 Meryem Banu Cavlak1*
Meryem Banu Cavlak1* Gagandeep Singh1Mohammed Alser1
Gagandeep Singh1Mohammed Alser1 Can Firtina1*Joël Lindegger1Mohammad Sadrosadati1Nika Mansouri Ghiasi1Can Alkan2Onur Mutlu1*
Can Firtina1*Joël Lindegger1Mohammad Sadrosadati1Nika Mansouri Ghiasi1Can Alkan2Onur Mutlu1*- 1SAFARI Research Group, Department of Information Technology and Electrical Engineering, ETH Zurich, Zurich, Switzerland
- 2Department of Computer Engineering, Bilkent University, Ankara, Türkiye
Basecalling is an essential step in nanopore sequencing analysis where the raw signals of nanopore sequencers are converted into nucleotide sequences, that is, reads. State-of-the-art basecallers use complex deep learning models to achieve high basecalling accuracy. This makes basecalling computationally inefficient and memory-hungry, bottlenecking the entire genome analysis pipeline. However, for many applications, most reads do not match the reference genome of interest (i.e., target reference) and thus are discarded in later steps in the genomics pipeline, wasting the basecalling computation. To overcome this issue, we propose TargetCall, the first pre-basecalling filter to eliminate the wasted computation in basecalling. TargetCall’s key idea is to discard reads that will not match the target reference (i.e., off-target reads) prior to basecalling. TargetCall consists of two main components: (1) LightCall, a lightweight neural network basecaller that produces noisy reads, and (2) Similarity Check, which labels each of these noisy reads as on-target or off-target by matching them to the target reference. Our thorough experimental evaluations show that TargetCall 1) improves the end-to-end basecalling runtime performance of the state-of-the-art basecaller by
1 Introduction
Genome sequencing, which determines the nucleotide sequence of an organism’s genome, plays a pivotal role in enabling many medical and scientific advancements (Alkan et al., 2009; Ashley, 2016; Chin et al., 2011; Ellegren, 2014; Alvarez-Cubero et al., 2017). Modern sequencing technologies produce increasingly large amounts of genomic data at low cost (Wang et al., 2021). Leveraging this genomic data requires fast, efficient, and accurate analysis tools.
Current sequencing machines are unable to determine an organism’s genome as a single contiguous sequence (Alser et al., 2021). Instead, they sequence fragments of a genome, called reads. The length of the reads depends on the sequencing technology and significantly affects the performance (i.e., speed or runtime) and accuracy of genome analysis. The use of long reads can provide higher accuracy and performance on many genome analysis steps (Treangen and Salzberg, 2011; Firtina and Alkan, 2016; Alkan et al., 2010; Lu et al., 2016; Magi et al., 2018; Firtina et al., 2023b).
Nanopore sequencing technology is one of the most prominent and widely used long read sequencing technologies (Wang et al., 2021; Branton et al., 2008; Gong et al., 2019; Jain et al., 2018; Logsdon et al., 2020; Amarasinghe et al., 2020). Nanopore sequencing relies on measuring the change in the electrical current when a nucleic acid molecule (DNA or RNA) passes through a pore of nanometer size (Zhang et al. 2021a). The measurement of the electrical current, called a raw signal, is converted to a nucleotide sequence, called a read, with a step called basecalling (Wang et al., 2021; Alser et al., 2021; Wick et al., 2019; Pagès-Gallego and de Ridder, 2023; Alser et al., 2022; Wan et al., 2021). Basecalling commonly uses computationally expensive deep neural network (DNN)-based architectures to achieve high basecalling accuracy (Senol Cali et al., 2019; Rang et al., 2018; Singh et al., 2024), which makes basecalling a computational bottleneck for genome analysis that consumes up to
Our goal in this work is to eliminate the wasted computation when basecalling the entire read, while maintaining high accuracy and applicability to a wide range of genome sequencing applications. To this end, we propose TargetCall, the first pre-basecalling filter. TargetCall is based on the key observation that the typical reason for discarding basecalled reads is that they do not match some target reference (e.g., a reference genome of interest) (Grumaz et al., 2016; Dunn et al., 2021). We call these off-target reads. Our key idea is to filter out off-target reads before basecalling by analyzing the entire read with a highly accurate and high-performance pre-basecalling filter to eliminate the wasted computation in basecalling off-target reads.
Prior works in targeted sequencing (Kovaka et al., 2021; Zhang et al., 2021a; Dunn et al., 2021; Bao et al., 2021; Firtina et al., 2023a; Lindegger et al., 2023; Firtina et al., 2024) propose adaptive sampling techniques to discard off-target reads during sequencing to better utilize sequencers. Sequencers provided by Oxford Nanopore Technologies (ONT) can enable adaptive sampling with a feature known as Read Until (Kovaka et al., 2021; Loose et al., 2016). ONT sequencers that support Read Until can selectively remove a read from the nanopore while the read is being sequenced. This requires a method of identifying which reads are off-target for further downstream analysis to decide which reads to remove from the nanopore. The state-of-the-art adaptive sampling methods can be classified into three groups based on the methodology used to label the read. The first group converts the target reference into a reference raw signal and performs raw signal-level alignment (Zhang et al., 2021a; Dunn et al., 2021; Loose et al., 2016). The second group generates noisy sequence representations of the raw signal to compare them with the target reference (Kovaka et al., 2021; Payne et al., 2020). The third group of works utilizes neural network classifiers to label the sequences (Bao et al., 2021; Noordijk et al., 2023). We provide a detailed background on different adaptive sampling approaches in Section 1 of the Supplementary Material.
Even though the labeling techniques of adaptive sampling can be repurposed for pre-basecalling filtering, the adaptive sampling problem is different from pre-basecalling filtering for three main reasons. First, in adaptive sampling, reads must be labeled during sequencing, requiring only the initial portion of the raw signal to classify reads as off-target or on-target. Analyzing a sub-region or raw signals in adaptive sampling methods often leads to low recall
TargetCall consists of two main components: 1) LightCall, a lightweight basecaller with a simple neural network model that outputs erroneous (i.e., noisy) reads with high performance and 2) Similarity Check to compute the similarity of the noisy read to the target reference where the similarity is determined by the conventional read mapping pipeline. LightCall’s model is
1.1 Key results
We evaluate the performance and accuracy impact of TargetCall on the state-of-the-art basecaller by Bonito (2022) and compare TargetCall with two state-of-the-art adaptive sampling methods, UNCALLED (Kovaka et al., 2021) and Sigmap (Zhang et al., 2021a), repurposed as pre-basecalling filters. TargetCall 1) improves the end-to-end runtime performance (i.e., runtime of all the steps, including basecalling and read mapping, used in an analysis) by
This article makes the following contributions:
2 Materials and methods
Our goal in this work is to eliminate the wasted computation in basecalling using an accurate pre-basecalling filtering technique. To this end, we propose TargetCall, which can perform pre-basecalling filtering in all genome sequencing applications accurately and efficiently without any additional overhead. To our knowledge, TargetCall is the first pre-basecalling filter that is applicable to a wide range of use cases and makes use of entire raw signal information to classify nanopore raw signals. TargetCall’s key idea is to quickly filter out off-target reads (i.e., reads that are dissimilar to the target reference) before the basecalling step to eliminate the wasted computation in basecalling. We present the high-level overview of TargetCall in Section 2.1 and explain its components in Sections 2.2, 2.3.
2.1 High level overview
Figure 1 shows TargetCall’s workflow. First, TargetCall performs noisy basecalling on the raw signal using LightCall (1). The output sequence of LightCall is highly accurate but erroneous compared to the reads basecalled using state-of-the-art basecallers. Second, Similarity Check compares the noisy read of LightCall to the target reference to label the read as an on-target or off-target read (2). TargetCall stops the analysis of off-target reads by removing them from the pipeline (3), whereas the analysis of the on-target reads continues with basecalling following the usual genomics pipeline to maintain basecalling accuracy1 (4).
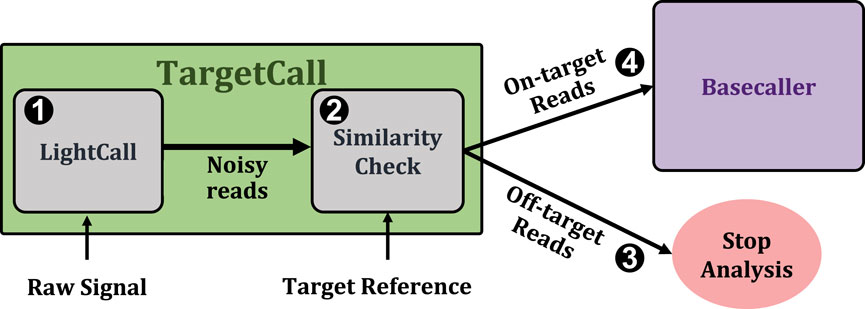
Figure 1. High-level overview of TargetCall.
The choice of the target reference depends on the specific genome sequencing application. The size of the target reference is a major constraint in most prior works, limiting the generality of the prior approaches. We design TargetCall such that its runtime performance scales well with the size of the target reference so that it is applicable to any genome sequencing application. We achieve our design goal because 1) the performance of LightCall is independent of the size of the target reference, and 2) the performance of the Similarity Check module scales well with the increasing target reference size.
2.2 LightCall
LightCall, the first component of TargetCall, is a lightweight neural network-based basecaller that produces noisy reads. Although LightCall is not designed to be as accurate as the state-of-the-art basecallers, its combination with Similarity Check is effective in determining if the read is an on-target read with respect to the target reference. We develop LightCall by modifying the state-of-the-art basecaller Bonito’s architecture in three ways: 1) reducing the channel sizes of convolution layers, 2) removing the skip connections, and 3) reducing the number of basic convolution blocks. Prior work (Singh et al., 2024) shows that Bonito’s model is over-provisioned, and we can maintain very high accuracy with reduced model sizes. Following prior work’s insight, we generate different neural network models by pruning the channel sizes of convolution and convolution blocks. The specific LightCall configurations are tested and designed based on our intuition. We select the neural network architecture for LightCall that balances basecalling accuracy with the pre-basecalling filtering performance.
Figure 2 shows the architecture of LightCall. Each block consists of grouped 1-dimensional convolution and pointwise 1-dimensional convolution. The convolution operation is followed by batch normalization (Batch Norm) (Ioffe and Szegedy, 2015) and a rectified linear unit (ReLU) (Agarap, 2018) activation function. The final output is passed through a connectionist temporal classification (CTC) (Graves et al., 2006) layer to produce the decoded sequence of nucleotides (A, C, G, and T). The CTC layer acts as the loss function by providing the correct alignment between the input and the output sequence. The CTC loss allows the model to handle the variable-length sequences by aligning the model’s output to the target sequence while ignoring the blank or padding symbols. Our LightCall architecture is composed of 18 convolution blocks containing
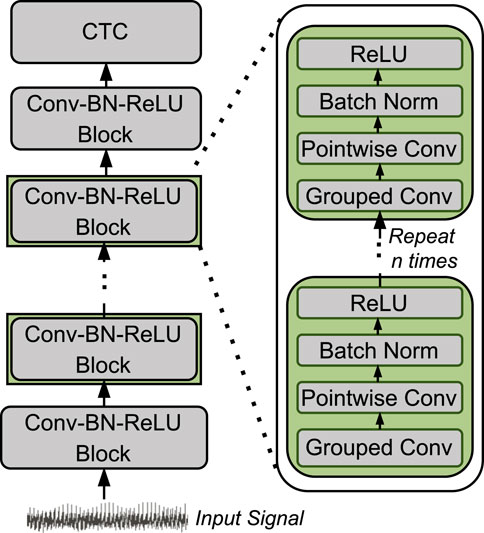
Figure 2. Overview of LightCall architecture.
Modern deep learning-based basecallers (Singh et al., 2024; Wick et al., 2019; Neumann et al., 2022; Konishi et al., 2021; Xu et al., 2021; Lou et al., 2020; Perešíni et al., 2021; Pagès-Gallego and de Ridder, 2023; Zhang et al., 2021b) incorporate skip connections to help mitigate the vanishing gradient and saturation problems (V and Kiran, 2022). Removing skip connections has a higher impact on basecalling accuracy. However, adding skip connections introduces the following three issues for performance (Shahroodi et al., 2023; Singh et al., 2024). First, the skip connections increase the data lifetime. The layers whose activations are reused in subsequent layers must wait for this activation reuse (or buffer the activations in memory) before accepting new input and continuing to compute. This leads to high resource and storage requirements due to data duplication. Second, skip connections introduce irregularity in neural network architecture as these connections span non-adjacent layers. Third, skip connections require additional computation to adjust the channel size to match the channel size at the non-consecutive layer’s input. Therefore, we remove the skip connections, as we can tolerate a lower LightCall accuracy to improve the performance of TargetCall.
LightCall works by splitting a long read in raw signal format (e.g., millions of samples) into multiple smaller chunks (e.g., thousands of samples per chunk) and basecalling these chunks. The CTC layer assigns a probability for all possible labels (i.e., A, C, G, and T) at each sequence position for a chunk. The label with the highest probability is selected as the final output for a sequence position. LightCall merges the outputs of each position to produce the basecalled chunk and merges the basecalled chunks to output the basecalled read. Because LightCall’s algorithm is independent of the target reference, LightCall’s performance does not depend on the target reference length.
2.3 Similarity check
After LightCall outputs the noisy read that approximately represents the raw signal, Similarity Check compares the noisy read to the target reference. For this task, we use a procedure common in genome analysis known as sequence alignment. Sequence alignment computes the similarity between a read and a reference. To provide a scalable and fast solution for Similarity Check, we use minimap2, a well-optimized sequence aligner designed by Li (2018). Similarity Check labels a read that is similar to the target reference, that is, a read that maps to the target reference as on-target. With the efficient index structure of minimap2, the performance of sequence alignment is almost independent of the length of the target reference genome. Hence, TargetCall is scalable to large target reference genomes of Gbp (i.e., Giga base pair) length.
The accuracy of the computed sequence alignments is not high enough to represent the true alignment between the read and the target reference because the reads computed by LightCall are noisy. However, the labeling accuracy of Similarity Check for determining the on-target/off-target reads is high enough to provide recall up to
3 Results
3.1 Experimental setup
3.1.1 Evaluated use cases
To show the applicability of TargetCall, we evaluate it on use cases with varying target reference sizes without compromising the accuracy. We describe three use cases we use to evaluate TargetCall: (1) COVID detection, (2) sepsis detection, and (3) viral detection. All three use cases contain a significant fraction of off-target reads that are eliminated using TargetCall to show the benefits of pre-basecalling filtering.
3.1.2 COVID detection
The first use case aims to accept reads coming from a small target reference. We choose SARS-CoV-2 detection as a sample biological application where the goal is to detect the reads coming from a SARS-CoV-2 reference genome (
3.1.3 Sepsis detection
The second use case aims to filter reads when the target reference is large (
3.1.4 Viral detection
The third use case aims to filter reads when the target reference contains a collection of reference genomes to show that TargetCall can correctly filter reads when the sample and the target reference have a wide variety of species. This is to test the specificity of TargetCall in filtering reads when the on-target and off-target reads resemble each other more than in previous use cases. We choose disease-causing viral read detection as a sample biological application where the goal is to detect the viral reads from a metagenomic sample of bacterial and viral reads, and the target reference contains a collection of viral reference genomes, as demonstrated by Mokili et al. (2012).
3.1.5 Evaluation system
We use NVIDIA TITAN V to train and evaluate the LightCall and Bonito baseline. For our evaluations, we increase the batch size maximally such that the entire GPU memory is occupied (Sections 3.2–3.5). We use NVIDIA A100 to evaluate TargetCall against prior work. We evaluate UNCALLED and Sigmap on a high-end server (AMD EPYC 7742 CPU with 1 TB DDR4 DRAM). For our evaluations, we optimize the number of threads (128) for UNCALLED/Sigmap and the batch size (128) for TargetCall such that all tools have the minimum execution time (Section 3.6). We use the state-of-the-art read mapper, minimap2 (Li, 2018), for the Similarity Check module of TargetCall with -a and -x map-ont flags. The -a flag is used to compute sequence alignments, and -x map-ont is used to configure minimap2 parameters for ONT data.
3.1.6 Training setting
We use the publicly available ONT dataset, Bonito (2022), sequenced using MinION Flow Cell (R9.4.1) (ONT, 2022) for the training and validation. The neural network weights are updated using the Adam optimizer (Kingma and Ba, 2014) with a learning rate of 2
3.1.7 Baseline techniques
We evaluate TargetCall’s runtime performance and accuracy as a pre-basecalling filter by integrating it as a pre-basecalling filter to Bonito (2022), which is one of the official basecalling tools developed by ONT. In Section 3.6, we evaluate two state-of-the-art, non-machine learning-based adaptive sampling methods, UNCALLED (Kovaka et al., 2021) and Sigmap (Zhang et al., 2021a), repurposed as pre-basecalling filters to compare against TargetCall. We repurposed UNCALLED and Sigmap by using their classification methods to classify the reads as on-target/off-target before the basecalling step without any change in their implementations. We identify the ground truth on-target and off-target by basecalling the reads using a high accuracy Bonito model followed by performing minimap2. We do not evaluate TargetCall against other adaptive sampling methods, such as SquiggleNet (Bao et al., 2021), that cannot be trivially used as pre-basecalling filters.
3.1.8 LightCall configurations evaluated
To determine the final architecture of TargetCall, we test
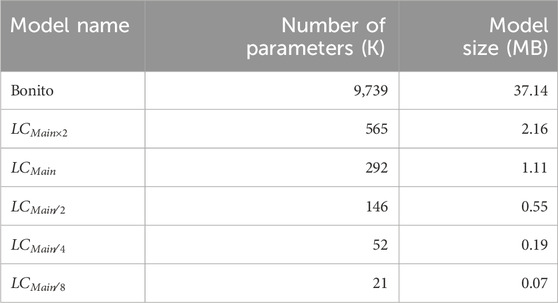
Table 1. Different LightCall configurations.
3.1.9 Evaluated datasets
We sampled 287,767 reads from prior work (Zook et al., 2019; cad, 2020; Wick et al., 2019) and simulated 35,000 reads using DeepSimulator (Li et al., 2018; Li et al., 2020) to evaluate TargetCall. We use four reference genomes to evaluate TargetCall on three different genome sequencing applications. The details of the exact read datasets and reference genomes used to produce all our results can be found in Section 2 of the Supplementary Material.
3.1.10 Evaluation metrics
We evaluate TargetCall using five different metrics: 1) filtering accuracy, 2) basecalling accuracy, 3) relative abundance (RA) estimation accuracy, 4) basecalling execution time, and 5) end-to-end execution time. For the basecalling execution time, we compare the wall-clock time spent on pre-basecalling filtering followed by basecalling of the reads that are accepted by the filter and conventional basecalling. For the end-to-end execution time, we compare the wall-clock time spent on the entire genome analysis pipeline of basecalling, read mapping, and variant calling with and without the use of pre-basecalling filtering. The index generation time of minimap2 is excluded from end-to-end execution time as this is a one-time task per reference genome. When comparing TargetCall with UNCALLED and Sigmap in terms of the end-to-end execution time, we acknowledge that TargetCall benefits from hardware acceleration as it uses GPUs while the other tools use CPUs. Implementing UNCALLED and Sigmap on GPUs could further improve their speed performance.
We evaluate the filtering accuracy of TargetCall by computing its precision and recall. We define precision as the number of reads that TargetCall correctly labels as on-target, divided by the total number of reads that TargetCall labels as on-target. We define recall as the number of reads that TargetCall correctly labels as on-target, divided by the overall number of on-target reads in the dataset. The ground truth on-target reads are determined by the conventional pipeline of basecalling with Bonito and read mapping. An ideal pre-basecalling filter should have 100% recall to maintain accuracy in the downstream analyses and 100% precision to provide the maximum possible runtime performance improvement.
For basecalling accuracy, we use Bonito’s training and evaluation procedure to extract the median identity as basecalling accuracy Bonito (2022). For relative abundance (RA) accuracy, we calculate the difference in relative abundances of viral species after 1) pre-basecalling filtering and 2) conventional basecalling. We compute how much RA deviates from the true RAs after pre-basecalling filtering. RA is defined as the proportion of reads corresponding to a particular species relative to the total number of reads. The true RA refers to the RA obtained after conventional basecalling, which is considered the benchmark. Equation 1 provides the calculation of the deviation in RAs where
3.2 Filtering accuracy
In Figures 3, 4, we assess the precision and recall of TargetCall with different LightCall configurations for all use cases explained in Section 3.1. We make four key observations. First, TargetCall’s precision and recall are between
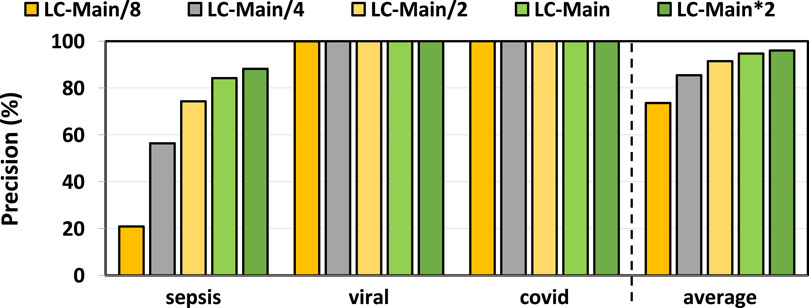
Figure 3. Precision of our evaluated use cases using TargetCall with different LightCall configurations.
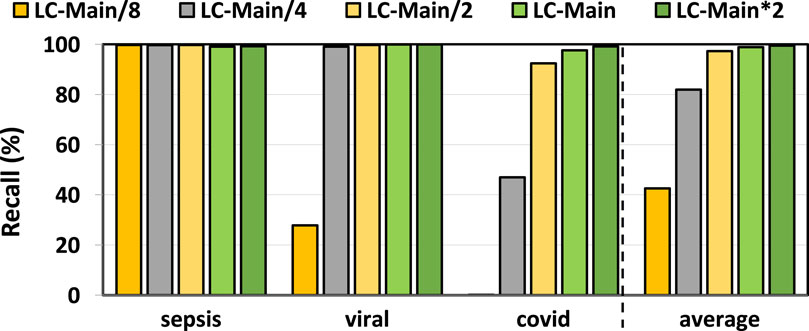
Figure 4. Recall of our evaluated use cases using TargetCall with different LightCall configurations.
3.3 Basecalling and relative abundance accuracy
Table 2 shows the basecalling accuracy and the RA deviation from the ground truth relative abundance estimation calculated for each LightCall configuration and using Bonito without pre-basecalling filtering. We evaluate the RA accuracy of TargetCall in the viral detection use case. We use Equation 1 to calculate the RA deviation as the RA accuracy metric.
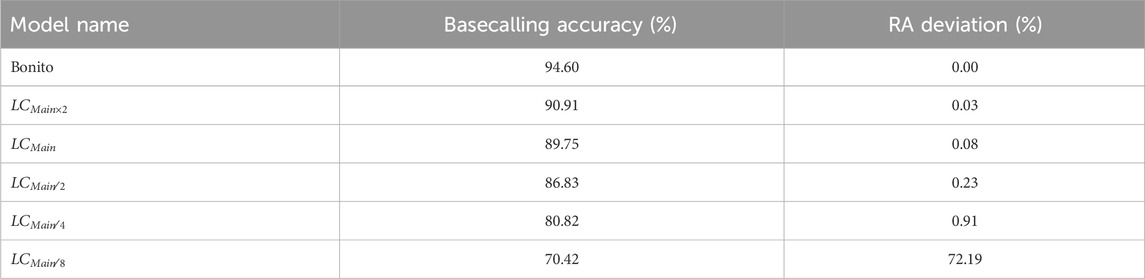
Table 2. Basecalling accuracy and relative abundance (RA) deviation.
We make the following two key observations. First, the RA deviation results are negligible (
Second, TargetCall’s minor inaccuracy is not biased toward any specific portion of the target reference. Otherwise, the deviation of the relative abundances would be higher. Losing a small number of on-target reads randomly enables sequencing depth-of-coverage to compensate for the loss of reads. We conclude that TargetCall’s high recall enables accurate estimation for relative abundance calculations.
3.4 Basecalling execution time
Figure 5 provides the total execution time of Bonito and Bonito with TargetCall. We make three key observations. First, TargetCall improves the runtime performance of Bonito by
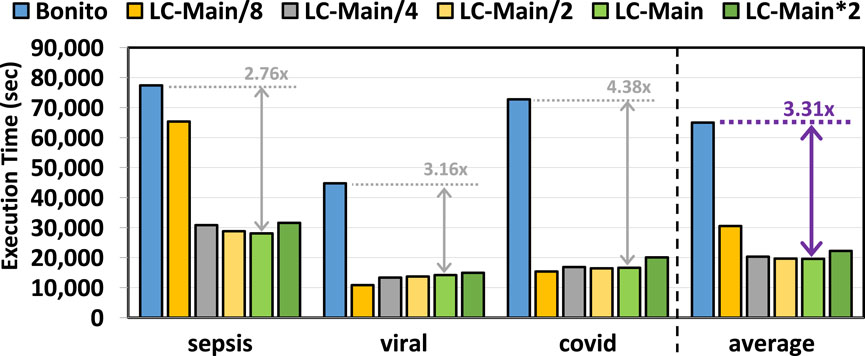
Figure 5. Basecalling execution time of our evaluated use cases using TargetCall with different LightCall configurations.
3.5 End-to-end execution time
Figure 6 provides the total time spent on the genome analysis pipeline of basecalling, read mapping, and variant calling with and without the use of pre-basecalling filtering. We used minimap2 (Li, 2018) and DeepVariant (Poplin et al., 2018) for read mapping and variant calling, respectively. We make three key observations. First, TargetCall improves the runtime performance of the entire genome sequence analysis pipeline by
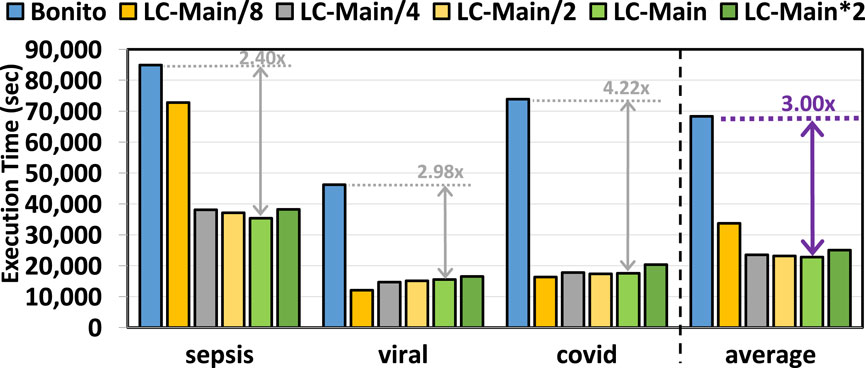
Figure 6. End-to-end execution time of our evaluated use cases using TargetCall with different LightCall configurations.
3.6 Comparison to prior work
We compare TargetCall with the best LightCall configuration (
We compare the recall of TargetCall with that of Sigmap and UNCALLED. The ground truth on-target reads are determined by the conventional pipeline. In our analysis, we observe that UNCALLED could not be executed on the hg38 reference genome. To ensure a fair assessment, we excluded hg38 from the performance evaluation of UNCALLED. Figure 7 shows the recall of Sigmap, UNCALLED, and TargetCall. We make two key observations. First, TargetCall provides significantly higher recall, +
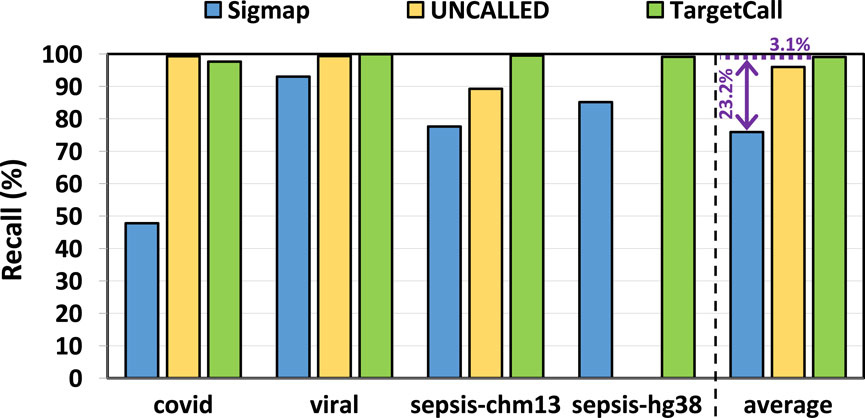
Figure 7. Recall of Sigmap, UNCALLED, and TargetCall.
We compare the precision of TargetCall with that of Sigmap and UNCALLED. Figure 8 shows the precision of Sigmap, UNCALLED, and TargetCall. We make two key observations. First, TargetCall provides significantly higher precision, +

Figure 8. Precision of Sigmap, UNCALLED, and TargetCall.
We compare the end-to-end execution time of TargetCall with that of Sigmap and UNCALLED, and the results are shown in Figure 9. We make two key observations. First, we observe that TargetCall outperforms Sigmap by
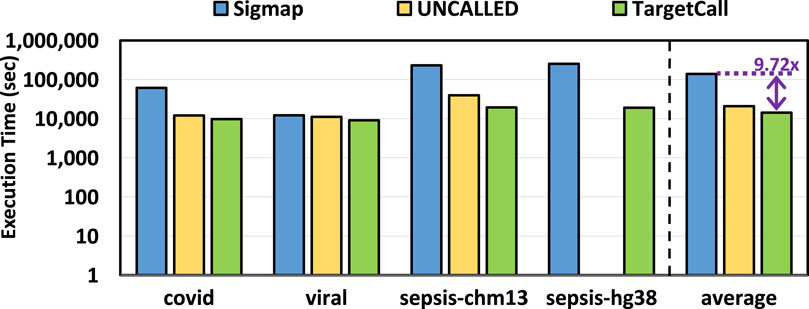
Figure 9. End-to-end execution time of Sigmap, UNCALLED, and TargetCall.
We evaluate the throughput of TargetCall and compare it with that of Sigmap and UNCALLED. Throughput shows the number of base pairs that the tools process per second. We use uncalled pafstats for evaluating the throughput of UNCALLED and use Sigmap and Bonito output for evaluating the throughput of LightCall. We only evaluate the throughput of the LightCall component of TargetCall, as it has a significantly lower throughput than Similarity Check and is the bottleneck of TargetCall. Figure 10 shows the throughput of Sigmap, UNCALLED, and LightCall. We make three key observations. First, we observe that LightCall improves the throughput of UNCALLED/Sigmap by
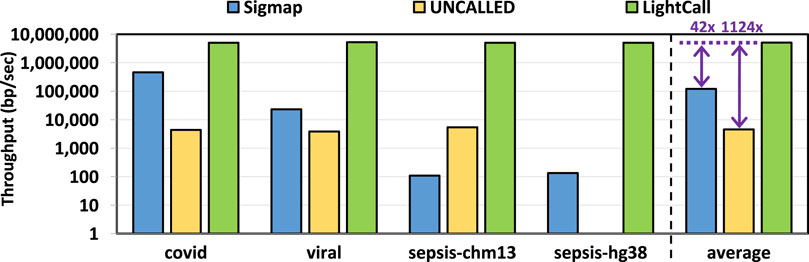
Figure 10. Throughput of Sigmap, UNCALLED, and LightCall.
Overall, we conclude that TargetCall 1) significantly improves the end-to-end execution time of basecalling compared to prior methods by filtering out a higher fraction of the off-target reads with its higher precision, 2) improves the recall of prior methods in filtering reads, 3) has a consistently higher throughput than all prior works, and 4) can be accurately applied to target reference lengths that prior methods are unable to be accurately applied while requiring much less (on average
3.7 TargetCall execution time breakdown
We analyzed the execution time breakdown of a pipeline that includes TargetCall as the pre-basecalling filter in Figure 11. We make four key observations. First, LightCall is the bottleneck of the new basecalling pipeline that includes pre-basecalling filtering by consuming
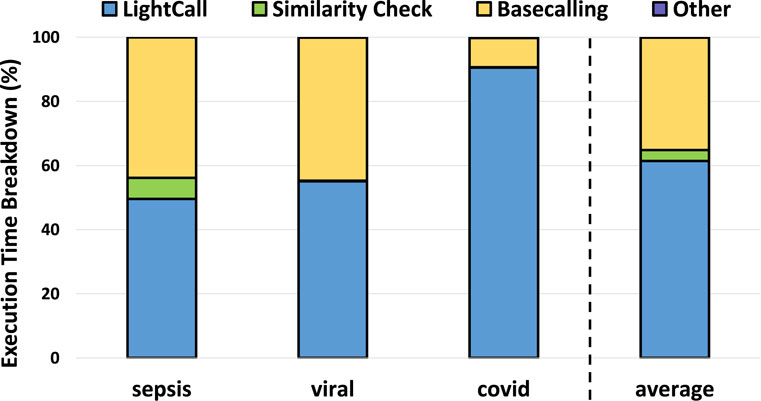
Figure 11. Execution time breakdown of TargetCall.
4 Discussion
We propose TargetCall, a pre-basecalling filtering mechanism for eliminating the wasted computation in basecalling. TargetCall performs lightweight basecalling to compute noisy reads using LightCall and labels these noisy reads as on-target/off-target using Similarity Check. TargetCall eliminates the wasted computation in basecalling by performing basecalling only on the on-target reads. We focus on convolution-based networks for TargetCall architecture for two reasons: (a) matrix multiplication, the core operation in these networks, is highly suitable for hardware acceleration, facilitating improved performance, and (b) the training and inference of RNN and LSTM models involve sequential computation tasks, which pose significant challenges for acceleration on modern hardware such as GPUs and field-programmable gate arrays (FPGAs) (Singh et al., 2024). We evaluate TargetCall for three different genome sequence analysis use cases of pre-basecalling filtering with varying requirements: covid detection, sepsis detection, and viral detection. We show that TargetCall reduces the execution time of basecalling by filtering out most off-target reads, and it is more applicable than the state-of-the-art adaptive sampling methods. We hope that TargetCall inspires future work in pre-basecalling filtering and real-time sequence classification that accelerate other bioinformatics workloads and emerging applications with its high throughput and recall. We explain future work and optimizations that can build upon TargetCall in Section 4 of the Supplementary Material.
Data availability statement
The data presented in the study are deposited in the Zenodo repository with the following DOI accession numbers: https://zenodo.org/records/7334648 (for D1 part 1), https://zenodo.org/records/7402342 (for D1 part 2), https://zenodo.org/records/7335539 (for D2), https://zenodo.org/records/7335525 (for D3), https://zenodo.org/records/7335517 (for D4), https://zenodo.org/records/7334592 (for D5), and https://zenodo.org/records/7335545 (for all the references we used). Further details about these datasets from D1 to D5 are included in the Supplementary Materials (Supplementary Section 2). TargetCall is fully open source at https://github.com/CMU-SAFARI/TargetCall.
Author contributions
MC: conceptualization, investigation, methodology, software, writing–original draft, and writing–review and editing. GS: investigation, software, and writing–review and editing. MA: conceptualization, supervision, and writing–review and editing. CF: supervision and writing–review and editing. JL: writing–review and editing. MS: writing–review and editing. NM: writing–review and editing. CA: supervision and writing–review and editing. OM: supervision and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is partially supported by the Semiconductor Research Corporation (SRC), the European Union’s Horizon Programme for Research and Innovation [101047160 - BioPIM], the Swiss National Science Foundation (SNSF) [200021_213084], and Open access funding by ETH Zurich.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2024.1429306/full#supplementary-material
Footnotes
1Basecalling accuracy is calculated as the ratio of the number of correctly identified bases to the total number of bases in the read, expressed as a percentage.
References
Agarap, A. F. (2018). Deep Learn. Using Rectified Linear Units (ReLU). arXiv. doi:10.48550/arXiv.1803.08375
Alkan, C., Kidd, J. M., Marques-Bonet, T., Aksay, G., Antonacci, F., Hormozdiari, F., et al. (2009). Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 41, 1061–1067. doi:10.1038/ng.437
Alkan, C., Sajjadian, S., and Eichler, E. E. (2010). Limitations of next-generation genome sequence assembly. Nat. Methods 8, 61–65. doi:10.1038/nmeth.1527
Alser, M., Lindegger, J., Firtina, C., Almadhoun, N., Mao, H., Singh, G., et al. (2022). From molecules to genomic variations: accelerating genome analysis via intelligent algorithms and architectures. Comput. Struct. Biotechnol. J. 20, 4579–4599. doi:10.1016/j.csbj.2022.08.019
Alser, M., Rotman, J., Deshpande, D., Taraszka, K., Shi, H., Baykal, P. I., et al. (2021). Technology dictates algorithms: recent developments in read alignment. Genome Biol. 22, 249. doi:10.1186/s13059-021-02443-7
Alvarez-Cubero, M. J., Saiz, M., Martínez-García, B., Sayalero, S. M., Entrala, C., Lorente, J. A., et al. (2017). Next generation sequencing: an application in forensic sciences? Ann. Hum. Biol. 44, 581–592. doi:10.1080/03014460.2017.1375155
Amarasinghe, S. L., Su, S., Dong, X., Zappia, L., Ritchie, M. E., and Gouil, Q. (2020). Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 21, 30. doi:10.1186/s13059-020-1935-5
Ashley, E. A. (2016). Towards precision medicine. Nat. Rev. Genet. 17, 507–522. doi:10.1038/nrg.2016.86
Bao, Y., Wadden, J., Erb-Downward, J. R., Ranjan, P., Zhou, W., McDonald, T. L., et al. (2021). SquiggleNet: real-time, direct classification of nanopore signals. Genome Biol. 22, 298. doi:10.1186/s13059-021-02511-y
Bonito (2022). Available at: https://github.com/nanoporetech/bonito.
Bowden, R., Davies, R. W., Heger, A., Pagnamenta, A. T., de Cesare, M., Oikkonen, L. E., et al. (2019). Sequencing of human genomes with nanopore technology. Nat. Commun. 10, 1869. doi:10.1038/s41467-019-09637-5
Branton, D., Deamer, D. W., Marziali, A., Bayley, H., Benner, S. A., Butler, T., et al. (2008). The potential and challenges of nanopore sequencing. Nat. Biotechnol. 26, 1146–1153. doi:10.1038/nbt.1495
Celik, I. H., Hanna, M., Canpolat, F. E., and Pammi, M. (2022). Diagnosis of neonatal sepsis: the past, present and future. Pediatr. Res. 91, 337–350. doi:10.1038/s41390-021-01696-z
Chin, L., Andersen, J. N., and Futreal, P. A. (2011). Cancer genomics: from discovery science to personalized medicine. Nat. Med. 17, 297–303. doi:10.1038/nm.2323
[Dataset] ONT (2022) MinION Flow Cell, R9.4.1. Available at: https://store.nanoporetech.com/flow-cell-r9-4-1.html.
Dunn, T., Sadasivan, H., Wadden, J., Goliya, K., Chen, K.-Y., Blaauw, D., et al. (2021). SquiggleFilter: an accelerator for portable virus detection. MICRO.
Ellegren, H. (2014). Genome sequencing and population genomics in non-model organisms. Trends Ecol. and Evol. 29, 51–63. doi:10.1016/j.tree.2013.09.008
Firtina, C., and Alkan, C. (2016). On genomic repeats and reproducibility. Bioinformatics 32, 2243–2247. doi:10.1093/bioinformatics/btw139
Firtina, C., Mansouri Ghiasi, N., Lindegger, J., Singh, G., Cavlak, M. B., Mao, H., et al. (2023a). RawHash: enabling fast and accurate real-time analysis of raw nanopore signals for large genomes. Bioinformatics 39, i297–i307. doi:10.1093/bioinformatics/btad272
Firtina, C., Park, J., Alser, M., Kim, J. S., Cali, D. S., Shahroodi, T., et al. (2023b). BLEND: a fast, memory-efficient and accurate mechanism to find fuzzy seed matches in genome analysis. NAR Genomics Bioinforma. 5, lqad004. doi:10.1093/nargab/lqad004
Firtina, C., Soysal, M., Lindegger, J., and Mutlu, O. (2024). RawHash2: mapping raw nanopore signals using hash-based seeding and adaptive quantization. Bioinformatics 40, btae478. doi:10.1093/bioinformatics/btae478
Frei, D., Veekman, E., Grogg, D., Stoffel-Studer, I., Morishima, A., Shimizu-Inatsugi, R., et al. (2021). Erratum to: ultralong Oxford nanopore reads enable the development of a reference-grade perennial ryegrass genome assembly. Genome Biol. Evol. 13, evab203. doi:10.1093/gbe/evab203
Geoghegan, J. L., Douglas, J., Ren, X., Storey, M., Hadfield, J., Silander, O. K., et al. (2021). Real-time genomics for tracking severe acute respiratory syndrome coronavirus 2 border incursions after virus elimination, New Zealand. Emerg. Infect. Dis. 27, 2361–2368. doi:10.3201/eid2709.211097
Gong, L., Wong, C.-H., Idol, J., Ngan, C. Y., and Wei, C.-L. (2019). Ultra-long read sequencing for whole genomic DNA analysis. JoVE. doi:10.3791/58954
Graves, A., Fernández, S., Gomez, F., and Schmidhuber, J. (2006). Connectionist temporal classification: labelling unsegmented sequence data with recurrent neural networks. ICML. doi:10.1145/1143844.1143891
Grumaz, S., Stevens, P., Grumaz, C., Decker, S. O., Weigand, M. A., Hofer, S., et al. (2016). Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 8, 73. doi:10.1186/s13073-016-0326-8
Hu, T., Chitnis, N., Monos, D., and Dinh, A. (2021). Next-generation sequencing technologies: an overview. Hum. Immunol. 82, 801–811. doi:10.1016/j.humimm.2021.02.012
Ioffe, S., and Szegedy, C. (2015). Batch normalization: accelerating deep network training by reducing internal covariate shift. ICML. doi:10.5555/3045118.3045167
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., Sasani, T. A., et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. doi:10.1038/nbt.4060
Kingma, D. P., and Ba, J. (2014). Adam: a method for stochastic optimization. arXiv. doi:10.48550/arXiv.1412.6980
Konishi, H., Yamaguchi, R., Yamaguchi, K., Furukawa, Y., and Imoto, S. (2021). Halcyon: an accurate basecaller exploiting an encoder-decoder model with monotonic attention. Bioinformatics 37, 1211–1217. doi:10.1093/bioinformatics/btaa953
Kovaka, S., Fan, Y., Ni, B., Timp, W., and Schatz, M. C. (2021). Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. Nat. Biotechnol. 39, 431–441. doi:10.1038/s41587-020-0731-9
Levy, S. E., and Myers, R. M. (2016). Advancements in next-generation sequencing. Annu. Rev. Genomics Hum. Genet. 17, 95–115. doi:10.1146/annurev-genom-083115-022413
Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Li, Y., Han, R., Bi, C., Li, M., Wang, S., and Gao, X. (2018). DeepSimulator: a deep simulator for nanopore sequencing. Bioinformatics 34, 2899–2908. doi:10.1093/bioinformatics/bty223
Li, Y., Wang, S., Bi, C., Qiu, Z., Li, M., and Gao, X. (2020). DeepSimulator1. 5: a more powerful, quicker and lighter simulator for nanopore sequencing. Bioinformatics 36, 2578–2580. doi:10.1093/bioinformatics/btz963
Lindegger, J., Firtina, C., Ghiasi, N. M., Sadrosadati, M., Alser, M., and Mutlu, O. (2023). RawAlign: accurate, fast, and scalable raw nanopore signal mapping via combining seeding and alignment. arXiv. doi:10.48550/arXiv.2310.05037
Logsdon, G. A., Vollger, M. R., and Eichler, E. E. (2020). Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614. doi:10.1038/s41576-020-0236-x
Loose, M., Malla, S., and Stout, M. (2016). Real-time selective sequencing using nanopore technology. Nat. Methods 13, 751–754. doi:10.1038/nmeth.3930
Lou, Q., Janga, S. C., and Jiang, L. (2020). “Helix: algorithm/architecture Co-design for accelerating nanopore genome base-calling,” in Proceedings of the ACM International Conference on Parallel architectures and Compilation techniques.
Lu, H., Giordano, F., and Ning, Z. (2016). Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinforma. 14, 265–279. doi:10.1016/j.gpb.2016.05.004
Magi, A., Semeraro, R., Mingrino, A., Giusti, B., and D’Aurizio, R. (2018). Nanopore sequencing data analysis: state of the art, applications and challenges. Brief. Bioinform 19, 1256–1272. doi:10.1093/bib/bbx062
Mokili, J. L., Rohwer, F., and Dutilh, B. E. (2012). Metagenomics and future perspectives in virus discovery. Curr. Opin. Virology 2, 63–77. doi:10.1016/j.coviro.2011.12.004
Munro, R., Wibowo, S., Payne, A., and Loose, M. (2024). Icarust, a real-time simulator for Oxford Nanopore adaptive sampling. Bioinformatics 40, btae141. doi:10.1093/bioinformatics/btae141
Neumann, D., Reddy, A. S., and Ben-Hur, A. (2022). RODAN: a fully convolutional architecture for basecalling nanopore RNA sequencing data. BMC Bioinforma. 23, 142. doi:10.1186/s12859-022-04686-y
Noordijk, B., Nijland, R., Carrion, V. J., Raaijmakers, J. M., de Ridder, D., and de Lannoy, C. (2023). baseLess: lightweight detection of sequences in raw MinION data. Bioinforma. Adv. 3. doi:10.1093/bioadv/vbad017
Pagès-Gallego, M., and de Ridder, J. (2023). Comprehensive benchmark and architectural analysis of deep learning models for nanopore sequencing basecalling. Genome Biol. 24, 71. doi:10.1186/s13059-023-02903-2
Payne, A., Holmes, N., Clarke, T., Munro, R., Debebe, B. J., and Loose, M. (2020). Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 39, 442–450. doi:10.1038/s41587-020-00746-x
Perešíni, P., Boža, V., Brejová, B., and Vinař, T. (2021). Nanopore base calling on the edge. Bioinformatics 37, 4661–4667. doi:10.1093/bioinformatics/btab528
Poplin, R., Chang, P.-C., Alexander, D., Schwartz, S., Colthurst, T., Ku, A., et al. (2018). A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987. doi:10.1038/nbt.4235
Quail, M. A., Smith, M., Coupland, P., Otto, T. D., Harris, S. R., Connor, T. R., et al. (2012). A tale of three next generation sequencing platforms: comparison of ion torrent, pacific biosciences and illumina MiSeq sequencers. BMC Genomics 13, 341. doi:10.1186/1471-2164-13-341
Rang, F. J., Kloosterman, W. P., and de Ridder, J. (2018). From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 19, 90. doi:10.1186/s13059-018-1462-9
Sands, K., Carvalho, M. J., Portal, E., Thomson, K., Dyer, C., Akpulu, C., et al. (2021). Characterization of antimicrobial-resistant gram-negative bacteria that cause neonatal sepsis in seven low- and middle-income countries. Nat. Microbiol. 6, 512–523. doi:10.1038/s41564-021-00870-7
Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C., and Mutlu, O. (2019). Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief. Bioinform 20, 1542–1559. doi:10.1093/bib/bby017
Shahroodi, T., Singh, G., Zahedi, M., Mao, H., Lindegger, J., Firtina, C., et al. (2023). “Swordfish: a framework for evaluating deep neural network-based basecalling using computation-in-memory with non-ideal memristors,” in Proceedings of the 56th Annual IEEE/ACM International Symposium on Microarchitecture, 1437–1452.
Sims, D., Sudbery, I., Ilott, N. E., Heger, A., and Ponting, C. P. (2014). Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 15, 121–132. doi:10.1038/nrg3642
Singh, G., Alser, M., Denolf, K., Firtina, C., Khodamoradi, A., Cavlak, M. B., et al. (2024). RUBICON: a framework for designing efficient deep learning-based genomic basecallers. Genome Biol. 25, 49. doi:10.1186/s13059-024-03181-2
Treangen, T. J., and Salzberg, S. L. (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. doi:10.1038/nrg3117
V, A., and Kiran, A. G. (2022). Synthnet: a skip connected depthwise separable neural network for novel view synthesis of solid objects. Results Eng. 13, 100383. doi:10.1016/j.rineng.2022.100383
Wan, Y. K., Hendra, C., Pratanwanich, P. N., and Göke, J. (2021). Beyond sequencing: machine learning algorithms extract biology hidden in nanopore signal data. Trends Genet. 38, 246–257. doi:10.1016/j.tig.2021.09.001
Wang, Y., Zhao, Y., Bollas, A., Wang, Y., and Au, K. F. (2021). Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365. doi:10.1038/s41587-021-01108-x
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford nanopore sequencing. Genome Biol. 20, 129. doi:10.1186/s13059-019-1727-y
Xu, Z., Mai, Y., Liu, D., He, W., Lin, X., Xu, C., et al. (2021). Fast-bonito: a faster deep learning based basecaller for nanopore sequencing. Artif. Intell. Life Sci. 1, 100011. doi:10.1016/j.ailsci.2021.100011
Zhang, H., Li, H., Jain, C., Cheng, H., Au, K. F., Li, H., et al. (2021a). Real-time mapping of nanopore raw signals. Bioinformatics 37, i477–i483. doi:10.1093/bioinformatics/btab264
Zhang, Z., Park, C. Y., Theesfeld, C. L., and Troyanskaya, O. G. (2021b). An automated framework for efficiently designing deep convolutional neural networks in genomics. Nat. Mach. Intell. 3, 392–400. doi:10.1038/s42256-021-00316-z
Keywords: nanopore sequencing, basecalling, deep learning, filtering, efficiency
Citation: Cavlak MB, Singh G, Alser M, Firtina C, Lindegger J, Sadrosadati M, Mansouri Ghiasi N, Alkan C and Mutlu O (2024) TargetCall: eliminating the wasted computation in basecalling via pre-basecalling filtering. Front. Genet. 15:1429306. doi: 10.3389/fgene.2024.1429306
Received: 07 May 2024; Accepted: 30 September 2024;
Published: 28 October 2024.
Edited by:
Nathan Olson, National Institute of Standards and Technology (NIST), United StatesReviewed by:
Yu-Wei Wu, Taipei Medical University, TaiwanXuechun Xu, Royal Institute of Technology, Sweden
Luotong Wang, Peking University, China
Copyright © 2024 Cavlak, Singh, Alser, Firtina, Lindegger, Sadrosadati, Mansouri Ghiasi, Alkan and Mutlu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meryem Banu Cavlak, bWJhbnVjYXZsYWtAZ21haWwuY29t; Can Firtina, ZmlydGluYWNAZXRoei5jaA==; Onur Mutlu, b211dGx1QGV0aHouY2g=