 Pan Hong1†
Pan Hong1† Ruikang Liu
Ruikang Liu Saroj Rai
Saroj Rai Jin Li
Jin Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet., 08 July 2022
Sec. Genetics of Common and Rare Diseases
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.920950
This article is part of the Research TopicGenetic and Environmental Factors in the Occurrence of Paediatric Disorders – Volume IIView all 10 articles
Background: Although certain genetic components have been reported as contributing factors for Perthes disease, its etiology remains unclear. We present a rare case of Perthes disease in a child with osteogenesis imperfecta (OI) caused by a mutation in the COL1A1 gene (NM_000088):exon25:c.1726C>T, (p.Gln576X).
Case presentations: A 7-year-old boy was initially treated at our medical facility in March 2016 with a history of chronic pain in right hip joint and limping for a year. He was diagnosed as Perthes disease in the right hip joint. He underwent acetabular osteotomy and ipsilateral proximal femoral varus osteotomy for better containment. During the follow-ups, the right hip demonstrated a normal range of motion without pain, and the pelvic X-ray demonstrated Stulberg Type II hip joint with a round femoral head. In the latest admission in 2022, he suffered from a right femoral shaft fracture after petty violence. After reviewing his medical history, he was suspected of having OI. The whole exome sequencing demonstrated a gene mutation in COL1A1 (OMIM 166200) and confirmed the diagnosis of OI. Telescopic nailing was used to treat the femoral shaft fracture. After the nailing of the right femur, the appearance of the lower extremity seemed normal and symmetrical.
Conclusion: This study revealed that there might be an association between OI and Perthes disease. Our case report enriches the phenotypes of osteogenesis imperfecta and provides insight into the pathogenesis of LCPD.
Osteogenesis imperfecta (OI) is a rare disease which is characterized by brittle bone, blue sclerae and short stature (Schindeler et al., 2022). Most of the OIs result from mutations in the COL1A1 and COL1A2 genes (Chen et al., 2022). Type I collagen is widely present in the bone, skin and tendon tissue, and nowadays, OI is recognized as a collagen-related disorder (Kaneto et al., 2014; Li et al., 2019).
Perthes disease, also known as Legg-Calve-Perthes disease (LCPD), is characterized by necrosis of the femoral head which results from interruption of blood supply to the femoral head in the pediatric population (Mörlin and Hailer, 2021). Although certain genetic components have been reported as contributing factors, its etiology remains elusive (Rodríguez-Olivas et al., 2022). Mutation in factor V Leiden, polymorphisms in prothrombin, and methylenetetrahydrofolate reductase have been attributed to increased risk of LCPD (Buendía-Pazarán et al., 2022; Yasa et al., 2007).
Here, We present a rare case of Perthes disease in a child with osteogenesis imperfecta (OI) caused by a mutation in the COL1A1 gene (NM_000088):exon25:c.1726C>T, (p.Gln576X). To the best of our knowledge, this is the first ever reported case genetically confirmed OI patient with Perthes disease. The genetic mutation in COL1A1 might be an underlying genetic cause of Perthes disease.
A 7-year-old boy was treated in our hospital in March 2016 with a history of chronic pain in the right hip joint and limping for a year. He was radiologically diagnosed as Perthes disease. He underwent right acetabular osteotomy and ipsilateral proximal femoral varus osteotomy in order to improve the femoral head containment in the acetabulum. Series radiographs in the subsequent follow-ups demonstrated the progression of Perthes disease (see Figure 1). In the past few years, he suffered from multiple fractures in his left upper extremity around elbow joint; however, he did not undergo any surgical treatment (see Figure 2). He suffered from right tibial shaft fracture in November 2019, and it was treated with closed reduction and elastic stable intramedullary nails (ESINs) at a local hospital. Unfortunately, he was presented to our hospital with his right femoral shaft fracture in February 2022. Prior to the fracture of his right femur, he had a normal range of motion (ROM) of the hip without limping, and the pelvic X-ray demonstrated Stulberg Type II hip joint with a round femoral head.
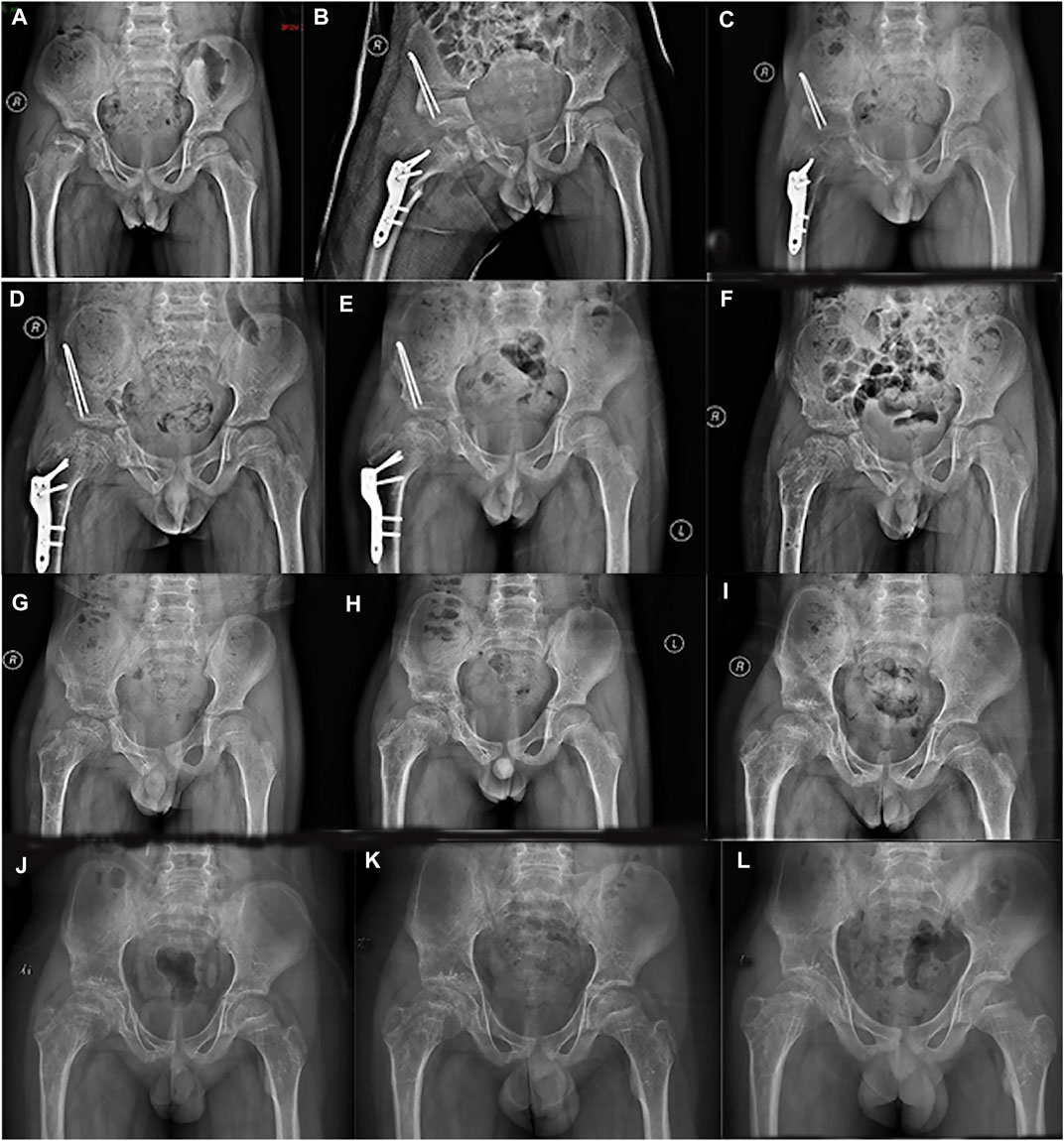
FIGURE 1. Series radiographs of pelvis from a boy with Perthes disease presentation. (A) Pelvic radiograph of a 7.5 years old boy (March 2016). (B) Postoperative pelvic radiograph (March 2016). (C). 4 months after operation (July 2016). (D). 7 months after operation (October 2016). (E) 11 months after operation (February 2017). (F). 12 months after primary operation, and 1 month after implant removal (March 2017). (G) 3 months after implant removal (April 2017). (H) 1 year after implant removal (March 2018). (I) 2 years after implant removal (January 2019). (J) 3 years after implant removal (November 2019). (K) 4 years after implant removal (October 2020). (L) 5 years after implant removal (January 2022).
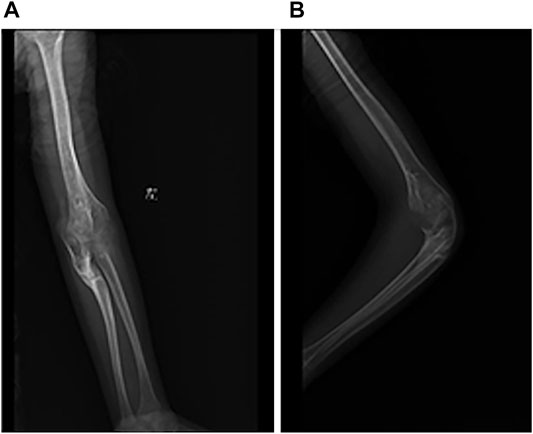
FIGURE 2. Radiograph of left elbow joint from conservative treatment. (A) AP view of elbow joint. (B) Latereal view of elbow joint.
Despite having multiple fractures over the period of time, he had neither undergone a complete evaluation in order to diagnose OI nor received any pharmacological intervention for fragility fracture. However, the Z-value on the DEXA (Dual Energy X-ray Absorptiometry) scan in the latest admission was -2.8.
In the latest admission, the patient’s height was 150 cm and body weight was 45 Kg. On inspection, a slightly bluish sclerae were noticed, and an abnormal appearance and limited ROM of the left elbow were evident. After reviewing his medical history, he was suspected of having OI. Subsequently, gene mutation in COL1A1 (OMIM 166200) was unraveled by whole exome sequencing (see Figure 3). Therefore, telescopic nailing was used to treat the femoral shaft fracture (see Figure 4). After the nailing of the right femur, the appearance of the lower extremity seemed normal and symmetrical.
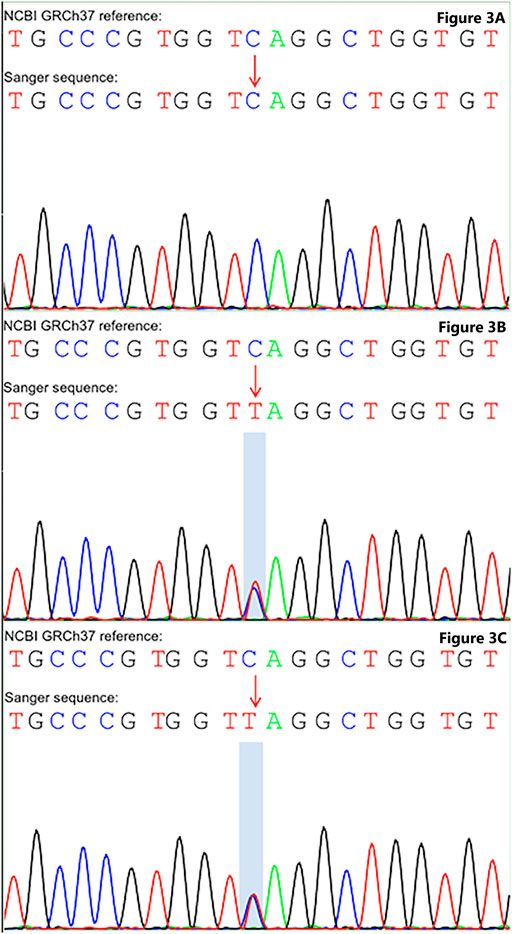
FIGURE 3. Sanger sequencing unravel gene mutation in COL1A1. (A) Wild type was detected in the COL1A1: NM_000088: exon25: c.C1726T: p.Q576X without mutation in the father. (B) Heterozygous mutation in the COL1A1: NM_000088: exon25: c.C1726T: p.Q576X of the mother. (C) Heterozygous mutation in the COL1A1: NM_000088: exon25: c.C1726T: p.Q576X of the patient.
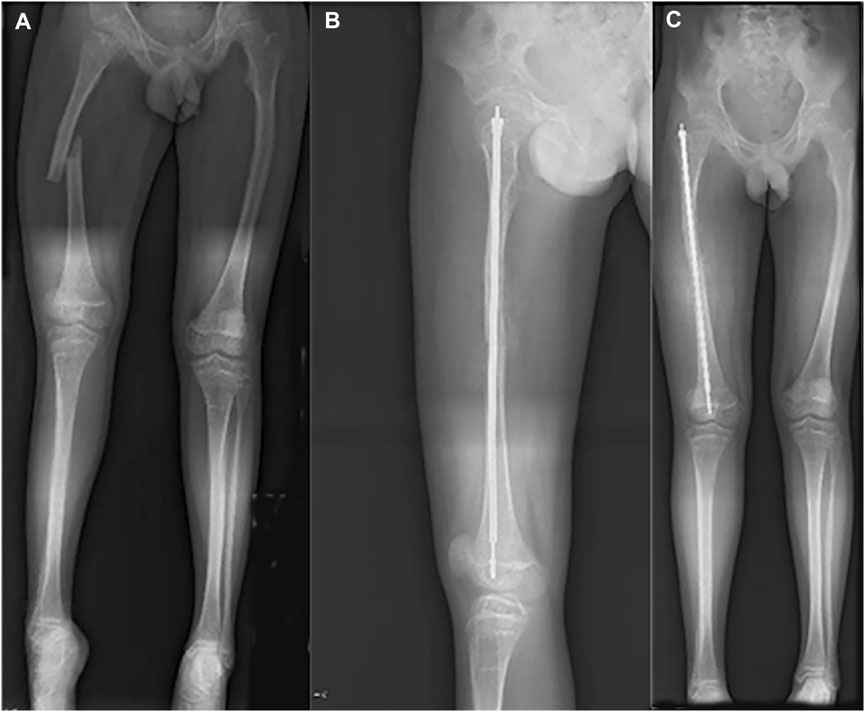
FIGURE 4. Radiograph of 13-year-old boy with right femoral fracture. (A) Full-length AP view of lower extremity before surgery. (B) Lateral of femur after surgery. (C) Full-length AP view of lower extremity after surgery.
As for the detailed methodology of genetic analysis, we collected 2 ml of whole blood (EDTA anticoagulation) of the children and their parents, and the samples were sequenced by Xiamen Jiyuan medical laboratory. Subsequently, we extracted the whole genomic DNA of peripheral blood leukocytes, built the DNA library, and carried out 150 bp double terminal sequencing. After analyzing the original data of sequencing by biological information, the detected single nucleotide polymorphism and indel variants were annotated by ANNOVAR software.
The annotation information includes the start and end positions of the chromosome, reference alleles, alternative alleles, gene function, population frequency in the public database (including thousand human genome, ExAC, gnomAD database, etc.), protein function prediction (including revert, ClinPred, SIFT, provian, mutation taster, polyphen2, dbscsnv11, etc.). Furthermore, we followed the classification standards and guidelines of genetic variation of the American College of medical genetics and genomics to evaluate the pathogenicity and genetic interpretation of candidate gene variation.
According to the whole exome sequencing analysis, we found the mutation c.1726C>T: p.Gln576X (NM_000088) in exon 25 of the COL1A1 gene. This was a heterozygous nonsense mutation that base 1726 changed from cytosine C to thymine T, resulting in the mutation of amino acid 576 from glutamine to stop the codon from forming the truncated protein. Besides, the detected variation had been reported in the OI & Ehlers-Danlos syndrome variant databases, but the variation has not been reported in the literature before. The upstream and downstream primers were designed for the candidate ectopic sites, and the polymerase chain reaction was carried out, which was verified by Sanger sequencing. Primer design: COL1A1-F, 5′-CTCCCAAGATGCCCTTCCAG-3’; COL1A1-R, 5′-TCTCCCCAAGTCCCACTCAT-3’. Sanger sequencing was performed on the patient and their parents, suggesting that the variation was inherited from the mother (see Figure 3). Combined with the results of whole exome sequencing and Sanger sequencing, the mutation was determined as a gene mutation with possible pathogenicity according to American College of medical genetics and genomics guidelines (the evidence level of pathogenicity was pvs1 + pm2_supporting).
We presented a boy with OI caused by a heterozygous mutation in the COL1A1 gene (NM_000088):exon25:c.1726C>T, (p.Gln576X). Series radiographs of the hip joint displayed the characteristics of Perthes disease.
OI is a genetic disorder caused by an abnormal production or structure of collagen type I (Nijhuis et al., 2022). The incidence of this rare condition is 1 in 15,000–20,000 births (Forlino and Marini, 2016). Autosomal dominant, recessive inheritance and X autosomal inheritance patterns have been reported. The severity of symptoms varies widely between different types of OI, ranging from mild symptoms with very few fractures with a normal quality of life to severe symptoms with frequent fractures, severe physical incompetence and decreased life span (Nijhuis et al., 2019; Robinson and Rauch, 2019). The patient had a normal height and relatively normal bone X-ray images in our study. Not only had the boy suffered multiple fractures in the past few years, but his mother, a heterozygous carrier, also experienced one fracture in adolescence and had a mild OI. Therefore, this mutation appeared to be pathogenic, leading to mild type I OI in this pedigree.
Perthes disease has been reported and studied for more than 100 years, but its etiology remains unknown (Pavone et al., 2019). Between 1909 and 1910, LCPD was described almost at the same time by different authors, Arthur Legg, Jacques Calve, Georg Perthes and Henning Waldernstrom (Hailer and Hailer, 2018). Several theories, including environmental, metabolic and genetic predilection, have been proposed as the causative factor for this disease (Leroux et al., 2018; Ibrahim and Little, 2016). Underprivileged social and economic status is associated with the incidence of LCPD in children (Perry et al., 2012a; Perry et al., 2012b), but this is not the scenario for our patient. Obesity has also been identified as a significant risk factor for LCPD (Neal et al., 2016), but this patient presented with average body weight and normal body mass index (BMI).
Presentation of Perthes disease has been reported in certain patients with increased susceptibility (Maleki et al., 2021; Miyamoto et al., 2017). Genetic mutation of COL2A1 has been reported to be involved in the genesis of a type II collagenopathy and a possible cause of LCPD (Kannu et al., 2011; Li et al., 2014). Besides, factor V Leiden has been reported to have an increased risk of Perthes disease, but the result was not significant (Szepesi et al., 2004; Glueck et al., 2007). Moreover, a child with Kienbock’s disease and factor V thrombophilia has been reported to develop LCPD (Baltzer et al., 2016). However, the available literature on substantial heterogeneity reached no unanimity on the etiology of LCPD (Woratanarat et al., 2014; Vosmaer et al., 2010; Balasa et al., 2004; Kenet et al., 2003).
The hip disorder has been reported in patients with OI, including hip dysplasia, acetabular protrusio, and pseudo-protrusio acetabular deformity (Kishta et al., 2017; Ahn et al., 2019; Mandel et al., 2020; Song et al., 2021). However, only one study reported a patient with OI associated with Perthes disease in 2003 (Petra and Benson, 2003). However, the follow-up of that patient did not fully demonstrate the progress of Perthes, and the diagnosis of OI was not genetically confirmed. In contrast, the patient in our study demonstrated classic progression of LCPD, and containment surgery was performed to maintain the femoral head within the acetabulum. In the latest follow-up, the radiograph of the hip joint demonstrated Stulberg type II. Moreover, the genetic test unraveled a mutation in the COL1A1 gene (NM_000088):exon25:c.1726C>T, (p.Gln576X), and confirmed the diagnosis of OI. Therefore, our study demonstrated the mutation leading to the abnormal structure or function of collagen type I, resulting in decreased mechanism strength, which should be further studied and clarified in the pathogenesis of the LCPD.
There are certain limitations in our study. Firstly, we did not perform genetic tests for the entire family members, including the grandparents. Besides, future investigations, including animal models, might be warranted to explore the significance of this genetic mutation. Moreover, Perthes disease might be a comorbidity of OI rather than a secondary disease.
In conclusion, we report an OI child with heterozygous c.1726C>T, (p.Gln576X) mutation in the COL1A1 gene who later had been diagnosed as having Perthes disease. This study revealed that there might be an association between OI and Perthes disease. Our case report enriches the phenotypes of osteogenesis imperfecta and provides insight into the pathogenesis of LCPD.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology (IORG No: IORG0003571) gave a final approval on 20 November 2019. Written consents were obtained from the legal guardians of patient for publication of this paper. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
JL and RX are in charge of the main idea and is the guarantor of integrity of the entire study; PH, XZ, and RL are in charge of the study concepts, design, manuscript preparation and editing; RL and YS are in charge of data extraction and statistical analysis. PH and SR are in charge of the language polishing and the grammar revision. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahn, J., Carter, E., Raggio, C. L., and Green, D. W. (2019). Acetabular Protrusio in Patients with Osteogenesis Imperfecta: Risk Factors and Progression. J. Pediatr. Orthop. 39 (10), e750–e754. doi:10.1097/BPO.0000000000001051
Balasa, V. V., Gruppo, R. A., Glueck, C. J., Wang, P., Roy, D. R., Wall, E. J., et al. (2004). Legg-calvé-perthes Disease and Thrombophilia. J. Bone Jt. Surgery-American 86 (12), 2642–2647. doi:10.2106/00004623-200412000-00009
Baltzer, H. L., Riester, S., and Moran, S. L. (2016). Bilateral Legg-Calve-Perthes Disease and Kienbock's Disease in a Child with Factor V Leiden Thrombophilia. Hand (New York, N,Y.) 11 (3), NP16–NP19. doi:10.1177/1558944715627274
Buendía-Pazarán, J. G., Hernández-Zamora, E., Rodríguez-Olivas, A. O., Casas-Ávila, L., Valdés-Flores, M., and Reyes-Maldonado, E. (2022). Association of MTHFR Rs1801133 and Homocysteine with Legg-Calvé-Perthes Disease in Mexican Patients. Orphanet. J. Rare Dis. 17 (1), 123. doi:10.1186/s13023-022-02264-2
Chen, P., Tan, Z., Shek, H. T., Zhang, J.-n., Zhou, Y., Yin, S., et al. (2022). Phenotypic Spectrum and Molecular Basis in a Chinese Cohort of Osteogenesis Imperfecta with Mutations in Type I Collagen. Front. Genet. 13, 816078. doi:10.3389/fgene.2022.816078
Forlino, A., and Marini, J. C. (2016). Osteogenesis Imperfecta. Lancet 387, 1657–1671. doi:10.1016/S0140-6736(15)00728-X
Glueck, C. J., Tracy, T., and Wang, P. (2007). Legg-Calve-Perthes Disease, Venous and Arterial Thrombi, and the Factor V Leiden Mutation in a Four-Generation Kindred. J. Pediatr. Orthop. 27 (7), 834–837. doi:10.1097/BPO.0b013e31815584bf
Hailer, Y. D., and Hailer, N. P. (2018). Is Legg-Calvé-Perthes Disease a Local Manifestation of a Systemic Condition? Clin. Orthop. Relat. Res. 476 (5), 1055–1064. doi:10.1007/s11999.0000000000000214
Ibrahim, T., and Little, D. G. (2016). The Pathogenesis and Treatment of Legg-Calvé-Perthes Disease. JBJS. Rev. 4 (7), e4. doi:10.2106/JBJS.RVW.15.00063
Kaneto, C. M., Lima, P. S., Zanette, D. L., Prata, K. L., Pina Neto, J. M., de Paula, F. J., et al. (2014). COL1A1and miR-29b Show Lower Expression Levels during Osteoblast Differentiation of Bone Marrow Stromal Cells from Osteogenesis Imperfecta Patients. Bmc. Med. Genet. 15, 45. doi:10.1186/1471-2350-15-45
Kannu, P., Irving, M., Aftimos, S., and Savarirayan, R. (2011). Two Novel COL2A1 Mutations Associated with a Legg-Calvé-Perthes Disease-like Presentation. Clin. Orthop. Relat. Res. 469 (6), 1785–1790. doi:10.1007/s11999-011-1850-x
Kenet, G., Hayek, S., Mor, M., Lubetsky, A., Miller, L., Rosenberg, N., et al. (2003). The 1226G (N370S) Gaucher Mutation Among Patients with Legg-Calve-Perthes Disease. Blood Cells, Mol. Dis. 31 (1), 72–74. doi:10.1016/s1079-9796(03)00121-9
Kishta, W., Abduljabbar, F. H., Gdalevitch, M., Rauch, F., Hamdy, R., and Fassier, F. (2017). Hip Dysplasia in Children with Osteogenesis Imperfecta: Association with Collagen Type I C-Propeptide Mutations. J. Pediatr. Orthop. 37 (7), 479–483. doi:10.1097/BPO.0000000000000644
Leroux, J., Abu Amara, S., and Lechevallier, J. (2018). Legg-Calvé-Perthes Disease. Orthop. Traumatology Surg. Res. 104 (1S), S107–S112. doi:10.1016/j.otsr.2017.04.012
Li, L., Cao, Y., Zhao, F., Mao, B., Ren, X., Wang, Y., et al. (2019). Validation and Classification of Atypical Splicing Variants Associated with Osteogenesis Imperfecta. Front. Genet. 10, 979. doi:10.3389/fgene.2019.00979
Li, N., Yu, J., Cao, X., Wu, Q.-Y., Li, W.-W., Li, T.-F., et al. (2014). A Novel P. Gly630Ser Mutation of COL2A1 in a Chinese Family with Presentations of Legg-Calvé-Perthes Disease or Avascular Necrosis of the Femoral Head. PLoS. One. 9 (6), e100505. doi:10.1371/journal.pone.0100505
Maleki, A., Qoreishy, S. M., and Bahrami, M. N. (2021). Surgical Treatments for Legg-Calvé-Perthes Disease: Comprehensive Review. Interact. J. Med. Res. 10 (2), e27075. doi:10.2196/27075
Mandel, M., Saloky, K., Mirenda, W., Seeley, A., and Seeley, M. (2020). Hip Dysplasia and Osteogenesis Imperfecta. JBJS. Case Connect. 10 (4), e2000369. doi:10.2106/JBJS.CC.20.00369
Miyamoto, Y., Matsuda, T., Kitoh, H., Haga, N., Ohashi, H., Nishimura, G., et al. (2007). A Recurrent Mutation in Type II Collagen Gene Causes Legg-Calvé-Perthes Disease in a Japanese Family. Hum. Genet. 121 (5), 625–629. doi:10.1007/s00439-007-0354-y
Mörlin, G. B., and Hailer, Y. D. (2021). High Blood Pressure and Overweight in Children with Legg-Calvé-Perthes Disease: a Nationwide Population-Based Cohort Study. Bmc. Musculoskelet. Disord. 22 (1), 32. doi:10.1186/s12891-020-03889-9
Neal, D. C., Alford, T. H., Moualeu, A., Jo, C.-H., Herring, J. A., and Kim, H. K. W. (2016). Prevalence of Obesity in Patients with Legg-Calvé-Perthes Disease. J. Am. Acad. Orthop. Surg. 24 (9), 660–665. doi:10.5435/JAAOS-D-16-00120
Nijhuis, W. H., Eastwood, D. M., Allgrove, J., Hvid, I., Weinans, H. H., Bank, R. A., et al. (2019). Current Concepts in Osteogenesis Imperfecta: Bone Structure, Biomechanics and Medical Management. J. Children's Orthop. 13, 1–11. doi:10.1302/1863-2548.13.180190
Nijhuis, W., Verhoef, M., van Bergen, C., Weinans, H., and Sakkers, R. (2022). Fractures in Osteogenesis Imperfecta: Pathogenesis, Treatment, Rehabilitation and Prevention. Children 9 (2), 268. doi:10.3390/children9020268
Pavone, V., Chisari, E., Vescio, A., Lizzio, C., Sessa, G., and Testa, G. (2019). Aetiology of Legg-Calvé-Perthes Disease: A Systematic Review. Wjo 10 (3), 145–165. doi:10.5312/wjo.v10.i3.145
Perry, D. C., Bruce, C. E., Pope, D., Dangerfield, P., Platt, M. J., and Hall, A. J. (2012a). Legg-Calvé-Perthes Disease in the UK: Geographic and Temporal Trends in Incidence Reflecting Differences in Degree of Deprivation in Childhood. Arthritis & Rheumatism 64 (5), 1673–1679. doi:10.1002/art.34316
Perry, D. C., Bruce, C. E., Pope, D., Dangerfield, P., Platt, M. J., and Hall, A. J. (2012b). Perthes' Disease of the Hip: Socioeconomic Inequalities and the Urban Environment. Arch. Dis. Child. 97 (12), 1053–1057. doi:10.1136/archdischild-2012-302143
Petra, M., and Benson, M. K. D. (2003). Perthes?? Disease Associated with Osteogenesis Imperfecta. J. Pediatr. Orthop. Part B 12 (5), 315–318. doi:10.1097/01.bpb.0000060281.96739.be
Robinson, M.-E., and Rauch, F. (2019). Mendelian Bone Fragility Disorders. Bone 126, 11–17. doi:10.1016/j.bone.2019.04.021
Rodríguez-Olivas, A. O., Hernández-Zamora, E., and Reyes-Maldonado, E. (2022). Legg-Calvé-Perthes Disease Overview. Orphanet. J. Rare Dis. 17 (1), 125. doi:10.1186/s13023-022-02275-z
Schindeler, A., Lee, L. R., O'Donohue, A. K., Ginn, S. L., and Munns, C. F. (2022). Curative Cell and Gene Therapy for Osteogenesis Imperfecta. J Bone & Mineral Res 37, 826–836. doi:10.1002/jbmr.4549
Song, M. H., Kamisan, N., Lim, C., Shin, C. H., Yoo, W. J., Song, H.-R., et al. (2021). Pseudo-Protrusio Acetabular Deformity in Osteogenesis Imperfecta Patients. J. Pediatr. Orthop. 41 (3), e285–e290. doi:10.1097/BPO.0000000000001739
Szepesi, K., Pòsán, E., Hársfalvi, J., Ajzner, É., Szücs, G., Gáspár, L., et al. (2004). The Most Severe Forms of Perthes' Disease Associated with the Homozygous Factor V Leiden Mutation. J. Bone Jt. Surg. Br. 86-B (3), 426–429. doi:10.1302/0301-620x.86b3.13442
Vosmaer, A., Pereira, R. R., Koenderman, J., Rosendaal, F., and Cannegieter, S. (2010). Coagulation Abnormalities in Legg-Calvé-Perthes Disease. J. Bone Jt. Surgery-American 92 (1), 121–128. doi:10.2106/JBJS.I.00157
Woratanarat, P., Thaveeratitharm, C., Woratanarat, T., Angsanuntsukh, C., Attia, J., and Thakkinstian, A. (2014). Meta-analysis of Hypercoagulability Genetic Polymorphisms in Perthes Disease. J. Orthop. Res. 32 (1), 1–7. doi:10.1002/jor.22473
Keywords: osteogenesis imperfecta, Perthes disease, Legg-Calve-Perthes Disease, COL1A1 gene, case report
Citation: Hong P, Zhao X, Liu R, Rai S, Song Y, Xu R and Li J (2022) Perthes Disease in a Child With Osteogenesis Imperfecta From a Rare Genetic Variant: A Case Report. Front. Genet. 13:920950. doi: 10.3389/fgene.2022.920950
Received: 15 April 2022; Accepted: 06 June 2022;
Published: 08 July 2022.
Edited by:
Fan Jin, Zhejiang University, ChinaReviewed by:
Silvia Izquierdo Alvarez, Hospital Universitario Miguel Servet, SpainCopyright © 2022 Hong, Zhao, Liu, Rai, Song, Xu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruijing Xu, eHVydWlqaW5nOTlAMTYzLmNvbQ==; Jin Li, bGlqaW4yMDAzd2h4aEBmb3htYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.