia (37). In contrast, the expression of miR-155 is decreased by anti-inflammatory cytokines, resolvins, glucocorticoids, and posttranscriptional negative regulators, e.g., tristetraprolin; and this decreased expression of miR-155 is an important part of the negative-feedback mechanism terminating immune responses. For example, IL-10 decreases miR-155 expression by inhibiting the transcription factor Ets2. Thus, LPS-induced miR-155 expression is attenuated in Ets2-deficient mice (38). Regulatory cytokines, e.g., TGFβ can induce or inhibit miR-155 expression depending on the cell type and tissue environment (39–41). Resolvins are lipid mediators produced, e.g., by tissue macrophages, upon activation of Tyrosine-protein kinase Mer (MerTK) by apoptotic inflammatory cells (42) and have broad anti-inflammatory effects. Resolvin D1 reduces inflammation in experimental corneal immunopathology by inhibiting miR-155 expression (43). Natural and synthetic glucocorticoids are highly effective at terminating acute inflammation, mediated in part by inhibition of miR-155 expression in a glucocorticoids receptor- and NF-κB-dependent manner (44, 45). Inflammation can be controlled by the short half-life of mRNA of pro-inflammatory mediators (e.g., TNF, GM-CSF, IL-8, and CCL2). The rapid elimination of these mRNAs is mediated by miRNAs; or by specific proteins, e.g., tristetraprolin (TTP/ZFP36) that recognizes adenine–uridine rich elements (AREs) in mRNA and orchestrate its degradation. TTP inhibits miR-155, albeit by an unusual mechanism. Cells that overexpress TTP show high levels of miR-1 that putatively prevents the processing of miR-155 precursor to the mature form (46). In addition, the functions of mature miR-155 are counterbalanced by other miRs and the best example is miR-146. This, induced by the same stimuli as miR-155, targets TRAF6 and IRAK1 of the TLR/IL-1R signaling pathway and the timing of this provides feedback inhibition of the inflammatory response driven by miR-155 (47). This is evident in miR-146-deficient mice that develop inflammatory syndrome, autoimmunity, and cancer. miR-146-deficient cells show an increased expression of miR-155 and their pro-inflammatory phenotype could be normalized by deletion of miR-155 (48), thus miR-155 and miR-146 can cross-regulate inflammatory responses.
The miR-155 gene contains binding sites for multiple transcription factors including sites localized downstream and upstream of exon 1, e.g., 2 NF-κB binding sites (−1,697 and −1,150 bp from the transcription initiation site, respectively), SMAD4 (−600 bp), interferon-sensitive response element ISRE (−311 bp), interferon regulatory factors IRF (−200 bp), and AP-1 (−40 bp). There are also two Ets binding sites within the transcription start site, two Foxp3 binding sites in intron 2, and three hypoxia-inducible factor-1 alpha binding sites in the promoter region (37, 49). This reflects the role of miR-155 in co-ordination of the cell response to changes in the tissue environment (49).
Macrophages can be polarized into different phenotypes in response to changes in tissue environments (50). For example, during bacterial infection, TLRL- or T-helper 1 (Th1)/ILC1-derived IFNγ polarizes the pro-inflammatory macrophage phenotype that drives the protective immune response. However, if uncontrolled, this leads to chronic inflammation (50). Homeostasis can be restored by IL-10 and TGFβ derived from regulatory T-cells and by glucocorticoids, that induce macrophage phenotypes that mediate resolution of the immune response and facilitate tissue repair. IL-4/13 derived from Th2/ILC2 induces alternative macrophage phenotypes that are key in eliminating the parasites (51). However, they are also involved in mediating tissue repair by producing collagen components (proline), TGFβ, and CCL18. These changes in macrophage phenotypes are co-ordinated by miRNA networks, including miR-155 (52).
miR-155 is induced in monocyte/macrophages upon TLR/IFNγ stimulation and drives their inflammatory response by epigenetic regulation of mRNA targets that are inhibitors of innate cell activation, e.g.,
(i) phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1 (SHIP-1); an inhibitor of TLR/PI3/Akt kinase pathways.
(ii) SOCS-1; a type 1 cytokine receptor/STAT pathway inhibitor.
(iii) Bcl6, an inhibitor of NF-κB (53–56).
Bone marrow-derived murine macrophages deficient for miR-155 express decreased levels of pro-inflammatory cytokines following LPS stimulation (56, 57), as do miR-155-deficient murine RAW264.7 macrophages (58) and human macrophages gene-silenced for miR-155 (57); each associated with commensurate upregulation of SHIP-1.
In contrast to pro-inflammatory activation of macrophages, miR-155 inhibits the polarization of anti-inflammatory and repair macrophage phenotypes. miR-155 targets translation of multiple molecules in the IL-13/IL-4 pathway, including IL-13Rα, CEBP/β (59), and Mafb (60) and thus prevents development of the STAT6-driven anti-inflammatory and helminth-expulsive macrophage phenotype (61). Furthermore, miR-155 regulates the TGF-β signaling pathway, which is crucial for wound healing and homeostatic remodeling (62, 63). Louafi and colleagues found that miR-155 represses the TGFβ signaling molecule Smad2 in macrophages, preventing TGFβ-induced expression of IL-4Rα, amplification of TGFβ1/2 production, and the development of repair macrophages (63). We recently extended these findings by showing that miR-155 negatively regulates remodeling pathways in alveolar macrophages by targeting the transcription factor LXRα. miR-155-deficient mice had exacerbated bleomycin-induced lung fibrosis compared to WT mice given bleomycin, and their alveolar macrophages showed an increased expression of Arginase 2, which is key for collagen production (36). Thus, miR-155 is a master-switch that determines the relative dynamics of TLR-induced inflammatory and IL-13/IL-4 or TGFβ-induced anti-inflammatory/repair phenotypes in macrophages (Figure 1). Inhibition of miR-155, e.g., by glucocorticoids or by regulatory T-cell-derived mediators (e.g., IL-10) allows the emergence of anti-inflammatory and tissue-repair macrophage phenotypes.
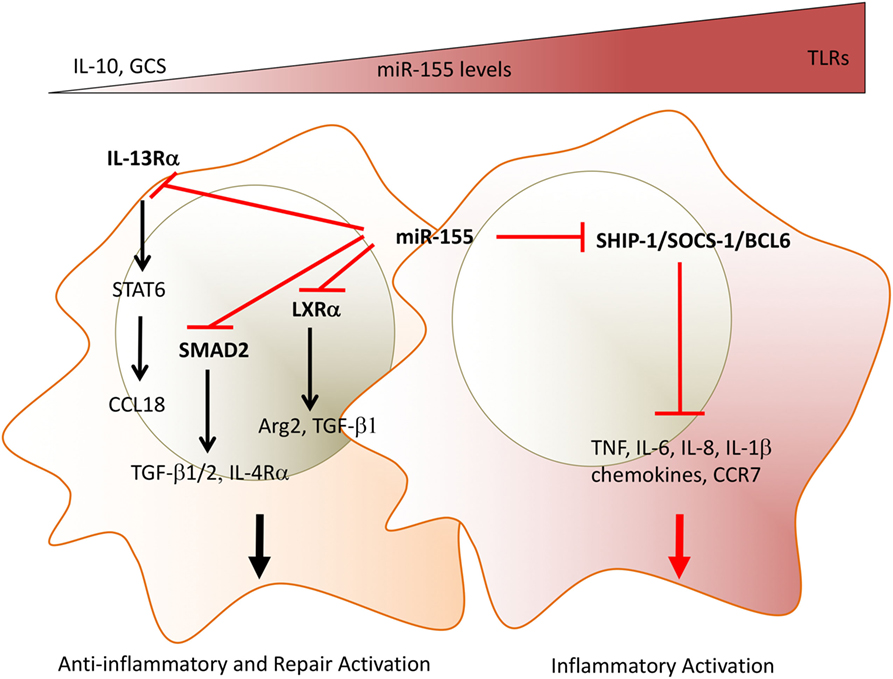
Figure 1. miR-155 acts as a molecular rheostat to determine the duration of inflammation and the initiation of repair programs in macrophages. Depending on the expression levels of miR-155, macrophages can show opposite phenotypes. TLRLs induce high expression levels of miR-155. MiR-155 targets inhibitors of TLR/cytokine receptor pathways and drive a pro-inflammatory phenotype as shown by the production of TNF, IL-1β, IL-6, and chemokines. In addition, high miR-155 levels prevent a phenotypic switch to anti-inflammatory/repair macrophages by targeting (i) IL-13Rα, thus decreasing STAT6 activation and expression of STAT6-dependent mediators, e.g., collagen production, inducing CCL18; (ii) TGFβ signaling molecule SMAD2, and the transcription factor LXRα, thus maintaining low levels of resolution/repair mediators, e.g., TGFβ, Arg2. Glucocorticoid and IL-10 override miR-155-induced phenotype by inhibiting its expression. The chronicity of synovial inflammation may be mediated by persistently high expression levels of miR-155 observed in synovial macrophages. Direct miR-155 targets are marked in bold font. TLR, toll-like receptors; IL-13Rα, interleukin-13 receptor alpha; TGFβ, transforming growth factor beta; SMAD2, small mothers against decapentaplegic homolog 2, LXRa, liver X receptor alpha; Arg2, arginase 2; GCS, glucocorticoids.
Animal studies show that miR-155 drives experimental models of arthritis and its comorbidity, atherosclerosis (56, 57, 64). In RA patients, blood monocytes have significantly higher copy-numbers of miR-155 than healthy controls, and the copy-number correlates strongly with disease activity score (DAS of 28 joints; DAS28) and with erythrocyte sedimentation rate; a non-specific biomarker of inflammation (65, 66). The expression levels of miR-155 in patients in drug-induced remission and with low DAS were similar to levels in healthy donors, whereas levels in patients with medium and high DAS were progressively increased. The expression of miR-155 in monocytes from patients with coronary heart disease is higher than normal and correlated with expression of TNF and IL-6 (67), suggesting that miR-155 expression in monocytes is closely associated with the degree of inflammatory activation (65).
Monocytes are recruited from the blood to the synovial fluid and to the synovial membrane where monocytes differentiate locally into pro-inflammatory macrophages (51, 68). This is an important step in RA progression and is mediated by locally produced chemokines (69). Both CD14+ monocytes from synovial fluid and CD68+ synovial tissue macrophages from RA patients with active disease have an activated phenotype (70–73) and produce pro-inflammatory cytokines, including TNF, IL-6, and GM-CSF that are targets for current successful RA therapies (74–76). Synovial fluid-derived monocytes can also differentiate toward osteoclasts that mediate bone erosion (77, 78) and promote production of the osteoclastogenic and stromal cell-activating cytokine IL-17 by CD4+ T-cells which further contributes to bone and cartilage activation (70–72). We found that miR-155 is constitutively upregulated in both RA synovial tissue macrophages and synovial fluid monocytes compared to RA blood monocytes and non-inflammatory osteoarthritis controls (57). We demonstrated that the increased expression of miR-155 in synovial fluid CD14+ cells and synovial tissue macrophages was commensurate with reduced expression of SHIP-1, which thereby enabled their inflammatory phenotype, e.g., TNF production (57). In monocytes, miR-155 supports the expression of the integrin CD11a (LFA-1) that interacts with intercellular adhesion molecule-1 on endothelium and facilitates migration of monocytes into inflamed tissue (79). Moreover, monocyte miR-155 drives the production of inflammatory chemokines, including CCL3, CCL4, CCL5, and CCL8, and downregulates the expression of CCR2 (65). These findings suggest that high levels of miR-155 in pro-inflammatory macrophages in synovial fluid and synovial tissue enables chemokine production and integrin expression that recruits precursors from the circulation, and then mediates the retention of these cells at sites of inflammation by downregulating chemokine receptor expression, and finally mediates local production of pro-inflammatory cytokines. Similarly, miR-155 supports the development of advanced atherosclerotic lesions. In experimental models, miR-155, induced by inflammation and oxidized lipids, targets Bcl6 in lesional macrophages and increases CCL2 production and recruitment of monocytes to vascular walls. Thus, miR-155 deficiency in macrophages ameliorated plaque formation (56).
Blood and synovial fluid monocytes from RA patients are resistant to spontaneous apoptosis and to apoptosis mediated by an agonistic anti-Fas antibody, as compared to blood monocytes of healthy donors (80–84). This resistance may contribute to the persistence of inflammatory monocytes and/or macrophages and their perpetuation of joint inflammation in RA. This resistance is due to the progressive inhibition of CASP10 and APAF1 by the increasing levels of miR-155 in RA cells. This was confirmed by miR-155 overexpression in healthy CD14+ cells that conferred resistance to spontaneous apoptosis (84). In vitro, mimicking the high expression levels of miR-155 in myeloid cells of patients with RA and cardiovascular disease by enforced expression of miR-155 in healthy human monocytes/macrophages demonstrated that miR-155 triggers the spontaneous production of several pro-inflammatory cytokines, including TNF, IL-6, IL-1β, and IL-8, and decreases anti-inflammatory IL-10 production (57, 65, 84). Thus, miR-155 overexpression promotes a pro-inflammatory phenotype in monocytes/macrophages, whereas in vitro treatment of RA patients’ synovial fluid CD14+ cells with miR-155 antagonist de-repressed SHIP-1, inhibited the production of TNF (57) and restored cellular homeostasis.
Synovitis is driven by cross talk between synovial macrophages with fibroblasts (10) and emerging evidence suggests that miRNAs packaged into macrophage-derived exosomes regulate the function of adjacent cells, including fibroblasts. This has yet to be formally established is human synovial macrophages and fibroblasts; however, an elegant paper by Wang et al. showed that, upon cardiac injury, macrophages secrete miR-155-enriched exosomes that are absorbed by cardiac fibroblasts. miR-155 is released into the fibroblasts cytoplasm, inhibits SOCS-1, and enhances production of pro-inflammatory mediators, including IL-6. Administration of macrophage-derived exosomes containing miR-155 into miR-155-deficient mice exacerbates myocardial infarction (85). Thus, it would be interesting to reveal the contribution of macrophage-derived miR-155 to the pathogenic phenotype of RA synovial fibroblasts.
In summary, sustained high levels of miR-155 in RA patients’ synovial monocytes and synovial tissue macrophages drive their pro-inflammatory activation and prevent the switch to anti-inflammatory/repair phenotypes, thus preclude resolution of inflammation and initiation of repair (Figure 2).
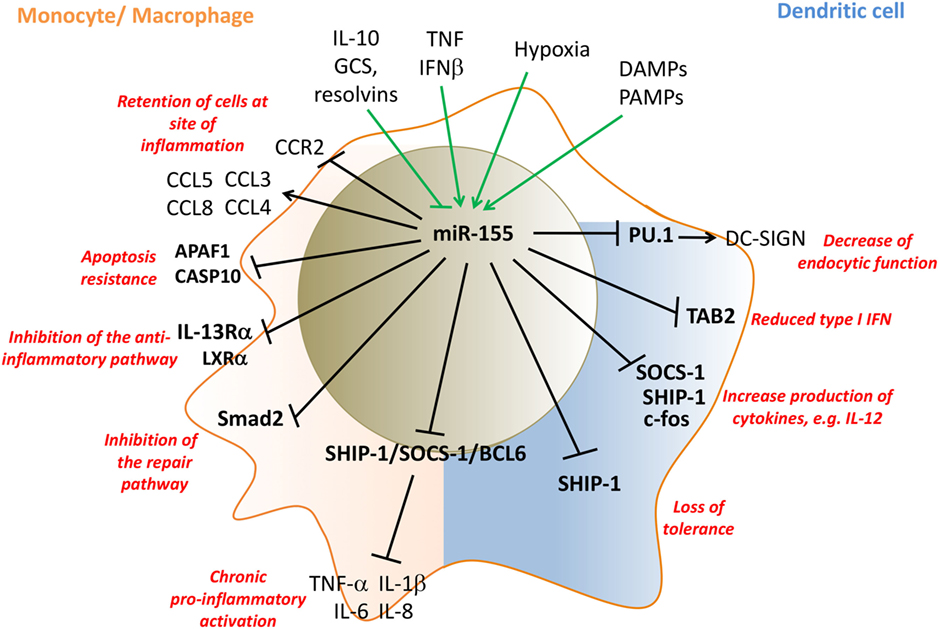
Figure 2. miR-155 regulates pro-inflammatory activation of monocytes/macrophages (pink) and dendritic cells (DCs) (blue). Inflammatory mediators that induce miR-155 expression in innate cells, include LPS, TNF, IFNβ, PAMPs, and DAMPs, while IL-10, GCS, and resolvins inhibit miR-155 expression. miR-155 promotes monocyte recruitment into inflamed tissue by the induction of appropriate chemokines, and then retains them by then downregulating CCR2 and inhibits their apoptosis by targeting APAF1 and CASP10. miR-155 drives inflammatory cytokine production by targeting SHIP-1, SOCS-1, and Bcl6. miR-155 represses the development of anti-inflammatory and repair macrophage phenotypes by targeting IL-13Rα, LXRα, and the TGFβ-signaling molecule SMAD2. In myeloid DCs, miR-155 targets PU.1 and decreases the endocytic function of DCs by downregulating DC-SIGN while supporting maturation of DCs and their T-cell stimulatory activity by targeting SOCS-1/SHIP-1 and c-fos. miR-155 limits plasmocytoid DC activation by decreasing type I IFN production by targeting TAB 2. Direct targets are marked in bold font. LPS, lipopolysaccharides; TNF, tumor necrosis factor; IFN, interferon; DAMPs, danger-associated molecular pattern; CCR2, C–C chemokine receptor type 2; PU.1: Spi-1 proto-oncogene; hematopoietic transcription factor PU.1; CCL, CC chemokine ligands; SHIP-1, phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1; SOCS-1, suppressor of cytokine signaling 1; TAB 2, TGF-beta activated kinase 1/MAP3K7 binding protein 2; APAF1, apoptotic peptidase activating factor 1; CASP10, caspase 10; TGF-β, transforming growth factor beta; IL, interleukin; DC-SIGN, DC-specific C-type lectin GCS, glucocorticoids.
Dendritic cells are key innate regulators of the adaptive immune response by activating antigen-specific T-cells. They have an important homeostatic function by either mounting an immune response against pathogens or by inducing tolerance to self-antigens. To induce tolerance, DCs activate regulatory T-cells and/or induce anergy of antigen-specific effector T-cells; thereby terminating an immune response and preventing autoimmunity (86, 87). Mouse and human DCs consist of developmentally and functionally distinct DC subsets regulated by a network of cytokines and transcriptional factors (88). Upon stimulation with PAMPs/DAMPs DCs undergo a maturation process which is characterized by functional changes including decreased pathogen binding/endocytic activity, increased cytokine production and enhanced antigen presentation (89, 90). miRNA networks act at checkpoints during the development, differentiation and activation of DC subsets (88). miR-155 is dispensable for DC differentiation but is an important regulator of DC activation; mice deficient in miR-155 have normal numbers of CD8a+cDCs, CD8a−cDCs, and pDCs (91); however, miR-155 is readily induced in mouse and human DCs following TLR stimulation (35, 92, 93). TLR stimulation activates two cellular pathways resulting in DC maturation; MyD88-IRAK-TRAF6 and TRIF-IRF3-IFNα, and both are required for optimal NF-κB activation, expression of co-stimulatory molecules, and the production of cytokines, such as IL-1β or IL-12 (94, 95). miR-155 regulates the switch from the endocytic function to co-stimulatory/activatory function during DC maturation. miR-155 reduces the capacity of DCs to bind pathogens by downregulation of DC-SIGN via inhibition of transcription factor PU.1 (93), while enhancing cytokine production (91, 96, 97). Studies show that miR-155−/− DCs exhibit lower levels of MHC class II and co-stimulatory molecules (CD80, CD40), and a decrease in IL1β, TNF, IL12, and IL6 expression upon TLR-induced maturation compared to wild-type DCs, and they demonstrate a significantly lower ability to induce effector T-cells (91, 98). This may be mediated by miR-155 inhibition of c-fos, continued expression of which was shown to be detrimental for DC maturation (91). Similarly, inhibition of miR-155 in human monocyte-derived DCs caused a reduction in IL-12p70 production (97) whereas overexpression of miR-155 in mature DCs enhanced IL12p70 expression, leading to higher levels of IFN-γ being released by cocultured natural killer cells (97). It was proposed that miR-155 mediated these activatory functions of DCs through epigenetic regulation of SHIP-1 and SOCS-1 in a similar manner as in macrophages (96, 97).
Interestingly, miR-155 may terminate some DC functions by targeting TAB 2, a key adaptor molecule in the TLR/IL-1 pathway. These include termination of type I IFN or IL-1β production induced by TLR7- or LPS-mediated human monocyte-derived DC activation, respectively (55, 92). This discrepancy requires further investigation, but the nature of the stimulatory factor and the abundance of specific target at precise time–points during DC activation may determine whether miR-155 supports or terminates DC activation.
Rheumatoid arthritis peripheral blood, synovial fluid and synovial tissue DCs, in particular CD1c+ and monocyte-derived populations, show a constitutively activated phenotype by their increased expression of co-stimulatory molecules, cytokines (e.g., IL6) and their ability to activate autologous T-cells (99–102). It was shown recently that high levels of miRNA-34a that lead to repression of Axl, an inhibitor of DC activation, contribute to the activated phenotype of circulating and synovial CD1c+ in RA (100). The involvement of miR-155 to this phenotype is unknown. However, a role for DC miR-155 in driving experimental autoimmunity was shown by Lind et al. (96) in a model of diabetes in which self-antigen-pulsed, TLR-matured DCs lacking miR-155 have an impaired ability to break immune tolerance, while the transfer of self-antigen-pulsed DCs overexpressing miR-155 was sufficient to break tolerance even in the absence of TLR stimuli (96).
Several lines of evidence support the role of miR-155 in T- and B-cell differentiation. miR-155 controls both effector and regulatory T-cell function (103) and regulates B-cell proliferation, germinal center formation, and antibody production (104). If aberrantly expressed, miR-155 contributes to initiation and progression of malignancies of B-cells and to autoimmunity.
miR-155 is highly expressed in human B-cell lymphomas, especially in large B-cell lymphomas, Hodgkin lymphomas, and certain types of Burkitt lymphomas (104). Transgenic mice expressing miR-155 under the control of Eμ enhancer and VH promoter (Eμ-miR-155) develop aberrant accumulation of pre-B-cells leading to the development of acute lymphoma. These cells have commensurately decreased expression of miR-155 targets, e.g., SHIP-1 and CCAAT enhancer-binding protein beta (C/EBPbeta), which in B-cells have anti-inflammatory and anti-proliferative functions (105, 106). miR-155 may promote survival and proliferation of naïve B cells in Eμ-miR-155 mice by targeting co-repressor partners Hdac4 (directly) and Bcl6 (indirectly), and their reduction leads to de-repression of Bcl6 targets, e.g., inhibitor of differentiation-2, IL-6, cMyc, Cyclin D1, and Mip1α/ccl3; all of which promote cell survival and proliferation. Importantly, meta-analysis of microarray data from diffuse large B-cell lymphoma patients found that miR-155 expression correlated negatively with Bcl6 and Hdac4 expression, strongly supporting the role of the miR-155/Bcl6/Hdac4 pathway in the pathogenesis of human leukemias (107). miR-155 also regulates proliferation of mature B-cells in germinal centers by interfering with TGF-β signaling. TGF-β1 secreted in secondary lymphoid organs limits mature B-cell proliferation to maintain immune-system homeostasis. miR-155 represses the TGF-β signaling molecule SMAD5, resulting in defective expression of the cell-cycle inhibitor p15/p21 (108) and allowing B-cells unhindered progress through the cell-cycle.
Systemic delivery of antisense miR-155, encapsulated in polymer nanoparticles, inhibited miR-155 and slowed the growth of pre-B-cell tumors in vivo, confirming that miR-155 is an important regulator of B-cell proliferation, and this may be a promising therapeutic option for lymphoma or leukemia (109).
A critical stage of effector B-cell development is represented by germinal center (GC) formation. These structures develop when mature naïve B-cells encounter cognate antigen in the secondary lymphoid organs during which the B-cells undergo somatic hyper-mutation of their immunoglobulin variable region genes and class-switch recombination. Emerging B-cells that express high-affinity antibody further differentiate into plasma cells and memory B-cells (110). B-cells also contribute to the immune response by acting as antigen-presenting cells and fuel inflammation by releasing a wide range of cytokine and chemokines (10). Thai and co-workers demonstrated that miR-155 plays an essential role in the regulation of GC formation. They found that miR-155-deficient mice had greatly reduced GC formation that was due to reduced production of TNF and LTα; obligate cytokines for GC neogenesis, by B-cells in response to antigen-ligation of the B-cell receptor (103). In support of a pivotal role for miR-155 in B-cell activation, chronic lymphocytic leukemia (CCL) B-cells that expressed high miR-155, demonstrated miR-155-dependent increased activation (calcium flux) induced by BCR ligation (111). The in vivo antibody response of miR-155-deficient mice showed significantly reduced concentration of IgM and isotype-switched antigen-specific antibody class production (98, 112). Mechanistic studies by Vigorito et al. (112) showed that miR-155 can directly target PU.1 mRNA via a predicted miR-155-binding site in the 3′ UTR, and that miR-155-deficient B-cells had increased PU.1 expression. Enforced expression of PU.1 in wild-type primary B-cells led to a reduction in the proportion of IgG1-expressing cells upon stimulation (112, 113). Mechanistically, miR-155 reduces PU.1 expression that leads to the downregulation of Pax5, a PU.1-dependent inhibitor of B-cell activation. This enables plasma cell differentiation along with the expression of other genes involved in B/T cellular interactions (e.g., Sema4a, Sema4b, Sema7a, and CD300If) facilitating antibody production (114).
miR-155 can also regulate the antigen-presenting function of B-cells. miR-155 targets the 3′UTR of CD1d in B-cells (115). Therefore, upregulation of miR-155 in B-cells, e.g., in chronic inflammation, can repress CD1d leading to impaired lipid-antigen presentation by B-cells to invariant natural killer cells. This causes a breach of tolerance as shown by CD1d deficiency exacerbating lupus nephritis in the pristine-induced model (115).
Of additional relevance to autoimmune disease, miR-155 modulates the B-cell response to immune complexes. The low-affinity IgG inhibitory receptor FcγRIIB (Fcγ receptor IIB) is key in terminating proliferative signals delivered by autoantigen-containing immune complexes by activating SHIP-1. Thai et al. showed that miR-155 can downregulate SHIP-1 and permit immune-complex-induced pathology in the Faslpr model of autoimmune lupus (113), and this may also be relevant to the signaling of the immune complexes in RA.
The onset of RA is preceded by preclinical immunological changes, e.g., the production of anti-citrullinated peptides antibodies (anti-CCP/ACPA) and rheumatoid factor, which reflect the loss of immunological tolerance to self-antigens (116), and this places B-cells at the center of RA pathogenesis. This is confirmed by the efficacy of B-cell depletion therapy in RA patients (117). The proportion of B-cell subsets is altered in the blood of RA patients (118, 119) with higher percentages and absolute numbers of naïve B-cells at the onset of disease, and increased proportions of double-negative (IgD−/CD27−) memory B-cells and plasmablasts in patients with established RA (120).
As previously stated, miR-155 was first described as an important epigenetic regulator of antibody synthesis (98, 112). In experimental models of arthritis, the production of anti-collagen antibodies was significantly decreased in miR-155-deficient mice compared to wild type (57, 64), and these mice did not develop arthritis. In RA patients, we found that the expression of miR-155 in B-cells is increased and is maximal during the early phase of disease. This increase is predominant in double-negative (IgD−/CD27−) memory B-cells (121), and occurs in ACPA+ but not ACPA− RA patients suggesting a role in autoantibody production. The IgD−/CD27− B-cell population is increased in other autoimmune diseases, including SLE (122, 123) and is important for IgG+ plasmablast generation (124). In a study of paired peripheral blood–synovial fluid samples from RA patients, miR-155 expression was higher in B-cells from the synovium compared with B-cells from blood. Mediators, including IL-6, BAFF, IgM, CD40L, and IL-21 were increased in synovial fluid, and these were able to upregulate miR-155 expression in B cells in vitro. In addition, in situ hybridization revealed that most of the resident synovial tissue CD20+ cells in germinal-center-like follicles were miR-155 positive. Commensurate with the increased miR-155 expression, synovial fluid-derived B-cells of RA patients showed reduced expression of the miR-155 target PU.1 in ACPA+ compared to ACPA− patients, and a reduced proportion of PU.1+ cells among the resident CD20+ cells in synovial follicles (121). Inhibition of endogenous miR-155 in RA B-cells led to the restoration of PU.1 expression and inhibition of antibody production (121) suggesting a key role of miR-155 in autoantibody production by B-cells in RA. Commensurate with this, the expression of miR-155 in circulating B-cells of ACPA+ RA is significantly higher than that of similarly inflamed patients with psoriatic arthritis (PsA), in which B-cells are minimally involved in pathogenesis (121). In addition, the expression of miR-155 in circulating B-cells is particularly high in RA patients with follicular synovitis; which is a prognostic factor for poor treatment response (125). Thus, miR-155 expression in RA circulating B-cells can serve as a biomarker of B cell activation and lymphoid/follicular synovitis among the three RA synovial pathotypes identified (myeloid, lymphoid/follicular, and fibroid) (126). A summary of miR-155 function in B-cells is presented on Figure 3.
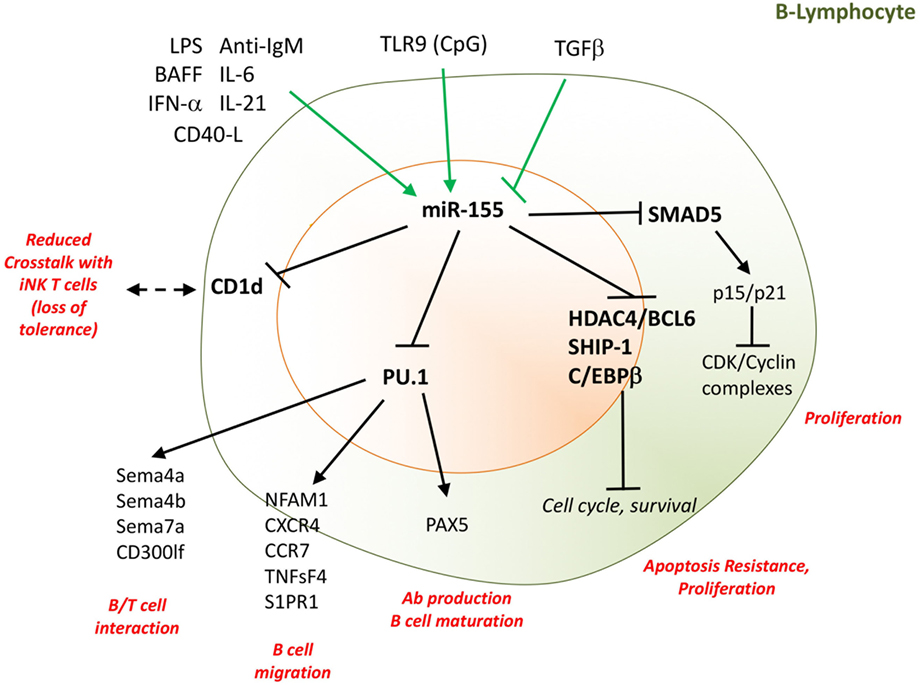
Figure 3. miR-155 supports B-cell proliferation and antibody production. Several cytokines and TLR-ligands can induce miR-155 expression in B-cells. These include LPS, IL-6, IL-21, CD40L, IFN-α, and BAFF, while TGFβ inhibits miR-155 expression. By targeting SHIP-1, C/EBPβ, the Hdac4/Bcl6 complex, and the TGFβ signaling molecule SMAD5, miR-155 drives proliferation and resistance to apoptosis of B-cells. miR-155, by epigenetic regulation of PU.1, is indispensable for B-cell maturation, migration, interaction with T-cells, and antibody production. Moreover, miR-155 regulates the surface expression of CD1d on B-cells, influencing the cellular cross talk with invariant NK T-cells. Direct targets are marked in bold font. LPS, lipopolysaccharides; IL, interleukin; TLR, toll-like receptor; Bcl, B-cell lymphoma; SMAD5, small mother against decapentaplegic 5; CD, cluster designation; PU.1, Spi-1 Proto-Oncogene; Sema, semaforin; NK, natural killer; BAFF, B-cell activating factor; CD40L, CD40 ligand. TGF-β, transforming growth factor beta; SHIP-1, phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1; Bcl6, B-cell lymphoma; HDAC4; histone deacetylase 4; C/EBPβ: CCAAT/enhancer-binding protein beta; NFAM1, NFAT activating protein with ITAM motif 1; CXCR4; C–X–C chemokine receptor type 4; CCR7, C–C chemokine receptor type 7; S1Pr1, sphingosine-1-phosphate receptor 1; TNFsF4, tumor necrosis factor (ligand) superfamily, member 4; CD300lf, CMRF35-like molecule 1.
In response to appropriate stimuli, CD4+ T-cells can differentiate into different phenotypes that include the Th1, Th2, Th17, follicular helper T (Tfh) cells, and T-regulatory (Treg) cell lineages with distinct functions (127). This differentiation is influenced by cytokines and by the expression and activation of phenotype-specific transcription factors (128). miRNAs have been implicated in CD4+ T-cell differentiation (129, 130) and miR-155 is central to this process (98, 131).
Rodriguez and colleagues examined the T-cell response of miR-155-deficient mice immunized with T-dependent antigen (tetanus toxin fragment C) and showed that splenocytes produced significantly reduced concentrations of IL-2 and IFN-γ compared to wild-type mice (98). An intrinsic requirement for miR-155 to Th1 development was confirmed in vitro by comparing the cytokine production by WT and miR-155−/− CD4+ cells in culture following anti-CD3 and anti-CD28 stimulation, which demonstrated reduced IFNγ production by miR-155−/− cells (98). In keeping with these observations, Banerjee and co-workers showed that overexpression of miR-155 in activated CD4+ T-cells promoted Th1 differentiation by targeting IFN-γRα thereby decreasing the sensitivity of Th1 cells to the anti-proliferative effects of IFN-γ (131).
miR-155 was initially described as a negative regulator of Th2 commitment through the repression of its target c-Maf, which is an IL-4 promoter transactivator (98). CD4+ T-cells from miR-155-deficient mice exhibit preferential Th2 differentiation upon activation (132–134). miR-155-deficient mice have an age-dependent increased number of Th2 cells and develop spontaneous airway remodeling due to the action of Th2 cytokines (98). In keeping with this, we recently reported that miR-155-deficient mice are susceptible to exacerbated bleomycin-induced lung fibrosis (36). In contrast, Malmhall et al., using an antigen-induced asthma model, observed that miR-155-deficient mice had decreased activation of Th2 cells and decreased production of IL-4, IL-5, and IL-13, decrease lung eosinophilia and fewer physiological changes compared with wild-type mice (135). The potential inhibition of Th2 development was not associated with c-Maf changes but with depression of PU.1 that negatively regulates the expression of GATA3, the key transcription factor for Th2 cell function. Similarly, Okoye et al. described a Th2-cell-selective role for miR-155 in the development of an allergic airway model. They demonstrated that miR-155 inhibits the expression of sphingosine-1-phosphate receptor 1, which is required for lymphocyte egress from lymphoid organs, thus mice with miR-155−/− T-cells had reduced airway pathology (136). This suggests that the miR-155 may highlight different roles for Th2 responses in acute allergic inflammation compared with fibrosis. It is possible that resident tissue memory T-cells are more involved in age-related and bleomycin-induced chronic lung remodeling, whereas recently activated recruited T-cells are more involved in acute airway inflammation.
Th17 lymphocytes have higher expression levels of miR-155 compared with other T-cell subset lineages (137). miR-155 is upregulated in Th17 cells by STAT3, IRF-4 and RORγt and supports the development of Th17 by inhibiting its epigenetic target Jarid2, which itself is an epigenetic suppressor of Il22, Il10, Il9, and Atf3 (137), and by suppressing SOCS-1, thereby facilitating IL-6/STAT3 signaling (138). Accordingly, miR-155-deficient mice have substantially reduced numbers of Th17 cells as demonstrated by reduced expression of IL-17A (139), IL-22, IL-6, and IL-23R (64, 139–141).
Follicular T-helper cells cells are key in GC formation and antibody production by providing help to B-cells in secondary lymphoid tissue. Elegant work led by Tang showed that miR-155 is indispensable in the development of Tfh. Using CD4+-specific miR-155-deficient mice, the authors showed reduced levels of Tfh (CXCR5+PD1+ Bcl6+) and consequently less-developed GCs and reduced antigen-specific antibody production in a variety of immunization schedules. Silencing of two proteins, Fosl2 (a component of AP1) and to a lesser extent Peli1 (E3 ubiquitin ligase) in miR-155−/− CD4+ T-cells, resulted in improved development of Tfh cells following immunization, suggesting that miR-155 mediates Tfh differentiation by downregulating these two targets (142).
There is abundant evidence that miR-155 can promote the development of Tregs. These cells are important to maintain immunological tolerance by limiting pathogenic Th-cell responses. Tregs are dependent on IL-2/STAT5 signaling and are characterized by the expression of Foxp3. This transcription factor induces miR-155 which then upregulates STAT5 phosphorylation by its epigenetic blockade of SOCS-1-mediated inhibition of STAT5. miR-155-deficient mice have reduced numbers of Tregs in the thymus and in peripheral blood due to attenuated IL-2 signaling by de-repressed SOCS-1 (143, 144). Cytotoxic T-lymphocyte antigen 4 (CTLA-4), which is an effector molecule of Tregs that binds to CD28 and limits the activation of effector T-cells, is a putative mRNA target of miR-155 (143, 145). However, Tregs from miR-155-deficient mice express normal levels of CTLA-4 suggesting that it is not under the epigenetic control of miR-155, at least in Tregs in mice. In contrast, Sonkoly et al. showed that miR-155 targets CTLA-4 mRNA in human effector T-cells. This group observed an increased level of miR-155 and decreased level of CTLA-4 in effector T-cells of patients with atopic dermatitis, suggesting that miR-155 inhibition of CTLA-4 contribute to activation of effector T-cells in the skin (146). Thus, miR-155/CTLA-4 interactions might be cell-type and species-specific.
Deletion of Dicer in cytotoxic CD8+ T-cells causes an impaired immune response to pathogens (147). Gracias et al. (148) found miR-155 upregulated in primary effector and effector memory CD8+ T-cells as compared to naive and central memory CD8+ cells. Antiviral CD8+ T-cell responses and viral clearance were impaired in miR-155-deficient mice, while miR-155 overexpression augmented antiviral CD8+ T-cell responses in vivo. Increased miR-155 inhibited the type I interferon response in CD8+ cells while miR-155-deficient CD8+ T-cells had enhanced type I interferon signaling and were more susceptible to IFNs antiproliferative effect. Inhibition of type I interferon-associated transcription factors STAT1 or IRF7 resulted in enhanced responses of miR-155-deficient CD8+ T-cells to viral infection in vivo (148). This suggests that miR-155 maintains proliferation of CD8+ in response to viral antigen by downregulation of STAT1/IRF7.
In experimental arthritis, miR-155-deficiency affects Th17 cell polarization. Production of IL-17 and IL-22 are significantly reduced in miR-155-deficient mice compared to wild type (64). Smigielska-Czepiel and colleagues provided important insight into the differential expression of specific miRNAs in human T-cell subsets, especially naive and memory T-cells in peripheral blood and synovial fluid in RA patients (149). Synovial fluid T-cells are predominantly memory T-cells (150) and are characterized by high expression of miR-155 compared with naive T-cells (149), thus miR-155 in this compartment may support local production of IL-17 and IFNγ by autoreactive T-cells.
Despite the presence of many Tregs in the synovial fluid of RA (151–153), joint inflammation persists in most patients regardless of the treatment regimen. One interpretation is that there is a functional defect in these Tregs, e.g., due to inhibition of Foxp3 by TNF (154) that is in high concentration in synovial fluid; or that the effector T-cells are resistant to the regulatory action of Tregs (155). Blood Tregs from RA patients have significantly lower expression of miR-155 than healthy controls (156). However, correcting this expression by enforcing exogenous miR-155 in RA Tregs in vitro enhanced their production of TNF, IL-17, and IFNγ after 24 h upon stimulation with anti-CD3/CD28 (156). This suggests that altering the expression of miR-155 alone is not sufficient to restore the immunosuppressive function of RA Tregs but promotes pathogenic cytokine production instead.
In summary, miR-155 is indispensable for activation of Th1, Th17, Tfh, CD8+, and Treg while inhibiting Th2 (Figure 4). Sustained high expression of miR-155 in synovial effector T-cells may contribute to chronic production of IFNγ, IL-17 and facilitate differentiation of T-cells toward Tfh cells in RA.
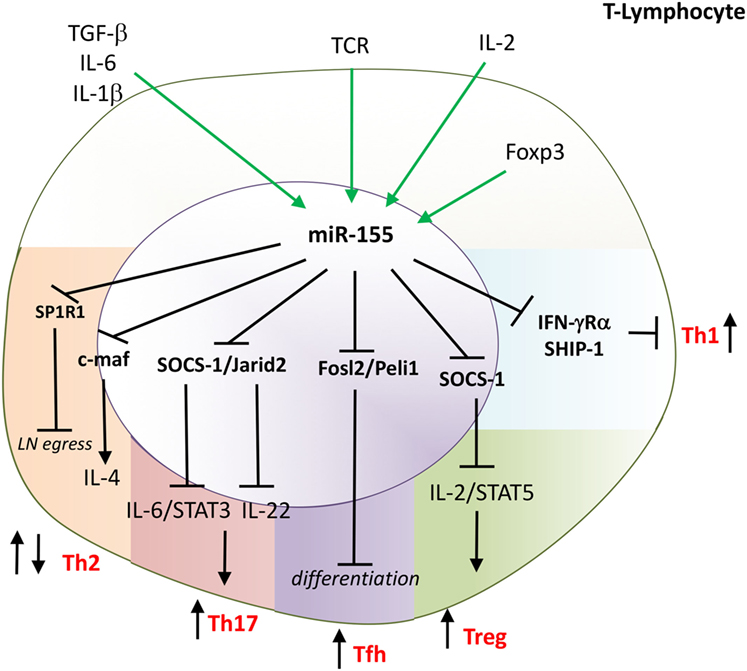
Figure 4. miR-155 drives differentiation of T-helper 1 (Th1), Th17, Tfh and is indispensable for the function of Tregs. miR-155 has multiple effects on T-cell function. miR-155 promotes: Th1 by targeting IFNRα; Th17 by targeting Jarid2 and SOCS-1; and follicular helper T-cells (Tfh) by targeting Fosl2 and Peli. Mir-155 supports the function of regulatory T-cells (Treg) by targeting SOCS-1 that enables IL-2 signaling. miR-155 regulates Th2 development and function; e.g., inhibiting development by targeting c-maf; a key transcription factor for IL-4 and regulating an egress of T cells from the lymph nodes by targeting S1PR1. Direct targets are marked in bold font. TGF, transforming growth factor; IL, interleukin; TCR, T cell receptor; IFN, interferon; SOCS-1, suppressor of cytokine signaling 1; STAT, signal transducer and activator of transcription; SHIP, phosphatidylinositol-3,4,5-trisphosphate 5-phosphatase 1; c-maf, transcription factor Maf; reg, regulatory; JARID2, Jumonji, AT rich interactive domain 2; SP1R1, sphingosine-1-phosphate receptor-1.
The literature and our own experimental findings summarized in this review indicate that miR-155 is a key mediator of chronic activation of innate and adaptive immunity in RA. Its contribution to RA pathogenesis might be more profound than in other forms of inflammatory arthritis such as PsA due to the involvement of miR-155 in driving autoantibody production in RA. A cross-sectional analysis of RA patients with different clinical responses to treatments revealed that miR-155 expression becomes reduced in the monocytes of RA patients who responded well to conventional disease-modifying anti-rheumatic drugs (e.g., methotrexate) or to biological DMARDs (65). Consistent with this, there are prospective studies in psoriasis patients showing that the expression levels of miR-155 in PBMCs decreased upon successful treatment with methotrexate as compared to pre-treatment levels (157). These observations suggest that a reduction of high to normal expression of miR-155 reflects a good clinical response to therapies that reduce inflammation (Figure 5). However, the effects of current anti-inflammatory (e.g., anti TNF, anti-IL-6R) or anti-adaptive immunity (e.g., CTLA-4) therapies on miR-155 expression in B-cells and Th17 cells still needs to be investigated in prospective studies.
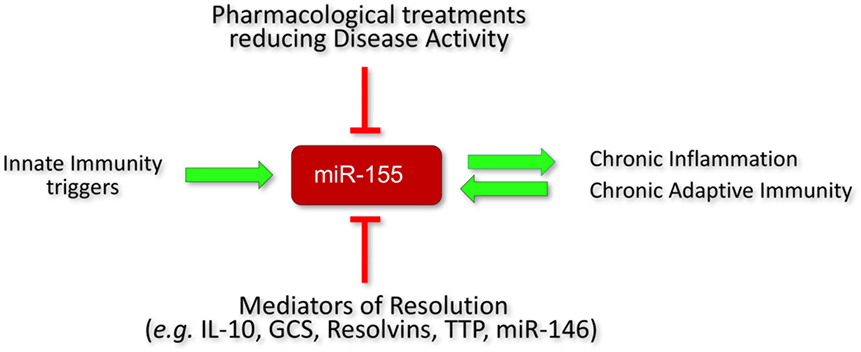
Figure 5. Regulation of miR-155 expression in RA. Initiators of inflammation, e.g., PAMPs and DAMPs increase miR-155 expression. Homeostatic resolution is controlled by anti-inflammatory mediators that act in part by decreasing miR-155 expression (e.g., IL-10, GCS, TTP, resolvins) or counterbalancing its function, e.g., miR-146. In rheumatoid arthritis (RA), there is persistent inflammation and activation of adaptive immunity which maintain high expression levels of miR-155 that lock the immune system in an activatory state. A successful response to treatment (reduced systemic and synovial inflammation) in RA patients is associated with a reduction of miR-155 expression (at least in monocytes). PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; GCS, glucocorticoids; TTP, tristetraproline; cDMARD, conventional disease-modifying anti-rheumatic drugs; TNFi, TNF inhibitors.
Chronic inflammation and continuous activation of adaptive immunity generate pro-inflammatory cytokines and autoantibodies that fuel high levels of miR-155 expression thus locking the immune system into an activatory state. Targeting miRNAs has proven to be a useful therapeutic option in infectious diseases, cancer and recently in tendinopathy (8, 9). The most clinically advanced example of targeting miRNA is neutralization of miR-122 to treat hepatitis C infections (HCV) that is currently in phase IIa clinical trials. miR-122 supports the replication of HCV (158) and treatment with antimiR-122 (Mirvirasen; Roche/Santaris and Regulus Therapeutics) achieved a reduction in viral titers in HCV-infected patients (8). Another miR-targeted therapeutic that reached clinical development is a mimic of the tumor suppressor miR-34. miR-34a targets pro-apoptotic proteins and cell-cycle inhibitors, and is downregulated in many human cancers (159). The miR-34 mimic MRX34 (Mirna Therapeutics), encapsulated in lipid nanoparticles, is currently being tested in several solid and hematological malignancies (8). We showed recently that chronic tendinopathy is driven by deregulation of the IL-33/miR-29 pathway leading to the production of pro-inflammatory mediators and collagen 3 with weak tensile strength, thus limiting effective tendon healing (160). The results from a randomized, blinded trial of local tendon miR-29a replacement therapy in an equine model, that closely mimics human disease, changed the collagen composition toward stronger collagen 1 and improved early tendon healing (9). Human trials will be conducted in the near future.
To date, the potential clinical outcome of miR-155 inhibition in arthritis is based on findings from animal models, and from ex vivo studies using RA patient-derived cells. miR-155-deficient mice are resistant to arthritis, and inhibition of miR-155 using specific antimiR in a collagen-induced arthritis model reduced the clinical onset and severity of disease (161). Thus, inhibiting miR-155 may represent an attractive therapeutic option. However, systemic inhibition of miR-155 may compromise its other physiological functions and lead to unwanted consequences. Experimental and clinical evidence suggest that miR-155 is indispensable for normal immunity against pathogens (55, 103). In addition, miR-155 in liver macrophages (Kupffer cells) is key in the regulation of lipid metabolism. Mir-155 regulates the expression levels of a target mRNA LXRα which is a transcription factor that controls lipid metabolism and lipid efflux, thus miR-155-deficient mice have an increased level of circulating cholesterol and develop fatty liver (liver steatosis) while on a high-fat diet (162). Furthermore, miR-155 controls lung interstitial remodeling by regulating the proliferation and collagen production of lung fibroblasts, the repair macrophage phenotype, and the production of type 2 cytokines (36, 98). miR-155-deficient mice develop spontaneous age-related lung fibrosis and exacerbated lung fibrosis associated with administration of bleomycin. The lack of miR-155 epigenetic control in these murine lung fibroblasts resembles the deregulated in vitro pro-fibrotic behavior of lung fibroblasts from patients with idiopathic pulmonary fibrosis (36, 98). This suggests that a therapeutic strategy in arthritis of systemic targeting of miR-155, or pan-macrophage-specific delivery of miR-155 inhibitors via liposomes might have detrimental effects. There may be an alternative method of delivery of a miR-155-inhibitor. An elegant study led by Cheng et al. (163) showed that delivery of anti-miR-155 could be specifically targeted to hypoxic tissues such as tumors or inflamed tissues with an ambient low pH created by anaerobic metabolism. The authors linked antimiR-155 with a low pH-induced transmembrane structure (pHLIP). At pH less than 7, the C-terminus of pHLIP inserts across lipid bilayers and facilitates delivery of the attached antimiR-155. Inside the cell, the disulfide bond between pHLIP and antimiR-155 is reduced in the cytosol and the intracellular antimiR-155 is free to inhibit miR-155. The inflamed synovial tissue environment is hypoxic (164), thus this novel delivery systems could inhibit miR-155 in arthritis and avoid off-target side effects such as reduced protective immunity, fibrosis, or liver steatosis. Alternatively, enhancing activation of inhibitors of cell activation that are repressed by miR-155, e.g., SHIP-1 by specific activators might be a safer option (165). SHIP-1 activator (AQX-1125) has generated positive clinical data from clinical trials in COPD and allergic asthma, demonstrating a favorable safety profile and anti-inflammatory activity (trial number: NCT01954628).
There are gaps in the identification and characterization of all direct miR-155 targets and the physiological impact of miR-155 inhibition on these targets would need to be revealed before anti-miR-155 therapy can be considered. Moreover, understanding the role of miR-155 that is naturally contained in vesicles transferred between cells in the synovium may provide insight into mechanisms of chronicity. Investigation of miR-155 in DCs and B-cells in the preclinical phase of RA (asymptomatic ACPA positive individuals) would help to dissect its role in the regulation of the breach of immunological tolerance. A biomarker for treatment responses in arthritis would be of considerable clinical value, therefore, prospective studies using cohorts of RA patients at different stages of disease would validate whether miR-155 could fulfill this additional role.
MK-S and SA selected the research and wrote the review. EG, CM, BT, GF, and IM selected the research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the ARUK Rheumatoid Arthritis Pathogenesis Centre of Excellence (RACE/20298).
1. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet (2016) 17:487–500. doi:10.1038/nrg.2016.59
2. Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nat Immunol (2008) 9:839–45. doi:10.1038/ni.f.209
3. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet (2004) 5:522–31. doi:10.1038/nrg1415
5. Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res (2009) 19:92–105. doi:10.1101/gr.082701.108
6. Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell (2006) 11:441–50. doi:10.1016/j.devcel.2006.09.009
7. Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol (2016) 16:279–94. doi:10.1038/nri.2016.40
8. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov (2017) 16:203–22. doi:10.1038/nrd.2016.246
9. Watts AE, Millar NL, Platt J, Kitson SM, Akbar M, Rech R, et al. MicroRNA29a treatment improves early tendon injury. Mol Ther (2017) 25:2415–26. doi:10.1016/j.ymthe.2017.07.015
10. Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity (2017) 46:183–96. doi:10.1016/j.immuni.2017.02.006
11. Nagy G, van Vollenhoven RF. Sustained biologic-free and drug-free remission in rheumatoid arthritis, where are we now? Arthritis Res Ther (2015) 17:181. doi:10.1186/s13075-015-0707-1
12. Ospelt C, Gay S, Klein K. Epigenetics in the pathogenesis of RA. Semin Immunopathol (2017) 39:409–19. doi:10.1007/s00281-017-0621-5
13. Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol (2009) 10:126–39. doi:10.1038/nrm2632
14. Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet (2010) 11:597–610. doi:10.1038/nrg2843
15. Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol (2005) 3:e85. doi:10.1371/journal.pbio.0030085
16. Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev (2004) 18:504–11. doi:10.1101/gad.1184404
17. Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell (2007) 27:91–105. doi:10.1016/j.molcel.2007.06.017
18. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell (2005) 120:15–20. doi:10.1016/j.cell.2004.12.035
19. Tam W, BenYehuda D, Hayward WS. bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol Cell Biol (1997) 17:1490–502. doi:10.1128/MCB.17.3.1490
20. Tam W. Identification and characterization of human BIC, a gene on chromosome 21 that encodes a noncoding RNA. Gene (2001) 274:157–67. doi:10.1016/S0378-1119(01)00612-6
21. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol (2002) 12:735–9. doi:10.1016/S0960-9822(02)00809-6
22. Zheng L, Xu CC, Chen WD, Shen WL, Ruan CC, Zhu LM, et al. MicroRNA-155 regulates angiotensin II type 1 receptor expression and phenotypic differentiation in vascular adventitial fibroblasts. Biochem Biophys Res Commun (2010) 400:483–8. doi:10.1016/j.bbrc.2010.08.067
23. Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell (2007) 129:1401–14. doi:10.1016/j.cell.2007.04.040
24. Masaki S, Ohtsuka R, Abe Y, Muta K, Umemura T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem Biophys Res Commun (2007) 364:509–14. doi:10.1016/j.bbrc.2007.10.077
25. Georgantas RW III, Hildreth R, Morisot S, Alder J, Liu CG, Heimfeld S, et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A (2007) 104:2750–5. doi:10.1073/pnas.0610983104
26. Griffiths-Jones S. The microRNA registry. Nucleic Acids Res (2004) 32:D109–11. doi:10.1093/nar/gkh023
27. Meijer HA, Smith EM, Bushell M. Regulation of miRNA strand selection: follow the leader? Biochem Soc Trans (2014) 42:1135–40. doi:10.1042/BST20140142
28. Ohanian M, Humphreys DT, Anderson E, Preiss T, Fatkin D. A heterozygous variant in the human cardiac miR-133 gene, MIR133A2, alters miRNA duplex processing and strand abundance. BMC Genet (2013) 14:18. doi:10.1186/1471-2156-14-18
29. Neilsen CT, Goodall GJ, Bracken CP. IsomiRs – the overlooked repertoire in the dynamic microRNAome. Trends Genet (2012) 28:544–9. doi:10.1016/j.tig.2012.07.005
30. Lee HY, Zhou K, Smith AM, Noland CL, Doudna JA. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res (2013) 41:6568–76. doi:10.1093/nar/gkt361
31. Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol (2007) 23:175–205. doi:10.1146/annurev.cellbio.23.090506.123406
32. Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet (2008) 9:102–14. doi:10.1038/nrg2290
33. Zhou H, Huang X, Cui H, Luo X, Tang Y, Chen S, et al. miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood (2010) 116:5885–94. doi:10.1182/blood-2010-04-280156
34. Tarassishin L, Loudig O, Bauman A, Shafit-Zagardo B, Suh HS, Lee SC. Interferon regulatory factor 3 inhibits astrocyte inflammatory gene expression through suppression of the proinflammatory miR-155 and miR-155. Glia (2011) 59:1911–22. doi:10.1002/glia.21233
35. O’Connell RM, Taganov KD, Boldin MP, Cheng GH, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A (2007) 104:1604–9. doi:10.1073/pnas.0610731104
36. Kurowska-Stolarska M, Hasoo MK, Welsh DJ, Stewart L, McIntyre D, Morton BE, et al. The role of microRNA-155/liver X receptor pathway in experimental and idiopathic pulmonary fibrosis. J Allergy Clin Immunol (2017) 139:1946–56. doi:10.1016/j.jaci.2016.09.021
37. Bruning U, Cerone L, Neufeld Z, Fitzpatrick SF, Cheong A, Scholz CC, et al. MicroRNA-155 promotes resolution of hypoxia-inducible factor 1alpha activity during prolonged hypoxia. Mol Cell Biol (2011) 31:4087–96. doi:10.1128/MCB.01276-10
38. Quinn SR, Mangan NE, Caffrey BE, Gantier MP, Williams BR, Hertzog PJ, et al. The role of Ets2 transcription factor in the induction of microRNA-155 (miR-155) by lipopolysaccharide and its targeting by interleukin-10. J Biol Chem (2014) 289:4316–25. doi:10.1074/jbc.M113.522730
39. Pottier N, Maurin T, Chevalier B, Puissegur MP, Lebrigand K, Robbe-Sermesant K, et al. Identification of keratinocyte growth factor as a target of microRNA-155 in lung fibroblasts: implication in epithelial-mesenchymal interactions. PLoS One (2009) 4:e6718. doi:10.1371/journal.pone.0006718
40. Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, et al. MicroRNA-155 is regulated by the transforming growth factor beta/smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol (2008) 28:6773–84. doi:10.1128/MCB.00941-08
41. Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F, et al. Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A (2010) 107:6982–7. doi:10.1073/pnas.1002472107
42. Triantafyllou E, Pop OT, Possamai LA, Wilhelm A, Liaskou E, Singanayagam A, et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut (2017). doi:10.1136/gutjnl-2016-313615
43. Rajasagi NK, Bhela S, Varanasi SK, Rouse BT. Frontline science: aspirin-triggered resolvin D1 controls herpes simplex virus-induced corneal immunopathology. J Leukoc Biol (2017) 102:1159–71. doi:10.1189/jlb.3HI1216-511RR
44. Zheng Y, Xiong S, Jiang P, Liu R, Liu X, Qian J, et al. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: a novel anti-inflammation mechanism. Free Radic Biol Med (2012) 52:1307–17. doi:10.1016/j.freeradbiomed.2012.01.031
45. Chinenov Y, Coppo M, Gupte R, Sacta MA, Rogatsky I. Glucocorticoid receptor coordinates transcription factor-dominated regulatory network in macrophages. BMC Genomics (2014) 15:656. doi:10.1186/1471-2164-15-656
46. Bhattacharyya S, Kumar P, Tsuchiya M, Bhattacharyya A, Biswas R. Regulation of miR-155 biogenesis in cystic fibrosis lung epithelial cells: antagonistic role of two mRNA-destabilizing proteins, KSRP and TTP. Biochem Biophys Res Commun (2013) 433:484–8. doi:10.1016/j.bbrc.2013.03.025
47. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med (2011) 208:1189–201. doi:10.1084/jem.20101823
48. Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, et al. An NF-kappaB-microRNA regulatory network tunes macrophage inflammatory responses. Nat Commun (2017) 8:851. doi:10.1038/s41467-017-00972-z
49. Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene (2013) 532:1–12. doi:10.1016/j.gene.2012.12.009
50. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi:10.1016/j.immuni.2014.06.008
51. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK III, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep (2014) 9:591–604. doi:10.1016/j.celrep.2014.09.032
52. Self-Fordham JB, Naqvi AR, Uttamani JR, Kulkarni V, Nares S. MicroRNA: dynamic regulators of macrophage polarization and plasticity. Front Immunol (2017) 8:1062. doi:10.3389/fimmu.2017.01062
53. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A (2009) 106:7113–8. doi:10.1073/pnas.0902636106
54. An H, Xu H, Zhang M, Zhou J, Feng T, Qian C, et al. Src homology 2 domain-containing inositol-5-phosphatase 1 (SHIP1) negatively regulates TLR4-mediated LPS response primarily through a phosphatase activity- and PI-3K-independent mechanism. Blood (2005) 105:4685–92. doi:10.1182/blood-2005-01-0191
55. Wang P, Hou J, Lin L, Wang C, Liu X, Li D, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol (2010) 185:6226–33. doi:10.4049/jimmunol.1000491
56. Nazari-Jahantigh M, Wei Y, Noels H, Akhtar S, Zhou Z, Koenen RR, et al. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. J Clin Invest (2012) 122:4190–202. doi:10.1172/JCI61716
57. Kurowska-Stolarska M, Alivernini S, Ballantine LE, Asquith DL, Millar NL, Gilchrist DS, et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc Natl Acad Sci U S A (2011) 108:11193–8. doi:10.1073/pnas.1019536108
58. Jing W, Zhang X, Sun W, Hou X, Yao Z, Zhu Y. CRISPR/CAS9-mediated genome editing of miRNA-155 inhibits proinflammatory cytokine production by RAW264.7 cells. Biomed Res Int (2015) 2015:326042. doi:10.1155/2015/326042
59. He M, Xu Z, Ding T, Kuang DM, Zheng L. MicroRNA-155 regulates inflammatory cytokine production in tumor-associated macrophages via targeting C/EBPbeta. Cell Mol Immunol (2009) 6:343–52. doi:10.1038/cmi.2009.45
60. Kim H. The transcription factor MafB promotes anti-inflammatory M2 polarization and cholesterol efflux in macrophages. Sci Rep (2017) 7:7591. doi:10.1038/s41598-017-07381-8
61. Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem (2011) 286:1786–94. doi:10.1074/jbc.M110.169367
62. Wahl SM, Allen JB, Costa GL, Wong HL, Dasch JR. Reversal of acute and chronic synovial inflammation by anti-transforming growth factor beta. J Exp Med (1993) 177:225–30. doi:10.1084/jem.177.1.225
63. Louafi F, Martinez-Nunez RT, Sanchez-Elsner T. MicroRNA-155 targets SMAD2 and modulates the response of macrophages to transforming growth factor-{beta}. J Biol Chem (2010) 285:41328–36. doi:10.1074/jbc.M110.146852
64. Bluml S, Bonelli M, Niederreiter B, Puchner A, Mayr G, Hayer S, et al. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum (2011) 63:1281–8. doi:10.1002/art.30281
65. Elmesmari A, Fraser AR, Wood C, Gilchrist D, Vaughan D, Stewart L, et al. MicroRNA-155 regulates monocyte chemokine and chemokine receptor expression in rheumatoid arthritis. Rheumatology (Oxford) (2016) 55:2056–65. doi:10.1093/rheumatology/kew272
66. Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum (2008) 58:1001–9. doi:10.1002/art.23386
67. Tian FJ, An LN, Wang GK, Zhu JQ, Li Q, Zhang YY, et al. Elevated microRNA-155 promotes foam cell formation by targeting HBP1 in atherogenesis. Cardiovasc Res (2014) 103:100–10. doi:10.1093/cvr/cvu070
68. Herenius MM, Thurlings RM, Wijbrandts CA, Bennink RJ, Dohmen SE, Voermans C, et al. Monocyte migration to the synovium in rheumatoid arthritis patients treated with adalimumab. Ann Rheum Dis (2011) 70:1160–2. doi:10.1136/ard.2010.141549
69. Asquith DL, Bryce SA, Nibbs RJ. Targeting cell migration in rheumatoid arthritis. Curr Opin Rheumatol (2015) 27:204–11. doi:10.1097/BOR.0000000000000150
70. Yoon BR, Yoo SJ, Choi Y, Chung YH, Kim J, Yoo IS, et al. Functional phenotype of synovial monocytes modulating inflammatory T-cell responses in rheumatoid arthritis (RA). PLoS One (2014) 9:e109775. doi:10.1371/journal.pone.0109775
71. Walter GJ, Evans HG, Menon B, Gullick NJ, Kirkham BW, Cope AP, et al. Interaction with activated monocytes enhances cytokine expression and suppressive activity of human CD4+CD45ro+CD25+CD127(low) regulatory T cells. Arthritis Rheum (2013) 65:627–38. doi:10.1002/art.37832
72. Evans HG, Gullick NJ, Kelly S, Pitzalis C, Lord GM, Kirkham BW, et al. In vivo activated monocytes from the site of inflammation in humans specifically promote Th17 responses. Proc Natl Acad Sci U S A (2009) 106:6232–7. doi:10.1073/pnas.0808144106
73. Firestein GS, Zvaifler NJ. Peripheral blood and synovial fluid monocyte activation in inflammatory arthritis. i. a cytofluorographic study of monocyte differentiation antigens and class ii antigens and their regulation by γgm-interferon. Arthritis Rheum (1987) 30:857–63. doi:10.1002/art.1780300803
74. Highton J, Carlisle B, Palmer DG. Changes in the phenotype of monocytes/macrophages and expression of cytokine mRNA in peripheral blood and synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol (2008) 102:541–6. doi:10.1111/j.1365-2249.1995.tb03850.x
75. De Rycke L, Baeten D, Foell D, Kruithof E, Veys EM, Roth J, et al. Differential expression and response to anti-TNFalpha treatment of infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J Pathol (2005) 206:17–27. doi:10.1002/path.1758
76. Chu CQ, Field M, Feldmann M, Maini RN. Localization of tumor-necrosis-factor-alpha in synovial tissues and at the cartilage pannus junction in patients with rheumatoid-arthritis. Arthritis Rheum (1991) 34:1125–32. doi:10.1002/art.1780340908
77. Danks L, Sabokbar A, Gundle R, Athanasou NA. Synovial macrophage-osteoclast differentiation in inflammatory arthritis. Ann Rheum Dis (2002) 61:916–21. doi:10.1136/ard.61.10.916
78. Adamopoulos IE, Sabokbar A, Wordsworth BP, Carr A, Ferguson DJ, Athanasou NA. Synovial fluid macrophages are capable of osteoclast formation and resorption. J Pathol (2006) 208:35–43. doi:10.1002/path.1891
79. Syed S, Amin M, Rabquer B. miR155 expression is increased by inflammation and modulates the expression of CD11a in monocytes. FASEB J (2015) 29(Suppl 1):634.6.
80. Perlman H, Pagliari LJ, Liu H, Koch AE, Haines GK, Pope RM. Rheumatoid arthritis synovial macrophages express the Fas-associated death domain-like interleukin-1?-converting enzyme-inhibitory protein and are refractory to Fas-mediated apoptosis. Arthritis Rheum (2001) 44:21–30. doi:10.1002/1529-0131(200101)44:1<21::AID-ANR4>3.0.CO;2-8
81. Liu H, Huang Q, Shi B, Eksarko P, Temkin V, Pope RM. Regulation of Mcl-1 expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum (2006) 54:3174–81. doi:10.1002/art.22132
82. Meusch U, Rossol M, Baerwald C, Hauschildt S, Wagner U. Outside-to-inside signaling through transmembrane tumor necrosis factor reverses pathologic interleukin-1beta production and deficient apoptosis of rheumatoid arthritis monocytes. Arthritis Rheum (2009) 60:2612–21. doi:10.1002/art.24778
83. Srivastava S, Macaubas C, Deshpande C, Alexander HC, Chang SY, Sun Y, et al. Monocytes are resistant to apoptosis in systemic juvenile idiopathic arthritis. Clin Immunol (2010) 136:257–68. doi:10.1016/j.clim.2010.04.003
84. Rajasekhar M, Olsson AM, Steel KJ, Georgouli M, Ranasinghe U, Brender Read C, et al. MicroRNA-155 contributes to enhanced resistance to apoptosis in monocytes from patients with rheumatoid arthritis. J Autoimmun (2017) 79:53–62. doi:10.1016/j.jaut.2017.01.002
85. Wang C, Zhang C, Liu L, Xi A, Chen B, Li Y, et al. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther (2017) 25:192–204. doi:10.1016/j.ymthe.2016.09.001
86. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature (2007) 449:419–26. doi:10.1038/nature06175
87. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
88. Zhou HB, Wu L. The development and function of dendritic cell populations and their regulation by miRNAs. Protein Cell (2017) 8:501–13. doi:10.1007/s13238-017-0398-2
89. Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol (2006) 6:476–83. doi:10.1038/nri1845
91. Dunand-Sauthier I, Santiago-Raber ML, Capponi L, Vejnar CE, Schaad O, Irla M, et al. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood (2011) 117:4490–500. doi:10.1182/blood-2010-09-308064
92. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci U S A (2009) 106:2735–40. doi:10.1073/pnas.0811073106
93. Martinez-Nunez RT, Louafi F, Friedmann PS, Sanchez-Elsner T. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN). J Biol Chem (2009) 284:16334–42. doi:10.1074/jbc.M109.011601
94. Shen H, Tesar BM, Walker WE, Goldstein DR. Dual signaling of MyD88 and TRIF is critical for maximal TLR4-induced dendritic cell maturation. J Immunol (2008) 181:1849–58. doi:10.4049/jimmunol.181.3.1849
95. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science (2007) 315:107–11. doi:10.1126/science.1136080
96. Lind EF, Millar DG, Dissanayake D, Savage JC, Grimshaw NK, Kerr WG, et al. miR-155 upregulation in dendritic cells is sufficient to break tolerance in vivo by negatively regulating SHIP1. J Immunol (2015) 195:4632–40. doi:10.4049/jimmunol.1302941
97. Lu C, Huang X, Zhang X, Roensch K, Cao Q, Nakayama KI, et al. miR-221 and miR-155 regulate human dendritic cell development, apoptosis, and IL-12 production through targeting of p27kip1, KPC1, and SOCS-1. Blood (2011) 117:4293–303. doi:10.1182/blood-2010-12-322503
98. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science (2007) 316:608–11. doi:10.1126/science.1139253
99. Jongbloed SL, Lebre MC, Fraser AR, Gracie JA, Sturrock RD, Tak PP, et al. Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis Res Ther (2006) 8:R15. doi:10.1186/ar1864
100. Kurowska-Stolarska M, Alivernini S, Melchor EG, Elmesmari A, Tolusso B, Tange C, et al. MicroRNA-34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis. Nat Commun (2017) 8:15877. doi:10.1038/ncomms15877
101. Moret FM, Bijlsma JWJ, Lafeber FPJG, van Roon JAG. The efficacy of abatacept in reducing synovial T cell activation by CD1c myeloid dendritic cells is overruled by the stimulatory effects of T cell-activating cytokines. Arthritis Rheumatol (2015) 67:637–44. doi:10.1002/art.38982
102. Moret FM, Hack CE, van der Wurff-Jacobs KM, de Jager W, Radstake TR, Lafeber FP, et al. Intra-articular CD1c-expressing myeloid dendritic cells from rheumatoid arthritis patients express a unique set of T cell-attracting chemokines and spontaneously induce Th1, Th17 and Th2 cell activity. Arthritis Res Ther (2013) 15:R155. doi:10.1186/ar4338
103. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science (2007) 316:604–8. doi:10.1126/science.1141229
104. Musilova K, Mraz M. MicroRNAs in B-cell lymphomas: how a complex biology gets more complex. Leukemia (2015) 29:1004–17. doi:10.1038/leu.2014.351
105. Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, et al. Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood (2009) 114:1374–82. doi:10.1182/blood-2009-05-220814
106. Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A (2006) 103:7024–9. doi:10.1073/pnas.0602266103
107. Sandhu SK, Volinia S, Costinean S, Galasso M, Neinast R, Santhanam R, et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the E mu-miR-155 transgenic mouse model. Proc Natl Acad Sci U S A (2012) 109:20047–52. doi:10.1073/pnas.1213764109
108. Jiang D, Aguiar RC. MicroRNA-155 controls RB phosphorylation in normal and malignant B lymphocytes via the noncanonical TGF-beta1/SMAD5 signaling module. Blood (2014) 123:86–93. doi:10.1182/blood-2013-07-515254
109. Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci U S A (2012) 109:E1695–704. doi:10.1073/pnas.1201516109
110. Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol (2008) 8:22–33. doi:10.1038/nri2217
111. Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood (2014) 124:546–54. doi:10.1182/blood-2014-03-559690
112. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity (2007) 27:847–59. doi:10.1016/j.immuni.2007.10.009
113. Thai TH, Patterson HC, Pham DH, Kis-Toth K, Kaminski DA, Tsokos GC. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Fas(lpr) mouse. Proc Natl Acad Sci U S A (2013) 110:20194–9. doi:10.1073/pnas.1317632110
114. Lu D, Nakagawa R, Lazzaro S, Staudacher P, Abreu-Goodger C, Henley T, et al. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. J Exp Med (2014) 211:2183–98. doi:10.1084/jem.20140338
115. Liu F, Fan H, Ren D, Dong G, Hu E, Ji J, et al. TLR9-induced miR-155 and Ets-1 decrease expression of CD1d on B cells in SLE. Eur J Immunol (2015) 45:1934–45. doi:10.1002/eji.201445286
116. van de Stadt LA, de Koning MH, van de Stadt RJ, Wolbink G, Dijkmans BA, Hamann D, et al. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum (2011) 63:3226–33. doi:10.1002/art.30537
117. Ferraccioli G, Tolusso B, Bobbio-Pallavicini F, Gremese E, Ravagnani V, Benucci M, et al. Biomarkers of good EULAR response to the B cell depletion therapy in all seropositive rheumatoid arthritis patients: clues for the pathogenesis. PLoS One (2012) 7:e40362. doi:10.1371/journal.pone.0040362
118. Fekete A, Soos L, Szekanecz Z, Szabo Z, Szodoray P, Barath S, et al. Disturbances in B- and T-cell homeostasis in rheumatoid arthritis: suggested relationships with antigen-driven immune responses. J Autoimmun (2007) 29:154–63. doi:10.1016/j.jaut.2007.07.002
119. Souto-Carneiro MM, Mahadevan V, Takada K, Fritsch-Stork R, Nanki T, Brown M, et al. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res Ther (2009) 11:R84. doi:10.1186/ar2718
120. Fedele AL, Tolusso B, Gremese E, Bosello SL, Carbonella A, Canestri S, et al. Memory B cell subsets and plasmablasts are lower in early than in long-standing rheumatoid arthritis. BMC Immunol (2014) 15:28. doi:10.1186/s12865-014-0028-1
121. Alivernini S, Kurowska-Stolarska M, Tolusso B, Benvenuto R, Elmesmari A, Canestri S, et al. MicroRNA-155 influences B-cell function through PU.1 in rheumatoid arthritis. Nat Commun (2016) 7:12970. doi:10.1038/ncomms12970
122. Mahmood Z, Muhammad K, Schmalzing M, Roll P, Dorner T, Tony HP. CD27-IgD- memory B cells are modulated by in vivo interleukin-6 receptor (IL-6R) blockade in rheumatoid arthritis. Arthritis Res Ther (2015) 17:61. doi:10.1186/s13075-015-0580-y
123. Rodriguez-Bayona B, Ramos-Amaya A, Perez-Venegas JJ, Rodriguez C, Brieva JA. Decreased frequency and activated phenotype of blood CD27 IgD IgM B lymphocytes is a permanent abnormality in systemic lupus erythematosus patients. Arthritis Res Ther (2010) 12:R108. doi:10.1186/ar3042
124. Tangye SG, Tarlinton DM. Memory B cells: effectors of long-lived immune responses. Eur J Immunol (2009) 39:2065–75. doi:10.1002/eji.200939531
125. Orr C, Najm A, Biniecka M, McGarry T, Ng CT, Young F, et al. Synovial immunophenotype and anti-citrullinated peptide antibodies in rheumatoid arthritis patients: relationship to treatment response and radiologic prognosis. Arthritis Rheumatol (2017) 69:2114–23. doi:10.1002/art.40218
126. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol (2013) 25:334–44. doi:10.1097/BOR.0b013e32835fd8eb
127. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol (2010) 28:445–89. doi:10.1146/annurev-immunol-030409-101212
128. Reiner SL. Development in motion: helper T cells at work. Cell (2007) 129:33–6. doi:10.1016/j.cell.2007.03.019
129. Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med (2005) 201:1367–73. doi:10.1084/jem.20050572
130. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med (2005) 202:261–9. doi:10.1084/jem.20050678
131. Banerjee A, Schambach F, DeJong CS, Hammond SM, Reiner SL. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur J Immunol (2010) 40:225–31. doi:10.1002/eji.200939381
132. Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A (2005) 102:3627–32. doi:10.1073/pnas.0500613102
133. Haasch D, Chen YW, Reilly RM, Chiou XG, Koterski S, Smith ML, et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto-oncogene, BIC. Cell Immunol (2002) 217:78–86. doi:10.1016/S0008-8749(02)00506-3
134. van den Berg A, Kroesen BJ, Kooistra K, de Jong D, Briggs J, Blokzijl T, et al. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer (2003) 37:20–8. doi:10.1002/gcc.10186
135. Malmhall C, Alawieh S, Lu Y, Sjostrand M, Bossios A, Eldh M, et al. MicroRNA-155 is essential for T(H)2-mediated allergen-induced eosinophilic inflammation in the lung. J Allergy Clin Immunol (2014) 133:1429–38, 1438.e1–7. doi:10.1016/j.jaci.2013.11.008
136. Okoye IS, Czieso S, Ktistaki E, Roderick K, Coomes SM, Pelly VS, et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc Natl Acad Sci U S A (2014) 111:E3081–90. doi:10.1073/pnas.1406322111
137. Escobar TM, Kanellopoulou C, Kugler DG, Kilaru G, Nguyen CK, Nagarajan V, et al. miR-155 activates cytokine gene expression in Th17 cells by regulating the DNA-binding protein Jarid2 to relieve polycomb-mediated repression. Immunity (2014) 40:865–79. doi:10.1016/j.immuni.2014.03.014
138. Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X, et al. MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS One (2012) 7:e46082. doi:10.1371/journal.pone.0046082
139. O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity (2010) 33:607–19. doi:10.1016/j.immuni.2010.09.009
140. Hu R, Huffaker TB, Kagele DA, Runtsch MC, Bake E, Chaudhuri AA, et al. MicroRNA-155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J Immunol (2013) 190:5972–80. doi:10.4049/jimmunol.1300351
141. Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol (2011) 187:2213–21. doi:10.4049/jimmunol.1003952
142. Hu R, Kagele DA, Huffaker TB, Runtsch MC, Alexander M, Liu J, et al. miR-155 promotes T follicular helper cell accumulation during chronic, low-grade inflammation. Immunity (2014) 41:605–19. doi:10.1016/j.immuni.2014.09.015
143. Kohlhaas S, Garden OA, Scudamore C, Turner M, Okkenhaug K, Vigorito E. Cutting edge: the Foxp3 target miR-155 contributes to the development of regulatory T cells. J Immunol (2009) 182:2578–82. doi:10.4049/jimmunol.0803162
144. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity (2009) 30:80–91. doi:10.1016/j.immuni.2008.11.010
145. Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB. MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components. Mol Immunol (2008) 45:1995–2006. doi:10.1016/j.molimm.2007.10.035
146. Sonkoly E, Janson P, Majuri ML, Savinko T, Fyhrquist N, Eidsmo L, et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J Allergy ClinImmunol (2010) 126:581–9.e1–20. doi:10.1016/j.jaci.2010.05.045
147. Zhang N, Bevan MJ. Dicer controls CD8+ T-cell activation, migration, and survival. Proc Natl Acad Sci U S A (2010) 107:21629–34. doi:10.1073/pnas.1016299107
148. Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol (2013) 14:593–602. doi:10.1038/ni.2576
149. Smigielska-Czepiel K, van den Berg A, Jellema P, van der Lei RJ, Bijzet J, Kluiver J, et al. Comprehensive analysis of miRNA expression in T-cell subsets of rheumatoid arthritis patients reveals defined signatures of naive and memory Tregs. Genes Immun (2014) 15:115–25. doi:10.1038/gene.2013.69
150. Dejaco C, Duftner C, Klauser A, Schirmer M. Altered T-cell subtypes in spondyloarthritis, rheumatoid arthritis and polymyalgia rheumatica. Rheumatol Int (2010) 30:297–303. doi:10.1007/s00296-009-0949-9
151. Mottonen M, Heikkinen J, Mustonen L, Isomaki P, Luukkainen R, Lassila O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin Exp Immunol (2005) 140:360–7. doi:10.1111/j.1365-2249.2005.02754.x
152. Raghavan S, Cao D, Widhe M, Roth K, Herrath J, Engstrom M, et al. FOXP3 expression in blood, synovial fluid and synovial tissue during inflammatory arthritis and intra-articular corticosteroid treatment. Ann Rheum Dis (2009) 68:1908–15. doi:10.1136/ard.2008.100768
153. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum (2004) 50:2775–85. doi:10.1002/art.20499
154. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood (2006) 108:253–61. doi:10.1182/blood-2005-11-4567
155. Wehrens EJ, Prakken BJ, van Wijk F. T cells out of control – impaired immune regulation in the inflamed joint. Nat Rev Rheumatol (2013) 9:34–42. doi:10.1038/nrrheum.2012.149
156. Zhou Q, Haupt S, Kreuzer JT, Hammitzsch A, Proft F, Neumann C, et al. Decreased expression of miR-146a and miR-155 contributes to an abnormal Treg phenotype in patients with rheumatoid arthritis. Ann Rheum Dis (2015) 74:1265–74. doi:10.1136/annrheumdis-2013-204377
157. Garcia-Rodriguez S, Arias-Santiago S, Blasco-Morente G, Orgaz-Molina J, Rosal-Vela A, Navarro P, et al. Increased expression of microRNA-155 in peripheral blood mononuclear cells from psoriasis patients is related to disease activity. J Eur Acad Dermatol Venereol (2017) 31:312–22. doi:10.1111/jdv.13861
158. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science (2005) 309:1577–81. doi:10.1126/science.1113329
159. Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, et al. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids (2014) 3:e194. doi:10.1038/mtna.2014.47
160. Millar NL, Gilchrist DS, Akbar M, Reilly JH, Kerr SC, Campbell AL, et al. MicroRNA29a regulates IL-33-mediated tissue remodelling in tendon disease. Nat Commun (2015) 6:6774. doi:10.1038/ncomms7774
161. Morton B, Neben S, Gibson N, McSharry C, McInnes IB, Kurowska-Stolarska M. Neutralisation of miR-155 ameliorates collagen-induced arthritis. Ann Rheum Dis (2015) 74:84–5. doi:10.1136/annrheumdis-2015-207259.194
162. Miller AM, Gilchrist DS, Nijjar J, Araldi E, Ramirez CM, Lavery CA, et al. MiR-155 has a protective role in the development of non-alcoholic hepatosteatosis in mice. PLoS One (2013) 8:e72324. doi:10.1371/journal.pone.0072324
163. Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature (2015) 518:107–10. doi:10.1038/nature13905
164. Ng CT, Biniecka M, Kennedy A, McCormick J, Fitzgerald O, Bresnihan B, et al. Synovial tissue hypoxia and inflammation in vivo. Ann Rheum Dis (2010) 69:1389–95. doi:10.1136/ard.2009.119776
Keywords: microRNA-155, inflammation, antibody production, arthritis rheumatoid, autoimmunity
Citation: Alivernini S, Gremese E, McSharry C, Tolusso B, Ferraccioli G, McInnes IB and Kurowska-Stolarska M (2018) MicroRNA-155—at the Critical Interface of Innate and Adaptive Immunity in Arthritis. Front. Immunol. 8:1932. doi: 10.3389/fimmu.2017.01932
Received: 31 August 2017; Accepted: 15 December 2017;
Published: 05 January 2018
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases, United StatesReviewed by:
Michael Bonelli, Medical University of Vienna, AustriaCopyright: © 2018 Alivernini, Gremese, McSharry, Tolusso, Ferraccioli, McInnes and Kurowska-Stolarska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariola Kurowska-Stolarska, bWFyaW9sYS5rdXJvd3NrYS1zdG9sYXJza2FAZ2xhc2dvdy5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.