 Xin Lai
Xin Lai Min Liu1
Min Liu1 Yuqian Liu
Yuqian Liu Jiayin Wang
Jiayin Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Genet. , 04 December 2024
Sec. Computational Genomics
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1400228
Open chromatin regions (OCRs) play a crucial role in transcriptional regulation and gene expression. In recent years, there has been a growing interest in using plasma cell-free DNA (cfDNA) sequencing data to detect OCRs. By analyzing the characteristics of cfDNA fragments and their sequencing coverage, researchers can differentiate OCRs from non-OCRs. However, the presence of noise and variability in cfDNA-seq data poses challenges for the training data used in the noise-tolerance learning-based OCR estimation approach, as it contains numerous noisy labels that may impact the accuracy of the results. For current methods of detecting OCRs, they rely on statistical features derived from typical open and closed chromatin regions to determine whether a region is OCR or non-OCR. However, there are some atypical regions that exhibit statistical features that fall between the two categories, making it difficult to classify them definitively as either open or closed chromatin regions (CCRs). These regions should be considered as partially open chromatin regions (pOCRs). In this paper, we present OCRClassifier, a novel framework that combines control charts and machine learning to address the impact of high-proportion noisy labels in the training set and classify the chromatin open states into three classes accurately. Our method comprises two control charts. We first design a robust Hotelling T2 control chart and create new run rules to accurately identify reliable OCRs and CCRs within the initial training set. Then, we exclusively utilize the pure training set consisting of OCRs and CCRs to create and train a sensitized T2 control chart. This sensitized T2 control chart is specifically designed to accurately differentiate between the three categories of chromatin states: open, partially open, and closed. Experimental results demonstrate that under this framework, the model exhibits not only excellent performance in terms of three-class classification, but also higher accuracy and sensitivity in binary classification compared to the state-of-the-art models currently available.
Open chromatin regions (OCRs) that are accessible to DNA regulatory elements, play a crucial role in cellular activities and gene expression (Thurman et al., 2012; Yao et al., 2015; Klemm et al., 2019). It is reported that OCR patterns are associated with many complex traits and diseases. For example, several cancers are observed unique patterns of open chromatin regions, which are considered valuable in underlying cancer development through epigenetic mechanisms. It is also suggested as potential biomarkers in cancer early-diagnosis (Ivanov et al., 2015; Flavahan et al., 2017). Thus, detecting OCRs and identifying their patterns is a basic computational problem in epigenetic research and their clinical applications.
The traditional methods for identifying OCRs (ChIP-Seq (Johnson et al., 2007), DNase-seq (Crawford et al., 2006), MNase-seq (Schones et al., 2008), FAIRE-seq (Giresi et al., 2007), ATAC-seq (Buenrostro et al., 2015)) are all complex experimental processes. Until recent decade, detecting OCRs from plasma cell-free DNA sequencing data becomes popular. Plasma cell-free DNA (cfDNA) molecules are short fragments generated through a non-random procedure (Ivanov et al., 2015; Gai and Sun, 2019; Van der Pol and Mouliere et al., 2019; Lo et al., 2021; An et al., 2023). Recent studies conducted by Snyder (Snyder et al., 2016) and Ulz (Ulz et al., 2016) have shown a correlation between the characteristics of cfDNA fragments and gene expression levels, which provide new insights on detecting OCRs. Snyder first proposed the windowed protection score (WPS) to established a whole-genome nucleosome landscape, which was able to transform the cfDNA sequencing data into waveforms whose peaks corresponded to nucleosomes (Snyder et al., 2016). Ulz presented EP (Expression Prediction) algorithm (Ulz et al., 2016), which focused on inferring gene expression levels by low sequencing coverage in OCRs. Sun found that the characteristics of cfDNA fragment distribution can be reflected by the differentially phased cfDNA fragment end signals (Sun et al., 2019). Based on these pioneering works, Wang proposed OCRDetector, which among the first approach introduces a machine learning framework (Wang et al., 2021). However, it could hardly overcome the limitations of artificially constructed features and it didn’t consider the noisy labels. Subsequently, Ren introduced OCRFinder (Ren et al., 2023), which first leverages deep learning for feature extraction and incorporates ensemble techniques and semi-supervised strategies to tackle noisy label learning. This approach has performed impressive results in various evaluations but the performance of the model also be affected by the high proportion of noisy labels in the training sets. Jin proposed utilizing OCF values and SVM classification model for predicting tissue injury (Jin et al., 2023), but this study relies on biological experiments to obtain known tissue-specific open chromatin regions, hence it is not possible to directly analyze the genome-wide chromatin accessibility using cfDNA-seq data. Clearly, current methods are limited to distinguishing between chromatin open and closed regions.
However, cfDNA data represents a mixed sample from multiple tissues, the chromatin states cannot be simplified as solely fully open or fully closed. Actually, in real sequencing data, we often observe the regions that seem partially open. We may call them partially open chromatin regions (pOCRs). For those pOCRs, none of the existing approach, whatever the machine learning-based ones or deep learning-based ones can achieve accurate detection. There are several reasons for this: the training data for the existing learning models only consists of two labels, where the genomic regions with active gene expression are categorized as open chromatin regions, while the regions with silent gene expression are categorized as non-open chromatin regions. It is a computational challenge to correctly mark the partially open chromatin regions, because those pOCRs are randomly assigned as either OCRs or non-OCRs. In addition, the threshold differentiate OCRs and pOCRs varies across samples, which may be influenced by sequencing coverage, local distribution of read depths, etc. Thus, a self-adaptive method is needed to identify three-class regions using training datasets that only contain two-class labels.
The patterns of cfDNA fragments is a random variable that varies along the genomic locus. If we map the reference genome to a X-axis, map the WPS scores to a Y-axis, then the patterns become a function on pseudo-time series. Statistical process control (SPC) is a family of methods that capture the statistical characteristics on time-series data which is a potential way of identifying the thresholds and differentiating the pOCRs from OCRs and non-OCRs. The three states of chromatin correspond to three distinct data distributions of cfDNA fragment features. Thus, let non-OCRs define as the normal state in the statistical process, then both the OCRs and pOCRs can be defined as abnormal states with different abnormal patterns, aligning precisely with the in-control and out-of-control states of a control chart. The computational problem switches to design a control chart that is able to efficiently report the out-of-control states. CUSUM is a control chart that has been widely used in a range of healthcare settings, from describing the learning curves of surgical or procedural skills (Waller and Connor, 2009; Je et al., 2015; Kim et al., 2015), to clinical audits (Cockings et al., 2006; Calsina et al., 2011), and quality-assurance studies (Novick et al., 2006; Hasegawa et al., 2017). Hotelling’s T2 is another control chart that has been widely used for measuring and monitoring biological and biomedical investigations (Lv et al., 2014; Ercan et al., 2015). To better use the control charts to detect open chromatin regions, we need to re-design it a little bit. For example, the T2 chart, assume that the in-control (IC) group is the sole population used for establishing a decision boundary. Nevertheless, this assumption has limited the advancement of more efficient control chart techniques that can leverage the available out-of-control (OC) information. A sensitized T2 chart (Park and Kim, 2019) is proposed later, wherein OC observations extracted from historical records were classified as “predefined OC,” while those arising from novel fault types not encountered previously were identified as “undefined OC.” Based on this, let OCR being the predefined OC and pOCR as undefined OC.
However, the classification of control charts is still subject to certain errors based on control limits. Therefore, to improve the recognition of multiple states of chromatin openness, this study then uses a CNN-based model under the framework of confident learning (Curtis et al., 2021). Nowadays, an increasing number of CNN-based models are being used in various fields. If an efficient encoding method is available, deep learning models have the capability to automatically extract the distinctive fragmentation patterns of cfDNA molecules at OCRs without requiring manual intervention. Consequently, employing a CNN-based model for OCR estimation is a viable and practical approach.
In this article, we proposed a novel computational approach, OCRClassfier, to accurately detect open chromatin regions together with the partially open chromatin regions from cfDNA sequencing data. A novel sensitized T2 control chart is designed to identify the candidate OCRs. This control chart integrates a MEWMA control chart with the OCRFinder noise-tolerance model. The control chart we designed considers two types of probabilities the first of which is derived from a MEWMA control chart, enabling the detection of mean shifts in any direction within the process. This aspect ensures robust monitoring capabilities. The second probability is based on a noise-tolerance machine learning method OCRFinder, effectively distinguishing between OC and IC data. Combining these probabilities enhances the accuracy and reliability of the control chart in accurately identifying the deviations in the process. This new control chart aims to effectively monitor chromatin states. By training the sensitized T2 control chart using data from OCRs and non-OCRs, we obtained a control chart capable of identifying OCRs, non-OCRs, and pOCRs. Thus, we transformed the original training dataset with only two labels into three categories. In order to obtain an improved control chart, we have also devised a robust Hotelling T2 control chart specifically designed to identify OCRs and non-OCRs. This helps in filtering out noisy labels from the training set and then can reduce the impact caused by such noisy labels. Additionally, it is worth mentioning that the obtained three-class training set still contains a small amount of noisy labels. Therefore, we conducted further training by using a neural network model under the confident learning strategy to minimize the impact of noisy labels.
In the WPS waveform, peaks represent nucleosome positions, whereas peaks disappear in OCRs. In terms of sequencing coverage, there was a significant decrease in coverage of OCRs. In the end signals, the head and tail peaks represent both sides of the nucleosomes, and the end signals are also lost in OCRs.
Here, we present a three-stage multiclass classification algorithm that introduces the idea of multivariate control charts and machine learning. The three stages are data pre-processing, control charts designing and model co-teaching. The second step consists of two control charts: robust Hotelling T2 control chart and sensitized T2 control chart. The data pre-processing converts cfDNA-seq data into vectors and matrixes suitable for control charts and two-dimensional images suitable for neural networks, respectively. The second stage first uses robust Hotelling T2 control chart with minimum regularized covariance determinant (MRCD) to reduce the scale of noisy labels on the training set and then designs sensitized T2 control chart that combines multivariate exponentially weighted moving average (MEWMA) and OCRFinder classification algorithm to give the model an initial recognition ability. In the last stage, we use confident learning strategy to avoid the impact of noisy labels and classify the three open states of chromatin in whole genome regions. The framework of OCRClassifier model is shown in Figure 1.
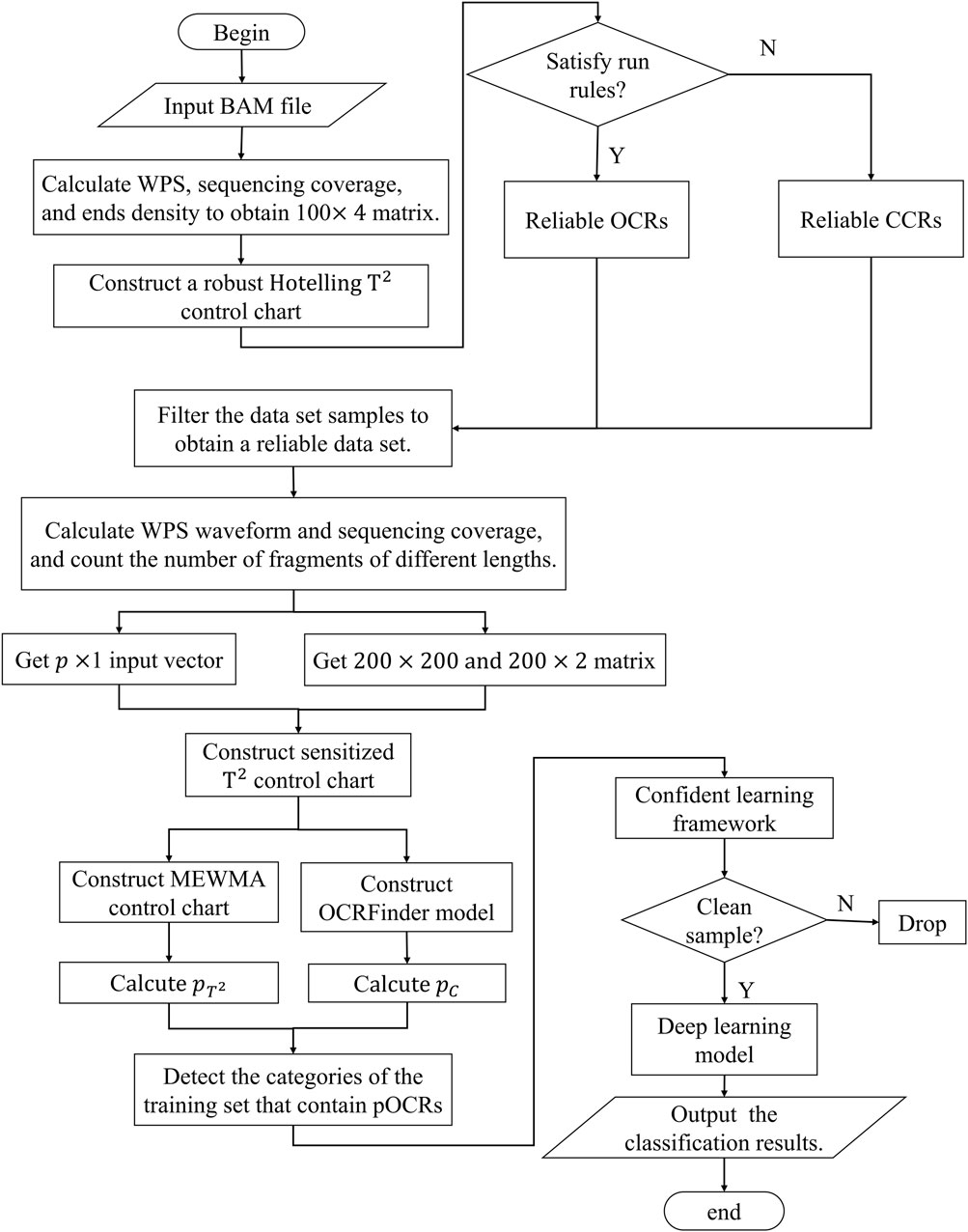
Figure 1. The flow chart of OCRclassifier.
The cfDNA-seq data in fastq format is processed by BWA and Samtools. The input data of the control charts is not the same as the input data of the deep learning model.
The human genome is divided into multiple intervals (Wang et al., 2021), each containing 20 kilobase pairs (kbp). Subsequently, for each of these intervals, the WPS waveform, sequencing coverage, Uend signal, and Dend signal are computed. As mentioned above, the size of a single nucleosome is about 167 base pairs (bp), while the linked DNA connecting the nucleosome is about 20 bp in size (Struhl and Segal, 2013). Features showed strong periodic patterns, with one nucleosome per period of approximately 190 bp in length (Snyder et al., 2016). Thus, a sliding window of 200 bp size was defined in each 20 kbp interval, and the averages of the four features within the window were calculated to obtain a 100 × 4 input matrix as the input data of Hotelling T2 control chart.
In order to apply the sensitized T2 control chart, the standard deviation of the windowed average values of the WPS and sequencing coverage features is calculated for each sample, the calculation process is shown in Figure 2. By doing so, the cfDNA-reads data is transformed into a vector representation. This enables us to assess the variability within the respective windows and gain insights into the fluctuation characteristics and dispersion patterns exhibited by these two specific attributes.
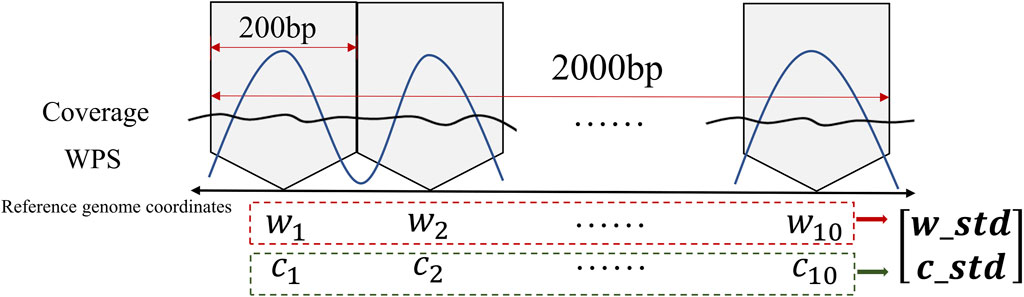
Figure 2. Illustration of input data processing for MEWMA control charts.
For deep learning model, the input data type is the same as OCRFinder. We convert cfDNA-reads data into two-dimensional matrixes
Let
Where
In the problem of detecting OCRs in training sets, the number of outliers in the observed values may be large, which may have a great impact on the mean vector and covariance matrix, resulting in biased results. Thus, we use the MRCD estimation to construct a robust Hotelling T2 statistic (Formula 2).
Here,
We assume that the distribution of
Thus
At this stage, we have derived robust Hotelling T2 statistics and established control limits based on the observations. When the statistic surpasses the control limit, an alarm signal is triggered. Conversely, if all the statistics fall within the control limit, the observations are considered to be in a normal state. Based on this, we can estimate the reliable OCRs and non-OCRs in the training set to reduce noise.
Because of the unique biological characteristics of biological data, OCRs cannot be assumed to occur once the robust Hotelling T2 control chart generates an alarm signal. Therefore, in order to effectively detect OCRs to obtain reliable training sets, we proposed three run rules for the detection of OCRs combined with Western Electric Rules (Cheng and Chou, 2008), which represent three possible cases of OCRs.
(1) Three or more consecutive observation points exceeded the control limit. After analyzing the length of the OCRs via ATAC-seq and DNase-seq, it was discovered that approximately 50% of the regions exceeded 300 bp in size, while over 25% exceeded 600 bp (Wang et al., 2021). For this study, a window size of 200 bp was selected, with each observation point representing that size. Specifically, two consecutive observations (400 bp in total) above the control limit were deemed to indicate an OCR. However, to account for noise interference in the original data, the error tolerance interval was set to one nucleosome size (approximately 167 bp). Consequently, OCR was defined as an area where three or more consecutive observations surpassed the control limits such as Figure 3A, based on the above considerations.
(2) The first or last point is outside the control limit. During data preprocessing, we truncated the reference genome into multiple segments. To detect OCRs that span across two segments, we established the criterion that if the first or last observation point in any monitored group exceeded the control limit, we would consider the region between the two observation points before and after that point (a total of 600 bp) as OCRs (Figure 3B).
(3) The number of points between two consecutive observations exceeding the control limit is less than three. The paper (Khoo, 2003) proposes two criteria for identifying out-of-control states: 1) If two out of the three most recent samples fall above the UCL or below the LCL on the control chart, the process is considered to be in an OC state. 2) If three out of the four most recent samples fall above the UCL or below the LCL on the control chart, the process is deemed to be in an OC state. Based on these criteria, we extended the analysis to include intervals with less than three points between adjacent points exceeding the control limits as indicative of an out-of-control state (Figure 3C).
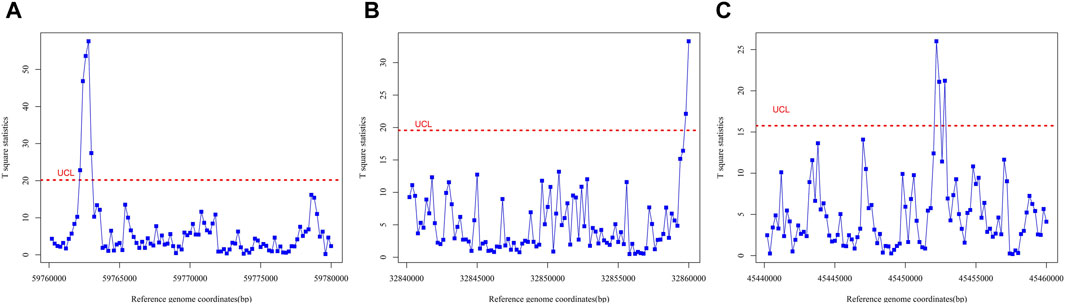
Figure 3. Hotelling T2 control charts on different reference genome fragments, representing each of the three cases in which open chromatin regions appear. Red lines are control limits. (A) Hotelling T2 control chart on reference genome fragment from 59760000 to 59780000, representing the occurrence of the OCR in run rule 1. (B) Hotelling T2 control chart on reference genome fragment from 32840000 to 32860000, representing the occurrence of the OCR in run rule 2. (C) Hotelling T2 control chart on reference genome fragment from 45440000 to 45460000, representing the occurrence of the OCR in run rule 3.
By adhering to the aforementioned run rules, the robust Hotelling T2 control chart can effectively identify chromatin open regions and closed regions across the entire genome, thereby achieving the objective of filtering out noise labels from the training datasets. However, it is important to note that the robust Hotelling T2 control chart can only identify two states, namely OCRs and non-OCRs, and is unable to distinguish pOCRs. In order to accurately classify samples that contain a mixture of partially open states, we propose to use a sensitized T2 control chart specifically tailored to this scenario.
The monitoring statistic of the sensitized T2 control chart combines the probabilities from a multivariate control chart and a noise-tolerance classification model respectively and can be calculated as Formula 6:
where
The first part of the monitoring statistic (i.e.,
where
Here,
where
The second part of the monitoring statistic (i.e.,
where
Now we return to Eq. 6. Here,
Because the distribution of the sensitized T2 monitoring statistic is unknown, the control limit (CL) is determined from a percentile value estimated by bootstrapping. To enhance the effectiveness of identifying pOCRs, we have fine-tuned the pre-determined control limits by multiplying them with the factor of mixture ratio. This approach allows for a more accurate classification of the sample categories. By accommodating the mixture ratio, we can fully utilize the characteristics of partially open states and thus improve the classification performance of the training set.
Although sensitized T2 control chart can classify OCRs, non-OCRs, and pOCRs to a certain extent in the training set, there may still be a few noisy labels in the classification results. In order to obtain more accurate classification results, we have introduced a machine learning model based on the confident learning framework (Curtis et al., 2021) after the sensitized T2 control chart. The confident learning framework can be divided into the following three steps: 1) estimating the joint distribution of noisy labels and true labels; 2) identifying erroneous samples and filtering them out; 3) adjusting the weights of the sample classes and retraining the model. In order to address the issue of noisy label overfitting, we have implemented a co-teaching approach (Han et al., 2018). Compared to training with noisy labels using a single model, this collaborative training method effectively reduces the overfitting impact of incorrect labels. This involves training two identical models and utilizing the clean data to guide each other during the back-propagation process. Specifically, in each iteration (e.g., the kth iteration), model A utilizes a clean dataset
In this section, we convert the cfDNA-seq data into image-based inputs. Our model architecture comprises several key components, including a one-dimensional convolutional layer, a max-pooling layer, a bidirectional LSTM layer, another one-dimensional convolutional layer, another max-pooling layer, and finally a fully connected layer. In this paper, the deep learning model is called ConvLSTM. The focus of this study is on how to design a noisy label learning algorithm based on deep learning for OCRs estimation rather than feature extraction, the selection of deep learning models is not considered in this paper.
The OCRClassifier algorithm is designed to accurately classify data that contains a mixture of the third category labels, while only two labels are marked. The training dataset, obtained after the application of the robust Hotelling T2 control chart filtering, exclusively consists of reliable OCRs and non-OCRs data. This dataset is used to train a sensitized T2 control chart that is capable of recognizing three categories: chromatin open regions, partially open regions, and closed regions. For dataset including pOCRs, the sensitized T2 control chart can reassign labels and then the co-teaching model will produce the final accurate classification results.
The cfDNA sequencing data of healthy individuals (IH01, IH02, BH01) used in this study were provided by Snyder (Snyder et al., 2016). The sequenced reads were aligned to the GRCh37 human reference genome. The sequencing coverage was 96-105x, with approximately 15–16 million sequencing fragments (Snyder et al., 2016). The OCRs of the hematopoietic lineage used in this study were obtained from the ENCODE database, specifically from the results of ATAC-seq and DNase-seq experiments. The housekeeping genes file, ATAC-seq experimental results, and DNase-seq experimental results were all obtained from Wang’s study (Wang et al., 2021).
In order to evaluate the performance of our algorithm, we adopt a similar approach as OCRFinder, relying on known gene expression levels or chromatin accessibility levels. For the initial training and test sets, we adopt the same selection approach as OCRFinder, chromosomes 2-7 are used for training, and chromosome 1 is used for testing. The OCR samples in initial training set are from housekeeping genes and non-OCRs samples are obtained by non-genetic regions. Specifically, the training set is filtered by the robust Hotelling T2 control charts based on MRCD estimation, and we get 800 OCR samples through this process. The principle of filtering is that if the OCR sample in the training set overlaps with the open area obtained by the robust Hotelling T2 control chart, the OCR sample is retained and others will be filtered out. The same filtering rules are applied to the non-OCR samples. Among these 800 OCR samples, we selected 400 to serve as OC information for training the sensitized T2 control chart. Additionally, we chose 1,000 samples from the filtered non-OCR samples to provide IC information for training the sensitized T2 control chart. Due to the unavailability of clearly labeled data for pOCRs, we simulate the required data
where
For test set, we also selected highly expressed housekeeping genes as OCR samples (class 1) and non-genetic regions without gene expression as non-OCR samples (class 0) and simulate the pOCR samples (class 2). To exclude gene-specific chance, the results of ATAC-seq and DNase-seq experiments in hematopoietic lineage cells are combined. We conducted comparison experiments on three test sets: HK_TSS, Hematopoietic_Lineage_ATAC, Hematopoietic_lineage_DNase.
We used the accuracy (ACC), recall (REC), precision (PRE), F1 score (F1), area under the receiver operating characteristic curve (AUC) and area under the precision-recall curve (AUPR) as the criteria for the performance evaluation.
During model training, we used the ConvLSTM network and selectd Adam optimizer (Kingma and Ba, 2014). The learning rate was set to 1e-4, the batch size was 128, and the training epoch was 150, with an additional warm-up training epoch of 10. It is important to note that the reported results in this paper are the average of five independent experiments, each initialized with a different random seed.
Due to the dynamic nature of open chromatin regions in tissue-specific contexts, it is challenging to obtain a large number of reliable labeled samples for model training. OCRFinder is a noise-tolerance approach and has performed impressive results in various evaluations. However, it is observed that the performance of current noisy label learning methods tends to diminish as the proportion of noisy labels in the training set increases. Thus, the performance of the OCRFinder is still attenuated by the proportion of noisy labels in the training sets. In this study, we utilize the robust Hotelling T2 control chart to further filter out the noisy labels in the initial training dataset and then enhance the performance of the noise-tolerance model.
As mentioned earlier, the original training dataset used for OCRFinder was filtered through robust Hotelling T2 control chart, resulting in 800 positive samples. To ensure a balanced dataset, an equal number of negative samples are also retained, resulting in a training set comprising a total of 1,600 samples. After retraining OCRFinder with the filtered training dataset, as shown in Figure 4, there has been an improvement in the model’s performance. Table 1 shows that the retrain model outperforms OCRFinder in both AUC and AUPR on each test set. From this perspective, it is evident that the robust Hotelling T2 control chart is capable of performing OCR recognition on the training dataset, thereby reducing noise percentage in the initial training set. This dual effect allows for an improved performance of the OCRFinder model in OCR recognition while also providing assurance for the training of a three-class sensitized T2 control chart recognition.
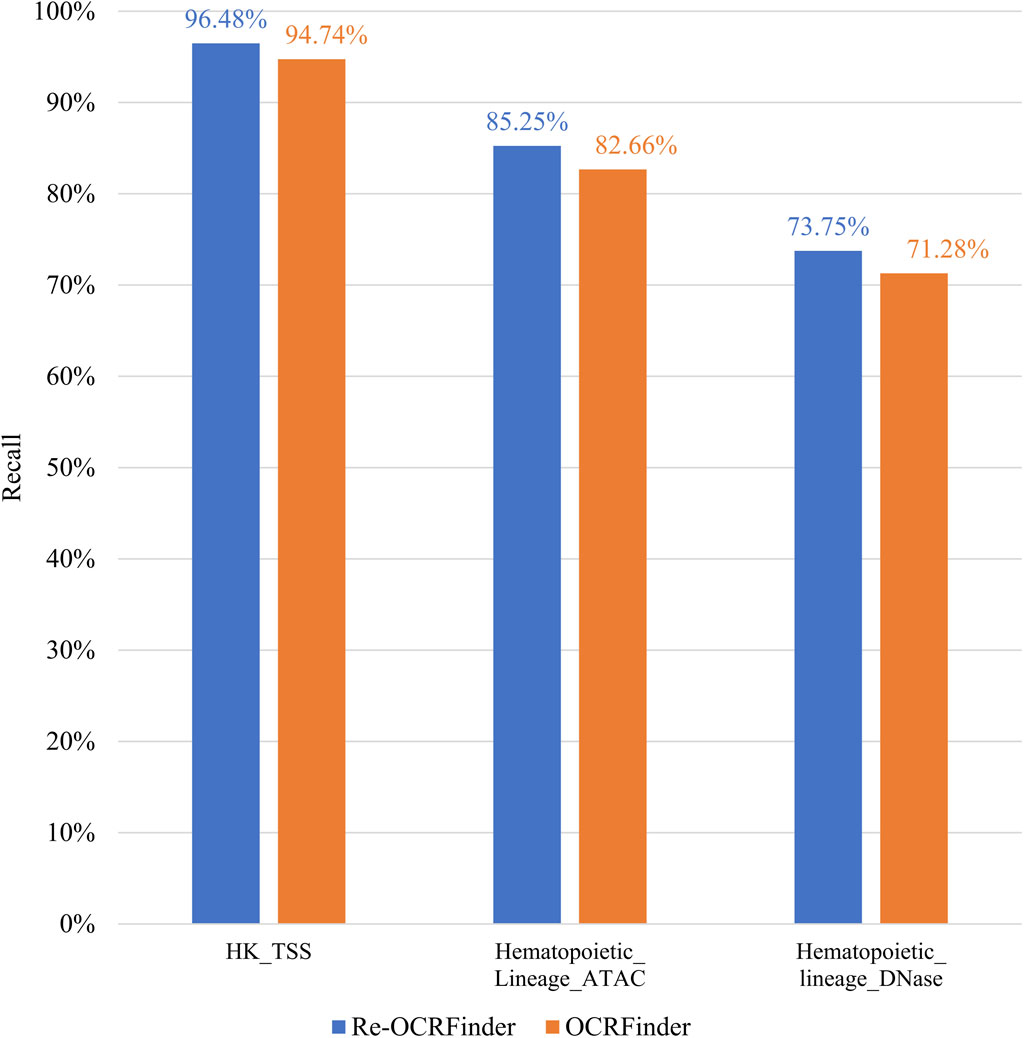
Figure 4. The comparison on sensitivity between retrained OCRFinder and OCRFinder on different testing sets.
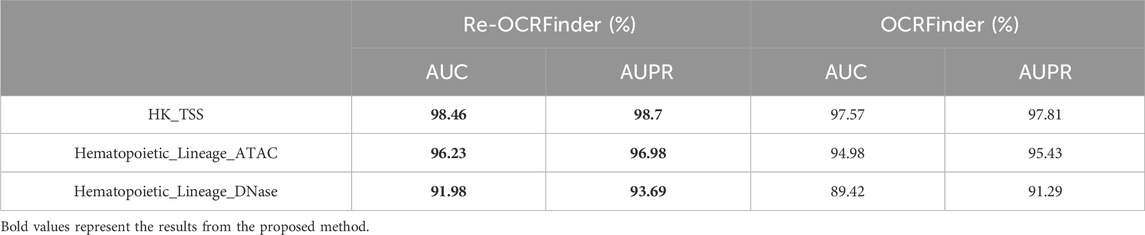
Table 1. The AUC and AUPR values of retrained OCRFinder and OCRFinder on different testing sets.
To verify the detection capability analysis of sensitized T2 control charts on partially open chromatin regions, we compared the classification results of EWMA, OCRFinder, and the designed sensitized T2 control chart. Traditional multivariate control charts can only identify two states: out-of-control and in-control. When the statistic exceeds the control limit, it is considered out-of-control, while it is considered in-control when the statistic is below the control limit. Similarly, the MEWMA multivariate control chart can only distinguish between OCRs (out-of-control) and non-OCRs (in-control), but it cannot differentiate pOCRs.
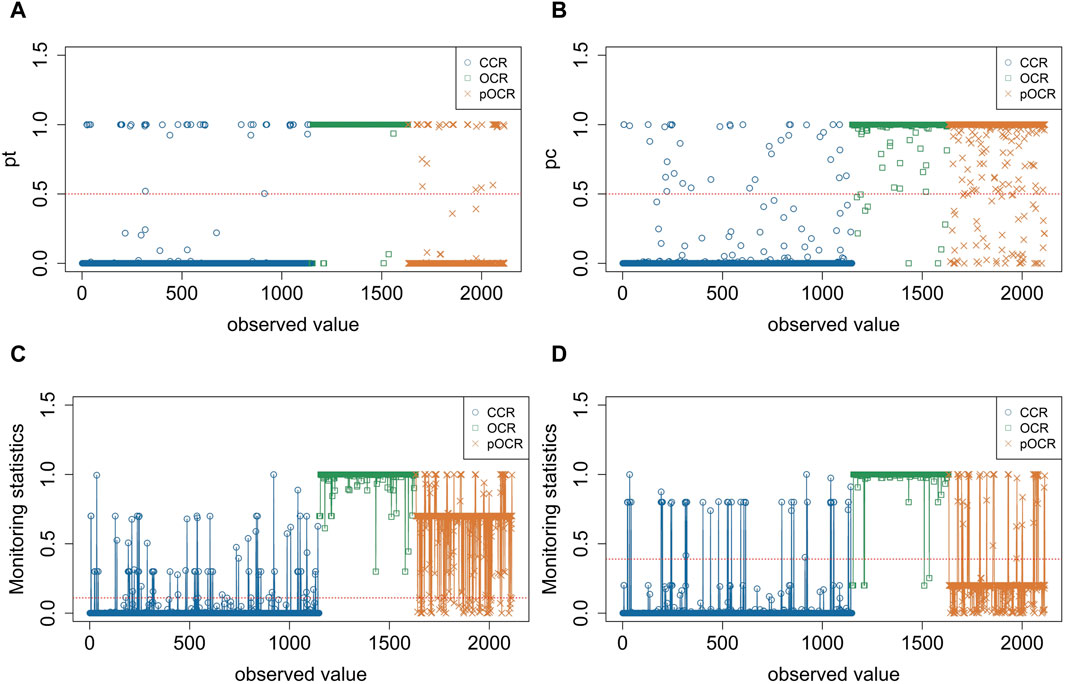
Figure 5. Performance of sensitized T2control chart. (A) MEWMA estimates the probability distribution of samples belonging to the open chromatin state. (B) OCRFinder estimates the probability distribution of samples belonging to the open chromatin state. (C) Statistical monitoring of sensitized T2 control chart (
To determine the optimal value of
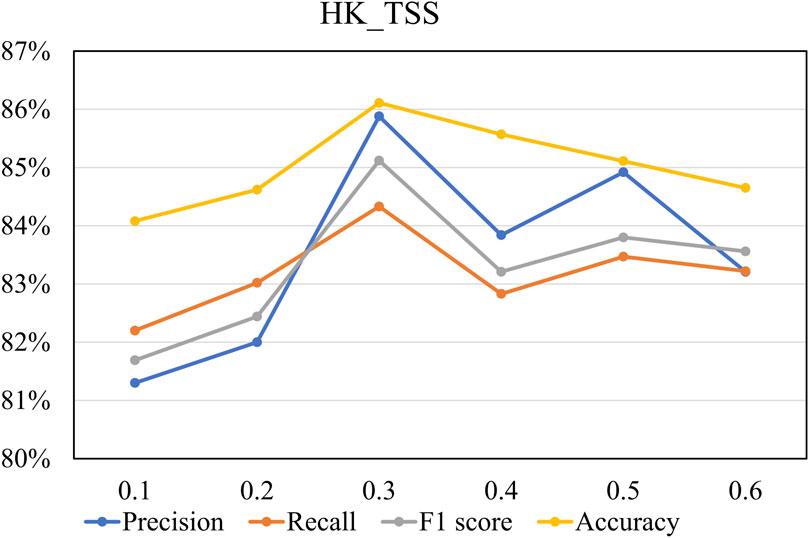
Figure 6. The performance on the HK_TSS dataset under different
The primary objective of our study is to thoroughly evaluate the performance of the OCRClassifier algorithm using diverse reference datasets. In Figure 7, we present an extensive analysis of the OCRClassifier’s performance specifically on the HK_TSS dataset. The results reveal that the OCRClassifier has exceptional performance in identifying three essential categories: open chromatin regions, partially open regions, and closed chromatin regions. In four evaluation criterions, the OCRClassifier achieves the level above 84%. Moreover, we also provide the results from the OCRClassifier without the confident learning (OCRClassifier_initinal). Notably, the OCRClassifier outperforms OCRClassifier_initinal, exhibiting improvements of up to 3 percentage points across all evaluation metrics, demonstrating that incorporating confident learning significantly enhances the overall performance of the algorithm.
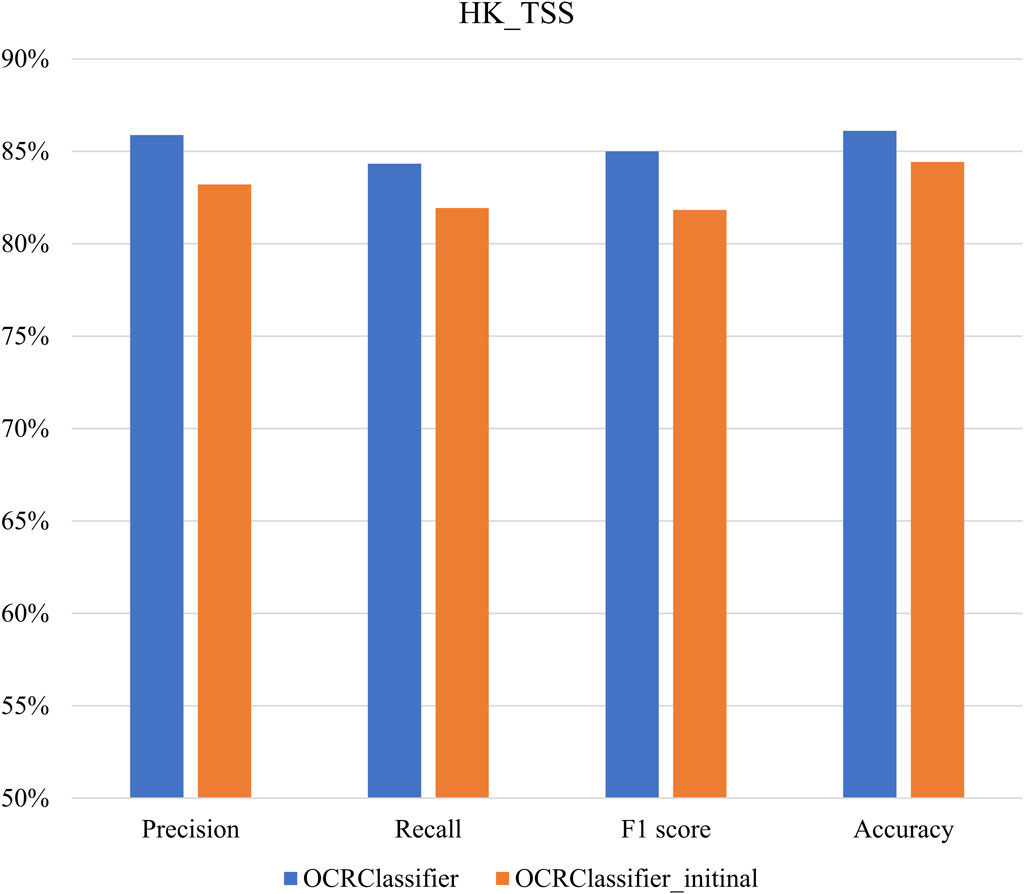
Figure 7. The performance of OCRClassifier on the HK_TSS dataset.
Table 2 presents a comprehensive overview of the performance of the OCRClassifier algorithm across different datasets. From the table, it is evident that the OCRClassifier consistently outperforms the OCRClassifier_initial in various metrics. Additionally, the HK_TSS dataset exhibits the highest model performance, followed by the Hematopoietic_Lineage_ATAC dataset, while the Hematopoietic_Lineage_DNase dataset lags behind. These results are consistent with established biological findings.
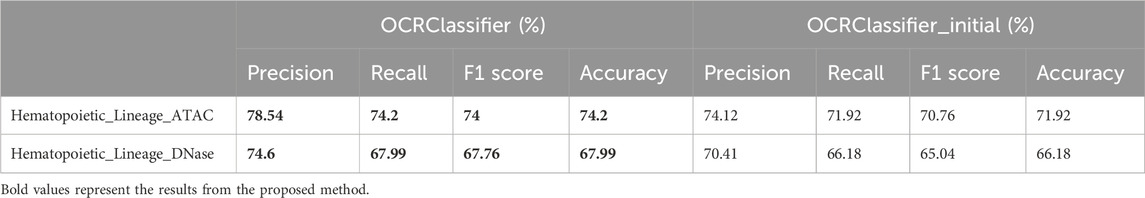
Table 2. The performance of the OCRClassifier algorithm across different datasets.
By varying the magnitude of the mixture ratio, the degree of openness of pOCRs can be altered. To investigate the impact of pOCRs openness on OCRClassifier model classification performance, we increased the values of the mixture ratio from 0.55 to 0.85. Then the relationship between pOCRs openness and model classification performance can be obtained. In Figure 8, the degree of openness of pOCRs is indicated by the mixture ratio. It could be observed that the variation of pOCR openness has no significant impact on OCRClassifier’s performance, suggesting the robustness of OCRClassifier. It should be noted that the model’s classification performance slightly decreased from 0.8 to 0.85. This could be the reason that the degree of openness of pOCRs at 0.85 becomes quite similar to that of OCRs, making it challenging for the sensitized T2 control chart to differentiate between these two states during the training set partitioning process.
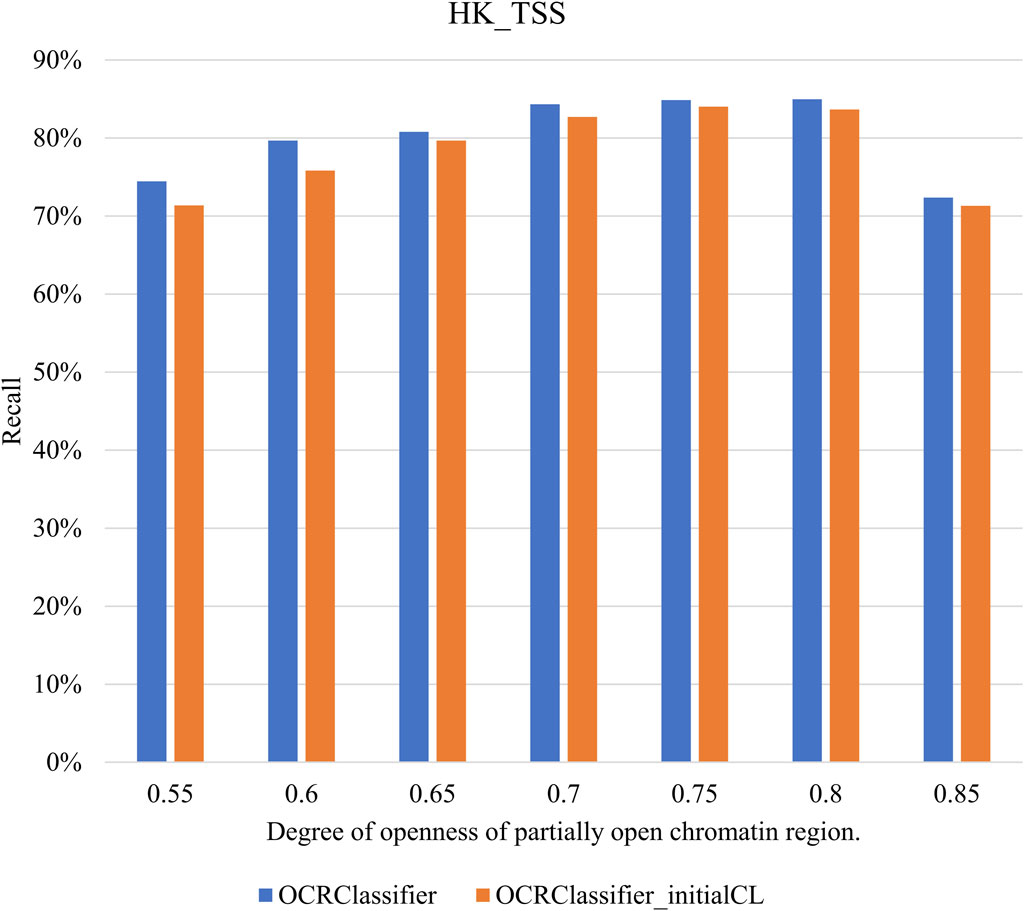
Figure 8. The classification performance under different mixture ratios on the HK_TSS dataset.
Furthermore, this study made fine adjustments to the control limits of the sensitized T2 control chart. As depicted in Table 3, OCRClassifier‘s performance improved after these adjustments. Because pOCRs are positioned in an intermediate state, the sensitized T2 control chart becomes more effective in distinguishing pOCRs by refining the control limits, thereby enhancing the overall performance of the model.
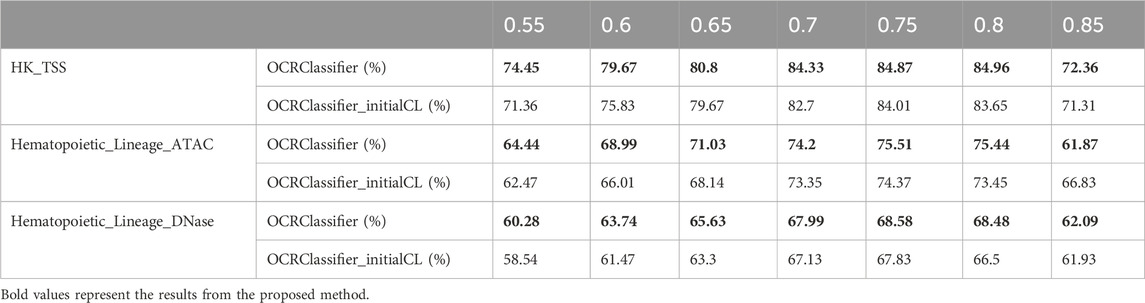
Table 3. The sensitivity of OCRClassifier before and after the adjustment of control limits in sensitized T2 control chart on different datasets.
The OCRFinder model is a binary classification model that can only identify two categories: open and closed regions of chromatin. However, the proposed model OCRClassifier in this study is a three-classification model that not only identifies open and closed chromatin regions but also recognizes partially open regions. Nevertheless, both models share similarities in terms of their training datasets, which consist of samples labeled as either 0 or 1. The difference lies in the fact that the OCRClassifier’s training dataset includes a mixture of samples labeled as 2 in practice. To evaluate the classification performance of the OCRClassifier, we conducted two comparative experiments.
Firstly, we compared its binary classification ability, specifically, the capability to identify OCRs and non-OCRs, with that of the OCRFinder model. The performance results of the OCRClassifier and OCRFinder for different datasets are presented in the Table 4. From the table, the OCRClassifier exhibits a slightly lower classification ability compared to the OCRFinder when the test dataset only includes OCRs and non-OCRs. This finding aligns with practical expectations because the OCRClassifier is designed for a three-classification. When restricted to binary classification, the OCRClassifier model may misclassify certain samples as a third category, leading to a weaker performance than the OCRFinder.
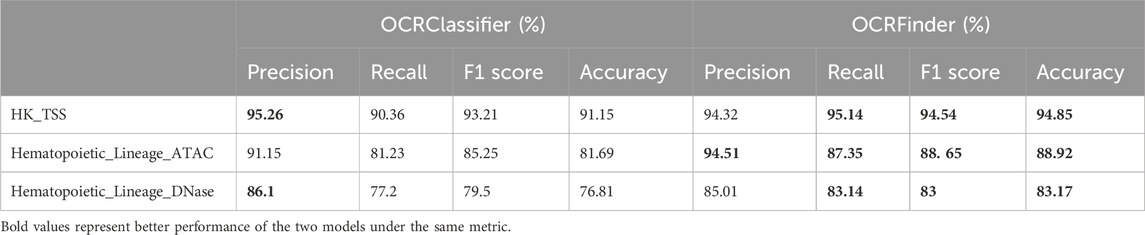
Table 4. The comprehensive performance of the OCRClassifier and OCRFinder models across different training sets.
Then, we validated the OCRClassifier’s ability in recognizing the pOCRs. We applied OCRFinder to classify a test dataset that contained pOCRs. The results of the classification generated only labels 0 and 1. Simultaneously, we employed OCRClassifier to classify the same test dataset, resulting in labels 0, 1, and 2. Analyzing the classification results of OCRFinder, we removed the intersection of labels 0 and 1 from the two sets (samples like Figures 9A, B). After this removal, we found that some samples (like Figures 9C, D) were classified as label 2 by OCRClassifier, while OCRFinder misclassified them as label 0 or label 1, as depicted in the waveform Figure 9. Hence, it is evident that OCRClassifier exhibits the capability to recognize the third category pOCRs.
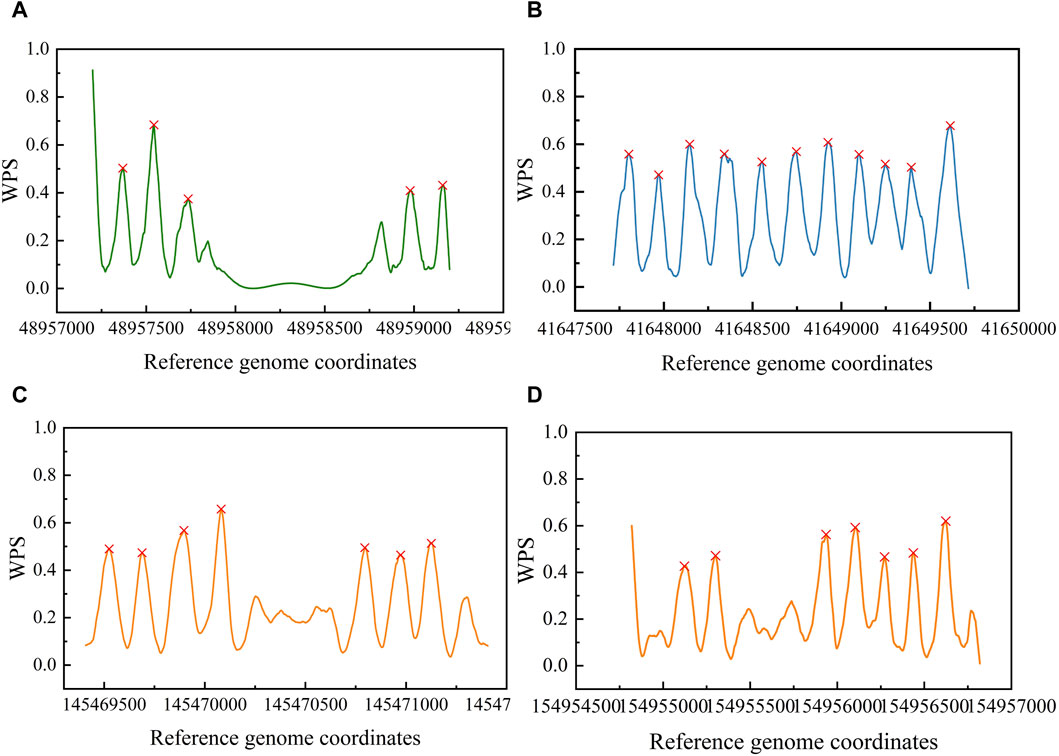
Figure 9. The classification results of different samples by OCRFinder and OCRClassifier models. (A) The OCR sample that was classified as OCR by both models. (B) The CCR sample that was classified as CCR by both models. (C) The pOCR sample that has been misclassified as OCR by OCRFinder. (D) The pOCR sample that has been misclassified as CCR by OCRFinder.
OCRs play a significant regulatory role in human biological activities and growth. The multi-class evaluation of the open state of chromatin regions can contribute to further cancer analysis research. Currently, methods for identifying chromatin open regions are limited to recognizing only two states: completely open and completely closed. For datasets that contain partially open chromatin regions, existing models may not be able to identify them. Additionally, training datasets based on biological experiments for OCRs and non-OCRs often include a high proportion of noisy labels, which substantially attenuates the model performance. By reducing the proportion of noisy labels in the training set, the identification ability of model could be further improved.
In this paper, we propose open chromatin region classifier (OCRClassifier), which can reduce the noisy labels in the initial training set and identify OCRs, non-OCRs, and pOCRs. The main task of OCRClassifier is to utilize robust Hotelling T2 control chart to filter the initial training set, which consists of only OCRs and non-OCRs, based on the different distributions of cfDNA-seq data features in different states. This filtering process aims to obtain more reliable samples for training, thereby enabling the development of a sensitized T2 control chart that can identify a third category of pOCRs. The experimental results demonstrate that OCRClassifier achieves a performance of over 84% in terms of accuracy, precision, recall, and F1-score, indicating its strong ability for three-class classification. Furthermore, the model exhibits high robustness in classification performance, which is unaffected by various openness levels of pOCRs. This framework allows for the improvement of OCRFinder in binary classification, by integrating the robust Hotelling T2 control chart with OCRFinder. Specifically, the OCRFinder is trained after filtering the training set by the robust Hotelling T2 control chart. The resulting model exhibits a 3-percentage point increase in sensitivity compared to the original OCRFinder. Additionally, the AUC and AUPR values of proposed method are also higher than original OCRFinder. The concept of OCRClassifier can be extrapolated to address label-unknown or label-ambiguous challenges in other bioinformatics domains as well, offering a promising approach to tackle the expensive annotation issues associated with biomedical samples.
The OCRClassifier is capable of identifying OCRs, non-OCRs, and pOCRs, where the data for pOCRs is simulated based on the other two categories. However, cfDNA is a composite of various cell types. Neutrophils, lymphocytes and liver are the primary contributors to cfDNA (Sun et al., 2015), resulting in the detection of a higher number of OCRs through cfDNA analysis, and our method can only detect these regions, independent of cell type. Therefore, the improvement of the OCRClassifier for predicting cell type-specific or tissue-specific OCRs and pOCRs is needed in future study.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
XL: Conceptualization, Investigation, Project administration, Writing–review and editing. ML: Methodology, Writing–original draft, Writing–review and editing. YL: Visualization, Writing–review and editing. XZ: Data curation, Methodology, Writing–review and editing. JW: Project administration, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (72274152) and Shaanxi’s Natural Science Basic Research Program, grant number 2020JC-01.
The authors are sincerely thankful to the editor, the associate editor and the reviewers for their helpful comments and suggestions on the earlier version of this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
An, Y., Zhao, X., Zhang, Z., Xia, Z., Yang, M., Ma, L., et al. (2023). DNA methylation analysis explores the molecular basis of plasma cell-free DNA fragmentation. Nat. Commun. 14 (1), 287. doi:10.1038/s41467-023-35959-6
Boudt, K., Rousseeuw, P. J., Vanduffel, S., and Verdonck, T. (2020). The minimum regularized covariance determinant estimator. Stat. Comput. 30, 113–128. doi:10.1007/s11222-019-09869-x
Buenrostro, J. D., Wu, B., Chang, H. Y., and Greenleaf, W. J. (2015). ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21. doi:10.1002/0471142727.mb2129s109
Calsina, L., Clará, A., and Vidal-Barraquer, F. (2011). The use of the CUSUM chart method for surveillance of learning effects and quality of care in endovascular procedures. Eur. J. Vasc. Endovasc. Surg. 41 (5), 679–684. doi:10.1016/j.ejvs.2011.01.003
Cheng, J., and Chou, C. (2008). A real-time inventory decision system using Western Electric run rules and ARMA control chart. Expert Syst. Appl. 35, 755–761. doi:10.1016/j.eswa.2007.07.019
Cockings, J. G., Cook, D. A., and Iqbal, R. K. (2006). Process monitoring in intensive care with the use of cumulative expected minus observed mortality and RiskAdjusted P charts. Crit. Care 10 (1), R28. doi:10.1186/cc3996
Crawford, G. E., Davis, S., Scacheri, P. C., Renaud, G., Halawi, M. J., Erdos, M. R., et al. (2006). DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods 3 (7), 503–509. doi:10.1038/nmeth888
Curtis, G., Northcutt, , Jiang, L., and Chuang, I. L. (2021). Confident learning: estimating uncertainty in dataset labels. arXiv:1911.00068. doi:10.48550/arXiv.1911.00068
Ercan, I., Sigirli, D., and Ozkaya, G. (2015). Examining the variations in the results of the hotelling t 2 test in case of changing baseline landmarks in the bookstein coordinates. Interdiscip. Sci. Comput. Life Sci. 7, 186–193. doi:10.1007/s12539-015-0025-y
Flavahan, W. A., Gaskell, E., and Bernstein, B. E. (2017). Epigenetic plasticity and the hallmarks of cancer. Sci. (New York, N.Y.) 357 (6348), eaal2380. doi:10.1126/science.aal2380
Gai, W., and Sun, K. (2019). Epigenetic biomarkers in cell-free DNA and applications in liquid biopsy. Genes (Basel) 10, 32. doi:10.3390/genes10010032
Giresi, P. G., Kim, J., McDaniell, R. M., Iyer, V. R., and Lieb, J. D. (2007). FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17 (6), 877–885. doi:10.1101/gr.5533506
Han, B., Yao, Q., Yu, X., et al. (2018). Circular RNA and its mechanisms in disease: from the bench to the clinic. Adv. neural Inf. Process. Syst. 187, 31–44. doi:10.1016/j.pharmthera.2018.01.010
Hasegawa, Y., Nitta, H., Takahara, T., Katagiri, H., Baba, S., Takeda, D., et al. (2017). Safely extending the indications of laparoscopic liver resection: when should we start laparoscopic major hepatectomy? Surg. Endosc. 31 (1), 309–316. doi:10.1007/s00464-016-4973-z
Hotelling, H. (1947). “Multivariate quality control illustrated by air testing of sample bombsights,” in Techniques of statistical analysis. Editors C. Eisenhart, M. W. Hastay, and W. A. Wallis (New York: McGraw Hill), 111–184.
Ivanov, M., Baranova, A., Butler, T., Spellman, P., and Mileyko, V. (2015). Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics 16 (Suppl. 13), S1. doi:10.1186/1471-2164-16-S13-S1
Je, S., Cho, Y., Choi, H. J., Kang, B., Lim, T., and Kang, H. (2015). An application of the learning curve-cumulative summation test to evaluate training for endotracheal intubation in emergency medicine. Emerg. Med. J. 32 (4), 291–294. doi:10.1136/emermed-2013-202470
Jin, X., Wang, Y., Xu, J., Li, Y., Cheng, F., Luo, Y., et al. (2023). Plasma cell-free DNA promise monitoring and tissue injury assessment of COVID-19. Mol. Genet. Genomics 298 (4), 823–836. doi:10.1007/s00438-023-02014-4
Johnson, D. S., Mortazavi, A., Myers, R. M., and Wold, B. (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316 (5830), 1497–1502. doi:10.1126/science.1141319
Khoo, M. B. C. (2003). Design of runs rules schemes. Qual. Eng. 16 (1), 27–43. doi:10.1081/QEN-120020769
Kim, C. W., Kim, W. R., Kim, H. Y., Kang, J., Hur, H., Min, B. S., et al. (2015). Learning curve for single-incision laparoscopic anterior resection for sigmoid colon cancer. J. Am. Coll. Surg. 221 (2), 397–403. doi:10.1016/j.jamcollsurg.2015.02.016
Kingma, D. P., and Ba, J. (2014). Adam: a method for stochastic optimization. arXiv Prepr. arXiv:1412.6980. doi:10.48550/arXiv.1412.6980
Klemm, S. L., Shipony, Z., and Greenleaf, W. J. (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20 (4), 207–220. doi:10.1038/s41576-018-0089-8
Lo, Y. M. D., Han, D. S. C., Jiang, P., and Chiu, R. W. K. (2021). Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science. 372, eaaw3616. doi:10.1126/science.aaw3616
Lv, D., Wang, H., Ma, W., Wang, S., and Gu, Z. (2014). Differential expression gene detection for biological pathway. J. Nat. ence Hlongjiang Univ. doi:10.13482/j.issn1001-7011.2014.03.253
Northcutt, C., Jiang, L., and Chuang, I. (2021). Confident learning: estimating uncertainty in dataset labels. J. Artif. Int. Res. 70 (May 2021), 1373–1411. doi:10.1613/jair.1.12125
Northcutt, C., Jiang, L., and Chuang, I. (2021). Confident learning: estimating uncertainty in dataset labels. J. Artif. Intell. Res. 70, 1373–1411. doi:10.1613/jair.1.12125
Novick, R. J., Fox, S. A., Stitt, L. W., Forbes, T. L., and Steiner, S. (2006). Direct comparison of RiskAdjusted and non-risk-adjusted CUSUM analyses of coronary artery bypass surgery outcomes. J. Thorac. Cardiovasc Surg. 132 (2), 386–391. doi:10.1016/j.jtcvs.2006.02.053
Park, S.Ho, and Kim, S. B. (2019). Multivariate control charts that combine the Hotelling T2 and classification algorithms. J. Operational Res. Soc. 70 (6), 889–897. doi:10.1080/01605682.2018.1468859
Ren, J., Liu, Y., Zhu, X., Wang, X., Li, Y., Liu, Y., et al. (2023). OCRFinder: a noise-tolerance machine learning method for accurately estimating open chromatin regions. Front. Genet. 14, 1184744. doi:10.3389/fgene.2023.1184744
Schones, D. E., Cui, K., Cuddapah, S., Roh, T. Y., Barski, A., Wang, Z., et al. (2008). Dynamic regulation of nucleosome positioning in the human genome. Cell 132 (5), 887–898. doi:10.1016/j.cell.2008.02.022
Snyder, M. W., Kircher, M., Hill, A. J., Daza, R. M., and Shendure, J. (2016). Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell 164 (1-2), 57–68. doi:10.1016/j.cell.2015.11.050
Struhl, K., and Segal, E. (2013). Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 20 (3), 267–273. doi:10.1038/nsmb.2506
Sun, K., Jiang, P., Chan, K. C., Wong, J., Cheng, Y. K., Liang, R. H., et al. (2015). Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc. Natl. Acad. Sci. U. S. A. 112 (40), E5503–E5512. doi:10.1073/pnas.1508736112
Sun, K., Jiang, P., Cheng, S. H., Cheng, T. H. T., Wong, J., Wong, V. W. S., et al. (2019). Orientation-aware plasma cell-free DNA fragmentation analysis in open chromatin regions informs tissue of origin. Genome Res. 29 (3), 418–427. doi:10.1101/gr.242719.118
Thurman, R. E., Rynes, E., Humbert, R., Vierstra, J., Maurano, M. T., Haugen, E., et al. (2012). The accessible chromatin landscape of the human genome. Nature 489 (7414), 75–82. doi:10.1038/nature11232
Ulz, P., Thallinger, G. G., Auer, M., Graf, R., Kashofer, K., Jahn, S. W., et al. (2016). Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 48 (10), 1273–1278. doi:10.1038/ng.3648
Van der Pol, Y., and Mouliere, F. (2019). Toward the early detection of cancer by decoding the epigenetic and environmental fingerprints of cell-free DNA. Cancer Cell 36, 350–368. doi:10.1016/j.ccell.2019.09.003
Waller, H. M., and Connor, S. J. (2009). Cumulative sum (cusum) analysis provides an objective measure of competency during training in endoscopic retrograde cholangio-pancreatography (ERCP). HPB 11 (7), 565–569. doi:10.1111/j.1477-2574.2009.00091.x
Wang, J., Chen, L., Zhang, X., Tong, Y., and Zheng, T. (2021). OCRDetector: accurately detecting open chromatin regions via plasma cell-free DNA sequencing data. Int. J. Mol. Sci. 22 (11), 5802. doi:10.3390/ijms22115802
Keywords: cell-free DNA, open chromatin region, sequencing data analysis, machine learning approach, multivariate control chart, noisy label
Citation: Lai X, Liu M, Liu Y, Zhu X and Wang J (2024) OCRClassifier: integrating statistical control chart into machine learning framework for better detecting open chromatin regions. Front. Genet. 15:1400228. doi: 10.3389/fgene.2024.1400228
Received: 13 March 2024; Accepted: 07 May 2024;
Published: 04 December 2024.
Edited by:
Junbai Wang, University of Oslo, NorwayCopyright © 2024 Lai, Liu, Liu, Zhu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiayin Wang, d2FuZ2ppYXlpbkBtYWlsLnhqdHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.