 Ivo Carrasco-Wong
Ivo Carrasco-Wong Javiera M. Sanchez
Javiera M. Sanchez Jaime A. Gutierrez
Jaime A. Gutierrez Delia I. Chiarello
Delia I. Chiarello- Escuela de Tecnología Médica, Facultad de Medicina y Ciencia, Universidad San Sebastián, Santiago, Chile
Preeclampsia (PE) is a complex pregnancy syndrome characterized by hypertension with or without proteinuria, affecting 2–6% of pregnancies globally. PE is characterized by excessive release of damage-associated molecular patterns (DAMPs) into the maternal circulation. This DAMP-rich milieu acts on innate immune cells, inducing a proinflammatory state characterized by elevated cytokines such as IL-1β and IL-18. This proinflammatory state in the mother and placenta results in the endothelial dysfunction strongly associated with cardiovascular disorders. While the immediate maternal and fetal risks of PE are well-documented, accumulating evidence indicates that PE also confers long-term cardiovascular risks to the mother, including hypertension, coronary heart disease, stroke, and heart failure. The underlying mechanisms connecting PE to these chronic cardiovascular conditions remain unclear. This article explores the potential role of trained innate immunity (TRIM) as a mechanistic link between PE and increased long-term cardiovascular risk. We propose that the persistent exposure to DAMPs during PE may epigenetically reprogram maternal innate immune cells and their progenitors, leading to TRIM. This reprogramming enhances the inflammatory response to subsequent stimuli, potentially contributing to endothelial dysfunction and chronic inflammation that predispose women to cardiovascular diseases later in life. Understanding the role of TRIM in PE could provide novel insights into the pathophysiology of PE-related cardiovascular complications and identify potential targets for therapeutic intervention. Further research is warranted to investigate the epigenetic and metabolic alterations in innate immune cells induced by PE and to determine how these changes may influence long-term maternal cardiovascular health.
Preeclampsia and maternal long-lasting effects
Preeclampsia (PE) is a serious syndrome of pregnancy, characterized by hypertension with or without proteinuria, which can lead to the severe condition of eclampsia (1). In patients without signs of proteinuria, the diagnosis of the syndrome considers the presence of thrombocytopenia or elevated liver transaminase levels (2). It is estimated that in general population the prevalence of preeclampsia is 2–6% (3). The syndrome is subclassified into either early-onset PE (EOPE) or late-onset PE (LOPE) (1), with the onset of pathological signs falling into <34th and >34th week of gestation, respectively, being EOPE the syndrome which presents the most severe additional symptoms and signs, such as proteinuria, hepatic damage or pulmonary edema, among others (1).
Increasing evidence shows that the syndrome has a long-lasting deleterious effect on their cardiovascular health. Thus, four meta-analysis made on 22 (4), 43 (5), 15 (6) and 21 studies (7) showed that in short-, medium- and long-term (i.e. follow-up from 1 month to 34 years (4–7)), women who had preeclampsia have increased risk of I) heart failure (adjusted risk ratio [aRR], 4.19; 95% confidence interval [CI], 2.09–8.38) (4); II) coronary heart disease (aRR, 2.50; 95% CI, 1.43–4.37) (4); III) coronary heart death (aRR, 2.10; 95% CI, 1.25–3.51) (4); IV) CV disease (aRR, 1.85; 95% CI, 0.80–4.29 (4) and odds ratio (OR), 2.28; 95% CI 1.87–2.77) (5); V) CV disease death [aRR, 2.21; 95% CI, 1.83–2.66 (4), OR, 2.89, 95% CI 1.71–4.89 (5) and RR, 2.29; 95% CI, 1.73-3.04) (6)]; VI) stroke (aRR, 1.81; 95% CI, 1.29–2.55) (4); VII) stroke death (aRR, 1.97; 95% CI, 0.80–4.88) (4); VIII) cerebrovascular disease (RR, 2.03; 95% CI, 1.54-2.67) (6); IX) peripheral arterial disease (RR, 1.87; 95% CI, 0.94-3.73) (6); and, X) hypertension [RR, 3.13, 95% CI 2.51–3.89) (5) and OR, 3.19, 95% CI, 1.52–6.70 (7)]. Thus, the effects of preeclampsia not only impact maternal health during the pregnancy but also induce subclinical alteration which can remain silent for years, increasing her cardiovascular risk. In this regard, increased microalbuminuria, a was found in association whit high hypertension risk in mothers who had PE 7 (8) and 10 (9) years before. Although, the pathophysiological mechanism of the findings is unknown, this slight alteration of the glomerular filtration is independently associated with high stroke (10) and coronary heart disease (11) risks. Nowadays, there is no knowledge about the pathophysiological mechanisms underlying the cardiovascular risk in mothers who had PE.
It has been agreed that the most likely etiology of PE is a poor remodeling of the spiral arteries and veins during early placentation. Meanwhile, LOPE appears to be linked to maternal factors, such as the inability of the cardiovascular system to meet the increasing metabolic needs of the fetoplacental unit, rather than issues with the placentation process (12). The PE condition maintains a pernicious low blood flow in a condition of high pressure (Jet-type), generating vascular mechanical stress, hypoxia (13), and syncytiotrophoblast (STB) dysfunction (14). STB is a multinucleated cell layer of fetal origin that covers the chorionic villi and is in direct contact with maternal blood (15). Thus, it has been reported that STB stress induced by the Jet-type blood flow can be characterized by: 1) STB damage markers, such as increased mitochondrial dysfunction, apoptotic markers, reticulum stress, oxidative stress, and inflammation; 2) An excessive release of microvesicles, exosomes, and cell fragments (16); and, 3) an increased release of ‘Damage-associated molecular patterns’ (DAMPs) (17). In PE, increased DAMPs activate the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, which promotes IL-1β and IL-18 maturation (18).
DAMPs are host to intracellular molecules that are not usually found in cell-free form. They can activate ‘Pattern Recognition Receptors’ (PRRs) mainly in innate immune cells, which are responsible for recognizing pattern molecules of microorganisms (19). PRRs are classified into several classes, including Toll-like receptors (TLR), nucleotide-binding oligomerization domain, Leucine-rich repeats, nucleotide-binding domain leucine-rich repeat containing receptors, retinoic acid-inducible gene 1 (RIG-1) -like receptors, and the C-type lectin receptors (20). PRRs are found in various cell types including monocytes/macrophages (21), neutrophils (22), and endothelial cells (23). The innate immune cells activated by DAMPs via TLR, switch from a tolerogenic, anti-inflammatory phenotype to a cytotoxic, pro-inflammatory phenotype (24). The activation of PRRs favors the proinflammatory status by inducing the secretion of proinflammatory cytokines (25). Under conditions of hypoxia and oxidative (17), reticulum (26), and mitochondrial (27) stress, all of them found in STB stress (16), the STB increases the releasing of several DAMPs, such as HMGB1 or cell-free fetal DNA (17). In this sense, the SBT stress contribute to the maternal pro-inflammatory milieu, which includes several increment level of circulating DAMPs and cytokines (28), are part of the pathophysiological signs of the syndrome and could activate and maintain the proinflammatory profile of innate immune cells and endothelial cell dysfunction observed in the syndrome (29). Both early-onset and late-onset preeclampsia appear to share systemic and placental inflammation as a common pathophysiological feature.
Proinflammatory status in preeclampsia
The immunological mechanisms underlying preeclampsia have been extensively explored (24, 30), highlighting the role of adaptive and innate immune pathways in the development of this condition. During the physiological pregnancy, the inflammatory profile of the maternal immune system changes, being proinflammatory during placentation. This is proposed as a requirement for an effective invasion of the placenta and remodeling of the spiral arteries (31). In the 2nd gestation trimester, the profile changes to anti-inflammatory which is the basis of tolerance toward the fetal-placental unit. At the time of delivery, the profile becomes proinflammatory contributing to labor (31). Therefore, a large part of the pregnancy takes place with strong maternal immunomodulation, which is manifested by: I) high levels of anti-inflammatory cytokines (e.g. IL-10), immunosuppressants (e.g. progesterone), suppressive (e.g. HLA-G), and tolerogenic molecules (e.g. TGF-β) (32); and by II) an increase of anti-inflammatory immune cells that including macrophages with the anti-inflammatory phenotype (M2), and a reduction ratio of helper T lymphocytes, Th1:Th2 and Th17:Tregs (33). In general, the placenta favors immunotolerance, either through the expression of human leukocyte antigen G (HLA-G) that reduces the reactivity of natural killer lymphocytes (34), or by releasing extracellular vesicles, which induce the secretion of anti-inflammatory cytokines from the macrophages that engulf them (35). Contrarily, PE is characterized by presenting a proinflammatory state in the mother and placenta (36), which includes an increment of circulating proinflammatory mediators (e.g. TNF-α and IL-6), diminishing of anti-inflammatory cytokines (e.g. IL-10) (36), decreased circulating levels of progesterone (37), decreased HLA-G expression (38), higher ratio lymphocytes Th1:Th2 and Th17:Tregs (36), and increased activation of monocytes, neutrophils, and macrophages (39). In PE, the unbalance toward the proinflammatory status is associated with endothelial activation, leading to endothelial dysfunction and high blood pressure (40).
Cardiovascular disorders and endothelial dysfunction/activation
Preeclampsia has been associated with microvascular dysfunction, which may contribute to the increased risk of obstructive coronary artery stenosis observed in women with a history of this condition, especially when preeclampsia is associated with preterm delivery or stillbirth (41). In physiological status, the endothelium shows a balance between vasodilator and vascular constrictor molecules, such as nitric oxide and Endothelin, respectively, which helps to maintain an anti-inflammatory and antithrombotic function (42). Cardiovascular disorders are strongly associated with endothelial dysfunction (43), characterized by an alteration in endothelium-dependent vascular relaxation, oxidative stress, and the inflammatory activation of endothelial cells (44). During endothelial activation, an overexpression of proinflammatory cytokines, chemokines, and adhesion molecules has been described in endothelial cells (43). Thus, systemic inflammation is associated with vascular diseases (45) and hypertension (46). Increasing evidence indicates that immune cells are directly involved in the onset of hypertension. In IFN-γ KO mice, a murine model of hypertension (DOCA+salt model) did not show the characteristic increment of the blood pressure of the model (47), being observed only when CD8+ T lymphocytes from hypertensive WT mice were transferred to knock-out (KO) individuals (47). However, not only are T cells implicated in the onset of hypertension but also innate immune cells, such as dendritic cells, monocytes/macrophages, natural killer cells, and neutrophils (48). Although adaptive immune cells have an important role in the onset of cardiovascular disorders, recently, innate cells have become relevant in the phenomenon due to the persistent activation of PRRs. It is proposed that cardiovascular diseases are related to a maladaptive inflammatory response of innate cells (49). In this sense, it was reported that circulating monocytes isolated from patients with coronary artery disease have increased NLRP3 and caspase-1 expression, both related to inflammasome formation and the elevated production of the downstream cytokines, such as IL-1β and IL-18 (50). Classically, it is described that inflammasome formation triggers pyroptosis (51). However, the neutrophils in a DAMP-rich inflammatory milieu are resistant to this type of cell death, becoming a permanent producer of IL-1β (52).
Monocytes and neutrophils and their association with cardiovascular pathologies
Meta-analysis of 27 studies evidenced that intermediate- and non-classical monocytes are increased in persons with cardiometabolic disorders and cardiovascular disease (53). On the other hand, a high neutrophil-to-lymphocyte ratio is associated with several cardiovascular conditions, as evidenced by a meta-analysis of 38 studies (54), being proposed as a biomarker of cardiovascular health. These innate immune cells are tightly involved in the magnitude of cardiovascular lesions or alterations. In the case of animals subjected to myocardial infarction and reperfusion, the depletion of neutrophils prior to the infarct showed a significant reduction in infarct size (55). During atherogenesis, neutrophils in the intima release reactive oxygen species and proteases, which alter the endothelium integrity, allowing the recruitment and extravasation of monocytes and its further differentiation to macrophages (Reviewed in (56)). The myeloperoxidase released by neutrophils oxidates LDL, enhancing the amount of oxLDL, which together with activated macrophages generates the foam cells (56). However, despite the pro-inflammatory profile of the cardiovascular event, high basal levels of pro-inflammatory cytokines precede the occurrence of cardiovascular pathologies. Thus, a meta-analysis showed that high levels of IL-6, IL-18, and TNFα increase the risk of non-fatal myocardial infarction or coronary heart disease death (57). All those cytokines can induce endothelial dysfunction (58, 59). The activated neutrophils secrete the pro-inflammatory cytokines TNFα, IL-1β and IL-12 (60), and activated monocytes secrete IL-1β, IL-6, TNFα (61). The latter suggests that chronic activation of monocytes and neutrophils can impact endothelial function increasing the risk of cardiovascular pathologies.
In the context of preeclampsia, the pro-inflammatory environment includes activated monocytes, showing increased expression of CD11b, ICAM-1, CD14, and TLR4, an overproduction of reactive-oxygen-species (ROS), and altered secretion of cytokines (62). In the same way, neutrophil concentration is increased in maternal blood (63) along with an increment of the neutrophil activation markers CD11b and CD62L (64). Considering that a proinflammatory milieu is shared characteristic between preeclampsia and cardiovascular disorder, then an alteration of the performance of the immune system could be part of the mechanisms that increase the long-lasting cardiovascular risk in women who had preeclampsia. In this sense, ‘Trained Innate Immunity’ (65) could be the mechanism since it has recently been involved as a player in the onset of cardiometabolic diseases (65, 66).
‘Trained Innate Immunity’ (TRIM)
TRIM is defined as ‘the long-term functional reprogramming of innate immune cells, which is evoked by exogenous or endogenous insults leading to an altered response toward a heterologous second challenge after the return to a non-activated state’ (67). TRIM can be triggered by DAMPs, including oxidized low-density lipoprotein (oxLDL) (66), lipoprotein(A) (68), catecholamines (69), aldosterone (70), heme (71), vimentin (72), uric acid (73), S100-alarmin (74) and HMGB1 (75). DAMPs can induce TRIM by signaling through PRRs, including the receptor for advanced glycation end products (RAGE) (76), CD36 (77), and the five types of PRRs (77). Even though there is no report about TRIM in preeclampsia, it is possible to propose that it could occur since several DAMP levels are increased in maternal blood (17, 78).
The effects of TRIM induction mediated by DAMPs trigger metabolic and epigenetic modification that will lead to memory. For instance, cellular metabolic changes are necessary for epigenetic reprogramming (79), including an overexpression of glycolytic enzymes after the first stimulus (80) and an increment of lactate due to higher glucose consumption (81). Fumarate and succinate are produced as intermediates in the tricarboxylic acid cycle and glycolysis, and those intermediate are increased in trained macrophages (82). Additionally, oxidative phosphorylation also in enriched in trained cells (83). The influence of cellular metabolism on epigenetic mechanisms is already known. In TRIM, the accumulation of fumarate inhibits the demethylase activity of KDM5, a lysine demethylase of histones (84).
There is no report about TRIM being induced in preeclampsia. However, in maternal preeclamptic monocytes showed basal intracellular reactive-oxygen-species and increased oxidative burst after stimulation, which is indicative of a potentiated oxidative phosphorylation (85), similar to the observation made in monocytes trained with oxLDL (83). Noteworthy, mothers with PE, exhibited an increased percentage of classical monocytes-2 (CD14++, CD16-, HLA-DR-) and a decreased percentage of non-classical monocytes (CD14+, CD16++) prior to delivery (86, 87). Since classical monocytes-2 are considered as pro-tolerogenic (88) while non-classical monocytes are associated with pro-inflammatory responses (88), the altered levels observed in PE-pregnancies are proposed to reflect a compensatory mechanism aimed at counterbalancing low-grade chronic inflammation (87). Interestingly, although monocyte-2, considered as monocyte-myeloid derived suppressive cell (89), mainly by its capacity of differentiate naïve CD4+ T cells to CD4+, CD25+, Foxp3+ regulatory T-cell (Treg) (90), in Psoriasis, the induced-Treg differentiated by monocytes-2 showed a deficient suppressive activity (91). The latter suggest that in preeclampsia, a similar phenomenon could be occurring, since circulatory Treg in preeclampsia shows reduced function, with reduced expression of FOXP3 and reduced IL-10 and TGF-β secretion (92). However, there is no data on the role of monocyte subpopulations in PE or their frequency during the postpartum period. Consequently, the potential permanent programming of monocyte subpopulations remains unknown.
Resident natural killer cells (NK) in decidua (dNK) is an essential cell type during the placentation due to its activity that include the induction of the remodeling of spiral arteries by the disruption of its vascular smooth muscle cells (93) and by the interaction with extravillous trophoblast cells (94) promoting its invasion activity an arterial remodeling through INF-γ and VEGFα secretion (95). Noteworthy, the dNK from multiparous mothers showed a higher response to trophoblast interaction characterized by enhanced INF-γ and VEGFα secretion, in association with an open state of chromatin of their locus, among other loci (95). Thus, it is proposed that the physiological pregnancy can promote epigenetically a tolerance to future pregnancies (95). In preeclampsia, dNK are increased in decidua but showing reduced activity (e.g. reduced INF-y secretion) (96), which impact in the spiral arteries remodeling. In this scenario, also, it is possible to propose a memory in dNK in PE, since the mothers that have a prior pregnancy with preeclampsia have the greatest relative risk (RR) of PE in a new pregnancy, with a RR of 8.4 (7.1 to 9.9, 95% CI) (97). Also, other conditions increase the risk of PE, such as chronic hypertension with a 5.1 of RR (4.0 to 6.5, 95% CI); pregestational diabetes with a RR of 3.7 (3.2 to 4.3, 95% CI); and, pre-pregnancy BMI>30 with a RR of 2.8 (2.6 to 3.1, 95% CI) (97). In all this pathologies the activity of NK is reduced (98–100). It is proposed that NK exhaustion can be produced by chronic inflammation (101) which is found in chronic hypertension (102), diabetes (103), obesity (104), and preeclampsia (described above). Then, chronic inflammation observed in several pathologies with high risk of PE may generate a pro-exhaustion memory in circulatory and decidual NKs favoring the onset of PE.
TRIM is associated with cardiovascular disorders (105), making it possible that PE-induced long-term TRIM could impact endothelial homeostasis. Then, as shows the Figure 1, we proposed the proinflammatory status of preeclampsia constituted at least by high concentration of pro-inflammatory cytokines and increased levels of several DAMPs (compiled in Table 1) is associated with the activation of innate cells, including monocytes and neutrophils. This context, as was discussed above, could be conducive to TRIM acquisition during the syndrome. Then, in a short-medium or long-term, the maternal trained innate cells could over respond to new challenges and generate a strong and fast proinflammatory status disturbing the cardiovascular physiology of women (see Figure 1). Regarding the moment during the pregnancy at which DAMPs could initiate in PE the challenge in innate immune cells is not clear. However, DAMPs seem to have permanent participation in the pathophysiology of the syndrome.
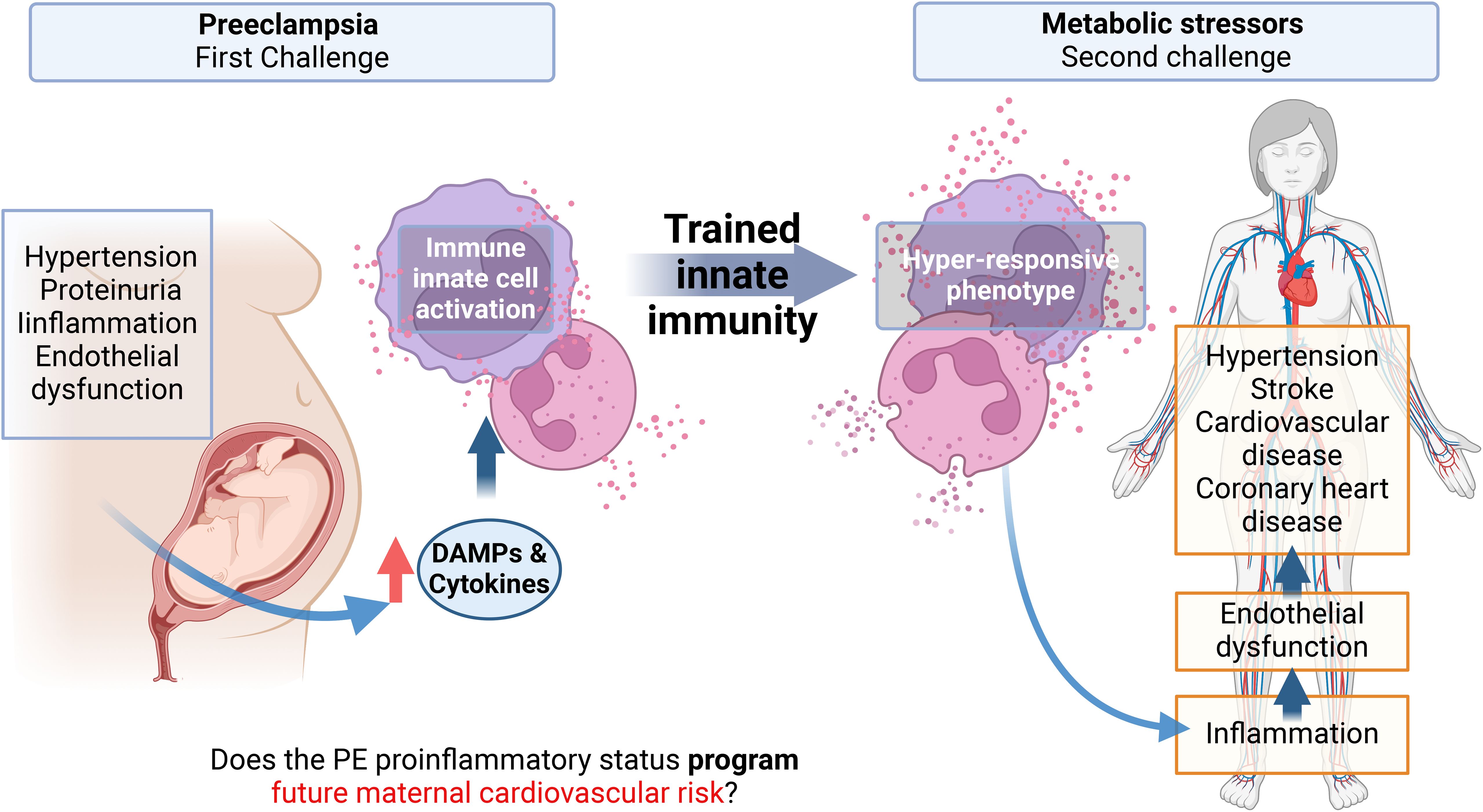
Figure 1. Graphical abstract. Preeclampsia is associated with an increased cardiovascular risk in the mother, observed months to years after the syndrome with no possible cure proposed yet. A characteristic of preeclampsia is a maternal systemic proinflammatory status with, among others, elevated levels of circulating cytokines and damage-associated molecular patterns (DAMPs). These molecules can activate innate immune cells and endothelial cells, inducing endothelial dysfunction, which is the basis of hypertension, the pathognomonic sign of Preeclampsia. Monocytes and neutrophils activated by DAMPs can result in a phenomenon called trained innate immunity (TRIM) by epigenetic mechanisms, characterized by a hyper-responsiveness of these cells to a second heterologous challenge. This memory can be maintained in myeloid precursors, for several cell generations. This project proposes that the proinflammatory state of EOPE can induce TRIM in monocytes and neutrophils during pregnancy. Then, in the maternal future, these trained monocytes will be hyper-responsive against new molecules associated with metabolic risk factors. This hyper-responsive phenotype could then activate endothelial cells generating endothelial dysfunction in the mother, favoring the appearance of cardiovascular disorders.
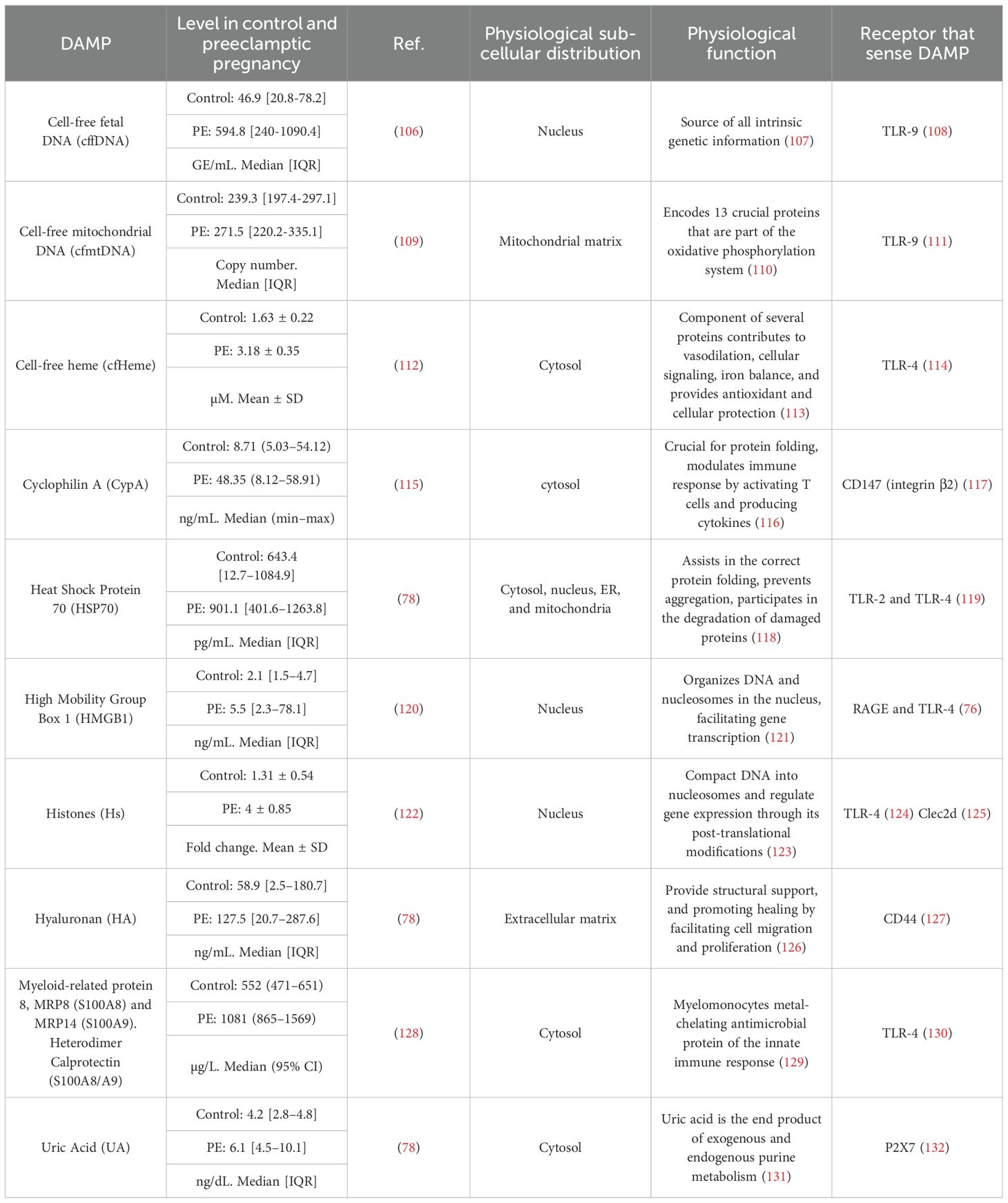
Table 1. Maternal circulatory DAMPs with increased levels in Preeclampsia.
DAMPs and the pathophysiology of preeclampsia
There is no clue as to whether DAMPs could be involved in the origin of PE, however it is possible to propose that DAMPs could maintain and even amplify the pro-inflammatory status of the syndrome. Exposure of control placental explants to PE serum increased HMGB1 release (133). Ex vivo induction of oxidative stress and hypoxia to control placenta increased liberation of several DAMPs, including HMGB1, HSP70, S100A8, S100A12 and Cell free-fetal DNA (18). The latter together with the findings that the receptors TLR-2,-3, -4, and -9 are increased in syncytiotrophoblast in PE (134) suggest a self-maintaining of the DAMP-induced proinflammatory status of the placenta.
DAMPs may enter maternal circulation, as several with high maternal plasma levels in PE are listed in Table 1, including cell-free DNA, crystals, and proteins. Although most of them can be released by the placenta it is not possible to determine the original source of them. However, DAMPs could induce alteration in endothelium. Thus, microvascular endothelial cells HMEC-1 treated with recombinant HMGB1 elevated the expression of the adhesion molecule ICAM-1 favoring the arrest of the monocyte cell line U937 on them (133). Cell-free mitochondrial DNA (cfmtDNA) also generated a similar effect, in this case cfmtDNA could increase macrovascular endothelial cell EA.hy926 permeability, together with the rising of ICAM and E-selectin expression which favored the arrest of primary polymorphonuclear leukocyte (135). In the other hand, the cfmtDNA released by hypoxic murine trophoblast reduced the endothelial-dependent vasodilation in abdominal aorta, partially mediated by NLRP3 since the effect on cfmtDNA diminished in NLRP3 -/- animals (136). The latter together with the fact that preeclampsia curses along endothelial dysfunction (137) suggests that elevated circulating DAMPs may be part of the syndrome.
Maternal plasma in PE shows elevated levels of the anti-angiogenic protein soluble fms-like tyrosine kinase 1 (sFLT-1) (138). sFLT-1 is proposed to be one of the responsible for endothelial dysfunction in PE by the sequestration of VEGF resulting in the endothelial expression of the adhesion proteins ICAM and VCAM and the vasoconstrictor peptide endothelin-1 (139). sFLT-1 can be released by THP-1-derived macrophages followed by the activation of inflammasome in a GSDMD dependent manner (140). In this sense the DAMPs hyaluronan and HSP70 (141), and uric Acid (142) generated a strong inflammasome activation in primary monocytes from mothers with PE probably contributing to the high levels of IL-1β and IL-18 found in PE-maternal plasma (142). The activation of inflammasomes also participates in the releasing of DAMPs since the induction of pyroptosis led to the liberation of HMGB1 (143).
In the most severe cases of preeclampsia, mothers have a higher risk of thrombotic events during pregnancy (144). In fact, the plasma from mothers with PE had fast and strong thrombin generation compared to control pregnancy plasma (122). The same study showed that plasma from preeclampsia patients strongly induced NETosis in neutrophils from healthy donors (122). NETosis is a neutrophil-specific activation characterized by the release of neutrophil extracellular traps (NETs), which consist of chromatin and antimicrobial proteins (145). In PE high levels of NETs were found in maternal circulation (146). The link between NETs thrombosis is based on the capacity of human nuclear-DNA and histones for inducing thrombin generation (147). Interestingly, intact-NETs or assembled histones are unable to induce thrombin generation, indicating that NETs must be dissembled to have coagulatory activity. Based on the latter, DAMPs may favor the elevated risk of thrombotic events in mothers-with-PE due to the high levels of circulatory histones (see Table 1), and to the induction of NETosis by HMGB1 (148).
In the context of PE, as Table 1 and Figure 2 show, several DAMPs have increased maternal circulatory concentration in PE, suggesting that TRIM could be induced in innate cells during the syndrome. The latter is supported by the evidenced ability of cell-free heme (151) and HMGB1 (75) to induce TRIM. In the case of S100A8/A9, the evidence indicates a possible dual role as pro-inflammatory molecules (153) but also as an immune modulator (152). The latter indicates that it will be necessary to evaluate not only if individual DAMPs with high levels in PE can induce TRIM but also how collectively high levels of different DAMPs affect TRIM acquisition.
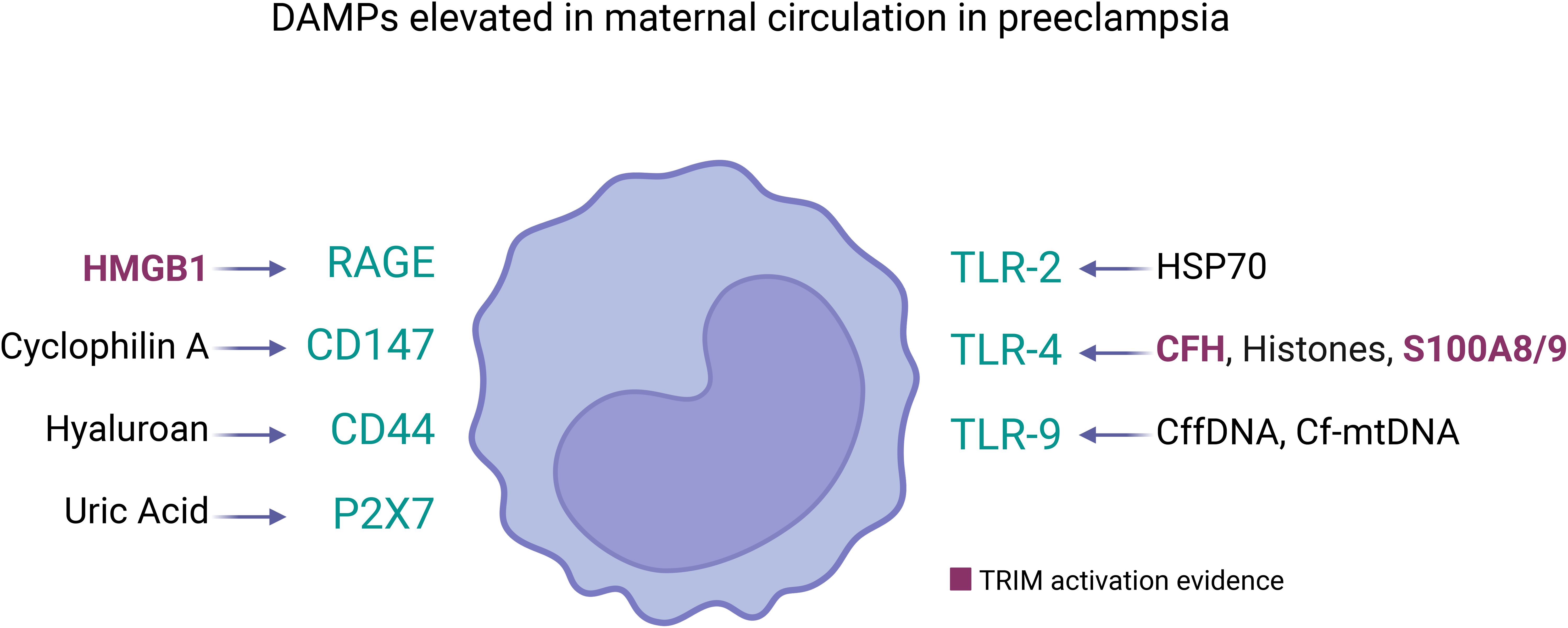
Figure 2. DAMPs elevated in maternal circulation in preeclampsia and its receptors in innate immune cells. Several DAMPs have high plasmatic concentration in mothers with preeclampsia, each of them can be sensed by pattern recognition receptors in cell membrane of innate immune cells (represented as monocyte). The DAMPs/Receptor are: High Mobility Group Box 1 (HMGB1) (120)/RAGE and TLR4 (114); Cyclophilin A/CD147 (117); Hyaluronan (78)/CD44 (127); Uric Acid (UA) (78)/P2X7 (132); Heat Shock Protein 70 (HSP70) (78)/TLR2 and TLR4 (119); Cell-free heme (CFH) (112)/TLR-4 (149); Histones (122)/TLR-4 (124) and Clec2d (125); Calprotectin (S100A8/A9) (128)/TLR-4 (123); Cell-free fetal DNA (cffDNA) (106)/TLR-9 (108); Cell-free mitochondrial DNA (cfmtDNA) (109)/TLR-9 (150). Among them, only HMBG1 (75), cell-free heme (151), and S100A8/9 (152) have been showed as TRIM inducers. However, the circulatory DAMPs in PE may act collectively to promote TRIM acquisition in innate immune cells during the syndrome.
TRIM on myeloid progenitors
The fact that mature innate immune cells are short-lived (i.e. Half-life 5-7 days (154, 155)), raise the unanswered question about how PE-induced TRIM could last years. In this scenario, it would be necessary that the precursors of innate cells also be involved. In this sense, human hematopoietic stem and progenitors cells (HSPCs) showed permanent alterations after in vivo Bacille Calmette-Guérin (BCG) vaccination (150), a classic TRIM inducer (156). The latter included, a permanent (i.e. at least 90 days post-vaccination) transcriptional reprogramming in HSPCs, leading to an upregulation of genes associated with myeloid and granulocytic cell lineage priming, generating a myeloid differentiation bias within HSPCs, and enhanced proinflammatory response to various stimuli of mature peripheral blood mononuclear cells (150). Regarding DAMPs, TRIM induced by heme in mice showed an increase in myeloid-biased long-term hematopoietic stem cells and multipotent progenitors with an expansion of myeloid-biased, associated to elevated chromatin accessibility in genes associated with myeloid differentiation of HSPCs, also there was a significant and permanent increase in mature myeloid cells (i.e. neutrophils and monocytes), with an enhanced phagocytic activity (151). HSPCs express TLR-2, - 4, and -9, and their activation induces the differentiation and expansion to macrophages (157) suggesting that DAMPs may reach bone marrow and promote TRIM-associated permanent modifications.
Conclusions
Preeclampsia is a severe multisystemic syndrome which manifest with different pathological characteristics (i.e. EOPE and LOPE). Additionally, it remains unclear what are the mechanisms that induce the syndrome. However, one fact is clear: mothers who had PE were at higher cardiovascular risk. Thus, mothers not only face a serious pregnancy pathology, but this syndrome will probably also affect their future health. Therefore, an understanding of the mechanisms that underlie higher cardiovascular risk is crucial. Trained innate immunity has recently changed the paradigm that adjudicated the immune memory only to T/B cells from adaptive immunity, and this type of epigenetic memory is a mechanism with a clear potential to impact cardiovascular physiology. This epigenetic memory could explain the increased cardiovascular risk observed in women who have experienced PE, potentially triggered by future health challenges throughout their lives. However, further research is required to explore this hypothesis, as no study has directly examined this possibility to date. If PE-related sterile inflammation can induce TRIM, testing seems to be mandatory since several research groups are focused on TRIM modulation (149). Thus, this offers a certain possibility to improve the future maternal health of women who have preeclampsia.
Author contributions
IC-W: Writing – review & editing, Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft. JS: Writing – review & editing, Conceptualization, Data curation, Investigation. JG: Writing – review & editing. DC: Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. PAI Convocatoria Nacional Subvención a Instalación en la Academia, Convocatoria Año 2021, Folio N°SA77210087 to IC-W. FONDECYT Regular 1221362 to JG (Universidad San Sebastián, Chile).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dimitriadis E, Rolnik DL, Zhou W, Estrada-Gutierrez G, Koga K, Francisco RPV, et al. Pre-eclampsia. Nat Rev Dis Prim. (2023) 9:1–22. doi: 10.1038/s41572-023-00417-6
2. Roberts JM. Preeclampsia epidemiology(ies) and pathophysiology(ies). Best Pract Res Clin Obstet Gynaecol. (2024) 94:102480. doi: 10.1016/J.BPOBGYN.2024.102480
3. Stuart JJ, Gray KJ, Rich-Edwards JW, Roberts JM. “Epidemiology of hypertensive disorders in pregnancy.,”. In: Chesley’s hypertensive disorders in pregnancy. Cambridge, Massachusetts, United States: Academic Press (2021). p. 21–43. doi: 10.1016/B978-0-12-818417-2.00023-3
4. Wu P, Haththotuwa R, Kwok CS, Babu A, Kotronias RA, Rushton C, et al. Preeclampsia and future cardiovascular health. Circ Cardiovasc Qual Outcomes. (2017) 10. doi: 10.1161/CIRCOUTCOMES.116.003497
5. Brown MC, Best KE, Pearce MS, Waugh J, Robson SC, Bell R. Cardiovascular disease risk in women with pre-eclampsia: Systematic review and meta-analysis. Eur J Epidemiol. (2013) 28:1–19. doi: 10.1007/S10654-013-9762-6
6. McDonald SD, Malinowski A, Zhou Q, Yusuf S, Devereaux PJ. Cardiovascular sequelae of preeclampsia/eclampsia: A systematic review and meta-analyses. Am Heart J. (2008) 156:918–30. doi: 10.1016/j.ahj.2008.06.042
7. Xu J, Li T, Wang Y, Xue L, Miao Z, Long W, et al. The association between hypertensive disorders in pregnancy and the risk of developing chronic hypertension. Front Cardiovasc Med. (2022) 9:897771. doi: 10.3389/fcvm.2022.897771
8. Nisell H, Lintu H, Lunell NO, Möllerström G, Pettersson E. Blood pressure and renal function seven years after pregnancy complicated by hypertension. BJOG Int J Obstet Gynaecol. (1995) 102:876–81. doi: 10.1111/j.1471-0528.1995.tb10874.x
9. Shammas AG, Maayah JF. Hypertension and its relation to renal function 10 years after pregnancy complicated by pre-eclampsia and pregnancy induced hypertension. Saudi Med J. (2000) 21:190–2.
10. Lee M, Saver JL, Chang KH, Liao HW, Chang SC, Ovbiagele B. Impact of microalbuminuria on incident stroke: A meta-analysis. Stroke. (2010) 41:2625–31. doi: 10.1161/STROKEAHA.110.581215
11. Perkovic V, Verdon C, Ninomiya T, Barzi F, Cass A, Patel A, et al. The relationship between proteinuria and coronary risk: A systematic review and meta-analysis. PloS Med. (2008) 5:1486–95. doi: 10.1371/journal.pmed.0050207
12. Ren Z, Gao Y, Gao Y, Liang G, Chen Q, Jiang S, et al. Distinct placental molecular processes associated with early-onset and late-onset preeclampsia. Theranostics. (2021) 11:5028–44. doi: 10.7150/thno.56141
13. Burton GJ, Woods AW, Jauniaux E, Kingdom JCP. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. (2009) 30:473–82. doi: 10.1016/j.placenta.2009.02.009
14. Chaiworapongsa T, Chaemsaithong P, Yeo L, Romero R. Pre-eclampsia part 1: current understanding of its pathophysiology. Nat Rev Nephrol. (2014) 10:466–80. doi: 10.1038/nrneph.2014.102
15. Lager S, Powell TL. Regulation of nutrient transport across the placenta. J Pregnancy. (2012), 1–14:179827. doi: 10.1155/2012/179827
16. Carrasco-Wong I, Aguilera-Olguín M, Escalona-Rivano R, Chiarello DI, Barragán-Zúñiga LJ, Sosa-Macías M, et al. Syncytiotrophoblast stress in early onset preeclampsia: The issues perpetuating the syndrome. Placenta. (2021) 113:57–66. doi: 10.1016/j.placenta.2021.05.002
17. Baker BC, Heazell AEP, Sibley C, Wright R, Bischof H, Beards F, et al. Hypoxia and oxidative stress induce sterile placental inflammation. vitro. Sci Rep. (2021) 11:7281. doi: 10.1038/s41598-021-86268-1
18. Garcia-Puente LM, Fraile-Martinez O, García-Montero C, Bujan J, De León-Luis JA, Bravo C, et al. Placentas from women with late-onset preeclampsia exhibit increased expression of the NLRP3 inflammasome machinery. Biomolecules. (2023) 13:1644. doi: 10.3390/biom13111644
19. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
20. Jentho E, Weis S. DAMPs and innate immune training. Front Immunol. (2021) 12:699563/BIBTEX. doi: 10.3389/FIMMU.2021.699563/BIBTEX
21. Billack B. Macrophage activation: Role of Toll-like receptors, nitric oxide, and nuclear factor kappa B. Am J Pharm Educ. (2006) 70. doi: 10.5688/aj7005102
22. Thomas CJ, Schroder K. Pattern recognition receptor function in neutrophils. Trends Immunol. (2013) 34:317–28. doi: 10.1016/j.it.2013.02.008
23. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun. (2015) 21:827–46. doi: 10.1177/1753425915606525
24. Bounds KR, Newell-Rogers MK, Mitchell BM. Four pathways involving innate immunity in the pathogenesis of preeclampsia. Front Cardiovasc Med. (2015) 2:20. doi: 10.3389/fcvm.2015.00020
25. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
26. Tannetta D, Masliukaite I, Vatish M, Redman C, Sargent I. Update of syncytiotrophoblast derived extracellular vesicles in normal pregnancy and preeclampsia. J Reprod Immunol. (2017) 119:98–106. doi: 10.1016/j.jri.2016.08.008
27. Walker OS, Ragos R, Wong MK, Adam M, Cheung A, Raha S. Reactive oxygen species from mitochondria impacts trophoblast fusion and the production of endocrine hormones by syncytiotrophoblasts. PloS One. (2020) 15. doi: 10.1371/journal.pone.0229332
28. Szarka A, Rigó J, Lázár L, Beko G, Molvarec A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol. (2010) 11. doi: 10.1186/1471-2172-11-59
29. Ann-Charlotte I. Inflammatory mechanisms in preeclampsia. Pregnancy Hypertens Int J Women’s Cardiovasc Heal. (2013) 3:58. doi: 10.1016/j.preghy.2013.04.005
30. Collier A ris Y, Smith LA, Karumanchi SA. Review of the immune mechanisms of preeclampsia and the potential of immune modulating therapy. Hum Immunol. (2021) 82:362–70. doi: 10.1016/J.HUMIMM.2021.01.004
31. Rambaldi MP, Weiner E, Mecacci F, Bar J, Petraglia F. Immunomodulation and preeclampsia. Best Pract Res Clin Obstet Gynaecol. (2019) 60:87–96. doi: 10.1016/j.bpobgyn.2019.06.005
32. Sargent IL, Borzychowski AM, Redman CW. Immunoregulation in normal pregnancy and pre-eclampsia: An overview. Reprod BioMed Online. (2006) 13:680–6. doi: 10.1016/S1472-6483(10)60659-1
33. Sasaki Y, Darmochwal-Kolarz D, Suzuki D, Sakai M, Ito M, Shima T, et al. Proportion of peripheral blood and decidual CD4+ CD25 bright regulatory T cells in pre-eclampsia. Clin Exp Immunol. (2007) 149:139–45. doi: 10.1111/j.1365-2249.2007.03397.x
34. Amodio G, Sales de Albuquerque R, Gregori S. New insights into HLA-G mediated tolerance. Tissue Antigens. (2014) 84:255–63. doi: 10.1111/tan.12427
35. Abumaree MH, Chamley LW, Badri M, El-Muzaini MF. Trophoblast debris modulates the expression of immune proteins in macrophages: A key to maternal tolerance of the fetal allograft? J Reprod Immunol. (2012) 94:131–41. doi: 10.1016/j.jri.2012.03.488
36. Geldenhuys J, Rossouw TM, Lombaard HA, Ehlers MM, Kock MM. Disruption in the regulation of immune responses in the placental subtype of preeclampsia. Front Immunol. (2018) 9:1659/BIBTEX. doi: 10.3389/FIMMU.2018.01659/BIBTEX
37. Wan J, Hu Z, Zeng K, Yin Y, Zhao M, Chen M, et al. The reduction in circulating levels of estrogen and progesterone in women with preeclampsia. Pregnancy Hypertens. (2018) 11:18–25. doi: 10.1016/j.preghy.2017.12.003
38. Yie SM, Li LH, Li YM, Librach C. HLA-G protein concentrations in maternal serum and placental tissue are decreased in preeclampsia. Am J Obstet Gynecol. (2004) 191:525–9. doi: 10.1016/j.ajog.2004.01.033
39. Faas MM, Spaans F, De Vos P. Monocytes and macrophages in pregnancy and pre-eclampsia. Front Immunol. (2014) 5:298. doi: 10.3389/fimmu.2014.00298
40. Boeldt DS, Bird IM. Vascular adaptation in pregnancy and endothelial dysfunction in preeclampsia. J Endocrinol. (2017) 232:R27–44. doi: 10.1530/JOE-16-0340
41. Ray JG, Austin PC, Park AL, Cohen E, Fang J, Chu A. Severity of obstructive coronary artery stenosis after pre-eclampsia. Heart. (2023) 109:449–56. doi: 10.1136/heartjnl-2022-321513
42. Heneghan HM, Miller N, McAnena OJ, O’Brien T, Kerin MJ. Differential miRNA expression in omental adipose tissue and in the circulation of obese patients identifies novel metabolic biomarkers. J Clin Endocrinol Metab. (2011) 96:E846–50. doi: 10.1210/jc.2010-2701
43. Boulanger CM. Endothelium. Arterioscler Thromb Vasc Biol. (2016) 36:e26–31. doi: 10.1161/ATVBAHA.116.306940
44. Ooi H, Chung W, Biolo A. Arterial stiffness and vascular load in heart failure. Congest Hear Fail. (2008) 14:31–6. doi: 10.1111/j.1751-7133.2008.07210.x
45. Henein MY, Vancheri S, Longo G, Vancheri F. The role of inflammation in cardiovascular disease. Int J Mol Sci. (2022) 23. doi: 10.3390/IJMS232112906
46. Vinh A, Drummond GR, Sobey CG. Immunity and hypertension: New targets to lighten the pressure. Br J Pharmacol. (2019) 176:1813–7. doi: 10.1111/BPH.14659
47. Benson LN, Liu Y, Wang X, Xiong Y, Rhee SW, Guo Y, et al. The IFNγ-PDL1 pathway enhances CD8T-DCT interaction to promote hypertension. Circ Res. (2022) 130:1550–64. doi: 10.1161/CIRCRESAHA.121.320373
48. Poller W, Heidecker B, Ammirati E, Kuss AW, Tzvetkova A, Poller WC, et al. Innate immunity in cardiovascular diseases—Identification of novel molecular players and targets. J Clin Med. (2023) 12:335. doi: 10.3390/JCM12010335
49. Jaén RI, Val-Blasco A, Prieto P, Gil-Fernández M, Smani T, López-Sendón JL, et al. Innate immune receptors, key actors in cardiovascular diseases. JACC Basic to Transl Sci. (2020) 5:735. doi: 10.1016/J.JACBTS.2020.03.015
50. Wang L, Qu P, Zhao J, Chang Y. NLRP3 and downstream cytokine expression elevated in the monocytes of patients with coronary artery disease. Arch Med Sci. (2014) 10:791–800. doi: 10.5114/aoms.2014.44871
51. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
52. Son S, Yoon SH, Chae BJ, Hwang I, Shim DW, Choe YH, et al. Neutrophils facilitate prolonged inflammasome response in the DAMP-rich inflammatory milieu. Front Immunol. (2021) 12:746032. doi: 10.3389/fimmu.2021.746032
53. Oh ES, Na M, Rogers CJ. The association between monocyte subsets and cardiometabolic disorders/cardiovascular disease: A systematic review and meta-analysis. Front Cardiovasc Med. (2021) 8:640124/FULL. doi: 10.3389/FCVM.2021.640124/FULL
54. Angkananard T, Anothaisintawee T, McEvoy M, Attia J, Thakkinstian A. Neutrophil lymphocyte ratio and cardiovascular disease risk: A systematic review and meta-analysis. BioMed Res Int. (2018) 2018. doi: 10.1155/2018/2703518
55. Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. (1983) 67:1016–23. doi: 10.1161/01.CIR.67.5.1016
56. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17:327–40. doi: 10.1038/s41569-019-0326-7
57. Kaptoge S, Seshasai SRK, Gao P, Freitag DF, Butterworth AS, Borglykke A, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J. (2014) 35:578–89. doi: 10.1093/EURHEARTJ/EHT367
58. Cheng M, Li Y, Wu J, Nie Y, Li L, Liu X, et al. IL-8 induces imbalances between nitric oxide and endothelin-1, and also between plasminogen activator inhibitor-1 and tissue-type plasminogen activator in cultured endothelial cells. Cytokine. (2008) 41:9–15. doi: 10.1016/j.cyto.2007.10.006
59. Bhagat K, Vallance P. Inflammatory cytokines impair endothelium-dependent dilatation in human veins. vivo. Circ. (1997) 96:3042–7. doi: 10.1161/01.CIR.96.9.3042
60. Tsioumpekou M, Krijgsman D, Leusen JHW, Olofsen PA. The role of cytokines in neutrophil development, tissue homing, function and plasticity in health and disease. Cells. (2023) 12. doi: 10.3390/cells12151981
61. Suzuki J, Hamada E, Shodai T, Kamoshida G, Kudo S, Itoh S, et al. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int Arch Allergy Immunol. (2013) 160:152–60. doi: 10.1159/000339857
62. Faas MM, de Vos P. Maternal monocytes in pregnancy and preeclampsia in humans and in rats. J Reprod Immunol. (2017) 119:91–7. doi: 10.1016/j.jri.2016.06.009
63. Barden A, Ritchie J, Walters B, Michael C, Rivera J, Mori T, et al. Study of plasma factors associated with neutrophil activation and lipid peroxidation in preeclampsia. Hypertension. (2001) 38:803–8. doi: 10.1161/hy1101.092969
64. Sabatier F, Bretelle F, D’Ercole C, Boubli L, Sampol J, Dignat-George F. Neutrophil activation in preeclampsia and isolated intrauterine growth restriction. Am J Obstet Gynecol. (2000) 183:1558–63. doi: 10.1067/mob.2000.108082
65. Fani L, van der Willik KD, Bos D, Leening MJG, Koudstaal PJ, Rizopoulos D, et al. The association of innate and adaptive immunity, subclinical atherosclerosis, and cardiovascular disease in the Rotterdam Study: A prospective cohort study. PloS Med. (2020) 17. doi: 10.1371/journal.pmed.1003115
66. Bekkering S, Quintin J, Joosten LAB, van der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887/-/DC1
67. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
68. Van Der Valk FM, Bekkering S, Kroon J, Yeang C, Van Den Bossche J, Van Buul JD, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134:611–24. doi: 10.1161/CIRCULATIONAHA.116.020838
69. Van Der Heijden CDCC, Groh L, Keating ST, Kaffa C, Noz MP, Kersten S, et al. Catecholamines induce trained immunity in monocytes. In Vitro In Vivo. Circ Res. (2020) 127:269–83. doi: 10.1161/CIRCRESAHA.119.315800
70. Van Der Heijden CDCC, Keating ST, Groh L, Joosten LAB, Netea MG, Riksen NP. Aldosterone induces trained immunity: the role of fatty acid synthesis. Cardiovasc Res. (2020) 116:317–28. doi: 10.1093/CVR/CVZ137
71. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourão-Sa D, Fortes GB, et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem. (2010) 285:32844–51. doi: 10.1074/jbc.M110.146076
72. Håversen L, Sundelin JP, Mardinoglu A, Rutberg M, Ståhlman M, Wilhelmsson U, et al. Vimentin deficiency in macrophages induces increased oxidative stress and vascular inflammation but attenuates atherosclerosis in mice. Sci Rep. (2018) 8. doi: 10.1038/S41598-018-34659-2
73. Crişan TO, Cleophas MCP, Novakovic B, Erler K, Van De Veerdonk FL, Stunnenberg HG, et al. Uric acid priming in human monocytes is driven by the AKT-PRAS40 autophagy pathway. Proc Natl Acad Sci U.S.A. (2017) 114:5485–90. doi: 10.1073/pnas.1620910114
74. Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T, et al. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol. (2017) 18:622–32. doi: 10.1038/NI.3745
75. Valdés-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Li J, et al. High-mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: Evidence from a murine model. Shock. (2013) 40:492–5. doi: 10.1097/SHK.0000000000000050
76. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. (2020) 11:484. doi: 10.3389/fimmu.2020.00484
77. Mineo C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc Res. (2020) 116:1254–74. doi: 10.1093/CVR/CVZ338
78. Romao-Veiga M, Ribeiro VR, Matias ML, Nunes PR, Romagnoli GG, Peracoli JC, et al. DAMPs are able to skew CD4+ T cell subsets and increase the inflammatory profile in pregnant women with preeclampsia. J Reprod Immunol. (2022) 149. doi: 10.1016/j.jri.2021.103470
79. Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. Immunometabolic crosstalk: an ancestral principle of trained immunity? Trends Immunol. (2019) 40:1–11. doi: 10.1016/j.it.2018.11.002
80. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U.S.A. (2014) 111:2247–52. doi: 10.1073/pnas.1322269111
81. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. (2016) 17:2562–71. doi: 10.1016/j.celrep.2016.11.011
82. Incalcaterra S, Dominguez JA. Trained Immunity at a Glance; A Review on the Innate Immune Memory and its Potential Role in Infections, Diseases and New Therapeutic Strategies. Adv J Grad Res. (2020) 8:68–81. doi: 10.21467/ajgr.8.1.68-81
83. Keating ST, Groh L, Thiem K, Bekkering S, Li Y, Matzaraki V, et al. Rewiring of glucose metabolism defines trained immunity induced by oxidized low-density lipoprotein. J Mol Med. (2020) 98:819–31. doi: 10.1007/S00109-020-01915-W/FIGURES/6
84. Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang SY, Oosting M, et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe. (2018) 23:89–100.e5. doi: 10.1016/j.chom.2017.12.010
85. Gervasi MT, Chaiworapongsa T, Pacora P, Naccasha N, Yoon BH, Maymon E, et al. Phenotypic and metabolic characteristics of monocytes and granulocytes in preeclampsia. Am J Obstet Gynecol. (2001) 185:792–7. doi: 10.1067/mob.2001.117311
86. Bernier E, Couture C, Borchers A, Brien M-E, Graham CH, Girard S. Circulating immune cells from early- and late-onset pre-eclampsia displays distinct profiles with differential impact on endothelial activation. J Immunol. (2024) 213:1292–304. doi: 10.4049/JIMMUNOL.2400196
87. Lodge-Tulloch NA, Paré JF, Couture C, Bernier E, Cotechini T, Girard S, et al. Maternal innate immune reprogramming after complicated pregnancy. Am J Reprod Immunol. (2024) 92:e13908. doi: 10.1111/aji.13908
88. Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J, et al. Monocytic myeloid-derived suppressor cells accumulate in renal transplant patients and mediate CD4+Foxp3+ treg expansion. Am J Transplant. (2013) 13:3123–31. doi: 10.1111/ajt.12461
89. Hegde S, Leader AM, Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. (2021) 54:875–84. doi: 10.1016/j.immuni.2021.04.004
90. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4+CD25+Foxp3+ T cells. Gastroenterology. (2008) 135:234–43. doi: 10.1053/j.gastro.2008.03.020
91. Soler DC, Young AB, Fiessinger L, Galimberti F, Debanne S, Groft S, et al. Increased, but functionally impaired, CD14+ HLA-DR–/low myeloid-derived suppressor cells in psoriasis: A mechanism of dysregulated T cells. J Invest Dermatol. (2016) 136:798–808. doi: 10.1016/j.jid.2015.12.036
92. Li J, Huang L, Wang S, Zhang Z. The prevalence of regulatory T and dendritic cells is altered in peripheral blood of women with pre-eclampsia. Pregnancy Hypertens. (2019) 17:233–40. doi: 10.1016/j.preghy.2019.07.003
93. Robson A, Harris LK, Innes BA, Lash GE, Aljunaidy MM, Aplin JD, et al. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J. (2012) 26:4876–85. doi: 10.1096/fj.12-210310
94. Wallace AE, Fraser R, Cartwright JE. Extravillous trophoblast and decidual natural killer cells: A remodelling partnership. Hum Reprod Update. (2012) 18:458–71. doi: 10.1093/humupd/dms015
95. Gamliel M, Goldman-Wohl D, Isaacson B, Gur C, Stein N, Yamin R, et al. Trained memory of human uterine NK cells enhances their function in subsequent pregnancies. Immunity. (2018) 48:951–962.e5. doi: 10.1016/j.immuni.2018.03.030
96. Zhang J, Dunk CE, Shynlova O, Caniggia I, Lye SJ. TGFb1 suppresses the activation of distinct dNK subpopulations in preeclampsia. EBioMedicine. (2019) 39:531–9. doi: 10.1016/J.EBIOM.2018.12.015
97. Bartsch E, Medcalf KE, Park AL, Ray JG, Al-Rubaie ZTA, Askie LM, et al. Clinical risk factors for pre-eclampsia determined in early pregnancy: Systematic review and meta-analysis of large cohort studies. BMJ. (2016) 353. doi: 10.1136/bmj.i1753
98. Viel S, Besson L, Charrier E, Marçais A, Disse E, Bienvenu J, et al. Alteration of Natural Killer cell phenotype and function in obese individuals. Clin Immunol. (2017) 177:12–7. doi: 10.1016/J.CLIM.2016.01.007
99. Kim JH, Park K, Lee SB, Kim MK, Nam JS, Kang S, et al. Relationship between NK cell activity and glucose regulation in type 2 diabetes patients. Diabetes. (2018) 67. doi: 10.2337/DB18-2414-PUB
100. Lee YK, Suh E, Oh H, Haam JH, Kim YS. Decreased natural killer cell activity as a potential predictor of hypertensive incidence. Front Immunol. (2024) 15:1376421. doi: 10.3389/fimmu.2024.1376421
102. Zhang Z, Zhao L, Zhou X, Meng X, Zhou X. Role of inflammation, immunity, and oxidative stress in hypertension: New insights and potential therapeutic targets. Front Immunol. (2023) 13:1098725. doi: 10.3389/FIMMU.2022.1098725
103. Calle MC, Fernandez ML. Inflammation and type 2 diabetes. Diabetes Metab. (2012) 38:183–91. doi: 10.1016/j.diabet.2011.11.006
104. Khanna D, Khanna S, Khanna P, Kahar P, Patel BM. Obesity: A chronic low-grade inflammation and its markers. Cureus. (2022) 14. doi: 10.7759/cureus.22711
105. Leentjens J, Bekkering S, Joosten LAB, Netea MG, Burgner DP, Riksen NP. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ Res. (2018) 122:664–9. doi: 10.1161/CIRCRESAHA.117.312465
106. Li N, He F, Gao H, Ge Y, Fan X, Zhang J, et al. Elevated cell-free fetal DNA contributes to placental inflammation and antiangiogenesis via AIM2 and IFI16 during pre-eclampsia. J Cell Physiol. (2020) 235:9577–88. doi: 10.1002/jcp.29766
107. Travers A, Muskhelishvili G. DNA structure and function. FEBS J. (2015) 282:2279–95. doi: 10.1111/febs.13307
108. Scharfe-Nugent A, Corr SC, Carpenter SB, Keogh L, Doyle B, Martin C, et al. TLR9 provokes inflammation in response to fetal DNA: mechanism for fetal loss in preterm birth and preeclampsia. J Immunol. (2012) 188:5706–12. doi: 10.4049/jimmunol.1103454
109. Pandey D, Yevale A, Naha R, Kuthethur R, Chakrabarty S, Satyamoorthy K. Mitochondrial DNA copy number variation – A potential biomarker for early onset preeclampsia. Pregnancy Hypertens. (2021) 23:1–4. doi: 10.1016/j.preghy.2020.10.002
110. Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. (2016) 85:133–60. doi: 10.1146/annurev-biochem-060815-014402
111. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. (2010) 464:104–7. doi: 10.1038/nature08780
112. Sandrim VC, Montenegro MF, Palei ACT, Metzger IF, Sertorio JTC, Cavalli RC, et al. Increased circulating cell-free hemoglobin levels reduce nitric oxide bioavailability in preeclampsia. Free Radic Biol Med. (2010) 49:493–500. doi: 10.1016/j.freeradbiomed.2010.05.012
113. Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: Implications for kidney disease. J Am Soc Nephrol. (2007) 18:414–20. doi: 10.1681/ASN.2006080894
114. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
115. Celik S, Soyer Çalışkan C. Predictive value of early second trimester maternal serum cyclophilin A concentrations in women complicated with preeclampsia: a preliminary case–control study. Hypertens Pregnancy. (2020) 39:387–92. doi: 10.1080/10641955.2020.1777301
116. Jin S, Zhang M, Qiao X. Cyclophilin A: promising target in cancer therapy. Cancer Biol Ther. (2024) 25. doi: 10.1080/15384047.2024.2425127
117. Bai X, Yang W, Zhao Y, Cao T, Lin R, Jiao P, et al. The extracellular cyclophilin A-integrin β2 complex as a therapeutic target of viral pneumonia. Mol Ther. (2024) 32:1510–25. doi: 10.1016/j.ymthe.2024.03.008
118. Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. (2010) 11:579–92. doi: 10.1038/nrm2941
119. Asea A, Rehli M, Kabingu E, Boch JA, Baré O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. (2002) 277:15028–34. doi: 10.1074/JBC.M200497200
120. Romão-Veiga M, Bannwart-Castro CF, Borges VTM, Golim MA, Peraçoli JC, Peraçoli MTS. Increased TLR4 pathway activation and cytokine imbalance led to lipopolysaccharide tolerance in monocytes from preeclamptic women. Pregnancy Hypertens. (2020) 21:159–65. doi: 10.1016/j.preghy.2020.06.002
121. Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): The prototypical endogenous danger molecule. Mol Med. (2015) 21:S6–S12. doi: 10.2119/molmed.2015.00087
122. Hu Y, Li H, Yan R, Wang C, Wang Y, Zhang C, et al. Increased neutrophil activation and plasma DNA levels in patients with pre-eclampsia. Thromb Haemost. (2018) 118:2064–73. doi: 10.1055/s-0038-1675788
123. Müller MM, Muir TW. Histones: At the crossroads of peptide and protein chemistry. Chem Rev. (2015) 115:2296–349. doi: 10.1021/cr5003529
124. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol. (2012) 23:1375–88. doi: 10.1681/ASN.2011111077/-/DCSUPPLEMENTAL
125. Wu H, Bao H, Liu C, Zhang Q, Huang A, Quan M, et al. Extracellular Nucleosomes Accelerate Microglial Inflammation via C-Type Lectin Receptor 2D and Toll-Like Receptor 9 in mPFC of Mice With Chronic Stress. Front Immunol. (2022) 13:854202. doi: 10.3389/fimmu.2022.854202
126. Stecco A, Bonaldi L, Fontanella CG, Stecco C, Pirri C. The effect of mechanical stress on hyaluronan fragments’ Inflammatory cascade: clinical implications. Life 2023 Vol 13 Page 2277. (2023) 13:2277. doi: 10.3390/LIFE13122277
127. Murakami T, Otsuki S, Okamoto Y, Nakagawa K, Wakama H, Okuno N, et al. Hyaluronic acid promotes proliferation and migration of human meniscus cells via a CD44-dependent mechanism. Connect Tissue Res. (2019) 60:117–27. doi: 10.1080/03008207.2018.1465053
128. Braekke K, Holthe MR, Harsem NK, Fagerhol MK, Staff AC. Calprotectin, a marker of inflammation, is elevated in the maternal but not in the fetal circulation in preeclampsia. Am J Obstet Gynecol. (2005) 193:227–33. doi: 10.1016/j.ajog.2004.11.055
129. Nakashige TG, Zhang B, Krebs C, Nolan EM. Human calprotectin is an iron-sequestering host-defense protein. Nat Chem Biol. (2015) 11:765–71. doi: 10.1038/nchembio.1891
130. Nishikawa Y, Kajiura Y, Lew JH, Kido JI, Nagata T, Naruishi K. Calprotectin induces IL-6 and MCP-1 production via toll-like receptor 4 signaling in human gingival fibroblasts. J Cell Physiol. (2017) 232:1862–71. doi: 10.1002/jcp.25724
131. Maiuolo J, Oppedisano F, Gratteri S, Muscoli C, Mollace V. Regulation of uric acid metabolism and excretion. Int J Cardiol. (2016) 213:8–14. doi: 10.1016/j.ijcard.2015.08.109
132. Eleftheriadis T, Pissas G, Karioti A, Antoniadi G, Golfinopoulos S, Liakopoulos V, et al. Uric acid induces caspase-1 activation, IL-1β secretion and P2X7 receptor dependent proliferation in primary human lymphocytes. Hippokratia. (2013) 17:141–5.
133. Shao J, Zhao M, Tong M, Wei J, Wise MR, Stone P, et al. Increased levels of HMGB1 in trophoblastic debris may contribute to preeclampsia. Reproduction. (2016) 152:775–84. doi: 10.1530/REP-16-0083
134. Pineda A, Verdin-Terán SL, Camacho A, Moreno-Fierros L. Expression of toll-like receptor TLR-2, TLR-3, TLR-4 and TLR-9 is increased in placentas from patients with preeclampsia. Arch Med Res. (2011) 42:382–91. doi: 10.1016/j.arcmed.2011.08.003
135. Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, et al. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PloS One. (2013) 8. doi: 10.1371/journal.pone.0059989
136. Lv Z, Lv DY, Meng JY, Sha XY, Qian XY, Chen YS, et al. Trophoblastic mitochondrial DNA induces endothelial dysfunction and NLRP3 inflammasome activation: Implications for preeclampsia. Int Immunopharmacol. (2023) 114:109523. doi: 10.1016/j.intimp.2022.109523
137. Bowyer L. Forearm blood flow in pre-eclampsia. BJOG Int J Obstet Gynaecol. (2003) 110:383–91. doi: 10.1016/s1470-0328(02)02930-0
138. Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. (2004) 350:672–83. doi: 10.1056/NEJMoa031884
139. Cindrova-Davies T, Sanders DA, Burton GJ, Charnock-Jones DS. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor-mediated signalling. Cardiovasc Res. (2011) 89:671–9. doi: 10.1093/cvr/cvq346
140. Tanaka H, Ozawa R, Henmi Y, Hosoda M, Karasawa T, Takahashi M, et al. Gasdermin D regulates soluble fms-like tyrosine kinase 1 release in macrophages. Reprod Biol. (2024) 24:100857. doi: 10.1016/j.repbio.2024.100857
141. Romão-Veiga M, Matias ML, Ribeiro VR, Nunes PR, Vera VT, Peraçoli JC, et al. Induction of systemic inflammation by hyaluronan and hsp70 in women with pre-eclampsia. Cytokine. (2018) 105:23–31. doi: 10.1016/j.cyto.2018.02.007
142. Matias ML, Romão M, Weel IC, Ribeiro VR, Nunes PR, Borges VT, et al. Endogenous and uric acid-induced activation of NLRP3 inflammasome in pregnant women with preeclampsia. PloS One. (2015) 10. doi: 10.1371/journal.pone.0129095
143. Phulphagar K, Kühn LI, Ebner S, Frauenstein A, Swietlik JJ, Rieckmann J, et al. Proteomics reveals distinct mechanisms regulating the release of cytokines and alarmins during pyroptosis. Cell Rep. (2021) 34. doi: 10.1016/j.celrep.2021.108826
144. Slesnick L, Birch MN, Holmgren C, Harrison R. Preterm preeclampsia and thromboembolism risk in pregnant and postpartum patients. Am J Obstet Gynecol. (2023) 228:S514. doi: 10.1016/j.ajog.2022.11.880
145. Vorobjeva NV, Chernyak BV. NETosis: molecular mechanisms, role in physiology and pathology. Biochem. (2020) 85:1178–90. doi: 10.1134/S0006297920100065
146. Guillotin F, Fortier M, Portes M, Demattei C, Mousty E, Nouvellon E, et al. Vital NETosis vs. suicidal NETosis during normal pregnancy and preeclampsia. Front Cell Dev Biol. (2023) 10:1099038. doi: 10.3389/fcell.2022.1099038
147. Noubouossie DF, Whelihan MF, Bin Y, Sparkenbaugh E, Pawlinski R, Monroe DM, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood. (2017) 129:1021–9. doi: 10.1182/blood-2016-06-722298
148. Zhan Y, Ling Y, Deng Q, Qiu Y, Shen J, Lai H, et al. HMGB1-mediated neutrophil extracellular trap formation exacerbates intestinal ischemia/reperfusion-induced acute lung injury. J Immunol. (2022) 208:968–78. doi: 10.4049/jimmunol.2100593
149. Wang J, Liu YM, Hu J, Chen C. Trained immunity in monocyte/macrophage: Novel mechanism of phytochemicals in the treatment of atherosclerotic cardiovascular disease. Front Pharmacol. (2023) 14:1109576. doi: 10.3389/fphar.2023.1109576
150. Cirovic B, de Bree LCJ, Groh L, Blok BA, Chan J, van der Velden WJFM, et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe. (2020) 28:322–334.e5. doi: 10.1016/j.chom.2020.05.014
151. Jentho E, Ruiz-Moreno C, Novakovic B, Kourtzelis I, Megchelenbrink WL, Martins R, et al. Trained innate immunity, long-lasting epigenetic modulation, and skewed myelopoiesis by heme. Proc Natl Acad Sci U.S.A. (2021) 118:e2102698118. doi: 10.1073/pnas.2102698118
152. von Wulffen M, Luehrmann V, Robeck S, Russo A, Fischer-Riepe L, van den Bosch M, et al. S100A8/A9-alarmin promotes local myeloid-derived suppressor cell activation restricting severe autoimmune arthritis. Cell Rep. (2023) 42:113006. doi: 10.1016/j.celrep.2023.113006
153. Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, et al. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. (2006) 8:1–12. doi: 10.1186/ar1939
154. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/J.IMMUNI.2012.12.001
155. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/ANNUREV-IMMUNOL-020711-074950
156. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille Calmette-Guérin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U.S.A. (2012) 109:17537–42. doi: 10.1073/PNAS.1202870109/SUPPL_FILE/PNAS.201202870SI.PDF
Keywords: preeclampsia, maternal cardiovascular health, long-lasting effects, trained immunity, DAMPs (damage-associated molecular pattern molecules)
Citation: Carrasco-Wong I, Sanchez JM, Gutierrez JA and Chiarello DI (2024) Trained innate immunity as a potential link between preeclampsia and future cardiovascular disease. Front. Endocrinol. 15:1500772. doi: 10.3389/fendo.2024.1500772
Received: 23 September 2024; Accepted: 27 November 2024;
Published: 16 December 2024.
Edited by:
Alicia E. Damiano, University of Buenos Aires, ArgentinaReviewed by:
Dimitris Grammatopoulos, University of Warwick, United KingdomBoris Novakovic, Royal Children’s Hospital, Australia
Andrea Canellada, University of Buenos Aires, Argentina
Copyright © 2024 Carrasco-Wong, Sanchez, Gutierrez and Chiarello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaime A. Gutierrez, amFpbWUuZ3V0aWVycmV6QHVzcy5jbA==; Delia I. Chiarello, ZGVsaWEuY2hpYXJlbGxvQHVzcy5jbA==