 Hong Yang
Hong Yang- 1Department of Neurology, The First Rehabilitation Hospital of Shanghai, Tongji University School of Medicine, Shanghai, China
- 2Department of Neurology, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 3Department of Neurology, Tongji Hospital, Tongji University School of Medicine, Shanghai, China
Background: N6-Methyladenosine (m6A) methylation is the most prevalent internal posttranscriptional modification on mammalian mRNA. But its role in neuromyelitis optica spectrum disorders (NMOSD) is not known.
Aims: To explore the mechanism of m6A in NMOSD patients.
Methods: This study assessed the m6A methylation levels in blood from two groups: NMOSD patients and healthy controls. Methylated RNA immunoprecipitation Sequencing (MeRIP-seq) and RNA-seq were performed to assess differences in m6A methylation between NMOSD patients and healthy controls. Ultra-high performance liquid chromatography coupled with triple quadruple mass spectrometry (UPLC-QQQ-MS) method was performed to check m6A level. Differential m6A methylation genes were validated by MeRIP-qPCR.
Results: Compared with that in the control group, the total m6A level was decreased in the NMOSD group. Genes with upregulated methylation were primarily enriched in processes associated with RNA splicing, mRNA processing, and innate immune response, while genes with downregulated methylation were enriched in processes associated with the regulation of transcription, DNA-templating, and the positive regulation of I-kappa B kinase/NF-kappa B signalling.
Conclusion: These findings demonstrate that differential m6A methylation may act on functional genes to regulate immune homeostasis in NMOSD.
Introduction
N6-Methyladenosine (m6A) is one of the most abundant internal modifications of eukaryotic messenger RNA (mRNA) and plays an important role in gene expression regulatory processes, including the maintenance of stability, splicing and translation (Chen et al., 2019a). Approximately 25% of mRNAs are estimated to contain at least one m6A nucleotide site, and most of them occur at the sixth position of adenosine. m6A was shown to be specifically catalysed at the consensus gene sequence DRACH (D = A/G/U, R = A/G, H = A/C/U). In addition to m6A, other types of posttranscriptional modifications exist, including 5-methylcytosine (m5C) and pseudouridine (ψ). To detect regions containing m6A peaks, researchers have developed an immunoprecipitation-based method combined with high-throughput sequencing, termed methylated RNA immunoprecipitation sequencing(MeRIP-seq) (Dominissini et al., 2012). To determine m6A level, researchers have developed the UPLC-QQQ-MS method, which has the advantages of short analysis time, good resolution and high sensitivity (Su et al., 2014).
m6A modification is dynamic and reversible and regulated by m6A methyltransferases (writers), demethylases (erasers) and reading proteins (readers). Writers include Methyltransferase Like 3 (METTL3), methyltransferase-like 14 (METTL4) and Wilms tumour 1-associated protein (WTAP). METTL3 catalyses the production of m6A; METTL4 forms a complex with METTL3, participates in interactions with target mRNA and recruits RNA, and WTAP stabilizes the complex (Liu et al., 2014; Ping et al., 2014; Lin et al., 2016). Erasers, including fat and obesity-associated protein (FTO) and alk B homologue 5 (ALKBH5), catalyse the demethylation of bases that have been modified by m6A (Jia et al., 2011; Zheng et al., 2013). Readers, including YTH N6 methyladenosine RNA-binding protein 1–3 (YTHDF1-3) and insulin-like growth factor 2 mRNA-binding protein 1–3 (IGF2BP1-3), are also necessary for this process (Wang et al., 2014; Wang et al., 2015; Hanniford et al., 2020). These proteins recognize sites of mRNA methylation and affect RNA metabolism directly or indirectly. Overall, m6A-modifying enzymes play important roles in tumorigenesis and in the development of the human central nervous system (Lin et al., 2017; Zhang et al., 2018; Heck et al., 2020; Xue et al., 2021). In recent years, studies have revealed that m6A participates in regulating the immune microenvironment (Chen et al., 2019b; Tang et al., 2020; Xu et al., 2020), but its involvement in NMOSD is unknown.
NMOSD is a kind of recurrent antibody-mediated neuroimmune disease, in which the optic nerve, spinal cord, and area postrema of the medulla are damaged, often causing severe symptoms (Wingerchuk et al., 2015). The specific diagnostic and pathogenic biomarker for NMOSD is aquaporin 4-immunoglobulin IgG (AQP4-IgG), which binds to the foot processes of astrocytes, activating complement and thereby recruiting neutrophils and eosinophils; this leads to astrocyte death, followed by axonal degeneration and oligodendrocyte injury (Lennon et al., 2004). The prevalence and incidence of NMOSD are 0.52–4.4 and 0.05–0.40 per 100,000, respectively (Pandit et al., 2015). Approximately 60% of NMOSD patients relapse within 1 year and approximately 90% within 5 years. Furthermore, approximately 50% of NMOSD patients require a wheelchair within 5 years of their diagnosis due to limb paralysis, and 62% become blind, affecting their quality of life and imposing a heavy burden on patients, families and society (Pittock and Lucchinetti, 2016). Currently, methylprednisolone and plasma exchange used in the acute stage as well as rituximab, mycophenolate mofetil and azathioprine used in the remission stage are only partially effective and do not substantially reduce the high disability associated with the disease or its recurrence. Therefore, it is necessary to further understand the pathogenesis of NMOSDs and explore more effective drugs.
Regardless, the m6A status has not been assessed in NMOSD. To investigate differences in m6A modification patterns between healthy controls (HCs) and NMOSD patients, we performed UPLC-QQQ-MS to assess the m6A level and MeRIP-seq to identify the first known transcriptome-wide m6A sites in NMOSD patients. In general, the total m6A level was decreased in patients compared to HCs. We also detected 3,371 differentially methylated peaks within mRNAs and 95 within long noncoding RNAs (lncRNCs, p < 0.05). Intriguingly, mRNAs harbouring differentially methylated peaks were shown to be involved in many important biological pathways associated with immune homeostasis.
Materials and Methods
Blood Collection From NMOSD Patients and HCs
(1.1) Inclusion criteria: 1) aged 18–75 years old; 2) meeting the diagnostic criteria for NMOSD (Wingerchuk et al., 2015); 3) within 30 days of onset in the acute phase and not receiving relevant drug treatment; and 4) provision of signed informed consent.
(1.2) Exclusion criteria: 1) severe mental symptoms precluding cooperation with treatment; 2) no informed consent signed; 3) receiving immunosuppressive therapy within 30 days of new clinical symptoms; 4) other autoimmune diseases or tumours; 5) new clinical symptoms and not receiving relevant treatment but with clinical symptoms for more than 30 days before seeing a doctor.
(1.3) On the second day after admission, 10 ml of blood was collected into a BD Paxgene blood RNA tube; the samples were stored at −4°C for 24 h and then at −80°C for preparation of RNA.
(1.4) In total, six healthy individuals with no acute or chronic illness from the Health Care Centre of the First Rehabilitation Hospital of Shanghai were enrolled in the study as a control group.
RNA Preparation and Quality Control
Total RNA was extracted from whole blood stored in Paxgene blood RNA tubes by using a Paxgene Blood RNA Extraction Kit (PreAnalytiX, Qiagen). A Qubit fluorometer was used to quantify RNA at 260/280 nm, and an Agilent 2,100 Bioanalyzer was employed to determine the RNA integrity number (RIN) and 28S/18S values for quality control (RIN ≥ 6.0 and 28S/18S ≥ 0.7) and confirmed by electrophoresis on a denaturing agarose gel.
UPLC-QQQ-MS Method
Analysis of m6A level was performed on the UPLC-QQQ-MS system, consisting of a Waters Acquity UPLC (Waters Corporation, Massachusetts, United States) and an AB SCIEX 5500 QQQ-MS instrument (AB Sciex LLC, Framingham, United States). As previously described (Su et al., 2014), we prepared a mixture containing all of the cofactors and enzymes needed for hydrolysis of the RNA into nucleosides. Then, 5 µl of the mixture was added to each sample of RNA, and the final volume was brought to 25 µl with RNase-free water. The samples were incubated at 37°C for 4 h, after which 75 µL of methanol was added to the system. Different dilutions were used for detection. UPLC separation was performed on an Acquity UPLC BEH amide column (1.7 µm, 2.1 mm × 100 mm, Waters Corporation, Massachusetts, United States) with a flow rate of 0.35 ml/min at 40°C. Formic acid in water (0.1%, v/v, solvent A, Aladdin, Shanghai) and methyl cyanides (v/v, solvent B, Sigma-Aldrich, Shanghai) were employed as the mobile phase. Mass spectrometry detection was performed in positive electrospray ionization mode, and multiple reaction monitoring (MRM) was used to monitor target nucleosides by using the following mass transitions: (precursor ions → product ions) of m6A (282.1 → 150.1), A (268 → 136), U (245 → 113), G (284 → 152), C (244 → 112), and I (269 →137). Quantification was performed using a standard curve originating from A and m6A. The calculated m6A/A ratio was used to indicate the m6A level. To achieve maximal detection sensitivity, we optimized the MRM parameters of all nucleosides.
RNA MeRIP-Seq Library Construction and Sequencing
MeRIP-seq was performed by Biotechnology Corporation (Shanghai, China). As previously described (Dominissini et al., 2012), GenSeq® m6A MeRIP KIT (GenSeq, China) was performed, and the protocol involved the following four steps. Step 1: RNA fragmentation. The RNA was randomly divided into approximately 200-nt segments by using fragmentation buffer and stop buffer. The sizes and concentrations of the RNA fragments were detected by an Agilent Bioanalyzer and an Agilent RNA 6000 Pico kit (Bioptic Inc., Taiwan, China). Three micrograms of fragmented RNA was used as the input group and stored at −80°C. The remaining fragmented RNA was used for subsequent immunoprecipitation experiments. Step 2: Preparation of immunoprecipitated magnetic beads. The porcine gastric mucine (PGM) magnetic beads were gently blown with a pipette to promote full suspension, after which they were washed with 1 × IP buffer, and an m6A antibody was added. Step 3: Immunoprecipitation. The MeRIP reaction solution, which included 50 µl of fragmented RNA, 150 µl of nuclease-free water, and 50 µl of 5 × IP buffer, was prepared. Next, 250 μL of the reaction solution was added to the magnetic beads prepared in step 2. The magnetic beads were washed with 1 × IP buffer, LB buffer, and HS buffer separately and repeatedly. Step 4: RNA purification. The magnetic beads were resuspended in RLT buffer, washed with 75% ethanol, and then completely resuspended in 11.25 µl of nuclease-free water. The eluted RNA was transferred to a new centrifuge tube and then immediately used for subsequent experiments or stored at −80°C. Both the input samples without immunoprecipitation and the m6A IP samples were used for RNA-seq library generation. The library quality was evaluated with a Bioptic Qsep100 Analyser (Bioptic Inc., Taiwan, China). Library sequencing was performed on an Illumina NovaSeq 6,000 (LC-Bio Technology) instrument with 150 bp paired-end reads.
FastQC was applied to analyse the quality of the sequencing data by assessing the sequencing quality distribution, base content distribution, and proportion of repeated sequencing fragments. HISAT2 software was used to compare the filtered clean reads with the human reference genome (GRCh38/hg38) and thereby obtain unique mapped reads for further analysis. Peak calling was carried out with exomePeak software. After obtaining the peak, the gene structure locations of the peaks and the overall distribution characteristics were determined, and peak annotation analysis was performed with HOMER software (http://homer.ucsd.edu/homer/ngs/peakMotifs.html).GO analysis is used to assess the function of genes from various aspects and is divided into three main categories: biological process (BP), molecular function (MF) and cellular component (CC). The mRNAs of genes modified by m6A were evaluated based on GO annotations in the BP, MF, and CC categories in the database, and Fisher’s test was used to determine the significance level (p-value) of each category to screen for significant GO terms. KEGG analysis was based on the gene annotations, and selected genes with mRNA m6A modification were annotated according to their associated KEGG pathways. The significance level (p-value) was determined by Fisher’s test to screen significant pathway terms related to m6A gene enrichment.
Methylated RNA ImmunoprecipitationeRIP-qPCR
MeRIP-qPCR was performed by ChoudSeq Biotech Inc. (Shanghai, China). Its analysis was used according to a previously reported method (Dominissini et al., 2012), as shown in the RNA MeRIP-seq library construction and sequencing section. m6A enrichment was determined by qPCR analysis. Reverse transcription was performed using a SuperScriptTM III Reverse Transcriptase kit (Invitrogen) for mRNA. For relative qRT-PCR, qPCR SYBR Green master mix (CloudSeq)was used to generate mRNA cDNA according to the manufacturer’s instructions. The reactions were performed on a QuantStudio five Real-Time PCR System (Thermo Fisher). The expression levels of mRNAs normalized to those of the endogenous control were calculated using the 2−ΔΔCT method and are presented as the fold change relative to the control group. % (IP/Input) was used to calculate the differences.The sequences of the primers utilized in this study are listed in Additional file 1: Supplementary Table S1.
Data Analysis
Data are presented as the mean ± SD. Significance differences between groups were determined by Student’s t-test using GraphPad Prism six sofeware. Significance was established at p < 0.05.
Results
Demographic and Clinical Features of the Patients With NMOSD
We enrolled three NMOSD patients and three HCs for MeRIP-seq, with mean ages of 47.33 ± 2.082 and 47.33 ± 3.055, respectively (p > 0.05). The female: male ratio was 3:0 in both groups (p > 0.05). The current disease duration at the time of recruitment was 5 ± 2 days (Table 1). We enrolled another three NMOSD patients and three HCs for MeRIP-qPCR validation, with mean ages of 59.00 ± 6.083 and 59.67 ± 6.807, respectively (p > 0.05). The female:male ratio was 3:0 in both groups (p > 0.05). The current disease duration at the time of recruitment was 5 ± 2 days (Table 1).

TABLE 1. Characteristics of subjects and m6A levels in NMOSD patients and healthy controls.
Levels of m6A in NMOSD Patients and HCs
The levels of m6A differed significantly between the NMOSD patients and HCs (0.09531 ± 0.009259 (%), 0.1399 ± 0.02533 (%); p < 0.05) (Table 1; Figure 1A). Additionally, R2 was greater than 0.99, indicating that the linear relationship of the standard curve is good (Figure 1B).
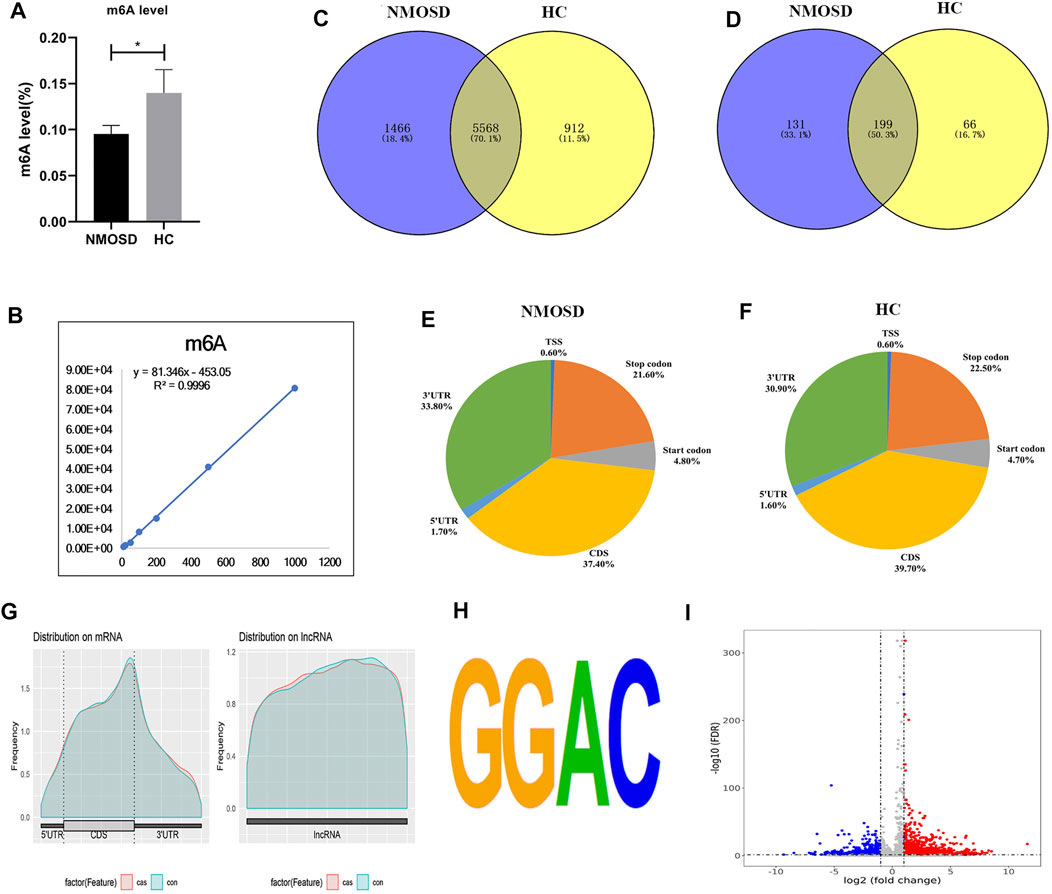
FIGURE 1. Overview of N6-methyladenosine methylation in NMOSD patients and HCs. (A) m6A levels in NMOSD and HCs,*p < 0.05,Student’s t-test. (B) R2 was greater than 0.99, indicating that the linear relationship of the standard curve is good. (C) Venn diagram showing the overlap of m6A peaks with mRNA in NMOSD patients and HCs. (D) We used a Venn diagram to depict the overlap of m6A peaks with lncRNAs in NMOSD and HCs. (E) We used pie charts to show the accumulation of m6A peaks along transcripts in NMOSD. (F) We used pie charts to display the accumulation of m6A peaks along transcripts in HCs. (G) Metagene plot of peak distribution in RNA structures (NMOSD and HCs). (H) The consensus motif for m6A modification is “GGAC”.(I) Volcano map of differential m6A modification. Diff.log2 FC < 0 indicates hypomethylation, indicating that the site was demethylated under treatment conditions; Diff.log2 FC > 0 indicates hypermethylation, indicating hypermethylation of this site under treatment conditions. Diff.lg.fdr: log10 (FDR) of differential methylation analysis is the log10 conversion value of the FDR value obtained from the differential analysis. Red indicates hypermethylation, and blue indicates demethylation. Red and blue represent genes with a more than 2-fold difference in m6A modification.
Sequencing Statistics and Quality Control
Some joint and low-quality sequences were abtained in the original data, and adaptor and low-quality data were removed to abtain clean reads. In the MeRIP-seq library, 6,308,338,650, 7,779,927,600 and 6,841,024,050 total bases and 6,125,183,549, 7,628,452,162 and 6,635,546,539 valid total bases were abtained, and the effective bases accounted for 97.10, 98.10 and 97.00% of the totals, respectively. In the HC blood samples, 6,495,933,300, 6,730,312,650 and 6,736,672,200 total bases and 6,292,021,712, 6,453,732,389 and 6,604,071,380 valid total bases were obtained, with the effective bases accounting for 96.90, 95.90 and 98.00% of the totals, respectively. In the RNA-seq library, the effective bases accounted for more than 90% of the total bases in both the NMOSD and HC groups. The results are shown in Table 2.
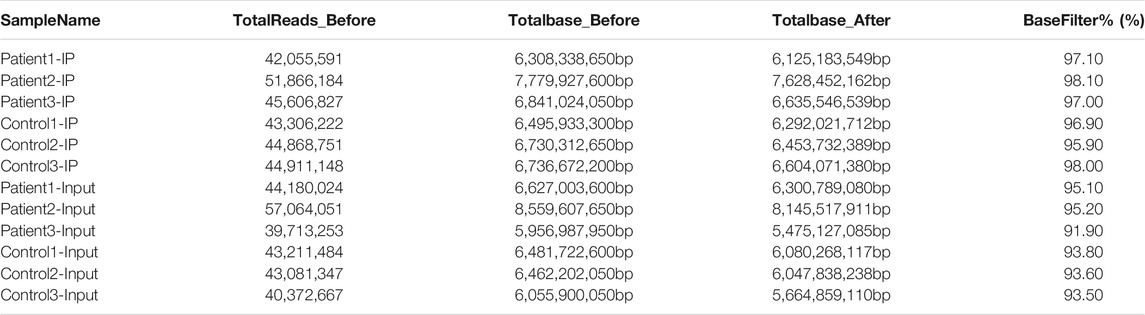
TABLE 2. Summary of reads quality control analysis.
Mapping Reads to the Reference Genome
We used HISAT2 to map reads to the GRCh38/hg38 genome with default parameters. Detailed statistical analyses were performed by comparing reads with reference sequences. In the MeRIP-seq library, the mapping ratios were 94.37, 87.19 and 91.55% in NMOSD patients, and 92.64, 93.87, 89.08% in HCs. In the RNA-seq library, the mapping ratios were 91.07, 89.26 and 86.16% in NMOSD patients and 89.31, 90.42, 88.04% in HCs. The ratios of unique mapped reads are shown in Table 3.
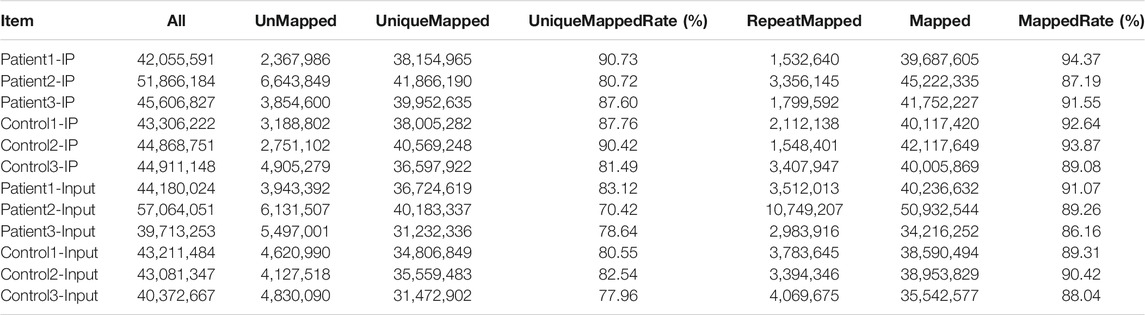
TABLE 3. Summary of reads mapped to the GRCh38/hg38 reference genome.
General Features of m6A Methylation in NMOSD Patients and HCs
MeRIP-seq analysis of RNA derived from whole blood revealed 14,444 nonoverlapping m6A peaks within 7,034 coding transcripts (mRNAs) and 440 nonoverlapping m6A peaks within 330 lncRNAs in the NMOSD group; there were three biological replicates. In the HC group, there were 12,806 nonoverlapping m6A peaks within 6,480 coding transcripts (mRNAs) and 369 nonoverlapping m6A peaks within 265 lncRNAs, with three biological replicates. Of these, 5,568 coding transcripts (70.1%) within mRNAs (Figure 1C) and 199 long noncoding transcripts (50.3%) within lncRNAs (Figure 1D) overlapped between the NMOSD patients and HCs. Of these, there were 3,600 differential peaks (p < 0.05), including 3,371 differentially methylated peaks within mRNAs and 95 within lncRNAs (p < 0.05) (Figure 1I).
To analyse the distribution profiles of m6A peaks within mRNAs, the peaks were categorized into five transcript segments: the 5’UTR; start codon (400 nucleotides centred on the start codon); coding sequence (CDS); stop codon (400 nucleotides centred on the stop codon); and 3’UTR. In our study, m6A was most often detected in the CDS, with some sites being observed near the 3’UTR and stop codon (Figures 1E–G).
Motif analysis of peaks within mRNAs with the highest scores (p-value = 1e-141) obtained from three biological replicates revealed a consensus sequence (GGAC) as well as other motifs in the NMOSD and HC samples (Figure 1H), indicating the reliability of the data.
Distribution of Differentially Methylated m6A Sites
In total, we identified 3,371 differentially methylated m6A sites (DMMSs) within mRNAs, of which 68.5% (2,310/3,371) showed significantly increased methylation and 31.5% (1,061/3,371) significantly reduced methylation (NMOSD vs. HCs; Table 4). We also identified 95 DMMSs for lncRNAs, of which 68.5% (71/95) exhibited significant increased methylation levels and 31.5% (24/95) exhibited significantly decreased methylation levels (Table 4). Tables 5, 6 provide the top ten m6A sites displaying increased and reduced methylation levels with the highest fold change values (Tables 5, 6).

TABLE 4. General numbers of differentially methylated peaks and genes.
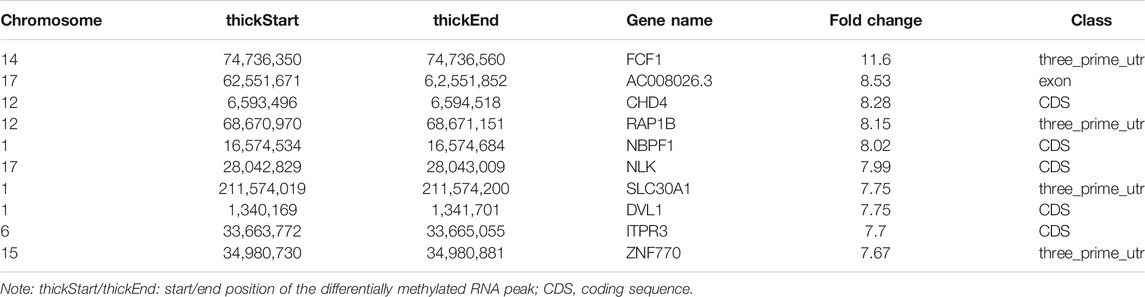
TABLE 5. Top ten increased methylation peaks.
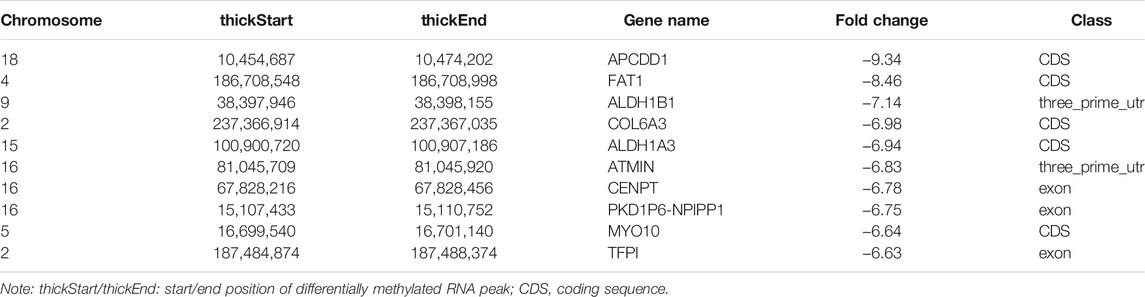
TABLE 6. Top ten reduced methylation peaks.
Differentially Methylated RNAs Are Involved in Important Biological Pathways
To elucidate the functions of m6A in NMOSD, we selected protein-coding genes containing DMMSs for GO and KEGG pathway analyses. In the BP category, genes with increased m6A site methylation were significantly (p < 0.05) enriched in RNA splicing, mRNA processing, and gene expression (Figure 2A); genes with decreased m6A methylation were highly enriched in the regulation of transcription, transcription, DNA templating, and viral processes (Figure 2B). In the MF category, poly(A) RNA binding and protein binding were enriched among mRNAs with increased m6A methylation (Figure 2A), and those with lower levels of methylation were enriched in protein binding and DNA binding (Figure 2B). With respect to the CC category, the nucleus and nucleolus showed enrichment for both increased (Figure 2A) and decreased m6A methylation (Figure 2B). We also used a GO bubble chart to illustrate the top twenty enriched GO terms, mainly involving transcription, DNA templating, regulation of transcription, and gene expression, among others (Figure 2C).
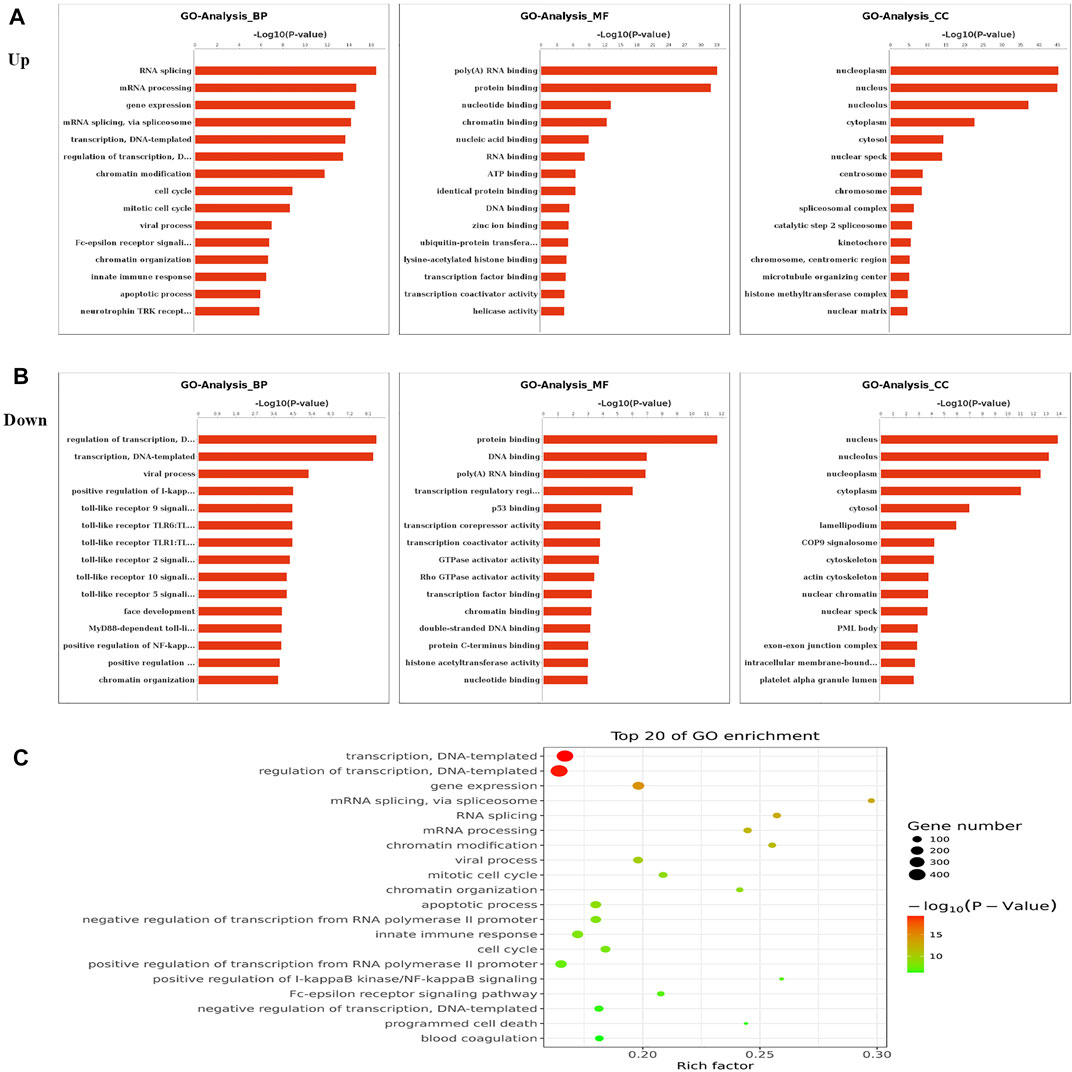
FIGURE 2. Gene ontology and Enrichment analysis of differential m6A peaks. (A) The top ten Gene Ontology terms related to mRNAs with increased methylation; terms in the biological process (BP), molecular function (MF), and cellular component (CC) categories are shown; (−log10p) for significance. The log-transformed results were used for visualization. (B) The top ten Gene Ontology terms related to mRNAs with decreased methylation, terms in the BP, MF and CC categories are shown; (−log10p) for significance. The log-transformed results were used for visualization. (C) The GO bubble shows the top 20 Gene Ontology terms corresponding to the methylated genes.
We performed KEGG pathway analysis of DMMSs and observed that genes with increased m6A methylation were significantly (p < 0.05) enriched in the mRNA surveillance pathway, neurotrophin signalling pathway, and Epstein-Barr virus infection, among others (Figure 3A). Genes with decreased m6A methylation were significantly (p < 0.05) enriched in the TNF signalling pathway, oestrogen signalling pathway, and focal adhesion (Figure 3B). We also used a KEGG bubble chart to show the top twenty enriched pathways, including Epstein-Barr virus infection and the mRNA surveillance pathway (Figure 3C). These results suggest that m6A may have many key roles in NMOSD.
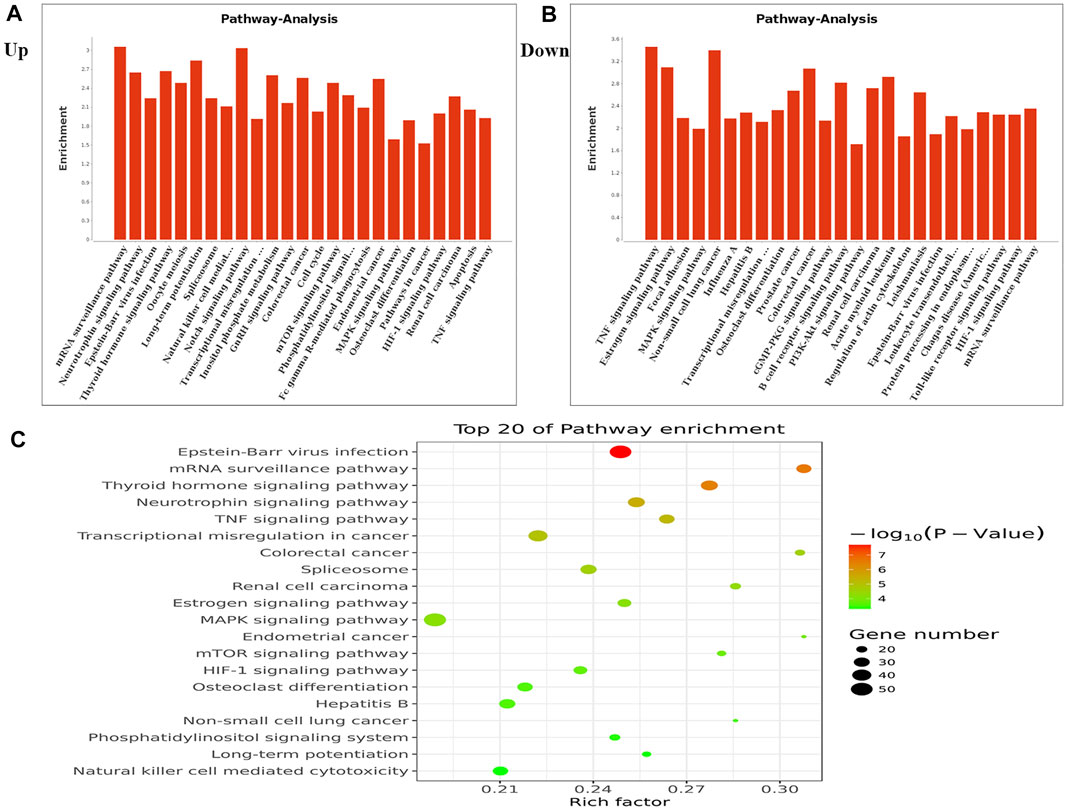
FIGURE 3. KEGG pathway and Enrichment analysis of differential m6A peaks. (A) The top ten enrichment pathway terms related to mRNAs with increased methylation are shown; (−log10p) for significance. The log-transformed results were used for visualization. (B) The top ten enrichment pathway terms related to mRNAs with decreased methylation; (−log10p) for significance. The log-transformed results were used for visualization. (C) The KEGG bubble shows the top 20 enrichment pathway terms related to methylated mRNAs.
Association Analysis of Differential RNA Modification and Differential Gene Expression
We drew a four-quadrant map based on two sets of data: RNA-seq, all gene differential expression table (including non-significant results); and RNA modification, all differential peak table (including nonsignificant results). The top 10 genes (the largest absolute values of diff.log2. fc are shown in purple) were determined to be MT-RNR1, C60rf203, XK, CD22, SEMA3A, HECW2, TIGD5, VISIG4, HLA-DQB1 and NOC2L. (Figure 4A).
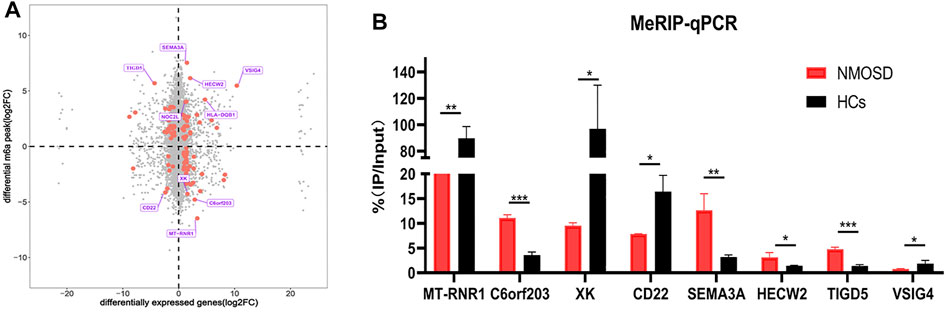
FIGURE 4. Differential RNA modification and validation. (A) Analysis of differential RNA modification and differential gene expression. The X-axis represents the RNA-seq differential genes, and the Y-axis represents the RNA-modified differential peak genes; the top 10 genes (largest absolute values of diff.log2. fc) are shown in purple. The red dots represent significant differences (both RNA-seq and RNA modification data meet p < 0.05); the grey dots indicate that the data did not meet the condition required for the red dots. (B) Validation of the top four hypomethylated genes and the top four hypermethylated genes by using MeRIP-qPCR.
Validation of Differential RNA Modifications by MeRIP-qPCR
In order to verify the reliability of MeRIP-seq results, MeRIP-qPCR was used to verify the modification levels of modification sites on the target genes.We chose the top four hypomethylated genes and the top four hypermethylated genes as target genes (Figure 4A). Among the eight target genes, MeRIP-qPCR results of MT-RNR1, XK, CD22, SEMA3A, HECW2 and TIGD5 were consistent with the sequencing results, and there were significant differences (p < 0.05) (Figure 4B). MeRIP-qPCR results of VISIG4 and C60rf203 were opposite with the sequencing results, although there were significant differences (p < 0.05) (Figure 4B). Nevertheless, it suggested that our sequencing results were credible.
Discussion
m6A modification is a dynamic and reversible RNA modification occurring in eukaryotes that is carried out by “writers”, “erasers”, and “readers”. In recent years, studies have found that m6A is involved in the regulation of the immune microenvironment (Chen et al., 2019b; Tang et al., 2020; Xu et al., 2020). NMOSD is a kind of autoimmune disease that is triggered by disruption of immune homeostasis. In this study, we assessed the m6A status in NMOSD patients and HCs, revealing differences between the groups that support the dynamic features of m6A modification. Indeed, we found that the m6A level was much lower in patients than in HCs, demonstrating that m6A modification is very important in the process of NMOSD. In addition, Zhang found that altering m6A levels might be a novel way for improving the efficacy of platinum in cancer treatment (Zhang et al., 2021a), and Zhao reported that m6A regulation could influence the progression of lung adenocarcinoma (Zhao and Xie, 2021). We hypothesized that increasing the level of m6A might relieve the symptoms of NMOSD, but further studies are needed to confirm this hypothesis.
In the present study, many important biological pathways were found to be related to differentially methylated mRNAs. Because of the construction of strand-specific libraries, we also predicted lncRNAs harbouring m6A peaks and identified DMMSs. While lncRNAs are known to play important roles in mediating transcriptional and posttranscriptional regulation (Cantile et al., 2021; Moison et al., 2021), their biological functions in regards to m6A modification remain unknown. In general, the m6A modification of lncRNAs is very common in cancers. For example, Wu found that m6A-induced lncRNA RP11 could cause the dissemination of colorectal cancer cells (Wu et al., 2019). In our study, we investigated the m6A modification of lncRNAs between NMOSD patients and HCs, which highlighted immune homeostasis. However, further analysis is needed to validate these results.
Evidence supports a strong relationship between immune homeostasis and m6A modification, with m6A modification playing important biological roles in NMOSD (Chen et al., 2020; Chong et al., 2021; Zhang et al., 2021b; Zhong et al., 2021). In our study, the GO and KEGG analyses of mRNAs harbouring DMMSs showed that those with increased methylation were mostly enriched in RNA splicing, mRNA processing and gene expression, which supports the importance of m6A modification in NMOSD. For example, the increased expression of phosphatase and tensin homolog deleted on chromosome Ten (PTEN) and an approximately twofold enhancement methylation were observed in NMOSD patients compared with the HCs, indicating PTEN serves as a key enzyme in NMOSD (Figure 5A) (Han et al., 2019). Furthermore, PTEN is involved in inositol phosphate metabolism. Gene expression is important in NMOSD, and our results suggest a link between NMOSD and the regulation of gene expression, subsequently resulting the disruption of immune homeostasis.
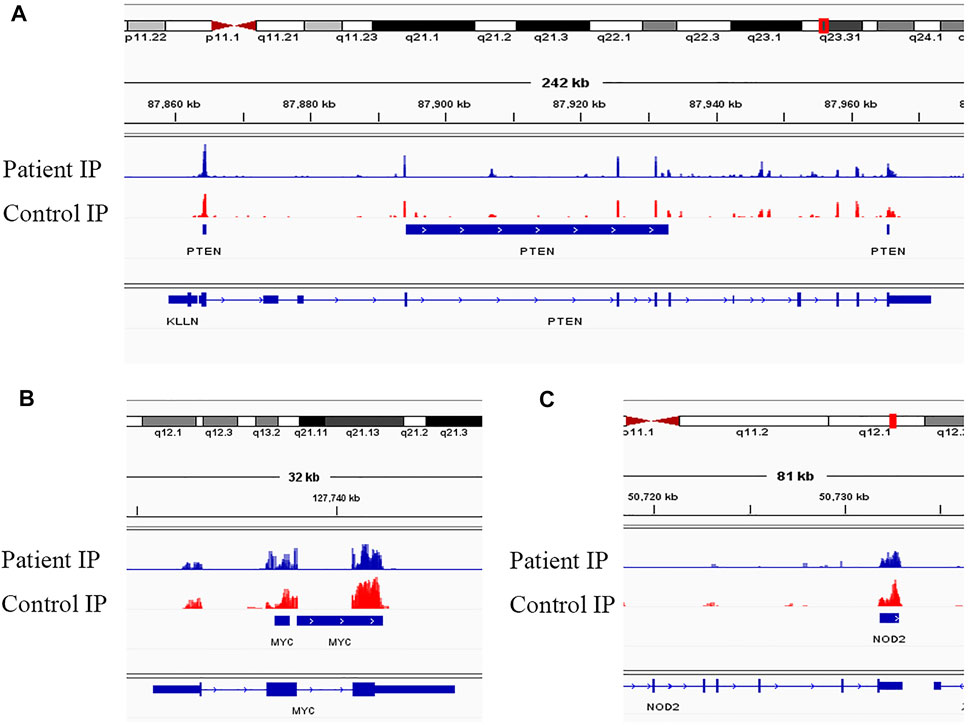
FIGURE 5. Visualization of representative genes. (A) Visualization of the m6A-modified gene PTEN in NMOSD patients and HCs. (B) Visualization of the m6A-modified gene MYC in NMOSD patients and HCs. (C) Visualization of the m6A-modified gene NOD2 in NMOSD patients and HCs.
In contrast to m6A sites with increased methylation, those with decreased methylation were mostly enriched in regulation of transcription, DNA templating, and positive regulation of I-kappa B kinase/NF-kappa B signalling pathways, which include genes such as v-myc avian myelocytomatosis viral oncogene homologue (MYC) (Figure 5B) and nucleotide-binding oligomerization domain containing 2 (NOD2) (Figure 5C). “Readers”, which include YTHDF1-3, recognize and bind to m6A modifications on mRNAs (Wang et al., 2014; Li et al., 2017; Jiang et al., 2019), thereby playing an important role in regulating translation. According to recent studies, we hypothesize that the methylation of mRNAs is associated with their translation, as it may influence their expression, leading to subsequent changes in global translation.
In our association analysis of differential RNA modification and differential gene expression, the top 10 genes included SEMA3A and CD22 (Figure 4A). Zhang also reported that SEMA3A may negatively regulate axonal regeneration in retinal ganglion cells (Zhang et al., 2020), and CD22 is an important inhibitory molecule on the surface of B cells that negatively regulates B cell activation (Gross et al., 2009). All these findings are very beneficial for future studies.
Although few studies have explored the roles of m6A modification in NMOSD, there are still some limitations to our study. For example, we may need to expand the number of enrolled patients. In addition, data from the same patient should be obtained before and after treatment.
Conclusion
Our study demonstrates the differential m6A methylome in NMOSD patients compared to healthy controls, suggesting a strong association between m6A methylation and the regulation of immune homeostasis in NMOSD. The findings fundamentally contribute to future studies on immune homeostasis in NMOSD.
Data Availability Statement
The raw data have been made publicly available under SRA accession number PRJNA737585.
Ethics Statement
The studies involving human participants were reviewed and approved by The Ethics Committee of Shanghai First Rehabilitation Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
HY contributed to the conception of the study and wrote the manuscript. Y-FW and WL contributed to the data collection. JD and D-SZ contributed to the data analyses. X-FS and Y-TG helped to guide this study.
Funding
This study was supported by the National Natural Science Foundation of China (81771295) and the Shanghai Municipal Health Commission Foundation (202140414).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The author would like to thank the NMOSD patients and HCs who provided blood for this article.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.735454/full#supplementary-material
References
Cantile, M., Di Bonito, M., Tracey De Bellis, M., and Botti, G. (2021). Functional Interaction Among lncRNA HOTAIR and MicroRNAs in Cancer and Other Human Diseases. Cancers 13, 570. doi:10.3390/cancers13030570
Chen, J., Fang, X., Zhong, P., Song, Z., and Hu, X. (2019a). N6-Methyladenosine Modifications: Interactions with Novel RNA-Binding Proteins and Roles in Signal Transduction. RNA Biol. 16, 991–1000. doi:10.1080/15476286.2019.1620060
Chen, Y. G., Chen, R., Ahmad, S., Verma, R., Kasturi, S. P., Amaya, L., et al. (2019b). N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 76, 96–109. doi:10.1016/j.molcel.2019.07.016
Chen, S., Zhang, N., Zhang, E., Wang, T., Jiang, L., Wang, X., et al. (2020). A Novel m6A Gene Signature Associated with Regulatory Immune Function for Prognosis Prediction in Clear-Cell Renal Cell Carcinoma. Front. Cell Dev. Biol. 8, 616972. doi:10.3389/fcell.2020.616972
Chong, W., Shang, L., Liu, J., Fang, Z., Du, F., Wu, H., et al. (2021). m6A Regulator-Based Methylation Modification Patterns Characterized by Distinct Tumor Microenvironment Immune Profiles in Colon Cancer. Theranostics 11, 2201–2217. doi:10.7150/thno.52717
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the Human and Mouse m6A RNA Methylomes Revealed by m6A-Seq. Nature 485, 201–206. doi:10.1038/nature11112
Gross, A. J., Lyandres, J. R., Panigrahi, A. K., Prak, E. T. L., and Defranco, A. L. (2009). Developmental Acquisition of the Lyn-CD22-SHP-1 Inhibitory Pathway Promotes B Cell Tolerance. J. Immunol. 182, 5382–5392. doi:10.4049/jimmunol.0803941
Han, J., Wang, J.-z., Yang, X., Yu, H., Zhou, R., Lu, H.-C., et al. (2019). METTL3 Promote Tumor Proliferation of Bladder Cancer by Accelerating Pri-miR221/222 Maturation in m6A-Dependent Manner. Mol. Cancer 18, 110. doi:10.1186/s12943-019-1036-9
Hanniford, D., Ulloa-Morales, A., Karz, A., Berzoti-Coelho, M. G., Moubarak, R. S., Sánchez-Sendra, B., et al. (2020). Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell 37, 55–70. doi:10.1016/j.ccell.2019.12.007
Heck, A. M., Russo, J., Wilusz, J., Nishimura, E. O., and Wilusz, C. J. (2020). YTHDF2 Destabilizes m6A-Modified Neural-specific RNAs to Restrain Differentiation in Induced Pluripotent Stem Cells. RNA 26, 739–755. doi:10.1261/rna.073502.119
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat. Chem. Biol. 7, 885–887. doi:10.1038/nchembio.687
Jiang, Q., Sun, B., Liu, Q., Cai, M., Wu, R., Wang, F., et al. (2019). MTCH2 Promotes Adipogenesis in Intramuscular Preadipocytes via an M 6 A‐YTHDF1‐Dependent Mechanism. FASEB j. 33, 2971–2981. doi:10.1096/fj.201801393RRR
Lennon, V. A., Wingerchuk, D. M., Kryzer, T. J., Pittock, S. J., Lucchinetti, C. F., Fujihara, K., et al. (2004). A Serum Autoantibody Marker of Neuromyelitis Optica: Distinction from Multiple Sclerosis. Lancet 364, 2106–2112. doi:10.1016/s0140-6736(04)17551-x
Li, A., Chen, Y.-S., Ping, X.-L., Yang, X., Xiao, W., Yang, Y., et al. (2017). Cytoplasmic m6A Reader YTHDF3 Promotes mRNA Translation. Cell Res. 27, 444–447. doi:10.1038/cr.2017.10
Lin, S., Choe, J., Du, P., Triboulet, R., and Gregory, R. I. (2016). The M(6) A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 62, 335–345. doi:10.1016/j.molcel.2016.03.021
Lin, Z., Hsu, P. J., Xing, X., Fang, J., Lu, Z., Zou, Q., et al. (2017). Mettl3-/Mettl14-Mediated mRNA N6-Methyladenosine Modulates Murine Spermatogenesis. Cell Res. 27, 1216–1230. doi:10.1038/cr.2017.117
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 Complex Mediates Mammalian Nuclear RNA N6-Adenosine Methylation. Nat. Chem. Biol. 10, 93–95. doi:10.1038/nchembio.1432
Moison, M., Pacheco, J. M., Lucero, L., Fonouni-Farde, C., Rodríguez-Melo, J., Mansilla, N., et al. (2021). The lncRNA APOLO Interacts with the Transcription Factor WRKY42 to Trigger Root Hair Cell Expansion in Response to Cold. Mol. Plant 14 (6), 937–948. doi:10.1016/j.molp.2021.03.008
Pandit, L., Asgari, N., Apiwattanakul, M., Palace, J., Paul, F., Leite, M., et al. (2015). Demographic and Clinical Features of Neuromyelitis Optica: A Review. Mult. Scler. 21, 845–853. doi:10.1177/1352458515572406
Ping, X.-L., Sun, B.-F., Wang, L., Xiao, W., Yang, X., Wang, W.-J., et al. (2014). Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cel Res. 24, 177–189. doi:10.1038/cr.2014.3
Pittock, S. J., and Lucchinetti, C. F. (2016). Neuromyelitis Optica and the Evolving Spectrum of Autoimmune Aquaporin-4 Channelopathies: a Decade Later. Ann. N.Y. Acad. Sci. 1366, 20–39. doi:10.1111/nyas.12794
Su, D., Chan, C. T. Y., Gu, C., Lim, K. S., Chionh, Y. H., Mcbee, M. E., et al. (2014). Quantitative Analysis of Ribonucleoside Modifications in tRNA by HPLC-Coupled Mass Spectrometry. Nat. Protoc. 9, 828–841. doi:10.1038/nprot.2014.047
Tang, R., Zhang, Y., Liang, C., Xu, J., Meng, Q., Hua, J., et al. (2020). The Role of m6A-Related Genes in the Prognosis and Immune Microenvironment of Pancreatic Adenocarcinoma. PeerJ 8, e9602. doi:10.7717/peerj.9602
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-Dependent Regulation of Messenger RNA Stability. Nature 505, 117–120. doi:10.1038/nature12730
Wang, X., Zhao, B. S., Roundtree, I. A., Lu, Z., Han, D., Ma, H., et al. (2015). N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. doi:10.1016/j.cell.2015.05.014
Wingerchuk, D. M., Banwell, B., Bennett, J. L., Cabre, P., Carroll, W., Chitnis, T., et al. (2015). International Consensus Diagnostic Criteria for Neuromyelitis Optica Spectrum Disorders. Neurology 85, 177–189. doi:10.1212/wnl.0000000000001729
Wu, Y., Yang, X., Chen, Z., Tian, L., Jiang, G., Chen, F., et al. (2019). m6A-induced lncRNA RP11 Triggers the Dissemination of Colorectal Cancer Cells via Upregulation of Zeb1. Mol. Cancer 18, 87. doi:10.1186/s12943-019-1014-2
Xu, S., Tang, L., Dai, G., Luo, C., and Liu, Z. (2020). Expression of m6A Regulators Correlated with Immune Microenvironment Predicts Therapeutic Efficacy and Prognosis in Gliomas. Front. Cell Dev. Biol. 8, 594112. doi:10.3389/fcell.2020.594112
Xue, L., Li, J., Lin, Y., Liu, D., Yang, Q., Jian, J., et al. (2021). m 6 A Transferase METTL3‐induced lncRNA ABHD11‐AS1 Promotes the Warburg Effect of Non‐small‐cell Lung Cancer. J. Cell Physiol. 236, 2649–2658. doi:10.1002/jcp.30023
Zhang, Z., Wang, M., Xie, D., Huang, Z., Zhang, L., Yang, Y., et al. (2018). METTL3-mediated N6-Methyladenosine mRNA Modification Enhances Long-Term Memory Consolidation. Cell Res 28, 1050–1061. doi:10.1038/s41422-018-0092-9
Zhang, J., Liu, W., Zhang, X., Lin, S., Yan, J., and Ye, J. (2020). Sema3A Inhibits Axonal Regeneration of Retinal Ganglion Cells via ROCK2. Brain Res. 1727, 146555. doi:10.1016/j.brainres.2019.146555
Zhang, M., Bai, M., Wang, L., Lu, N., Wang, J., Yan, R., et al. (2021a). Targeting SNHG3/miR-186-5p Reverses the Increased m6A Level Caused by Platinum Treatment through Regulating METTL3 in Esophageal Cancer. Cancer Cell Int. 21, 114. doi:10.1186/s12935-021-01747-9
Zhang, X., Zhang, S., Yan, X., Shan, Y., Liu, L., Zhou, J., et al. (2021b). m6A Regulator‐mediated RNA Methylation Modification Patterns Are Involved in Immune Microenvironment Regulation of Periodontitis. J. Cell Mol Med. 25, 3634–3645. doi:10.1111/jcmm.16469
Zhao, W., and Xie, Y. (2021). KIAA1429 Promotes the Progression of Lung Adenocarcinoma by Regulating the m6A Level of MUC3A. Pathol. - Res. Pract. 217, 153284. doi:10.1016/j.prp.2020.153284
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C.-M., Li, C. J., et al. (2013). ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 49, 18–29. doi:10.1016/j.molcel.2012.10.015
Keywords: neuromyelitis optica spectrum disorder, immune homeostasis, N6-methyladenosine (m6A) methylation, differential methylation peaks, MeRIP-seq, UPLC-QQQ-MS
Citation: Yang H, Wu Y-F, Ding J, Liu W, Zhu D-S, Shen X-F and Guan Y-T (2021) Comprehensive Analysis of N6-Methyladenosine (m6A) Methylation in Neuromyelitis Optica Spectrum Disorders. Front. Genet. 12:735454. doi: 10.3389/fgene.2021.735454
Received: 02 July 2021; Accepted: 25 October 2021;
Published: 11 November 2021.
Edited by:
Jia Meng, Xi’an Jiaotong-Liverpool University, ChinaReviewed by:
Omid Mirmosayyeb, Isfahan University of Medical Sciences, IranPiyush Khandelia, Birla Institute of Technology and Science, India
Copyright © 2021 Yang, Wu, Ding, Liu, Zhu, Shen and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia-Feng Shen, shenxiafeng@aliyun.com; Yang-Tai Guan, yangtaiguan@hotmail.com
†These authors have contributed equally to this work