 Haixia Zhong1†
Haixia Zhong1† Fuchun Zhang1†Xiaoming Zhou1Mingqi Pan1Juan Xu1Jingzhe Hao2Shouan Han1Chuang Mei1He Xian3Min Wang1Jiahui Ji1Wei Shi4,5*Xinyu Wu1*
Fuchun Zhang1†Xiaoming Zhou1Mingqi Pan1Juan Xu1Jingzhe Hao2Shouan Han1Chuang Mei1He Xian3Min Wang1Jiahui Ji1Wei Shi4,5*Xinyu Wu1*- 1Institute of Horticulture Crops, Xinjiang Academy of Agricultural Sciences, Urumqi, China
- 2Research Institute of Plant Protection, Xinjiang Academy of Agricultural Sciences, Urumqi, China
- 3Comprehensive Experiment Station of Xinjiang Academy of Agricultural Sciences, Urumqi, China
- 4State Key Laboratory of Desert and Oasis Ecology, Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences, Urumqi, China
- 5Turpan Eremophytes Botanic Garden, Chinese Academy of Sciences, Turpan, China
The Munake grape cultivar produces uniquely flavored and high-quality fruits. Despite the numerous beneficial agronomic traits of Munake, there are few genetic resources available for this cultivar. To address this knowledge gap, the entire genome was sequenced using whole-genome sequencing approaches and compared with a Vitis vinifera L. reference genome. This study describes more than 3 million single nucleotide polymorphism (SNP), 300,000 insertion and deletion (InDel), 14,000 structural variation (SV), and 80,000 simple sequence repeat (SSR) markers (one SSR per 4.23 kb), as well as their primers. Among the SSRs, 44 SSR primer pairs were randomly selected and validated by polymerase chain reaction (PCR), allowing discrimination between the different Munake cultivar genotypes. The genetic data provided allow a deeper understanding of Munake cultivar genomic sequence and contribute to better knowledge of the genetic basis behind its key agronomic traits.
Introduction
In recent years, genomic studies on grapevine cultivars have provided a significant quantity of genomic information, including evolution and domestication processes (Zhou et al., 2019), molecular markers and transferability (Li et al., 2017; Zou et al., 2020), sex determination (Massonnet et al., 2020), and population genetics (Zhou et al., 2019). Furthermore, V. vinifera cultivar genomic information has been utilized as evidence to support ancestral hexaploidization in major angiosperm phyla. This information has been applied both to scientific research and biotechnological development of new grapevine cultivars. China is one of the largest grape-producing countries in the world, with a cultivation area of 809,600 hm2 that mainly produces table grapes. There are many table grape, high quality grapevine cultivars with a long history of cultivation in China, in particular the Munake cultivar, which is native to Xinjiang Province (Zhang et al., 2010). This cultivar germinates in early April, blossoms in mid-May, and ripens in early October. It produces uniquely flavored, thin-skinned, sweet fruits with a crispy texture (Pang et al., 2015; Keriman et al., 2016; Xu et al., 2018; Supplementary Figure 1), with a berry weight of 5 to 10 g and grape cluster weight from 600 to 1,000 g.
Several unsuccessful attempts have been made to introduce the Munake cultivar to other Chinese provinces. With the exception of a few areas in the Hexi Corridor of Gansu province, it is mainly cultivated and narrowly distributed around the Atushi area of Xinjiang province. This is because the Munake cultivar requires particular weather conditions to grow, such as an average temperature of 12 to 13°C, a frost-free period of more than 220 days, an effective accumulated temperature greater than 4,200°C, and less than 120 mm rainfall per year (Keriman et al., 2016). Despite its numerous, desirable agronomic traits, this cultivar is not widely grown (Biagini et al., 2016; Zhang et al., 2016), creating challenges associated with its storage and transport (Yang et al., 2009; Zhang et al., 2016; Xu et al., 2018). Breeding programs thus aim to transfer the beneficial agronomic traits of the Munake cultivar into other hardier cultivars, or breed a more climatically versatile Munake cultivar to overcome the storage and transport problems. However, there are currently few genetic resources available for this cultivar, which hinders such breeding programs.
The completion of genome sequencing of the Munake cultivar is necessary for comparative and functional genomics studies of table grapes, and the resulting genetic and genomic data will be a valuable resource for Munake cultivar breeding programs (Zhang et al., 2017; Karastan et al., 2018; Minio et al., 2019). This study describes whole-genome sequencing of the Munake cultivar and compares it with a V. vinifera L. reference genome (Jaillon et al., 2007). A significant number of effective DNA markers were developed for exploring agronomic trait-related genes by comparing the sequences obtained with the reference genome of V. vinifera, as well as some specific simple sequence repeat (SSR) markers. The SSR markers have the advantages of accurate and rapid cultivar detection even with the existence of co-dominance, multiple alleles, high polymorphism, and variability, and hence can be used for species identification, establishment of well-known varieties of germplasm resources, and determination of differences between varieties (Patzak et al., 2019; Prysiazhniuk et al., 2019). The knowledge gained will create a large tag library and the foundation for future genetic studies, enabling molecular breeding and species resource protection of the Munake cultivar and its relatives.
Materials and Methods
Sample Preparation, DNA Isolation, and Genome Sequencing
A Munake cultivar sample was selected from its primary production area of Artux City, Xinjiang, China. Genomic DNA was extracted from young leaves using the modified cetyl trimethylammonium bromide method (Doyle and Doyle, 1990). The extracted DNA was randomly iron fragmented at 95°C, and 350-bp fragments were gel-purified. These fragments were used to construct DNA libraries by cutting the genomic DNA using a Covaris M220 Focused-ultrasonicator. The HiSeq X-Ten platform (Illumina, Inc., San Diego, CA, United States) was used for adaptor ligation and DNA cluster preparation. Only high-quality data were used for mapping by filtering the low-quality reads (single reads <20 bp)1, in order to reduce the error probability in the mapping process. The clean reads were used for subsequent analyses and were stored in the National Center for Biotechnology Information database under the BioProject accession number PRJNA632683.
Mapping Reads to the Referenced Genome
The Burrows-Wheeler alignment (BWA) software was used to align the paired-end (PE) sequences of the Munake cultivar to V. vinifera reference genomes2 based on default parameters (Li and Durbin, 2010; Table 1). Reads that could be aligned to more than one position in the referenced genome were filtered (Subbaiyan et al., 2012). The alignment results were used to calculate the average sequencing depth and coverage of mapped reads (Li et al., 2012). Single nucleotide polymorphisms (SNPs), insertions and deletions (InDels), and structural variation (SV) polymorphisms were detected in the mapped reads.

Table 1. Summary of original re-sequencing data of Munake cultivar.
Detection of SNPs, InDels, and SVs
Variants were identified using the Unified Genotyper application of the Genome Analysis Toolkit (DePristo et al., 2011), programmed to detect SNPs and InDels. Based on the nucleotide substitutions, the SNPs detected were classified as transitions (Ti) (C/T and G/A) or transversions (Tv) (C/G, T/A, A/C, and G/T) (Table 2). SAM files generated by BWA were converted to BAM format using SAMtools (Li et al., 2009), duplicates were marked first, and local realignment and base recalibration were completed using the Picard tool to ensure correction of the clean reads. The Haplotype Caller method was then used to detect SNPs and InDels. All results were shown as gVCF files, which were prepared for joint-genotype analysis and reads filtering. The filtered parameters were: varFilter -w 5 -W 10 (vcfutils. pl), QUAL < 30, QD < 2.0, MQ < 40, and FS > 60.0 (Fisher test); the other parameters were executed using the default values.
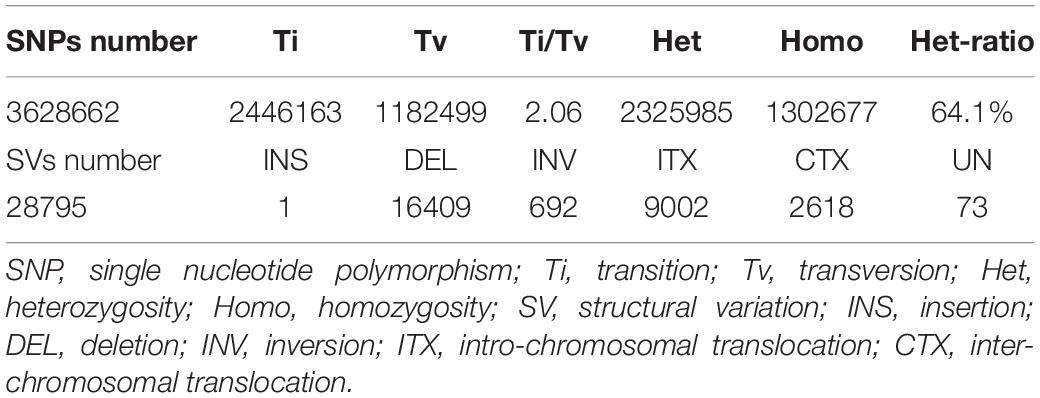
Table 2. The classification of nucleotide substitutions in the SNPs detected and summary of SVs identified in Munake cultivar aligned with the reference genomes.
SV detection was performed using the Breakdancer Max. pl software with default parameters (Chen et al., 2009). To obtain reliable SVs, the detected SVs must be returned to the PE alignments between the Munake cultivar and the reference genome. Six SV types were detected: insertion (INS), deletion (DEL), interchromosomal translocation (CTX), deletion including insertion, intrachromosomal translocation (ITX), and inversion (INV).
Annotation of SNPs
Single nucleotide polymorphisms were detected using the SnpEff software (Cingolani et al., 2012). The polymorphisms in the genes and other genomic regions were annotated as: Intergenic, Intragenic (without transcript information), Intron, Upstream, Downstream, Utr_5_prime (within 5′UTR), Utr_3_prime (within 3′UTR), Splice_site_acceptor (<2 bp, before the exon), Splice_site_donor (<2 bp, after exon), Splice_site_region (1–3 bp in an exon or 3–8 bp in an intron), Start_gained, Start_lost, and coding sequence region (CDS). SNPs in the CDS were separated into Synonymous_start, Non-_ synonymous_start, Synonymous_coding, Non-_synonymous _coding, Synonymous_stop, Stop_gained, and Stop_lost.
The positions of small InDels in the Munake cultivar were also located using the SnpEff software, and described using the same main polymorphism types as annotated for SNPs, except that the InDels in the CDS were separated into Start_Lost, Frame_Shift, Codon_Deletion, Exon_Deleted, Codon_Insertion, Codon_Change_Plus_Codon_Deletion, Codon_Change_Plus_Codon_Insertion, Stop_Gained, and Stop_Lost. The genic SVs were classified as either CDS, untranslated regions (UTR), or introns, according to their localization (Birney et al., 2004). The gene ontology (GO)/Pfam annotation data were further used to functionally annotate each gene, including non-synonymous SNPs −10 to 10-bp in length.
Designation of SSR Markers
In the Microsatellite identification tool3, clean reads were used to identify and localize microsatellite motifs. Only the SSRs, including mono-, di-, tri-, tetra-, penta-, and hexanucleotide motifs with numbers of uninterrupted repeat units exceeding 10, 7, 6, 5, and 4 were selected. The selected SSR loci for developing genetic markers should include a perfect repeat motif and two unique flanking sequences with 150 bp on each side of the repeat (Varshney et al., 2005). Primer 3 software4 was used to design unique flanking sequences with three parameters: (1) primer length ranging from 20 to 26 bases with an optimal size of 23 bp; (2) melting temperature between 58 and 63°C, with an optimum annealing temperature of 6 0°C; (3) a guanine-cytosine content of 40–60%, with an optimum content of 50%. To validate the new SSRs, a few primer pairs were chosen for polymerase chain reaction (PCR) amplification.
A DNA Engine thermal cycler (Bio-Rad, Hercules, CA, United States) was used for amplification with the following program: 94°C for 4 min; 35 cycles of 94°C for 45 s, 52°C for 1 min, and 72°C for 45 s; and a final extension at 72°C for 10 min. The PCR products were checked by electrophoresis in 1.5% agarose gels containing 1:20 GoldView in our lab and analyzed with fluorescent primers on ABI instrumentation (Shanghai SBS, Biotech Ltd., China), and photographed with a Photoprint 215 SD (Vilber Lourmat Co., Marne la Vallée, France).
PCR products were sequenced to obtain the fluorescence absorption peak pattern and peak value. GenALEx 6.5 was used to calculate the number of alleles, effective allele number, observed heterozygosity (Het), expected heterozygosity, Shannon’s index, primer polymorphism information at the locus level, and polymorphic information content (PIC) (Mahmood et al., 2019).
Results
Mapping of Re-sequencing Reads to the Reference Genome
A total of 111,846,066 short reads of 111 nucleotides (1.12 × 107 reads) were generated in this study. 1.06 × 108 short reads were successfully matched to V. vinifera (accession GCF_000003745.3 at the International Grape Genome Program and GenBank2). The average sequencing depth was approximately 22.71× coverage of the reference genome, and the resulting consensus sequences covered approximately 94.79% of the reference genome (Table 1). Among them, 5.59 × 107 PE reads and 5.01 × 107 single-end (SE) reads were mapped to chromosomes corresponding to 111.8 Mb of the reference genome (Table 1 and Figure 1). Although the percentage of sequenced reads from the Munake cultivar aligned with the reference genome was above 90%, the PE reads aligned with V. vinifera exceeded the SE reads. Furthermore, a total of 9.5 × 106 reads were uniquely (85.4%) mapped to the reference genome, and the remaining 1.63 × 107 reads were mapped to multiple locations (Table 1).
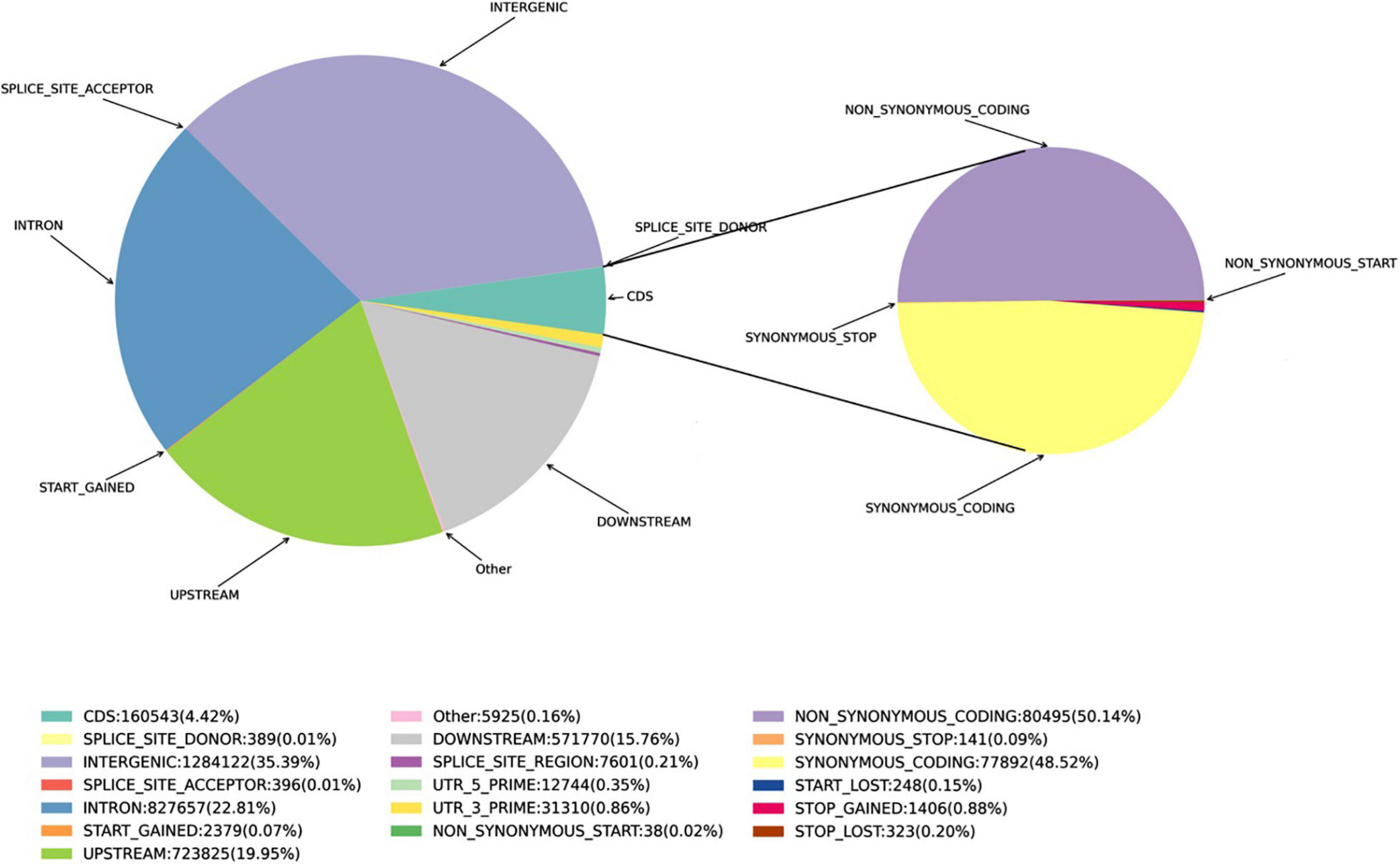
Figure 1. Proportions of the various SNPs in the Munake cultivar and the reference genome of V. vinifera.
Detection and Distribution of Variation
A total of 3,628,662 SNPs were detected, including 2,446,163 Ti, 1,182,499 Tv, and a 64.1% Het-ratio (Heterozygosity/homozygosity (Homo) × 100%). A total of 26,802 InDels of <10-bp in length were detected. This included 9,551 InDels in the CDS, and the main concentration of InDels were −1 and 1-bp in length both genome-wide and in the CDS. The proportion of SNPs is shown in Figure 1.
Among the variations detected, the highest numbers of SNPs (n = 397,613), InDels (n = 81,801), and SVs (n = 1,473) were observed in chromosome 14. Contrarily, the lowest numbers of SNPs (n = 216,848), InDels (n = 43,668), and SVs (n = 1,287) were observed in chromosome 17. The average density of detected polymorphisms was 7,462.2 SNPs per Mb (Table 2). The density of SNPs in the different chromosomes is shown in Figure 2. Chromosome 14 had the highest density of SNPs (3,251.7 SNPs per Mb), and chromosome 10 had the lowest SNP density (1,014.9 SNPs per Mb) (Table 2). The distribution of polymorphisms was uneven within chromosomes. All chromosomes comprised a mixture of dense and sparse SNP regions (Figure 2).
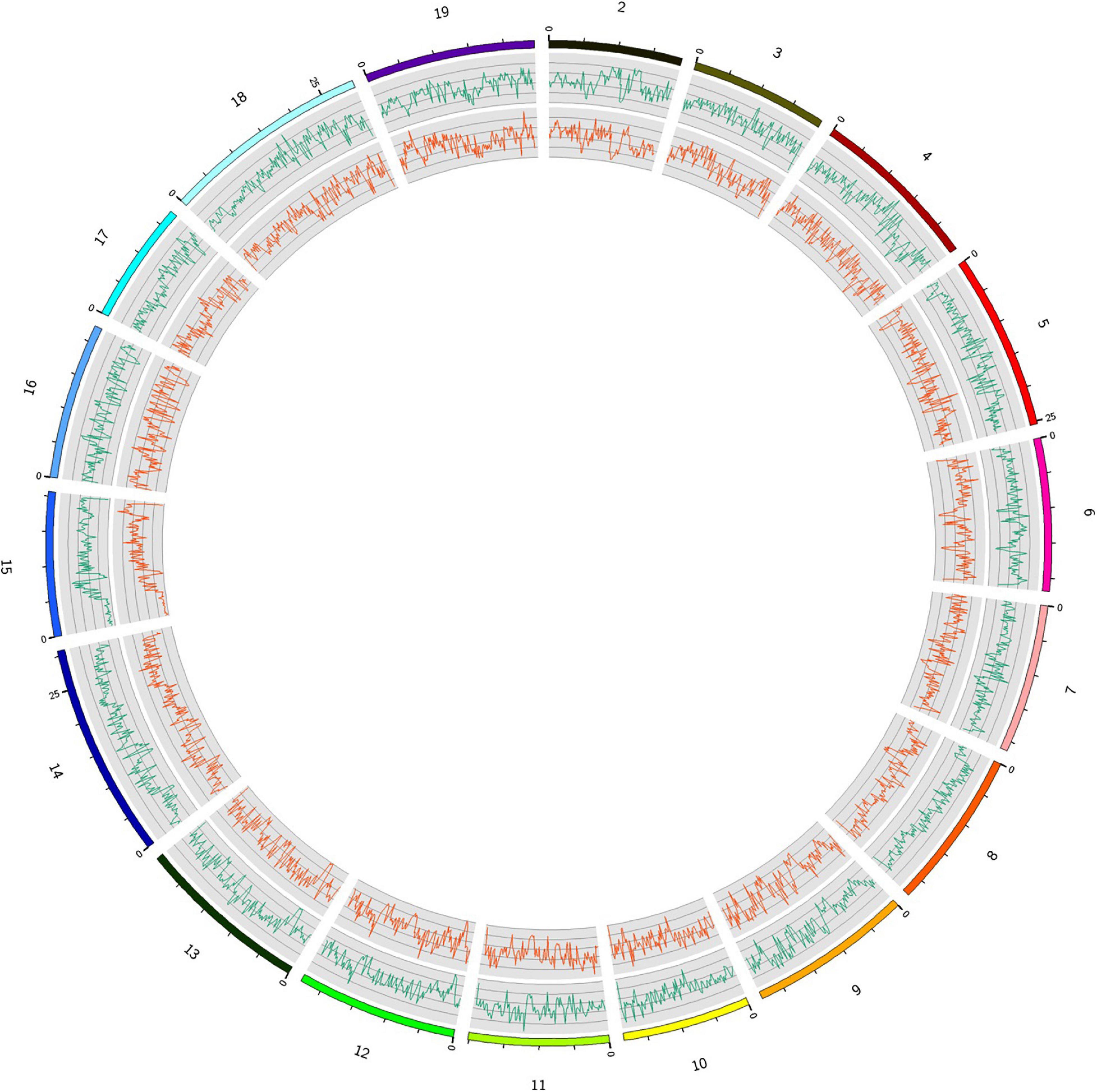
Figure 2. Distribution of SNPs and InDels detected in the Munake cultivar and the reference genome of V. vinifera L. in the 19 chromosomes (outside, green lines = number of SNPs; inside, red lines = InDels).
The distribution patterns of InDels and SVs densities were similar to those of SNPs (Table 2). The proportion of Ti was larger than that of Tv, with a Ti/Tv ratio of 2.06. A total of 772,885 InDels were detected, with the Genome-Deletion type (52.3%) more abundant than the Genome-Insertion type (47.7%), and the genome Het number (66.5%) was significantly higher than the genome Homo number (33.5%) (Figure 3).
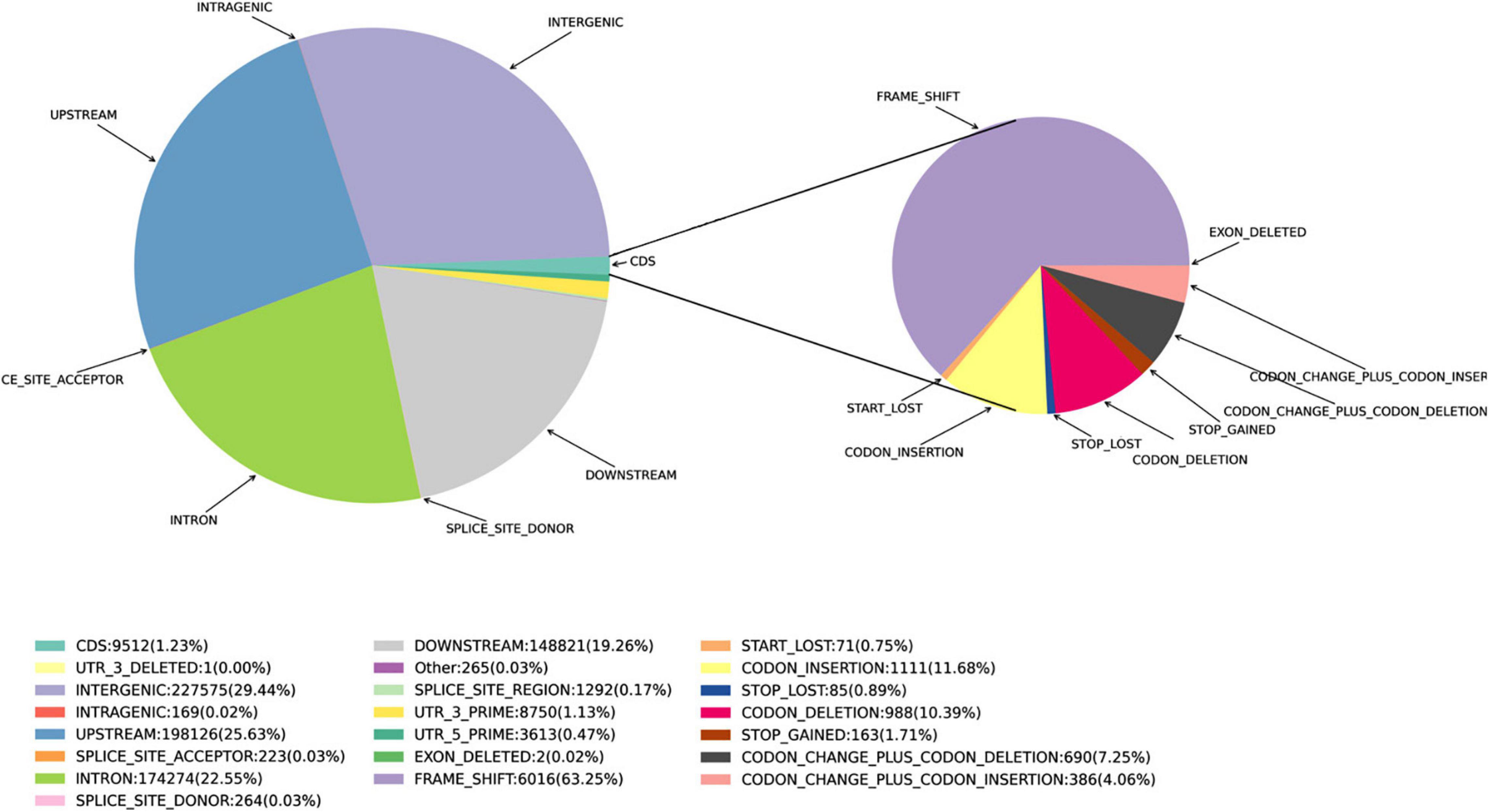
Figure 3. Proportions of the various types of InDels in the Munake cultivar and the reference genome of V. vinifera.
A total of 28,795 SVs were detected, including 16,409 DEL (57.0%), 9,002 ITX (31.3%), 692 INV (2.4%), and 2,618 CTX (9.1%), and one insertion (Table 2). The majority of DEL were less than 1,000 bp (65.6%), and 25.6% of them were between 1,000 and 10,000 bp. The largest DEL was 927,523 bp. Numerous ITXs were detected, at up to 984,059 bp in length, while most of them were less than 1,000 bp (79.6%). Although there were numerous SNPs (3,628,662 bp), InDels (n = 772,885), and SVs (n = 28,795) located in gene regions, only 160,543 SNPs, 9,512 InDels, and 9,798 SVs were found in the CDS (Figures 2, 3 and Table 2).
Annotation and Effect of SNPs
The ratio of non-synonymous-to-synonymous SNPs (1.01) was calculated based on all gene models. There were 6,828 InDels and 21,153 SNPs annotated according to the two gene functional databases (GO and Cluster of Orthologous Groups of proteins).
Using the GO database (Figure 4), the SNPs were classified as those involved in biological processes, cellular components, or molecular functions. The majority of SNPs (n = 3,067) appeared to be involved in a biological process, including reproduction, metabolic processes, cellular processes, and response to a stimulus. Many SNPs (n = 2,353) annotated as involved in a molecular function were detected, mainly annotated as having catalytic activity and involved in molecular binding. There were only 558 SNPs classified as being associated with a cellular component, in which the gene participates as a component of the cell, cell membrane, or organelle.
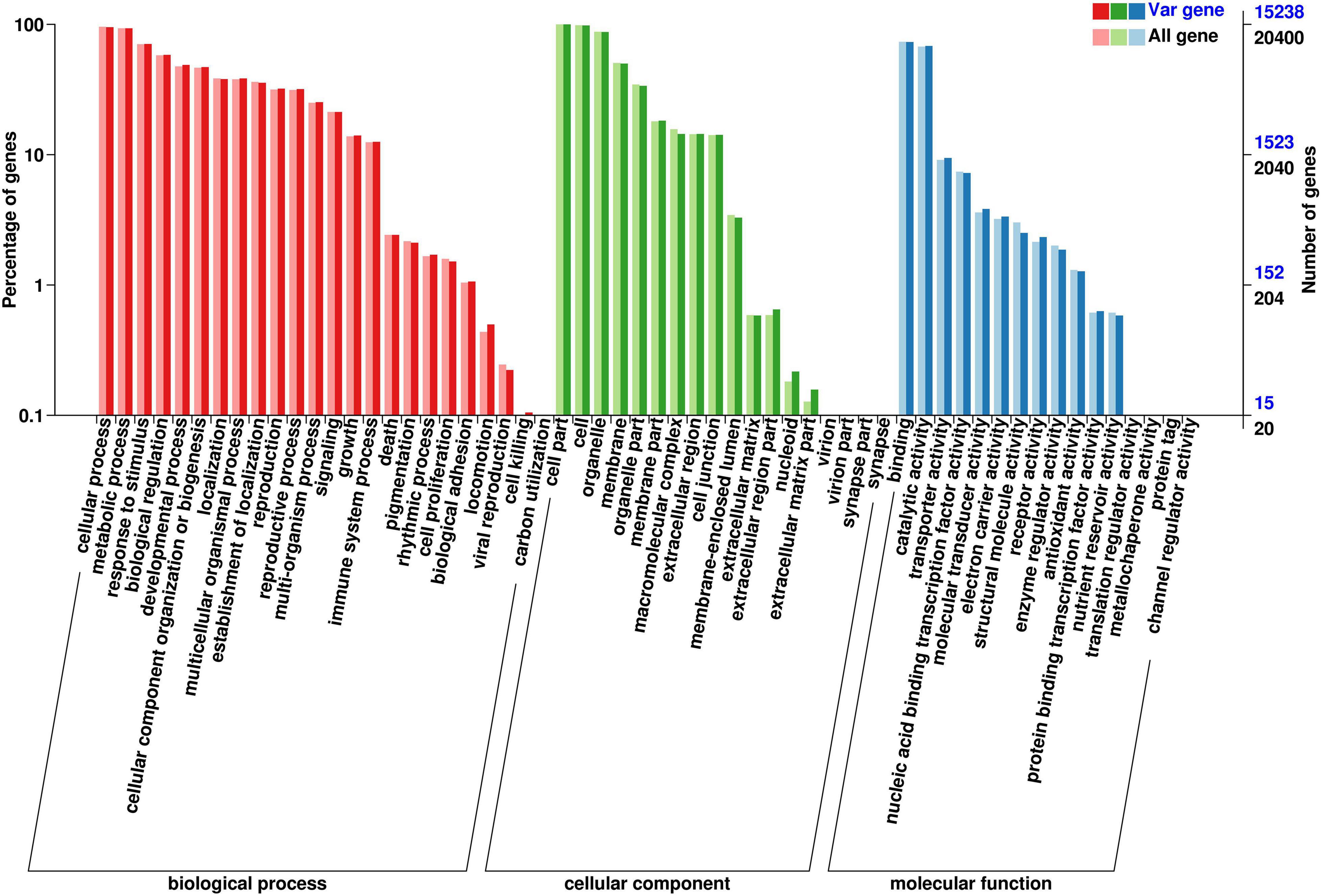
Figure 4. Genetic diversity of 44 genomic SSR loci in four or six Munake cultivar accessions.
According to the Cluster of Orthologous Groups of proteins database (Figure 5), the gene annotations were mainly concentrated on general function prediction only (n = 2,510), gene transcription (n = 1,287), and replication, recombination, and repair (n = 1,202). By comparing the SNPs in the Kyoto Encyclopedia of Genes and Genomes database (Figure 6), 2,733 were annotated, mainly referencing the plant hormone signal transduction (n = 146), RNA transport (n = 136), and oxidative phosphorylation (n = 125).
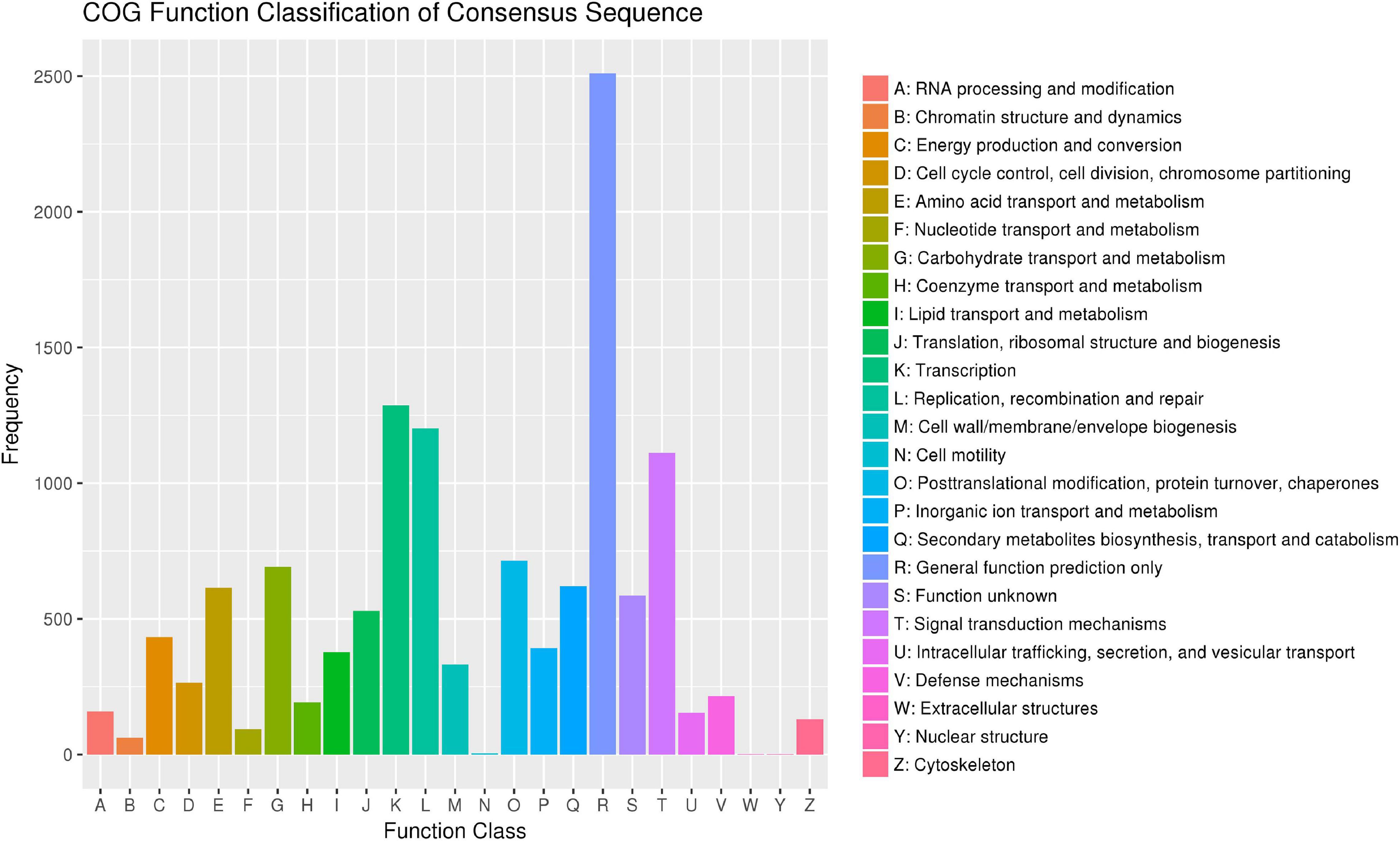
Figure 5. The SNPs of Munake grape annotated according to the COG database, the x-axis shows the taxonomical content of COG data. The y-axis shows the number of genes.
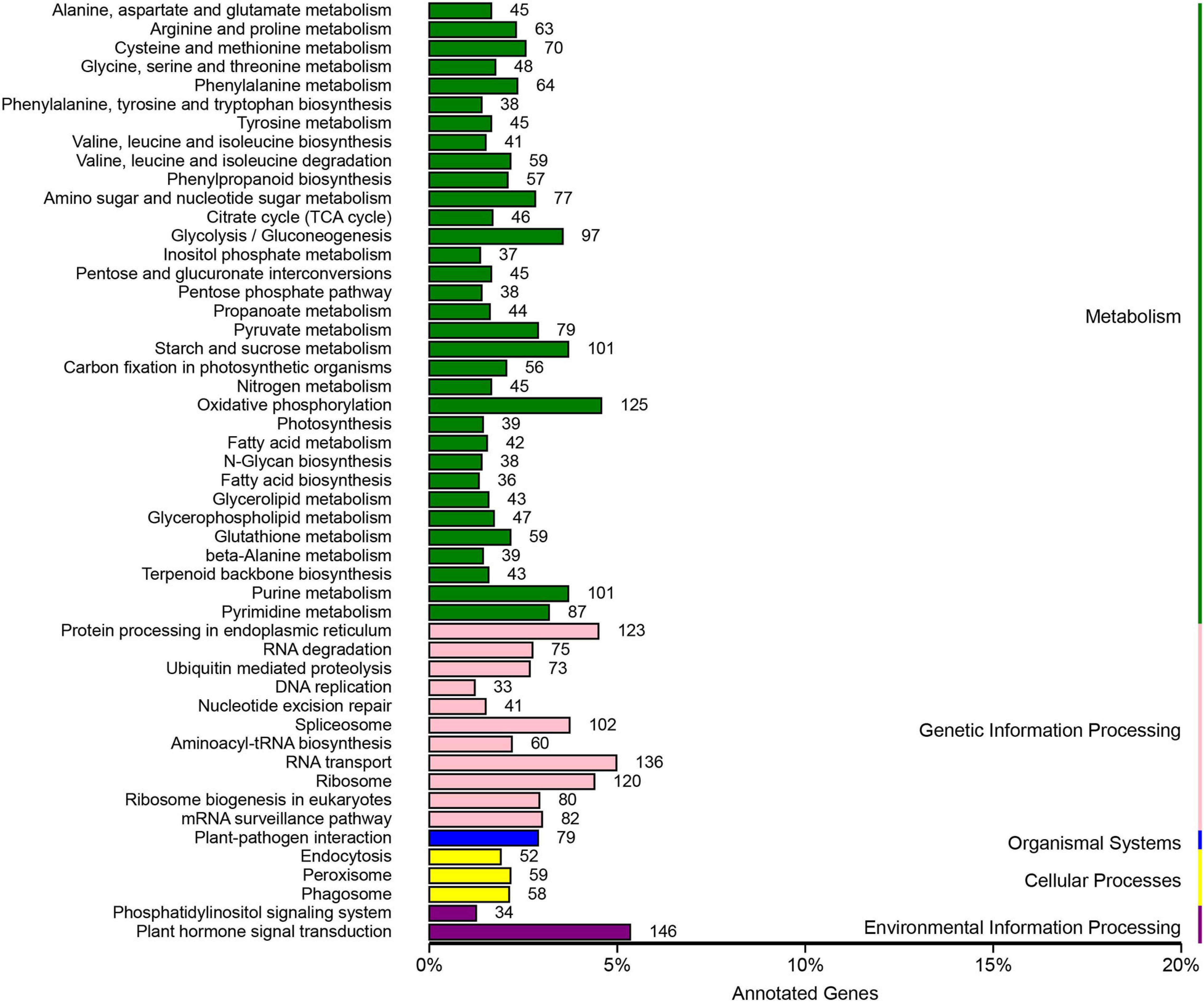
Figure 6. The SNPs of Munake grape annotated according to the KEGG database, the x-axis shows the taxonomical content of KEGG data. The y-axis shows the number of genes.
Development and Potential Applications of the New SSR Markers
A total of 87,872 SSRs with a genome frequency of one SSR per 4.32 kb were identified. Di-nucleotide repeats were the most abundant type followed by tri-nucleotide repeats. AG/CT was the most frequent di-nucleotide motif, while AAG/CTT was the most abundant (5.40%) tri-nucleotide motif.
Forty-four pairs of primers were designed based on the sequences flanking the SSRs to validate the markers by the PCR experiments (Supplementary Table S1), and clear bands were produced (Figure 7). Using 12 of the 44 pairs of SSR primers to analyze the genetic diversity of 57 Munake cultivar samples, a total of 168 alleles were detected with an average of 44 for each pair of primers. In this study, only MNG15, MNG18, and MNG1314 showed low variation (PIC < 0.25), MNG6666 and MNG9999 had medium variation (0.25 < PIC < 0.5), while in MNG03, MNG10, MNG14, MNG23, MNG26, MNG29, and MNG35 the degree of variation was high (PIC > 0.5). The PIC of MNG29 was the highest, reaching 0.842 (Table 3). The SSR markers have the advantages of codominance, multiple alleles, high polymorphism and variability, accurate and rapid detection, which can be used for species identification, establishment of well-known varieties germplasm resources and determination of differences between varieties.
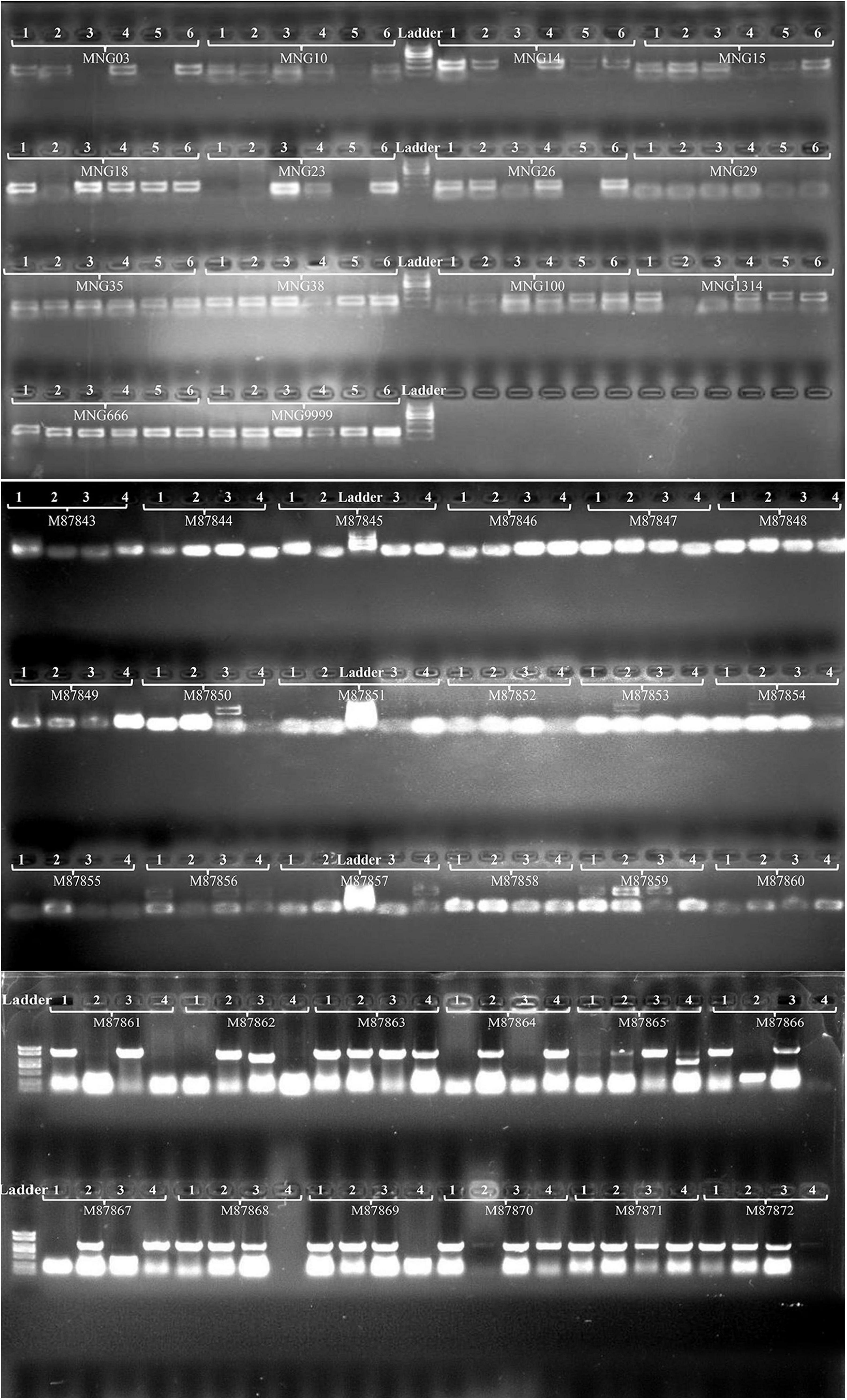
Figure 7. Genetic diversity of 44 genomic SSR loci in four or six Munake cultivar accessions.
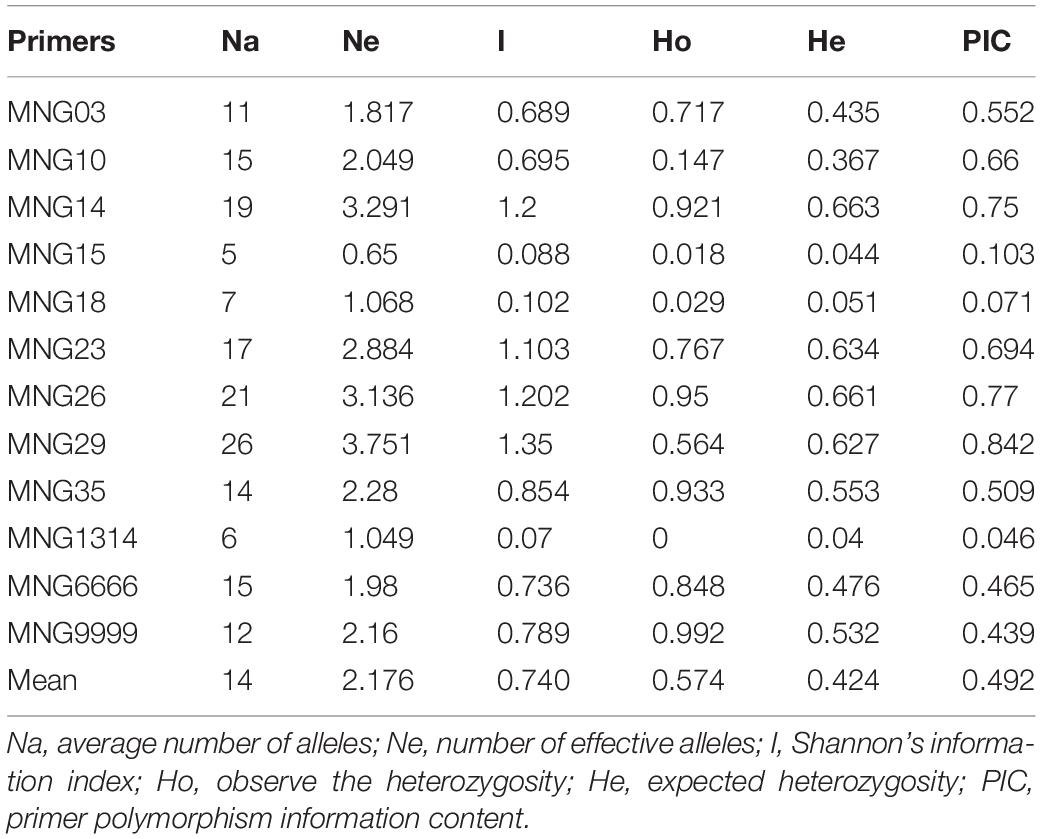
Table 3. Genetic diversity of 12 genomic simple sequence repeat SSR loci in 57 Munake cultivar samples.
Discussion
There are numerous genomic resources available for grapevines. Publication of the V. vinifera genome accelerated the genetic research of this valuable crop, including analyses of its genetic structure, construction of genetic maps, assessment of genetic diversity, detection of genotype/phenotype associations, and marker-assisted breeding (Chen et al., 2015; Mercati et al., 2016; Money et al., 2017). Due to its clonal mode of propagation and genomic complexity, there was limited genomic information available for grapevines before the publication of the V. vinifera reference genome (Lijavetzky et al., 2007; Guo et al., 2015; Xu et al., 2016, Zhou Y. et al., 2017). Massonnet et al. (2020) studied the structure and evolution of the sex-determining region in Vitis species and reported an improved, chromosome-scale Cabernet Sauvignon genome sequence. Zou et al. (2020) added shotgun genome sequences from 40 grapevine accessions to enable the identification of conserved core PCR primer binding sites flanking polymorphic haplotypes with high information content. From these target regions, Zou et al. developed 2,000 rhAmpSeq markers as a PCR multiplex and validated the panel in four biparental populations, spanning the diversity of the Vitis genus and showing transferability increases to 91.9%. Munake grape, as a specific cultivars with high superior for table (Wu et al., 2011), were still kept in nothing referring its genomic studies (Guo et al., 2014; Ma et al., 2018; Xu et al., 2018). Its germplasm reservation and other applications were depending on the traditional agriculture as far (Keriman et al., 2016), the availability of its whole genome sequence can allow a positional selection of DNA fragments to be re-sequenced, enhancing the usefulness of the discovered and efficient implementation on its SNPs and other information (Magris et al., 2019).
Multiple whole-genome sequencing projects of grapevines have contributed to the efficient implementation of SNP discovery in key cultivars (Tanksley et al., 1981; Berry et al., 1992; Wang et al., 2015), such as the Franco-Italian sequencing project and the IASMA sequencing project, both of which focused on the Pinot Noir (PN40024) cultivar (Roach et al., 2018). However, there have been few such projects conducted in China (Bai et al., 2013; Zhou J. et al., 2017; Mu et al., 2018). SSR markers have been applied to all Chinese grapevine landraces, which confirmed that the Munake cultivar was clustered in the Oriental cultivars group (Li et al., 2017), which includes the most ancient Chinese accessions. However, the origin of the Munake cultivar still requires further evidence. Here, the whole-genome sequence of the Munake cultivar was described with more than 3 million SNPs, more than 300,000 InDels, and more than 14,000 SVs reported. Li et al. (2017) analyzed 61 Chinese grapevine cultivars and 33 foreign grapevine cultivars using nine SSR markers and found that Munake and Lvmunage cultivars distributed in the Xinjiang Province (China) are the same cultivar. The present study also described more than 80,000 SSR markers. Of these, 44 SSR pairs were randomly selected and validated by PCR; all tested SSR pairs were able to discriminate between Munake cultivar genotypes.
The SNP and InDel molecular markers are useful alternatives to SSR markers in high density marker studies, such as quantitative trait locus identification, genetic map construction, and fine genetic mapping (Song et al., 2015; Nicolas et al., 2016). The statistical findings of the detected SNPs and InDels showed that there are more than 3 million SNPs, 300,000 InDels, and 10,000 SVs (Table 2). These variations constitute useful genomic resources for future studies of genetic differentiation. SSR markers were widely and randomly distributed throughout the genome, presenting several advantages (i.e., co-dominance, hypervariability, polymorphism, and ease and reliability of scoring) (Figure 7). SNP markers serve as effective markers for high-throughput mapping and for studying complex genetic traits. Our study contributes a considerable amount of genomic resources, including SNPs, InDels, SVs and genomic SSRs for the Munake grape, which all identification in our study can offer some genetic methods for the geneticists and breeders. The improvement application of the Munake grape can be facilitated on the utilization of this genomic information in linkage mapping, comparative genomics and molecular breeding based on the considerable efforts in future.
Conclusion
The Munake grape cultivar is a prime example of a species that is critically endemic because of invasive cultivar, diseases and habitat degradation. As a typical late-matured and unique tasted variety, its genetic analyses are an essential prerequisite for implementing effective management strategies. The provided genomic resources (SNPs, InDels, SVs, and genomic SSRs) for the Munake cultivar in here will be useful for geneticists and grape breeders to construct linkage mapping, apply genomic comparisons, and assist in molecular breeding for grape, and will expedite plant breeding programs of the Munake cultivar. In its exist populations, despite the threats the species faces, with adequate management, it is still possible to prevent its genetic resources in future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA632683.
Author Contributions
HZ and FZ: conceptualization, validation, and writing – original draft. HZ, FZ, MP, and XW: data curation. HZ, FZ, XW, JX, HX, and WS: formal analysis. HZ and XW funding acquisition and project administration. HZ, FZ, and WS: methodology. FZ, JH, XZ, SH, CM, and MW: resources. XW and WS: supervision. HZ, FZ, JJ, and WS: writing – review and editing. All authors read and approved the final manuscript.
Funding
This research was financed by the Basic Scientific Research Funding Project of Autonomous Region Public Welfare Scientific Research Institutes (Development and analysis of SNP markers in Munake grape and its progeny based on whole-genome resquencing), Forestry Reform and Development from the Central Government [Xin(2021) TG04], China Agriculture Research System (CARS-29-ZP-08), and Tianshan Youth-Excellence Youth Project (No. 2018Q037) funds project.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Genesis Technology Communication (Beijing), Co., Ltd., for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.664835/full#supplementary-material
Supplementary Table 1 | The primers for detecting 44 microsatellite loci.
Footnotes
- ^ http://hannonlab.cshl.edu/fastx_toolkit/
- ^ http://ftp.ensemblgenomes.org/pub/plants/release-25/fasta/vitis_vinifera/
- ^ http://pgrc.ipk-gatersleben.de/misa
- ^ http://primer3.sourceforge.net/
References
Bai, H., Cao, Y. H., Quan, J. Z., Dong, L., Li, Z. Y., Zhu, Y. B., et al. (2013). Identifying the genome-wide sequence variations and developing new molecular markers for genetics research by re-sequencing a Landrace cultivar of foxtail millet. PLoS One 8:12. doi: 10.1371/journal.pone.0073514
Berry, S. Z., Wiese, K. L., and Gould, W. A. (1992). Ohio 7983’ processing tomato. HortScience 27, 939. doi: 10.21273/HORTSCI.27.8.939
Biagini, B., Imazio, S., Scienza, A., Failla, O., and De Lorenzis, G. (2016). Renewal of wild grapevine (Vitis vinifera L. subsp. sylvestris (Gmelin) Hegi) populations through sexual pathway: Some Italian case studies. Flora 219, 85–93. doi: 10.1016/j.flora.2016.01.003
Birney, E., Clamp, M., and Durbin, R. (2004). Genewise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Chen, J., Wang, N., Fang, L. C., Liang, Z. C., Li, S. H., and Wu, B. H. (2015). Construction of a high-density genetic map and QTLs mapping for sugars and acids in grape berries. BMC Plant Biol. 15:28. doi: 10.1186/s12870-015-0428-2
Chen, K., Wallis, J. W., McLellan, M. D., Larson, D. E., Kalicki, J. M., Pohl, C. S., et al. (2009). BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681. doi: 10.1038/nmeth.1363
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Tung, N., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Doyle, J. J. T., and Doyle, J. L. (1990). Isolation of plant DNA from fresh tissue. Focus 12, 13–15.
Guo, C., Guo, R., Xu, X., Gao, M., Li, X., Song, J., et al. (2014). Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J. Exp. Bot. 65, 1513–1528. doi: 10.1093/jxb/eru007
Guo, Y., Shi, G., Liu, Z., Zhao, Y., Yang, X., Zhu, J., et al. (2015). Using specific length amplified fragment sequencing to construct the high-density genetic map for Vitis (Vitis vinifera L. x Vitis amurensis Rupr.). Front. Plant Sci. 6:393. doi: 10.3389/fpls.2015.00393
Jaillon, O., Aury, J., Noel, B., Policriti, A., Clepet, C., Casagrande, A., et al. (2007). The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467. doi: 10.1038/nature06148
Karastan, O. M., Muliukina, N. A., and Papina, O. S. (2018). Verification of grape pedigree by microsatellite analysis. Cytol. Genet. 52, 331–342. doi: 10.3103/S0095452718050031
Keriman, S., Yue, C., Bahar, G., Liu, A., Ayixiamu, and Yang, J. (2016). Fruit quality and mineral element content of ‘Munake’ grape under different site conditions. Northern Horticulture 40, 9–13.
Li, B., Jiang, J., Fan, X., Zhang, Y., Sun, H., Zhang, G., et al. (2017). Molecular characterization of Chinese grape landraces (Vitis L.) using microsatellite DNA markers. HortScience 52, 533–540. doi: 10.21273/HORTSCI11802-17
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, S., Wang, S., Deng, Q., Zheng, A., Zhu, J., Liu, H., et al. (2012). Identification of genome-wide variations among three elite restorer lines for hybrid-rice. PLoS One 7:e30952. doi: 10.1371/journal.pone.0030952
Lijavetzky, D., Cabezas, J. A., Ibáñez, A., Rodríguez, V., and Martínez-Zapater, J. M. (2007). High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genomics 8:424. doi: 10.1186/1471-2164-8-424
Ma, Z. Y., Wen, J., Ickert-Bond, S. M., Nie, Z. L., Chen, L. Q., and Liu, X. Q. (2018). Phylogenomics, biogeography, and adaptive radiation of grapes. Mol. Phylogenet. Evol. 129, 258–267. doi: 10.1016/j.ympev.2018.08.021
Magris, G., Di Gaspero, G., Marroni, F., Zenoni, S., Tornielli, G. B., Celii, M., et al. (2019). Genetic, epigenetic and genomic effects on variation of gene expression among grape varieties. Plant J. 99, 895–909. doi: 10.1111/tpj.14370
Mahmood, A. M., Ali, S. H., and Ai hawezy, S. (2019). Molecular characterization of autochthonous grapevine (Vitis vinifera L.) cultivars in Kurdistan using simple sequence repeats (SSRs). Journal of Garmian university 3, 328–335.
Massonnet, M., Cochetel, N., Minio, A., Vondras, A. M., Lin, J., Muyle, A., et al. (2020). The genetic basis of sex determination in grapes. Nat. Commun. 11, 1–12. doi: 10.1038/s41467-020-16700-z
Mercati, F., De Lorenzis, G., Brancadoro, L., Lupini, A., Abenavoli, M. R., Barbagallo, M. G., et al. (2016). High-throughput 18K SNP array to assess genetic variability of the main grapevine cultivars from Sicily. Tree Genet. Genomes 12, 59. doi: 10.1007/s11295-016-1021-z
Minio, A., Massonnet, M., Figueroa-Balderas, R., Castro, A., and Cantu, D. (2019). Diploid genome assembly of the wine grape Carménère. G3-Genes Genom. Genet. 9, 1331–1337. doi: 10.1534/g3.119.400030
Money, D., Migicovsky, Z., Gardner, K., and Myles, S. (2017). LinkImputeR: user-guided genotype calling and imputation for non-model organisms. BMC Genomics 18:523. doi: 10.1186/s12864-017-3873-5
Mu, X. Y., Shen, X. L., Wu, Y. M., Zhu, Y. X., Dong, S. B., Xia, X. F., et al. (2018). Plastid phylogenomic study of grape species and its implications for evolutionary study and conservation of Vitis. Phytotaxa 364, 71–80. doi: 10.11646/phytotaxa.364.1.4
Nicolas, S. D., Péros, J. P., Lacombe, T., Launay, A., Le Paslier, M. C., Bérard, A., et al. (2016). Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L.) diversity panel newly designed for association studies. BMC Plant Biol. 16:74. doi: 10.1186/s12870-016-0754-z
Pang, H., Liao, K., Wu, S., Aisikaier, M., Zhao, S., Xu, G., et al. (2015). Analysis of fruit structure difference of Munake and other grape varieties. Journal of Xinjiang Agricultural University 38, 370–375.
Patzak, J., Henychová, A., Paprštein, F., and Sedlák, J. (2019). Evaluation of genetic variability within sweet cherry (Prunus avium L.) genetic resources by molecular SSR markers. Acta Sci. Pol. Hortorum Cultus 18, 157–165.
Prysiazhniuk, L., Shytikova, Y., Dikhtiar, I., and Mizerna, N. (2019). Evaluation of genetic and morphological distances between soybean (Glycine max L.) cultivars. Zemdirbyste 106, 117–122. doi: 10.13080/z-a.2019.106.015
Roach, M. J., Johnson, D. L., Bohlmann, J., van Vuuren, H. J. J., Jones, S. J. M., Pretorius, I. S., et al. (2018). Population sequencing reveals clonal diversity and ancestral inbreeding in the grapevine cultivar Chardonnay. PLoS Genet. 14:e1007807. doi: 10.1371/journal.pgen.1007807
Song, X., Ge, T., Li, Y., and Hou, X. (2015). Genome-wide identification of SSR and SNP markers from the non-heading Chinese cabbage for comparative genomic analyses. BMC Genomics 16:328. doi: 10.1186/s12864-015-1534-0
Subbaiyan, G. K., Waters, D. L. E., Katiyar, S. K., Sadananda, A. R., Vaddadi, S., and Henry, R. J. (2012). Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 10, 623–634. doi: 10.1111/j.1467-7652.2011.00676.x
Tanksley, S. D., Zamir, D., and Rick, C. M. (1981). Evidence for extensive overlap of sporophytic and gametophytic gene expression in lycopersicon esculentum. Science 213, 453–455. doi: 10.1126/science.213.4506.453
Varshney, R. K., Graner, A., and Sorrells, M. E. (2005). Genic microsatellite markers in plants: features and applications. Trends Biotechnol. 23, 48–55. doi: 10.1016/j.tibtech.2004.11.005
Wang, K., Zhuang, J. Y., Huang, D. R., Ying, J. Z., and Fan, Y. Y. (2015). Genome-wide polymorphisms between the parents of an elite hybrid rice and the development of a novel set of PCR-based InDel markers. Genet. Mol. Res. 14, 3209–3222. doi: 10.4238/2015.April.10.33
Wu, X. Y., Luo, S. P., Xia, P. B., and Zhu, Y. (2011). Identification of seedless characters of embryo rescue seedlings of Xinjiang Munake grape by RAPD markers. Hortic. Plant J. 38, 2487.
Xu, J., Zhang, L., Wei, J., Zhang, Z., Wu, B., and Li, X. P. (2018). Effects of N2O fumigation on postharvest quality of Munake grapes. Modern Food Science and Technology 34, 56–61 and 46. doi: 10.13982/j.mfst.1673-9078.2018.05.008
Xu, Y., Gao, Z., Tao, J., Jiang, W., Zhang, S., Wang, Q., et al. (2016). Genome-wide detection of SNP and SV variations to reveal early ripening-related genes in grape. PLoS One 11:e0147749. doi: 10.1371/journal.pone.0147749
Yang, L., Xiao, K., Alimu, Nisha, K. B., and Yang, S. (2009). Occurrence regularity and control techniques of leafhopper in Xinjiang Munake grape. Northwest Horticulture (special issue on fruit trees) 23–24. doi: 10.3969/j.issn.1004-4183-B.2009.08.012
Zhang, J. W., Yue, C. Y., Jiao, S. P., Zhang, X. P., Nuer, G. L., Niu, J. L., et al. (2016). Investigation and Analysis of Nutrient Status of Munage Grapes Leaf in Atushi. Southwest China J. Agric. Sci. 29, 1425–1429. doi: 10.16213/j.cnki.scjas.2016.06.034
Zhang, Y., Feng, L., Fan, X., Jiang, J., Zheng, X., Sun, H., et al. (2017). Genome-wide assessment of population structure, linkage disequilibrium and resistant QTLs in Chinese wild grapevine. Sci. Hortic. 215, 59–64. doi: 10.1016/j.scienta.2016.12.014
Zhang, Z., Wang, Y., Zhang, J., Wang, J., Tian, B., Yu, J., et al. (2010). Preliminary studies on diversity of fruit characteristics of Munage grapevine in Akesu region of Xinjiang. Sin-Overseas Grapevine and Wine 4–10. doi: 10.13414/j.cnki.zwpp.2010.01.003
Zhou, J., Fan, X., Dong, Y., Zhang, Z., Ren, F., Hu, G., et al. (2017). Complete nucleotide sequence of a new variant of grapevine fanleaf virus from northeastern China. Arch. Virol. 162, 577–579. doi: 10.1007/s00705-016-3101-7
Zhou, Y., Massonnet, M., Sanjak, J. S., Cantu, D., and Gaut, B. S. (2017). Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication. Proc. Natl. Acad. Sci. U. S. A. 114, 11715–11720. doi: 10.1073/pnas.1709257114
Zhou, Y., Minio, A., Massonnet, M., Solares, E., Lv, Y., Beridze, T., et al. (2019). The population genetics of structural variants in grapevine domestication. Nat. Plants 5, 965–979. doi: 10.1038/s41477-019-0507-8
Keywords: genome-wide identification, sequence variations, SSR marker development, munake grape, cultivar
Citation: Zhong H, Zhang F, Zhou X, Pan M, Xu J, Hao J, Han S, Mei C, Xian H, Wang M, Ji J, Shi W and Wu X (2021) Genome-Wide Identification of Sequence Variations and SSR Marker Development in the Munake Grape Cultivar. Front. Ecol. Evol. 9:664835. doi: 10.3389/fevo.2021.664835
Received: 06 February 2021; Accepted: 11 March 2021;
Published: 12 April 2021.
Edited by:
Yongfeng Zhou, University of California, Irvine, United StatesReviewed by:
Yuejin Wang, Northwest A&F University, ChinaChonghuai Liu, Zhengzhou Fruit Research Institute (CAAS), China
Copyright © 2021 Zhong, Zhang, Zhou, Pan, Xu, Hao, Han, Mei, Xian, Wang, Ji, Shi and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Shi, d2F0ZXI1MTE2QDE2My5jb20=; Xinyu Wu, NDU0NjkxNjI3QHFxLmNvbQ==
†These authors have contributed equally to this work