 Qingming Wang
Qingming Wang Fang Zhang1
Fang Zhang1 Haiming Yuan
Haiming Yuan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 19 March 2025
Sec. Human and Medical Genomics
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1564824
This article is part of the Research Topic Advances in molecular genetics of Marfan syndrome and related disorders View all articles
Marfan syndrome (MFS MIM#154700), due to pathogenic variants in the FBN1 gene, is an autosomal dominant connective tissue disorder, typically involving the skeletal, cardiovascular and ocular systems. Currently, over 3000 MFS patients were reported, and approximately 1800 pathogenic variants in FBN1 were identified. However, the molecular diagnosis still remains challenging for 8%–10% of patients with clinical features suggestive of MFS. In this study, we reported a 2-month-old Chinese female patient whose clinical features were compatible with the MFS. Whole-exome sequencing (WES) identified a novel de novo deep intronic variant, c.4943-8_4943-7insTATGTGATATTCAT TCAC in intron 40 of FBN1 that was predicted to affect the RNA splicing. Minigene analysis showed that this variant causes skipping of exon 41, leading to the deletion of 41 amino acids (c.4943_5065del, p.Val1649_Asp1689del). It confirmed the pathogenic nature of the variant and established the genotype-phenotype relationship. Our study expands the mutation spectrum of FBN1 and emphasizes the importance of deep intronic variant interpretation and the need for additional functional studies to verify the pathogenicity of these variants.
Marfan syndrome is a clinically recognized genetic disorder involving multiple congenital anomalies. Three cardinal manifestations are frequently seen in MFS patients and can be used as diagnostic clues, including skeletal anomalies (tall, thin stature, disproportionately long limbs and digits, anterior chest deformity and scoliosis), cardiovascular problems (mitral valve prolapse, mitral regurgitation, dilatation of the aortic root, and aortic regurgitation), and ocular anomalies (ectopia lentis and myopia). Aneurysm of the aorta and aortic dissection are the major life-threatening cause. MFS is caused by pathogenic variants in FBN1 (encoding fibrillin-1) (MIM # 134797) (Pyeritz, 2000). To date, more than 3,000 patients with MFS have been reported in the literatures and 1800 pathogenic variants in FBN1 have been identified (Collod-Béroud et al., 2003; Landrum et al., 2018; Arnaud et al., 2021); HGMD database. Pathogenic or likely pathogenic variants in coding exons and canonical splicing sites in FBN1 could account for the majority of individuals with clinical features suggestive of MFS (Dietz et al., 2001). However, it was found that 8%–10% of individuals with suspected MFS remain genetically unexplained (Loeys et al., 2004; Baetens et al., 2011; Zeigler et al., 2021). It is partly attributed to some cryptic variants, such as noncanonical splicing variants, that may be missed during variant interpretation because they were generally considered to be non-deleterious effect on protein products. Furthermore, it is laborious to carry out functional studies in the lab on variants suspected of being a potential genetic cause of patients with MFS phenotypes.
In this study, we reported a 2-month-old female patient who displayed characteristics typical for MFS. Whole exome sequencing (WES) was performed for the patient and identified a novel de novo deep intronic variant (NM_000138.4: c.4943-8_4943-7insTATGTGATATTCATTCAC) in FBN1.
This study was approved by the Ethics Committee of Dongguan Maternal and Child Health Hospital (DMCH 202307) and was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from the legal guardian for the release of any potentially identifiable image or data contained in this paper.
Genomic DNA was extracted using nucleic acid extraction reagents according to the kit instructions. Whole exome sequencing (WES) was used to screen for causal variants in this patient. Sequencing was performed with an Illumina NovaSeq 6,000 (Illumina, San Diego,CA, United States). The bcl2fastq2 Conversion Software (v2.20) was applied for extracting Fastq files, and all reads were mapped to the human genome (GRCh37/hg19) by using BWA (v0.2.10) with default parameters. The Genome Analysis Toolkit (GATK; v.3.7) HaplotypeCaller was performed for identifying variants. The aligned reads were visualized by using the Integrated Genome Viewer (IGV). Common variants were filtered based on their frequencies in the databases of the Genome Aggregation Database (https://gnomad.broadinstitute.org/) and our internal database. The suspected variant was verified by Sanger sequencing. The pathogenicity of the sequence variants was interpreted according to ACMG/AMP guidelines (Richards et al., 2015).
Wild type and mutant minigene plasmids were constructed for the FBN1 variant (c.4943-8_4943-7insTATGTGATATTCATTCAC) using the exon trap vectors pcMINI. The sequence of exon 41 (123 bp), part of intron 40 (463 bp), and part of intron 41 (496 bp) were amplified from the proband’s or her mother’s genomic DNA, using the following primer pairs: forward 5‘-GGTAGGTACCGAGTGCAATGGCATGATCTT-3’ and reverse 5‘-TGCAGAATTC TACCTATGCTGCTACAAGAT-3’. The amplified products were inserted into the pcMINI vector. Then, plasmids were constructed and transfected into human embryonic kidney 293T (HEK 293T) and human breast cancer cells (MCF-7) respectively, in triplicates using Lipofectamine 2000 (Invitrogen, USA). Cell cultures were operated according to the literature previously published (Li et al., 2021). After 48 h of transfection of cells, the total RNA was extracted using TRIzol reagent (Cowin Biotech Co., Jiangsu, China). For RT-PCR, a pair of primers was designed to amplify the target sequence originated from the expressed minigenes: forward 5‘-CTAGAGAACCCACTGCTTAC-3’ and reverse 5’-TAGAAGGCACAGTCGAGG-3’. Finally, the PCR product was verified by Sanger sequencing and visualized with electrophoresis on a 1.2% agarose gel.
The Chinese female patient was the third-born child of a nonconsanguineous couple, and her siblings were unaffected. She was born at 39 weeks of gestational age by spontaneous vaginal delivery. She had normal birth measurements: her weight was 3.1 kg, her length was 50 cm and her head circumference was 34 cm. She was referred to the clinic at 2 months of age because of pneumonia and congestive heart failure. She displayed distinctive facial features including bilateral temporal skull flattening, enophthalmos and retrognathia (Figures 1A, B), and skeletal anomalies including arachnodactyly, pes planus, long, narrow feet, hammer toes, skin striae (Figures 1C, D), scoliosis and pectus excavatum. Positive wrist and thumb signs, reduced extension at elbows and joint hypermobility were observed. X-ray showed significant enlargement of the heart shadow with a cardiothoracic ratio of 0.67 (normal value < 0.6) (Figure 1E). Echocardiography showed atrial septal defect, mitral regurgitation, tricuspid regurgitation and dilatation of the aortic root, 15 mm with Z-score of 4.48 (Figures 1F–H). The combination of aortic root dilatation and 10 points of systemic features resulted in the clinical diagnosis of MFS, based on a set of manifestations from the revised Ghent nosology (Loeys et al., 2010).
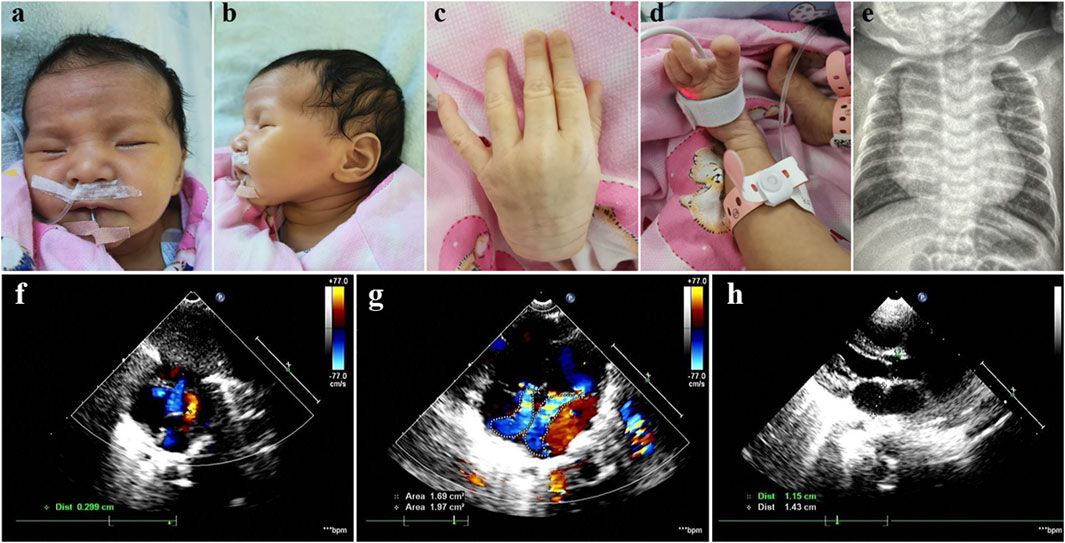
Figure 1. Photographs of our patient with MFS. Note bilateral temporal skull flattening, enophthalmos, retrognathia, arachnodactyly, pes planus, long, narrow feet, hammer toes and skin striae. (A–D) X-ray showed significant enlargement of the heart shadow with a cardiothoracic ratio of 0.67 (normal value < 0.6) (E). Echocardiography showed atrial septal defect, mitral regurgitation, tricuspid regurgitation and dilatation of the aortic root, 15 mm with Z-score of 4.48 (F–H).
WES identified a novel deep intronic variant in intron 40 of FBN1, (NM_000138.4:c.4943-8_ 4943-7insTATGTGATATTCATTCAC) in the patient. Sanger sequencing confirmed the variant and the variant was absent from the parents, thus it was a de novo event (Figure 2) (PS2). In addition, the variant was not present in the Genome Aggregation Database, the 1000 Genomes Project or our internal database (PM2_supporting). Four computational splicing tools (Human Splicing Finder, SpliceAI, ESE Finder v3.0, NetGene2) predicted that this deep intronic variant could lead to use of a new splice acceptor site within intron 40 of the FBN1 transcript. To further assess the impact of this variant, minigene study was performed to investigate the transcriptional outcome of the variant identified. An FBN1-pcMINI minigene was constructed and the cDNA of the wild-type and mutant mRNAs was obtained via RT-PCR. We observed that the variant affected splicing and caused exon 41 skipping. The final annotation was an infame deletion variant: c.4943_5065del, p.Val1649_Asp1689del. (PM4_strong) (Figure 3). Moreover, the patient’ manifestations were highly consistent with that of MFS (PP4). Furthermore, WES did not identify any other variants in FBN1 and also excluded other possible known genetic causes. Thus, this variant was evaluated as clinical pathogenic according to the ACMG/AMP guidelines (PS2 + PM2_supporting + PM4_strong + PP4) (PS: pathogenic strong; PM: pathogenic moderate; PP: pathogenic supporting).
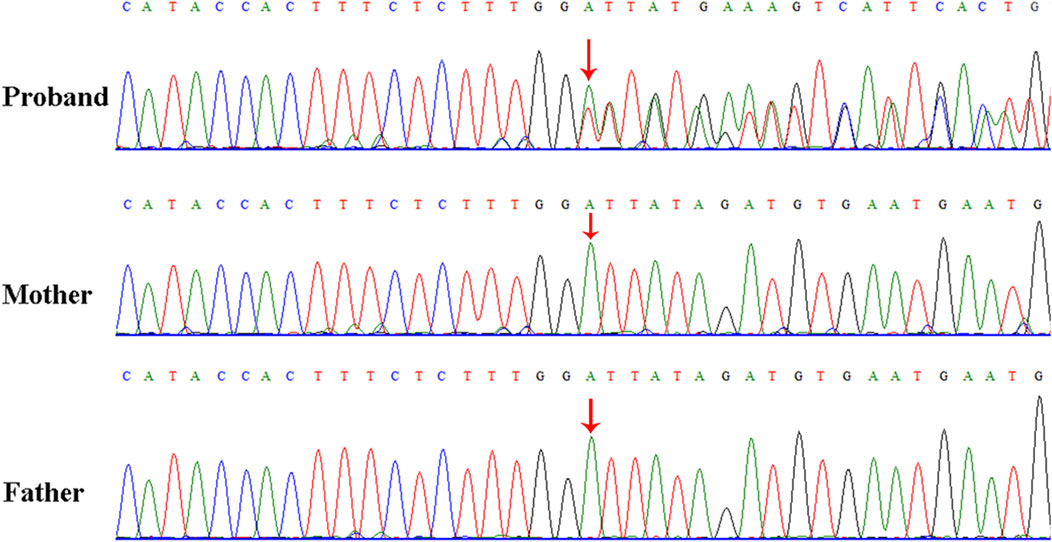
Figure 2. Sanger sequencing results for the patient, and the patient’s father and mother. The analysis demonstrated the presence of a deep intronic variant in FBN1 (c.4943-8_4943-7insTATGTGATATTCATTCAC) in the patient and the absence of the variant in her parents. The red arrow indicates the variant site.
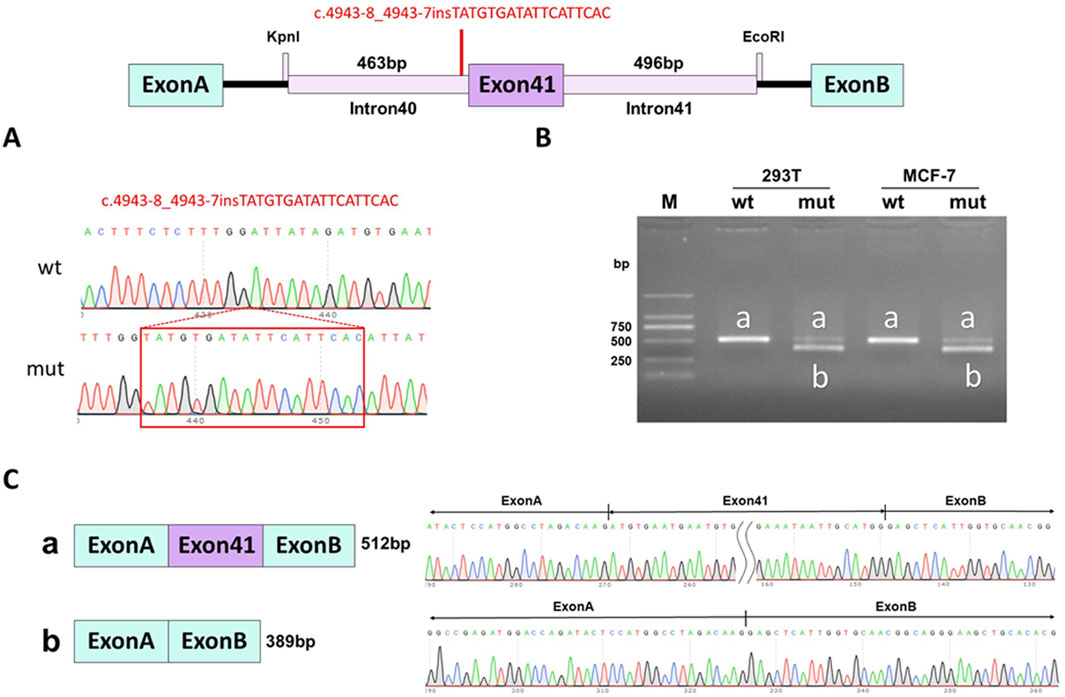
Figure 3. Minigene assay for FBN1 c.4943-8_4943-7insTATGTGATATTCATTCAC variant and schematic diagram of the splicing pattern. (A) The construction of FBN1-pcMINI minigene plasmid; (B) Gel electrophoresis of RT-PCR revealed a single band for wild-type (wt) and two bands for mutant-type (mut); (C) minigene product sequencing demonstrated that the wild-type minigene formed normal mRNA, but the c.4943-8_4943-7insTATGTGATATTCATTCAC variant in FBN1 caused a splicing abnormality, which abrogates the canonical splice site of intron 40, resulting in exon 41 skipping.
The FBN1 gene is located on 15q21.1, consists of 66 exons and encodes the large molecule fibrillin-1 of 2,871 amino acids. Fibronectin-1 is widespread in connective tissues, and the protein contains 47 cysteine-rich epidermal growth factor EGF-like repeats and seven transforming growth factor-β1 binding protein-like domains (Ramirez and Dietz, 2007). FBN1 mutations cause MFS through haploinsufficiency or dominant-negative effects mechanism (Aoyama et al., 1994; Whiteman et al., 2001; Faivre et al., 2007; Mátyás et al., 2007). Interestingly, studies have shown that missense mutations in exon 41 or 42 of FBN1 cause geleophysic dysplasia (GD, MIM #614185) or acromicric dysplasia (AD, MIM #102370) through a gain-of-function mechanism, which is characterized by severe short stature, short hands and feet, joint stiffness, and skin thickening, but without cardiac involvement or early death (Le Goff et al., 2011; Passarge et al., 2016).
Many different types of FBN1 variants have been identified. FBN1 null variants (frameshift, nonsense and canonical variants) and missense variants are frequently detected in MFS patients (Robinson et al., 2006). However, deep intronic variants are rarely reported in MFS patients because these variants were generally considered to have no deleterious effect on protein product, thus they were easily to be missed during variant interpretation. Here, we identified a novel de novo deep intronic variant in FBN1 (c.4943-8_4943-7insTATGTGATATTCATTCAC) in a 2-month-old female patient with clinical features suggestive of MFS. This variant was initially considered as a variant of unknown significance according to ACMG/AMP guideline. Since the patient met the clinical diagnostic criteria for MFS, the deep intronic variant was considered as a candidate variant. Multiple computational splicing tools predicted that the variant could cause use of a new splice acceptor site within intron 40 of the FBN1 transcript. In order to seek the convincing evidence, in vitro minigene testing was performed and showed that the variant abrogates the canonical splice site of intron 40, resulting in exon 41 skipping. Eventually, the variant was annotated as an inframe deletion variant (c.4943_5065del, p.Val1649_Asp1689del), which was evaluated as clinical pathogenic according to the ACMG/AMP guidelines. Thus, the patient was clinically and molecularly diagnosed with MFS.
Then, we systematically reviewed and analyzed deep intronic variants in FBN1 (Biggin et al., 2004; Gillis et al., 2014; Xiong et al., 2015; Groth et al., 2017; Fusco et al., 2019; Wai et al., 2020; Guo et al., 2023; Bai et al., 2024); [HGMD database]. Currently, a total of 85 deep intronic variants, including the novel variant identified in our study, were identified (Figure 4). Among these variants, 28 of 86 (32.6%) have been verified to be pathogenic through functional analysis such as patient’s mRNA expression or in vitro minigene assays. Thus, additional investigations are needed to determine the pathogenicity of the other deep intronic variants. No obvious mutation spots were observed. Certainly, it is also necessary to collect more cases with deep intronic variants in FBN1 to enrich mutation spectrum of FBN1.
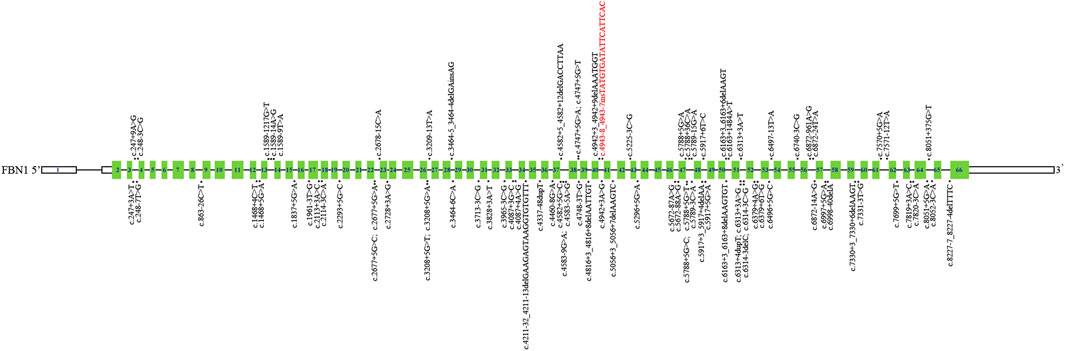
Figure 4. Schematic representation of FBN1 deep intronic variants identified to date. The structure of FBN1 contained 66 exons (blue rectangles), introns (black horizontal line); The localization of variants identified is depicted with dots. The dots above FBN1 indicate deep intronic variants determined by functional analysis, and those below FBN1 indicate deep intronic variants not determined by functional analysis. Red: Novel variants identified in this study.
In conclusion, we identified a novel deep intronic variant in FBN1 in a Chinese patient diagnosed with MFS. Our findings expanded FBN1 mutation spectrum, and highlighted that deep intronic variants should not be neglected in the interpretation of variants, and may be a potential cause of disease. Additional functional studies are necessary to verify the pathogenicity of deep intronic variants.
The data presented in the study are deposited in NODE (https://www.biosino.org/node) with the accession number OEP00006104 or through the URL: https://www.biosino.org/node/project/detail/OEP00006104.
The studies involving humans were approved by the Ethics Committee of Dongguan Maternal and Child Health Hospital (DMCH 202307). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
QW: Writing–original draft. FZ: Writing–original draft, Data curation, Methodology. XZ: Investigation, Resources, Writing–original draft. HL: Resources, Writing–original draft. JZ: Resources, Data curation, Writing–review and editing. HY: Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by Hospital Scientific Research Fund Project of Huadu District People’s Hospital (Grant No. 2021A02) and District and Hospital Joint Funding Project by the Huadu District Basic and Applied Basic Research (Grant No.23HDQYLH15). The funding body participated in the design of the project and interpretation of whole exome sequencing.
We would like to express our sincere gratitude to the pedigree for their cooperation. Furthermore, we extend our thanks to all the colleagues who actively participated in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MFS, Marfan syndrome; HEK 293T, human embryonic kidney 293T; GD, geleophysic dysplasia; AD, acromicric dysplasia; WES, Whole-exome sequencing.
Aoyama, T., Francke, U., Dietz, H. C., and Furthmayr, H. (1994). Quantitative differences in biosynthesis and extracellular deposition of fibrillin in cultured fibroblasts distinguish five groups of Marfan syndrome patients and suggest distinct pathogenetic mechanisms. J. Clin. Invest 94 (1), 130–137. doi:10.1172/JCI117298
Arnaud, P., Milleron, O., Hanna, N., Ropers, J., Ould Ouali, N., Affoune, A., et al. (2021). Clinical relevance of genotype-phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 23 (7), 1296–1304. doi:10.1038/s41436-021-01132-x
Baetens, M., Van Laer, L., De Leeneer, K., Hellemans, J., De Schrijver, J., Van De Voorde, H., et al. (2011). Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum. Mutat. 32 (9), 1053–1062. doi:10.1002/humu.21525
Bai, Y., Sun, Y., Yu, C., Xia, Y., Wu, J., Wang, L., et al. (2024). Causative role of a novel intronic indel variant in FBN1 and maternal germinal mosaicism in Marfan syndrome. Orphanet J. Rare Dis. 19 (1), 209. doi:10.1186/s13023-024-03139-4
Biggin, A., Holman, K., Brett, M., Bennetts, B., and Adès, L. (2004). Detection of thirty novel FBN1 mutations in patients with Marfan syndrome or a related fibrillinopathy. Hum. Mutat. 23 (1), 99. doi:10.1002/humu.9207
Collod-Béroud, G., Le Bourdelles, S., Ades, L., Ala-Kokko, L., Booms, P., Boxer, M., et al. (2003). Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum. Mutat. 22 (3), 199–208. doi:10.1002/humu.10249
Dietz, H. (2001). “FBN1-Related marfan syndrome,” in GeneReviews. Editors M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, L. J. H. Beanet al. (University of Washington, Seattle), 1993–2024. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK1335/.
Faivre, L., Collod-Beroud, G., Loeys, B. L., Child, A., Binquet, C., Gautier, E., et al. (2007). Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am. J. Hum. Genet. 81 (3), 454–466. doi:10.1086/520125
Fusco, C., Morlino, S., Micale, L., Ferraris, A., Grammatico, P., and Castori, M. (2019). Characterization of two novel intronic variants affecting splicing in FBN1-related disorders. Genes (Basel) 10 (6), 442. doi:10.3390/genes10060442
Gillis, E., Kempers, M., Salemink, S., Timmermans, J., Cheriex, E. C., Bekkers, S. C., et al. (2014). An FBN1 deep intronic mutation in a familial case of Marfan syndrome: an explanation for genetically unsolved cases? Hum. Mutat. 35 (5), 571–574. doi:10.1002/humu.22540
Groth, K. A., Von Kodolitsch, Y., Kutsche, K., Gaustadnes, M., Thorsen, K., Andersen, N. H., et al. (2017). Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet. Med. 19 (7), 772–777. doi:10.1038/gim.2016.181
Guo, D. C., Duan, X., Mimnagh, K., Cecchi, A. C., Marin, I. C., Yu, Y., et al. (2023). An FBN1 deep intronic variant is associated with pseudoexon formation and a variable Marfan phenotype in a five generation family. Clin. Genet. 103 (6), 704–708. doi:10.1111/cge.14322
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062-D1067–D1067. doi:10.1093/nar/gkx1153
Le Goff, C., Mahaut, C., Wang, L. W., Allali, S., Abhyankar, A., Jensen, S., et al. (2011). Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am. J. Hum. Genet. 89 (1), 7–14. doi:10.1016/j.ajhg.2011.05.012
Li, Z., Ma, F., Li, R., Xiao, Z., Zeng, H., and Wang, D. W. (2021). Case report: a novel LAMP2 splice-altering mutation causes cardiac-only danon disease. Front. Cardiovasc Med. 8, 763240. doi:10.3389/fcvm.2021.763240
Loeys, B., De Backer, J., Van Acker, P., Wettinck, K., Pals, G., Nuytinck, L., et al. (2004). Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum. Mutat. 24 (2), 140–146. doi:10.1002/humu.20070
Loeys, B. L., Dietz, H. C., Braverman, A. C., Callewaert, B. L., De Backer, J., Devereux, R. B., et al. (2010). The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 47 (7), 476–485. doi:10.1136/jmg.2009.072785
Mátyás, G., Alonso, S., Patrignani, A., Marti, M., Arnold, E., Magyar, I., et al. (2007). Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum. Genet. 122 (1), 23–32. doi:10.1007/s00439-007-0371-x
Passarge, E., Robinson, P. N., and Graul-Neumann, L. M. (2016). Marfanoid-progeroid-lipodystrophy syndrome: a newly recognized fibrillinopathy. Eur. J. Hum. Genet. 24 (9), 1244–1247. doi:10.1038/ejhg.2016.6
Pyeritz, R. E. (2000). The Marfan syndrome. Annu. Rev. Med. 51, 481–510. doi:10.1146/annurev.med.51.1.481
Ramirez, F., and Dietz, H. C. (2007). Fibrillin-rich microfibrils: structural determinants of morphogenetic and homeostatic events. J. Cell Physiol. 213, 326–330. doi:10.1002/jcp.21189
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Robinson, P. N., Arteaga-Solis, E., Baldock, C., Collod-Béroud, G., Booms, P., De Paepe, A., et al. (2006). The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet. 43 (10), 769–787. doi:10.1136/jmg.2005.039669
Wai, H. A., Lord, J., Lyon, M., Gunning, A., Kelly, H., Cibin, P., et al. (2020). Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet. Med. 22 (6), 1005–1014. doi:10.1038/s41436-020-0766-9
Whiteman, P., Smallridge, R. S., Knott, V., Cordle, J. J., Downing, A. K., and Handford, P. A. (2001). A G1127S change in calcium-binding epidermal growth factor-like domain 13 of human fibrillin-1 causes short range conformational effects. J. Biol. Chem. 276 (20), 17156–17162. doi:10.1074/jbc.M006547200
Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K., et al. (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347 (6218), 1254806. doi:10.1126/science.1254806
Keywords: deep intronic variant, FBN1, Marfan syndrome, minigene, RNA splicing
Citation: Wang Q, Zhang F, Zhou X, Li H, Zhao J and Yuan H (2025) Functional analysis of a novel FBN1 deep intronic variant causing Marfan syndrome in a Chinese patient. Front. Genet. 16:1564824. doi: 10.3389/fgene.2025.1564824
Received: 22 January 2025; Accepted: 28 February 2025;
Published: 19 March 2025.
Edited by:
Georgia Damoraki, National and Kapodistrian University of Athens, GreeceReviewed by:
Yongxiang Jiang, Fudan University, ChinaCopyright © 2025 Wang, Zhang, Zhou, Li, Zhao and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Zhao, bHVsdTE5ODR6aGFvQGFsaXl1bi5jb20=; Haiming Yuan, aGFpbWluZ3l1YW5Ac2luYS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.