 Ying Wang
Ying Wang Fei Xiong1*
Fei Xiong1* Zhaobin Song
Zhaobin Song- 1Hubei Engineering Research Center for Protection and Utilization of Special Biological Resources in the Hanjiang River Basin, College of Life Sciences, Jianghan University, Wuhan, China
- 2Sichuan Key Laboratory of Conservation Biology on Endangered Wildlife, College of Life Sciences, Sichuan University, Chengdu, China
- 3Key Laboratory of Bio-Resources and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China
Salmonids are composed of anadromous and freshwater fishes, which is an important model for studying adaptive evolution. Herein, 49 salmonid complete mitochondrial genomes and those of two outgroups were used to infer a robust phylogeny for the family Salmonidae. The BI and RAxML phylogenetic trees based on 13 concatenated mitochondrial protein-coding genes showed well-supported nodes, and topologies were highly congruent. The concatenated 13 mitochondrial protein-coding genes, ND2, ND3, and ND5 genes were shown to have significantly larger dN/dS ratios in anadromous species than in freshwater species of Salmonidae, but the CYTB gene had significantly smaller dN/dS in anadromous species. The FEL analysis identified positively selected sites and negatively selected sites in each mitochondrial protein-coding gene separately. The RELAX program revealed that the ATP8 and CYTB genes supported intensified selection of the anadromous lineages. Our results demonstrated the phylogeny of Salmonidae and explored the mitochondrial DNA evolution pattern between anadromous and freshwater salmonids.
Introduction
The family Salmonidae consists of three subfamilies with 11 genera and about 66 species: Coregoninae (whitefish, cisco, and inconnu), Thymallinae (grayling), and Salmoninae (lenok, huchen, salmon, trout, and char) (Gosline, 1963; Nelson, 2006). Salmonids have been intensively studied due to their high value in sport and commercial fisheries and their importance in a wide range of evolutionary and ecological questions of fishes (Groot and Margolis, 1991; Elliott, 1994; Hendry and Stearns, 2004). The most noteworthy feature of salmonids is that they could overcome the tremendous obstacles to return from distant oceans to natal streams for spawning. This notable migratory behavior of salmonids has raised many researchers’ attention to the evolution of migrations and the origin of this life history strategy (Mcdowall, 2001).
Mitochondria provide a great deal of energy via oxidative phosphorylation. Through comparison of the ratios of the rate of nonsynonymous nucleotide substitution to that of synonymous substitution (dN/dS), natural selection acting on protein-coding genes was ever characterized (Nei, 2005). A negative correlation was found between the dN/dS ratio and locomotive speed in birds and was also found for the dN/dS ratio and locomotive speed by examining 214 complete mammalian mitogenomes (Shen et al., 2009). Similarly, the teleost migratory ability has been reflected by selection patterns observed in mitochondrial genomes (Sun et al., 2011). Most species of Salmonidae are distributed throughout the Northern Hemisphere, including freshwater and anadromous species (Nelson, 2006). In particular, within Salmonidae, whitefish in the genus Prosopium, grayling (subfamily Thymallinae), lenok (Brachmystax spp.), and huchen/taimen (Hucho spp.) are exclusively freshwater species, while other salmonid groups are anadromous (Myers, 1949; McDowall, 1988; Davidson, 2013). Therefore, the dN/dS ratios of salmonid mitochondrial genomes could be estimated to test whether the mitochondrial protein-coding genes of the anadromous and freshwater salmonids have undergone different selective constraints.
Molecular systematics and phylogenetic investigations of salmonids have been elaborated in the previous studies (Crespi and Fulton, 2004; Phillips et al., 2004; Wilson and Turner, 2009; Shedko et al., 2012; Shedko et al., 2013; Macqueen and Johnston, 2014). Crespi and Fulton (2004) addressed the phylogeny of salmonid fishes based on 269 GenBank sequences of mitochondrial DNA and nuclear DNA, but they could not resolve the phylogenetic relationships including the genus-level and species-level relationships substantially since the freshwater species, Hucho bleekeri, Hucho taimen, and Brachymystax lenok tsinlingensis were not included. The recent study focused on elucidating the systematic relationships among salmonid fishes, but the phylogenetic tree topology was labeled with low support values (Crête-Lafrenière et al., 2012). Macqueen and Johnston (2014) combined protein (7222 AA), or nucleotide of truly orthologous nuclear genes and protein data (3790 AA) of mitogenome to conduct a phylogenetic analysis to reveal whole-genome duplication is decoupling from species diversification. This study recovered a Thymallinae–Coregoninae sister relationship with maximal support. Jacobsen et al. (2015) utilized 107 full mitogenomes of recently diverged species and lineages of whitefish to search for signals of positive selection at the mitogenome level. However, the evolution acting at the mitogenome level between freshwater and anadromous salmonids was unknown so far.
In the present study, 49 salmonid complete mitochondrial genomes, as well as those of two outgroups, were chosen to reconstruct the phylogeny for the family Salmonidae. At the same time, we investigated the potential selective pressure changes at the mitochondrial genome level between freshwater and anadromous salmonids. The purpose was to address the phylogenetic relationships for the family Salmonidae and explore the adaptive evolution patterns between freshwater and anadromous salmonids.
Materials and Methods
Data Collection and Analysis
All available complete mitogenomes of salmonids (n = 49) plus two outgroups Esox lucius (NC_004593) and Osmerus mordax (NC_015246) were downloaded from the National Center for Biotechnology Information (http://www.ncbi.nih.gov/). The corresponding accession numbers for the newly determined sequences and previously published sequences are listed in Supplementary Table S1. The nucleotide sequences of the 13 protein-coding genes (PCGs) were first extracted using purpose-built Perl scripts based on annotations and then separately aligned according to their corresponding amino acid translations by the software application TranslatorX (Abascal et al., 2010). The concatenated nucleotide sequence alignment from 13 PCGs (total = 11,355 bp) without stop codons was generated with our in-house scripts to conduct the phylogenetic analysis. Prior to the phylogenetic reconstruction, the extent of substitution saturation was estimated separately for entire codons and for the first, second, and third codon positions of the concatenated alignment using DAMBE 6.4.107 (Xia, 2017). The pairwise nucleotide differences (transitions and transversions) were plotted against the GTR genetic distance (Supplementary Figure S1).
Phylogenetic Analysis
To improve the reliability of the phylogenetic analysis, the best-fit partitioning scheme across each gene and codon position was determined for each data set under the Bayesian information criterion by PartitionFinder software (Lanfear et al., 2012). Both partitioned maximum likelihood (ML) and Bayesian Inference (BI) approaches with the selected partition scheme were employed to reconstruct the phylogenetic relationships among salmonids. RAxML 7.0.3 (Stamatakis, 2006) with 1,000 nonparametric bootstrap replicates and MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003) were used to construct maximum likelihood (ML) and Bayesian inference (BI) trees, respectively. The ML trees were inferred using RAxML with the GTRGAMMA model. The Bayesian posterior probabilities were estimated using the Markov Chain Monte Carlo (MCMC) method with one cold chain and three heated chains for 20,000,000 generations, with every 1000th sample being retained. Twenty-five percent of the samples (5000 samples) from the burn-in were discarded, and the resulting trees were used to generate a 50% majority consensus tree with posterior probabilities. All MCMC runs were repeated twice to confirm a consistent approximation of the posterior parameter distributions.
Analyses of Selection
Based on the aforementioned optimized tree topology from the concatenated 13 protein-coding genes, the ratio of nonsynonymous (dN) to synonymous (dS) substitutions rates (dN/dS) for concatenated gene sequences and each of the 13 mitochondrial protein-coding genes were computed separately to provide an indication of a change in selective pressure (Yang, 2007). The CODEML program of the PAML4.4 package (Yang, 2007) was implemented to assess whether the mitochondrial protein-coding genes of salmonids have undergone different selective constraints among freshwater and anadromous salmonids. Herein, model 1 with a free ω (dN/dS) ratio was estimated separately for each branch of the robust tree topology. The Wilcoxon rank-sum test was assigned to compute the level of statistical significance. All the statistical analysis was performed with SPSS 13.0 statistical package (SPSS Inc., Chicago, IL). To explore the difference in selective constraints between freshwater and anadromous salmonids, we applied the branch-site test from BUSTED (branch-site unrestricted statistical test for episodic diversification) (Murrell et al., 2015) to identify the branches under positive selection, which was implemented in the HyPhy package (Pond et al., 2005). Additionally, the fixed effects likelihood (FEL) approach in HyPhy software, which is more powerful than the CODEML program for detecting the individual sites subjected to episodic diversifying selection, was used to detect site-specific selection pressure. The FEL approach was run using the best-fitting nucleotide substitution model for each gene that was identified by jModelTest 2.1.3 (Darriba et al., 2012) on the ML phylogenetic tree. The subtree consisting of the 31 anadromous salmonids was specified as the foreground branch and tested, while the rest of the branches shared an arbitrary dN/dS ratio. Two models were nested in this method: H0, dN = dS (the neutral model), and HA, where dN and dS are estimated independently (the selection model). When the LRT is significant, if dN > dS, the site is declared to be under positive selection, otherwise the site is under negative selection. A nominal significance level of 0.1 for the likelihood ratio test was chosen based on the desired power of this analysis. RELAX is most useful for identifying trends and/or shifts in the stringency of natural selection on a given gene (Wertheim et al., 2015). Therefore, we used the program RELAX in HyPhy to distinguish intensified selection from the relaxed selection using 31 anadromous salmonids as the test lineages. RELAX then was used to test for the relaxed/intensified selection by introducing the parameter k. A significant result of k > 1 would indicate an intensified selection on test lineages, and a significant result of k < 1 would indicate a relaxed selection on test lineages.
Results
Characteristics of Salmonid Mitochondrial Genomes
The mitochondrial genome sizes of salmonids ranged from 16,526 to 16,997 bp. The mitochondrial genome contains 13 protein-coding genes, two ribosomal RNA genes, 22 transfer RNA genes, and a putative control region (CR) which is similar to those of other teleosts (Broughton et al., 2001; Saitoh et al., 2006). The length variations resulted from the control regions. The concatenated data set consisted of 11,355 bp from 13 PCGs of 51 mitochondrial genomes. There were 5,058 variable sites and 4,153 parsimony informative sites among the identified sites. The saturation test of the concatenated alignments was derived using DAMBE 6.4.107. There was no saturation for any codon position in the concatenated alignments (Supplementary Figure S1). The best-fit partitioning scheme across each gene and codon position is determined and shown in Table 1.
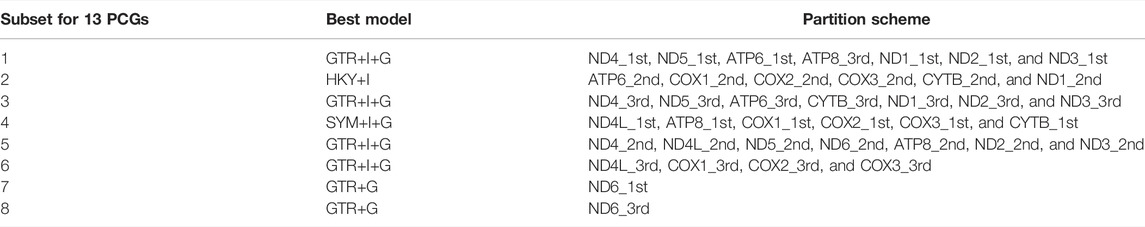
TABLE 1. Partition scheme of 13 PCGs used in this study.
Phylogenetic Analysis From Mitochondrial Genes
The BI and ML trees obtained from 13 mitochondrial protein-coding genes (total = 11,355 bp) revealed a consistent topology with high bootstrap values and posterior probabilities (Figure 1). Both freshwater and anadromous salmonids were mapped onto the phylogenetic topology. The phylogenetic results indicated that anadromous salmonids clustered as one monophyletic group in Coregoninae and clustered as a polyphyletic group in Salmoninae. Within Salmonidae, the freshwater Thymallinae emerged as a sister group to all other salmonids with high support values (0.80/100). There was a high support value for a sister relationship between both Coregoninae and Salmoninae subfamilies. Within the subfamily Coregoninae, three genera Coregonus, Prosopium, and Stenodus exhibited the evolutionary relationships [Prosopium (Coregonus, Stenodus)]. The phylogenetic analysis of the data sets produced six distinct and well-supported evolutionary groups for the subfamily Salmoninae as follows: Brachymystax, Hucho, Salmo, Parahucho, Salvelinus, and Oncorhynchus, which were supported by high support values. Brachymystax and Hucho were retained with strong nodal support (1.00/100) in the basal position of the trees orderly followed by the genera Salmo, Parahucho, Salvelinus, and Oncorhynchus. When we plotted the behaviors on the phylogeny of salmonids, we found there were three transitions from freshwater salmonids to anadromous salmonids (Figure 1). The results suggested the migratory behaviors of salmonids resulted from the multiple evolutionary origins.
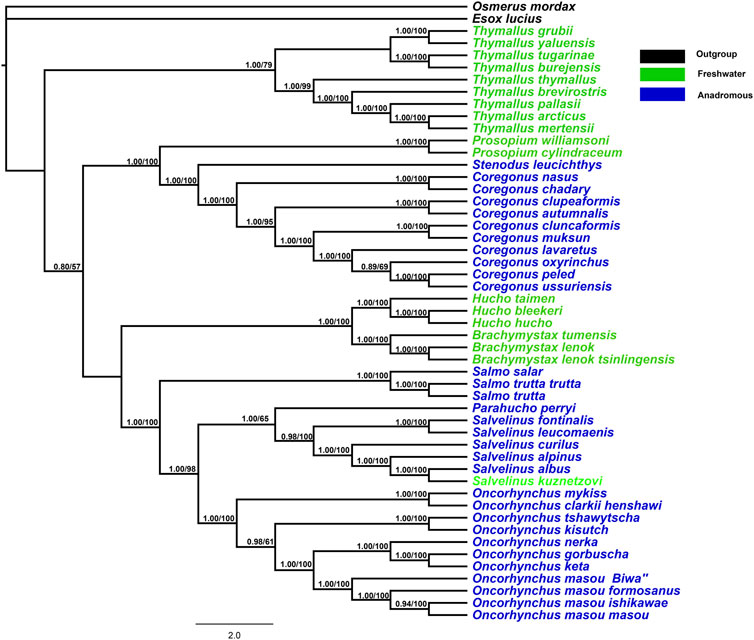
FIGURE 1. Phylogenetic tree constructed for combining 13 mitochondrial protein-coding gene orthologs using RAxML and Bayesian inference methods. MrBayes with GTR+I+G model was used. The Bayesian posterior probabilities and bootstrap support values are indicated at the nodes.
Analyses of Selection on Mitochondrial DNA
The salmonids can be grouped into two categories: anadromous salmonids (marine\freshwater) and freshwater salmonids. The freshwater salmonids comprised 18 species, while the remaining 31 species are anadromous. We found that the overall dN/dS ratio for the concatenated 13 protein-encoding genes of the anadromous group was significantly larger than that of the freshwater group. Also, with respect to each gene separately, most mitochondrial genes of the anadromous group had larger dN/dS ratios than that of the freshwater group (Supplementary Table S2). Especially, ND2, ND3, and ND5 genes possessed significantly larger dN/dS ratios in the anadromous group than that in the freshwater group (Figure 2). But the CYTB gene had significantly smaller dN/dS in the anadromous group.
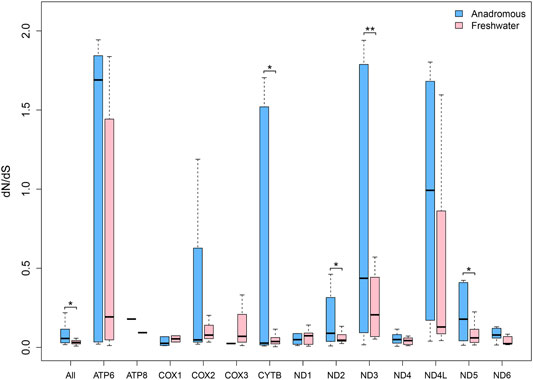
FIGURE 2. dN/dS ratios for each of 13 mitochondrial protein-coding genes of freshwater and anadromous salmonids.
When 31 anadromous salmonids were specified as the foreground branch, the FEL analysis identified positively selected sites and negatively selected sites in each mitochondrial protein-coding gene separately (Table 2). The RELAX program was used to compare the selective pressures on different parts of the tree, and the ATP8 and CYTB genes supported intensified selection on the anadromous lineages among the 13 mitochondrial protein-coding genes (Table 3). BUSTED provided evidence that at least one site on at least one test branch has experienced diversifying selection. The results showed that the ND5 gene evolved under positive selection in freshwater lineages, while the ND5 gene and ND6 gene evolved under positive selection in anadromous lineages. Therefore, it is reasonable to assume that the ND6 gene might play an important role in the evolution of the migratory behavior of salmonids.
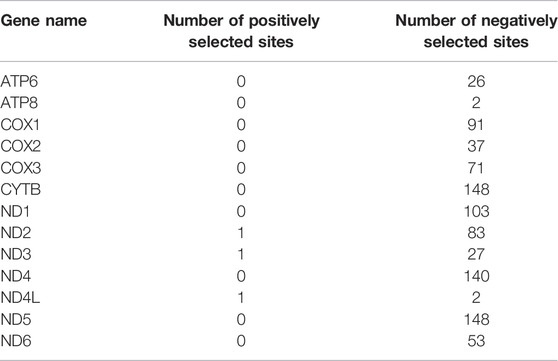
TABLE 2. Statistics of positively selected sites and negatively selected sites derived from the FEL (fixed effects likelihood) method.
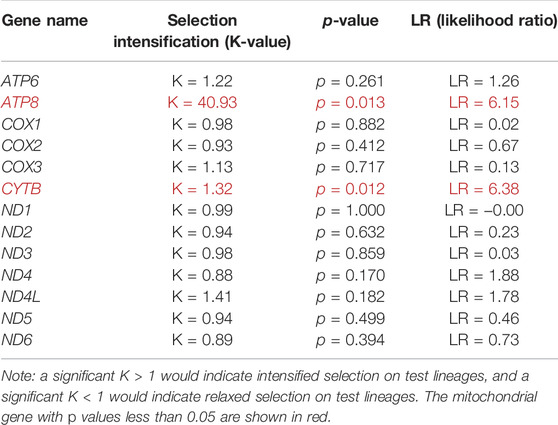
TABLE 3. Intensified and relaxed selection for mitochondrial protein-coding genes on the anadromous branches.
Discussion
The phylogenetic trees based on 13 concatenated protein-coding genes showed well-supported evolutionary relationships of the Salmonidae family, covering more genera (n = 10), compared to the previous salmonid mitogenome studies (Yasuike et al., 2010; Wang et al., 2011; Jacobsen et al., 2012; Campbell et al., 2013). Our present analyses yielded strong support for the basal position of Thymallinae, followed by a sister relationship between Coregoninae and Salmoninae, which is congruent with the recent molecular phylogenetic analyses based on whole mitochondrial genome sequences at the subfamily level (Li et al., 2010; Campbell et al., 2013; Macqueen and Johnston, 2014). However, the previous studies have revealed the inconsistency of the subfamily relationships that Coregoninae is in an ancestral phylogenetic position within Salmonidae and the sister relationship of Thymallinae and Salmoninae using the few mitochondrial genomes or a single nuclear locus (Yasuike et al., 2010; Wang et al., 2011; Shedko et al., 2012). The inconsistency is mainly resulting from the inclusion of only one Coregonus genus in phylogenetic analyses. Additionally, the phylogenetic analysis based on 78 nuclear gene sets suggested that the subfamily Thymallinae is in an ancestral phylogenetic position within Salmonidae, and the Coregoninae is the sister group to Salmoninae (Koop et al., 2008). Thymallinae and Salmoninae were clustered as a clade that is the sister group to the Coregoninae in the morphologically-based analysis of salmonid phylogenetic relationships (Wilson and Williams, 2010). The disagreements in subfamily relationships within the family Salmonidae that occurred in the previous studies might have resulted from the lack of the representative genera, the choice of outgroups, and different methodologies. In addition, the characteristics of mitochondria including linkage, absence of recombination, and small effective populations might limit their applicability in phylogenetic reconstruction (Birky et al., 1989; Tanya and Kumar, 2010). The evolutionary relationship is {[Thymallus, (Prosopium, (Coregonus, Stenodus))], [(Hucho, Brachymystax), (Salmo, (Oncorhynchus, (Parahucho, Salvelinus)))]} among these genera. Within the subfamily Coregoninae, the genus Prosopium occupied a basal position followed by a sister clade of Coregonus and Stenodus, which is in agreement with the early phylogenetic studies (Stearley and Smith, 1993; Macqueen and Johnston, 2014). The subfamily Thymallinae includes a single genus Thymall, which is generally accepted (Nelson, 2006; Yasuike et al., 2010). An extended dataset of salmonid mitochondrial genomes provided good resolution for the phylogeny of the family Salmonidae. Although some limitations occurred in mtDNA-based phylogeny, this study would keep the door open for subsequent work to address with additional datasets.
With respect to the subfamily Salmoninae, the evolutionary relationship based on the mitochondrial genes was consistent with the previous studies (Crespi and Fulton, 2004; Phillips et al., 2004; Yasuike et al., 2010). The results supported a sister relationship between the genus Salvelinus and Oncorhynchus, instead of Salmo and Oncorhynchus as expected from the morphology and ribosomal ITS1 sequences (Phillips and Oakley, 1997), which agreed with the suggestions by Oakley and Phillips (1999) and Koop et al. (2008). The valid genus of the family Salmonidae was modified as Coregonus, Stenodus, Prosopium, Thymallus, Hucho, Brachymystax, Salmo, Parahucho, Salvelinus, and Oncorhynchus. As reported in the previous studies, the genera Prosopium, Thymallus, and Hucho are strictly freshwater species while other genera are anadromous species (Davidson, 2013). However, the freshwater and anadromous species were not clustered as monophyly on the phylogenetic trees, respectively, which reflected that anadromous species evolved at least twice from the freshwater salmonid ancestors (in Coregoninae and Salmoninae, respectively).
Sun et al. (2011) concluded the mitochondrial protein-coding genes of migratory fishes accumulate fewer nonsynonymous mutations than nonmigratory fishes. It suggested that the functional constraints act on mitochondria due to energy metabolism and influences the evolution of mitochondrial-encoded proteins in teleosts. At the same time, the analysis results suggested that CYTB, ND1, ND3, ND5, and COX3 genes may have more important roles in energy production. Garvin et al. (2011) examined the codon sites of 12 protein-coding genes to identify what role natural selection may have played in the divergence of the mitochondrial-coding proteins among salmonids. Therefore, we investigated the adaptive evolution of salmonids and explored the selective patterns of freshwater and anadromous salmonids from mitogenomic perspectives. The concatenated 13 mitochondrial protein-coding genes were shown to have significantly larger dN/dS in the anadromous species than in the freshwater species of Salmonidae. Moreover, the ND2, ND3, and ND5 genes were shown to have significantly larger dN/dS in the anadromous species than in the freshwater species of Salmonidae. But the CYTB gene had significantly smaller dN/dS in the anadromous fishes. This result is inconsistent with the conclusion drawn by 401 teleost complete mitochondrial genomes in the previous study (Sun et al., 2011). This inconsistency might result from the complex migratory behavior of salmonids or the small sample size (n = 51) available for this test in this study. Taken together, our results demonstrated that anadromous salmonids accumulated more nonsynonymous mutations in the mtDNA than freshwater salmonids. The result means migratory ability has significant effects on the mitochondrial dN/dS variation, which is reflected by the mitochondrial-encoded gene evolution. In conclusion, the adaptive mtDNA evolution pattern associated with complex migratory behavior was reflected in the specific group of salmonids. Through the branch-site model in HyPhy software, we detected the positively selected genes and sites, which suggested that mitochondrial protein-coding genes evolved at the molecular level to cope with the complex anadromous behaviors.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
ZS and YW conceived and designed the research. YW performed the computational analyses and wrote the manuscript. ZS and FX revised the manuscript.
Funding
This research was funded by the Program for New Century Excellent Talents in University (NCET-11-0347) to ZS and by the grant from the National Natural Science Foundation of China (31702016) to YW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.903240/full#supplementary-material
Supplementary Figure S1 | Saturation tests for any codon position in the concatenated alignment.
References
Abascal, F., Zardoya, R., and Telford, M. J. (2010). TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic Acids Res. 38, W7–W13. doi:10.1093/nar/gkq291
Birky, C. W., Fuerst, P., and Maruyama, T. (1989). Organelle Gene Diversity under Migration, Mutation, and Drift: Equilibrium Expectations, Approach to Equilibrium, Effects of Heteroplasmic Cells, and Comparison to Nuclear Genes. Genetics 121, 613–627. doi:10.1093/genetics/121.3.613
Broughton, R. E., Milam, J. E., and Roe, B. A. (2001). The Complete Sequence of the Zebrafish (Danio rerio) Mitochondrial Genome and Evolutionary Patterns in Vertebrate Mitochondrial DNA. Genome Res. 11, 1958–1967. doi:10.1101/gr.156801
Campbell, M. A., López, J. A., Sado, T., and Miya, M. (2013). Pike and Salmon as Sister Taxa: Detailed Intraclade Resolution and Divergence Time Estimation of Esociformes+Salmoniformes Based on Whole Mitochondrial Genome Sequences. Gene 530, 57–65. doi:10.1016/j.gene.2013.07.068
Crespi, B. J., and Fulton, M. J. (2004). Molecular Systematics of Salmonidae: Combined Nuclear Data Yields a Robust Phylogeny. Mol. phylogenetics Evol. 31, 658–679. doi:10.1016/j.ympev.2003.08.012
Crête-Lafrenière, A., Weir, L. K., and Bernatchez, L. (2012). Framing the Salmonidae Family Phylogenetic Portrait: a More Complete Picture from Increased Taxon Sampling. PloS one 7, e46662.
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 9, 772. doi:10.1038/nmeth.2109
Davidson, W. S. (2013). Understanding Salmonid Biology from the Atlantic Salmon Genome. Genome 56, 548–550. doi:10.1139/gen-2013-0163
Garvin, M. R., Bielawski, J. P., and Gharrett, A. J. (2011). Positive Darwinian Selection in the Piston that Powers Proton Pumps in Complex I of the Mitochondria of Pacific Salmon. Plos One 6, e24127. doi:10.1371/journal.pone.0024127
Gosline, W. A., and Norden, C. R. (1963). Comparative Osteology of Representative Salmonid Fishes, with Particular Reference to the Grayling (Thymallus arcticus) and its Phylogeny. Copeia 1963, 209. doi:10.2307/1441325
Jacobsen, M. W., da Fonseca, R. R., Bernatchez, L., and Hansen, M. M. (2015). Comparative Analysis of Complete Mitochondrial Genomes Suggests that Relaxed Purifying Selection Is Driving High Nonsynonymous Evolutionary Rate of the NADH2 Gene in Whitefish (Coregonus ssp.). Mol. Phylogenet Evol. 95, 161–170. doi:10.1016/j.ympev.2015.11.008
Jacobsen, M. W., Hansen, M. M., Orlando, L., Bekkevold, D., Bernatchez, L., Willerslev, E., et al. (2012). Mitogenome Sequencing Reveals Shallow Evolutionary Histories and Recent Divergence Time between Morphologically and Ecologically Distinct European Whitefish (Coregonusspp.). Mol. Ecol. 21, 2727–2742. doi:10.1111/j.1365-294x.2012.05561.x
Koop, B. F., Von Schalburg, K. R., Leong, J., Walker, N., Lieph, R., Cooper, G. A., et al. (2008). A Salmonid EST Genomic Study: Genes, Duplications, Phylogeny and Microarrays. BMC genomics 9, 545. doi:10.1186/1471-2164-9-545
Lanfear, R., Calcott, B., Ho, S. Y. W., and Guindon, S. (2012). Partitionfinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Mol. Biol. Evol. 29, 1695–1701. doi:10.1093/molbev/mss020
Li, J., Xia, R., McDowall, R. M., López, J. A., Lei, G., and Fu, C. (2010). Phylogenetic Position of the Enigmatic Lepidogalaxias Salamandroides with Comment on the Orders of Lower Euteleostean Fishes. Mol. Phylogenetics Evol. 57, 932–936. doi:10.1016/j.ympev.2010.07.016
Macqueen, D. J., and Johnston, I. A. (2014). A Well-Constrained Estimate for the Timing of the Salmonid Whole Genome Duplication Reveals Major Decoupling from Species Diversification. Proc. R. Soc. B 281, 20132881. doi:10.1098/rspb.2013.2881
McDowall, R. M. (1988). Diadromy in Fishes: Migrations between Freshwater and Marine Environments. Croom Helm 38.
Mcdowall, R. M. (2001). The Origin of the Salmonid Fishes: Marine, freshwater. Or Neither? Rev. Fish Biol. Fish. 11, 171–179. doi:10.1023/a:1020370328194
Murrell, B., Weaver, S., Smith, M. D., Wertheim, J. O., Murrell, S., Aylward, A., et al. (2015). Gene-wide Identification of Episodic Selection. Mol. Biol. Evol. 32, 1365–1371. doi:10.1093/molbev/msv035
Myers, G. S. (1949). Usage of Anadromous, Catadromous and Allied Terms for Migratory Fishes. Copeia 1949, 89–97. doi:10.2307/1438482
Nei, M. (2005). Selectionism and Neutralism in Molecular Evolution. Mol. Biol. Evol. 22, 2318–2342. doi:10.1093/molbev/msi242
Oakley, T. H., and Phillips, R. B. (1999). Phylogeny of Salmonine Fishes Based on Growth Hormone Introns: Atlantic (Salmo) and Pacific (Oncorhynchus) Salmon Are Not Sister Taxa. Mol. Phylogenet Evol. 11, 381–393. doi:10.1006/mpev.1998.0599
Phillips, R. B., Matsuoka, M. P., Konkol, N. R., and McKay, S. (2004). Molecular Systematics and Evolution of the Growth Hormone Introns in the Salmoninae. Environ. Biol. Fishes 69, 433–440. doi:10.1023/b:ebfi.0000022873.32682.3f
Pond, S. L. K., Frost, S. D. W., and Muse, S. V. (2005). HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 21, 676–679. doi:10.1093/bioinformatics/bti079
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian Phylogenetic Inference under Mixed Models. Bioinformatics 19, 1572–1574. doi:10.1093/bioinformatics/btg180
Saitoh, K., Sado, T., Mayden, R. L., Hanzawa, N., Nakamura, K., Nishida, M., et al. (2006). Mitogenomic Evolution and Interrelationships of the Cypriniformes (Actinopterygii: Ostariophysi): the First Evidence toward Resolution of Higher-Level Relationships of the World's Largest Freshwater Fish Clade Based on 59 Whole Mitogenome Sequences. J. Mol. Evol. 63, 826–841. doi:10.1007/s00239-005-0293-y
Shedko, S. V., Miroshnichenko, I. L., and Nemkova, G. A. (2013). Phylogeny of Salmonids (Salmoniformes: Salmonidae) and its Molecular Dating: Analysis of mtDNA Data. Russ. J. Genet. 49, 623–637. doi:10.1134/s1022795413060112
Shedko, S. V., Miroshnichenko, I. L., and Nemkova, G. A. (2012). Phylogeny of Salmonids (Salmoniformes: Salmonidae) and its Molecular Dating: Analysis of Nuclear RAG1 Gene. Russ. J. Genet. 48, 575–579. doi:10.1134/s1022795412050201
Shen, Y.-Y., Shi, P., Sun, Y.-B., and Zhang, Y.-P. (2009). Relaxation of Selective Constraints on Avian Mitochondrial DNA Following the Degeneration of Flight Ability. Genome Res. 19, 1760–1765. doi:10.1101/gr.093138.109
Stamatakis, A. (2006). RAxML-VI-HPC: Maximum Likelihood-Based Phylogenetic Analyses with Thousands of Taxa and Mixed Models. Bioinformatics 22, 2688–2690. doi:10.1093/bioinformatics/btl446
Stearley, R. F., and Smith, G. R. (1993). Phylogeny of the Pacific Trouts and Salmons (Oncorhynchus) and Genera of the Family Salmonidae. Trans. Am. Fish. Soc. 122, 1–33. doi:10.1577/1548-8659(1993)122<0001:potpta>2.3.co;2
Sun, Y.-B., Shen, Y.-Y., Irwin, D. M., and Zhang, Y.-P. (2011). Evaluating the Roles of Energetic Functional Constraints on Teleost Mitochondrial-Encoded Protein Evolution. Mol. Biol. Evol. 28, 39–44. doi:10.1093/molbev/msq256
Tanya, C., and Kumar, R. (20102010). Molecular Markers and Their Applications in Fisheries and Aquaculture. Adv. Biosci. Biotechnol. 1.
Wang, Y., Guo, R., Li, H., Zhang, X., Du, J., and Song, Z. (2011). The Complete Mitochondrial Genome of the Sichuan Taimen (Hucho Bleekeri): Repetitive Sequences in the Control Region and Phylogenetic Implications for Salmonidae. Mar. genomics 4, 221–228. doi:10.1016/j.margen.2011.06.003
Wertheim, J. O., Murrell, B., Smith, M. D., Kosakovsky Pond, S. L., and Scheffler, K. (2015). RELAX: Detecting Relaxed Selection in a Phylogenetic Framework. Mol. Biol. Evol. 32, 820–832. doi:10.1093/molbev/msu400
Wilson, M., and Williams, R. R. G. (2010). “Frege's Mathematical Setting,” in Origin and Phylogenetic Interrelationships of Teleosts. Editors J. S. Nelson, and H. P. Schultze (München: Verlag Dr. Friedrich Pfeil), 379379–412409. doi:10.1017/ccol9780521624282.010
Wilson, W. D., and Turner, T. F. (2009). Phylogenetic Analysis of the Pacific Cutthroat Trout (Oncorhynchus clarki ssp.: Salmonidae) Based on Partial mtDNA ND4 Sequences: A Closer Look at the Highly Fragmented Inland Species. Mol. phylogenetics Evol. 52, 406–415. doi:10.1016/j.ympev.2009.03.018
Xia, X. (2017). DAMBE6: New Tools for Microbial Genomics, Phylogenetics, and Molecular Evolution. J. Hered. 108, 431–437. doi:10.1093/jhered/esx033
Yang, Z. (2007). PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 24, 1586–1591. doi:10.1093/molbev/msm088
Keywords: Salmonidae, phylogenetic analyses, mitochondrial DNA, dN/dS, adaptive evolution
Citation: Wang Y, Xiong F and Song Z (2022) Molecular Phylogeny and Adaptive Mitochondrial DNA Evolution of Salmonids (Pisces: Salmonidae). Front. Genet. 13:903240. doi: 10.3389/fgene.2022.903240
Received: 24 March 2022; Accepted: 10 May 2022;
Published: 17 June 2022.
Edited by:
Zuogang Peng, Southwest University, ChinaReviewed by:
Renyi Zhang, Guizhou Normal University, ChinaChenguang Feng, Northwestern Polytechnical University, China
Copyright © 2022 Wang, Xiong and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Xiong, eGlvbmdmZWlAamh1bi5lZHUuY24=; Zhaobin Song, emJzb25nQHNjdS5lZHUuY24=