 Zhuo Wei
Zhuo Wei Liying Yao3†
Liying Yao3† Meiyi Xu
Meiyi Xu Dan Wu
Dan Wu Wen Li
Wen Li Ying Chang
Ying Chang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 12 March 2025
Sec. Genetics of Common and Rare Diseases
Volume 16 - 2025 | https://doi.org/10.3389/fgene.2025.1463485
Introduction: Intellectual disability, autosomal dominant 29 is a rare disorder resulting from pathogenic variants of SETBP1 gene with no specific mutation hotspot identified. Systematic descriptions of new cases are crucial for understanding the genotypic and phenotypic spectrums of the disease.
Case presentation: A pregnant woman was referred to the prenatal diagnosis center at our hospital because she has an intellectual disability and has previously given birth to a child with intellectual disabilities. Karyotype, CNV-seq and whole-exome sequencing (WES) were employed to investigate the potential genetic issues in the family. The SETBP1 NM_015559.2: c.2425C>T (p.Gln809*) nonsense variant was found in the proband and mother, who were diagnosed with MRD29. Amniocentesis and genetic analysis (CNV-seq and sanger sequencing for mutation site) were performed as fetal cortical abnormalities and subependymal cystic area presented by ultrasonic examination at 25 + 5 gestational weeks. The genetic analysis confirmed the SETBP1 c.2425C>T (p.Gln809*) nonsense mutation in the fetus. The parents terminated the pregnancy at 30 + 4 gestational weeks.
Conclusion: The SETBP1 NM_015559.2: c.2425C>T (p.Gln809*) nonsense variant is pathogenic and SETBP1 haploinsufficiency may be associated with fatal cortical abnormalities. More prenatal clinical data is helpful for a better productive decision making and patient management.
Intellectual disability, autosomal dominant 29 (MRD29, MIM #616078) is a rare disorder that commonly associated with speech impairment, mild motor developmental delay and intellectual disability as reported in small case series. Additionally, hypotonia, vision impairment, concentration deficits, and hyperactivity have been documented in several cases. Prior to Jansen et al.’s delineation of the clinical spectrum of MRD29 among 34 individuals in 2021, there were no systematic descriptions of the disorder’s phenotypic and genotypic spectrums (Jansen et al., 2021). Subsequently, Morgan et al. emphasized the centrality of speech and language deficits among 31 individuals (Morgan et al., 2021). Nevertheless, the underlying mechanisms remain unclear.
With the advancement of next-generation sequencing (NGS), SET binding protein 1(SETBP1) has been identified as the disease-causing gene for MRD29. SETBP1 gene is located at 18q12.3 and encodes a protein with molecular mass of ∼170 kDa in most tissues. The SETBP1 protein possesses multiple functional domains, including a SET-binding region, an oncoprotein SKI homologous region, three bipartite NLS (nuclear localization signal) motifs, three AT hook domains, six PEST sequences, three sequential proline-rich repeats, four KxKHKxK, eight LSxxL and ten PxxPS repeated sentences (Minakuchi et al., 2001). The SKI-homology domain shares homology with the nuclear oncoprotein SKI and contains a degron motif that is recognized by the proteasome for protein degradation (Minakuchi et al., 2001).
MRD29 is believed associated with heterozygous gene deletion or loss-of-function (LoF) variants of SETBP1, without clear mutation hotspots (Jansen et al., 2021). In contrast, gain-of-function mutations in the SKI domain lead to the more sever Schinzel-Giedion syndrome (SGS, OMIM ID: 269150) characterized by recognizable facial characteristics, severe intellectual disability, and various congenital anomalies (Hoischen et al., 2010). These observations indicate a dose-dependent effect of SETBP1. However, the underlying mechanism of how altered SETBP1 protein dosage affects brain development remains elusive. Research utilizing human embryonic stem cells (hESCs) has demonstrated that SETBP1 deficiency affects forebrain progenitor expansion and neurogenic differentiation (Cardo et al., 2023). Nevertheless, few cases have reported abnormal brain MRI findings, implying that there may be issues with the timing of brain development detection.
In this report, clinical and molecular findings in a Chinese family with MRD29 are presented. Whole-exome sequencing (WES) analysis identified a nonsense variant. To discuss the prenatal diagnosis of MRD29 disease and improve understanding of the disease, previously reported cases were reviewed.
A 29-year-old pregnant woman, gravida 3, para 1, was referred to Tianjin Central Hospital of Obstetrics and Gynecology due to intellectual disability and a history of intellectual disability childbirth at 19+5 weeks of gestation. The pregnant woman exhibited intellectual disability, delayed language development, and could not use complete sentences before the age of 14 years old. She and her partner were un-related, and no disorder was reported about her partner. Their first child, a 6-year-old son (proband), presented with intellectual disability (Wechsler Intelligence Scale for Children-IQ test score of 52) and an inability to use complete sentences.
Peripheral blood samples of the parents and proband were collected at 20 weeks gestational age for karyotype analysis and chromosome copy number variation sequencing (CNV-seq) initially. As no abnormality was detected but nonnegligible genetic predisposition, trio-exome sequencing was then employed. Written informed consent was obtained from patients clarifying the benefits and risks of clinical whole-exome sequencing testing. As expected, the mother and proband were found to have a heterozygous SETBP1 c.2425C>T (p.Gln809*) nonsense mutation (Figures 1A, B). While, no pathogenic variant of the SETBP1 gene was detected in the father (Figure 1C). Considering the clinical features presented and potential genetic mechanism, the proband and mother was diagnosed with MRD29.
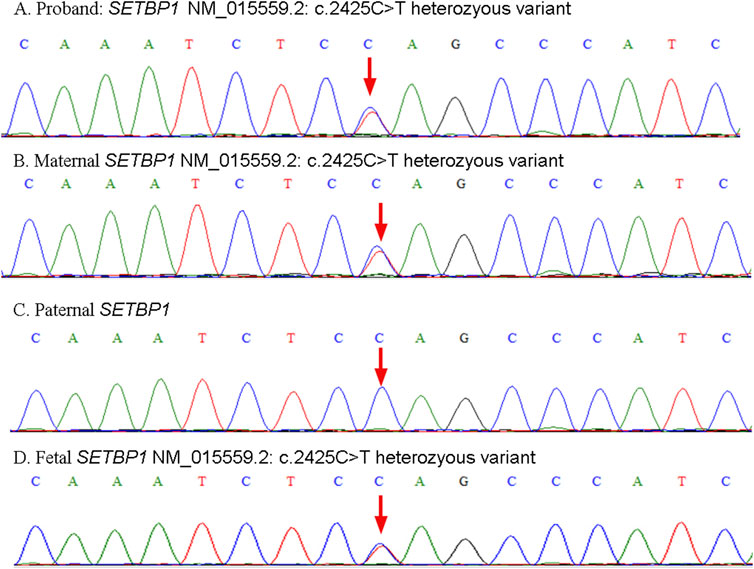
Figure 1. Schematic representation of validation results by Sanger sequencing. The heterozyous variants presented in the proband (A), mother (B) and fetus (D). The wild type SETBP1 presented in the father (C).
At 25+5 weeks of gestation, fetal cortical abnormalities and subependymal cystic area were detected by ultrasonic examination (Figure 2). Subsequently, amniocentesis was performed at 26+2 weeks of gestation for genetic analysis (CNV-seq and Sanger sequencing). A heterozygous SETBP1 c.2425C>T (p.Gln809*) nonsense mutation was detected (Figure 1D), and the fetus was subsequently diagnosed with MRD29 prenatally. The parents chose to terminate the pregnancy at 30+4 weeks of gestation and declined a post-mortem examination of the fetus. The patient’s general condition was good at discharge.
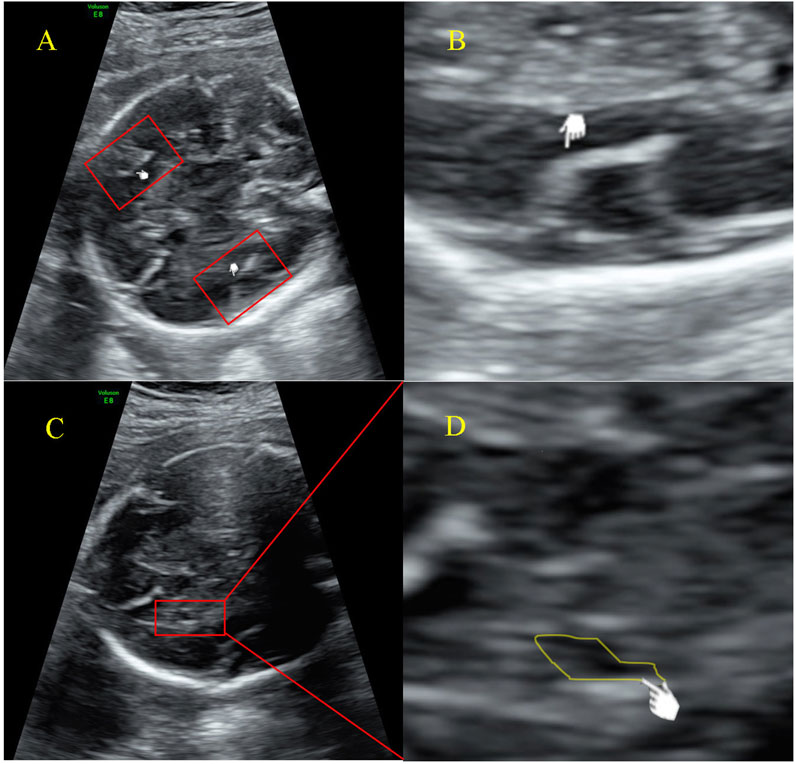
Figure 2. Ultrasound examination image of the fetal (A) Malformation of cortical development indicated by an increased Sylvian fissure angle; (B) Measurement of the Sylvian fissure angle; (C) Subependymal cyst; (D) Magnified image of the indicated cystic area, presented by ultrasonic examination at 25+5 weeks of gestation.
To summarize the clinical phenotype of MRD29 disorder, “MRD29 and SETBP1” were used as the formula for literature retrieval in the PubMed database. All variants and their positions are summarized in Figure 3. The clinical spectrum of individuals, including prenatal and brain MRI findings, is systematically outlined in Supplementary Table 1.
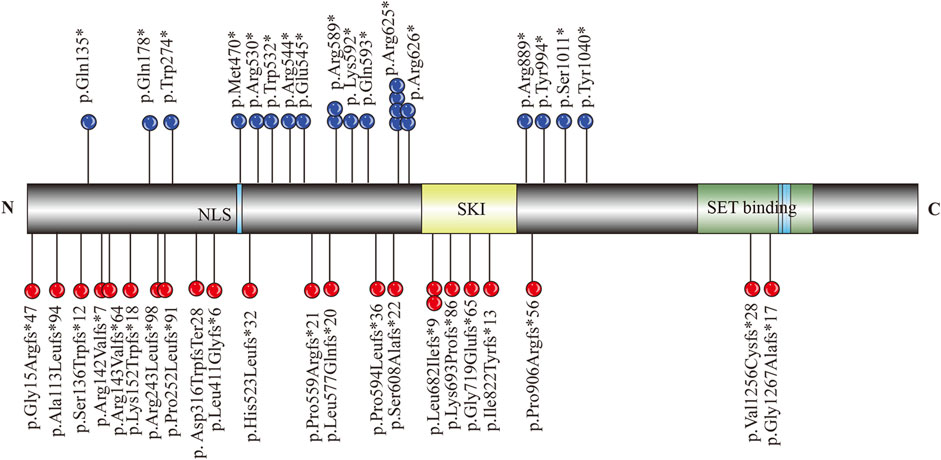
Figure 3. Schematic diagram of SETBP1 functional domains and variants identified in relation to MRD29.
We report a SETBP1 c.2425C>T variant here, and the mutation results in the 809th amino acid, glutamine, in the protein SKI domain replaced by a stop codon and causes termination of the SETBP1 protein. This is the first time a fetus with SETBP1 haploinsufficiency has been reported. Given the limited number of MRD29 cases reported to date, it is critical to focus on the phenotypic features of individuals with different variants. We systematically evaluated the phenotypes in 59 individuals reported to date: speech delay was reported in almost all cases evaluated (55/56, 98.21%); motor development delay (51/57, 89.47%) and intellectual disability (49/51, 96.08%) were also reported in almost all cases; 22 out of 42 (52.38%) cases had a history of feeding difficulties; 19 out of 36 (52.78%) cases were reported with vision impairments, including hypermetropia (9 cases), astigmatism (4 cases), strabismus (6 cases), myopia (3 cases), amblyopia (1 case), color blindness (1 case), and lack of binocular vision (1 case); hypotonia (19/30, 63.33%) and attention/concentration deficit (29/41, 70.73%) were also commonly reported, as shown in Supplementary Table 1 (Jansen et al., 2021; Morgan et al., 2021; Marseglia et al., 2012; Hamdan et al., 2014; Miolo et al., 2024; Kaspi et al., 2023; Vrkic Boban et al., 2022; Wang et al., 2023; Zhou et al., 2022; Rauch et al., 2012; Filges et al., 2011; Coe et al., 2014; Eising et al., 2019; Hildebrand et al., 2020; Alesi et al., 2024). The clinical findings in this family align with current knowledge on the spectrum of MRD29, including speech problems and intellectual disability.
We have reported for the first time the delayed development of the Sylvian fissure in the fetus as well as subependymal cysts. As we illustrated in Figures 2A, B, the development of Sylvian fissure was delayed according to works conducted by Chen et al. (2017) and Pooh et al. (2019). These works summarized the changing appearance on prenatal ultrasound of the sylvian fissure and determined sylvian fissure changes as important part of fetal cortical development. Interestingly, most individuals were previously reported to have normal brain MRI scans, with the exception of three cases under 4 years old who were identified with delayed myelination (Jansen et al., 2021; Morgan et al., 2021; Hamdan et al., 2014; Filges et al., 2011; Coe et al., 2014). In line with these reports, the mother and proband also presented normal MRI scans in this Chinese family. This finding underscores the need for further investigation into the role of SETBP1 in neurological phenotypes during early brain development, as well as its potential association with speech and language disorders at an early stage of life. However, our understanding of the prenatal characteristics of the MRD29 disorder remains limited, with only a few reported cases involving amniotic fluid abnormalities, fetal heart arrhythmia, fetal heart bradycardia, dysmaturity, hypotonia, fetal distress, and the presence of a single uterine artery. Further research is warranted to elucidate the full prenatal profile of this disorder and to establish a correlation analysis between prenatal and postnatal phenotypes, enabling personalized management strategies for patients.
Mechanistically, Lucia F. et al. have revealed that SETBP1-deficiency affects forebrain progenitor expansion and neurogenic differentiation by CRISPR/Cas9 genome editing hESC lines (Cardo et al., 2023). However, the precise role of SETBP1 in aggravating brain pathology remains unclear. Specifically, the cerebral cortex, particularly the posterior regions surrounding the Sylvian fissure, is crucial for regulating speech and language functions. Recently, Cabet S. et al. found that a prenatal lack of opercularization of the Sylvian fissure, without any other extracranial anomalies, is associated with speech delay (Cabet et al., 2024). Given our observation of delayed development of the Sylvian fissure in certain cases, we hypothesize that SETBP1 plays a role in the development of this fissure, which in turn regulates language and speech abilities. To clearly explore the influence of SETBP1 mutation on the development of the Sylvian fissure, animal experiments should be conducted. It is also important to note that more high-quality cases describing prenatal findings are needed, given the potential for significant heterogeneity in the manifestation of SETBP1 disorders.
The original contributions presented in the study are publicly available. This data can be found here: ClinVar repository, accession number SCV005442721, https://www.ncbi.nlm.nih.gov/clinvar/variation/807682/?oq=SCV005442721&m=NM_015559.3(SETBP1):c.2425C%3ET%20(p.Gln809Ter).
The studies involving humans were approved by Ethics Committee of Tianjin Central Hospital of Obstetrics and Gynecology. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study and publication of this case report was provided by the participants’ legal guardians/next of kin.
ZW: Data curation, Funding acquisition, Investigation, Project administration, Writing–original draft, Writing–review and editing, Formal Analysis, Visualization. LY: Conceptualization, Data curation, Project administration, Writing–original draft, Resources. LZ: Data curation, Project administration, Writing–original draft, Resources. SL: Formal Analysis, Methodology, Writing–original draft, Project administration. MX: Data curation, Project administration, Writing–original draft. DW: Data curation, Project administration, Writing–original draft. WL: Methodology, Project administration, Supervision, Writing–review and editing. YC: Conceptualization, Resources, Supervision, Writing–original draft.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Natural Science Foundation of Tianjin (Grant No. 21JCQNJC00040, 22JCQNJC00460); Tianjin Health Research Project (Grant No. TJWJ2021QN052).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2025.1463485/full#supplementary-material
SUPPLEMENTARY TABLE 1 | SETBP1 variants and clinical characteristic related to MRD29.
Alesi, V., Genovese, S., Roberti, M. C., Sallicandro, E., Di Tommaso, S., Loddo, S., et al. (2024). Structural rearrangements as a recurrent pathogenic mechanism for SETBP1 haploinsufficiency. Hum. Genomics 18 (1), 29. doi:10.1186/s40246-024-00600-0
Cabet, S., Putoux, A., Lesca, G., Lesage, A., Massoud, M., Guibaud, L., et al. (2024). Prenatal diagnosis of microcephaly with simplified gyral pattern: series of eight cases. Ultrasound Obstet. Gynecol. 63 (2), 271–275. doi:10.1002/uog.27450
Cardo, L. F., de la Fuente, D. C., and Li, M. (2023). Impaired neurogenesis and neural progenitor fate choice in a human stem cell model of SETBP1 disorder. Mol. Autism 14 (1), 8. doi:10.1186/s13229-023-00540-x
Chen, X., Li, S. L., Luo, G. Y., Norwitz, E. R., Ouyang, S. Y., Wen, H. X., et al. (2017). Ultrasonographic characteristics of cortical sulcus development in the human fetus between 18 and 41 Weeks of gestation. Chin. Med. J. Engl. 130 (8), 920–928. doi:10.4103/0366-6999.204114
Coe, B. P., Witherspoon, K., Rosenfeld, J. A., van Bon, B. W. M., Vulto-van Silfhout, A. T., Bosco, P., et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 46 (10), 1063–1071. doi:10.1038/ng.3092
Eising, E., Carrion-Castillo, A., Vino, A., Strand, E. A., Jakielski, K. J., Scerri, T. S., et al. (2019). A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 24 (7), 1065–1078. doi:10.1038/s41380-018-0020-x
Filges, I., Shimojima, K., Okamoto, N., Röthlisberger, B., Weber, P., Huber, A. R., et al. (2011). Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J. Med. Genet. 48 (2), 117–122. doi:10.1136/jmg.2010.084582
Hamdan, F. F., Srour, M., Capo-Chichi, J. M., Daoud, H., Nassif, C., Patry, L., et al. (2014). De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10 (10), e1004772. doi:10.1371/journal.pgen.1004772
Hildebrand, M. S., Jackson, V. E., Scerri, T. S., Van Reyk, O., Coleman, M., Braden, R. O., et al. (2020). Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology 94 (20), e2148–e2167. doi:10.1212/WNL.0000000000009441
Hoischen, A., van Bon, B. W. M., Gilissen, C., Arts, P., van Lier, B., Steehouwer, M., et al. (2010). De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 42 (6), 483–485. doi:10.1038/ng.581
Jansen, N. A., Braden, R. O., Srivastava, S., Otness, E. F., Lesca, G., Rossi, M., et al. (2021). Clinical delineation of SETBP1 haploinsufficiency disorder. Eur. J. Hum. Genet. 29 (8), 1198–1205. doi:10.1038/s41431-021-00888-9
Kaspi, A., Hildebrand, M. S., Jackson, V. E., Braden, R., van Reyk, O., Howell, T., et al. (2023). Genetic aetiologies for childhood speech disorder: novel pathways co-expressed during brain development. Mol. Psychiatry 28 (4), 1647–1663. doi:10.1038/s41380-022-01764-8
Marseglia, G., Scordo, M. R., Pescucci, C., Nannetti, G., Biagini, E., Scandurra, V., et al. (2012). 372 kb microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur. J. Med. Genet. 55 (3), 216–221. doi:10.1016/j.ejmg.2012.01.005
Minakuchi, M., Kakazu, N., Gorrin-Rivas, M. J., Abe, T., Copeland, T. D., Ueda, K., et al. (2001). Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia-associated protein SET. Eur. J. Biochem. 268 (5), 1340–1351. doi:10.1046/j.1432-1327.2001.02000.x
Miolo, G., Colavito, D., Della Puppa, L., and Corona, G. (2024). Delayed bone age in a child with a novel loss-of-function variant in SETBP1 gene sheds light on the potential role of SETBP1 protein in skeletal development. Mol. Syndromol. 15 (2), 167–174. doi:10.1159/000535057
Morgan, A., Braden, R., Wong, M. M. K., Colin, E., Amor, D., Liégeois, F., et al. (2021). Speech and language deficits are central to SETBP1 haploinsufficiency disorder. Eur. J. Hum. Genet. 29 (8), 1216–1225. doi:10.1038/s41431-021-00894-x
Pooh, R. K., Machida, M., Nakamura, T., Uenishi, K., Chiyo, H., Itoh, K., et al. (2019). Increased Sylvian fissure angle as early sonographic sign of malformation of cortical development. Ultrasound Obstet. Gynecol. 54 (2), 199–206. doi:10.1002/uog.20171
Rauch, A., Wieczorek, D., Graf, E., Wieland, T., Endele, S., Schwarzmayr, T., et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380 (9854), 1674–1682. doi:10.1016/S0140-6736(12)61480-9
Vrkic Boban, I., Sekiguchi, F., Lozić, M., Miyake, N., Matsumoto, N., and Lozić, B. (2022). A novel SETBP1 gene disruption by a de novo balanced translocation in a patient with speech impairment, intellectual, and behavioral disorder. J. Pediatr. Genet. 11 (2), 135–138. doi:10.1055/s-0040-1715639
Wang, L., Wang, X. D., Yang, B., Wang, X. M., Peng, Y. Q., Tan, H. J., et al. (2023). Novel SETBP1 mutation in a Chinese family with intellectual disability. BMC Med. Genomics 16 (1), 233. doi:10.1186/s12920-023-01649-x
Keywords: MRD29, SETBP1, prenatal diagnosis, WES, cortical abnormalities
Citation: Wei Z, Yao L, Zhang L, Li S, Xu M, Wu D, Li W and Chang Y (2025) Prenatal diagnosis of intellectual disability, autosomal dominant 29 with a nonsense pathogenic variant in SETBP1: a case report and literature review. Front. Genet. 16:1463485. doi: 10.3389/fgene.2025.1463485
Received: 12 July 2024; Accepted: 13 February 2025;
Published: 12 March 2025.
Edited by:
Mara Marongiu, National Research Council (CNR), ItalyReviewed by:
Tayyab Ali, University of Agriculture, Faisalabad, PakistanCopyright © 2025 Wei, Yao, Zhang, Li, Xu, Wu, Li and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen Li, NTAyMDIwMDI5MEBuYW5rYWkuZWR1LmNu; Ying Chang, Y2hhbmd5aW5nNDQ3MEBzaW5hLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.