
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 04 March 2024
Sec. Genetics of Common and Rare Diseases
Volume 15 - 2024 | https://doi.org/10.3389/fgene.2024.1341272
This article is part of the Research Topic Recent Advances in Understanding The Genetics of Immunological Disorders View all 12 articles
 Nicholas Kim-Wah Yeo1,2†
Nicholas Kim-Wah Yeo1,2† Che Kang Lim2,3†
Che Kang Lim2,3† Katherine Nay Yaung1,2
Katherine Nay Yaung1,2 Nicholas Kim Huat Khoo1
Nicholas Kim Huat Khoo1 Thaschawee Arkachaisri1,2,4‡
Thaschawee Arkachaisri1,2,4‡ Salvatore Albani1,2,4‡
Salvatore Albani1,2,4‡ Joo Guan Yeo1,2,4*‡
Joo Guan Yeo1,2,4*‡Early-onset systemic lupus erythematosus presents with a more severe disease and is associated with a greater genetic burden, especially in patients from Black, Asian or Hispanic ancestries. Next-generation sequencing techniques, notably whole exome sequencing, have been extensively used in genomic interrogation studies to identify causal disease variants that are increasingly implicated in the development of autoimmunity. This Review discusses the known casual variants of polygenic and monogenic systemic lupus erythematosus and its implications under certain genetic disparities while suggesting an age-based sequencing strategy to aid in clinical diagnostics and patient management for improved patient care.
Systemic lupus erythematosus (SLE, or lupus) is an autoimmune disease characterized by autoantibody formation targeting nucleic components like double-stranded DNA (dsDNA) and RNA (Caielli et al., 2023). The vast spectrum of clinical manifestations ranges from mild skin rashes to widespread destructive multi-organ inflammation, which in some cases, could result in death. The pathogenesis of SLE is complex and multi-factorial (Tsokos, 2011), with genetic and environmental contributions to the disease. It has also been observed that various autoimmune diseases are more common in women (Vinuesa et al., 2023), and in SLE, individuals from Black, Asian or Hispanic ethnicities have an increased disease burden, with patients presenting with a more severe phenotype (Lewis and Jawad, 2017).
SLE can be grouped according to the age of disease onset into adult- and childhood-onset SLE (cSLE); the latter referring to those diagnosed before the age of 18 years and generally presents with greater severity especially in children under 5 years old (Bundhun et al., 2017; Alperin et al., 2018). This early onset of SLE has been associated with an increased genetic burden, highlighting the contribution of one or several risk alleles to disease (Webb et al., 2011). And within this patient group, around 3%–10% of patients carry a single disease-causing variant (Almlof et al., 2019; Belot et al., 2020; Charras et al., 2023), thus being increasingly recognized and termed as monogenic SLE (Harley and Sawalha, 2022; Vinuesa et al., 2023). Pinpointing the disease-causing variant will contribute greatly to our current knowledge of lupus pathogenesis, and this can be achieved through the use of next-generation sequencing (NGS) techniques (Sanger et al., 1977; Slatko et al., 2018; You et al., 2018; Yaung et al., 2023). As such, a focused strategy is needed together with prioritizing NGS and research efforts towards cSLE patients (Mina and Brunner, 2013).
Knowing that SLE has a strong genetic component to disease (Lewis and Jawad, 2017), multiple susceptibility loci have since been identified, following the advent of genome-wide association studies (GWAS) (Deng and Tsao, 2014). Further diving into genomic studies of SLE through NGS techniques has brought to light the utility of whole exome (WES) and whole genome sequencing (WGS). Our colleagues have also reviewed various technologies that could be employed to elucidate disease mechanisms (Yaung et al., 2023), such as Sanger sequencing (Sanger et al., 1977), single nucleotide polymorphism (SNP) array (You et al., 2018), WES and WGS (Slatko et al., 2018). In this Review, we expound further into the use of NGS techniques, notably WES, across the current genomic landscape of polygenic and monogenic SLE, discussing its potential in reconciling disease risk variants and copy number variations (CNVs) and evaluating the identification of such variants.
Sequencing technologies have been fundamental for researchers due to their high-throughput capabilities and more recently, their cost-effectiveness (Goodwin et al., 2016). This has allowed for comprehensive genomic studies (i.e., point mutations, small indels, CNVs) and paved the way for multi-omics studies (Levy and Myers, 2016; Lee et al., 2022; Satam et al., 2023; Yaung et al., 2023). In the context of systemic autoimmune diseases like SLE, multiple susceptibility loci identified by GWAS cumulatively contribute risk towards its development but carry a relatively low disease risk individually (Sestak et al., 2011; Wahren-Herlenius and Dorner, 2013).
Several methods have been employed in SLE genomics, including WGS, WES and targeted sequencing (Table 1). Briefly, WGS allows for comprehensive interrogation of the entire human genome and has contributed significantly to the genomic landscape via the 1000 Genomes project since 2010 (Genomes Project et al., 2010; Genomes Project et al., 2015; Sudmant et al., 2015). However, around 85% of disease-related mutations are concentrated in the exome, which constitutes about 2% of the whole genome (Majewski et al., 2011). WES then involves the selection of protein-coding regions (exons) in the genome for sequencing to identify any changes that could impact protein sequences (Ng et al., 2009). This has led to its increased use due to the significant reduction vis-à-vis the starting material, cost and data management (Petersen et al., 2017). In addition, mutations in the exonic region have been shown to be a major contributor to the development of monogenic diseases (Kuhlenbaumer et al., 2011). With the knowledge obtained from the above-mentioned methods, sequencing panels could be generated to target certain regions of interest that harbor pathogenic mutations, hence the utility of targeted sequencing for potential clinical care (Gulilat et al., 2019).
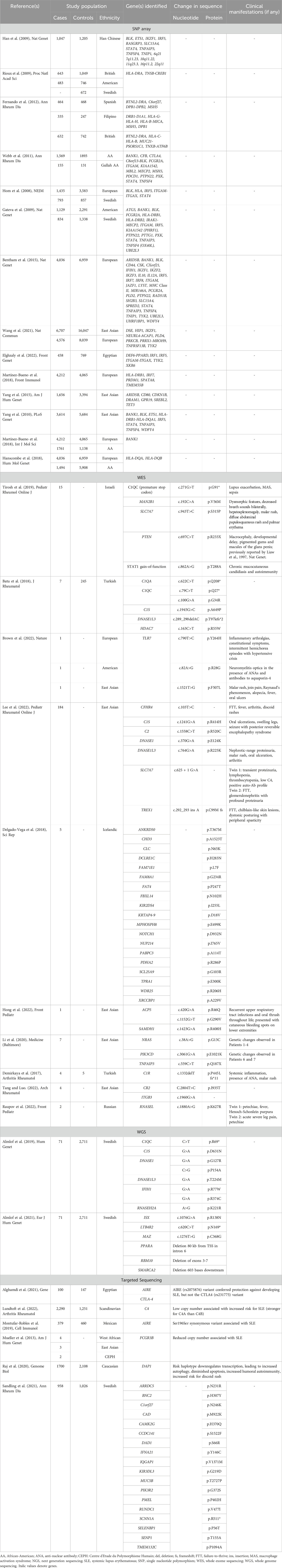
TABLE 1. NGS techniques and respective applications in SLE studies.
Autoimmune diseases have been known to arise from an accumulation of genetic and environmental factors across one’s lifetime, as in the case of adult-onset SLE (Goodnow et al., 2005). More than 100 loci associated with SLE have been identified through GWAS (Wang et al., 2021), such as regions in the Human Leukocyte Antigen (HLA) locus (Hanscombe et al., 2018), STAT4 (Remmers et al., 2007; Han et al., 2009), TNFSF4 (Han et al., 2009), BANK1 (Kozyrev et al., 2008; Martinez-Bueno et al., 2018), TNFAIP3 (Graham et al., 2008; Musone et al., 2008; Han et al., 2009), BLK (Hom et al., 2008; Han et al., 2009), IRF5 (Jones et al., 2019), ETS1 (Han et al., 2009; Yang et al., 2010; Jones et al., 2019), WDFY4 (Yang et al., 2010) and TNIP1 (Han et al., 2009; Yang et al., 2010; Jones et al., 2019). However, these variants are unlikely to contribute significantly to SLE pathogenesis individually, unless coupled either with variants in certain regulatory regions or in other genes that maintain immune tolerance (Jones et al., 2019). Importantly, epistatic interaction between genes may contribute in part to the development of complex diseases such as lupus (Hughes et al., 2012; Wei et al., 2014).
It has been recently suggested that polygenic risk scores (PRS) could be utilized to identify and stratify potential SLE patients for early intervention, if needed (Khunsriraksakul et al., 2023). Briefly, GWAS-identified risk variants are statistically compiled to predict disease incidence in a population and risk for developing SLE in individuals (Khunsriraksakul et al., 2022). An association between a high PRS and poorer prognosis in SLE has been observed (Chen et al., 2020; Reid et al., 2020; Sandling et al., 2021), with one study going further to delineate T cell differentiation and innate immunity as the two key axes of SLE association mediated by HLA and interferons (IFNs) respectively (Sandling et al., 2021). The strong involvement of HLA and IFNs has also been described for SLE pathogenesis (Chen et al., 2017; Villarino et al., 2017; Alunno et al., 2019; Crow and Ronnblom, 2019). Despite its utility, PRS has yet to be generalizable beyond the specific population being studied, which further emphasizes the need for larger, diverse and well-represented datasets in order to draw meaningful conclusions (Torkamani et al., 2018). In addition, data generated from GWAS is primarily based on SNP arrays which can be limited by its inability to identify causal variants and ultra-rare mutations, particularly in ethically under-represented populations (Tam et al., 2019). NGS techniques thus provide an answer to interrogating such variants, which might aid in enriching our knowledge of SLE pathogenesis, the clinical diagnosis and management of polygenic SLE together with the potential use of PRS.
Single gene defects are part of the diverse heterogenous etiologies for lupus, where about 1%–3% of SLE patients carry a single mutation that leads to disease development (Costa-Reis and Sullivan, 2017). Albeit rare, monogenic SLE is characterized by a more severe phenotype early in life (Webb et al., 2011; Vinuesa et al., 2023). Several gene sets involved in the complement pathway, IFN responses, nucleic acid sensing and immune tolerance have been implicated in the pathogenesis of monogenic SLE (Alperin et al., 2018; Vinuesa et al., 2023).
Most complement-related SLE defects are found in the C1 (C1QA, C1QB, C1QC, C1R, C1S) (Lood et al., 2009; Bienaime et al., 2010; Demirkaya et al., 2017; Almlof et al., 2019) or C4 (C4A, C4B) compartments (Blanchong et al., 2001; Vinuesa et al., 2023). Other affected regions include the C2 and C3 genes (Miller and Atkinson, 2012; Vinuesa et al., 2023). Complement-deficient patients tend to have an impaired clearance of cellular apoptotic fragments, which in turn facilitates autoimmunity (Costa-Reis and Sullivan, 2017; Vinuesa et al., 2023). Next, studies have observed an elevated IFN signature in SLE patients (Baechler et al., 2003; Reynier et al., 2011), and the association of variants in ADAR1 (Crow and Ronnblom, 2019), TREX1 (Rice et al., 2015), SAMHD1 (Abdel-Salam et al., 2010; Ravenscroft et al., 2011) and IFIH1 (Rice et al., 2014; Almlof et al., 2019) genes to disease (Abdel-Salam et al., 2010; Ravenscroft et al., 2011; Rice et al., 2012; Rice et al., 2014; Crow et al., 2015). ADAR1, TREX1 and SAMHD1 are involved in nucleic acid metabolism while IFIH1 is involved in nucleic acid sensing. Lastly, defects in nucleic acid sensing and degradation genes DNASE1 (Yasutomo et al., 2001; Almlof et al., 2019), DNASE1L3 (Al-Mayouf et al., 2011; Almlof et al., 2019) and TLR7 (Giltiay et al., 2013; Brown et al., 2022) have been found in SLE patients. This causes an accumulation of extracellular nucleic acids, leading to TLR7 activation and downstream type I IFN production. Type I IFN further upregulates TLR7 expression, creating a positive feedback loop that eventuates into autoantibody production (Caielli et al., 2023). With this, it has been recently found that two TLR7 variants (Y264H and F507L) were recently identified to cause SLE, with the Y264H variant presenting with an increased sensing of guanosine and 2′,3′-cGMP (Shibata et al., 2016; Zhang et al., 2016; Zhang et al., 2018; Brown et al., 2022).
Across autoimmune diseases, a hallmark of its development is the loss of tolerance to self-antigens, with AIRE and CTLA-4 being implicated in SLE (Pullmann et al., 1999; Ahmed et al., 2001; Hudson et al., 2002; Lee et al., 2005; Cunninghame Graham et al., 2006; Lovewell et al., 2015; Montufar-Robles et al., 2019; Alghamdi et al., 2021). AIRE, or autoimmune regulator is essential for maintaining central immune tolerance by controlling the negative thymic selection of hyper-reactive T lymphocytes against self-antigens (Yang et al., 2015). Mutations in this gene region have been observed in Norwegian patients with autoimmune polyendocrine syndrome type I (APS-1; (Oftedal et al., 2023)) and Japanese patients with rheumatoid arthritis (RA; (Terao et al., 2011)). More recently, an AIRE Ser196Ser synonymous variant was associated with SLE in a recent targeted sequencing study in a Mexican cohort (Montufar-Robles et al., 2019). However, when extended to GWAS performed on a larger European SLE cohort, no association was found (Bentham et al., 2015). Next, cytotoxic T-lymphocyte associated protein 4 (CTLA-4, or CD152) is an important checkpoint inhibitor in peripheral immune tolerance via negative signaling in regulating autoreactive T cells (Liu and Zhang, 2013; Van Coillie et al., 2020). Though several reports have identified certain polymorphisms contributing to SLE development (Pullmann et al., 1999; Ahmed et al., 2001; Hudson et al., 2002; Lee et al., 2005; Cunninghame Graham et al., 2006; Jury et al., 2010), a meta-analysis has highlighted no association of said variants to lupus (Liu and Zhang, 2013; Alghamdi et al., 2021). In some cases, specific CTLA-4 variants could even contribute to protection against SLE (Barreto et al., 2004), suggesting that only certain variants within the CTLA-4 gene region have an association with SLE development.
Recent studies have described several novel genes associated with SLE following WES analysis in an Asian population, such as the decreased expression of cell division cycle 27 (CDC27) in patients (Shang et al., 2022), and novel variants in genes encoding for complement receptor 2 (CR2) (Tang and Luo, 2022), C1R (Demirkaya et al., 2017), NRAS, TNFAIP3 and PIK3CD (Li et al., 2020), WNT16 and ERVW-1 (Chen et al., 2022), ACP5 and SAMHD1 (Hong et al., 2022). This list of genes contributing to monogenic SLE continues to grow with increased usage of WES over the past few years, further enriching our knowledge about the genetic contribution to SLE.
Copy number variation (CNV) is a phenomenon where repeated genomic sequences occur and arise from the process of genomic rearrangement, which can manifest as translocations, inversions, insertions and deletions (Feuk et al., 2006; Human Genome Structural Variation Working et al., 2007). However, the total number of gene copies and its downstream effects may vary between individuals (Usher and McCarroll, 2015). In the past two decades, several CNVs associated with SLE development have been identified, such as C4 (C4A, C4B) (Yang et al., 2007; Pereira et al., 2019; Kamitaki et al., 2020; Lundtoft et al., 2022), FCGR3A, FCGR3B (Willcocks et al., 2008; Niederer et al., 2010), CCL3L1 (Gonzalez et al., 2005), RABGAP1L (Kim et al., 2013), TLR7 (Garcia-Ortiz et al., 2010) and HSP90 (Zhang et al., 2019).
As previously mentioned, defects in complement genes have been observed to be a monogenic cause of SLE. Of note, C4, or complement compartment protein 4, is usually present in most individuals as two copies of C4A and C4B respectively. In some cases, SLE patients may carry a range of zero to five copies of C4A and zero to four copies of C4B (Yang et al., 2007; Pereira et al., 2019). A recent study has described an association between a low C4A copy number and an increased risk of developing SLE (Kamitaki et al., 2020). Though C4 genes are highly homologous and are usually excluded from variant calling analysis, Lundtoft et al. performed a focused analysis into C4 CNVs via targeted sequencing and found Scandinavian SLE patients with a low C4A copy number and carrying a common loss-of-function (LoF) variant presenting with lowered plasma C4 levels (Lundtoft et al., 2022). Whether this phenomenon can be extended to other ancestral populations remains unknown and warrants further investigation.
Other genes like FCG3RA and FCGR3B encode for low-affinity Fc gamma (Fcγ) receptors of IgG and are crucial in the binding and clearing of immune complexes (Willcocks et al., 2008; Niederer et al., 2010), while CCL3L1 (C-C chemokine ligand 3 like-1) translates into a ligand that binds to C-C chemokine receptor 5 (CCR5) (Gonzalez et al., 2005). Healthy individuals carry two copies of each respective gene, but SLE risk increases when there are either lower or higher copy numbers of said genes (Willcocks et al., 2008). Increased SLE susceptibility was also observed with low RASGAP1L and high TLR7 copy numbers respectively. RASGAP1L encodes for a Rab GTPase-activating protein (Kim et al., 2013), while TLR7 is a key receptor in innate immunity that recognizes single-stranded RNA (Lund et al., 2004; Takeda and Akira, 2005). Lastly, abnormal CNVs in heat shock proteins 90 (HSP90), especially in its AB1 isoform, were identified to correlate with SLE in the Han Chinese (Zhang et al., 2019). This highlights the importance of CNVs in SLE and autoimmunity and thus the need for more traction toward implementing a pipeline to include them in future genetic screens (Zhao et al., 2020).
Genetic testing using NGS techniques has identified potential disease-causing variants and led to better preventative risk management of diseases (Shaw et al., 2023). However, given its complexity, the labeling of variants as potentially pathogenic should be done with caution to prevent misdiagnoses. A misdiagnosis of a pathogenic variant can result in unnecessary medical interventions and cause undue psychological distress to both patients and their families (Manrai et al., 2016; Shaw et al., 2023). Such detrimental consequences have occurred in diseases like hypertrophic cardiomyopathy and cancers, where variants that were thought to be pathogenic were subsequently found to be benign due to the under-representation of certain ancestries in reference control groups (Manrai et al., 2016; Shaw et al., 2023).
To prevent such genetic misclassifications, the American College of Medical Genetics and Genomics has introduced a standardized framework for variant interpretation (Richards et al., 2015). In the case of SLE and other autoimmune diseases, pathogenic variants can be better identified prior to further functional validation through this framework, thus reducing the occurrence of false positives as the number of sequencing studies continues to rise (Vinuesa et al., 2023). In addition, various consortia like the Clinical Genome Resource (ClinGen; (Rehm et al., 2015)), Rheumatologic Autoimmune Clinical Domain Working Group under ClinGen, Lupus in Minority Populations, Nature versus Nurture (LUMINA; (Alarcon et al., 2001)) have been established to aggregate all available genomic data and concentrate global research efforts. Crucially, the consolidation of genomic data overcomes the major limitation of genome-wide studies of requiring large sample sizes due to the need to adopt a high level of significance to account for multiple testing (Tam et al., 2019). With this framework for variant interpretation and genomic data from various consortia, this can be potentially applied to the dysmorphic syndromes associated with SLE, specifically genes of the Ras/mitogen-activated protein kinase (Ras/MAPK) pathway to identify with greater certainty the potential pathogenic genetic variants within this pathway that contribute to SLE (Amoroso et al., 2003; Lisbona et al., 2009; Leventopoulos et al., 2010; Hanaya et al., 2017; Uehara et al., 2018). However, further investigations would be needed to delineate the underlying mechanism with functional studies of the different genes in the Ras/MAPK pathway as these are currently described in case reports and series.
Our current understanding has informed us that certain ancestral groups have an increased predilection towards developing SLE (Lewis and Jawad, 2017), which requires controlling for in future sequencing studies to prevent any potential misclassification of disease-causing variants due to the unavailability of an adequate ancestry-specific reference genome. Past research has been largely focused on European ancestry (Yang et al., 2007; Lewis and Jawad, 2017; Hanscombe et al., 2018), resulting in an under-representation of data from other ancestries to draw meaningful generalizations about the disease. This can be resolved by tapping on several biobanks that have been consolidated over the years to provide greater depth and insights into the genetic differences within and across various ancestries. These include, and are not limited to, the Tohoku Biobank (150,000 participants; (Minegishi et al., 2019)), Mexican Biobank (6,057 participants; (Sohail et al., 2023)), Biobank Japan (BBJ, 260,000 participants; (Kanai et al., 2018)), China Kadoorie Biobank (500,000 participants; (Chen et al., 2011)), H3Africa (70,000 participants; (Consortium et al., 2014; Mulder et al., 2018)), UK Biobank (500,000 participants; (Bycroft et al., 2018; Van Hout et al., 2020; Gaynor et al., 2023)), Michigan Genomic Initiative (MGI, 91,000 participants; (Zawistowski et al., 2023)), Vanderbilt University Biobank (BioVU, 300,000 participants; (Khunsriraksakul et al., 2023)), and SG10K (9,051 participants; (Chan et al., 2022)). It should be noted that SG10K has since been expanded to SG100K, whereby data from 70,000 participants across four national cohort studies will be pooled together with the additional recruitment of 30,000 individuals (Begum, 2022).
In this Review, we have provided an overview of various susceptibility genes contributing to the development of SLE either through a polygenic or monogenic route identified via NGS techniques, highlighted the involvement and importance of CNVs and urged for the inclusiveness of control groups to account for ancestral differences to prevent any potential variant misclassification.
The introduction of WGS and WES has resulted in faster genomic interrogation, allowing for one’s entire genome to be generated in a matter of days to weeks (Bourchany et al., 2017; Duncavage et al., 2021). The data generated from WGS provides comprehensive information on both intronic (non-coding) and exonic (protein-coding) regions. However, the contributions of non-coding variants towards disease have yet to be thoroughly elucidated and the downstream analyses of such intronic regions remain complex and highly challenging (Zhao et al., 2020). As such, WES has become increasingly popular in clinical diagnostics and research due to its utility (∼95% capture of exonic and splice site regions (Field, 2021; Zhang et al., 2021)), ease of analysis (Yaung et al., 2023) and lower cost (one-third that of WGS (Goodwin et al., 2016; Field, 2021))
In addition, structural variants like CNV are relatively common across the whole genome (with a frequency of around 12%; (Iafrate et al., 2004; Sebat et al., 2004; Tuzun et al., 2005; Conrad et al., 2006; McCarroll et al., 2006)), and can influence gene expression (Somerville et al., 2005; Lee et al., 2006; McCarroll et al., 2006). As we have alluded to the growing importance of CNV in SLE immunogenetics, the coupling of WES with CNV detection addresses the need for a holistic interrogation of the genetic contribution to SLE through the dual identification of variations in exonic sequences and gene copy numbers. This is achievable with tools such as CoNIFER (Krumm et al., 2012), exomeCopy (Love et al., 2011), CNVkit (Talevich et al., 2016), cn. MOPS (Klambauer et al., 2012), CNest (Fitzgerald and Birney, 2022), CNVind (Kusmirek and Nowak, 2022), CoverageMaster (Rapti et al., 2022) and EXCAVATOR2 (D’Aurizio et al., 2016; D’Aurizio et al., 2018). More recently, Olfe et al. have demonstrated CTLA-4 insufficiency due to a novel CTLA-4 deletion using ClinCNV (German and Stephan, 2019; Olfe et al., 2023), further highlighting the synergy of CNV calling with WES analysis. Beyond the scope of autoimmune diseases, NGS techniques have also been extensively utilized in identifying causal variants (including CNVs) contributing to cancer (van Dijk et al., 2014; Papp et al., 2021; Satam et al., 2023), congenital (Lai et al., 2021; Li et al., 2022; Liu et al., 2022; Wang et al., 2022; Wu et al., 2022; Refeat et al., 2023), cardiovascular (Hu et al., 2023) and hematological diseases (Hassan et al., 2023).
Though the method of WES has been well-established over the years, notable limitations persist in WES-based CNV analyses. The technique primarily targets coding regions, leading to a restricted view of the genome and potentially missing important regulatory components within non-coding regions such as intergenic or intronic regions (Mandelker et al., 2016; Royer-Bertrand et al., 2021). This significantly impacts the sensitivity of CNV detection. In addition, it is susceptible to biases, such as GC content bias, which can impact the reliability of CNV calls (Lelieveld et al., 2015). Furthermore, a relatively higher false positive rate and the limitation of achieving homogeneous coverage of sequencing reads restrict its inclusion as a gold-standard method for CNV detection (Marchuk et al., 2018; Burdick et al., 2020). These limitations emphasize the necessity of integrating WES with other omics approaches for more accuracy in CNV detection (Gabrielaite et al., 2021). Nonetheless, with ongoing upgrades to sequencing libraries, capture kits and bioinformatics pipelines, it is anticipated that the existing limitations will be alleviated (Zhou et al., 2021). Future applications of third-generation sequencing (TGS) techniques such as long-read sequencing hold promise in addressing these constraints and provide additional possibilities in detecting structural variations (SVs) (Xiao and Zhou, 2020).
Though SLE is known to have a strong genetic predilection, its typical development is usually due to polygenic contributions coupled with an environmental trigger (Harley and Sawalha, 2022); the latter of which must not be ignored. Research into the host-environment interplay has yielded physical/chemical factors (smoking, chemical exposure (Kilburn and Warshaw, 1992; Speyer and Costenbader, 2018; Akhil et al., 2023)), Epstein-Barr virus (EBV) infections (Poole et al., 2006; Jog and James, 2020), gut microbiota (Neuman and Koren, 2017) and obesity (Kang et al., 2020) as contributors to the development of SLE (Parks et al., 2017; Gulati and Brunner, 2018; Akhil et al., 2023). Such environmental triggers can influence methylation patterns in genes related to B and T cells, which are associated with SLE pathology (Akhil et al., 2023). These include observations of hypomethylation in CD40L (Vordenbaumen et al., 2021) and CD70 (Keshavarz-Fathi et al., 2022), as well as hypermethylation of FOXP3 (Hanaei et al., 2020) and CTLA-4 (Nosrat zehi et al., 2021).
Elucidating the pathogenesis of autoimmune diseases like SLE remains complex, and studies have called for the need for a multi-omics approach to furnish our current understanding of the disease (Fang et al., 2016; Hedrich, 2017; Kwon et al., 2019; Yaung et al., 2023). Thus far, transcriptomic signatures obtained from blood and tissues have shown an enrichment of genes involved in the IFN response (Banchereau et al., 2016; Der et al., 2019), which corroborates with previous genetic data (Baechler et al., 2003; Reynier et al., 2011). Epigenetic modifications in the genome such as methylation (Ballestar, 2011; Hedrich, 2017), non-coding RNAs (Taheri et al., 2020) and post-translational histone modifications (i.e., methylation, acetylation; (Hu et al., 2008)) have also been associated with the development of SLE. Proteomic studies have proven difficult to isolate biomarkers for diagnosis, management and monitoring due to the heterogeneity of the disease and its involvement across multiple organs (ref), but current efforts continue to show some promise (Huang et al., 2022; Fasano et al., 2023). Indeed, more needs to be done to reconcile multi-omics and genetic data of SLE in the future.
Up to 10% of patients below the age of 18 years can carry a significant disease-causing variant which manifests as severe SLE, alluding to a monogenic etiology and highlights the value of doing NGS in children with a very early onset of disease (Alperin et al., 2018; Charras et al., 2021). Previous studies have shown the utility of WES in unraveling novel rare variants and determining its respective contribution(s) to disease (Pullabhatla et al., 2018; Almlof et al., 2019; Tirosh et al., 2019; Almlof et al., 2021). However, genetic variation across ancestries should not be overlooked to prevent variant misclassification and downstream misdiagnoses. This can be controlled via the inclusion of gene datasets across various biobanks, consortia and databases. With that, establishing a pipeline where WES and CNV detection are coupled together will allow for the timely and pinpoint clinical diagnosis of SLE to allow for better clinical management and intervention.
We searched PubMed between 30 August 2023 and 7 February 2024, using the terms “systemic lupus erythematosus (SLE)”, “next-generation sequencing (NGS)”, “genomics”, “copy number variation” in articles published from 1 Jan 2013 until 7 February 2024. Articles were also identified through references from articles identified through the search. Only papers published in English were reviewed and the final reference list was generated based on the relevance to the scope of this Review.
NK-WY: Data curation, Writing–original draft, Writing–review and editing. CL: Data curation, Writing–original draft, Writing–review and editing. KN: Writing–review and editing. NK: Writing–review and editing. TA: Writing–review and editing. SA: Writing–review and editing. JY: Conceptualization, Data curation, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research is supported by the Singapore Ministry of Health’s National Medical Research Council (NMRC) under its Centre Grant Programme (MOH-000988) and other NMRC grants: CSAINV22jul-0008 (JY), CIRG21nov-0031 (JY), NMRC/MOHIAFCAT2/005/2015 (SA), NMRC/TCR/0015-NCC/2016 (SA), NMRC/OFLCG/002/2018 (SA) and CIRG19may0052 (SA), is gratefully acknowledged. We also gratefully acknowledge the SingHealth Duke-NUS Academic Medicine Grant, Special Category (PRISM, AM/PRM002/2018).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel-Salam, G. M., El-Kamah, G. Y., Rice, G. I., El-Darouti, M., Gornall, H., Szynkiewicz, M., et al. (2010). Chilblains as a diagnostic sign of aicardi-goutieres syndrome. Neuropediatrics 41 (1), 18–23. doi:10.1055/s-0030-1255059
Ahmed, S., Ihara, K., Kanemitsu, S., Nakashima, H., Otsuka, T., Tsuzaka, K., et al. (2001). Association of CTLA-4 but not CD28 gene polymorphisms with systemic lupus erythematosus in the Japanese population. Rheumatol. Oxf. 40 (6), 662–667. doi:10.1093/rheumatology/40.6.662
Akhil, A., Bansal, R., Anupam, K., Tandon, A., and Bhatnagar, A. (2023). Systemic lupus erythematosus: latest insight into etiopathogenesis. Rheumatol. Int. 43 (8), 1381–1393. doi:10.1007/s00296-023-05346-x
Alarcon, G. S., McGwin, G., Bartolucci, A. A., Roseman, J., Lisse, J., Fessler, B. J., et al. (2001). Systemic lupus erythematosus in three ethnic groups. IX. Differences in damage accrual. Arthritis Rheum. 44 (12), 2797–2806. doi:10.1002/1529-0131(200112)44:12<2797::aid-art467>3.0.co;2-9
Alghamdi, S. A., Kattan, S. W., Toraih, E. A., Alrowaili, M. G., Fawzy, M. S., and Elshazli, R. M. (2021). Association of AIRE (rs2075876), but not CTLA4 (rs231775) polymorphisms with systemic lupus erythematosus. Gene 768, 145270. doi:10.1016/j.gene.2020.145270
Al-Mayouf, S. M., Sunker, A., Abdwani, R., Abrawi, S. A., Almurshedi, F., Alhashmi, N., et al. (2011). Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 43 (12), 1186–1188. doi:10.1038/ng.975
Almlof, J. C., Nystedt, S., Leonard, D., Eloranta, M. L., Grosso, G., Sjowall, C., et al. (2019). Whole-genome sequencing identifies complex contributions to genetic risk by variants in genes causing monogenic systemic lupus erythematosus. Hum. Genet. 138 (2), 141–150. doi:10.1007/s00439-018-01966-7
Almlof, J. C., Nystedt, S., Mechtidou, A., Leonard, D., Eloranta, M. L., Grosso, G., et al. (2021). Contributions of de novo variants to systemic lupus erythematosus. Eur. J. Hum. Genet. 29 (1), 184–193. doi:10.1038/s41431-020-0698-5
Alperin, J. M., Ortiz-Fernandez, L., and Sawalha, A. H. (2018). Monogenic lupus: a developing paradigm of disease. Front. Immunol. 9, 2496. doi:10.3389/fimmu.2018.02496
Alunno, A., Padjen, I., Fanouriakis, A., and Boumpas, D. T. (2019). Pathogenic and therapeutic relevance of JAK/STAT signaling in systemic lupus erythematosus: integration of distinct inflammatory pathways and the prospect of their inhibition with an oral agent. Cells 8 (8), 898. doi:10.3390/cells8080898
Amoroso, A., Garzia, P., Vadacca, M., Galluzzo, S., Del Porto, F., Mitterhofer, A. P., et al. (2003). The unusual association of three autoimmune diseases in a patient with Noonan syndrome. J. Adolesc. Health 32 (1), 94–97. doi:10.1016/s1054-139x(02)00364-6
Baechler, E. C., Batliwalla, F. M., Karypis, G., Gaffney, P. M., Ortmann, W. A., Espe, K. J., et al. (2003). Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. U. S. A. 100 (5), 2610–2615. doi:10.1073/pnas.0337679100
Ballestar, E. (2011). Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 7 (5), 263–271. doi:10.1038/nrrheum.2011.16
Banchereau, R., Hong, S., Cantarel, B., Baldwin, N., Baisch, J., Edens, M., et al. (2016). Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165 (6), 1548–1550. doi:10.1016/j.cell.2016.05.057
Barreto, M., Santos, E., Ferreira, R., Fesel, C., Fontes, M. F., Pereira, C., et al. (2004). Evidence for CTLA4 as a susceptibility gene for systemic lupus erythematosus. Eur. J. Hum. Genet. 12 (8), 620–626. doi:10.1038/sj.ejhg.5201214
Batu, E. D., Koşukcu, C., Taşkıran, E., Sahin, S., Akman, S., Sözeri, B., et al. (2018). Whole exome sequencing in early-onset systemic lupus erythematosus. J. Rheumatol. 45 (12), 1671–1679. doi:10.3899/jrheum.171358
Begum, S. (2022). Project SG100K: DNA of 100,000 Singaporeans to be mapped to identify new ways to prevent diseases. Straits Times.
Belot, A., Rice, G. I., Omarjee, S. O., Rouchon, Q., Smith, E. M. D., Moreews, M., et al. (2020). Contribution of rare and predicted pathogenic gene variants to childhood-onset lupus: a large, genetic panel analysis of British and French cohorts. Lancet Rheumatol. 2 (2), e99–e109. doi:10.1016/S2665-9913(19)30142-0
Bentham, J., Morris, D. L., Graham, D. S. C., Pinder, C. L., Tombleson, P., Behrens, T. W., et al. (2015). Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47 (12), 1457–1464. doi:10.1038/ng.3434
Bienaime, F., Quartier, P., Dragon-Durey, M. A., Fremeaux-Bacchi, V., Bader-Meunier, B., Patey, N., et al. (2010). Lupus nephritis associated with complete C1s deficiency efficiently treated with rituximab: a case report. Arthritis Care Res. Hob. 62 (9), 1346–1350. doi:10.1002/acr.20163
Blanchong, C. A., Chung, E. K., Rupert, K. L., Yang, Y., Yang, Z., Zhou, B., et al. (2001). Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, Slp and C4. Int. Immunopharmacol. 1 (3), 365–392. doi:10.1016/s1567-5769(01)00019-4
Bourchany, A., Thauvin-Robinet, C., Lehalle, D., Bruel, A. L., Masurel-Paulet, A., Jean, N., et al. (2017). Reducing diagnostic turnaround times of exome sequencing for families requiring timely diagnoses. Eur. J. Med. Genet. 60 (11), 595–604. doi:10.1016/j.ejmg.2017.08.011
Brown, G. J., Canete, P. F., Wang, H., Medhavy, A., Bones, J., Roco, J. A., et al. (2022). TLR7 gain-of-function genetic variation causes human lupus. Nature 605 (7909), 349–356. doi:10.1038/s41586-022-04642-z
Bundhun, P. K., Soogund, M. Z., and Huang, F. (2017). Impact of systemic lupus erythematosus on maternal and fetal outcomes following pregnancy: a meta-analysis of studies published between years 2001-2016. J. Autoimmun. 79, 17–27. doi:10.1016/j.jaut.2017.02.009
Burdick, K. J., Cogan, J. D., Rives, L. C., Robertson, A. K., Koziura, M. E., Brokamp, E., et al. (2020). Limitations of exome sequencing in detecting rare and undiagnosed diseases. Am. J. Med. Genet. A 182 (6), 1400–1406. doi:10.1002/ajmg.a.61558
Bycroft, C., Freeman, C., Petkova, D., Band, G., Elliott, L. T., Sharp, K., et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562 (7726), 203–209. doi:10.1038/s41586-018-0579-z
Caielli, S., Wan, Z., and Pascual, V. (2023). Systemic lupus erythematosus pathogenesis: interferon and beyond. Annu. Rev. Immunol. 41, 533–560. doi:10.1146/annurev-immunol-101921-042422
Chan, S. H., Bylstra, Y., Teo, J. X., Kuan, J. L., Bertin, N., Gonzalez-Porta, M., et al. (2022). Analysis of clinically relevant variants from ancestrally diverse Asian genomes. Nat. Commun. 13 (1), 6694. doi:10.1038/s41467-022-34116-9
Charras, A., Haldenby, S., Smith, E. M. D., Egbivwie, N., Olohan, L., Kenny, J. G., et al. (2023). Panel sequencing links rare, likely damaging gene variants with distinct clinical phenotypes and outcomes in juvenile-onset SLE. Rheumatol. Oxf. 62 (SI2), SI210–SI225. doi:10.1093/rheumatology/keac275
Charras, A., Smith, E., and Hedrich, C. M. (2021). Systemic lupus erythematosus in children and young people. Curr. Rheumatol. Rep. 23 (3), 20. doi:10.1007/s11926-021-00985-0
Chen, J., Zhang, P., Chen, H., Wang, X., He, X., Zhong, J., et al. (2022). Whole-genome sequencing identifies rare missense variants of WNT16 and ERVW-1 causing the systemic lupus erythematosus. Genomics 114 (3), 110332. doi:10.1016/j.ygeno.2022.110332
Chen, L., Morris, D. L., and Vyse, T. J. (2017). Genetic advances in systemic lupus erythematosus: an update. Curr. Opin. Rheumatol. 29 (5), 423–433. doi:10.1097/BOR.0000000000000411
Chen, L., Wang, Y. F., Liu, L., Bielowka, A., Ahmed, R., Zhang, H., et al. (2020). Genome-wide assessment of genetic risk for systemic lupus erythematosus and disease severity. Hum. Mol. Genet. 29 (10), 1745–1756. doi:10.1093/hmg/ddaa030
Chen, Z., Chen, J., Collins, R., Guo, Y., Peto, R., Wu, F., et al. (2011). China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int. J. Epidemiol. 40 (6), 1652–1666. doi:10.1093/ije/dyr120
Conrad, D. F., Andrews, T. D., Carter, N. P., Hurles, M. E., and Pritchard, J. K. (2006). A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 38 (1), 75–81. doi:10.1038/ng1697
Consortium, H. A., Rotimi, C., Abayomi, A., Abimiku, A., Adabayeri, V. M., Adebamowo, C., et al. (2014). Research capacity. Enabling the genomic revolution in Africa. Science 344 (6190), 1346–1348. doi:10.1126/science.1251546
Costa-Reis, P., and Sullivan, K. E. (2017). Monogenic lupus: it's all new. Curr. Opin. Immunol. 49, 87–95. doi:10.1016/j.coi.2017.10.008
Crow, M. K., and Ronnblom, L. (2019). Type I interferons in host defence and inflammatory diseases. Lupus Sci. Med. 6 (1), e000336. doi:10.1136/lupus-2019-000336
Crow, Y. J., Chase, D. S., Lowenstein Schmidt, J., Szynkiewicz, M., Forte, G. M., Gornall, H. L., et al. (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 167A (2), 296–312. doi:10.1002/ajmg.a.36887
Cunninghame Graham, D. S., Wong, A. K., McHugh, N. J., Whittaker, J. C., and Vyse, T. J. (2006). Evidence for unique association signals in SLE at the CD28-CTLA4-ICOS locus in a family-based study. Hum. Mol. Genet. 15 (21), 3195–3205. doi:10.1093/hmg/ddl395
D’Aurizio, R., Pippucci, T., Tattini, L., Giusti, B., Pellegrini, M., and Magi, A. (2016). Enhanced copy number variants detection from whole-exome sequencing data using EXCAVATOR2. Nucleic Acids Res. 44 (20), e154. doi:10.1093/nar/gkw695
D’Aurizio, R., Semeraro, R., and Magi, A. (2018). Using XCAVATOR and EXCAVATOR2 to identify CNVs from WGS, WES, and TS data. Curr. Protoc. Hum. Genet. 98 (1), e65. doi:10.1002/cphg.65
Delgado-Vega, A. M., Martínez-Bueno, M., Oparina, N. Y., López Herráez, D., Kristjansdottir, H., Steinsson, K., et al. (2018). Whole exome sequencing of patients from multicase families with systemic lupus erythematosus identifies multiple rare variants. Sci. Rep. 8 (1), 8775. doi:10.1038/s41598-018-26274-y
Demirkaya, E., Zhou, Q., Smith, C. K., Ombrello, M. J., Deuitch, N., Tsai, W. L., et al. (2017). Brief report: deficiency of complement 1r subcomponent in early-onset systemic lupus erythematosus: the role of disease-modifying alleles in a monogenic disease. Arthritis Rheumatol. 69 (9), 1832–1839. doi:10.1002/art.40158
Deng, Y., and Tsao, B. P. (2014). Advances in lupus genetics and epigenetics. Curr. Opin. Rheumatol. 26 (5), 482–492. doi:10.1097/BOR.0000000000000086
Der, E., Suryawanshi, H., Morozov, P., Kustagi, M., Goilav, B., Ranabothu, S., et al. (2019). Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat. Immunol. 20 (7), 915–927. doi:10.1038/s41590-019-0386-1
Duncavage, E. J., Schroeder, M. C., O'Laughlin, M., Wilson, R., MacMillan, S., Bohannon, A., et al. (2021). Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N. Engl. J. Med. 384 (10), 924–935. doi:10.1056/NEJMoa2024534
Elghzaly, A. A., Sun, C., Looger, L. L., Hirose, M., Salama, M., Khalil, N. M., et al. (2022). Genome-wide association study for systemic lupus erythematosus in an egyptian population. Front. Genet. 13, 948505. doi:10.3389/fgene.2022.948505
Fang, M., Abolhassani, H., Lim, C. K., Zhang, J., and Hammarstrom, L. (2016). Next generation sequencing data analysis in primary immunodeficiency disorders - future directions. J. Clin. Immunol. 36 (Suppl. 1), 68–75. doi:10.1007/s10875-016-0260-y
Fasano, S., Milone, A., Nicoletti, G. F., Isenberg, D. A., and Ciccia, F. (2023). Precision medicine in systemic lupus erythematosus. Nat. Rev. Rheumatol. 19 (6), 331–342. doi:10.1038/s41584-023-00948-y
Fernando, M. M., Freudenberg, J., Lee, A., Morris, D. L., Boteva, L., Rhodes, B., et al. (2012). Transancestral mapping of the MHC region in systemic lupus erythematosus identifies new independent and interacting loci at MSH5, HLA-DPB1 and HLA-G. Ann. Rheum. Dis. 71 (5), 777–784. doi:10.1136/annrheumdis-2011-200808
Feuk, L., Carson, A. R., and Scherer, S. W. (2006). Structural variation in the human genome. Nat. Rev. Genet. 7 (2), 85–97. doi:10.1038/nrg1767
Field, M. A. (2021). Detecting pathogenic variants in autoimmune diseases using high-throughput sequencing. Immunol. Cell Biol. 99 (2), 146–156. doi:10.1111/imcb.12372
Fitzgerald, T., and Birney, E. (2022). CNest: a novel copy number association discovery method uncovers 862 new associations from 200,629 whole-exome sequence datasets in the UK Biobank. Cell Genom 2 (8), 100167. doi:10.1016/j.xgen.2022.100167
Gabrielaite, M., Torp, M. H., Rasmussen, M. S., Andreu-Sanchez, S., Vieira, F. G., Pedersen, C. B., et al. (2021). A comparison of tools for copy-number variation detection in germline whole exome and whole genome sequencing data. Cancers (Basel). 13 (24), 6283. doi:10.3390/cancers13246283
Garcia-Ortiz, H., Velazquez-Cruz, R., Espinosa-Rosales, F., Jimenez-Morales, S., Baca, V., and Orozco, L. (2010). Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann. Rheum. Dis. 69 (10), 1861–1865. doi:10.1136/ard.2009.124313
Gateva, V., Sandling, J. K., Hom, G., Taylor, K. E., Chung, S. A., Sun, X., et al. (2009). A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41 (11), 1228–1233. doi:10.1038/ng.468
Gaynor, S. M., Joseph, T., Bai, X., Krasheninina, O., Boutkov, B., Maxwell, E., et al. (2023). Yield of genetic association signals from genomes, exomes, and imputations in the UK biobank. medRxiv.
Genomes Project, C., Abecasis, G. R., Altshuler, D., Auton, A., Brooks, L. D., Durbin, R. M., et al. (2010). A map of human genome variation from population-scale sequencing. Nature 467 (7319), 1061–1073. doi:10.1038/nature09534
Genomes Project, C., Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526 (7571), 68–74. doi:10.1038/nature15393
German, D., and Stephan, O. (2019). ClinCNV: novel method for allele-specific somatic copy-number alterations detection. bioRxiv.
Giltiay, N. V., Chappell, C. P., Sun, X., Kolhatkar, N., Teal, T. H., Wiedeman, A. E., et al. (2013). Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. J. Exp. Med. 210 (12), 2773–2789. doi:10.1084/jem.20122798
Gonzalez, E., Kulkarni, H., Bolivar, H., Mangano, A., Sanchez, R., Catano, G., et al. (2005). The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307 (5714), 1434–1440. doi:10.1126/science.1101160
Goodnow, C. C., Sprent, J., Fazekas de St Groth, B., and Vinuesa, C. G. (2005). Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 435 (7042), 590–597. doi:10.1038/nature03724
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17 (6), 333–351. doi:10.1038/nrg.2016.49
Graham, R. R., Cotsapas, C., Davies, L., Hackett, R., Lessard, C. J., Leon, J. M., et al. (2008). Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 40 (9), 1059–1061. doi:10.1038/ng.200
Gulati, G., and Brunner, H. I. (2018). Environmental triggers in systemic lupus erythematosus. Semin. Arthritis Rheum. 47 (5), 710–717. doi:10.1016/j.semarthrit.2017.10.001
Gulilat, M., Lamb, T., Teft, W. A., Wang, J., Dron, J. S., Robinson, J. F., et al. (2019). Targeted next generation sequencing as a tool for precision medicine. BMC Med. Genomics 12 (1), 81. doi:10.1186/s12920-019-0527-2
Han, J. W., Zheng, H. F., Cui, Y., Sun, L. D., Ye, D. Q., Hu, Z., et al. (2009). Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 41 (11), 1234–1237. doi:10.1038/ng.472
Hanaei, S., Sanati, G., Zoghi, S., Gharibzadeh, S., Ziaee, V., and Rezaei, N. (2020). The status of FOXP3 gene methylation in pediatric systemic lupus erythematosus. Allergol. Immunopathol. Madr. 48 (4), 332–338. doi:10.1016/j.aller.2020.03.014
Hanaya, A., Miyamae, T., Kishi, T., Sahara, M., Tani, Y., Yamanaka, H., et al. (2017). Systemic lupus erythematosus associated with RASopathy. Mod. Rheumatol. Case Rep. 1 (2), 94–98. doi:10.1080/24725625.2017.1337310
Hanscombe, K. B., Morris, D. L., Noble, J. A., Dilthey, A. T., Tombleson, P., Kaufman, K. M., et al. (2018). Genetic fine mapping of systemic lupus erythematosus MHC associations in Europeans and African Americans. Hum. Mol. Genet. 27 (21), 3813–3824. doi:10.1093/hmg/ddy280
Harley, I. T. W., and Sawalha, A. H. (2022). Systemic lupus erythematosus as a genetic disease. Clin. Immunol. 236, 108953. doi:10.1016/j.clim.2022.108953
Hassan, S., Bahar, R., Johan, M. F., Mohamed Hashim, E. K., Abdullah, W. Z., Esa, E., et al. (2023). Next-generation sequencing (NGS) and third-generation sequencing (TGS) for the diagnosis of thalassemia. Diagn. (Basel) 13 (3), 373. doi:10.3390/diagnostics13030373
Hedrich, C. M. (2017). Epigenetics in SLE. Curr. Rheumatol. Rep. 19 (9), 58. doi:10.1007/s11926-017-0685-1
Hom, G., Graham, R. R., Modrek, B., Taylor, K. E., Ortmann, W., Garnier, S., et al. (2008). Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 358 (9), 900–909. doi:10.1056/NEJMoa0707865
Hong, S. M., Chen, W., Feng, J., Dai, D., and Shen, N. (2022). Novel mutations in ACP5 and SAMHD1 in a patient with pediatric systemic lupus erythematosus. Front. Pediatr. 10, 885006. doi:10.3389/fped.2022.885006
Hu, H., Geng, Z., Zhang, S., Xu, Y., Wang, Q., Chen, S., et al. (2023). Rare copy number variation analysis identifies disease-related variants in atrioventricular septal defect patients. Front. Genet. 14, 1075349. doi:10.3389/fgene.2023.1075349
Hu, N., Qiu, X., Luo, Y., Yuan, J., Li, Y., Lei, W., et al. (2008). Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 35 (5), 804–810.
Huang, X., Luu, L. D. W., Jia, N., Zhu, J., Fu, J., Xiao, F., et al. (2022). Multi-platform omics analysis reveals molecular signatures for pathogenesis and activity of systemic lupus erythematosus. Front. Immunol. 13, 833699. doi:10.3389/fimmu.2022.833699
Hudson, L. L., Rocca, K., Song, Y. W., and Pandey, J. P. (2002). CTLA-4 gene polymorphisms in systemic lupus erythematosus: a highly significant association with a determinant in the promoter region. Hum. Genet. 111 (4-5), 452–455. doi:10.1007/s00439-002-0807-2
Hughes, T., Adler, A., Kelly, J. A., Kaufman, K. M., Williams, A. H., Langefeld, C. D., et al. (2012). Evidence for gene-gene epistatic interactions among susceptibility loci for systemic lupus erythematosus. Arthritis Rheum. 64 (2), 485–492. doi:10.1002/art.33354
Human Genome Structural Variation Working, G., Eichler, E. E., Nickerson, D. A., Altshuler, D., Bowcock, A. M., Brooks, L. D., et al. (2007). Completing the map of human genetic variation. Nature 447 (7141), 161–165. doi:10.1038/447161a
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y., et al. (2004). Detection of large-scale variation in the human genome. Nat. Genet. 36 (9), 949–951. doi:10.1038/ng1416
Jog, N. R., and James, J. A. (2020). Epstein barr virus and autoimmune responses in systemic lupus erythematosus. Front. Immunol. 11, 623944. doi:10.3389/fimmu.2020.623944
Jones, S. A., Cantsilieris, S., Fan, H., Cheng, Q., Russ, B. E., Tucker, E. J., et al. (2019). Rare variants in non-coding regulatory regions of the genome that affect gene expression in systemic lupus erythematosus. Sci. Rep. 9 (1), 15433. doi:10.1038/s41598-019-51864-9
Jury, E. C., Flores-Borja, F., Kalsi, H. S., Lazarus, M., Isenberg, D. A., Mauri, C., et al. (2010). Abnormal CTLA-4 function in T cells from patients with systemic lupus erythematosus. Eur. J. Immunol. 40 (2), 569–578. doi:10.1002/eji.200939781
Kamitaki, N., Sekar, A., Handsaker, R. E., de Rivera, H., Tooley, K., Morris, D. L., et al. (2020). Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 582 (7813), 577–581. doi:10.1038/s41586-020-2277-x
Kanai, M., Akiyama, M., Takahashi, A., Matoba, N., Momozawa, Y., Ikeda, M., et al. (2018). Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 50 (3), 390–400. doi:10.1038/s41588-018-0047-6
Kang, J. H., Xu, H., Choi, S. E., Park, D. J., Lee, J. K., Kwok, S. K., et al. (2020). Obesity increases the incidence of new-onset lupus nephritis and organ damage during follow-up in patients with systemic lupus erythematosus. Lupus 29 (6), 578–586. doi:10.1177/0961203320913616
Keshavarz-Fathi, M., Sanati, G., Sadr, M., Mohebbi, B., Ziaee, V., and Rezaei, N. (2022). DNA methylation of CD70 promoter in juvenile systemic lupus erythematosus. Fetal Pediatr. Pathol. 41 (1), 58–67. doi:10.1080/15513815.2020.1764681
Khunsriraksakul, C., Li, Q., Markus, H., Patrick, M. T., Sauteraud, R., McGuire, D., et al. (2023). Multi-ancestry and multi-trait genome-wide association meta-analyses inform clinical risk prediction for systemic lupus erythematosus. Nat. Commun. 14 (1), 668. doi:10.1038/s41467-023-36306-5
Khunsriraksakul, C., Markus, H., Olsen, N. J., Carrel, L., Jiang, B., and Liu, D. J. (2022). Construction and application of polygenic risk scores in autoimmune diseases. Front. Immunol. 13, 889296. doi:10.3389/fimmu.2022.889296
Kilburn, K. H., and Warshaw, R. H. (1992). Prevalence of symptoms of systemic lupus erythematosus (SLE) and of fluorescent antinuclear antibodies associated with chronic exposure to trichloroethylene and other chemicals in well water. Environ. Res. 57 (1), 1–9. doi:10.1016/s0013-9351(05)80014-3
Kim, J. H., Jung, S. H., Bae, J. S., Lee, H. S., Yim, S. H., Park, S. Y., et al. (2013). Deletion variants of RABGAP1L, 10q21.3, and C4 are associated with the risk of systemic lupus erythematosus in Korean women. Arthritis Rheum. 65 (4), 1055–1063. doi:10.1002/art.37854
Klambauer, G., Schwarzbauer, K., Mayr, A., Clevert, D. A., Mitterecker, A., Bodenhofer, U., et al. (2012). cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res. 40 (9), e69. doi:10.1093/nar/gks003
Kozyrev, S. V., Abelson, A. K., Wojcik, J., Zaghlool, A., Linga Reddy, M. V., Sanchez, E., et al. (2008). Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 40 (2), 211–216. doi:10.1038/ng.79
Krumm, N., Sudmant, P. H., Ko, A., O'Roak, B. J., Malig, M., Coe, B. P., et al. (2012). Copy number variation detection and genotyping from exome sequence data. Genome Res. 22 (8), 1525–1532. doi:10.1101/gr.138115.112
Kuhlenbaumer, G., Hullmann, J., and Appenzeller, S. (2011). Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum. Mutat. 32 (2), 144–151. doi:10.1002/humu.21400
Kusmirek, W., and Nowak, R. (2022). CNVind: an open source cloud-based pipeline for rare CNVs detection in whole exome sequencing data based on the depth of coverage. BMC Bioinforma. 23 (1), 85. doi:10.1186/s12859-022-04617-x
Kwon, Y. C., Chun, S., Kim, K., and Mak, A. (2019). Update on the genetics of systemic lupus erythematosus: genome-wide association studies and beyond. Cells 8 (10), 1180. doi:10.3390/cells8101180
Lai, W., Feng, X., Yue, M., Cheung, P. W. H., Choi, V. N. T., Song, Y. Q., et al. (2021). Identification of copy number variants in a southern Chinese cohort of patients with congenital scoliosis. Genes (Basel) 12 (8), 1213. doi:10.3390/genes12081213
Lee, J. A., Madrid, R. E., Sperle, K., Ritterson, C. M., Hobson, G. M., Garbern, J., et al. (2006). Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann. Neurol. 59 (2), 398–403. doi:10.1002/ana.20732
Lee, J. Y., Kannan, B., Lim, B. Y., Li, Z., Lim, A. H., Loh, J. W., et al. (2022). The multi-dimensional biomarker landscape in cancer immunotherapy. Int. J. Mol. Sci. 23 (14), 7839. doi:10.3390/ijms23147839
Lee, Y. H., Harley, J. B., and Nath, S. K. (2005). CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Hum. Genet. 116 (5), 361–367. doi:10.1007/s00439-004-1244-1
Lelieveld, S. H., Spielmann, M., Mundlos, S., Veltman, J. A., and Gilissen, C. (2015). Comparison of exome and genome sequencing technologies for the complete capture of protein-coding regions. Hum. Mutat. 36 (8), 815–822. doi:10.1002/humu.22813
Leventopoulos, G., Denayer, E., Makrythanasis, P., Papapolychroniou, C., and Fryssira, H. (2010). Noonan syndrome and systemic lupus erythematosus in a patient with a novel KRAS mutation. Clin. Exp. Rheumatol. 28 (4), 556–557.
Levy, S. E., and Myers, R. M. (2016). Advancements in next-generation sequencing. Annu. Rev. Genomics Hum. Genet. 17, 95–115. doi:10.1146/annurev-genom-083115-022413
Lewis, M. J., and Jawad, A. S. (2017). The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatol. Oxf. 56 (Suppl. l_1), i67–i77. doi:10.1093/rheumatology/kew399
Li, D., Gao, H., Zheng, W., Jin, C., Huang, Y., and Pan, S. (2022). Case report: fetal cervical immature teratoma and copy number variations. Front. Oncol. 12, 843268. doi:10.3389/fonc.2022.843268
Li, G., Li, Y., Liu, H., Shi, Y., Guan, W., Zhang, T., et al. (2020). Genetic heterogeneity of pediatric systemic lupus erythematosus with lymphoproliferation. Med. Baltim. 99 (20), e20232. doi:10.1097/MD.0000000000020232
Liaw, D., Marsh, D. J., Li, J., Dahia, P. L., Wang, S. I., Zheng, Z., et al. (1997). Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 16 (1), 64–67. doi:10.1038/ng0597-64
Lisbona, M. P., Moreno, M., Orellana, C., Gratacos, J., and Larrosa, M. (2009). Noonan syndrome associated with systemic lupus erythematosus. Lupus 18 (3), 267–269. doi:10.1177/0961203308094996
Liu, J., and Zhang, H. X. (2013). CTLA-4 polymorphisms and systemic lupus erythematosus: a comprehensive meta-analysis. Genet. Test. Mol. Biomarkers 17 (3), 226–231. doi:10.1089/gtmb.2012.0302
Liu, Y., Lei, C., Wang, R., Yang, D., Yang, B., Xu, Y., et al. (2022). Case report: whole-exome sequencing-based copy number variation analysis identified a novel DRC1 homozygous exon deletion in a patient with primary ciliary dyskinesia. Front. Genet. 13, 940292. doi:10.3389/fgene.2022.940292
Lood, C., Gullstrand, B., Truedsson, L., Olin, A. I., Alm, G. V., Ronnblom, L., et al. (2009). C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. 60 (10), 3081–3090. doi:10.1002/art.24852
Love, M. I., Mysickova, A., Sun, R., Kalscheuer, V., Vingron, M., and Haas, S. A. (2011). Modeling read counts for CNV detection in exome sequencing data. Stat. Appl. Genet. Mol. Biol. 10 (1), 52. doi:10.2202/1544-6115.1732
Lovewell, T. R., McDonagh, A. J., Messenger, A. G., Azzouz, M., and Tazi-Ahnini, R. (2015). The AIRE -230Y polymorphism affects AIRE transcriptional activity: potential influence on AIRE function in the thymus. PLoS One 10 (5), e0127476. doi:10.1371/journal.pone.0127476
Lund, J. M., Alexopoulou, L., Sato, A., Karow, M., Adams, N. C., Gale, N. W., et al. (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. U. S. A. 101 (15), 5598–5603. doi:10.1073/pnas.0400937101
Lundtoft, C., Pucholt, P., Martin, M., Bianchi, M., Lundstrom, E., Eloranta, M. L., et al. (2022). Complement C4 copy number variation is linked to SSA/ro and SSB/La autoantibodies in systemic inflammatory autoimmune diseases. Arthritis Rheumatol. 74 (8), 1440–1450. doi:10.1002/art.42122
Majewski, J., Schwartzentruber, J., Lalonde, E., Montpetit, A., and Jabado, N. (2011). What can exome sequencing do for you? J. Med. Genet. 48 (9), 580–589. doi:10.1136/jmedgenet-2011-100223
Mandelker, D., Schmidt, R. J., Ankala, A., McDonald Gibson, K., Bowser, M., Sharma, H., et al. (2016). Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next-generation sequencing. Genet. Med. 18 (12), 1282–1289. doi:10.1038/gim.2016.58
Manrai, A. K., Funke, B. H., Rehm, H. L., Olesen, M. S., Maron, B. A., Szolovits, P., et al. (2016). Genetic misdiagnoses and the potential for Health disparities. N. Engl. J. Med. 375 (7), 655–665. doi:10.1056/NEJMsa1507092
Marchuk, D. S., Crooks, K., Strande, N., Kaiser-Rogers, K., Milko, L. V., Brandt, A., et al. (2018). Increasing the diagnostic yield of exome sequencing by copy number variant analysis. PLoS One 13 (12), e0209185. doi:10.1371/journal.pone.0209185
Martinez-Bueno, M., Oparina, N., Dozmorov, M. G., Marion, M. C., Comeau, M. E., Gilkeson, G., et al. (2018). Trans-Ethnic mapping of BANK1 identifies two independent SLE-risk linkage groups enriched for Co-transcriptional splicing marks. Int. J. Mol. Sci. 19 (8), 2331. doi:10.3390/ijms19082331
McCarroll, S. A., Hadnott, T. N., Perry, G. H., Sabeti, P. C., Zody, M. C., Barrett, J. C., et al. (2006). Common deletion polymorphisms in the human genome. Nat. Genet. 38 (1), 86–92. doi:10.1038/ng1696
Miller, E. C., and Atkinson, J. P. (2012). Overcoming C2 deficiency. Clin. Immunol. 144 (3), 269–271. doi:10.1016/j.clim.2012.07.005
Mina, R., and Brunner, H. I. (2013). Update on differences between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Res. Ther. 15 (4), 218. doi:10.1186/ar4256
Minegishi, N., Nishijima, I., Nobukuni, T., Kudo, H., Ishida, N., Terakawa, T., et al. (2019). Biobank establishment and sample management in the Tohoku medical megabank project. Tohoku J. Exp. Med. 248 (1), 45–55. doi:10.1620/tjem.248.45
Montufar-Robles, I., Robles-Garnica, J. C., Cadena-Sandoval, D., Barbosa-Cobos, R. E., Gonzalez-Castillo, D. D., Romero-Diaz, J., et al. (2019). The AIRE Ser196Ser synonymous variant is a risk factor for systemic lupus erythematosus. Cell Immunol. 346, 103986. doi:10.1016/j.cellimm.2019.103986
Mueller, M., Barros, P., Witherden, A. S., Roberts, A. L., Zhang, Z., Schaschl, H., et al. (2013). Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am. J. Hum. Genet. 92 (1), 28–40. doi:10.1016/j.ajhg.2012.11.013
Mulder, N., Abimiku, A., Adebamowo, S. N., de Vries, J., Matimba, A., Olowoyo, P., et al. (2018). H3Africa: current perspectives. Pharmgenomics Pers. Med. 11, 59–66. doi:10.2147/PGPM.S141546
Musone, S. L., Taylor, K. E., Lu, T. T., Nititham, J., Ferreira, R. C., Ortmann, W., et al. (2008). Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat. Genet. 40 (9), 1062–1064. doi:10.1038/ng.202
Neuman, H., and Koren, O. (2017). The gut microbiota: a possible factor influencing systemic lupus erythematosus. Curr. Opin. Rheumatol. 29 (4), 374–377. doi:10.1097/BOR.0000000000000395
Ng, S. B., Turner, E. H., Robertson, P. D., Flygare, S. D., Bigham, A. W., Lee, C., et al. (2009). Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461 (7261), 272–276. doi:10.1038/nature08250
Niederer, H. A., Clatworthy, M. R., Willcocks, L. C., and Smith, K. G. (2010). FcgammaRIIB, FcgammaRIIIB, and systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 1183, 69–88. doi:10.1111/j.1749-6632.2009.05132.x
Nosrat zehi, S., Nosrat zehi, M., Atighi, S., Dianat, T., and Kord Tamandani, K. (2021). Promoter methylation and expression status of cytotoxic T-lymphocyte-associated antigen-4 gene in patients with lupus. J. Epigenetics 2 (1), 31–38. doi:10.22111/jep.2020.30124.1019
Oftedal, B. E., Berger, A. H., Bruserud, O., Goldfarb, Y., Sulen, A., Breivik, L., et al. (2023). A partial form of AIRE deficiency underlies a mild form of autoimmune polyendocrine syndrome type 1. J. Clin. Invest. 133 (21), e169704. doi:10.1172/JCI169704
Olfe, L., von Hardenberg, S., Hofmann, W., Auber, B., Baumann, U., Beier, R., et al. (2023). CTLA-4 insufficiency due to a novel CTLA-4 deletion, identified through copy number variation analysis. Int. Arch. Allergy Immunol. 184 (1), 76–84. doi:10.1159/000527051
Papp, O., Doma, V., Gil, J., Marko-Varga, G., Karpati, S., Timar, J., et al. (2021). Organ specific copy number variations in visceral metastases of human melanoma. Cancers (Basel) 13 (23), 5984. doi:10.3390/cancers13235984
Parks, C. G., de Souza Espindola Santos, A., Barbhaiya, M., and Costenbader, K. H. (2017). Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best. Pract. Res. Clin. Rheumatol. 31 (3), 306–320. doi:10.1016/j.berh.2017.09.005
Pereira, K. M. C., Perazzio, S., Faria, A. G. A., Moreira, E. S., Santos, V. C., Grecco, M., et al. (2019). Impact of C4, C4A and C4B gene copy number variation in the susceptibility, phenotype and progression of systemic lupus erythematosus. Adv. Rheumatol. 59 (1), 36. doi:10.1186/s42358-019-0076-6
Petersen, B. S., Fredrich, B., Hoeppner, M. P., Ellinghaus, D., and Franke, A. (2017). Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet. 18 (1), 14. doi:10.1186/s12863-017-0479-5
Poole, B. D., Scofield, R. H., Harley, J. B., and James, J. A. (2006). Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 39 (1), 63–70. doi:10.1080/08916930500484849
Pullabhatla, V., Roberts, A. L., Lewis, M. J., Mauro, D., Morris, D. L., Odhams, C. A., et al. (2018). De novo mutations implicate novel genes in systemic lupus erythematosus. Hum. Mol. Genet. 27 (3), 421–429. doi:10.1093/hmg/ddx407
Pullmann, R., Lukac, J., Skerenova, M., Rovensky, J., Hybenova, J., Melus, V., et al. (1999). Cytotoxic T lymphocyte antigen 4 (CTLA-4) dimorphism in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 17 (6), 725–729.
Raj, P., Song, R., Zhu, H., Riediger, L., Jun, D. J., Liang, C., et al. (2020). Deep sequencing reveals a DAP1 regulatory haplotype that potentiates autoimmunity in systemic lupus erythematosus. Genome Biol. 21 (1), 281. doi:10.1186/s13059-020-02184-z
Rapti, M., Zouaghi, Y., Meylan, J., Ranza, E., Antonarakis, S. E., and Santoni, F. A. (2022). CoverageMaster: comprehensive CNV detection and visualization from NGS short reads for genetic medicine applications. Brief. Bioinform 23 (2), bbac049. doi:10.1093/bib/bbac049
Raupov, R. K., Suspitsin, E. N., Imelbaev, A. I., and Kostik, M. M. (2022). Simultaneous Onset of Pediatric Systemic Lupus Erythematosus in Twin Brothers: Case Report. Front. Pediatr. 10, 929358. doi:10.3389/fped.2022.929358
Ravenscroft, J. C., Suri, M., Rice, G. I., Szynkiewicz, M., and Crow, Y. J. (2011). Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am. J. Med. Genet. A 155A (1), 235–237. doi:10.1002/ajmg.a.33778
Refeat, M. M., Naggar, W. E., Saied, M. M. E., and Kilany, A. (2023). Whole exome screening of neurodevelopmental regression disorders in a cohort of Egyptian patients. Neurogenetics 24 (1), 17–28. doi:10.1007/s10048-022-00703-7
Rehm, H. L., Berg, J. S., Brooks, L. D., Bustamante, C. D., Evans, J. P., Landrum, M. J., et al. (2015). ClinGen--the clinical genome resource. N. Engl. J. Med. 372 (23), 2235–2242. doi:10.1056/NEJMsr1406261
Reid, S., Alexsson, A., Frodlund, M., Morris, D., Sandling, J. K., Bolin, K., et al. (2020). High genetic risk score is associated with early disease onset, damage accrual and decreased survival in systemic lupus erythematosus. Ann. Rheum. Dis. 79 (3), 363–369. doi:10.1136/annrheumdis-2019-216227
Remmers, E. F., Plenge, R. M., Lee, A. T., Graham, R. R., Hom, G., Behrens, T. W., et al. (2007). STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 357 (10), 977–986. doi:10.1056/NEJMoa073003
Reynier, F., Petit, F., Paye, M., Turrel-Davin, F., Imbert, P. E., Hot, A., et al. (2011). Importance of correlation between gene expression levels: application to the type I interferon signature in rheumatoid arthritis. PLoS One 6 (10), e24828. doi:10.1371/journal.pone.0024828
Rice, G. I., Del Toro Duany, Y., Jenkinson, E. M., Forte, G. M., Anderson, B. H., Ariaudo, G., et al. (2014). Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 46 (5), 503–509. doi:10.1038/ng.2933
Rice, G. I., Kasher, P. R., Forte, G. M., Mannion, N. M., Greenwood, S. M., Szynkiewicz, M., et al. (2012). Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 44 (11), 1243–1248. doi:10.1038/ng.2414
Rice, G. I., Rodero, M. P., and Crow, Y. J. (2015). Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol. 35 (3), 235–243. doi:10.1007/s10875-015-0147-3
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Rioux, J. D., Goyette, P., Vyse, T. J., Hammarström, L., Fernando, M. M., Green, T., et al. (2009). Mapping of multiple susceptibility variants within the MHC region for 7 immune-mediated diseases. Proc. Natl. Acad. Sci. U S A 106 (44), 18680–18685. doi:10.1073/pnas.0909307106
Royer-Bertrand, B., Cisarova, K., Niel-Butschi, F., Mittaz-Crettol, L., Fodstad, H., and Superti-Furga, A. (2021). CNV detection from exome sequencing data in routine diagnostics of rare genetic disorders: opportunities and limitations. Genes (Basel). 12 (9), 1427. doi:10.3390/genes12091427
Sandling, J. K., Pucholt, P., Hultin Rosenberg, L., Farias, F. H. G., Kozyrev, S. V., Eloranta, M. L., et al. (2021). Molecular pathways in patients with systemic lupus erythematosus revealed by gene-centred DNA sequencing. Ann. Rheum. Dis. 80 (1), 109–117. doi:10.1136/annrheumdis-2020-218636
Sanger, F., Nicklen, S., and Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74 (12), 5463–5467. doi:10.1073/pnas.74.12.5463
Satam, H., Joshi, K., Mangrolia, U., Waghoo, S., Zaidi, G., Rawool, S., et al. (2023). Next-generation sequencing technology: current trends and advancements. Biol. (Basel) 12 (7), 997. doi:10.3390/biology12070997
Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P., et al. (2004). Large-scale copy number polymorphism in the human genome. Science 305 (5683), 525–528. doi:10.1126/science.1098918
Sestak, A. L., Furnrohr, B. G., Harley, J. B., Merrill, J. T., and Namjou, B. (2011). The genetics of systemic lupus erythematosus and implications for targeted therapy. Ann. Rheum. Dis. 70 (Suppl. 1), i37–i43. doi:10.1136/ard.2010.138057
Shang, S., Zhou, Y., Chen, K., Chen, L., Li, P., Li, D., et al. (2022). A novel gene CDC27 causes SLE and is associated with the disease activity. Front. Immunol. 13, 876963. doi:10.3389/fimmu.2022.876963
Shaw, T., Fok, R., Courtney, E., Li, S. T., Chiang, J., and Ngeow, J. (2023). Missed diagnosis or misdiagnosis: common pitfalls in genetic testing. Singap. Med. J. 64 (1), 67–73. doi:10.4103/singaporemedj.SMJ-2021-467
Shibata, T., Ohto, U., Nomura, S., Kibata, K., Motoi, Y., Zhang, Y., et al. (2016). Guanosine and its modified derivatives are endogenous ligands for TLR7. Int. Immunol. 28 (5), 211–222. doi:10.1093/intimm/dxv062
Slatko, B. E., Gardner, A. F., and Ausubel, F. M. (2018). Overview of next-generation sequencing technologies. Curr. Protoc. Mol. Biol. 122 (1), e59. doi:10.1002/cpmb.59
Sohail, M., Palma-Martinez, M. J., Chong, A. Y., Quinto-Cortes, C. D., Barberena-Jonas, C., Medina-Munoz, S. G., et al. (2023). Mexican Biobank advances population and medical genomics of diverse ancestries. Nature 622, 775–783. doi:10.1038/s41586-023-06560-0
Somerville, M. J., Mervis, C. B., Young, E. J., Seo, E. J., del Campo, M., Bamforth, S., et al. (2005). Severe expressive-language delay related to duplication of the Williams-Beuren locus. N. Engl. J. Med. 353 (16), 1694–1701. doi:10.1056/NEJMoa051962
Speyer, C. B., and Costenbader, K. H. (2018). Cigarette smoking and the pathogenesis of systemic lupus erythematosus. Expert Rev. Clin. Immunol. 14 (6), 481–487. doi:10.1080/1744666X.2018.1473035
Sudmant, P. H., Rausch, T., Gardner, E. J., Handsaker, R. E., Abyzov, A., Huddleston, J., et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526 (7571), 75–81. doi:10.1038/nature15394
Taheri, M., Eghtedarian, R., Dinger, M. E., and Ghafouri-Fard, S. (2020). Exploring the role of non-coding RNAs in the pathophysiology of systemic lupus erythematosus. Biomolecules 10 (6), 937. doi:10.3390/biom10060937
Takeda, K., and Akira, S. (2005). Toll-like receptors in innate immunity. Int. Immunol. 17 (1), 1–14. doi:10.1093/intimm/dxh186
Talevich, E., Shain, A. H., Botton, T., and Bastian, B. C. (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12 (4), e1004873. doi:10.1371/journal.pcbi.1004873
Tam, V., Patel, N., Turcotte, M., Bosse, Y., Pare, G., and Meyre, D. (2019). Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20 (8), 467–484. doi:10.1038/s41576-019-0127-1
Tang, Y., and Luo, Y. (2022). Identification of a novel mutation in complement receptor 2 in Chinese familial systemic lupus erythematosus. Arch. Rheumatol. 37 (4), 566–573. doi:10.46497/ArchRheumatol.2022.9167
Terao, C., Yamada, R., Ohmura, K., Takahashi, M., Kawaguchi, T., Kochi, Y., et al. (2011). The human AIRE gene at chromosome 21q22 is a genetic determinant for the predisposition to rheumatoid arthritis in Japanese population. Hum. Mol. Genet. 20 (13), 2680–2685. doi:10.1093/hmg/ddr161
Tirosh, I., Spielman, S., Barel, O., Ram, R., Stauber, T., Paret, G., et al. (2019). Whole exome sequencing in childhood-onset lupus frequently detects single gene etiologies. Pediatr. Rheumatol. Online J. 17 (1), 52. doi:10.1186/s12969-019-0349-y
Torkamani, A., Wineinger, N. E., and Topol, E. J. (2018). The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 19 (9), 581–590. doi:10.1038/s41576-018-0018-x
Tsokos, G. C. (2011). Systemic lupus erythematosus. N. Engl. J. Med. 365 (22), 2110–2121. doi:10.1056/NEJMra1100359
Tuzun, E., Sharp, A. J., Bailey, J. A., Kaul, R., Morrison, V. A., Pertz, L. M., et al. (2005). Fine-scale structural variation of the human genome. Nat. Genet. 37 (7), 727–732. doi:10.1038/ng1562
Uehara, T., Hosogaya, N., Matsuo, N., and Kosaki, K. (2018). Systemic lupus erythematosus in a patient with Noonan syndrome-like disorder with loose anagen hair 1: more than a chance association. Am. J. Med. Genet. A 176 (7), 1662–1666. doi:10.1002/ajmg.a.38834
Usher, C. L., and McCarroll, S. A. (2015). Complex and multi-allelic copy number variation in human disease. Brief. Funct. Genomics 14 (5), 329–338. doi:10.1093/bfgp/elv028
Van Coillie, S., Wiernicki, B., and Xu, J. (2020). Molecular and cellular functions of CTLA-4. Adv. Exp. Med. Biol. 1248, 7–32. doi:10.1007/978-981-15-3266-5_2
van Dijk, E. L., Auger, H., Jaszczyszyn, Y., and Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends Genet. 30 (9), 418–426. doi:10.1016/j.tig.2014.07.001
Van Hout, C. V., Tachmazidou, I., Backman, J. D., Hoffman, J. D., Liu, D., Pandey, A. K., et al. (2020). Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586 (7831), 749–756. doi:10.1038/s41586-020-2853-0
Villarino, A. V., Kanno, Y., and O'Shea, J. J. (2017). Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 18 (4), 374–384. doi:10.1038/ni.3691
Vinuesa, C. G., Shen, N., and Ware, T. (2023). Genetics of SLE: mechanistic insights from monogenic disease and disease-associated variants. Nat. Rev. Nephrol. 19 (9), 558–572. doi:10.1038/s41581-023-00732-x
Vordenbaumen, S., Rosenbaum, A., Gebhard, C., Raithel, J., Sokolowski, A., Dusing, C., et al. (2021). Associations of site-specific CD4<b>+</b>-T-cell hypomethylation within CD40-ligand promotor and enhancer regions with disease activity of women with systemic lupus erythematosus. Lupus 30 (1), 45–51. doi:10.1177/0961203320965690
Wahren-Herlenius, M., and Dorner, T. (2013). Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 382 (9894), 819–831. doi:10.1016/S0140-6736(13)60954-X
Wang, Q., Qin, T., Wang, X., Li, J., Lin, X., Wang, D., et al. (2022). Whole-exome sequencing and copy number analysis in a patient with warburg micro syndrome. Genes (Basel) 13 (12), 2364. doi:10.3390/genes13122364
Wang, Y. F., Zhang, Y., Lin, Z., Zhang, H., Wang, T. Y., Cao, Y., et al. (2021). Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat. Commun. 12 (1), 772. doi:10.1038/s41467-021-21049-y
Webb, R., Kelly, J. A., Somers, E. C., Hughes, T., Kaufman, K. M., Sanchez, E., et al. (2011). Early disease onset is predicted by a higher genetic risk for lupus and is associated with a more severe phenotype in lupus patients. Ann. Rheum. Dis. 70 (1), 151–156. doi:10.1136/ard.2010.141697
Wei, W. H., Hemani, G., and Haley, C. S. (2014). Detecting epistasis in human complex traits. Nat. Rev. Genet. 15 (11), 722–733. doi:10.1038/nrg3747
Willcocks, L. C., Lyons, P. A., Clatworthy, M. R., Robinson, J. I., Yang, W., Newland, S. A., et al. (2008). Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J. Exp. Med. 205 (7), 1573–1582. doi:10.1084/jem.20072413
Wu, C. W., Lim, T. Y., Wang, C., Seltzsam, S., Zheng, B., Schierbaum, L., et al. (2022). Copy number variation analysis facilitates identification of genetic causation in patients with congenital anomalies of the kidney and urinary tract. Eur. Urol. Open Sci. 44, 106–112. doi:10.1016/j.euros.2022.08.004
Xiao, T., and Zhou, W. (2020). The third generation sequencing: the advanced approach to genetic diseases. Transl. Pediatr. 9 (2), 163–173. doi:10.21037/tp.2020.03.06
Yang, S., Fujikado, N., Kolodin, D., Benoist, C., and Mathis, D. (2015). Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348 (6234), 589–594. doi:10.1126/science.aaa7017
Yang, W., Shen, N., Ye, D. Q., Liu, Q., Zhang, Y., Qian, X. X., et al. (2010). Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 6 (2), e1000841. doi:10.1371/journal.pgen.1000841
Yang, Y., Chung, E. K., Wu, Y. L., Savelli, S. L., Nagaraja, H. N., Zhou, B., et al. (2007). Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am. J. Hum. Genet. 80 (6), 1037–1054. doi:10.1086/518257
Yasutomo, K., Horiuchi, T., Kagami, S., Tsukamoto, H., Hashimura, C., Urushihara, M., et al. (2001). Mutation of DNASE1 in people with systemic lupus erythematosus. Nat. Genet. 28 (4), 313–314. doi:10.1038/91070
Yaung, K., Yeo, J., Kumar, P., Wasser, M., Chew, M., Ravelli, A., et al. (2023). Artificial intelligence and high-dimensional technologies in the theragnosis of systemic lupus erythematosus. Lancet Rheumatology 5, e151–e165. doi:10.1016/S2665-9913(23)00010-3
You, Q., Yang, X., Peng, Z., Xu, L., and Wang, J. (2018). Development and applications of a high throughput genotyping tool for polyploid crops: single nucleotide polymorphism (SNP) array. Front. Plant Sci. 9, 104. doi:10.3389/fpls.2018.00104
Zawistowski, M., Fritsche, L. G., Pandit, A., Vanderwerff, B., Patil, S., Schmidt, E. M., et al. (2023). The Michigan Genomics Initiative: a biobank linking genotypes and electronic clinical records in Michigan Medicine patients. Cell Genom 3 (2), 100257. doi:10.1016/j.xgen.2023.100257
Zhang, M., Gu, Y., Huang, S., Lou, Q., Xie, Q., Xu, Z., et al. (2019). Copy number variations and polymorphisms in HSP90AB1 and risk of systemic lupus erythematosus and efficacy of glucocorticoids. J. Cell Mol. Med. 23 (8), 5340–5348. doi:10.1111/jcmm.14410
Zhang, Q., Qin, Z., Yi, S., Wei, H., Zhou, X. Z., and Su, J. (2021). Clinical application of whole-exome sequencing: a retrospective, single-center study. Exp. Ther. Med. 22 (1), 753. doi:10.3892/etm.2021.10185
Zhang, Z., Ohto, U., Shibata, T., Krayukhina, E., Taoka, M., Yamauchi, Y., et al. (2016). Structural analysis reveals that toll-like receptor 7 is a dual receptor for guanosine and single-stranded RNA. Immunity 45 (4), 737–748. doi:10.1016/j.immuni.2016.09.011
Zhang, Z., Ohto, U., Shibata, T., Taoka, M., Yamauchi, Y., Sato, R., et al. (2018). Structural analyses of toll-like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. 25 (12), 3371–3381. doi:10.1016/j.celrep.2018.11.081
Zhao, L., Liu, H., Yuan, X., Gao, K., and Duan, J. (2020). Comparative study of whole exome sequencing-based copy number variation detection tools. BMC Bioinforma. 21 (1), 97. doi:10.1186/s12859-020-3421-1
Keywords: systemic lupus erythematosus, genomics, next-generation sequencing, whole exome sequencing, monogenic, copy number variation
Citation: Yeo NK-W, Lim CK, Yaung KN, Khoo NKH, Arkachaisri T, Albani S and Yeo JG (2024) Genetic interrogation for sequence and copy number variants in systemic lupus erythematosus. Front. Genet. 15:1341272. doi: 10.3389/fgene.2024.1341272
Received: 20 November 2023; Accepted: 20 February 2024;
Published: 04 March 2024.
Edited by:
Seik-Soon Khor, Nanyang Technological University, SingaporeReviewed by:
Emanuele Micaglio, IRCCS San Donato Polyclinic, ItalyCopyright © 2024 Yeo, Lim, Yaung, Khoo, Arkachaisri, Albani and Yeo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joo Guan Yeo, Z21zeWVvQGR1a2UtbnVzLmVkdS5zZw==
†These authors have contributed equally to this work
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.