 Yinbo Liu
Yinbo Liu Yingying Shen†
Yingying Shen† Xiaolei Zhu
Xiaolei Zhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 30 March 2022
Sec. Computational Genomics
Volume 13 - 2022 | https://doi.org/10.3389/fgene.2022.853258
As one of the most important post-transcriptional modifications of RNA, 5-cytosine-methylation (m5C) is reported to closely relate to many chemical reactions and biological functions in cells. Recently, several computational methods have been proposed for identifying m5C sites. However, the accuracy and efficiency are still not satisfactory. In this study, we proposed a new method, m5Cpred-XS, for predicting m5C sites of H. sapiens, M. musculus, and A. thaliana. First, the powerful SHAP method was used to select the optimal feature subset from seven different kinds of sequence-based features. Second, different machine learning algorithms were used to train the models. The results of five-fold cross-validation indicate that the model based on XGBoost achieved the highest prediction accuracy. Finally, our model was compared with other state-of-the-art models, which indicates that m5Cpred-XS is superior to other methods. Moreover, we deployed the model on a web server that can be accessed through http://m5cpred-xs.zhulab.org.cn/, and m5Cpred-XS is expected to be a useful tool for studying m5C sites.
RNA modification plays pivotal roles in many biological processes (Tang et al., 2001; Matzke et al., 2004; Xu et al., 2013; Jespersen et al., 2017; Xue Chen et al., 2020). Until now, about 170 types of RNA modifications have been discovered (Xuan et al., 2018), among which, 5-methylcytosine (m5C) is one of the most abundant post-transcriptional modifications (PTCM). In this modification, a methyl group is transferred to the fifth carbon atom of cytosine by RNA methyl-transferase (Jespersen et al., 2017). The m5C modification plays important roles in many biochemical reactions (Catania and Fairweather 1991; Fasolino et al., 2017; Yang et al., 2017; He et al., 2020; Xue MiaoMiao et al., 2020; Zhang et al., 2020), such as the pathogenesis of various cancers (He et al., 2020; Xue MiaoMiao et al., 2020; Zhang et al., 2020), rRNA assembly (Zhang et al., 2020), and cellular aging (Catania and Fairweather 1991), etc. Thus, it is meaningful to pinpoint m5C sites in RNA sequences.
Several experimental methods have been developed to identify m5C sites, including miCLIP-seq (Hussain et al., 2013), Aza-IP-seq (Khoddami and Cairns 2013), bisulfite sequencing (Agris 2008; Schaefer et al., 2010), and m5C-RIP-seq (Khoddami et al., 2019). However, these methods have their own shortcomings (Fu et al., 2012). For example, bisulfite sequencing cannot detect m5C sites in low-abundance RNA. Moreover, these existing experimental methods are time-consuming and expensive. In recent years, with the development of computer technology, several computational methods, especially those machine-learning based methods, have been developed for RNA m5C site identification (Feng et al., 2016; Qiu et al., 2017; Sabooh et al., 2018; Zhang et al., 2018).
The computational methods are mainly classified into two categories: random forest (RF)-based models and support vector machine (SVM)-based models. Based on RF, Qiu et al. (2017) proposed iRNAm5C-PseDNC based on pseudo dinucleotide composition (PseDNC) feature encoding, and Li et al. (2018) constructed RNAm5Cfinder by using mononucleotide binary encoding (MNBE) to encode the RNA sequences. Based on these two feature encodings and K-tuple nucleotide frequency component (KNFC), Song et al. (2018) developed a predictor named PEA-m5C. By using SVM as the classifier, Feng et al. (2016) developed m5C-PseDNC based on features of PseDNC. Fang et al. (2019) built RNAm5CPred based on the features of PseDNC, KNFC, and MNBE. By integrating multiple SVM methods, Zhang et al. (2018) developed an ensemble model, m5C-HPCR, by incorporating different physical–chemical properties into PseDNC. Chen Xiao et al. (2020) proposed another SVM-based model, m5CPred-SVM, which uses six sequence-based features, including k-nucleotide frequency (KNF), K-spaced nucleotide pair frequency (KSNPF), position-specific nucleotide propensity (PSNP), K-spaced position-specific dinucleotide propensity (KSPSDP), PseDNC, and chemical property with density (CPD).
As mentioned above, different kinds of features have been generated for predicting m5C sites, and the dimension of these features can be very high; however, not all the features are relevant for building machine learning models. Moreover, the features with ultrahigh dimensions also pose a great challenge to computer performance (Li et al., 2021). Selecting the optimal feature subset by appropriate feature selection methods can not only improve the accuracy of the prediction model, but also effectively reduce the huge computing power required for model training.
Recently, different feature selection methods have been used in developing models for predicting the RNA modification sites. Wang et al. (2018) used a minimum redundancy maximum (mRMR) correlation algorithm to select discriminative features from the features encoded based on RNA sequences. Sabooh et al. (2018) developed a new computational method pm5CS-Comp-mRMR by also using mRMR for selecting the discriminate features. Furthermore, Visentini et al. (2016) first sorted the features according to the F-score obtained in the eXtreme gradient boosting (XGBoost) (Chen, 2016) package and then selected the top 50 features based on the incremental feature selection (IFS) strategy as the optimal feature subset. To reduce the dimension of features, Chai et al. (2021a) proposed an efficient m5C sites prediction approach, Staem5, based on features selected by F-score. The SHapley Additive ExPlanations (SHAP) (Wang and Gribskov 2019; Bi et al., 2020) method, which can interpret the importance of features, is another effective method for selecting relevant features. The method was also used in several recent works (Bi et al., 2020; Pathy et al., 2020; Effrosynidis and Arampatzis 2021).
In this study, we established a new method to predict m5C sites by using XGBoost based on features selected by SHAP. We named this method m5Cpred_XS, which can be used to predict m5C sites in multiple species. Extensive experiments demonstrated that the proposed predictor, m5Cpred_XS, outperformed other existing prediction methods. Finally, a web server (http://m5cpred-xs.zhulab.org.cn/) was deployed for the users.
For building our model reasonably, we conducted our study in six steps. I) A benchmark data set was collected. The benchmark data set was divided into the training set and the independent test set. II) The features were extracted from RNA sequences. III) The SHAP-based feature selection was carried out to select the optimal feature subset. IV) The XGBoost was used to train the model. V) The comparison and analysis of different models was conducted. VI) A web server for predicting m5C sites was developed for the community. The overall flow chart of our study is shown in Figure 1.
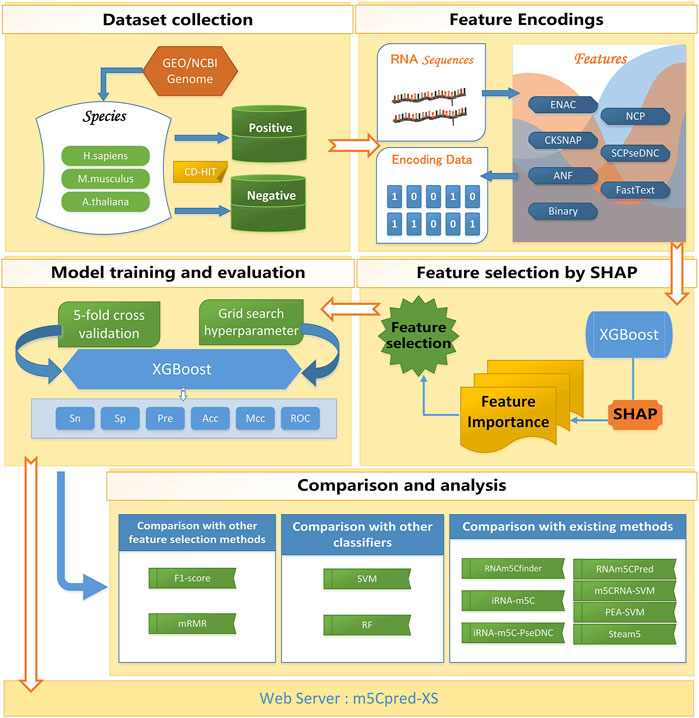
FIGURE 1. The flowchart of m5Cpred_XS.
For fair comparison, we used the same data sets as in Chen Xiao et al. (2020). In their work, they collected data for three species: H. sapiens, M. musculus, and A. thaliana. As shown in Table 1, the data sets contain 269, 5563, and 6289 positive samples for the three species, respectively, and the numbers of negative samples are the same as positive samples. The positive samples of H. sapiens, M. musculus, and A. thaliana were collected from the work of Yang et al. (2017), Khoddami et al. (2019), and Cui et al. (2017), respectively. For the details about how the data sets were obtained, please refer to Chen Xiao et al. (2020).
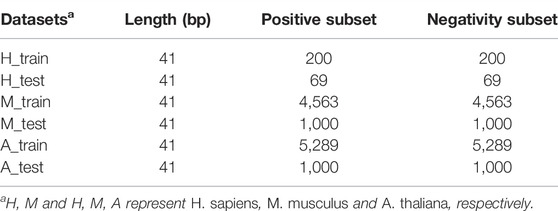
TABLE 1. Training and test data sets of three species.
To build and evaluate the models, the benchmark data sets were divided into two parts: the training data sets and the independent test sets. The training data sets were used for the model construction, cross-validation, and the determination of the hyperparameters of machine learning algorithms, whereas the independent test sets were used for testing the prediction performance and generalization ability of the models. For A. thaliana, 1000 positive and 1000 negative samples were randomly selected from the data set as the independent test data set, and the remaining 5298 positive and 5298 negative samples were selected as the training data set. Similarly, 1000 positive and 1000 negative samples from M. musculus’ benchmark data set were selected as the independent test set, and the remaining 4563 positive and 4563 negative samples were selected as the training data set. For H. sapiens, 69 positive and 69 negative samples were randomly selected as the independent test set, and the remaining 200 positive and 200 negative samples were selected as the training data set. The specific partitioning of the data sets is shown in Table 1.
For each RNA segment, it can be expressed in the following form:
In this formula,
ENAC encoding (Ahmad and Shatabda 2019) is used for feature extraction in equal-length RNA sequences. It first determines a fixed length window, and then the window is slid from the 5-terminal to the 3-terminal of the RNA segment without interval. The features of ENAC are expressed as follows (Han et al., 2019):
In this formula,
The CKSNAP feature encoding scheme (Cui et al., 2017; Ju and Wang 2020) is based on the frequency of k-spaced nucleotide pairs (k = 0, 1, 2, 3, 4, 5). For example, when k = 1, the nucleotide pairs corresponding to k-spaced 16 possible nucleotide pairs (i.e., “A∗A″, “A∗C″, “A∗G″, …, “C∗G″, “G∗A″, …, “G∗C″, “U∗U″, “U∗A″, “U∗C″, “U∗G″), CKSNAP can be expressed as the following formula:
where
ANF, also known as nucleotide density (ND), fully considers the distribution and nucleotide frequency information of each nucleotide in the RNA sequence (Chen Zhen, et al., 2020). The density of a nucleotide
where
Adenine (A), guanine (G), cytosine (C), and uracil (U) are the four types of nucleotides in RNA, each of which has unique chemical properties and physical structure. According to different chemical properties, these four nucleotides can be divided into three categories (Chen et al., 2016). The details are shown in Table 2.
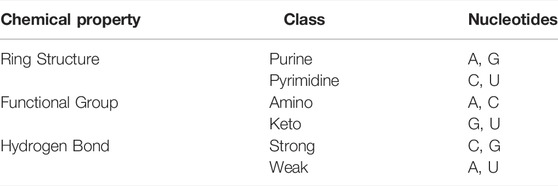
TABLE 2. Chemical structure of each nucleotide.
Based on the three types of chemical properties, A, C, U, and G can be expressed as (1, 1, 1), (0, 1, 0), (1, 0, 0), and (0, 0, 1), respectively. The feature dimension generated by NCP is 123.
The method of using a four-dimensional binary vector to encode the nucleotide is called the binary encoding scheme (Foster et al., 2003) by which A, C, G, and U are encoded as (1, 0, 0, 0), (0, 1, 0, 0), (0, 0, 1, 0), and (0, 0, 0, 1), respectively. Thus, we obtained a 164-dimensional feature vector for an RNA segment containing 41 nucleotides.
The expression of SCPseDNC (Chen et al., 2014) coding is as follows:
where
Here,
where the correlation function
where
FastText is a natural language model released by Facebook (Joulin et al., 2017). By considering the RNA segments as sentences, we used the FastText program to build a word2vec model and then to encode the RNA segments as word vectors. Both skipgram and cbow models can be trained in FastText; we, thus, trained a cbow model to generate word embeddings for RNA segments. A total of 100-dimensional feature data was generated by using FastText.
Feature selection is an important step in building effective machine learning models when high-dimensional features were generated. In this study, three different feature selection methods were employed to select the optimal feature subsets. As one of the frameworks for explaining the prediction model, the SHAP algorithm was proposed to characterize feature importance and assess feature behavior (Swann et al., 2011). The contribution of each feature can be evaluated by the SHAP value, which is calculated by
where
The F-score (Polat and Guenes 2009) is another feature selection method that measures the discriminatory ability of two sets of real values. The F-score value of each feature in the data set can be calculated by the following equation:
where
The third feature selection method used in this study is maximum relevance minimum redundancy (mRMR), which was developed by Peng et al. (Hanchuan et al., 2005). In this method, mutual information (MI) is used to evaluate the relationships among the features and the labels, and the goal of the method is to identify features that can maximize the relevance between features and labels and simultaneously minimize the relevance between the features. The following equation is used to select features recursively:
where
where
The XGBoost was a distributed gradient enhancement library that was widely used in classification scenarios (Ji et al., 2019; Zhao et al., 2019; Ding et al., 2020; Samat et al., 2020). It has many advantages, such as flexibility, efficiency, and portability. The basic principle of this algorithm is to assign quantitative weight to each leaf node of a series of decision trees. The parallel enhanced trees are provided by XGBoost. This algorithm has very good ability to process sparse and high-dimensional data, and it also inherits the high accuracy of the original boosting algorithm. Some researchers apply this algorithm in bioinformatics, such as the prediction of m6A (Qiang et al., 2018; Zhao et al., 2019) and m7G sites (Bi et al., 2020). In this paper, we used a python package to build the XGBoost model and used a grid search method to optimize hyperparameters, max_depth, learning_rate, and n_estimators. The ranges of these three hyperparameters are (2, 4, 6, 8,10, 12, 14.16), (0.005, 0.01, 0.02, 0.05, 0.1), and (1,600,1800,2000, 2,200, 2,400, 2,600, 2,800), respectively. Finally, we obtained different optimal hyperparameters for different species. The optimal hyperparameters for three species are shown in Table 3.

TABLE 3. The optimal hyperparameters of XGBoost for three species.
Cross-validation is often used to evaluate the performance and generalization ability of machine learning models. In this paper, five-fold cross-validation was used to evaluate the models, and the random sampling method was used to divide the training data set into five subsets with very close data volume (Fushiki 2011). In each training, one of the five subsets was used as validation data set, and the other four were used for training the model. Thus, a total of five m5C site prediction models were obtained. Finally, the prediction results of these five models were evaluated, and the five evaluation values were averaged as the ultimate evaluation indices. Similarly, this five-fold cross-validation was also adopted for hyperparameter selection, algorithm comparison, etc.
Different evaluation metrics are used in bioinformatics classification. In this study, we selected the accuracy (Acc), sensitivity (Sen), specificity (Spe), precision (Pre), Matthews correlation coefficient (Mcc), and F1-score as the main evaluation metrics (Zhang et al., 2019; Lv et al., 2020). Counts of true positive, true negative, false positive, and false negative predictions were recorded as TP, TN, FP, and FN, respectively. Thus, the six metrics can be represented as follows:
In addition to the above evaluation indicators, the precision recall curve (PRC curve) (Keilwagen et al., 2014; Saito and Rehmsmeier 2017) and receiver operating characteristic curve (ROC curve) (Fawcett 2006; Li et al., 2019) were also used to evaluate the model. These two curves have the ability to evaluate the prediction performance of the proposed method in the whole decision value range, and the areas under the curves (AUPRC and AUROC) are often used to quantify the performance of the models. We quantify the performance of the model by plotting these two kinds of curves and calculating the areas under the ROC and PRC curves.
Seven kinds of features were generated from the RNA segments of the three species of which the dimension is 808 in total. Considering the redundancy between the features, SHAP was used to select the optimal feature subsets by which the scores of importance of the 808-dimensional features were calculated based on XGBoost ensemble algorithm. Figure 2 shows the cross-validation AUROC values of models based on the top n features. The highest AUROCs were obtained when the top 48, 228, and 208 features were used for H. sapiens, M. musculus, and A. thaliana, respectively. The corresponding AUROC values are 0.935, 0.834, and 0.787, for the three species, respectively.
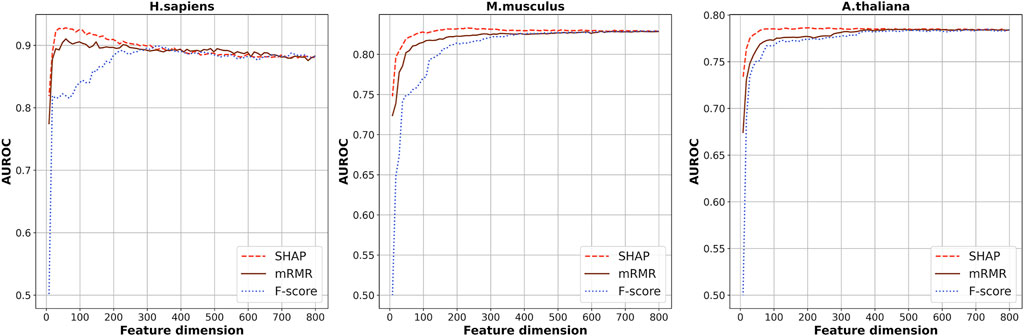
FIGURE 2. The cross-validation AUROC values of models based on the top n features selected by SHAP, mRMR, and f-score.
In addition, Table 4 shows all the evaluation metrics for the models based on features selected by SHAP and the models based on the original 808 features. It indicates that the models based on features selected by SHAP achieved higher values than the model based on the original 808 features for most of the metrics, which demonstrates the advantages of using SHAP for feature selection.
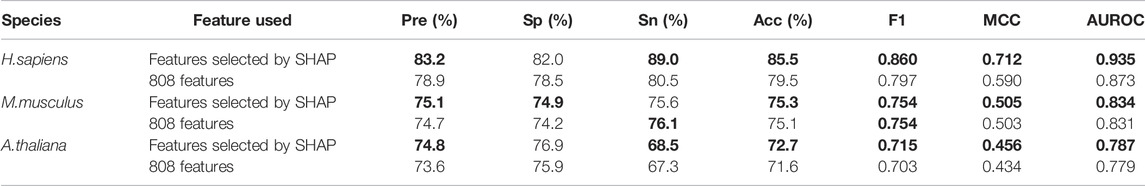
TABLE 4. The five-fold cross-validation results for models based on features selected by SHAP or the original 808 features.
Besides this, another two kinds of feature-selection methods, F-score (Polat and Guenes 2009) and mRMR (Li et al., 2017; Bugata and Drotar 2020), were also used to select the optimal feature subsets. The cross-validation AUROCs of the models based on the top n features selected by these two methods are also plotted in Figure 2. As shown in Figure 2, generally, the models based on features selected by SHAP are superior to the models based on features selected by the other two methods. Thus, we used the feature subsets selected by SHAP as the optimal feature subsets.
To verify the effectiveness of the XGBoost algorithm in m5C site prediction, two other learning algorithms, random forests (Biau 2012; Ziegler and Konig 2014; Li et al., 2018) and support vector machine (Boopathi et al., 2019; Chen et al., 2019; Liu et al., 2020), were also used to build models based on the optimal feature subsets selected by SHAP. The hyperparameters of RF and SVM were also optimized by grid search.
Table 5 shows the five-fold cross-validation performances for the models based on the three different learning algorithms. For A. thaliana, the AUROC value of the model based on XGBoost is 0.787, which is higher than the models based on RF (0.780) and SVM (0.768). For M. musculus, the AUROC value of the model based on XGBoost is 0.834, which is also higher than the models based on RF (0.795) and SVM (0.824). For H. sapiens, the AUROC value of the model based on XGBoost is 0.935, which is also higher than the models based on RF (0.911) and SVM (0.903). The ROC and PRC curves for three species are shown in Figure 3. As shown in Figure 3, for H. sapiens, the AUPRC of the model based on XGBoost is 0.942, which is higher than the models based on RF (0.910) and SVM (0.897). Similarly, for A. thaliana, the AUPRC of the model based on XGBoost is 0.794, which is higher than that based on RF (0.784) and SVM (0.771). In addition, for M. musculus, the AUPRC of the model based on XGBoost is 0.827, which is higher than the models based on SVM (0.812) and RF (0.791). Thus, the models built by using XGBoost were selected as our final models.
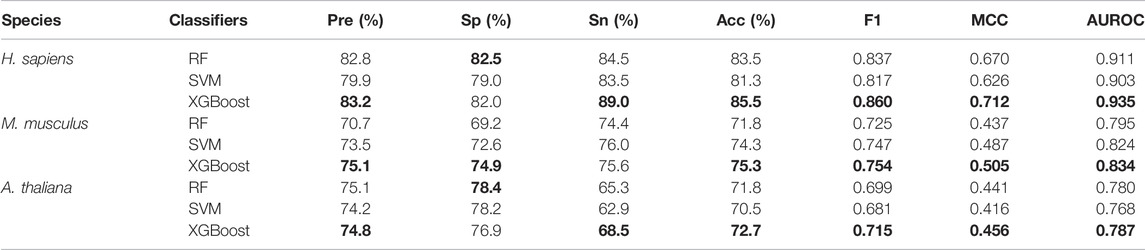
TABLE 5. The five-fold cross-validation performance of models built based on different classifiers with the features selected by SHAP.
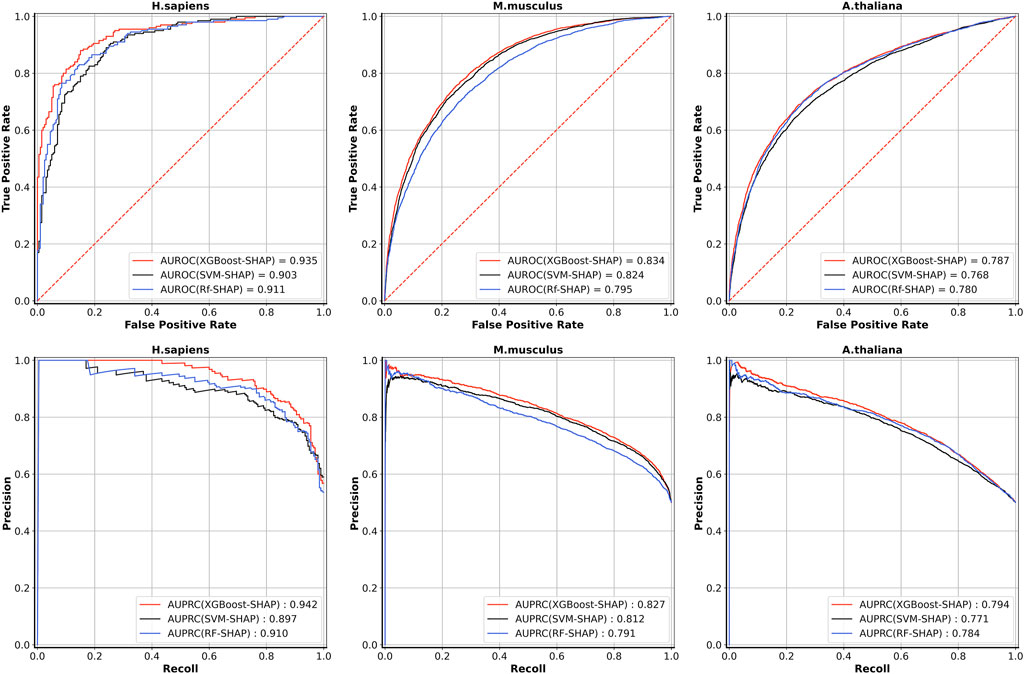
FIGURE 3. The ROC curves and PRC curves of five-fold cross-validation results based on three learning algorithms for the three species.
To further evaluate the generalization of our models, the predictive results of our models on the independent test sets were compared with other existing methods, iRNA-m5C (Lv et al., 2020), m5CPred-SVM (Chen Xiao et al., 2020), RNAm5Cfinder (Li et al., 2018), iRNAm5C-PseDNC (Qiu et al., 2017), RNAm5CPred (Fang et al., 2019), PEA-m5C (Song et al., 2018), and Staem5 (Chai et al., 2021b). However, not all of these methods can predict m5C sites in all three species. For example, RNAm5Cfinder (Li et al., 2018) can predict m5C sites for H. sapiens and M. musculus but not for A. thaliana. iRNAm5C-PseDNC (Qiu et al., 2017) and RNAm5CPred (Fang et al., 2019) can only predict the m5C sites of H. sapiens, and PEA-m5C (Song et al., 2018) can only be used for prediction of A. thaliana. By using the default decision threshold, Table 6 shows that our model achieved the highest performance for all seven evaluation metrics except specificity for H. sapiens compared with other state-of-the-art methods. For M. musculus, our model obtained the best AUROC, MCC, accuracy, and FOR (false omission rate). For A. thaliana, our model achieved the highest values for all seven evaluation metrics. Thus, we prove the superiority of our m5Cpred_XS model for predicting the m5C sites for three species. By using other decision thresholds as shown in Table 6, the precisions, specificities, accuracies, and MCCs of our models can be improved; however, other evaluation metrics, such as sensitivities and F1 scores drop away.
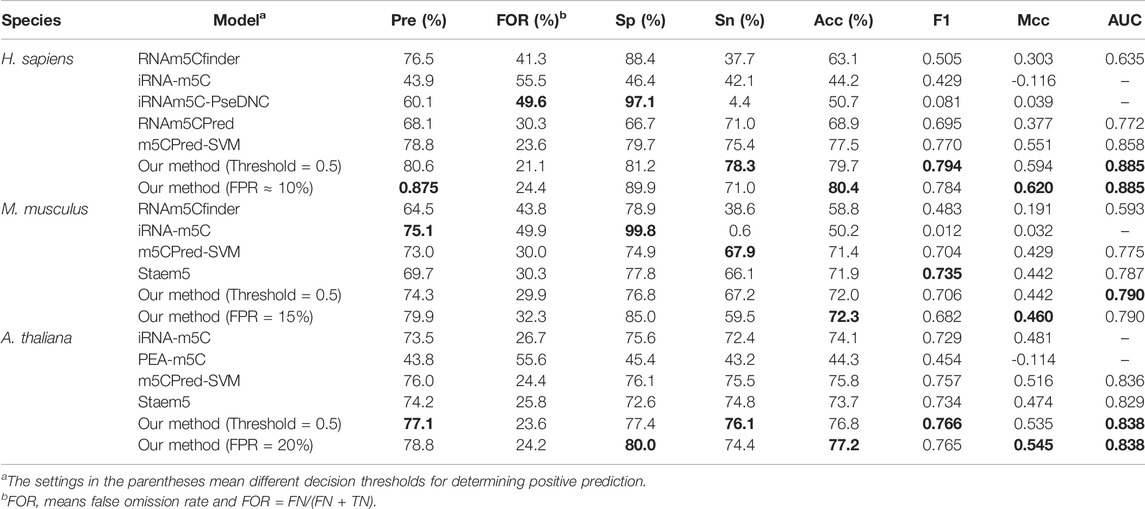
TABLE 6. Comparison with other existing models on the independent test sets.
It is noted that the predictive accuracies of iRNA-5mC and PEA-m5C on the independent test sets are even less than 0.50. The possible reason is that the corresponding training data sets for building these models are small. For example, the model of iRNA-m5C for homo sapiens is based on a data set that only contains 120 positive samples, and PEA-m5C is based on a data set that contains 1196 positive samples. Both data sets were smaller than the data sets used in this study. The small size of the data set limits the generalization of the model on the independent test set. In addition, the model was not evaluated on an independent test set in the original paper of iRNA-m5C and the redundancy of the data set used for PEA-m5C was not removed.
To facilitate the use of our model, we built a web server that is freely available at http://m5cpred-xs.zhulab.org.cn/. The server was implemented using flask, docker, and nginx. The users can easily carry out the prediction by the following procedures: First, users can type the query RNA sequences into the input box or upload a FASTA format file. (Note that the input sequence should be in FASTA format, and the length of each query sequence should be longer than 41 bp.) After that, one of the three species, H. sapiens, M. musculus, and A. thaliana, should be chosen. Users can provide their email address as a way to obtain the query results. Then, by clicking the “submit” button, the server generates a unique task ID and do the calculation until the final result is reached. During this process, you can query the task status by task ID. When the task was done, the results would be sent back to the users as an email attachment.
To further analyze the features selected by SHAP, the most important top 20 features for the three species are shown in Figure 4, in which the horizontal axis shows the distribution of the SHAP values and the vertical axis shows the features. If the SHAP values are positive, it will help to predict the m5C sites. Otherwise, it means the prediction tends to be of the negative class.
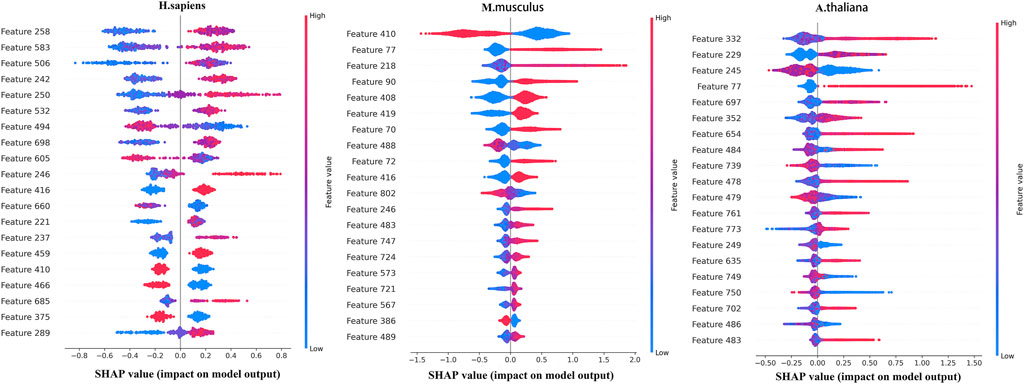
FIGURE 4. Top 20 features sorted by SHAP for the three species.
Figure 5 shows the distribution of the top 20 features in the seven types of features for three species. Overall, the top 20 most important features are not evenly distributed in the seven types of features for the three species. ENAC and SCPseDNC are the two types of features that appear in the top 20 features of all three species. ENAC represents the detailed distribution of nucleotides in each slide window. SCPseDNC represents the detailed distribution of dinucleotides and the distribution of its physical–chemical properties. Our results indicate that the distribution of nucleotides and their properties are related to the modification. Specifically, when identifying m5C sites of H. sapiens, features belonging to ENAC account for the largest proportion of the top 20 most important features, including a total of seven features. The three types of features, binary, ANF, and word2vec, are not included in the top 20 most important features, which indicates that these features contribute little to the prediction m5C sites of H. sapiens. For M. musculus, five features from NCP and SCPseDNC appeared in the top 20 features, and ANF and CKSNAP did not appear. For A. thaliana, five features of SCPseDNC and FastText appeared in top 20 features, and NCP was not included. These results indicate that the relevant features are related to the data sets, and feature selection is helpful for building high-performance models.
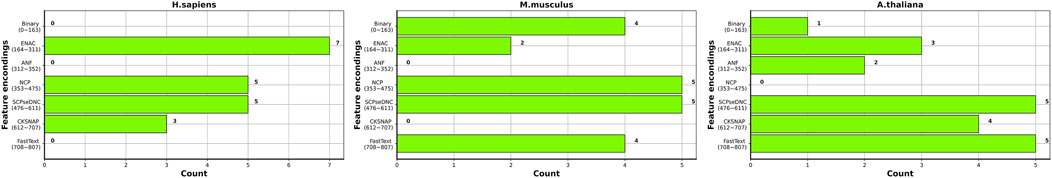
FIGURE 5. Distribution of top 20 features in the seven types of features for the three species.
Moreover, the principal component analysis was used to visualize the effectiveness of the selected features. Figure 6 shows that the boundaries between positive and negative samples for the three species are a little bit clearer in the features selected by SHAP than the original 808 dimensional features.
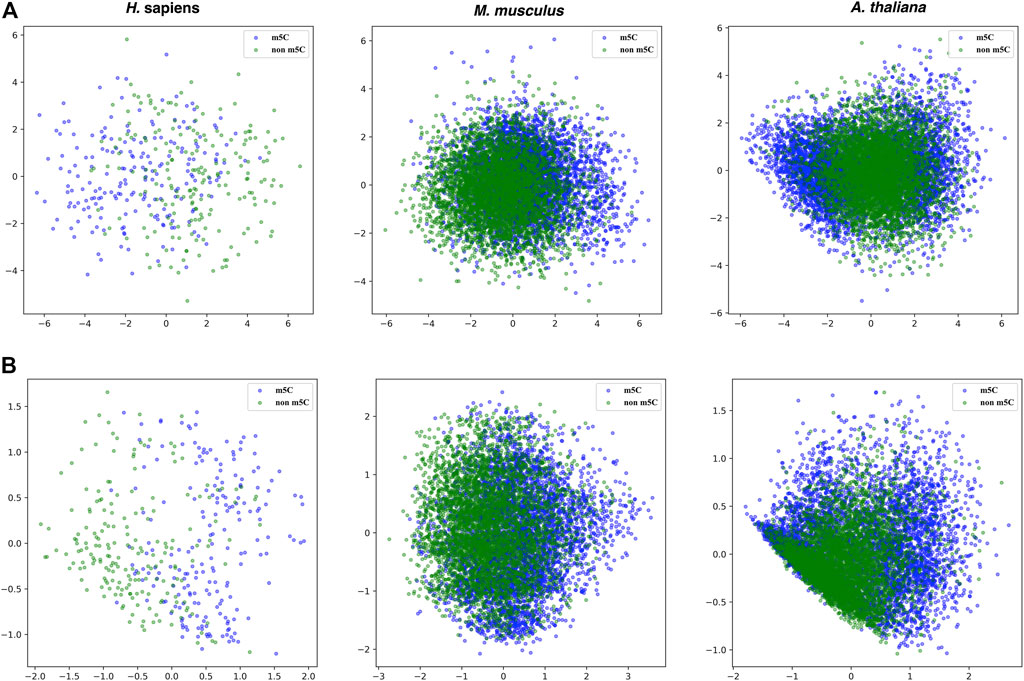
FIGURE 6. PCA plots for the original 808 dimensional features and features selected by SHAP for the three species. Upper panel: the original 808 dimensional features; Lower panel: the features selected by SHAP.
To further evaluate the generalization of our models, we conducted the cross-species validation to analyze the species-specificity and transferability of the models that were tested on the three independent test sets of the three species. Figure 7 shows that the models of all three species performs well (AUROC>0.7) on the independent test set of H. sapiens. However, the model of H. sapiens does not performs well on the independent test sets of the other two species. Figure 7 also shows that the model of M. musculus performs on the independent set of H. sapiens even better than that of M. musculus. In addition, the model of A. thaliana performs worse on the independent test set of M. musculus. We thought the small size of the benchmark data set of H. sapiens was one of the possible reasons for the results. The other reason is that both M. musculus and H. sapiens are mammals.
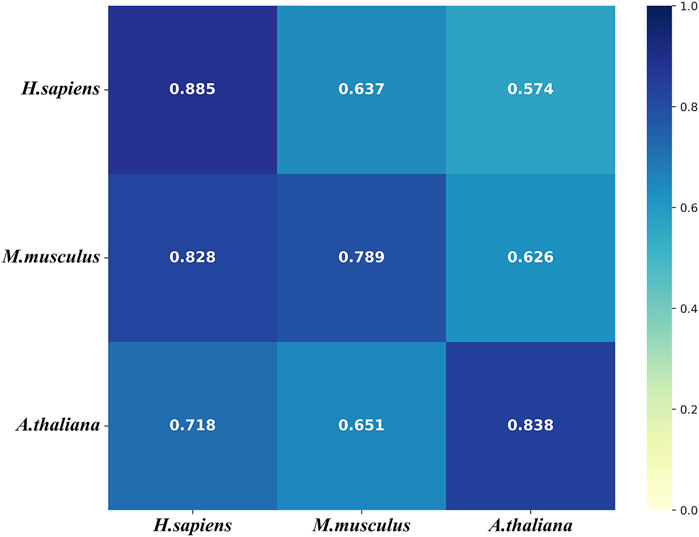
FIGURE 7. The heat map for the cross species predictive AUROCs. The models (y-axis) were tested on the three independent test sets (x-axis).
In this study, we proposed a new computational model, m5Cpred_XS, for predicting m5C sites. Three different feature-selection methods were used to select the optimal subset from 808 dimensional data of seven kinds of features. It turns out that the features selected by SHAP are more relevant compared with the features selected by the other two methods. The selected feature subsets were used to build our models. Our results show that the models based on XGBoost are superior to the models trained with RF and SVM. The m5Cpred_XS was further compared with other existing methods on the independent test sets, which demonstrates that our model outperforms the other methods according to AUROC values.
Publicly available data sets were analyzed in this study. This data can be available at: https://github.com/yinboliu-git/m5Cpred-XS.
XZ and YZ conceived the study; XZ and YL designed the experiments; YL and YS performed the experiments. YL, YS and HW analyzed the data. YL, XZ and YZ wrote the paper. All authors have read and agreed to the published version of the manuscript.
This work was supported in part by National Natural Science Foundation of China (grant numbers: 21403002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agris, P. F. (2008). Bringing Order to Translation: the Contributions of Transfer RNA Anticodon‐domain Modifications. EMBO Rep. 9, 629–635. doi:10.1038/embor.2008.104
Ahmad, A., and Shatabda, S. (2019). EPAI-NC: Enhanced Prediction of Adenosine to Inosine RNA Editing Sites Using Nucleotide Compositions. Anal. Biochem. 569 (569), 16–21. doi:10.1016/j.ab.2019.01.002
Bi, Y., Xiang, D., Ge, Z., Li, F., Jia, C., and Song, J. (2020). An Interpretable Prediction Model for Identifying N7-Methylguanosine Sites Based on XGBoost and SHAP. Mol. Ther. - Nucleic Acids 22 (22), 362–372. doi:10.1016/j.omtn.2020.08.022
Boopathi, V., Subramaniyam, S., Malik, A., Lee, G., Manavalan, B., and Yang, D. C. (2019). mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 20, 20. doi:10.3390/ijms20081964
Bugata, P., and Drotar, P.(2020). On Some Aspects of Minimum Redundancy Maximum Relevance Feature Selection. Sci. China Inform. Sci. Jan;63. doi:10.1007/s11432-019-2633-y
Catania, J., and Fairweather, D. S. (1991). DNA Methylation and Cellular Ageing. Mutat. Research/DNAging 256, 283–293. doi:10.1016/0921-8734(91)90019-8
Chai, D., Jia, C., Zheng, J., Zou, Q., and Li, F. J. M. T-N. A. (2021b). Staem5: A Novel Computational Approach for Accurate Prediction of m5C Site. Mol. Therapy-Nucleic Acids 26, 1027–1034. doi:10.1016/j.omtn.2021.10.012
Chai, D., Jia, C., Zheng, J., Zou, Q., and Li, F. (2021a). Staem5: A Novel Computational Approach for Accurate Prediction of m5C Site. Mol. Ther. - Nucleic Acids 26 (26), 1027–1034. doi:10.1016/j.omtn.2021.10.012
Chen, T. G. C. (2016). “XGBoost: A Scalable Tree Boosting System,” in the 22nd ACM SIGKDD International Conference, 785–794.
Chen, W., Tang, H., Ye, J., Lin, H., and Chou, K. C. (2016). iRNA-PseU: Identifying RNA Pseudouridine Sites. Mol. Ther. Nucleic Acids 5, e332. doi:10.1038/mtna.2016.37
Chen, W., Lei, T.-Y., Jin, D.-C., Lin, H., and Chou, K.-C. (2014). PseKNC: A Flexible Web Server for Generating Pseudo K-Tuple Nucleotide Composition. Anal. Biochem. 456 (456), 53–60. doi:10.1016/j.ab.2014.04.001
Chen, X., Xiong, Y., Liu, Y., Chen, Y., Bi, S., and Zhu, X. (2020). m5CPred-SVM: a Novel Method for Predicting m5C Sites of RNA. BMC Bioinformatics 21, 489. doi:10.1186/s12859-020-03828-4
Chen, Y. T., Xiong, J., Xu, W. H., and Zuo, J. W. (2019). A Novel Online Incremental and Decremental Learning Algorithm Based on Variable Support Vector Machine. Cluster Comput. May 22, S7435–S7445. doi:10.1007/s10586-018-1772-4
Chen, Z., Zhao, P., Li, F., Marquez-Lago, T. T., Leier, A., Revote, J., et al. (2020). iLearn: an Integrated Platform and Meta-Learner for Feature Engineering, Machine-Learning Analysis and Modeling of DNA, RNA and Protein Sequence Data. May 21, 1047–1057. doi:10.1093/bib/bbz041
Cui, X., Liang, Z., Shen, L., Zhang, Q., Bao, S., Geng, Y., et al. (2017). 5-Methylcytosine RNA Methylation in Arabidopsis Thaliana. Mol. Plant 10 (10), 1387–1399. doi:10.1016/j.molp.2017.09.013
Ding, Z., Nguyen, H., Bui, X.-N., Zhou, J., and Moayedi, H. (2020). Computational Intelligence Model for Estimating Intensity of Blast-Induced Ground Vibration in a Mine Based on Imperialist Competitive and Extreme Gradient Boosting Algorithms. Nat. Resour. Res. 29, 751–769. doi:10.1007/s11053-019-09548-8
Effrosynidis, D., and Arampatzis, A.(2021). An Evaluation of Feature Selection Methods for Environmental Data. Ecol. Inform.Mar;61.doi:10.1016/j.ecoinf.2021.101224
Fang, T., Zhang, Z., Sun, R., Zhu, L., He, J., Huang, B., et al. (2019). RNAm5CPred: Prediction of RNA 5-Methylcytosine Sites Based on Three Different Kinds of Nucleotide Composition. Mol. Ther. - Nucleic Acids 18 (18), 739–747. doi:10.1016/j.omtn.2019.10.008
Fasolino, M., Liu, S., Wang, Y., and Zhou, Z. (2017). Distinct Cellular and Molecular Environments Support Aging-Related DNA Methylation Changes in the Substantia Nigra. Epigenomics 9, 21–31. doi:10.2217/epi-2016-0084
Fawcett, T. (2006). An Introduction to ROC Analysis. Pattern Recognition Lett. 27, 861–874. doi:10.1016/j.patrec.2005.10.010
Feng, P., Ding, H., Chen, W., and Lin, H. (2016). Identifying RNA 5-methylcytosine Sites via Pseudo Nucleotide Compositions. Mol. Biosyst. 12, 3307–3311. doi:10.1039/c6mb00471g
Foster, P. G., Nunes, C. R., Greene, P., Moustakas, D., and Stroud, R. M. (2003). The First Structure of an RNA m5C Methyltransferase, Fmu, Provides Insight into Catalytic Mechanism and Specific Binding of RNA Substrate. Structure 11, 1609–1620. doi:10.1016/j.str.2003.10.014
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: Accelerated for Clustering the Next-Generation Sequencing Data. Bioinformatics 28 (28), 3150–3152. doi:10.1093/bioinformatics/bts565
Fushiki, T. (2011). Estimation of Prediction Error by Using K-fold Cross-Validation. Stat. Comput. 21, 137–146. doi:10.1007/s11222-009-9153-8
Han, S., Liang, Y., Ma, Q., Xu, Y., Zhang, Y., Du, W., et al. (2019). LncFinder: an Integrated Platform for Long Non-coding RNA Identification Utilizing Sequence Intrinsic Composition, Structural Information and Physicochemical Property. Nov 20, 2009–2027. doi:10.1093/bib/bby065
Hanchuan Peng, P., Fuhui Long, L., and Ding, C. (2005). Feature Selection Based on Mutual Information Criteria of max-dependency, max-relevance, and Min-Redundancy. IEEE Trans. Pattern Anal. Machine Intell. 27, 1226–1238. doi:10.1109/tpami.2005.159
He, Y., Shi, Q., Zhang, Y., Yuan, X., and Yu, Z. (2020). Transcriptome-Wide 5-Methylcytosine Functional Profiling of Long Non-coding RNA in Hepatocellular Carcinoma. Cmar Vol. 12, 6877–6885. doi:10.2147/cmar.s262450
Hussain, S., Sajini, A. A., Blanco, S., Dietmann, S., Lombard, P., Sugimoto, Y., et al. (2013). NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines its Processing into Regulatory Small RNAs. Cel Rep. 4, 255–261. doi:10.1016/j.celrep.2013.06.029
Jespersen, M. C., Peters, B., Nielsen, M., and Marcatili, P. (2017). BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. Jul 45, W24–W29. doi:10.1093/nar/gkx346
Ji, X., Tong, W., Liu, Z., and Shi, T. (2019). Five-Feature Model for Developing the Classifier for Synergistic vs. Antagonistic Drug Combinations Built by XGBoost. Front. Genet. 10, 600. doi:10.3389/fgene.2019.00600
Joulin, A., Grave, E., Bojanowski, P., and Mikolov, T. (2017). “Bag of Tricks for Efficient text Classification,” in 15th Conference of the European Chapter of the Association for Computational Linguistics, EACL 2017 - Proceedings of Conference (Valencia, Spain: Association for Computational Linguistics (ACL) 2, 427–431. doi:10.18653/v1/e17-2068
Ju, Z., and Wang, S.-Y. (2020). Prediction of Lysine Formylation Sites Using the Composition of K-Spaced Amino Acid Pairs via Chou's 5-steps Rule and General Pseudo Components. Genomics 112, 859–866. doi:10.1016/j.ygeno.2019.05.027
Keilwagen, J., Grosse, I., and Grau, J. (2014). Area under Precision-Recall Curves for Weighted and Unweighted Data. PLoS One 9, e92209. doi:10.1371/journal.pone.0092209
Khoddami, V., Yerra, A., Mosbruger, T. L., Fleming, A. M., Burrows, C. J., and Cairns, B. R. (2019). Transcriptome-wide Profiling of Multiple RNA Modifications Simultaneously at Single-Base Resolution. Proc. Natl. Acad. Sci. U S A. 116 (116), 6784–6789. doi:10.1073/pnas.1817334116
Khoddami, V., and Cairns, B. R. (2013). Identification of Direct Targets and Modified Bases of RNA Cytosine Methyltransferases. Nat. Biotechnol. 31, 458–464. doi:10.1038/nbt.2566
Li, F., Zhang, Y., Purcell, A. W., Webb, G. I., Chou, K. C., Lithgow, T., et al. (2019). Positive-unlabelled Learning of Glycosylation Sites in the Human Proteome. BMC Bioinformatics 20, 112. doi:10.1186/s12859-019-2700-1
Li, J., Huang, Y., Yang, X., Zhou, Y., and Zhou, Y. (2018). RNAm5Cfinder: A Web-Server for Predicting RNA 5-methylcytosine (m5C) Sites Based on Random Forest. Sci. Rep. 8, 17299. doi:10.1038/s41598-018-35502-4
Li, Y. X., Chai, Y., Zhou, H., and Yin, H. P.(2021). A Novel Dimension Reduction and Dictionary Learning Framework for High-Dimensional Data Classification. Pattern Recogn. Apr;112.doi:10.1016/j.patcog.2020.107793
Li, Y., Yang, Y., Li, G., Xu, M., and Huang, W. (2017). A Fault Diagnosis Scheme for Planetary Gearboxes Using Modified Multi-Scale Symbolic Dynamic Entropy and mRMR Feature Selection. Mech. Syst. Signal Process. 91, 295–312. doi:10.1016/j.ymssp.2016.12.040
Liu, B., Li, C.-C., and Yan, K. (2020). DeepSVM-fold: Protein Fold Recognition by Combining Support Vector Machines and Pairwise Sequence Similarity Scores Generated by Deep Learning Networks. Sep 21, 1733–1741. doi:10.1093/bib/bbz098
Lv, H., Zhang, Z.-M., Li, S.-H., Tan, J.-X., Chen, W., and Lin, H. (2020). Evaluation of Different Computational Methods on 5-methylcytosine Sites Identification. May 21, 982–995. doi:10.1093/bib/bbz048
Matzke, M., Aufsatz, W., Kanno, T., Daxinger, L., Papp, I., Mette, M. F., et al. (2004). Genetic Analysis of RNA-Mediated Transcriptional Gene Silencing. Biochim. Biophys. Acta 1677 (1677), 129–141. doi:10.1016/j.bbaexp.2003.10.015
Pathy, A., Meher, S., and Balasubramanian, P.(2020). Predicting Algal Biochar Yield Using eXtreme Gradient Boosting (XGB) Algorithm of Machine Learning Methods. Algal Res. Sep;50:102006. doi:10.1016/j.algal.2020.102006
Polat, K., and Güneş, S. (2009). A New Feature Selection Method on Classification of Medical Datasets: Kernel F-Score Feature Selection. Expert Syst. Appl. 36, 10367–10373. doi:10.1016/j.eswa.2009.01.041
Qiang, X., Chen, H., Ye, X., Su, R., and Wei, L. (2018). M6AMRFS: Robust Prediction of N6-Methyladenosine Sites with Sequence-Based Features in Multiple Species. Front. Genet. 9, 495. doi:10.3389/fgene.2018.00495
Qiu, W. R., Jiang, S. Y., Xu, Z. C., Xiao, X., and Chou, K. C. (2017). iRNAm5C-PseDNC: Identifying RNA 5-methylcytosine Sites by Incorporating Physical-Chemical Properties into Pseudo Dinucleotide Composition. Oncotarget 8 (8), 41178–41188. doi:10.18632/oncotarget.17104
Sabooh, M. F., Iqbal, N., Khan, M., Khan, M., and Maqbool, H. F. (2018). Identifying 5-methylcytosine Sites in RNA Sequence Using Composite Encoding Feature into Chou's PseKNC. J. Theor. Biol. 452 (452), 1–9. doi:10.1016/j.jtbi.2018.04.037
Saito, T., and Rehmsmeier, M. (2017). Precrec: Fast and Accurate Precision-Recall and ROC Curve Calculations in R. Bioinformatics 33 (33), 145–147. doi:10.1093/bioinformatics/btw570
Samat, A., Li, E. Z., Wang, W., Liu, S. C., Lin, C., and Abuduwaili, J.(2020). Meta-XGBoost for Hyperspectral Image Classification Using Extended MSER-Guided Morphological Profiles. Remote Sens-Basel. Jun;12.
Schaefer, M., Pollex, T., Hanna, K., Tuorto, F., Meusburger, M., Helm, M., et al. (2010). RNA Methylation by Dnmt2 Protects Transfer RNAs against Stress-Induced Cleavage. Genes Dev. 24 (24), 1590–1595. doi:10.1101/gad.586710
Song, J., Zhai, J. J., Bian, E. Z., Song, Y. J., Yu, J. T., and Ma, C. (2018). Transcriptome-Wide Annotation of M(5)C RNA Modifications Using Machine Learning. Front. Plant Sci. 9, 519. Nov 30;9. doi:10.3389/fpls.2018.00519
Swann, S. L., Brown, S. P., Muchmore, S. W., Patel, H., Merta, P., Locklear, J., et al. (2011). A Unified, Probabilistic Framework for Structure- and Ligand-Based Virtual Screening. J. Med. Chem. 54 (54), 1223–1232. doi:10.1021/jm1013677
Tang, W., Luo, X. Y., and Sanmuels, V. (2001). Gene Silencing: Double-Stranded RNA Mediated mRNA Degradation and Gene Inactivation. Cell Res 11, 181–186. doi:10.1038/sj.cr.7290084
Visentini, I., Snidaro, L., and Foresti, G. L. (2016). Diversity-aware Classifier Ensemble Selection via F-Score. Inf. Fusion 28, 24–43. doi:10.1016/j.inffus.2015.07.003
Wang, J., and Gribskov, M. (2019). IRESpy: an XGBoost Model for Prediction of Internal Ribosome Entry Sites. BMC Bioinformatics 20, 409. doi:10.1186/s12859-019-2999-7
Wang, S., Kong, W., Aorigele, Deng. J., Deng, J., Gao, S., and Zeng, W. (2018). Hybrid Feature Selection Algorithm mRMR-ICA for Cancer Classification from Microarray Gene Expression Data. Cchts 21, 420–430. doi:10.2174/1386207321666180601074349
Xu, C., Tian, J., and Mo, B. (2013). siRNA-mediated DNA Methylation and H3K9 Dimethylation in Plants. Protein Cell 4, 656–663. doi:10.1007/s13238-013-3052-7
Xuan, J. J., Sun, W. J., Lin, P. H., Zhou, K. R., Liu, S., Zheng, L. L., et al. (2018). RMBase v2.0: Deciphering the Map of RNA Modifications from Epitranscriptome Sequencing Data. Nucleic Acids Res. 46, D327–D334. doi:10.1093/nar/gkx934
Xue, C., Zhao, Y., and Li, L. (2020). Advances in RNA Cytosine-5 Methylation: Detection, Regulatory Mechanisms, Biological Functions and Links to Cancer. Biomark Res. 8, 43. doi:10.1186/s40364-020-00225-0
Xue, M. M., Shi, Q. M., Zheng, L., Li, Q. B., Yang, L. Y., and Zhang, Y. Y. (2020). Gene Signatures of m5C Regulators May Predict Prognoses of Patients with Head and Neck Squamous Cell Carcinoma. Am. J. Transl Res. 12, 6841–+.:
Yang, X., Yang, Y., Sun, B.-F., Chen, Y.-S., Xu, J.-W., Lai, W.-Y., et al. (2017). 5-methylcytosine Promotes mRNA export - NSUN2 as the Methyltransferase and ALYREF as an m5C Reader. Cel Res 27, 606–625. doi:10.1038/cr.2017.55
Zhang, M., Li, F., Marquez-Lago, T. T., Leier, A., Fan, C., Kwoh, C. K., et al. (2019). MULTiPly: a Novel Multi-Layer Predictor for Discovering General and Specific Types of Promoters. Bioinformatics 35 (35), 2957–2965. doi:10.1093/bioinformatics/btz016
Zhang, M., Xu, Y., Li, L., Liu, Z., Yang, X., and Yu, D.-J. (2018). Accurate RNA 5-methylcytosine Site Prediction Based on Heuristic Physical-Chemical Properties Reduction and Classifier Ensemble. Anal. Biochem. 550 (550), 41–48. doi:10.1016/j.ab.2018.03.027
Zhang, Q., Zheng, Q., Yu, X., He, Y., and Guo, W. (2020). Overview of Distinct 5-methylcytosine Profiles of Messenger RNA in Human Hepatocellular Carcinoma and Paired Adjacent Non-tumor Tissues. J. Transl Med. 18, 245. doi:10.1186/s12967-020-02417-6
Zhao, X., Zhang, Y., Ning, Q., Zhang, H., Ji, J., and Yin, M. (2019). Identifying N6-Methyladenosine Sites Using Extreme Gradient Boosting System Optimized by Particle Swarm Optimizer. J. Theor. Biol. 467 (467), 39–47. doi:10.1016/j.jtbi.2019.01.035
Keywords: 5-cytosine-methylation, XGBoost, machine learning, shap, feature selection
Citation: Liu Y, Shen Y, Wang H, Zhang Y and Zhu X (2022) m5Cpred-XS: A New Method for Predicting RNA m5C Sites Based on XGBoost and SHAP. Front. Genet. 13:853258. doi: 10.3389/fgene.2022.853258
Received: 12 January 2022; Accepted: 16 February 2022;
Published: 30 March 2022.
Edited by:
Fengfeng Zhou, Jilin University, ChinaReviewed by:
Leyi Wei, Shandong University, ChinaCopyright © 2022 Liu, Shen, Wang, Zhang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaolei Zhu, eGx6aHVfbWRsQGhvdG1haWwuY29t; Yong Zhang, eW9uZ3poYW5nQGFoYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.