
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 15 March 2021
Sec. Genomic Medicine
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.595702
This article is part of the Research TopicThe Path towards Precision Health: Prospects and ChallengesView all 10 articles
 Valentina J. Ngo-Bitoungui1,2,3
Valentina J. Ngo-Bitoungui1,2,3 Suzanne Belinga4Khuthala Mnika2Tshepiso Masekoameng2
Suzanne Belinga4Khuthala Mnika2Tshepiso Masekoameng2 Victoria Nembaware1René G. Essomba5,6Francoise Ngo-Sack7
Victoria Nembaware1René G. Essomba5,6Francoise Ngo-Sack7 Gordon Awandare1
Gordon Awandare1 Gaston K. Mazandu2,8
Gaston K. Mazandu2,8 Ambroise Wonkam2*
Ambroise Wonkam2*Background: Renal dysfunctions are associated with increased morbidity and mortality in sickle cell disease (SCD). Early detection and subsequent management of SCD patients at risk for renal failure and dysfunctions are essential, however, predictors that can identify patients at risk of developing renal dysfunction are not fully understood.
Methods: In this study, we have investigated the association of 31 known kidney dysfunctions-related variants detected in African Americans from multi-ethnic genome wide studies (GWAS) meta-analysis, to kidney-dysfunctions in a group of 413 Cameroonian patients with SCD. Systems level bioinformatics analyses were performed, employing protein-protein interaction networks to further interrogate the putative associations.
Results: Up to 61% of these patients had micro-albuminuria, 2.4% proteinuria, 71% glomerular hyperfiltration, and 5.9% had renal failure. Six variants are significantly associated with the two quantifiable phenotypes of kidney dysfunction (eGFR and crude-albuminuria): A1CF-rs10994860 (P = 0.02020), SYPL2-rs12136063 (P = 0.04208), and APOL1 (G1)-rs73885319 (P = 0.04610) are associated with eGFR; and WNT7A-rs6795744 (P = 0.03730), TMEM60-rs6465825 (P = 0.02340), and APOL1 (G2)-rs71785313 (P = 0.03803) observed to be protective against micro-albuminuria. We identified a protein-protein interaction sub-network containing three of these gene variants: APOL1, SYPL2, and WNT7A, connected to the Nuclear factor NF-kappa-B p105 subunit (NFKB1), revealed to be essential and might indirectly influence extreme phenotypes. Interestingly, clinical variables, including body mass index (BMI), systolic blood pressure, vaso-occlusive crisis (VOC), and haemoglobin (Hb), explain better the kidney phenotypic variations in this SCD population.
Conclusion: This study highlights a strong contribution of haematological indices (Hb level), anthropometric variables (BMI, blood pressure), and clinical events (i.e., vaso-occlusive crisis) to kidney dysfunctions in SCD, rather than known genetic factors. Only 6/31 characterised gene-variants are associated with kidney dysfunction phenotypes in SCD samples from Cameroon. The data reveal and emphasise the urgent need to extend GWAS studies in populations of African ancestries living in Africa, and particularly for kidney dysfunctions in SCD.
Sickle Cell Disease (SCD) is a monogenic disease with high prevalence and high mortality rates in Africa. Globally, SCD is estimated to affect more than 300,000 births per year, with nearly two-thirds occurring in sub-Sahara Africa (Piel et al., 2013). Cameroon is a sub-Saharan African country with an estimated SCD carrier frequency rate between 8 and 34% (Weatherall and Clegg, 2001). Although Cameroon declared SCD as a public health priority, access to care and treatment is still limited due to lack of a national medical insurance, leaving SCD patients to self-fund or depend on familial financial support. Therefore, medical care costs are often not met (Wonkam et al., 2014) and patients frequently suffer from severe SCD complications such as kidney dysfunction (Geard et al., 2017).
Renal failure caused by recurrent episodes of ischemia-reperfusion injury and haemolytic anaemia, occurs in 5–18% of SCD patients and is associated with an increased risk of early mortality (Platt et al., 1994; Gladwin, 2017). The prevention of renal failure relies on early detection and management of kidney dysfunction. In SCD patients, renal failure can be caused by gradual infiltration of glomerulus, which leads to glomerular sclerosis or promotes progression of micro-albuminuria to macro-albuminuria/proteinuria and finally to nephrotic-range proteinuria (Nath and Hebbel, 2015). Micro-albuminuria is prevalent in 26–68% of adult patients (Ataga et al., 2014; Gosmanova et al., 2014) and is the most sensitive early clinical marker for glomerular damage and other types of kidney dysfunction. Recent studies have demonstrated that the co-inheritance of alpha-thalassemia with SCD and/or specific variants in the HbF promoting loci can delay the clinical progression of kidney disease in African American SCD patients (Saraf et al., 2017). In addition, genetic variations in two coding regions of Apolipoprotein L1 (APOL1) and Heme oxygenase 1 (HMOX1) genes have been associated to chronic kidney disease (Genovese et al., 2010; Tzur et al., 2010), and to SCD nephropathy (Saraf et al., 2015; Schaefer et al., 2016).
In a previous study, we showed that variants in APOL1 and HMOX1 variants are associated with kidney dysfunctions using a targeted SNP based approach. Further investigations reveal that these variants are associated with albumin creatinine ratio, micro-/macro-albuminuria and eGFR in a group of SCD patients in Cameroon (Geard et al., 2017). Given the high rate of renal dysfunction in SCD patients in Africa with its high genetic diversity, there is need to explore possible novel genetic variants associated with kidney dysfunction. Ideally, whole genome sequencing and other large-scale gene discovery approaches should be use, expanding the targeted genetic discovery to other variants known to be associated with renal dysfunctions. Given the limited number of gene and variant discovery research in SCD patients, a plausible strategy is using renal dysfunctions associated SNPs from non-SCD affected populations.
Several kidney dysfunction genome-wide association studies (GWAS) have been conducted in many non SCD-affected populations (Kottgen et al., 2009, 2010; Chambers et al., 2010; Pattaro et al., 2012). Furthermore, a recent GWAS meta-analysis integrated 15 GWAS studies of 133,413 individuals from multiple ethnicities and uncovered 53 SNPs associated with renal dysfunction, including 26 SNPs found in individuals of African descent (Pattaro et al., 2016). In this study, we investigated the associations of these 26 SNPs in addition to four previously characterised kidney dysfunction-related variants, including APOL1 (G1 or G2) for rs60910145, rs73885319 and rs71785313, and HMOX1 for rs3074372 and rs743811, relevant to populations of African ancestry (Pattaro et al., 2016), e.g., SCD patients from Cameroon.
Several studies, including a previous study from our group (Geard et al., 2017), have shown that, in addition to genetic variants, clinical, and biological factors also contribute to glomerular damage (Audard et al., 2017). This highlights the need to employ a multi-factorial approach in investigating factors associated to renal abnormalities in SCD patients. Therefore, in addition to investigating the contribution of the selected 31 SNPs to renal dysfunctions in SCD patients from Cameroon, we also explored the contribution of clinical factors: socio-demographic, anthropometric, clinical and haematological variables, and employed multifactorial regression models for associating the variable to kidney dysfunction parameters. Finally, systems level bioinformatics analyses were performed, employing protein-protein interaction networks to further interrogate the putative associations.
The study was performed following the Declaration of Helsinki. This study was approved by the Faculty of Health Sciences Human Research Ethics Committee of the University of Cape Town, South Africa (HREC REF: 661/2015), and the National Ethics Committee of the Ministry of Public Health, Yaoundé, Republic of Cameroon (No. 193/CNE/SG/10). Patients older than 18 years self-consented into the study and informed consent was given by the parents or guardians for participants younger than 18 years old with a requirement for children older than 7 years to also sign assent forms.
Cameroonians living with SCD were prospectively recruited at the Yaoundé Central Hospital and Laquintinie Hospital in Douala, between January 2010 and December 2012. Only patients older than 2 years of age who had not received a blood transfusion in the past 6 months were included. None of the patients were receiving hydroxycarbamide treatment. Community-based recruitments were also conducted through two SCD patients’ associations who were engaged for collaboration. Additional patients were subsequently recruited during the SCD patient associations’ monthly meetings. No incentive was provided for participation in the study. Socio-demographic and clinical events were collected by means of a structured questionnaire administered to parents/guardians or adult patients. Body mass index (BMI) and blood pressure (BP) were also measured. Patients’ clinical records were extracted from their medical records covering the past 3 years. These clinical records include blood transfusion history, the occurrence of vaso-occlusive crisis (VOC) and hospitalisation rates per year.
Complete blood counts and foetal haemoglobin (HbF) quantifications were conducted during hospital visits. Two methods of HbF detection were employed in this study for patients: initially using the alkali denaturation test in 55% of the cohort and subsequently High-Performance Liquid Chromatography when it became available at the haematological laboratory of the Centre Pasteur of Cameroon (CPC).
Urinary albumin quantifications were performed using either the Siemens Clinitek Status test or the Hemocue Albumin 20 system on the first morning urine samples during planned hospital visits, when patients were not experiencing VOC, as previously reported by Geard et al. (2017). The presence of albumin in the urine is defined as normal when the concentration is <30 mg/dl, micro-albuminuria (30–300 mg/dl) or macro-albuminuria >300 mg/dl. The glomerular filtration rate (GFR) is estimated (eGFR) using the Chronic Kidney Disease-Epidemiology Collaboration (CKD-EPI-creatinine) formula. Kidney failure is defined as an eGFR <90 ml/min/1.73 m2, renal hyperfiltration as an eGFR > 130 ml/min/1.73 m2 for women and >140 ml/min/1.73 m2 for men, and normal filtration as an eGFR between 90 ml/min/1.73 m2 and 130/140 ml/min/1.73 m2 (Haymann et al., 2010).
DNA was extracted from peripheral blood in EDTA following the manufacturer’s instructions (Puregene Blood Kit) at CPC, and Genotype analyses were performed at the Division of Human Genetics, University of Cape Town, South Africa.
Molecular analysis to confirm the presence of the sickle mutation w carried out on 200 ng of DNA by polymerase chain reaction (PCR) to amplify a 770 bp segment of HBB, followed by a digestion with DdeI restriction enzyme on the PCR product (Saiki et al., 1985).
Five restriction fragment length polymorphism (RFLP) sites in the HBB cluster were amplified using published primers and methods to analyse the HBB haplotype background (Bitoungui et al., 2015).
The 3.7 kb HBA1/HBA2 deletion was successfully screened using the expand-long template PCR as previously published (Rumaney et al., 2014).
Twenty-six African American specific kidney dysfunction-related gene variants were genotyped in this SCD cohort after being mapped to 53 single nucleotide polymorphisms (SNPs) identified in a GWAS meta-analysis of kidney diseases (Pattaro et al., 2016). Five additional gene variants in APOL1 and HMOX1 from the literature were also considered (Genovese et al., 2010; Tzur et al., 2010; Wonkam et al., 2014; Geard et al., 2017; Saraf et al., 2017). Thus, all targeted SNPs in A1CF-rs10994860, WNT7A-rs6795744, PTPRO-rs7956634, UMOD-rs4293393, LRP2 -rs4667594 ANXA9-rs267734, GCKR-rs1260326, TFDP2-rs3476 85, DAB2-rs11959928, SLC22A2-rs2279463, TMEM60-rs64658 25, SLC6A13-rs10774021, BCAS1-rs17216707, SKIL-rs9682041, UNCX-rs10277115, KBTBD2-rs3750082, CNQ1-rs163160, AP5B 1-rs4014195, NFKB1-rs228611, CACNA1S-rs3850625, SYPL2-rs1 2136063, ETV5-rs10513801, DPEP1-s164748, SIPA1L3-rs1166649 7, NFATC1-rs8091180 and IGFBP5-rs2712184, and in APOL1 (G1)-rs60910145, APOL1 (G1)-rs73885319, APOL1 (G2)-rs71 785313, HMOX1-rs3074372, HMOX1-rs743811, were genotyped with the iPLEX Gold Sequenom Mass Genotyping Array. Thereafter, the validation of the genotyping results was done by Sanger sequencing using BigDye terminator mix in 10% subset of sample (Supplementary Figure S1).
We performed association analyses between kidney dysfunction outcomes (characterised by eGFR and crude-albuminuria scores), socio-demographic and clinical variables, and 31 known kidney dysfunction-related variants among this group of SCD patients. First, to ensure genotypic quality of the data, we ran PLINK 1.9 (Purcell et al., 2007), performing a Hardy-Weinberg Equilibrium (HWE) test with significant level, minor allele frequency (MAF), and missing genotype data thresholds of 0.001, 0.05, and 0.1, respectively. A total of 13 out of 31 SNPs in the dataset did not pass quality control (QC) filters, of which 8 were removed due to missing genotype data and 5 others do not meeting the set minor allele thresholds. With R software, we first performed descriptive statistics to provide a general summary of different parameters to be considered in the analysis. Thereafter, two regression analyses were performed: (1) multi-variable regressions for each kidney dysfunction phenotype with different socio-demographic, anthropometric, haematogical, clinical variables and each genetic variant after adjusting or transforming phenotype values to approximate a symmetric (normal) distribution based on their Fisher-Pearson skewness coefficient scores; (2) logistic regressions for each kidney dysfunction phenotype and all variables and genetic variants under consideration after mapping phenotypes to 0 (controls) or 1 (cases: micro- and macro-albuminuria for crude-albuminuria, then renal failure and hyperfiltration for eGFR). Finally, we performed functional and protein–protein interaction network enrichment analyses, using Gene Ontology (GO) process (The Gene Ontology Consortium, 2019), the protein GO Annotation (GOA) mapping (Mi et al., 2019) and the Kyoto Encyclopeadia of Genes and Genomes (KEGG) pathway (Kanehisa et al., 2019) datasets, to identify potential enriched biological processes and pathways in which identified candidate genes are involved. A significance level of 0.05 was considered after adjusting p-values (P) for Bonferroni multiple corrections and gene functional annotations were retrieved from the Ensembl database (Yates et al., 2020).
A total of 413 SCD steady state Cameroonian patients were included in the study. The participants’ characteristics are described in Table 1. There are roughly equal numbers of males and females (M/F = 210/203), with a median age of 15.5 years. The median number of VOC per year was 2 (range: 0–80). 41% (n = 168) of patients had (3 VOC per year and 28.1% (n (=114) had 2 hospitalizations hospitalisations per year. All participants were homozygous HbSS, Benin being the most prevalent (-globin-like gene cluster haplotype (55%; n = 195). It is worth noting that, in this study, we used the modified annotation protocol suggested by Crawford et al. (2002) and Hanchard et al. (2007) for determining β-globin gene cluster haplotypes Benin, Cameroon, Bantu, Senegal and Arab-Indian. Haplotypes that are not conform to these are considered to be “Atypical.” We refer to “Other haplotypes,” all underrepresented haplotypes in the cohort, covering Bantu, Senegal, and Arab-Indian. 32 and 11% co-inherited a single or double 3.7 kb HBA1/HBA2 deletion, respectively.
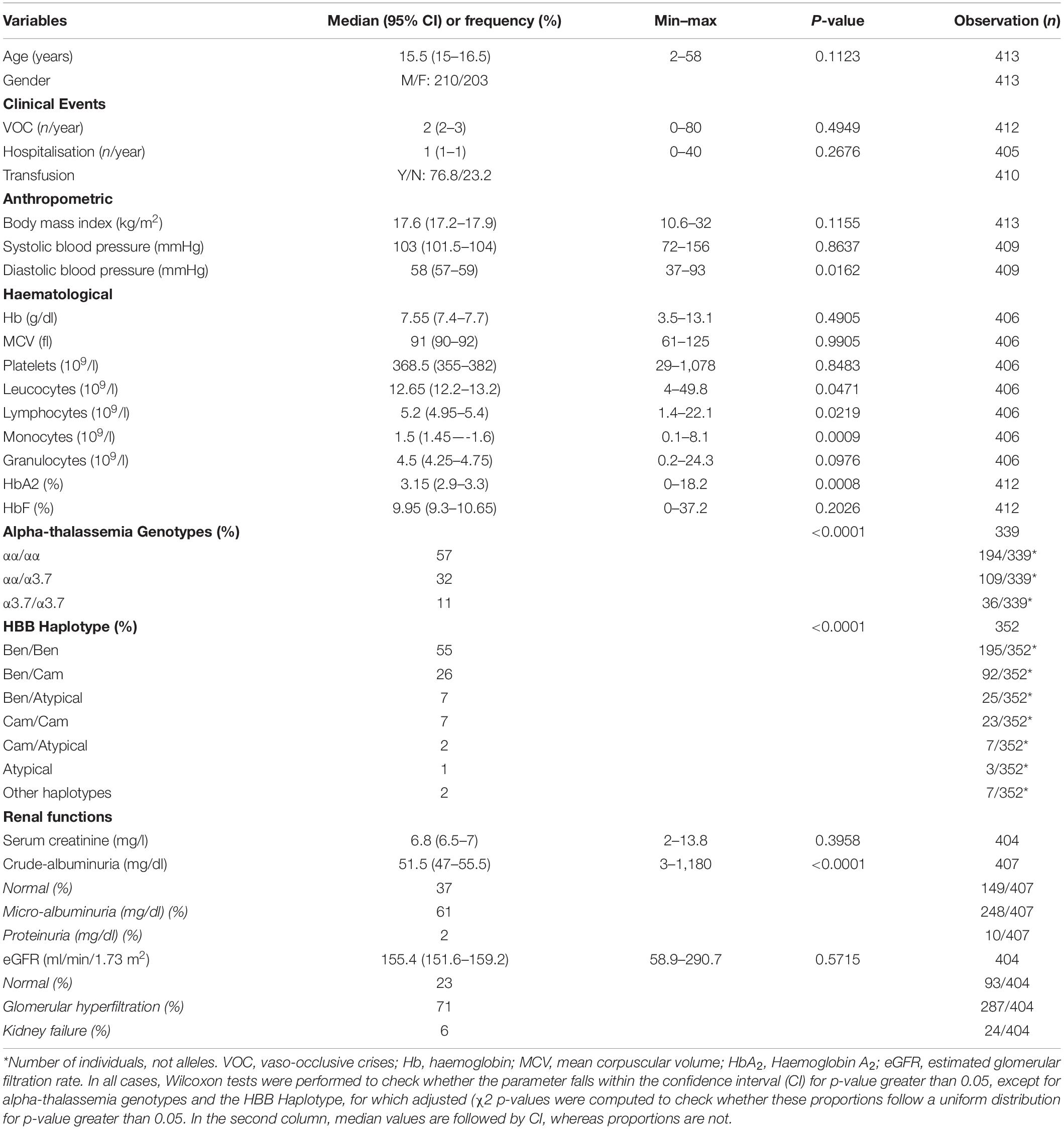
Table 1. Description of the Cameroonian SCD cohort.
For the genotype dataset and two renal phenotypes under consideration, a prior QC and value adjustment processes were performed, respectively. Only 18 out the 31 genetic variants (highlighted in Table 2) were considered for further analyses: eight variants were removed due to missing genotype data (genotype call rate 0.9: for cut-off of 0.1) and 5 had minor allele frequency of less than 5%. For each phenotype to approximate a symmetric or normal distribution, we computed the Fisher-Pearson skewness coefficients to select the type of transformation required. For eGFR, no value adjustment was required as its skewness coefficient was 0.27581, comprised in the range of -0.5 and 0.5 fitting a symmetric distribution. For crude-albuminuria, the skewness coefficient is 6.45456 ≥ 1, in which case, the log10 transformation was applied to ensure that crude-albuminuria dataset approximate a symmetric distribution (Figure 1).

Table 2. Allele frequencies of kidney dysfunction-related gene variants.
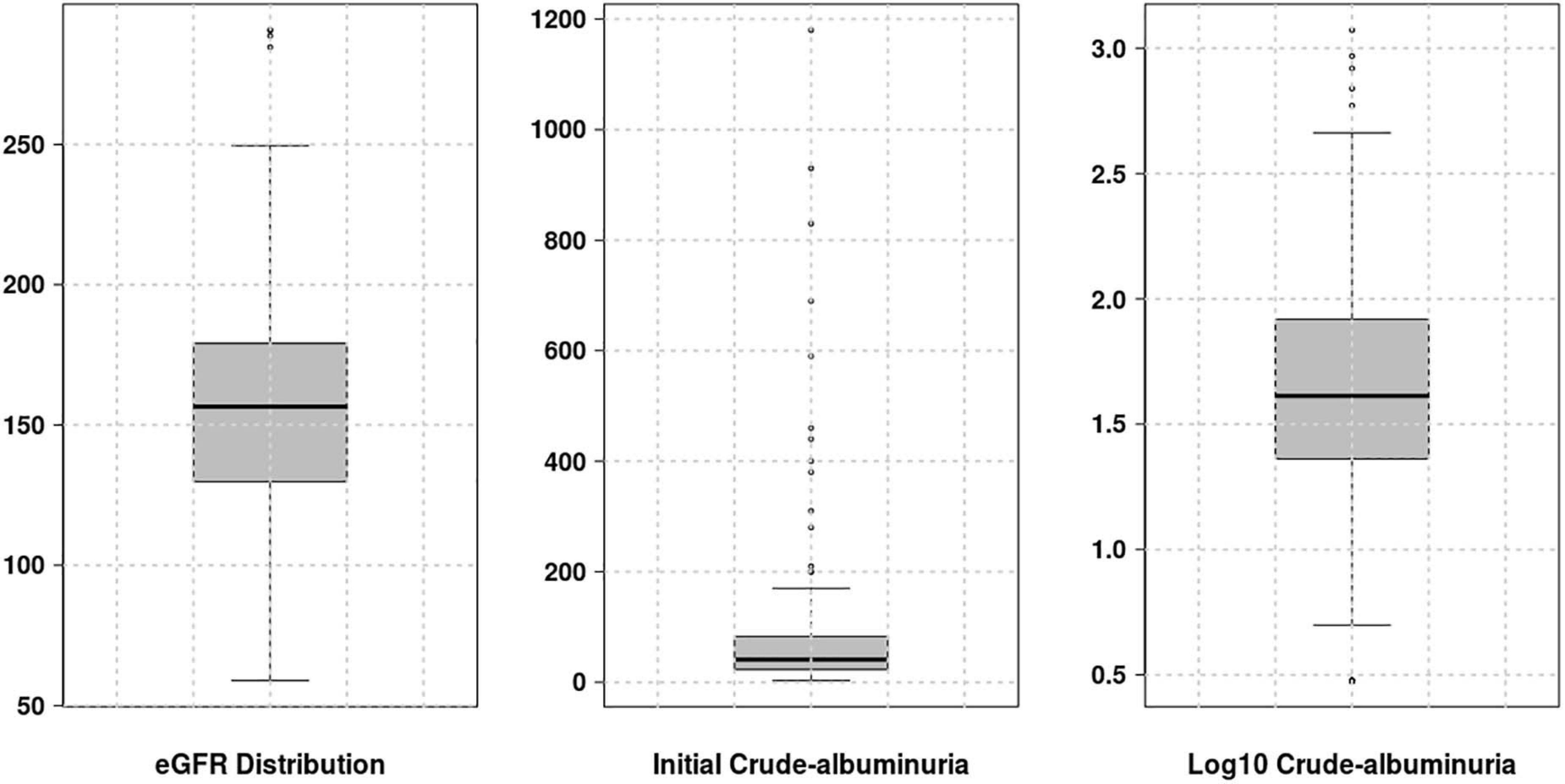
Figure 1. Distribution of different phenotype values. For eGFR, no transformation was required, however, for crude-albuminuria, the initial distribution is highly skewed and log10 transformation was applied to approximate a normal distribution.
After the transformation, we performed linear regression models and factors associated with crude albuminuria and eGFR in Cameroonian SCD patients are presented in Table 3. Age and gender as well as creatinine were not considered in the eGFR-based regression models to prevent biases in outputs as these two factors are confounders, mixing up with eGFR as an outcome (because age, gender and creatinine contribute to the eGFR calculation).
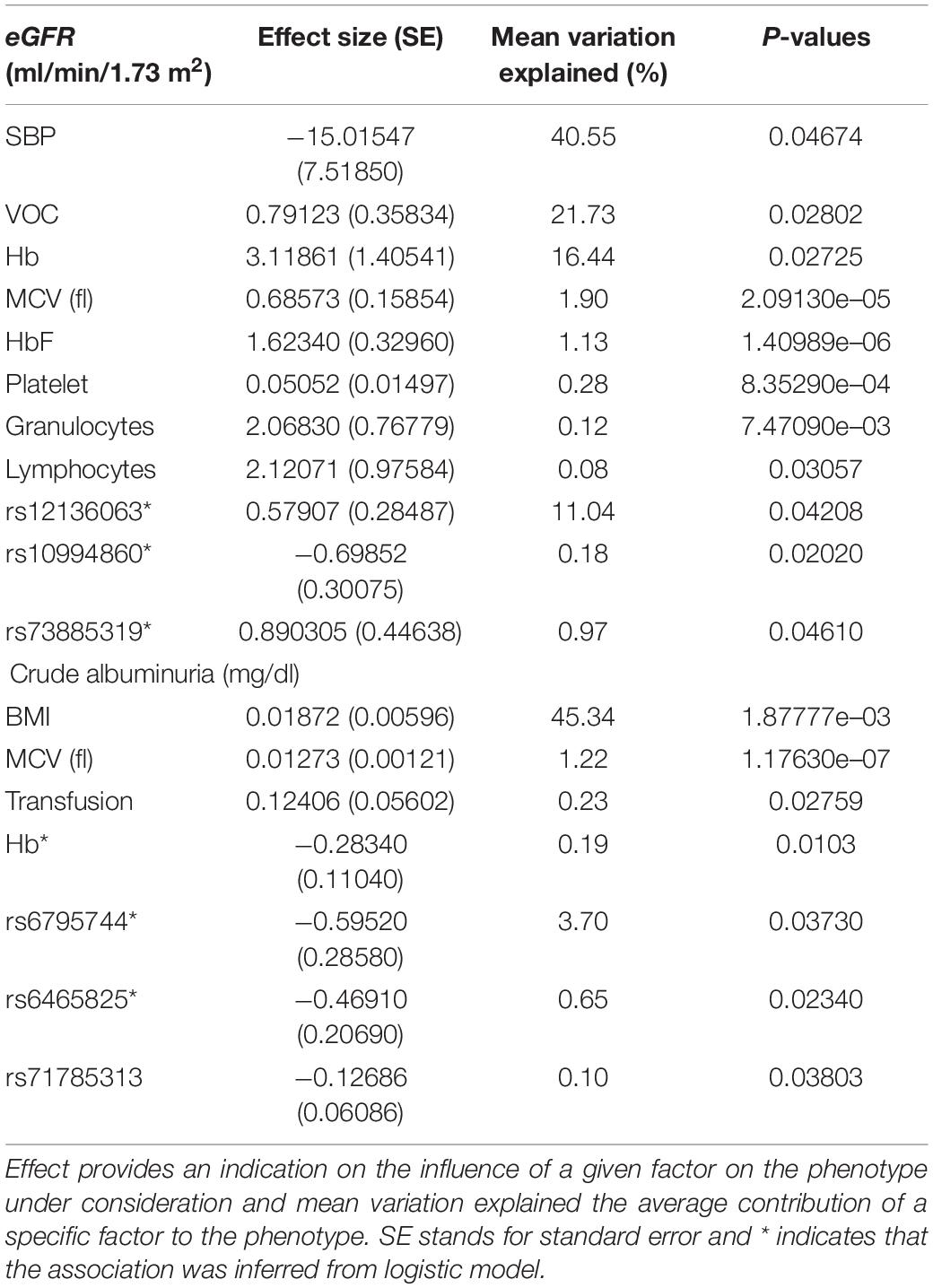
Table 3. Blood pressure, clinical, and haematological variables, and genetic variants associated eGFR and Crude albuminuria.
the level of serum creatinine used to estimate GFR had a median of 6.8 mg/l and the eGFR median was 155.4 ml/min/1.73 m2 (Table 1). Up to 71% of the patients had glomerular hyperfiltration and 5.9% renal failure (see overall age-based population distributions in Figure 2A). Figure 2A suggests that the prevalence of glomerular hyperfiltration is high amongst patients under 21 years old and kidney failure is relatively high up to 30 years, decreasing after 30 years. This agrees with another Canadian study which highlighted that children with SCD between 4 and 11 years have a significantly higher mean eGFR (Mammen et al., 2017). The eGFR was significantly increased in male patients (P = 4.65297e–10). Isolated hyperfiltration was present in 25.8% (n = 105) of patients, while 41.5% (n = 169) and 2.2% (n = 9) were experiencing glomerular hyperfiltration with micro- and macro-albuminuria, respectively. Haemoglobin, HbF, MCV, platelet, lymphocytes, and granulocytes presented highly significant p-values and positive correlation with eGFR, explaining some proportion of eGFR variations (Table 3). This suggested that there was a reduced protection of kidney from haemoglobin mediated toxicity. Though high HbF level as well as lymphocytes and granulocytes have beneficial clinical effect on SCD patients and results indicated that an increased level of these variables beyond the steady state likely increase the risk of clinical complications. Finally, eGFR is also correlated to systolic BP (P = 0.04674) and VOC frequencies (P = 0.02802), explaining 40.55 and 21.73% of the eGFR variation, respectively (Table 3), with stable systolic BP being highly protective against glomerular hyperfiltration.
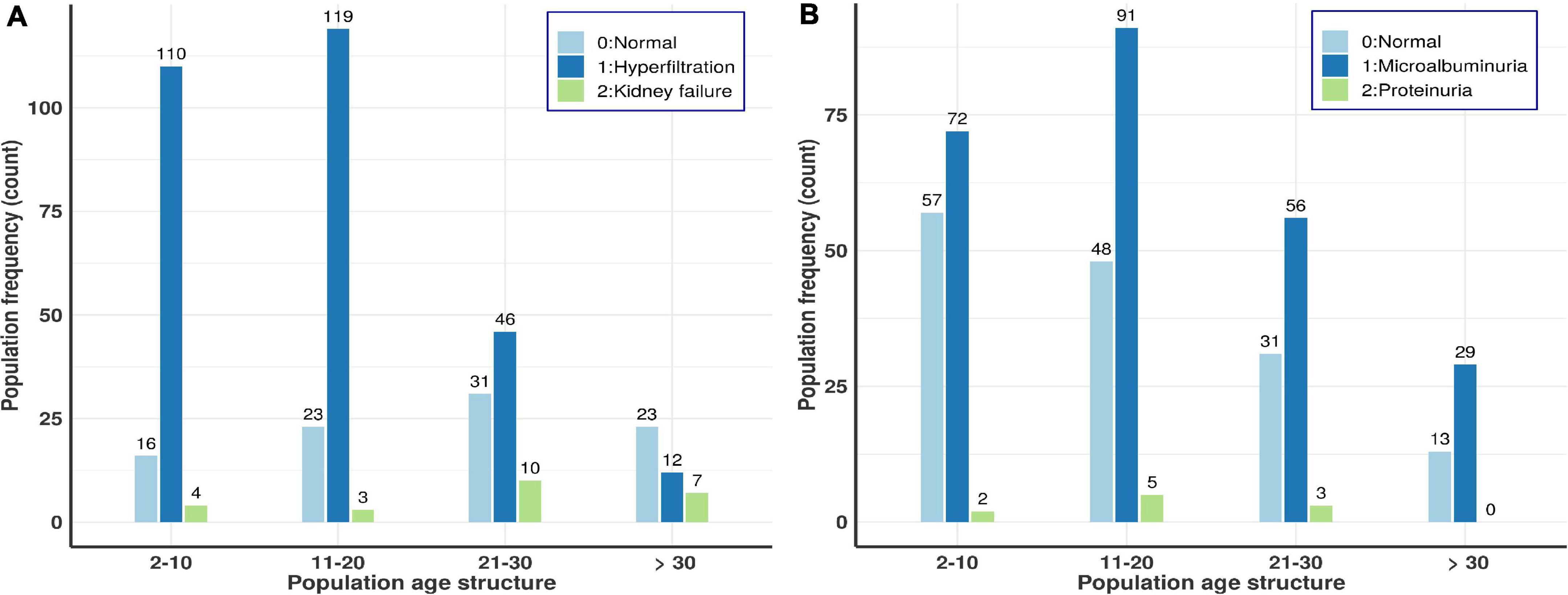
Figure 2. Age-based population distributions for the two kidney dysfunction indicators, i.e., crude-albuminuria and eGFR scores. (A) Age-based population distribution using eGFR scores. (B) Age-based population distribution using microalbuminuria scores.
The median of crude-albuminuria was 51.5 mg/dl ranging from 3 to 1,180 mg/dl. The prevalence of micro and macro-albuminuria was 61 and 2%, respectively. All age ranges have nearly equal proportions of micro-albuminuria, while proteinuria is shown to increase with age (Figure 2B). Figure 2B shows similar profile as for eGFR in Figure 2A and indicates that the prevalence of microalbuminurea is high amongst patients under 21 years old, with some proteinurea cases that vanish after 30 years of ages. This also agrees with another cohort study in Ghana (Anto et al., 2019), which indicated that the prevalence of renal complications, such as proteinuria, is high in young patients aged between 5 and 12 years. BMI, MCV, haemoglobin and transfusion are significantly associated with crude albuminuria with the highest phenotypic variation explained by BMI (45.34%) and Haemoglobin with reduced risk of micro-albuminuria, though with small effect size (Table 3).
Three genetic variants: Synaptophysin-like protein 2 (SYPL2-rs12136063), APOBEC1 complementation factor (A1CF-rs10994860) and Apolipoprotein L1 (APOL1 (G1)-rs73885319) are significantly associated with eGFR with respective P-values of 0.04208, 0.02020, and 0.04610. Figure 3 shows the eGFR values for three gene variants distributed in homozygous dominant, recessive, and heterozygous genotypes. These distribution values suggested that the two allele changes (homozygous recessive) of A1CF-rs10994860 is protective against renal dysfunction (Table 3). On the other hand, one allele change or heterozygous genotype showed a protective effect against prevalent hyperfiltration considering the negative correlation with eGFR (conferring about 2 times more protection as compared to a patient with no copy). SYPL2-rs12136063 confers increased risk of progressing to the renal dysfunction and APOL1 (G1)-rs73885319 to prevalent hyperfiltration and ultimately to the renal dysfunction (approximately 2 times more likely to progress to renal dysfunction).
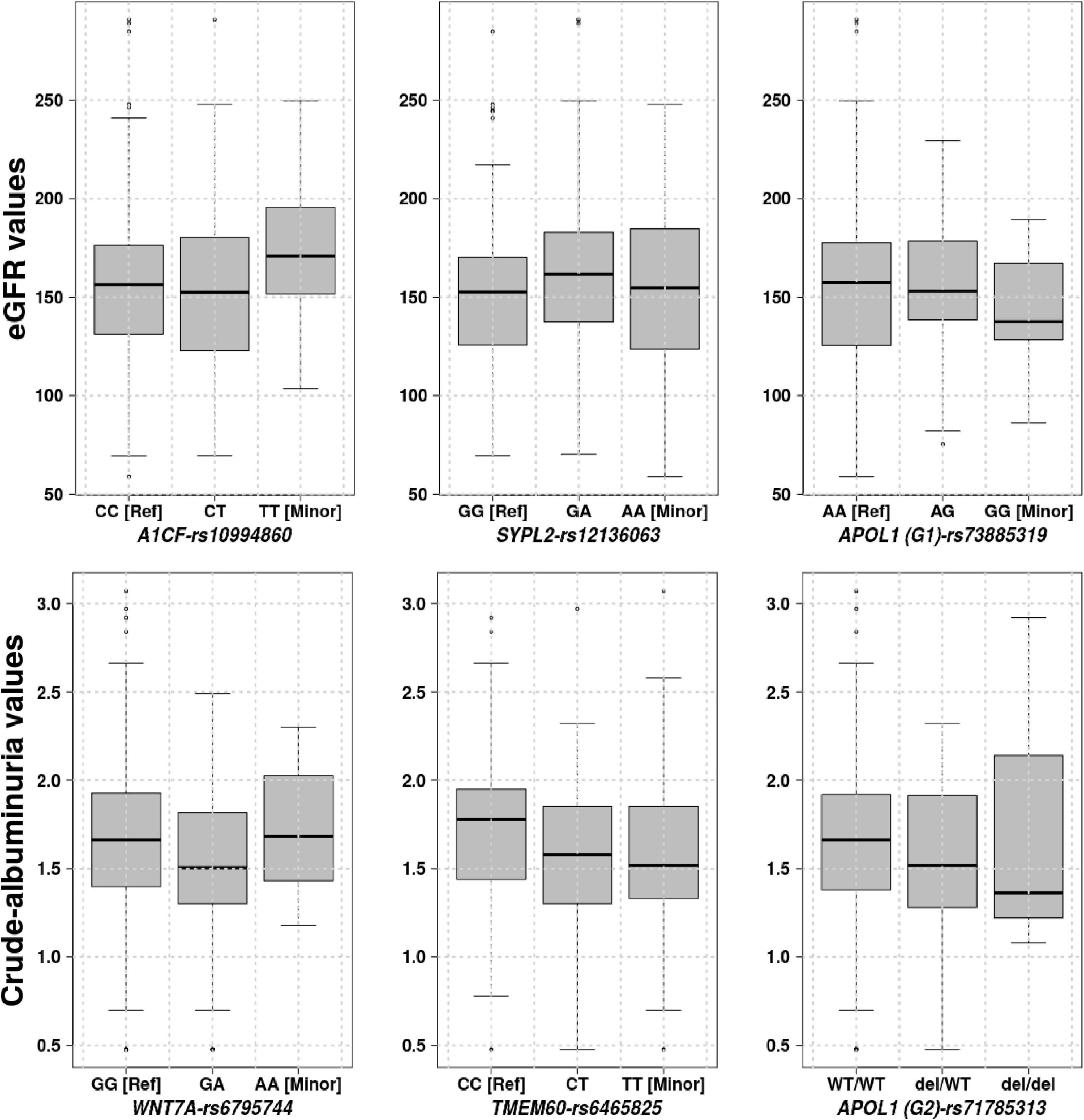
Figure 3. Phenotype values, eGFR and crude-albuminuria values based on the significant gene variants distributed over homozygous dominant, recessive, and heterozygous genotypes.
Three genetic variants were also identified to be significantly associated with crude-albuminuria: Protein Wnt-7a (WNT7A-rs6795744), Transmembrane protein 60 (TMEM60-rs6465825), APOL1 (G2)-rs71785313 with P-values of 0.03730, 0.02340, and 0.03803, respectively. With knowledge of the crude-albuminuria distribution as shown in Figure 3, a change in allele or WT deletion in the case of APOL1 provides a protective effect against prevalent micro-albuminuria (Table 3). These results indicate that patients with WNT7A-rs6795744 and TMEM60-rs6465825 changes are approximately 2 times less likely to progress to micro-albuminuria state, and a single and double WT APOL1 (G2) deletions decrease crude-albuminuria value by 0.12686 and 0.24732 ml/min/1.73/m2, respectively. Though this phenotype level change is negligible due to the effect size, it can be essential for crude-albuminuria extreme values (e.g., values on the border line of state changes).
HbF is a major SCD modifier, which is known to modulate the SCD phenotype (Coleman and Inusa, 2007), to ameliorates pathophysiological and clinical manifestations of the sickling process (Adekile, 2020). There is accumulating evidence indicating that this major disease modifier is influenced by β-globin gene cluster haplotypes (Lakkakula et al., 2017; Piel et al., 2017; Adekile, 2020) and (α-globin gene deletions (Piel et al., 2017). Thus, we have looked at distribution of fetal haemoglobin levels vs. representative β-globin gene cluster haplotypes and α-globin gene deletions and results are shown in Figure 4. These results indicate that HbF levels vary in the population based on β-globin gene cluster haplotypes and α-globin gene deletions. Individuals with Benin haplotyte and double 3.7 kb α-globin gene deletions having significantly higher HbF levels with P = 0.0813e–2 and 0.0203, respectively, accepting the alternative hypothesis that the HbF level median is greater than 9.3%, the lower bound of the HbF level confidence interval (Table 1), which is slightly higher than the minimal level (8.6%) indicated to improve SCD patient survival (Coleman and Inusa, 2007). Furthermore, we checked whether there is any association between exposure to Benin haplotype or double 3.7 kb α-globin gene deletions and outcomes, namely renal dysfunction phenotypes: eGFR and albuminurea. Results obtained have revealed an association between double 3.7 kb α-globin gene deletions and eGRF showing a protective effect against prevalent hyperfiltration/kidney failure with odds ratio = 0.36320 (95%CI: 0.16695–0.80532, P = 0.01007) under the null hypothesis that odds ratio is equal to 1.
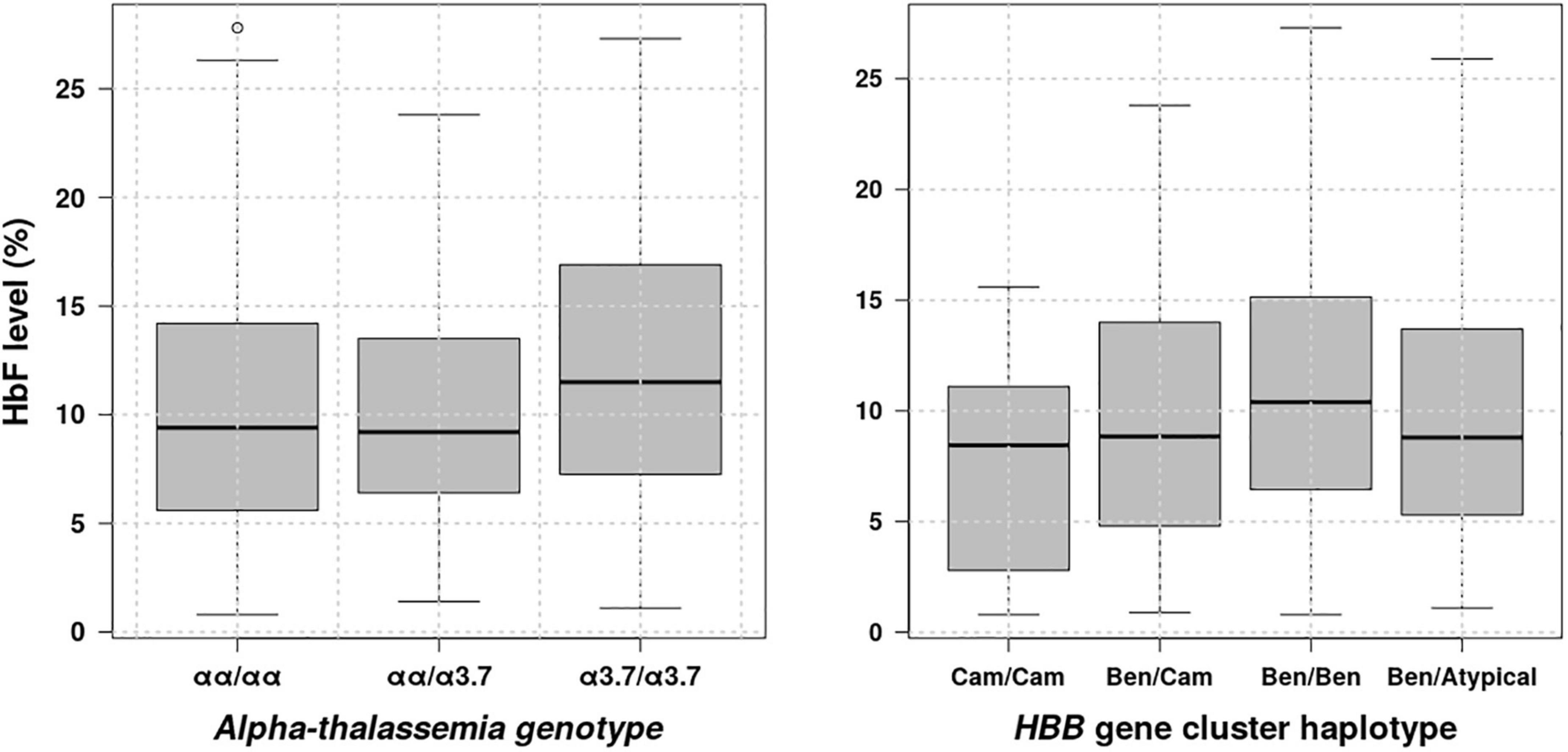
Figure 4. Distribution of HbF levels vs. β-globin gene cluster haplotypes and α-globin gene deletions.
Using the eGFR- and crude-albuminuria-based logistic regression model, we compute areas under the receiver operating characteristic (ROC) and Precision-Recall (PR) curves, as well as accuracy to identify the best phenotype proxy for predicting kidney dysfunction in Cameroonian SCD patients. Different areas are shown in Figure 5, with eGFR-based model achieving the area under ROC of 0.76 and an accuracy score of 0.78 vs. the area under ROC of 0.73 with an accuracy score of 0.70 for crude-albuminuria-based model. This suggests that it is more effective to use eGFR phenotype as a proxy for predicting kidney dysfunction in Cameroonian SCD population. This is also in agreement with the Akaike’s Information Criterion (AIC) scores produced by the two models, 284.33 for eGFR-based model vs. 361.32 for crude-albuminuria-based model, indicating that eGFR-based model fits data better than crude-albuminuria phenotype proxy. This suggests that a simple classification learning algorithm can be designed, taking as inputs, patient age, gender, and level of serum creatinine and predicting kidney dysfunction in patients.
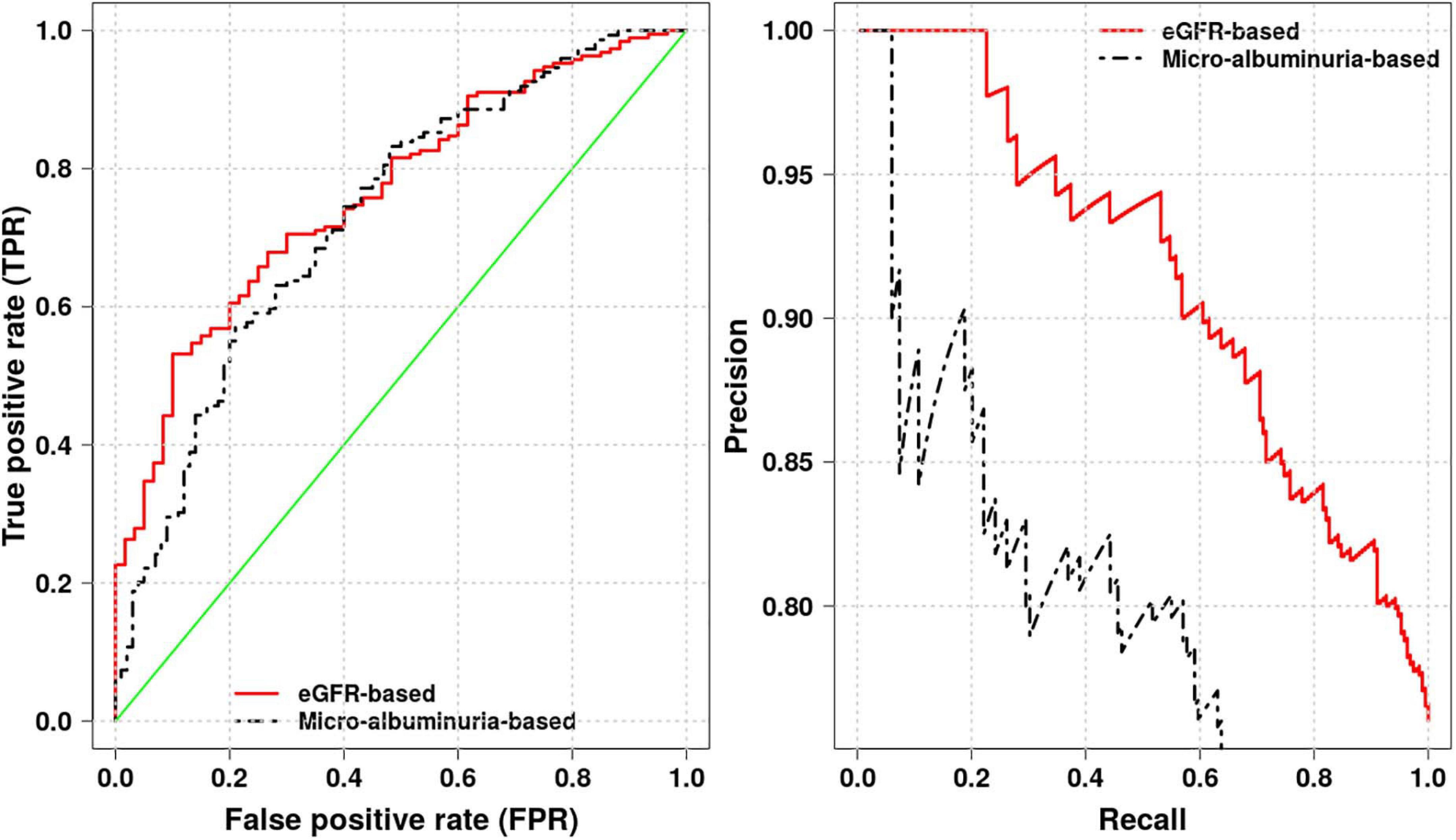
Figure 5. Receiver operating characteristic (ROC) and Precision-Recall curves for eGFR- and crude-albuminuria-based logistic models.
We analysed how interactive genes from knowledge-based Protein-Protein Interaction (PPI) interacted with the 18 genes used in different analyses, focusing specifically on six genes identified to be associated with kidney dysfunction phenotypes. Mapping these 18 genes to a comprehensive human Protein-Protein Interaction (PPI) network (Wu et al., 2009; Mazandu et al., 2018), we identify sub-networks containing these gene variants: APOL1, SYPL2, WNT7A, IGFBP5, UNCX, NFKB1, UMOD, and SKIL. Three gene variants within this sub-network, namely APOL1 (G1)-rs73885319, as well as APOL1 (G2)-rs71785313, WNT7A-rs6795744, and SYPL2-rs12136063, have been identified to influence variation in renal dysfunction phenotypes in SCD patients. These variant genes are connected to NFKB1, identified to be essential or a hub based on the network centrality measures within the sub-network via some specific intermediate genes (Figure 6 and Table 4), following a small world property of human PPI network (Mazandu et al., 2020). This NFKB1 gene might indirectly influence extreme phenotype levels. Moreover, these gene variants are enriched with the cartilage condensation process (P = 0.02976) in which WNT7A, revealed to be likely implicated in SCD patient renal dysfunction, is involved. This process is possibly involved in the development of VOC complications, resulting in proteinuria and glomerular hyperfiltration, and ultimately in kidney damage.
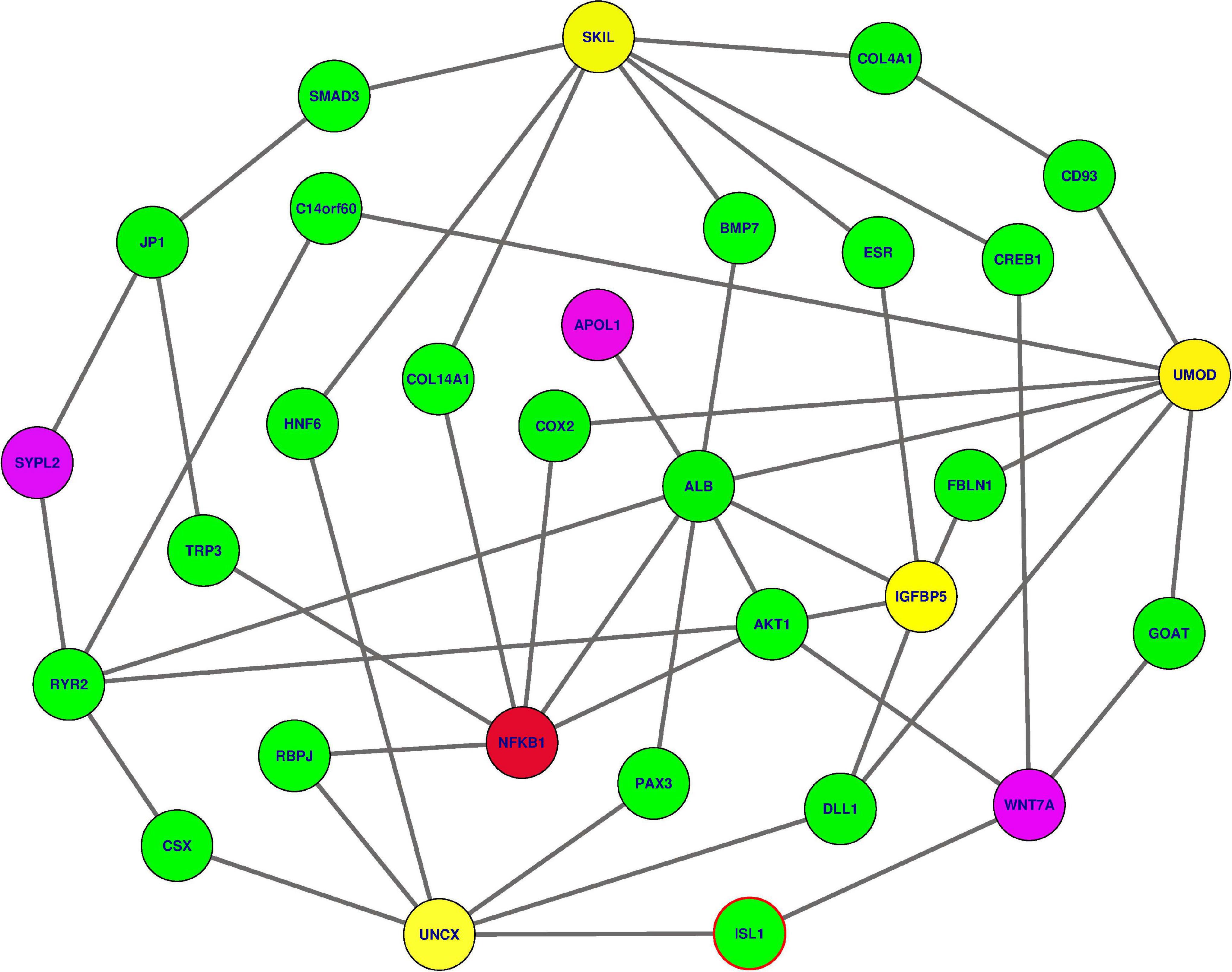
Figure 6. The subnetwork extracted from the human PPI network revealing how predicted gene variants interact together to influence the kidney dysfunction (refer to Table 4 for gene descriptions). In yellow and magenda are gene variants previously shown to be associated with kidney-dysfunction, three in magenda color have been confirmed. In green color, are intermediate nodes used by kidney-dysfunction gene variants to reach the one in red color (NFKB1) indicated to be essential in the PPI network.

Table 4. The description of the different genes displayed in Figure 6 extracted from the UniProt database (https://www.uniprot.org/).
This is the first study to investigate the relevance of kidney dysfunction-related variants identified through a GWAS meta-analysis as well as functional enrichment and protein-protein interaction network analyses in SCD patients. The results highlighted the high prevalence of micro-albuminuria as presented in a previous study in Cameroon by Geard in 2017 (Geard et al., 2017). This prevalence is much higher than the values of 18.5 and 27% observed in paediatric cohorts from several sub-Saharan African countries (Ranque et al., 2014; Aloni et al., 2017), the 13.2% in the multicentric study of children with SCD in the United States (Schaefer et al., 2016) and the 44% in adults from Nigerian and the United States (Bolarinwa et al., 2012; Drawz et al., 2016). These differences likely reflect the lack of appropriate care of SCD or the manifestation of the most severe SCD phenotype in Cameroon. The low proportion of macro-albuminuria found in this study is distinct from 15.1% reported in a cohort of SCD patients in the United Kingdom (UK) (Brewin et al., 2017). This could be due to the difference in age structure between the two cohorts. This study replicated a positive association of crude-albuminuria with increasing age as presented by a multi-center African study (Ranque et al., 2014) and a Nigeria-based study (Brewin et al., 2017). Some haematological variables, such as MCV and Hb level (Table 3), influence crude albuminuria among SCD patients from Cameroon, Hb level observed to be protective against micro- albuminuria. This is in accordance with studies in Jamaica and the United States (Aban et al., 2017; Niss et al., 2020) which revealed that lower concentration of Hb is associated with development of micro-albuminuria, leading to relative renal ischemia, ischemia-reperfusion injury, and increased medullary sickling (Aban et al., 2017). BMI provides the highest mean variation explained for crude-albuminuria and may be a major anthropometric factor leading to renal dysfunction. This is likely related to the nutrition of SCD patients who struggle to maintain an adequate quality of life. We have not observed any significant association of crude-albuminuria with WBC counts or BP as previously observed in a Jamaican cohort (Asnani et al., 2016).
The prevalence of glomerular hyperfiltration was similar to the 76% found among SCD children in the United States (Aygun et al., 2011), but higher than that previously reported in France (51%) (2010) (Haymann et al., 2010) and DRC (2017) (Aloni et al., 2017). It was lower than the 98% presented in the United Kingdom (Drawz et al., 2016). These differences may be explained by the variability in the median ages of participants in the other studies; they may also be due to the method used to calculate eGFR, as the Schwartz formula tends to underestimate GFR in children compared to the CKD-EPI used in the present study. Unlike the studies of Asnani in Jamaica (Aban et al., 2017) and Vazquez in the United States (Aygun et al., 2011), no significant associations between eGFR and crude albuminuria were identified in this study. However, 41 and 2% of patients had glomerular hyperfiltration associated with micro-albuminuria and proteinuria, respectively. These results are different from the values highlighted by Haymann et al. (2010) and from the 22% of patients who had both glomerular hyperfiltration and micro-albuminuria in DRC (Aloni et al., 2017). The eGFR was highly associated with haemoglobin, HbF and SBP, as previously observed in patients from the United Kingdom (Wu et al., 2009). The strong association between SBP and eGFR is also consistent with data reported in the study by Niss et al. (2020) in Jamaica confirming the highly protective role of stable SBP against glomerular hyperfiltration (Table 3). VOC frequencies are also highly correlated to eGFR, explaining the morbidity due to renal dysfunction among SCD patients in Cameroon. Associations between eGFR with Hb level and WBC (granulocytes and lymphocytes) were previously highlighted by Aban et al. (2017), revealing that a low Hb level and increased WBC are associated with renal failure. The correlations between eGFR with age, BMI and creatinine were observed in patients from the United Kingdom (Drawz et al., 2016), Jamaica (Eke et al., 2012), Nigeria (Ajite et al., 2019), and the United States (Becker et al., 2014). However, age and creatinine were not confirmed by this study as they were confounding factors, mixing up with eGFR. This suggests that previous observations were biased, and results obtained by these studies may be flawed. Our analysis agreed with publications within the Taiwanese (Chang et al., 2018) and Caucasian (Brown et al., 2012) which found no association between BMI and renal dysfunctions.
Results highlighted that only three replicated genetic variants are associated to renal dysfunction: A1CF-rs10994860 with reduced risk of renal dysfunction for two allele changes and a protective effect against prevalent glomerular hyperfiltration; It confers about 2 times more protection to SCD individuals with one allele change or heterozygous genotype as compared to a patient with no copy, and explaining only 0.18% of status change to the glomerular hyperfiltration or renal dysfunction. However, SYPL2-rs12136063 as well as APOL1 (G1)-rs73885319 conferred increased risk of progressing to the renal dysfunction or to prevalent glomerular hyperfiltration, explaining 11.04 and 0.97% of status changes, respectively. Like eGFR, three gene variants are also identified for crude-albuminuria: WNT7A-rs6795744, TMEM60-rs6465825, and APOL1 (G2)-rs71785313 providing a protective effect against prevalent micro-albuminuria (Table 3), explaining less than 4% of status changes. These relative low contributions to status changes suggests that the targeted SNPs may not be relevant to the ancestral African populations, or it is possible SCD specific kidney dysfunctions associated variants are still to be found. Indeed, there is bias in polygenic risk scores (PRSs) regarding usability, and transferability for complex trait, as most PRSs do not account for multiple alleles that are either limited or of high frequency among Africans, due highest genomic variations (Gurdasani et al., 2019). A genome-wide association study (GWAS) on genetic susceptibility to type 2 diabetes (T2D) identified a previously unreported African-specific significant locus, while showing transferability of 32 established T2D loci (Adeyemo et al., 2019). Alternatively, kidney dysfunction in SCD may be mostly driven by the pathophysiology of the disease itself rather than genetic factors. For eGFR, SBP explained this status changes by nearly 40.55%, followed by the number of VOC (21.73%), Hb (16.55%), and the upper out range level of HbF would explain the status changes for nearly 1.13%. The HbF level distribution results indicate that HbF levels vary with β-globin gene cluster haplotypes, as well as with α-globin gene deletions in agreement with the current knowledge. The HbF level in patients with Benin haplotyte and double 3.7 kb α-globin gene deletions is significantly higher as compared to the HbF level cutoff observed to improve the patient survival. Thus, these two genetic events may confer a relatively favorable clinical manifestation (Steinberg, 2009; Bitoungui et al., 2015) and patients with double 3.7 kb α-globin gene deletions are about 3 times less likely to progress to the glomerular hyperfiltration or kidney failure state as compared to a patient with no or one deletion (odds ratio = 0.36320). For crude-albuminuria, the status change is mainly explained by BMI with approximately 45.34% of variations, followed by MCV with 1.22%, transfusion (0.23%), and Hb (0.19%).
The first limitation of the present study is the cross-sectional design. A longitudinal study would give more precise data on kidney dysfunction in SCD in Cameroon. Another limitation is the use of the CKD-EPI-creatinine equation to estimate GFR. Recent reports indicate that the CKD-EPI original formula overestimates GFR values. The reliability of the CKD-EPI equation was recently adjusted by inclusion of a molecular weight protein, Cystatin C (CysC), which is eliminated exclusively by glomerular filtration (Yee et al., 2017). However, the protein was not quantified for this cohort. The imbalanced distribution of individuals without kidney dysfunction in this group of SCD patients likely affects the performance of the different regression models (Jedrzejowicz et al., 2018), tending to be biased toward the normal ranges (KrishnaVeni and Sobha, 2011) and potentially failing to identify possible signals.
This study has replicated APOL1 gene variants: (G1)-rs73885319 and (G2)-rs71785313, shown to be strongly associated with renal dysfunction in SCD patients, as well as A1CF-rs10994860, SYPL2-rs12136063, WNT7A-rs6795744, and TMEM60-rs6465825 in Cameroonian SCD patients. Though the protein-protein interaction network and enrichment analyses have revealed the sub-network, which may influence extreme phenotype levels, enriched with the cartilage condensation process, which likely contributes to the development of VOC complications and possibly to the renal dysfunction, these gene variants only explain a small proportion of status changes. The results also suggest that haematological indices, clinical events, anthropometric and socio-demographic variables, are major contributors to the pathophysiology of kidney dysfunction in SCD. This elicits the need for further research to investigate new genetic biomarkers which account for kidney dysfunction risk factors in SCD patients in the African setting.
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
The studies involving human participants were reviewed and the study was performed following the Declaration of Helsinki. This study was approved by the Faculty of Health Sciences Human Research Ethics Committee of the University of Cape Town, South Africa (HREC REF: 661/2015), and the National Ethics Committee of the Ministry of Public Health, Yaoundé, Republic of Cameroon (No. 193/CNE/SG/10). Patients older than 18 years self-consented into the study and informed consent was given by the parents or guardians for participants younger than 18 years old with a requirement for children older than 7 years to also sign assent forms. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
AW, VN-B, KM, TM, and GA conceived and designed the experiments. VN-B, SB, KM, and TM performed the experiments. VN-B, SB, RE, FN-S, TM, KM, and AW needed patient recruitment, samples, and clinical data collection. GM, VN-B, VN, KM, and AW processed and analysed the data. AW, GA, GM, and VN contributed to reagents, materials, and analysis tools. VN-B, KM, GM, and AW wrote the manuscript. GM, VN-B, SB, KM, TM, RE, VN, FN-S, GA, and AW revised and approved the manuscript. All the authors approved the manuscript and agreed to be accountable for all aspects of the presented work.
The data collection, molecular experiments and analysis of the study were supported by following Awards: Welcome Trust, Developing Excellence in Leadership, Training and Science (DELTAS) Africa, Award 107755Z/15/Z to GA, and AW; the NIH/H3Africa, U54HG009790 and U01HG009716 to AW, and NIH/NHLBI U24HL135600 to AW. VN-B was supported by a WACCBIP DELTAS postdoctoral fellowship (DEL-15-007: Awandare). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.595702/full#supplementary-material
SCD, sickle cell disease; eGFR, estimated glomerular filtration rate; VOC, vaso-occlusive crisis; GWAS, genome-wide association studies; BMI, Body mass index; BP, blood pressure; HbF, foetal haemoglobin; HBB, beta-globin gene; HBA, alpha-globin gene; PCR, polymerase chain reaction; RFLP, Restriction fragment length polymorphism; WBC, white blood cell; Hb, haemoglobin; MCV, mean corpuscular volume; CKD-EPI, Chronic Kidney Disease-Epidemiology.
Aban, I., Baddam, S., Hilliard, L. M., Howard, T. H., Feig, D. I., and Lebensburger, J. D. (2017). Severe anemia early in life as a risk factor for sickle-cell kidney disease. Blood 129, 385–387. doi: 10.1182/blood-2016-09-738104
Adekile, A. (2020). The genetic and clinical significance of fetal hemoglobin expression in sickle cell disease. Med. Princ. Pract. doi: 10.1159/000511342 [Epub ahead of print].
Adeyemo, A. A., Zaghloul, N. A., Chen, G., Doumatey, A. P., Leitch, C. C., Hostelley, T. L., et al. (2019). ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Nat. Commun. 10, 3195.
Ajite, A., Ogundare, E., Oluwayemi, O., Olatunya, O., Oke, O., Tolorunju, K., et al. (2019). The pattern of blood pressure and renal function among children with Sickle cell anaemia presenting in a tertiary health institution in Nigeria. J. Clin. Nephrol. 3, 083–092. doi: 10.29328/journal.jcn.1001031
Aloni, M. N., Mabidi, J.-L. L., Ngiyulu, R. M., Ekulu, P. M., Mbutiwi, F. I., Makulo, J. R., et al. (2017). Prevalence and determinants of microalbuminuria in children suffering from sickle cell anemia in steady state. Clinical Kidney Journal 10, 479–486. doi: 10.1093/ckj/sfx058
Anto, E. O., Obirikorang, C., Acheampong, E., Adua, E., Donkor, S., Afranie, B. O., et al. (2019). Renal abnormalities among children with sickle cell conditions in highly resource-limited setting in Ghana. PLoS One. 14:e0225310. doi: 10.1371/journal.pone.0225310
Asnani, M., Serjeant, G., Royal-Thomas, T., and Reid, M. (2016). Predictors of renal function progression in adults with homozygous sickle cell disease. British journal of haematology 173, 461–468. doi: 10.1111/bjh.13967
Ataga, K. I., Derebail, V. K., and Archer, D. R. (2014). The glomerulopathy of sickle cell disease. American journal of hematology 89, 907–914.
Audard, V., Bartolucci, P., and Stehle, T. (2017). Sickle cell disease and albuminuria: recent advances in our understanding of sickle cell nephropathy. Journal of Clinical Nephrology 10, 475–478. doi: 10.1093/ckj/sfx027
Aygun, B., Mortier, N. A., Smeltzer, M. P., Hankins, J. S., and Ware, R. E. (2011). Glomerular hyperfiltration and albuminuria in children with sickle cell anemia. Pediatric nephrology 26, 1285–1290. doi: 10.1007/s00467-011-1857-2
Becker, A. M., Goldberg, J. H., Henson, M., Ahn, C., Tong, L., Baum, M., et al. (2014). Blood pressure abnormalities in children with sickle cell anemia. Pediatric blood & cancer 61, 518–522. doi: 10.1002/pbc.24843
Bitoungui, V. J., Pule, G. D., Hanchard, N., Ngogang, J., and Wonkam, A. (2015). Beta-globin gene haplotypes among cameroonians and review of the global distribution: is there a case for a single sickle mutation origin in Africa? Omics 19, 171–179. doi: 10.1089/omi.2014.0134
Bolarinwa, R. A., Akinlade, K. S., Kuti, M. A., Olawale, O. O., and Akinola, N. O. (2012). Renal disease in adult Nigerians with sickle cell anemia: a report of prevalence clinical features and risk factors. Saudi journal of kidney diseases and transplantation 23, 171–175.
Brewin, J., Tewari, S., Hannemann, A., Al-Balushi, H., Sharpe, C., Gibson, J. S., et al. (2017). Early markers of sickle nephropathy in children with sickle cell anemia are associated with red cell cation transport activity. HemaSphere 1:11e2.
Brown, R. N. K. L., Mohsen, A., Green, D., Green, D., Hoefield, R. A., Summers, L. K. M., et al. (2012). Body mass index has no effect on rate of progression of chronic kidney disease in non-diabetic subjects. Nephrol. Dial. Transplant. 27, 2776–2780. doi: 10.1093/ndt/gfr757
Chambers, J. C., Zhang, W., Lord, G. M., Van der Harst, P., Lawlor, D. A., Sehmi, J. S., et al. (2010). Genetic loci influencing kidney function and chronic kidney disease. Nat. Genet. 42, 373–375.
Chang, T. J., Zheng, C. M., and Wu, M. Y. (2018). Relationship between body mass index and renal function deterioration among the Taiwanese chronic kidney disease population. Sci. Rep. 8:6908.
Coleman, E., and Inusa, B. (2007). Sickle cell anemia: targeting the role of fetal hemoglobin in therapy. Clin. Pediatr. (Phila.) 46, 386–391. doi: 10.1177/0009922806297751
Crawford, D. C., Caggana, M., Harris, K. B., Lorey, F., Nash, C., Pass, K. A., et al. (2002). Characterization of beta-globin haplotypes using blood spots from a population-based cohort of newborns with homozygous HbS. Genet. Med. 4, 328–335. doi: 10.1097/00125817-200209000-00003
Drawz, P., Ayyappan, S., Nouraie, M., Saraf, S., Gordeuk, V., Hostetter, T., et al. (2016). Kidney disease among patients with sickle cell disease haemoglobin SS and SC. Clin. J. Am. Soc. Nephrol. 11, 207–215. doi: 10.2215/cjn.03940415
Eke, C. B., Okafor, H. U., and Ibe, B. C. (2012). Prevalence and correlates of microalbuminuria in children with sickle cell anaemia: experience in a tertiary health facility in Enugu Nigeria. Int. J. Nephrol. 2021:240173.
Geard, A., Pule, G. D., Chetcha-Chemegni, B., Ngo Bitoungui, V. J., Kengne, A. P., Chimusa, E. R., et al. (2017). Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br. J. Haematol. 178, 629–639. doi: 10.1111/bjh.14724
Genovese, G., Friedman, D. J., Ross, M. D., Lecordier, L., Uzureau, P., Freedman, B. I., et al. (2010). Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845. doi: 10.1126/science.1193032
Gladwin, M. T. (2017). Cardiovascular complications in patients with sickle cell disease. Am. Soc. Hematol. Educ. Program 2017, 423–430. doi: 10.1182/asheducation-2017.1.423
Gosmanova, E. O., Zaidi, S., Wan, J. Y., and Adams-Graves, P. E. (2014). Prevalence and progression of chronic kidney disease in adult patients with sickle cell disease. J. Investig. Med. 62, 804–807. doi: 10.1097/01.jim.0000446836.75352.72
Gurdasani, D., Barroso, I., Zeggini, E., and Manjinder, S. S. (2019). Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 20, 520–535. doi: 10.1038/s41576-019-0144-0
Hanchard, N., Elzein, A., Trafford, C., Rockett, K., Pinder, M., Jallow, M., et al. (2007). Classical sickle beta-globin haplotypes exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC Genet 8:52.
Haymann, J. P., Stankovic, K., Levy, P., Avellino, V., Tharaux, P. L., Letavernier, E., et al. (2010). Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin. J. Am. Soc. Nephrol. 5, 756–761. doi: 10.2215/cjn.08511109
Jedrzejowicz, J., Kostrzewski, R., Neumann, J., and Zakrzewska, M. (2018). Imbalanced data classification using MapReduce and relief. J. Inform. Telecommun. 2, 217–230. doi: 10.1080/24751839.2018.1440454
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K., and Tanabe, M. (2019). New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590—-D595.
Kottgen, A., Glazer, N. L., Dehghan, A., Hwang, S. J., Katz, R., Li, M., et al. (2009). Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 41, 712–717.
Kottgen, A., Pattaro, C., Boger, C. A., Fuchsberger, C., Olden, M., Glazer, N. L., et al. (2010). New loci associated with kidney function and chronic kidney disease. Nat. Genet. 42, 376–384.
KrishnaVeni, C. V., and Sobha, R. T. (2011). On the classification of imbalanced datasets international. J. Comput. Sci. Technol. 2, 145–148.
Lakkakula, B. V. K. S., Verma, H. K., Choubey, M., Patra, S., Khodiar, P. K., and Patra, P. K. (2017). Assessment of renal function in Indian patients with sickle cell disease. Saudi J. Kidney Dis. Transpl. 28, 524–531. doi: 10.4103/1319-2442.206440
Mammen, C., Bissonnette, M. L., and Matsell, D. G. (2017). Acute kidney injury in children with sickle cell disease—compounding a chronic problem. Pediatr. Nephrol. 32, 1287–1291. doi: 10.1007/s00467-017-3650-3
Mazandu, G. K., Chimusa, E. R., Rutherford, K., Zekeng, E. G., Gebremariam, Z. Z., Onifade, M. Y., et al. (2018). Large-scale data-driven integrative framework for extracting essential targets and processes from disease-associated gene data sets. Brief. Bioinform. 19, 1141–1152.
Mazandu, G. K., Hooper, C., Opap, K., Makinde, F., Nembaware, V., Thomford, N. E., et al. (2020). IHP-PING—generating integrated human protein–protein interaction networks on-the-fly. Brief. Bioinform. bbaa277. doi: 10.1093/bib/bbaa277 [Epub ahead of print].
Mi, H., Muruganujan, A., Ebert, D., Huang, X., and Thomas, P. D. (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D41–D426.
Nath, K. A., and Hebbel, R. P. (2015). Sickle cell disease: renal manifestations and mechanisms. Nat. Rev. Nephrol. 11, 161–171. doi: 10.1038/nrneph.2015.8
Niss, O., Lane, A., Asnani, M. R., Yee, M. E., Raj, A., Creary, S., et al. (2020). Progression of albuminuria in patients with sickle cell anemia: a multicenter, longitudinal study. Blood Adv. 4, 1501–1511. doi: 10.1182/bloodadvances.2019001378
Pattaro, C., Kottgen, A., Teumer, A., Garnaas, M., Boger, C. A., Fuchsberger, C., et al. (2012). Genome-wide association and functional follow-up reveals new loci for kidney function. PLoS Genet. 8:e1002584.
Pattaro, C., Teumer, A., Gorski, M., Chu, A. Y., Li, M., Mijatovic, V., et al. (2016). Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun. 7:10023.
Piel, F. B., Patil, A. P., Howes, R. E., Nyangiri, O. A., Gething, P. W., Dewi, M., et al. (2013). Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 381, 142–151. doi: 10.1016/s0140-6736(12)61229-x
Piel, F. B., Steinberg, M. H., and Rees, D. C. (2017). Sickle cell disease. N Engl J Med. 376, 1561–1573.
Platt, O. S., Brambilla, D. J., Rosse, W. F., Milner, P. F., Castro, O., Steinberg, M. H., et al. (1994). Mortality in sickle cell disease life expectancy and risk factors for early death. N. Engl. J. Med. 330, 1639–1644. doi: 10.1056/nejm199406093302303
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Ranque, B., Menet, A., Diop, I. B., Thiam, M. M., Diallo, D., Diop, S., et al. (2014). Early renal damage in patients with sickle cell disease in sub-Saharan Africa: a multinational prospective cross-sectional study. Lancet Haematol. 1, e64–e73.
Rumaney, M. B., Ngo Bitoungui, V. J., Vorster, A. A., Ramesar, R., Kengne, A. P., Ngogang, J., et al. (2014). The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One 9:e100516. doi: 10.1371/journal.pone.0100516
Saiki, R. K., Scharf, S., Faloona, F., Mullis, K. B., Horn, G. T., Erlich, H. A., et al. (1985). Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230, 1350—-1354.
Saraf, S. L., Shah, B. N., Zhang, X., Han, J., Tayo, B. O., Abbasi, T., et al. (2017). APOL1 alpha-thalassemia and BCL11A variants as a genetic risk profile for progression of chronic kidney disease in sickle cell anemia. Haematologica 102, e1–e6.
Saraf, S. L., Zhang, X., Shah, B., Kanias, T., Gudehithlu, K. P., Kittles, R., et al. (2015). Genetic variants and cell-free haemoglobin processing in sickle cell nephropathy. Haematologica 100, 1275–1284. doi: 10.3324/haematol.2015.124875
Schaefer, B. A., Flanagan, J. M., Alvarez, O. A., Nelson, S. C., Aygun, B., Nottage, K. A., et al. (2016). Genetic modifiers of white blood cell count albuminuria and glomerular filtration rate in children with sickle cell anemia. PLoS One 11:e0164364. doi: 10.1371/journal.pone.0164364
Steinberg, M. H. (2009). Genetic etiologies for phenotypic diversity in sickle cell anemia. Sci. World J. 9, 46–67. doi: 10.1100/tsw.2009.10
The Gene Ontology Consortium (2019). The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338.
Tzur, S., Rosset, S., Shemer, R., Selig, S., Tarekegn, A., Bekele, E., et al. (2010). Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum. Genet. 128, 345–350. doi: 10.1007/s00439-010-0861-0
Weatherall, D. J., and Clegg, J. B. (2001). Inherited haemoglobin disorders: an increasing global health problem. Bul. World Health Organ. 79, 704–712.
Wonkam, A., Ngo Bitoungui, V. J., Vorster, A. A., Ramesar, R., Cooper, R. S., Tayo, B., et al. (2014). Association of variants at BCL11A and HBS1L-MYB with haemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PLoS One 9:e92506. doi: 10.1371/journal.pone.0092506
Wu, J., Vallenius, T., Ovaska, K., Westermarck, J., Mäkelä, T. P., and Hautaniemi, S. (2009). Integrated network analysis platform for protein-protein interactions. Nat. Methods 6, 75–77. doi: 10.1038/nmeth.1282
Yates, A. D., Achuthan, P., Akanni, W., Allen, J., Allen, J., Alvarez-Jarreta, J., et al. (2020). Ensembl 2020. Nucleic Acids Res. 48, D682–D688.
Keywords: sickle cell disease, kidney dysfunctions, gene variants, cameroon, Africa
Citation: Ngo-Bitoungui VJ, Belinga S, Mnika K, Masekoameng T, Nembaware V, Essomba RG, Ngo-Sack F, Awandare G, Mazandu GK and Wonkam A (2021) Investigations of Kidney Dysfunction-Related Gene Variants in Sickle Cell Disease Patients in Cameroon (Sub-Saharan Africa). Front. Genet. 12:595702. doi: 10.3389/fgene.2021.595702
Received: 17 August 2020; Accepted: 19 February 2021;
Published: 15 March 2021.
Edited by:
Maria G. Stathopoulou, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Leon Mutesa, University of Rwanda, RwandaCopyright © 2021 Ngo-Bitoungui, Belinga, Mnika, Masekoameng, Nembaware, Essomba, Ngo-Sack, Awandare, Mazandu and Wonkam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ambroise Wonkam, YW1icm9pc2Uud29ua2FtQHVjdC5hYy56YQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.