 Qingchun Wang
Qingchun Wang Haiying Bao1,2*
Haiying Bao1,2*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 13 July 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fgene.2023.1221491
This article is part of the Research Topic Genetics, Genomics, and Breeding of Edible Mushrooms in Asia View all 10 articles
Inonotus hispidus mainly growing in broad-leaved trees, including Morus alba, Fraxinus mandshurica, and Ulmus macrocarpa etc. The fruiting body of I. hispidus growing in M. alba (hereafter as MA) is used as a traditional Chinese medicine “Sanghuang”. However, differences between the genetic material basis of I. hispidus growing in other tree species have not been reported. Therefore, in this paper, the genomic comparison between MA and I. hispidus growing in F. mandshurica (hereafter as FM) were studied. The whole genome of MA monokaryon was sequenced by Illumina combined with Pac Bio platform. Next, genome assembly, genome component prediction and genome functional annotation were performed. Comparative genomics analysis was performed between FM monokaryon and MA monokaryon, using MA as the reference. The results showed that, MA had 24 contigs with a N50 length of 2.6 Mb. Specifically, 5,342, 6,564, 1,595, 383 and 123 genes were annotated from GO, KEGG, KOG, CAZymes and CYP450, respectively. Moreover, comparative genomics showed that, the coding genes and total number of genes annotated in different databases of FM were higher than that of MA. This study provides a foundation for the medicinal application of FM as MA from the perspective of genetic composition.
Edible and medicinal fungus Inonotus hispidus (Bull.: Fr.) P. Karst belongs to Basidiomycota, Agaricomycetes, Hymenochaetales, Hymenochaetaceae, and Inonotus. It is distributed in China, Korea, Japan, Germany, Russia, Canada and other countries. Recent international medical studies reported that its pharmacological activities include anti-tumor, anti-oxidation, anti-inflammatory, liver protection, bacteriostasis, anti-virus, hypolipidemic and immune enhancement (Awadh et al., 2003; Benarous et al., 2015; Gründemann et al., 2016; Angelini et al., 2019; Li et al., 2019; Liu et al., 2019; Yang et al., 2019; Yang et al., 2020; Li and Bao, 2022a; Zhang et al., 2022a; Yang et al., 2022; Machado-Carvalho et al., 2023; Saka et al., 2023; Tang et al., 2023). I. hispidus has an excellent development space as medical and functional food in future. Its main hosts were M. alba, F. mandshurica, U. macrocarpa, Z. jujuba and M. pumila (Dai et al., 2000). FM is mainly distributed in Northeast China (Dai et al., 2000), whereas MA is prevalent in Northeast, Northwest, North, East and Southwest of China (Bao et al., 2020). According to the morphological characteristics, habitat and herbal research of MA, some Chinese scholars believe that the MA was positive source of traditional Chinese medicine “Sanghuang” (Bao et al., 2020). Historically, MA has been used as “Sanghuang” in both “The Ancient Course of the Yellow River” and southern Xinjiang regions. The types and enrichment degrees of chemical metabolites in the fruiting bodies of I. hispidus growing in five different tree species were reported to be significantly different using non-targeted metabolomics (Li and Bao, 2022b).
Previous research on I. hispidus has primarily focused on exploring the chemical composition, pharmacological action and domesticated culture, with little emphasis on whole genome sequencing. The rapid development of sequencing technology has greatly helped to solve the complex problems in the fungi biology. As of June 2023, 2,426 fungal genome projects had been completed and published on the JGI website (https://mycocosm.jgi.doe.gov/fungi/fungi.info.html), as well as S. sanghuang (Jiang et al., 2021), I. hispidus (Zhang et al., 2022b; Tang et al., 2022) and G. lucidum (Liu et al., 2012). These findings reveal novel insights for understanding gene network control, metabolite synthesis pathways, edible and medicinal fungal cultivation and breeding, contributing to large-scale fungi industrialization development bottleneck. As a result, there is no comprehensive explanation for the whole genome sequence and functional gene coding of I. hispidus growing in diverse tree species. This makes it difficult to elucidate the internal mechanism of different hosts at the molecular and genetic levels.
In order to establish a high-quality reference genome of MA and to facilitate subsequent analyses of its genome function and genetic material, as well as to determine whether FM has the value of development and utilization like MA. Therefore, in this study, the whole genome sequencing of MA would be carried out based on the SMRT (https://www.pacb.com/support/sofware-downloads/) (Ardui et al., 2018) and Illumina sequencing. The resulting genomic data of MA would be compared with the sequencing assembly, genome prediction, functional annotation and comparative genome analysis of FM. Furthermore, the similarities and differences in genetic material and metabolites of MA and FM would be explored, which would provide an important theoretical basis to research for transcriptomics, proteomics, metabolomics and development of I. hispidus growing in various tree species.
In this study, I. hispidus growing in two different trees were collected from the field. MA (Figure 1A) was collected from Xiajin Ancient Mulberry Garden, Xiajin County, Shandong Province, China, 10 August 2020 (36°59′N, 115°11′E). FM (Figure 1B) was collected from Jingyuetan National Forest Park, Changchun City, Jilin Province, China, on 11 August 2020 (43°79′N, 125°46′E). The voucher specimen is deposited in the Herbarium of Mycology of Jilin Agricultural University (HMJAU), MA under No.58767 and FM under No.58769. We get the monokaryons of MA and FM by single spore isolation and used for whole genome sequencing. Then, we cultured the MA and FM monokaryon in PDA medium (Potato 200 g, Dextrose 20 g, Agar 20 g) in the dark at 28°C for 14 days. We verified isolation of MA and FM monokaryon were using an Axio Imager A2 fluorescence microscope (Zeiss). Subsequently, all samples were stored at −80°C until they were ready for genome-wide analysis.

FIGURE 1. Fruiting bodies of I. hispidus. growing in two different trees. (A) Fruiting bodies of MA. (B) Fruiting bodies of FM.
Genomic DNA of MA and FM were extracted with the GP1 extraction method (Zhong et al., 2016). Added 25 mL GP1 lysate to the monokaryon of MA and FM, and immediately added 500 µL β-mercaptoethanol, mixed upside down, and lysed in a 65°C water bath for 30 min. Centrifuging at 12,000 rpm for 10 min, drawing the supernatant into a new 50 mL centrifuge tube; the equal volume of phenol, chloroform, isoamyl alcohol (25:24:1) was added to each tube, and the mixture was shaken and mixed for 5 min. Then centrifugation at 10°C, 12,500 rpm for 8 min, transfer the uppermost aqueous phase to a new 50 mL centrifuge tube. And added a double volume of chloroform, isoamyl alcohol and the mixture was inverted or shaken for 5 min; and the supernatant was transferred to a new 50 mL centrifuge tube after centrifugation at 10°C and 12,500 rpm for 8 min. The total volume of 3/4 volume of precooled isopropanol was added, mixed upside down for 10 times, incubated in dry ice at −80°C for at least 20 min, then centrifuged at 10°C, 12,000 rpm for 10 min, and the supernatant was discarded; each tube was added with 5 mL of new 75% alcohol to wash the precipitate, and centrifuged at 10°C, 12,000 rpm for 5 min to discard the supernatant carefully. Then 5 mL of 75% ethanol was added to wash the precipitate, and centrifuged at 10°C and 12,000 rpm for 3 min to discard the supernatant carefully. And centrifuge at 12,000 rpm for 2 min, absorb the liquid in the tube, and dry the fume hood for 3–5 min; then 200–400 µL of EB was added to dissolve the DNA, and 2 µL of RNase A was digested at 37°C for 25 min. Finally, the DNA sample of MA and FM were purified with the PowerClean Pro DNA, respectively. To ensure that the harvested DNA was detected by agarose gel electrophoresis and quantified by Qubit® 2.0 fluorescence meter (Thermo Scientific).
Libraries for SMRT (Ardui et al., 2018) sequencing was constructed with an insert size of 20 kb using the SMRT bell TM Template kit (version 1.0.). Briefly, the process was that fragment and concentrate DNA, repair DNA damage and ends, prepare blunt ligation reaction, purify SMRT bell Templates with 0.45X AMPure PB Beads, size-selection using the BluePippin System, repair DNA damage after size-selection. Finally, the library quality was assessed using the Qubit® 2.0 Fluorometer (Thermo Scientific) and the insert fragment size was determined using the Agilent 2100 (Agilent Technologies).
A total amount of 1 µg DNA per sample was used as input material for the DNA sample preparations. Sequencing libraries were generated using NEBNext® Ultra™ DNA Library Prep Kit for Illumina (NEB, United States) following manufacturer’s protocol and index codes were added to attribute sequences to each sample. Briefly, the DNA sample was fragmented by sonication to a size of 350 bp, then DNA fragments were end-polished, a-tailed, and ligated with the full-length adaptor for Illumina sequencing with further PCR amplification. Finally, PCR products were purified (AMPure XP system) and libraries were analyzed for size distribution by Agilent2100 Bioanalyzer and quantified using real-time PCR.
The whole genome of MA was sequenced using Illumina Nova Seq PE150 and Pac Bio Sequel, and FM using Illumina Nova Seq PE150. All of the above were sequenced at the Beijing Novogene Bioinformatics Technology Co., Ltd., China.
The genome of MA and FM were obtained by assembling the clean data from the second-generation Illumina Nova Seq PE150 system by SOAP de novo software (Li et al., 2008). The gap close software was used to fill the gap in preliminary assembly results and removed the same lane pollution by filtering the reads with low sequencing depth (less than 0.35 of the average depth) to obtain the final assembly result of MA and FM. Then, fragments below 500 bp were filtered out and the final result was counted for genome prediction of MA and FM.
All of the data obtained from the Illumina Nova Seq PE150 system will pave the way for the Pac Bio Sequel genomic information of MA. In order to ensure the accuracy of the subsequent analysis results of MA, after filtering the low-quality reads (less than 500 bp) to obtain clean data, which from the Pac Bio Sequel platform were preliminary assembled using SMRT Link version 5.0.1 (Ardui et al., 2018). The long reads of MA were selected (more than 6,000 bp) as the seed sequence, and the other shorter reads were aligned to the seed sequence by Blasr. Then, by the variant Caller module of the SMRT Link software, the arrow algorithm was used to correct and count the variant sites in the initial genome sequence of MA. Finally, the integrity of sequence assembly of MA was evaluated by BUSCO (Benchmarking Universal Single Copy Orthologs, version 2.0) (Simão et al., 2015).
Following the assembly process, bioinformatics software was employed to analyzed the genome components of MA and FM. Genome prediction predicts the number of coding genes, repeat sequences and non-coding RNAs. The obtained MA and FM genome-wide data were subjected to Augustus 2.7 program (Keller et al., 2011) ab initio prediction. The interspersed repetitive sequences, tandem repeat sequences, transfer RNA (tRNA) and rRNA were predicted by Repeat Masker (Saha et al., 2008) (http://www.repeatmasker.org/), TRF (Maddi et al., 2022) (Tandem repeats finder), tRNAscan-SE (Chan et al., 2021) and rRNAmmer (Lagesen et al., 2007) software, respectively. The sRNA, snRNA and miRNA were predicted by BLAST against the Rfam (Kalvari et al., 2018) database.
Genome function annotation results of MA and FM were compared using functional databases, including Gene Ontology (GO) (http://www.ebi.ac.uk/GOA, accessed on 7 August 2021) (Ashburner et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp/, accessed on 7 August 2021) (Kanehisa et al., 2006), Clusters of Orthologous Groups (KOG) (http://www.ncbi.nlm.nih.gov/COG, accessed on 7 August 2021) (Galperin et al., 2015), Non-Redundant Protein Database (NR) (ftp://ftp.ncbi.nlm.nih.gov/blast/db/, accessed on 7 August 2021) (Li et al., 2002), Transporter Classification Database (TCDB) (http://www.tcdb.org, accessed on7 August 2021) (Saier et al., 2021), Cytochromes P450 (CYP450) (http://drnelson.utmem.edu/CytochromeP450.html, accessed on 7 August 2021) (Kohler et al., 2015), Swiss-Prot (http://www.expasy.org/sprot/and http://www.ebi.ac.uk/swissprot/, accessed on 7 August 2021) (Bairoch and Apweiler, 2000) and Protein Families Database of Alignments and Hidden Markov Models (Pfam) (http://pfam.xfam.org/, accessed on 7 August 2021) (Eddy, 1998). A whole genome Blast (Reiner et al., 2018) search (E-value less than 1e−5, minimal alignment length percentage larger than 40%) was performed against above eight databases. The predict secretory protein, gene cluster of secondary metabolites and carbohydrate-active enzyme were predicted using the Carbohydrate-active Enzyme Database (CAZymes) (http://www.cazy.org/, accessed on 7 August 2021) (Cantarel et al., 2008), Signal P database (Petersen et al., 2011) and antiSMASH (Blin et al., 2021) database.
Genomic alignment between the sample of FM genome and reference genome of MA were performed by MUMmer (Marçais et al., 2018) and LASTZ (Aljouie and Zhong, 2020) tools. Genomic synteny was analyzed based on the alignment results. InDels, SNPs and SVs were found by the genomic alignment results among samples by the MUMmer and LASTZ. Comparative analysis of genome sequence, component predictions and genome function annotations were the same as Sections 2.2–2.4.
Through the Pac Bio Sequel platform, it was found that MA generated 817,989 Mb Pac Bio-data. The 34.14 Mb genome sequence was assembled from 24 contigs with N50 length was 2.6 Mb. Of the 24 contigs, the longest one was 4.5 Mb in length. The analysis of GC content and read coverage depth of the MA assembly sequence revealed no significant contamination of the sample DNA. However, scattered points were observed in the region with a GC content ranging from 20% to 40%. These anomalies could potentially be attributed to the presence of mitochondrial DNA in the fungal genomic samples (Figure 2A). The sequence and feature summary of MA de novo assembly results demonstrated that high-quality sequence have been obtained (Supplementary Table S1). Using BUSCO software, we identified 94.8% (277/290) of well-known fungal complete and single-copy in MA assembly, which further illustrated a high accuracy and integrity of MA monokaryon (Supplementary Table S2). This Whole Genome Shotgun project has been deposited in GenBank under the accession number JASXRQ000000000. The final genome assembly results and related data of MA were submitted to the NCBI Bio Project PRJNA973857, Bio Sample SAMN35152529 and SRA accession number was SUB13375186, respectively.
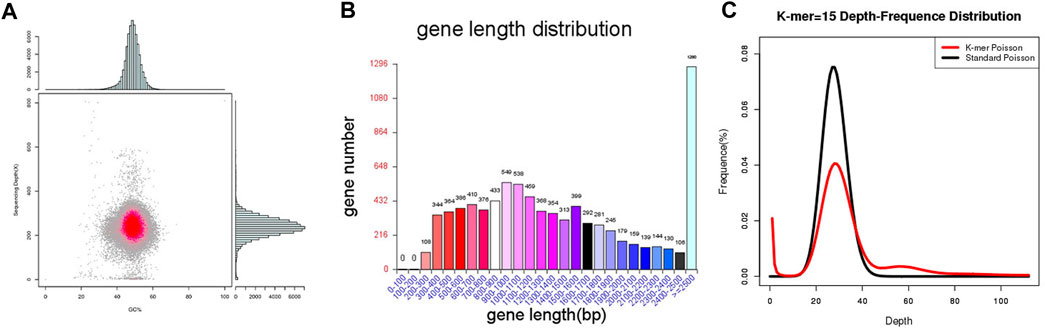
FIGURE 2. Genome sequencing, assembly and prediction of MA. (A) Statistical chart of correlation analysis between MA GC content and sequencing depth. The horizontal axis represents the GC content, the vertical axis represents the sequencing depth, the right is the sequencing depth distribution, and the upper is the GC content distribution. (B) The gene length distribution of MA. The horizontal axis shows the gene length, and the vertical axis shows the corresponding gene number. (C) K-mer analysis diagram of MA.
MA had a total number of 8,356 protein-coding genes were predicted with a total gene length of 12,839,852 bp, accounting for 37.61% of the whole genome size (34,136,716 bp); and an average gene length of coding genes was 1,537 bp, most of which ranged from 300 bp to 1,700 bp (Table 1; Figure 2B). In the prediction of repeat sequences, the interspersed and repeats were 2,761 and 5,626, respectively. For interspersed, the LTRs, DNAs, LINEs, SINEs and rolling circles contained 1,901, 493, 3,146 and 33, respectively. Moreover, 14 unknowns with a total length of 1,371 bp are to be further explored. For repeats, the TRs, minisatellite DNAs and microsatellite DNAs contained 3,045, 1,985 and 596, respectively (Supplementary Table S3). Regarding RNAs, 88 tRNAs, 9 rRNAs and 13 snRNAs were predicted (Supplementary Table S4). Based on the aforementioned information, the detailed circular genome diagram information of MA was visually represented using Circos (Krzywinski et al., 2009) software (Figure 3).
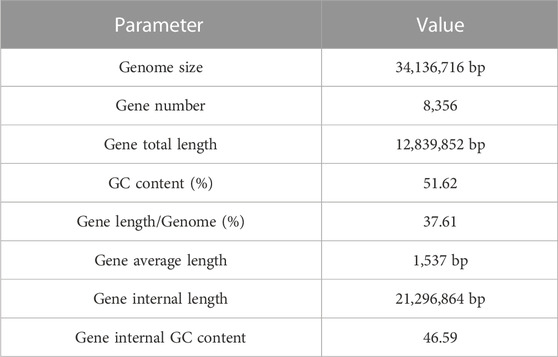
TABLE 1. Statistics of gene prediction results of MA.
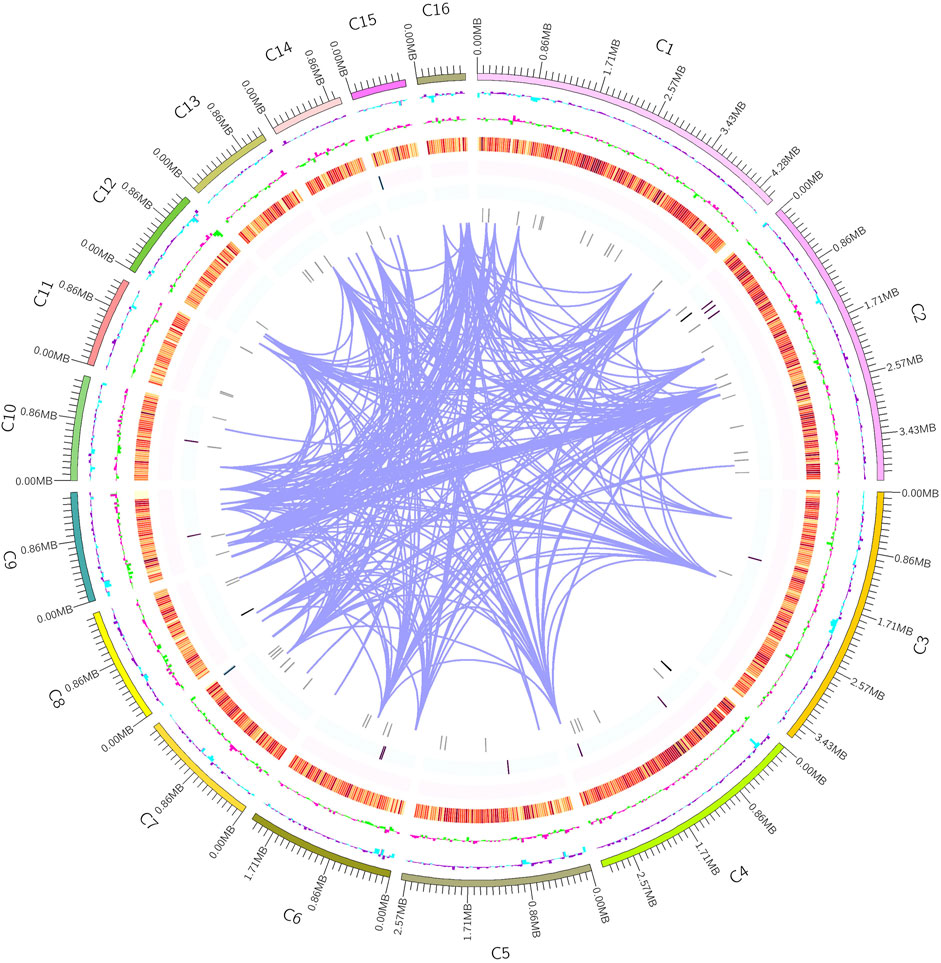
FIGURE 3. Circular whole genome diagram of MA. The outermost layer is the position coordinates. From the outer circle to the inner circle is GC content (purple: > mean value, blue: < mean value). GC skew (the specific algorithm= (G−C)/(G + C); pink: > 1, green: < 1). Gene density (four circles were taken inward from orange, representing the numerical value of coding genes, rRNA, snRNA, and tRNA, respectively). Genome chromosome duplication (regions with similarity greater than 90% of 8 kb sequences were connected by purple lines).
Different public databases were used to predict the number of genes enriched in each functional category (Supplementary Table S5). According to the GO database, MA obtained 5,342 genes accounted for 17.60% of the genome and were classified into 3 categories and 48 subcategories. There were mainly distributed in six functional subcategories, binding (2,877), metabolic process (2,790), cellular process (2,781), catalytic activity (2,403), cell (1,724) and cell part (1,724) (Figure 4A).
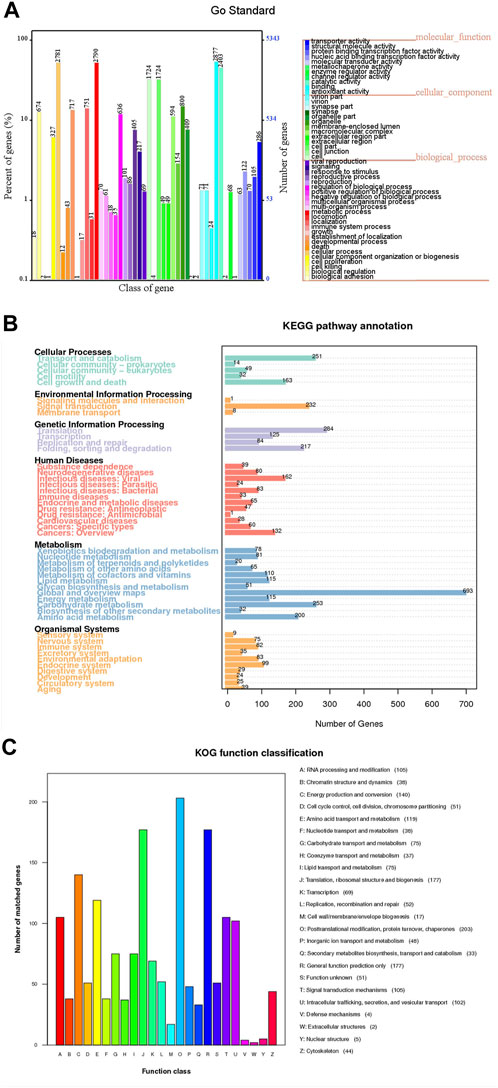
FIGURE 4. Different public database gene annotation map of MA. (A) GO annotation. The horizontal axis represents the GO functional classification on the sample annotation, the right vertical axis represents the number of genes on the annotation, and the left vertical axis represents the percentage of the number of genes on the annotation to all coding genes. (B) KEGG (Level 1) annotation. The number on the bar graph represents the number of genes on the annotation. (C) KOG annotation. The horizontal axis represents the KOG functional type, and the vertical axis represents the number of genes on the annotation.
MA had 6,564 genes were annotated in the KEGG database, among them 1,864 genes were enriched in 366 KEGG pathways (Figure 4B). The most and least enriched functional annotations were metabolism (1,813) and environmental information processing (241), respectively; with the top three coding genes being global and overview maps (693), translation (284) and carbohydrate metabolism (253) (Supplementary Table S6). There were 107 metabolism pathways, with purine metabolism (59), oxidative phosphorylation (56), pyrimidine metabolism (51), amino sugar and nucleotide sugar metabolism (46), starch and sucrose metabolism (43), pyruvate metabolism (37), glycolysis/gluconeogenesis (36), cysteine and methionine metabolism (34) and arginine and proline metabolism (33) and so on. Furthermore, the main enriched signaling pathways were MAPK signaling pathway-yeast (54), mTOR signaling pathway (43), PI3K-Akt signaling pathway (29), Ras signaling pathway (27), MAPK signaling pathway (25), FoxO signaling pathway (24) and other effective pathways (Supplementary Table S7).
The data with the KOG protein database to obtain the phylogenetic relationship classification of fungal complete genome-encoded proteins (Figure 4C). There were 1,595 genes annotated in the KOG database. Posttranslational modification, protein conversion, and chaperones (203) coding genes were the most abundant. The abundant number of classes in the KOG groups were translation, ribosomal structure and biogenesis (177), general function prediction only (177), energy production and conversion (140) and amino acid transport and metabolism (119).
We used multiple databases, including NR, Pfam, Swiss-prot, TCDB, CAZymes, secretory proteins, CYPs and gene clusters of secondary metabolites to further elucidated the genomic functions of MA. In MA, 7,177 (23.67%) genes were assigned to the NR database. The top three species were S. baumii, F. mediterranea and P. pouzarii (Supplementary Table S8). A total of 5,342 genes of MA were annotated to the Pfam database. Moreover, 6.89% (2,090) of the predicted genes were annotated to the Swiss-prot database. There were 365 (4.37%) genes annotated in TCDB database. Among TCDB, the order of proportion from large to small were the primary active transporters (122, 33.42%), electrochemical potential-driven transporters (119, 32.60%), incompletely characterized transport systems (45, 12.32%), channels/pores (42, 11.50%), accessory factors involved in transport (29, 7.95%), group transporters (8, 2.19%) and transmembrane electron carriers were not included.
The genome of macrofungus contains CAZymes that play an essential role in facilitating the acquisition of complex carbohydrate metabolism. There were 418 genes annotated in CAZymes database (Table 2). Most of these genes encode glycoside hydrolases (GHs) (185, 44.26%), glycosyltransferases (GTs) (79, 18.90%), auxiliary activity (AAs) (63, 15.07%), carbohydrate-binding modules (CBMs) (47, 11.24%), carbohydrate esterases (CEs) (33, 7.89%) and polysaccharide lyases (PLs) (11, 2.63%). From the CAZymes database, a total of 148 CAZymes families were detected in MA monokaryon (Supplementary Table S9). In the MA genome, GHs were distributed in 65 families; among the main enzymes of lignocellulose degradation, cellulase identified 12 families with GH1-GH3, GH-7, GH9, GH10, GH12, GH51, GH74, AA9 and AA16. Hemicellulose was mainly distributed in 15 families with GH1-3, GH6-7, GH9, GH12, GH17, GH27, GH29-31, GH35, GH51 and GH74. Pectinase was mainly distributed in GH2-3, GH9, GH12, GH28, GH53, GH74, GH78, PL14, CE8 and other 10 families. Ligninase was mainly distributed in AA1-2 2 families. CBMs include CBM1, CBM12, CBM13, CBM20, CBM21 and CBM50 families. CEs were divided into CE1, CE4, CE8-9, CE12 and CE15-16 8 families. PLs were mainly distributed in 8 families with PL14, PL35 and PL38 (Supplementary Table S10).
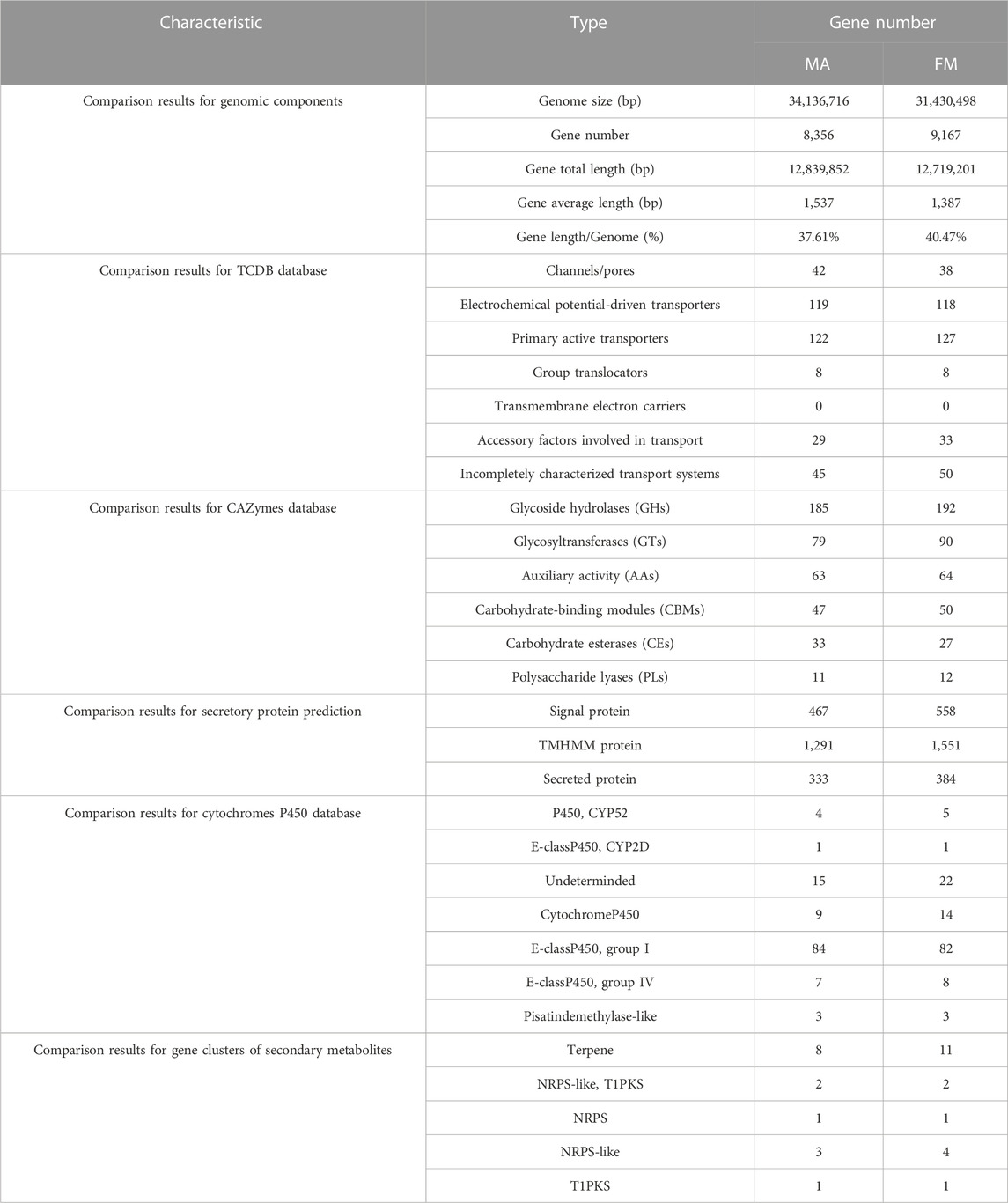
TABLE 2. Comparative analysis of genomic components and comparison of different databases for MA and FM.
Next, 467, 1,291 and 333 proteins with signal peptide structure, transmembrane structure and signal peptide structure without transmembrane structure were obtained of MA, respectively. A total of 123 genes from MA were assigned to encode CYPs. The highest (84, 68.29%) had E-class P450 group I annotations (Table 2). Moreover, 20 genes were involved in the “Metabolism of xenobiotics by cytochrome P450” (Supplementary Table S11) and 18 genes were involved in the KEGG sub-pathway “Drug metabolism-cytochrome P450” (Supplementary Table S12). A total of 15 gene clusters were obtained from MA, which were 8 terpenes, 1 NRPS, 1 T1PKS and 5 other clusters. Additionally, we detected the MA gene in the “Terpenoid backbone biosynthesis (map 00900)” pathway and found 12 key enzymes distributed in the MVA pathway (Table 3). Hexaprenyl-diphosphate synthase, prenylcysteine oxidase/farnesylcysteine lyase, farnesyl diphosphate synthase and protein farnesyltransferase/geranylgeranyltransferase type-1 subunit alpha was encoded by double-copy genes. In contrast, the other 8 enzymes were encoded by a single-copy gene (Supplementary Table S13).
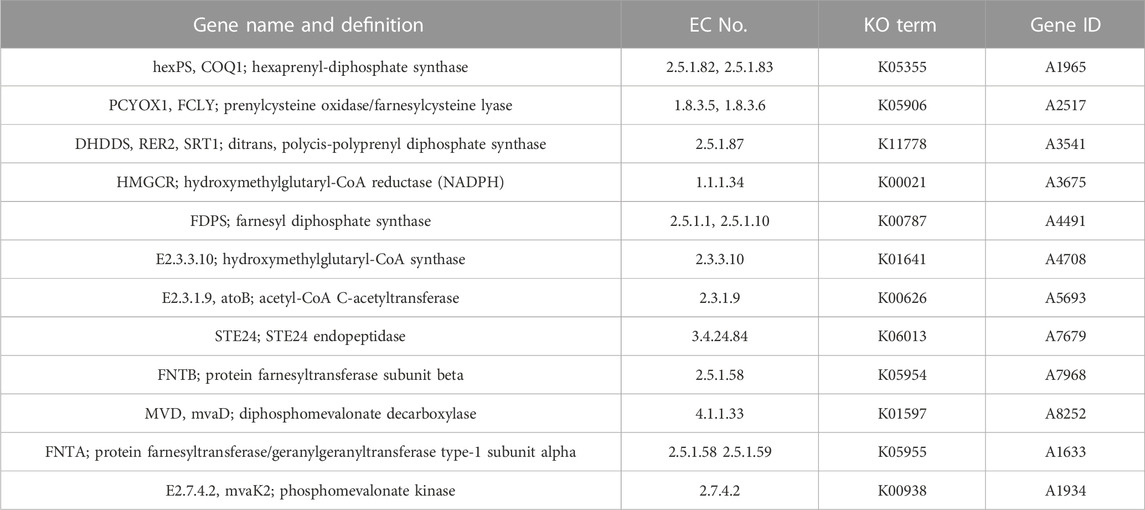
TABLE 3. Core enzymes involved in terpenoid biosynthesis of MA.
Comparative genomic analysis of genome sequence, it was found that raw reads, clean reads and filtered reads of FM were 5,595, 4,507 Mb and 46.14%, respectively, above numbers were all higher than ones of MA. Then, the number and the depth of K-mer of FM were also higher than those of MA (Figure 2C), and the former number was 885,330,899 and the latter number was 22.92. But the number of revised size and repeat rate of FM were lower than ones of MA (Supplementary Table S1). By comparing the genome component analysis, it was demonstrated that the genome size, gene total length and gene average length of FM were lower than those of MA, which were 31,430,498, 12,719,201, and 1,387 bp, respectively. Genome component prediction comparison showed that protein-coding genes of FM were higher than MA, reaching 9,167 (Table 2). However, the repeat sequences results of FM were lower than those of MA (Supplementary Table S3), contained only 71 tRNAs (Supplementary Table S4).
The numerical comparison of MA and FM in the GO functional database showed that more genes were enriched in binding, metabolic process and cellular process, and they were annotated to three types of biological functions, indicating high similarity. Compared with MA, FM lacked three subcategories such as growth, synapse and synapse part. And a total number of 5,555 genes were obtained of FM, accounting 17.55% for all the genes, and it was found that cellular component and molecular function were higher than MA, while biological process was lower than MA (Supplementary Figure S1A). Compared with KEGG annotations of MA and FM, the results indicated that the total number of FM (6,943, 21.93%) genes were higher than that of MA and also categorized into 6 levels and 46 categories; and the number of FM genes associated with most genome functions was not significantly different from the number of genes for MA. In addition, its metabolism is also mainly related to global and overview maps (724), carbohydrate metabolism (256), amino acid metabolism (203), energy metabolism (125) and lipid metabolism (123) (Supplementary Figure S1B). However, due to the preliminary nature of the FM measurement, a more in-depth exploration is required to elucidate specific metabolic pathways and identify enriched genes, which can be further compared with MA. In view of MA and FM in the KOG functional database also have high similarity. The number of genes annotated by MA were less than that of FM genome (1,641), but there was no difference in KOG types (Supplementary Figure S1C).
Given that the TCDB database annotation, the total number of FM genes were higher than MA, reaching 374. There was no markedly difference in types (Table 2). Like MA, primary active transporters (127, 33.95%) were the most enriched (Table 2). FM contained more CAZymes genes, reaching 394. Among them, glycoside hydrolases (GHs) (192, 48.73%), glycosyltransferases (GTs) (90, 22.84%), auxiliary activity (AAs) (64, 16.24%), carbohydrate-binding modules (CBMs) (50, 12.69%) and polysaccharide lyases (PLs) (12, 3.05%) were higher than MA, but carbohydrate esterases (CEs) (33, 8.38%) was lower than MA (Table 2). In addition, the total number of FM secretory protein prediction were higher than that of MA (Table 2).
Cytochromes P450 database comparison showed that, E-class P450 group I (82, 60.74%) were high expression enrichment in FM genome, followed by Undeterminded (22, 16.30%) and Cytochrome P450 (14, 10.37%) (Table 2). There were 19 gene clusters of secondary metabolites annotated in FM, including 11 terpenes and 4 NRPS-likes. Both samples contained five kinds of gene clusters of secondary metabolites, further revealing high similarity; it also contains two NRPS-like, T1PKS genes, one NRPS gene and one T1PKS gene. The comparative analysis of the above genome functional annotations will provide a potential reference for whether FM is as medicinal as MA (Table 2).
The results of collinearity analysis between MA and FM showed that, the total base length of the MA sequence was 29,119,278 bp, which accounted for 92.65% of the total gene length of 31,430,498 bp. There were 6,499 collinearity blocks in MA and FM. MA and FM had many similar translocations, translocation and inversion alignments, indicating that high collinearity relationship between them (Figure 5A; Supplementary Table S14). There were 8,356 InDels, including 275 gene coding regions, 124 insertions and 151 deletions identified between FM and MA. These InDels affected 11 CDS regions, 3 of which led to frameshifts, and 1 InDel resulted in a premature stop (Supplementary Tables S15, S16). There were 228,256 SNPs found between FM and MA, of which 66,280 SNPs occurred in the gene coding regions. The CDS SNPs and intergenic FM were 0, 57, 62, 63, 227, 41,037, 24,834, 66,280 and 161,976, respectively. These values represented 0%, 0.0250%, 0.0272%, 0.0276%, 0.0994%, 17.9785%, 10.8799%, 29.0376% and 70.9624% of the total SNPs (Supplementary Tables S17, S18). FM had 9,020 SVs, of which 3,799 translocations account for 42.1% of the total SVs; 179 inversions account for 1.9%; 3,462 translocations and inversions account for 38.3%; 336 insertions account for 3.7%; 1,143 deletions account for 12.6%; and 98 complex InDels contribute for 1% (Figure 5B; Supplementary Table S19).
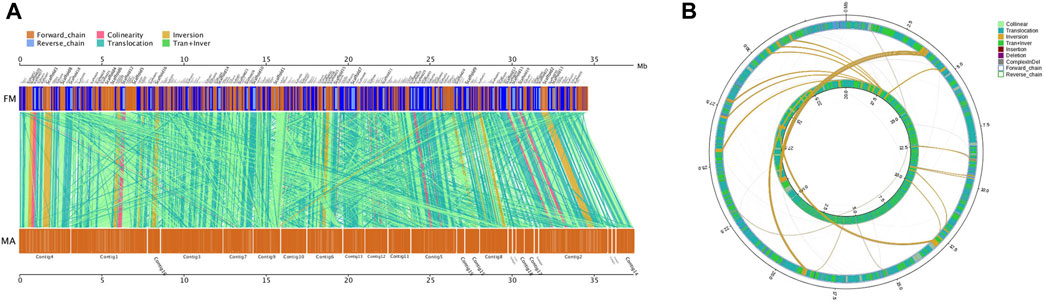
FIGURE 5. Comparative genomics analysis of MA and FM. (A) Parallel collinearity comparison between MA and FM. (B) Genome-wide structural variation type pairing map. The inner ring and the outer ring were FM and MA, respectively.
Fungi has been used as functional food and traditional medicine for a long history in China. Among them, “Sanghuang” is a well-known edible and medicinal fungus, which is used as health preservation and treatment of diseases. It was recorded in several ancient traditional Chinese herbal books, including Shennong’s Classic of Materia Medica and Compendium of Materia Medica (Bao et al., 2020). At present, the whole genome sequencing of I. hispidus growing in different tree species has not been reported. Hence, in this paper, two strains of I. hispidus growing in M. alba and F. mandshurica were chosen to reveal differences of genome sequence and epigenetics. The whole genome of MA monokaryon was sequenced by the combination of the Illumina and Pac Bio platform. Then using MA as a reference, comparative genomics between MA monokaryon and FM monokaryon was analyzed. The result demonstrated that, MA had 8,356 protein-coding genes, 24 contigs with a contig N50 size of 2.6 Mb and a genome-wide map of 34.14 Mb was drawn. The average genome size of most fungi is about 40 Mb (Duan et al., 2022), and the assembled genome size obtained by MA is basically in line with the expected results.
According to the results of genome annotation, MA obtained 30,321 genes from different public databases, but a large number of predicted genes could not match the public database. As we know, KEGG is the essential public database about pathways. On the basis of KEGG functional annotation, MA were primarily connected with carbohydrate metabolism and amino acid metabolism. In light of pathways analysis, the most significant biochemical metabolic pathways and signal transduction pathways of MA were relevant to metabolites. By further analysis of metabolic pathways, MA were found that principally involved in purine metabolism, oxidative phosphorylation, pyrimidine metabolism, amino sugar and nucleotide sugar metabolism, starch and sucrose metabolism, pyruvate metabolism and some other secondary metabolic pathways. There were 31 signal transduction pathways, including MAPK signaling pathway-yeast, mTOR signaling pathway, PI3K-Akt signaling pathway and so on. MA contains 14 xenobiotics biodegradation and metabolisms. It was testified that MA has a strongly primary metabolic process and powerful ability to degrade aromatic compounds. Furthermore, CAZymes as a matrix-degrading enzyme that plays an extremely critical role in maintaining the metabolism and circulation of natural substances. MA had a great deal of genes encoding cellulase, hemicellulase, glucan degradation related enzymes, chitinase (EC 3.2.1.14), α-amylase (EC 3.2.1.-) and peptidoglycan degradation related enzymes. It was announced that MA can not only degrade plant polysaccharides, but also degrade N-acetylamino polysaccharides, and can destroy the integrity of cell wall structure by cooperating with glucanase, cellulase, peptidoglycanase and chitinase to maintain the growth and senescence of pileus (Ding et al., 2007; Tao et al., 2013). Simultaneously, other cell wall structure-related enzyme genes also further denoted that MA has the potential to specifically degrade the host cell wall to achieve the effect of biocontrol.
In addition, CYPs belong to the heme-containing monooxygenase superfamily. The functional and evolutionary diversity of fungal CYPs makes the classification of fungal CYPs more complex, mainly associated with the biosynthesis and metabolism of fungal endogenous substances such as fatty acids, sterols, terpenes. In the field of pharmacology and therapeutics, the secondary metabolites of medicinal fungi have been reported to have anti-inflammatory and anti-viral effects (Alves et al., 2012; Alves et al., 2013), usually located in the adjacent gene clusters (Keller et al., 2005), and can be found through gene mining. Gene clusters of secondary metabolites with TS, NRPS and PKS. It was found that all known fungal siderophores were synthesized by NRPS (Haas et al., 2008). Like most basidiomycetes, MA contains a small amount of NRPS and T1pks genes, demonstrated that MA has the ability to resist the stress of the external environment and maintain its own growth and metabolism. Among fungi, one of the largest groups of corroborate bioactive natural products were terpenoids, which were composed of isoprene, mainly derived from the mevalonate pathway (Miziorko, 2011; Quin et al., 2014; Adamek et al., 2017), and had a variety of pharmacological activities. For example, the triterpenoid ganoderic acids has been reported to have anti-tumor and immunomodulatory effects (Shi et al., 2010). Twelve key enzymes involved in the MVA pathway were identified from the “Terpenoid backbone biosynthesis (map 00900)” pathway, revealing that the biosynthesis of MA terpenoids is mainly formed by the MVA pathway like most fungi. Among them, lanosterol synthase (LSS) can catalyze the cyclization of (3S)-2,3-0xidosqualene to produce lanosterol. Various enzymes act downstream to convert precursors into ergosterol and other sterols in the fungus. Notably, the catalytic derivatization of these compounds is heavily reliant on the involvement of CYPs enzymes, which play a pivotal role in modifying the lanosterol skeleton (Benveniste, 2004). In the pursuit of identifying potential triterpenoid biosynthesis genes in MA, a single copy of the LSS gene (ERG7; K01852; EC 5.4.99.7) was investigated. Consequently, it is postulated that diverse structural types of sterols and triterpenes may be produced through the action of different enzymes acting upon the LSS gene, as evidenced by the presence of various families of MA CYPs and eight triterpene-related gene clusters.
Moreover, comparative genomics as a research method to comprehend the function, expression mechanism and species evolution of genes by comparing the gene structure and distribution of different genome. During the evolutionary trajectory of fungi, genetic variations such as gene deletions and acquisitions can occur as a result of environmental selection, ultimately influencing the fungal adaptive capacity. In this study, we conducted a comparative analysis of the genome structure and function of FM, utilizing MA as the reference genome. It was worth noting that, the genome composition of FM exhibited distinctions from MA in terms of genome size, the number of coding genes, repeat sequences and non-coding RNAs. These differences suggest that FM may possess enhanced environmental adaptability, implying a potential expansion of its adaptive repertoire throughout its evolutionary history. Comparison of GO, KEGG, KOG and CAZymes results showed that, there was no apparently difference in the function of genes enriched with MA. In accordance with gene clusters, FM contains 11 terpenes. Although, FM and MA types of genome function annotation were high similarity, the specific intrinsic active substances of FM need further exploration. As two common InDels and SNPs markers for genetic polymorphism analysis, it can thorough understand the genetic polymorphism of different species (Santos et al., 2018). A significant number of InDels and SNPs sites were identified between MA and FM, which provided a basis for the subsequent genetic map construction and phylogenetic research of I. hispidus. Since accurate screening and functional verification of candidate genes for FM are currently lacking, it is crucial to prioritize comprehensive functional verification of candidate sites. This will enable us to gain clues into the regulatory mechanisms underlying these genes and enhance our understanding of FM genetic characteristics. Through the implementation of gene knock-out experiments, we can examine alterations in the intrinsic activity and ascertain potential significant modifications in the target peak of the active product. The primary objective of the research is to understand the genetic composition of MA and to determine whether FM has the value of development and utilization like MA by comparative genomics analysis. In the future, we would continue to collect I. hispidus growing in other tree species and analyze the influence of different tree species on these strains by comparative genomics and transcriptomics.
In this study, a comprehensive overview of MA was conducted at the genetic level, and a comparative genomics analysis was performed for FM to compare differences in genetic materials and whether it has potential medicinal value as MA. A 34.14 Mb genome-wide map of MA was drawn by sequencing and assembly. Additionally, 9 major secondary metabolic pathways and 31 signal transduction pathways were obtained in MA. Although, comparative genomics analysis showed that FM had high similarity with MA, the total number of genes annotated in different databases of FM were higher than MA, including 11 terpenes, which provided new insights into the correlation of FM phenotypic characteristics and genetic mechanisms. It also provided a foundation for the development and utilization of I. hispidus growing in different tree species from the aspect of genomics research.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI, BioProject ID: PRJNA973857.
QW, HB, and ZL conceived experiments and collected literature. HB survey of resource and shooting sample. QW and ZL data collation and analysis. QW wrote the first draft of this study. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (No. 32070021).
We thank the National Natural Science Foundation for funding. We also thank the samples provided by Xiajin DeBai Sanghuang Biotechnology Co., Ltd., and the experimental platform and technical support provided by the Key Laboratory of Northern Edible Fungi Resources Utilization, Ministry of Agriculture and Rural Affairs of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2023.1221491/full#supplementary-material
Adamek, M., Spohn, M., Stegmann, E., and Ziemert, N. (2017). Mining bacterial genomes for secondary metabolite gene clusters. Methods Mol. Biol. 1520, 23–47. doi:10.1007/978-1-4939-6634-9_2
Aljouie, A., and Zhong, L. (2020). High scoring segment selection for pairwise whole genome sequence alignment with the maximum scoring subsequence and gpus. Int. J. Comput. Biol. Drug Des. 13, 71. doi:10.1504/IJCBDD.2020.10026787
Alves, M. J., Ferreira, I. C., Dias, J., Teixeira, V., Martins, A., and Pintado, M. (2013). A review on antifungal activity of mushroom (basidiomycetes) extracts and isolated compounds. Curr. Top. Med. Chem. 13, 2648–2659. doi:10.2174/15680266113136660191
Alves, M. J., Ferreira, I. C., Martins, A., and Pintado, M. (2012). Antimicrobial activity of wild mushroom extracts against clinical isolates resistant to different antibiotics. J. Appl. Microbiol. 113, 466–475. doi:10.1111/j.1365-2672.2012.05347.x
Angelini, P., Girometta, C., Tirillini, B., Moretti, S., Venanzoni, R., Cipriani, M., et al. (2019). A comparative study of the antimicrobial and antioxidant activities of Inonotus hispidus fruit and their mycelia extracts. Int. J. Food Prop. 22, 768–783. doi:10.1080/10942912.2019.1609497
Ardui, S., Ameur, A., Vermeesch, J. R., and Hestand, M. S. (2018). Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 46, 2159–2168. doi:10.1093/nar/gky066
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: Tool for the unification of biology. The gene Ontology consortium. Nat. Genet. 25, 25–29. doi:10.1038/75556
Awadh, A. N., Mothana, R. A., Lesnau, A., Pilgrim, H., and Lindequist, U. (2003). Antiviral activity of Inonotus hispidus. Fitoterapia 74, 483–485. doi:10.1016/s0367-326x(03)00119-9
Bairoch, A., and Apweiler, R. (2000). The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48. doi:10.1093/nar/28.1.45
Benarous, K., Bombarda, I., Iriepa, I., Moraleda, I., Gaetan, H., Linani, A., et al. (2015). Harmaline and hispidin from Peganum harmala and Inonotus hispidus with binding affinity to Candida rugosa lipase: In silico and in vitro studies. Bioorg Chem. 62, 1–7. doi:10.1016/j.bioorg.2015.06.005
Benveniste, P. (2004). Biosynthesis and accumulation of sterols. Annu. Rev. Plant Biol. 55, 429–457. doi:10.1146/annurev.arplant.55.031903.141616
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van, W. G., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi:10.1093/nar/gkab335
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2008). The carbohydrate-active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. doi:10.1093/nar/gkn663
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi:10.1093/nar/gkab688
Dai, Y., Qin, G., and Xu, M. (2000). The forest pathogens of root and butt rot on northeast China. For. Res. 13, 15–22. doi:10.13275/j.cnki.lykxyj.2000.01.003
Ding, S., Ge, W., and Buswell, J. A. (2007). Molecular cloning and transcriptional expression analysis of an intracellular beta-glucosidase, a family 3 glycosyl hydrolase, from the edible straw mushroom, Volvariella volvacea. FEMS Microbiol. Lett. 267, 221–229. doi:10.1111/j.1574-6968.2006.00550.x
Duan, Y., Han, H., Qi, J., Gao, J., Xu, Z., Wang, P., et al. (2022). Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genomics 23, 314. doi:10.1186/s12864-022-08511-x
Eddy, S. R. (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. doi:10.1093/bioinformatics/14.9.755
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi:10.1093/nar/gku1223
Gründemann, C., Arnhold, M., Meier, S., Bäcker, C., Garcia-Käufer, M., Grunewald, F., et al. (2016). Effects of Inonotus hispidus extracts and compounds on human immunocompetent cells. Planta Med. 82, 1359–1367. doi:10.1055/s-0042-111693
Haas, H., Eisendle, M., and Turgeon, B. G. (2008). Siderophores in fungal physiology and virulence. Annu. Rev. Phytopathol. 46, 149–187. doi:10.1146/annurev.phyto.45.062806.094338
Jiang, J., Wu, S., and Zhou, L. (2021). The first whole genome sequencing of Sanghuangporus sanghuang provides insights into its medicinal application and evolution. J. Fungi (Basel). 7, 787. doi:10.3390/jof7100787
Kalvari, I., Nawrocki, E. P., Argasinska, J., Quinones-Olvera, N., Finn, R. D., Bateman, A., et al. (2018). Non-coding RNA analysis using the Rfam database. Curr. Protoc. Bioinforma. 62, e51. doi:10.1002/cpbi.51
Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F., Itoh, M., Kawashima, S., et al. (2006). From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 34, D354–D357. doi:10.1093/nar/gkj102
Keller, N. P., Turner, G., and Bennett, J. W. (2005). Fungal secondary metabolism - from biochemistry to genomics. Nat. Rev. Microbiol. 3, 937–947. doi:10.1038/nrmicro1286
Keller, O., Kollmar, M., Stanke, M., and Waack, S. (2011). A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 27, 757–763. doi:10.1093/bioinformatics/btr010
Kohler, A., Kuo, A., Nagy, L. G., Morin, E., Barry, K. W., Buscot, F., et al. (2015). Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat. Genet. 47, 410–415. doi:10.1038/ng.3223
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi:10.1101/gr.092759.109
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi:10.1093/nar/gkm160
Li, Q., Bao, H., Bau, T., Li, Y., Chen, X., and Cao, G. (2019). Chemical composition and anti-inflammatory activity of Inonotus hispidus. J. Jilin Univ. Sci. Ed. 57, 177–201. (In Chinese). doi:10.1142/S0192415X19500095
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: Short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi:10.1093/bioinformatics/btn025
Li, W., Jaroszewski, L., and Godzik, A. (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82. doi:10.1093/bioinformatics/18.1.77
Bao, H., Yang, S., Li, Q., Bau, T., and Li, Y. (2020). Supplementary textual research on “Sanghuang”. Journal of Fungal Research. 15, 264–270. doi:10.13341/j.jfr.2014.1171
Li, Z., and Bao, H. (2022b). Comparative analysis of metabolic compositions and trace elements of Inonotus hispidus mushroom grown on five different tree species. ACS Omega 7, 9343–9358. doi:10.1021/acsomega.1c06226
Li, Z., and Bao, H. (2022a). Deciphering key regulators of Inonotus hispidus petroleum ether extract involved in anti-tumor through whole transcriptome and proteome analysis in H22 tumor-bearing mice model. J. Ethnopharmacol. 296, 115468. doi:10.1016/j.jep.2022.115468
Liu, D., Gong, J., Dai, W., Kang, X., Huang, Z., Zhang, H., et al. (2012). The genome of Ganoderma lucidum provides insights into triterpenes biosynthesis and wood degradation [corrected]. PLoS One 7, e36146. doi:10.1371/journal.pone.0036146
Liu, X., Hou, R., Yan, J., Xu, K., Wu, X., Lin, W., et al. (2019). Purification and characterization of Inonotus hispidus exopolysaccharide and its protective effect on acute alcoholic liver injury in mice. Int. J. Biol. Macromol. 129, 41–49. doi:10.1016/j.ijbiomac.2019.02.011
Machado-Carvalho, L., Martins, T., Aires, A., and Marques, G. (2023). Optimization of phenolic compounds extraction and antioxidant activity from Inonotus hispidus using ultrasound-assisted extraction technology. Metabolites 13, 524. doi:10.3390/metabo13040524
Maddi, A. M., Kavousi, K., Arabfard, M., Ohadi, H., and Ohadi, M. (2022). Tandem repeats ubiquitously flank and contribute to translation initiation sites. BMC Genom Data 23, 59. doi:10.1186/s12863-022-01075-5
Marçais, G., Delcher, A. L., Phillippy, A. M., Coston, R., Salzberg, S. L., and Zimin, A. (2018). MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944. doi:10.1371/journal.pcbi.1005944
Miziorko, H. M. (2011). Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 505, 131–143. doi:10.1016/j.abb.2010.09.028
Petersen, T. N., Brunak, S., von, H. G., and Nielsen, H. (2011). SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi:10.1038/nmeth.1701
Quin, M. B., Flynn, C. M., and Schmidt-Dannert, C. (2014). Traversing the fungal terpenome. Nat. Prod. Rep. 31, 1449–1473. doi:10.1039/c4np00075g
Reiner, J., Pisani, L., Qiao, W., Singh, R., Yang, Y., Shi, L., et al. (2018). Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet-Biedl Syndrome 9 (BBS9) deletion. NPJ Genom Med. 3, 3. doi:10.1038/s41525-017-0042-3
Saha, S., Bridges, S., Magbanua, Z. V., and Peterson, D. G. (2008). Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294. doi:10.1093/nar/gkn064
Saier, M. H., Reddy, V. S., Moreno-Hagelsieb, G., Hendargo, K. J., Zhang, Y., Iddamsetty, V., et al. (2021). The transporter classification database (TCDB): 2021 update. Nucleic Acids Res. 49 (1), D461–D467. doi:10.1093/nar/gkaa1004
Saka, V. P., Chitra, V., and Narayanasamy, D. (2023). Protective role of hispolon and its derivatives against apoptosis in cortical neurons induced by electromagnetic radiation from 4G mobile phone. J. Biochem. Mol. Toxicol. 30, e23351. doi:10.1002/jbt.23351
Santos, L. A., Adhikarla, H., Yan, X., Wang, Z., Fouts, D. E., Vinetz, J. M., et al. (2018). Genomic comparison among global isolates of L. interrogans serovars copenhageni and icterohaemorrhagiae identified natural genetic variation caused by an Indel. Front. Cell Infect. Microbiol. 8, 193. doi:10.3389/fcimb.2018.00193
Shi, L., Ren, A., Mu, D., and Zhao, M. (2010). Current progress in the study on biosynthesis and regulation of ganoderic acids. Appl. Microbiol. Biotechnol. 88, 1243–1251. doi:10.1007/s00253-010-2871-1
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi:10.1093/bioinformatics/btv351
Tang, S., Jin, L., Lei, P., Shao, C., Wu, S., Wang, Y., et al. (2022). Whole-genome assembly and analysis of a medicinal fungus: Inonotus hispidus. Front. Microbiol. 13, 967135. doi:10.3389/fmicb.2022.967135
Tang, S., Shao, C., Yang, Y., Ren, R., Jin, L., Hu, D., et al. (2023). The antitumor effect of mycelia extract of the medicinal macrofungus Inonotus hispidus on HeLa cells via the mitochondrial-mediated pathway. J. Ethnopharmacol. 311, 116407. doi:10.1016/j.jep.2023.116407
Tao, Y., Xie, B., Yang, Z., Chen, Z., Chen, B., Deng, Y., et al. (2013). Identification and expression analysis of a new glycoside hydrolase family 55 exo-β-1,3-glucanase-encoding gene in Volvariella volvacea suggests a role in fruiting body development. Gene 527, 154–160. doi:10.1016/j.gene.2013.05.071
Wang, Z., Feng, X., Liu, C., Gao, J., and Qi, J. (2022). Diverse metabolites and pharmacological effects from the Basidiomycetes Inonotus hispidus. Antibiotics (Basel) 11, 1097. doi:10.3390/antibiotics11081097
Yang, H., Li, S., Qu, Y., Li, L., Li, Y., and Wang, D. (2022). Anti-colorectal cancer effects of Inonotus hispidus (bull: Fr) P. Karst. Spore powder through regulation of gut microbiota-mediated JAK/STAT signaling. Nutrients 14, 3299. doi:10.3390/nu14163299
Yang, S., Bao, H., Wang, H., and Li, Q. (2019). Anti-tumour effect and pharmacokinetics of an active ingredient isolated from Inonotus hispidus. Biol. Pharm. Bull. 42, 10–17. doi:10.1248/bpb.b18-00343
Zhang, R., Feng, X., Wang, Z., Xie, T., Duan, Y., Liu, C., et al. (2022b). Genomic and metabolomic analyses of the medicinal fungus Inonotus hispidus for its metabolite's biosynthesis and medicinal application. J. Fungi (Basel). 8, 1245. doi:10.3390/jof8121245
Zhang, Y., Hao, J., Liu, Z., Li, Z., Teng, L., and Wang, D. (2022a). Inonotus hispidus protects against hyperlipidemia by inhibiting oxidative stress and inflammation through Nrf2/NF-κB signaling in high fat diet fed mice. Nutrients 14, 3477. doi:10.3390/nu14173477
Keywords: Inonotus hispidus, whole genome sequencing, comparative genomic, CAZymes, functional annotation
Citation: Wang Q, Bao H and Li Z (2023) Genomic comparison between two Inonotus hispidus strains isolated from growing in different tree species. Front. Genet. 14:1221491. doi: 10.3389/fgene.2023.1221491
Received: 12 May 2023; Accepted: 03 July 2023;
Published: 13 July 2023.
Edited by:
Chenyang Huang, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Bing Wen Gong, Huazhong Agricultural University, ChinaCopyright © 2023 Wang, Bao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiying Bao, YmFvaGFpeWluZ0BqbGF1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.