 Amelia Hurley-Novatny
Amelia Hurley-Novatny David Chang3
David Chang3 Ling Wang
Ling Wang Hongshuai Li
Hongshuai Li- 1Department of Orthopedics and Rehabilitation, University of Iowa, Iowa City, IA, United States
- 2Department of Anatomy and Cell Biology, University of Iowa, Iowa City, IA, United States
- 3Medical Scientist Training Program, Carver College of Medicine, University of Iowa, Iowa City, IA, United States
Duchenne muscular dystrophy (DMD) is a progressive, fatal muscle wasting disease caused by X-linked mutations in the dystrophin gene. Alongside the characteristic muscle weakness, patients face a myriad of skeletal complications, including osteoporosis/osteopenia, high susceptibility to vertebral and long bone fractures, fat embolism post-fracture, scoliosis, and growth retardation. Those skeletal abnormalities significantly compromise quality of life and are sometimes life-threatening. These issues were traditionally attributed to loss of ambulation and chronic corticosteroid use, but recent investigations have unveiled a more intricate etiology. Factors such as vitamin D deficiency, hormonal imbalances, systemic inflammation, myokine release from dystrophic muscle, and vascular dysfunction are emerging as significant contributors as well. This expanded understanding illuminates the multifaceted pathogenesis underlying skeletal issues in DMD. Present therapeutic options are limited and lack specificity. Advancements in understanding the pathophysiology of bone complications in DMD will offer promising avenues for novel treatment modalities. In this review, we summarize the current understanding of factors contributing to bone problems in DMD and delineate contemporary and prospective multidisciplinary therapeutic approaches.
1 Introduction
Duchenne Muscular Dystrophy (DMD) is a debilitating disease characterized by progressive muscle wasting due to X-linked recessive mutations in the dystrophin (DMD) gene (1). While the decline in motor function is well-documented, DMD patients face a myriad of other challenges, including intellectual disabilities, depression, delayed puberty, gastrointestinal complications, and skeletal abnormalities (2). Among these, skeletal issues such as osteoporosis, fragility fractures, scoliosis, short stature, and fracture-associated fat embolism (3) significantly impact patients’ quality of life (1, 2).
Poor skeletal health is one of the most overlooked aspects of this disease despite causing considerable morbidity. Low bone mineral density (BMD) (osteoporosis/osteopenia) is prevalent in DMD boys, especially those on glucocorticoid treatment, which poses a significant risk of pathological fractures (2, 4). Up to 60% of patients experience low-trauma extremity fractures (usually the distal femur, tibia, or fibula) by age 15 (5), and vertebral fractures occur in 30% to 60% patients (6). If left untreated, vertebral fractures can lead to chronic back pain and spine deformity, while leg fractures can cause premature, permanent loss of ambulation (2, 7, 8). Death due to fat embolism syndrome after long-bone fractures has also been reported in DMD boys (1, 3, 9). Moreover, reduced growth and short stature are also common (10, 11), further adding to the burden of skeletal issues in DMD.
The cause of poor bone health in DMD is complicated. It was previously thought that corticosteroid use and decreased mechanical stimulus from loss of ambulation (12), both of which are well-known to independently affect bone health (13), are the primary drivers of bone loss in DMD. These factors both substantially contribute to osteoporosis and increased fracture risk but do not completely explain the pathogenesis. Patients still develop osteoporosis and experience fractures without corticosteroid treatment and before loss of ambulation (12, 14). Thus, poor bone health cannot be explained solely by corticosteroid use or loss of ambulation, indicating that there are other mechanisms contributing to these aspects of disease. Indeed, recent evidence suggests that the pathogenesis of poor bone health is far beyond corticosteroid usage and muscle weakness, implicating nutritional deficiencies, hormonal imbalances, systemic inflammation, myokine release from dystrophic muscle, and vascular dysfunction in contributing to the deterioration of bone health to varying degrees, as summarized in Figure 1 (15–18). These factors, to varying degrees, disturb bone homeostasis by preventing bone deposition by osteoblasts and promoting bone resorption by osteoclasts, thus disrupting BMD and bone microarchitecture in DMD patients (18–21). Further understanding of these mechanisms will help us better understand DMD’s impact on bone health, identify novel therapeutic targets, develop therapeutic approaches, and better manage this debilitating issue.
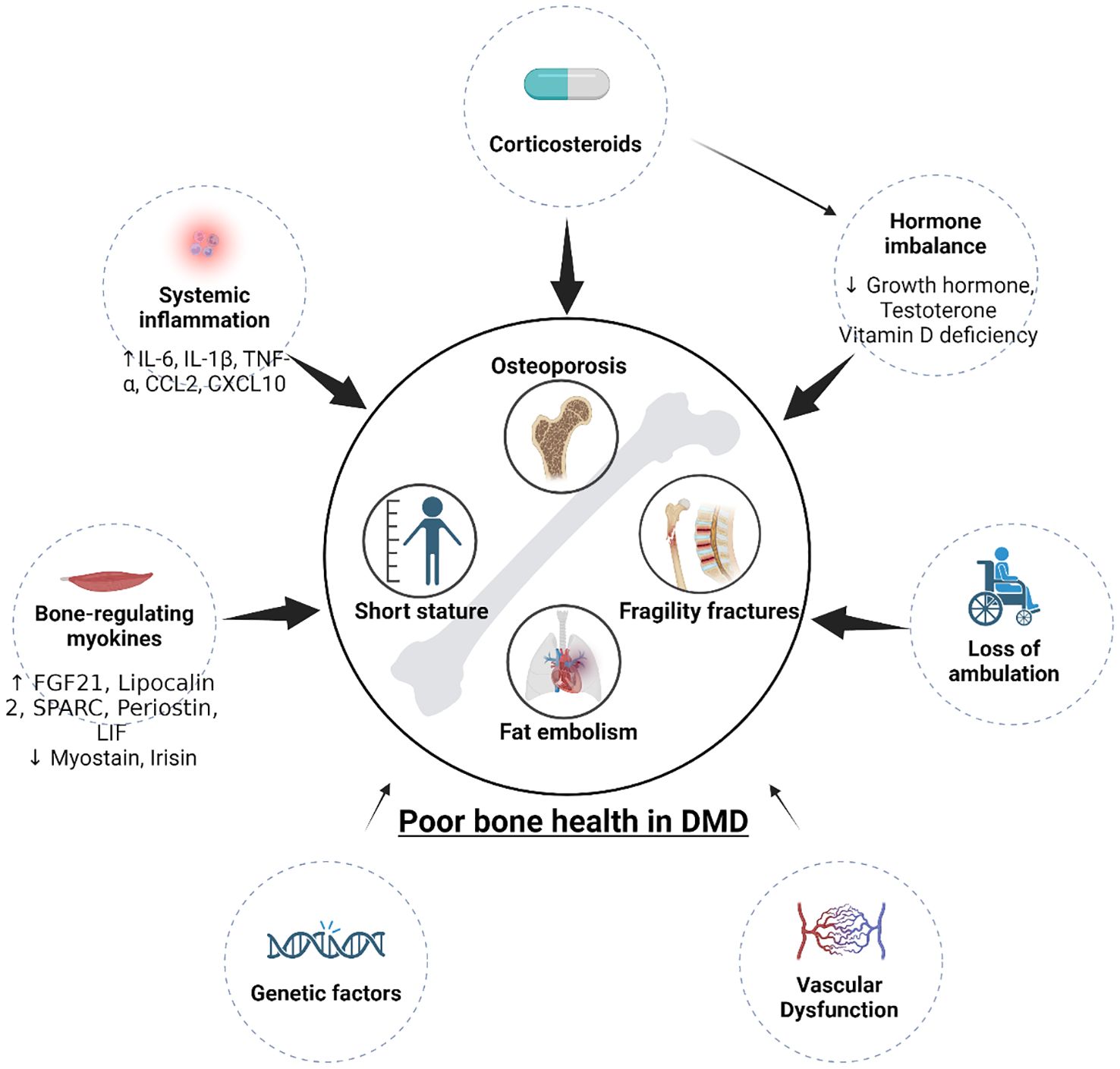
Figure 1. Pathogenesis (contributors) of poor bone health in DMD. Multiple factors contribute to short stature and osteoporosis. Corticosteroids, loss of ambulation, vascular dysfunction, hormone imbalance (GH and testosterone deficiencies), systemic inflammation, and dystrophic myokines all contribute to the poor bone health in DMD. Genetic factors that may contribute to poor bone health has not been explored.
We currently lack comprehensive guidelines for managing bone problems in DMD. The DMD Care Considerations Working Group (DCCWG) recommends lateral spine radiographs to detect vertebral fractures every 2-3 years in corticosteroid-naïve patients and every 1-2 years for those treated with corticosteroids (22). However, monitoring of bone mineral density (BMD) via dual energy X-ray absorptiometry (DEXA) scanning is not recommended as routine management since there is no established correlation between BMD and fracture risk for this population (2, 23). Therapeutic intervention in the form of intravenous bisphosphonates is primarily recommended after detection of severe pathological fractures (2). Excitingly, emerging strategies, including physical therapy (PT), innovative bone-sparing corticosteroids, anti-resorptive agents, hormone replacement therapy, and targeted antibodies offer promise for improving bone health in DMD.
Despite the significant impact on patient’s lives, skeletal health in DMD remains an understudied area. In this review, we aim to critically analyze existing evidence, providing an up-to-date assessment of risk factors, pathogenic mechanisms, and emerging therapeutic avenues for addressing skeletal issues in DMD.
2 Risk factors and mechanisms of poor bone health in DMD
2.1 Immobilization and reduced mechanical loading
As DMD progresses, patients experience a gradual loss of mobility, often leading to wheelchair use by the second decade of life. Bone health is closely linked to mechanical stimulus (13), with increased loading promoting bone deposition while chronic unloading favors bone resorption (24). As DMD advances, there is diminished force on the skeleton. This is especially pronounced in the transition from the ambulatory to non-ambulatory phase, after which there is a marked decrease in BMD (12). This reduction in mechanical loading has long been considered a primary driver of impaired bone health in DMD patients.
2.2 Corticosteroid side effects
Corticosteroids, namely prednisone and deflazacort, are mainstay therapies in DMD, albeit non-curative. Their use has revolutionized DMD treatment and significantly improved survival rates by delaying the need for mechanical ventilation and preserving cardiac function (6, 25–27). They prevent muscle deterioration, which is thought to be by improving sarcolemma healing and reducing inflammation (27–29). However, their long-term use is associated with significant adverse effects, including osteoporosis, pan-hormonal suppression, mood disturbances, and metabolic dysfunction (6, 30–33). Numerous studies have demonstrated increased fracture risk and growth retardation in corticosteroid-treated patients (5, 25, 30, 32, 34–37), with earlier initiation and longer treatment durations correlating with higher risk (36, 38, 39).
Long-term corticosteroid use is known to cause osteoporosis through both direct and indirect mechanisms. Corticosteroids directly inhibit bone formation and increase bone resorption by acting on osteoblasts and osteoclasts (40, 41), leading to impaired bone microarchitecture and low BMD (19). On average, 6-12% of BMD is lost in the first year of corticosteroid treatment and 3% is lost every year after that (41). Beyond their direct effects on osteoblasts and osteoclasts, corticosteroids impair intestinal calcium absorption (42) and contribute to vitamin deficiency (4, 43), which indirectly affect bone homeostasis.
Corticosteroids also significantly contribute to growth retardation in DMD patients. Although growth retardation occurs in corticosteroid-naïve DMD patients through mechanisms which are not fully understood (10, 44), corticosteroid usage significantly worsens this manifestation of the disease (39, 45–47). Corticosteroids exert direct toxicity to the growth plate by inducing chondrocyte apoptosis (48, 49). They further affect growth plate physiology by interfering with the growth hormone (GH) – insulin-like growth factor-1 (IGF-1) axis. Corticosteroids inhibit endogenous GH production (42, 50) and antagonize the peripheral action of GH and IGF-1 (51). Furthermore, GH and testosterone act synergistically to promote bone growth during puberty (52). Corticosteroid-induced hypogonadotropic hypogonadism and consequent testosterone deficiency further impair the partnership of the GH-IGF-1 axis and testosterone, which substantially contributes to corticosteroid-induced short stature if used before puberty (47, 52).
2.3 Vitamin D deficiency
Studies have shown that up to 84% of DMD patients have a documented vitamin D deficiency at some point, even with supplementation (5, 38). Multiple aspects of disease may contribute to this phenomenon. Loss of ambulation is linked to lower vitamin D levels, possibly due to reduced sunlight exposure (14). Corticosteroids may be a major contributor to vitamin D deficiency and resistance to supplementation by increasing breakdown (4, 43). DMD patients also have elevated fat mass, which increases vitamin D distribution into fat and reduces its availability (53–55). Additionally, lower serum vitamin D binding protein levels were observed in DMD patients, which reduces vitamin D stores, further explaining resistance to supplementation (56, 57). Despite a high prevalence of vitamin D deficiency in DMD patients, the relative impact of vitamin D supplementation on bone health in this patient population is still elusive. Retrospective and cohort studies have not consistently correlated supplementation with improved BMD or reduced fracture risk (5, 16), questioning the importance of vitamin D deficiency as a contributor to poor bone health in DMD. However, resistance to supplementation demonstrates a need for more effective strategies to reach adequate vitamin D levels in this population.
2.4 Systemic inflammation
Muscle degeneration and systemic inflammation are characteristic features of DMD (58, 59), leading to increased circulating levels of pro-inflammatory cytokines, which in turn negatively impact bone metabolism (60). Elevated levels of inflammatory cytokines, including interleukin (IL)-1β, IL-6, tumor necrosis factor α (TNF-α), chemokine ligand 2 (CCL2), and CXC motif chemokine ligand 10 (CXCL10) have been reported in DMD animal models and patients, which may contribute to bone loss in DMD (summarized in Table 1) (59, 61, 62). IL-1β, IL-6, and TNF-α all have been shown to increase bone resorption by stimulating osteoclast activity and differentiation (63). Likewise, CCL2 and CXCL10 also affect osteoclastogenesis and increase bone resorption (64–66). So far, the effects of these cytokines on bone metabolism have been mostly studied in inflammatory, autoimmune and cancer conditions. Only IL-6 has been directly studied in DMD, showing increased osteoclast formation in ex vivo murine calvaria bone cultures treated with mdx mouse serum, which was reduced following treatment with IL-6 neutralization antibody (7).
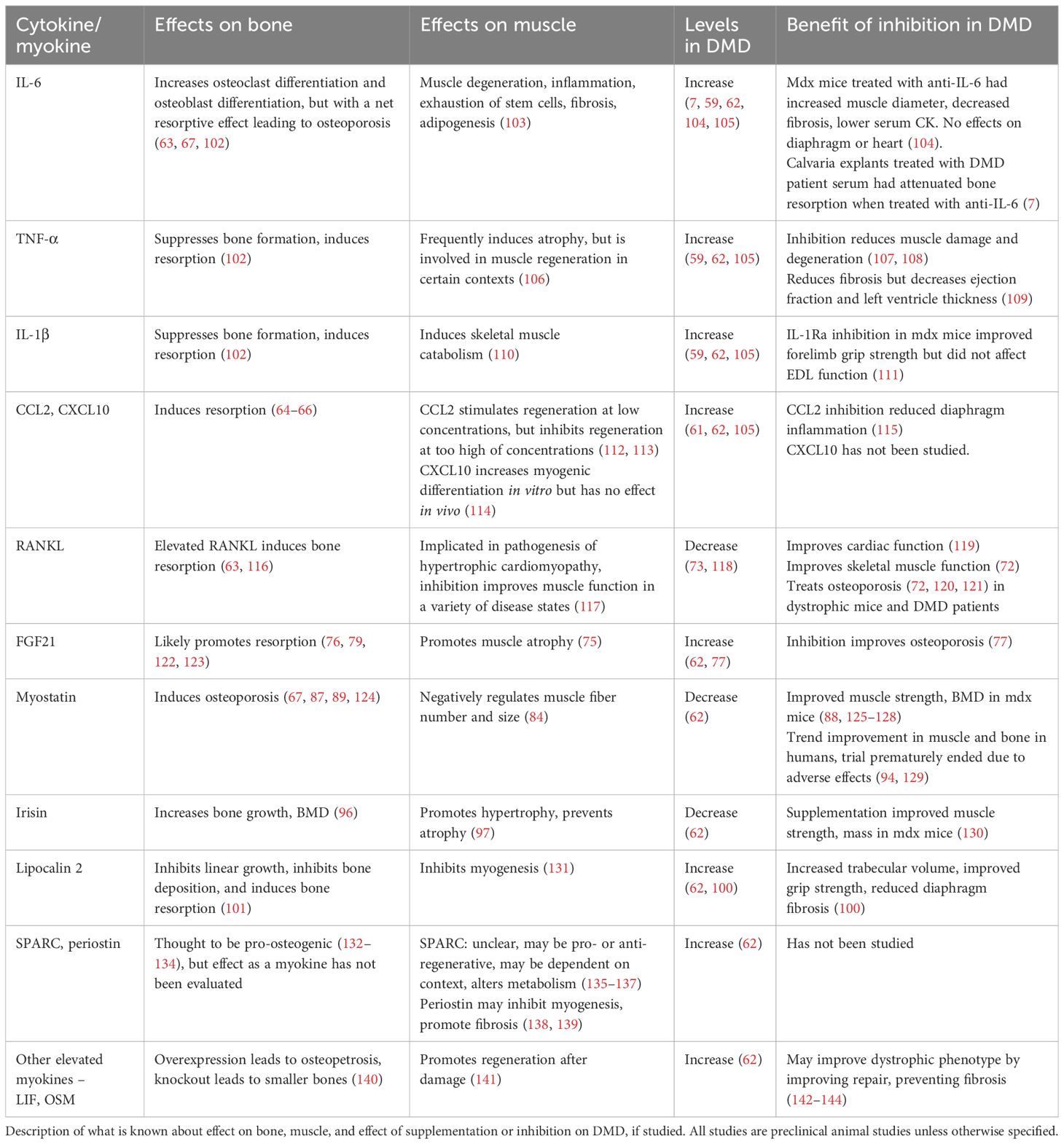
Table 1. Cytokines and myokines involved in bone loss in DMD.
2.5 Myokines
Recent studies have highlighted that myokines secreted from skeletal muscle exert regulatory effects on bone metabolism (67, 68). While the precise composition of myokines from dystrophic muscle remains to be fully elucidated, several myokines have been implicated in modulating bone turnover in DMD, including receptor activator of nuclear factor к B ligand (RANKL), fibroblast growth factor (FGF) 21, myostatin, lipocalin 2, and irisin (62). Understanding the roles of these myokines in DMD-associated osteoporosis is essential for exploring them as potential therapeutic targets. The current understanding of the roles of those myokines is summarized in Table 1.
2.5.1 RANKL
RANKL, in conjunction with its membrane receptor RANK and the soluble decoy receptor osteoprotegerin (OPG), plays a pivotal role in osteoclast differentiation and bone remodeling (69). Notably, the expression of RANK, RANKL, and OPG is not confined to bone; all three are also expressed in muscle (70, 71). Skeletal muscle in dystrophic mouse models has been shown to exhibit elevated RANKL expression (62). While the precise function of this axis in skeletal muscle remains elusive, systemic neutralization of RANKL in dystrophic mice improved both skeletal muscle function (70) and bone strength in mdx mice (21, 72), which suggests dual function of RANKL on skeletal muscle and bone. Despite one study finding paradoxically lower levels of RANKL, OPG, and RANKL/OPG ratio in patients compared to healthy controls (73), the concept that the RANKL/RANK/OPG system may interconnect bone and muscle in DMD warrants further investigation.
2.5.2 FGF21
FGF21 is a member of the endocrine FGF19 subfamily, an atypical group of FGFs secreted into systemic circulation, acting as endocrine hormones (74, 75). While FGF21 is primarily expressed by the liver and adipose tissue, it can also be secreted in large quantities from muscle tissue under pathological conditions, including mitochondrial dysfunction and muscular dystrophy (62, 76–78). In dystrophic mice, considerable amounts of FGF21 are expressed in muscle tissue and contribute to the elevated circulating levels of FGF21 (62). Blockade of FGF21 via neutralizing antibodies has shown promise in improving bone phenotype in a severe DMD mouse model (77). The impact of FGF21 on musculoskeletal system is largely unknown, with emerging evidence implicating that it may play important roles in homeostasis of both skeletal muscle and bone [as reviewed recently in (75)]. Although not fully understood, it is likely that FGF21 regulates bone homeostasis via multiple direct and indirect mechanisms. Recent studies have demonstrated that bone is a direct target of FGF21 since osteoclasts, bone marrow adipocytes, and mesenchymal stem cells express the obligate FGF21 co-receptor β-klotho (77, 79). Furthermore, FGF21 can interact with the GH/IGF-1 signaling pathway (80), which plays a crucial role in protein synthesis and bone homeostasis (81). FGF21 inhibits the action of GH/IFG-1 on proliferation and differentiation of chondrocytes directly at the growth plate (82, 83), potentially implicating its pathological role in growth retardation as well.
2.5.3 Myostatin
Myostatin, also known as growth differentiation factor 8 (GDF-8), is a member of the transforming growth factor-beta (TGF-β) superfamily and is known for its inhibitory effects on both muscle and bone growth (84–86). Myostatin acts as a catabolic stimulus, resulting in muscle fiber atrophy (17). In addition to its anti-myogenic effects, it negatively regulates bone homeostasis by inducing bone resorption (85, 87) by promoting osteoclastogenesis and inhibiting osteogenesis (86, 88). Notably, the effect of myostatin on bone loss are potentiated in the context of decreased mechanical loading (87, 89, 90). Despite downregulation of myostatin in mdx mice (62) and in DMD patients (91–93), myostatin inhibition has long been considered a promising intervention to reverse pathology in muscle-wasting diseases, including DMD (94). However, there has been a failure to translate positive results of myostatin inhibition in animal models to humans (94), which will be discussed in the treatment section (Section 3).
2.5.4 Irisin
Irisin, an exercise-induced myokine, is a cleaved product of secreted fibronectin type II domain containing 5 (FNDC5) (95). Unlike myostatin, irisin serves as an anabolic regulator, influencing both muscle growth and bone health (96, 97). However, the expression levels of irisin in dystrophic muscle, particularly in DMD, remains unclear. While one study reported mild decreases in serum irisin levels in Becker muscular dystrophy (98), others have found increased levels in exercised dystrophic mice (99). Another study noted reduced Fndc5 mRNA levels in skeletal muscle of dystrophic mice, but increased protein levels, indicating intricate posttranslational regulation of FNDC5/irisin in DMD (62). The function of irisin in regulating muscle and bone homeostasis in DMD is still unknown.
2.5.5 Lipocalin 2
Originally identified as an adipokine, elevated levels of lipocalin 2 have been reported in the muscles of mdx mice (100). Transgenic mice overexpressing lipocalin exhibit decreased BMD and short stature due to growth plate abnormalities (101), suggesting a potential role for lipocalin 2 in the development of osteoporosis and short stature in DMD. Inhibition of lipocalin in mdx mice via either Lcn2 knockout or the administration of lipocalin monoclonal antibody has shown increased trabecular volume, enhanced grip strength, and decreased diaphragm fibrosis (100), indicating that it may play an important role in both skeletal muscle and bone pathologies in DMD.
2.5.6 Other myokines
A plethora of other bone-regulating myokines have been identified as upregulated in dystrophic muscle. In addition to the previously mentioned myokines, leukemia inhibiting factor (LIF), secreted protein acidic and rich in cysteine (SPARC), and periostin have been found to be elevated at both the transcript and protein levels (62). However, their roles in regulating bone homeostasis in DMD or other conditions have not been studied.
Together, these findings collectively suggest that altered levels of multiple potent myokines contribute to the pathogenesis of DMD-associated bone loss. Notably, increased/decreased expression of both pro- and anti-osteogenic factors indicate a complex interplay within the skeletal muscle and bone microenvironment. Despite the complexity, the net effect of these factors is bone loss in DMD. Further research is warranted to elucidate the exact effects, underlying mechanisms, and relative contributions of these myokines, as well as to validate their clinical relevance. Understanding the interplay between these myokines and their impact on muscle function and bone health is crucial for developing targeted therapeutic interventions for DMD.
2.6 Vascular dysfunction
Bone is intricately connected to vascular function throughout its development, growth, maintenance, and healing processes (145). Vasculature is essential for adequate delivery of oxygen and nutrients, thus shaping the metabolic environment. Additionally, significant cross-talk exists between vascular endothelial cells and adjacent bone cells, with mutual influence on osteogenesis, bone resorption, and angiogenesis (145). Notably, impaired angiogenesis has been associated with osteoporosis (145).
Vascular dysfunction in skeletal muscle is well-documented in the context of DMD. Dystrophin, known for its structural role in cytoskeletal stabilization, binds to neuronal nitric oxide synthase (nNOS) in skeletal muscle fibers, facilitating nitric oxide (NO) synthesis for vasorelaxation (146). In DMD, the absence of dystrophin results in the loss of nNOS from the sarcolemma and its significant downregulation in the cytoplasm, leading to functional ischemia contributing to muscle damage and impaired regeneration (147). Dystrophic muscles exhibit not only vascular dysfunction, but also low vessel density and impaired angiogenesis (148–152). In addition to skeletal muscle fibers, smooth muscle cells (152–154) and endothelial cells (148, 150) also express full-length dystrophin, suggesting its crucial role in regulating vasodilation, vasocontraction, and angiogenesis [refer to recent review (155)]. However, little is known regarding the functional dysregulation of dystrophin expression out of skeletal muscle fibers in DMD. Furthermore, no studies have directly evaluated vasculature in dystrophic bone, even though it is reasonable to hypothesize that dystrophin mutations may affect bone vasculature via NOS. One study evaluating fracture healing in mdx mice observed decreased vessel density in early repair stages, correlating with delayed healing (18). The study also noted abnormally high inflammation after fracture (18), which brings to question whether impaired fracture healing was due to angiogenic dysfunction, excessive inflammation, or both. The question of whether bone exhibits vascular anomalies in DMD remains unanswered. A deeper understanding of the role of angiogenesis, vascular function, and NO• signaling in DMD pathogenesis in both muscle and bone may unveil potential targets for enhancing muscular, cardiovascular, and bone health.
3 Treatments: current and future
Current DMD treatment is negligent to bone health. While vitamin D is often used to prevent osteoporosis, most patients still experience osteoporosis and pathological fractures (5). Bisphosphonate use is typically reserved for use following pathological fractures. Disappointing statistics regarding rates of fracture, especially those which lead to loss of ambulation, indicate that current management of bone health is inadequate in DMD patients. Furthermore, patient age is important to consider in managing bone health in DMD. In pediatric patients, growth retardation is of great concern, while in adult patients, osteoporosis and pathological fracture are of most concern. Optimal timing of therapeutic intervention requires more research.
The intricate relationship between muscle and bone along with the impact of long-term corticosteroid usage necessitate an integrated, multi-factorial approach to the management of DMD bone issues. Combining strategies to address multiple aspects of disease while minimizing off-target effects of current treatment hold the most promise. As the field gains more understanding of disease pathogenesis, more effective and targeted therapeutic avenues can be explored. Below, we will discuss potential therapeutics at varying states of investigation, which are also summarized in Table 2. As research continues to advance, a more comprehensive toolkit of interventions is expected to emerge.
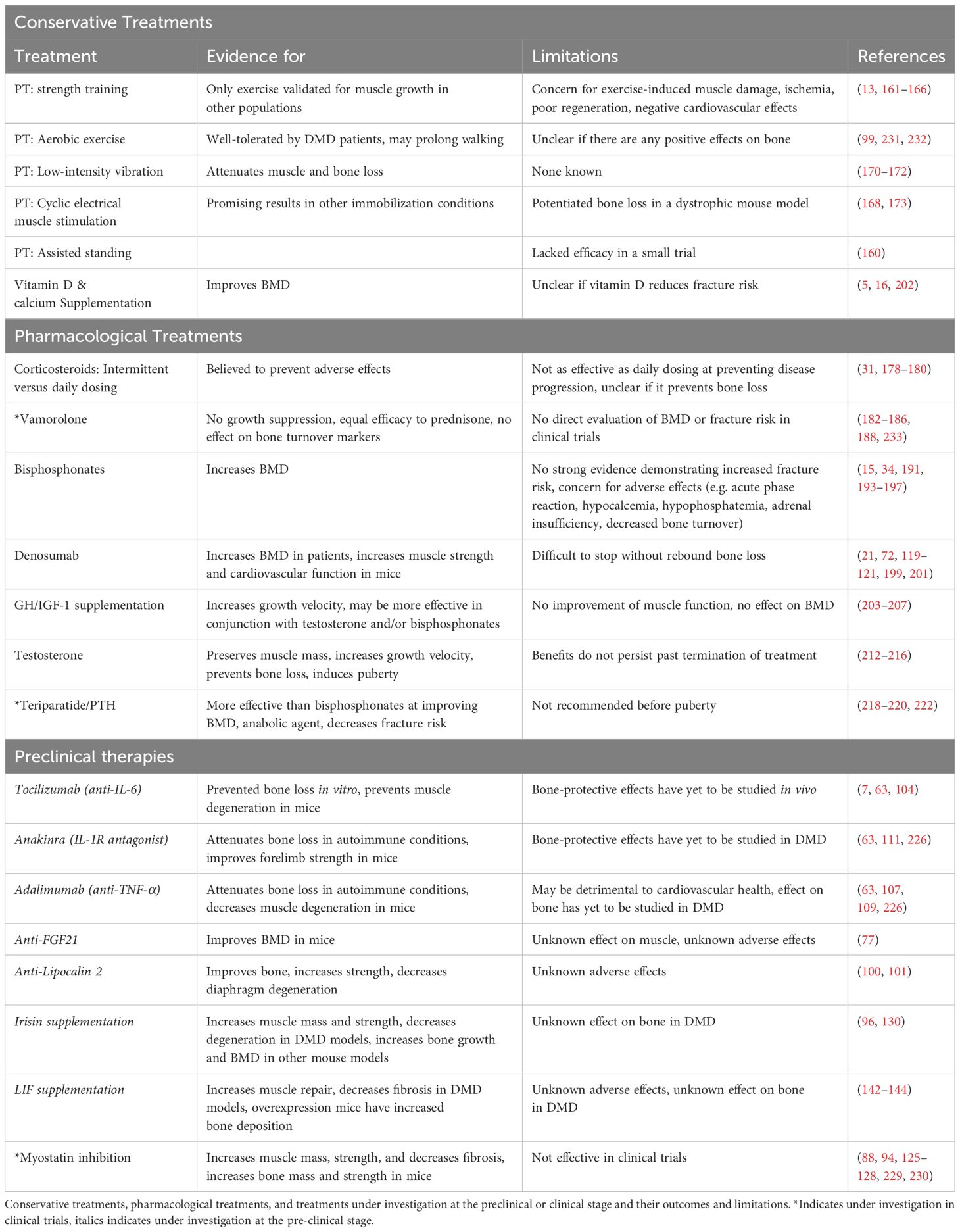
Table 2. Treatments for bone comorbidities in DMD.
3.1 Monitoring bone health and early detection of fractures
Due to the high incidence of bone issues in DMD patients, frequent monitoring of bone health in this patient population is important for early detection and intervention. Vertebral fracture affects 30-60% of patients, many of which are asymptomatic (2). Current guidelines emphasize early fracture detection, and thus recommend routine lateral spine radiographs at an interval of every 1-2 years in corticosteroid-treated patients and every 2-3 years in those who are corticosteroid-naïve (2). DEXA scans may be used as an adjacent method of monitoring BMD but are not of particular emphasis since there is no established cutoff for what may warrant intervention (1, 8). Additionally, BMD Z-score may be underestimated in DMD patients since they often have a younger bone age than chronological age, which could alter estimation of fracture risk (23). Early detection of fracture, however, does not prevent fracture. Further research should be done to correlate BMD with fracture risk to establish guidelines for when to start prophylactic treatment.
3.2 Physical therapy
Current guidelines published by the DCCWG recommend PT as an integral part of DMD management (22). However, there is no consensus on protocols, leading to inconsistent recommendations between physicians and physical therapists based solely on provider experience and anecdotal evidence (156). Moreover, the current focus of PT is to improve or maintain muscle function and is not focused on improving bone health. Currently, only low-intensity and aerobic exercise are recommended (156–158). Investigation into modalities such as low-intensity vibration and assisted standing have been explored with some benefit but are not yet standard of care (159, 160). All of them show some benefit to maintain muscle function in clinical studies [A more detailed summary of these studies can be found in a recent review by Spaulding and Selsby (157)]. Unfortunately, no studies have evaluated any type of exercise for improving bone health in DMD patients (156).
When discussing PT to improve bone health in healthy populations, high-load resistance training or repetitive high-impact movements are optimal to increase muscle mass and increase BMD (13). However, for DMD patients, there are several concerns, including exercise-induced muscle damage, ischemia, regenerative ability of muscle, and effects on cardiovascular function. It is recommended to provide sub-maximal loading to reduce the risk of muscle damage (156). However, the scientific basis of this recommendation is not definitive. Numerous studies have demonstrated that dystrophic muscle is more prone to damage following exercise than healthy muscle (161–163), and satellite cells in dystrophic muscle have impaired regenerative capacity (164–166). However, some studies also indicate that dystrophic muscle has a high functional regenerative capacity and recovers function well after contractile injury (162, 167), indicating that higher intense exercise may not lead to a functional deficiency as we thought. There is certainly more to investigate regarding exercise regimens and regenerative capacity of dystrophic muscle and how that may affect bone.
In addition to exercise, some modalities, such as low-intensity vibration, assisted standing, repetitive electrical muscle stimulation, and high-dose compressive loading, have shown benefits in other muscle-wasting and immobility related bone conditions (168, 169). Of these, electrical muscle stimulation and low-intensity vibration can also attenuate muscle loss (170–172), thus potentially addressing both muscle and bone problems in DMD. So far, electrical muscle stimulation has been evaluated in mice, while low-intensity vibration and assisted standing have been evaluated in humans. One study found that electrical muscle stimulation, which provides cyclic loading, worsened bone loss in mdx mice, potentially by increasing release of harmful myokines (173). This has not been evaluated in humans with DMD, so it is unclear if this result would hold. In patients, assisted standing, meant to return body-weight stimulus to bone, did not prevent bone loss in four wheelchair-bound DMD patients (160). Low-intensity vibration is FDA-approved to treat osteoporosis in adults and is thought to increase BMD by stimulating osteocytes and mesenchymal stem cells, although the exact mechanism is unclear (170, 171). It has also shown some efficacy in increasing BMD in DMD patients and in other pediatric populations and is generally well-tolerated (13, 159, 174), warranting further exploration of this modality.
The optimal PT protocols to promote maintenance or gain of muscle function and bone still require much more mechanistic and clinical exploration. The complexities of muscle damage and regeneration, its crosstalk with bone, differences in the response of muscle and bone to different types of exercise, and potential confounders of vascular dysfunction, leave many unanswered questions. The optimal protocol may not be intuitive, and likely should include additional modalities targeting bone.
3.3 Optimal corticosteroid treatment and alternatives
Corticosteroids play a pivotal role in preserving muscle function by mitigating inflammation; however, their usage is accompanied by a range of adverse effects, such as excessive weight gain, adrenal insufficiency, behavioral changes, Cushing’s syndrome, stunted growth, and osteoporosis. The optimization of corticosteroid administration including the choice of agent, dosing, timing, and frequency remains a dynamic area of research in the field, as recently reviewed by Biggar et al. (175).
Intermittent dosing of corticosteroids has been explored as an alternative to daily administration with hopes of mitigating side effects while maintaining efficacy. However, results have been inconsistent. Studies have found that intermittent dosing is at least better than placebo (176, 177), but it is not as effective at slowing disease progression as daily dosing (31, 178–180). One RCT of weekend versus daily dosing found a statistically significant increase in growth and BMD in the weekend group compared to the daily group, but patients’ heights and BMD were not statistically significantly different at the end of the study (179). Despite this being a theoretically appealing option, evidence suggests that intermittent dosing is inferior for disease progression and does not substantially reduce the risk of bone loss.
Ongoing efforts in treatment development focus on generating more selective corticosteroids to balance efficacy and side effects. Vamorolone is a synthetic corticosteroid which is more selective for the glucocorticoid receptor while antagonizing the mineralocorticoid receptor, which should theoretically reduce side effects, especially bone-related side effects (181). In preclinical studies, vamorolone improved skeletal muscle repair with minimal impact on hormonal regulation, growth, and immune suppression (181–183). Phase III clinical trials have shown comparable or superior efficacy to prednisone in retaining muscle function while preventing growth suppression (184–186). Vamorolone’s potential bone-sparing properties are also noteworthy, as evidenced by its neglectable effects on bone growth and trabecular thickness in mice (181, 187) and decreased markers of bone turnover in patients (186, 188). However, further clinical investigations are required to ascertain whether it is truly less detrimental to bone than prednisone.
3.4 Anti-resorptive therapy
Anti-resorptive therapies, such as bisphosphonates and denosumab, are widely used for osteoporosis treatment (189, 190). There is currently no consensus on the use of antiresorptive therapies to manage osteoporosis in DMD, but bisphosphonates are commonly used following severe fracture (2, 191). Bisphosphonates inhibit osteoclast attachment to the bone surface and induce cell apoptosis (190), while denosumab, a monoclonal antibody targeting RANKL, prevents osteoclast formation (192).
DCCWG recommends intravenous bisphosphonate treatment following a pathological fracture (vertebral Genant grade 2 or 3 or low-trauma long bone fracture) (2). However, it is still unclear whether bisphosphonates are beneficial as a prophylactic treatment. Landfeldt et al. recently published a systematic review on bisphosphonate use in corticosteroid-treated DMD patients (191). Notably, there is good quality evidence that bisphosphonate use increases BMD in DMD patients, but it is unclear if this translates to a lower fracture risk (15, 34, 191, 193, 194). Unfortunately, the rate of adverse effects in DMD patients is high, with acute phase reaction, hypocalcemia and hypophosphatemia, and precipitation of adrenal insufficiency of particular concern (191, 195). Of further concern, histomorphometric analyses indicate that bisphosphonates globally decrease bone turnover (1, 196, 197), which is problematic during bone development (198). Currently, bisphosphonates are one of the few options for treating osteoporosis but may not be the best option for young DMD patients.
Denosumab has also been used in pediatric populations (189) and has shown promising results in DMD. Case reports demonstrate improved BMD without notable side effects, indicating its relative effectiveness and tolerability in children (120, 121, 189). Notably, denosumab and other anti-RANKL therapies may also improve muscle strength and cardiac function, as reported in mouse models of DMD (21, 72, 119, 199). One consideration when starting denosumab treatment is that it cannot be stopped abruptly; its effects on BMD decline rapidly following termination of treatment (72, 200, 201). This is easily remedied by a short course of bisphosphonates (72, 200, 201). Given the favorable outcomes observed in both muscle and bone in DMD patients and animal models, further investigation into the use of denosumab for this population is warranted.
3.4 Vitamin D and calcium supplementation
Calcium and vitamin D supplementation are strongly recommended for DMD patients (22). DMD patients are particularly susceptible to vitamin D deficiency because of corticosteroid use, obesity, and low levels of vitamin D binding protein, meaning supplementation needs to be aggressive (2000 IU/day) to reach adequate levels in most patients (53). Cohort studies do not find an association between vitamin D supplementation and fracture risk, but an RCT found that adequate supplementation improved BMD (5, 16, 202). Although vitamin D supplementation alone does not fully prevent osteoporosis in DMD (5), due to its simplicity, low risk, and possible benefit, vitamin D supplementation should be a pillar of maintaining bone health in DMD patients.
3.5 Hormone therapy
Endocrine management is an important aspect of DMD treatment given the hormonal imbalances induced by corticosteroids. GH and testosterone therapy have been investigated to restore hormonal levels to normal ranges and promote linear growth. Additionally, to improve bone health, PTH therapy via teriparatide, a PTH analogue, has been explored as a strategy specifically to improve bone quality and prevent osteoporosis.
The role of GH in bone problems in DMD has been extensively studied, particularly due to growth retardation observed in DMD patients. Although there is no clear evidence of GH dysregulation in DMD patients who are not treated with corticosteroids (44), corticosteroid use is well-known to impair the GH axis and GH secretion (42, 51). Both GH supplementation and inhibition have been tested in DMD patients, but yielded controversial and inconclusive results. Supplementation, either via recombinant human GH (rhGH) or IGF-1, improved in growth velocity but failed to demonstrate motor function improvement (203–205). Moreover, there has been no documented improvement in BMD with GH or IGF-1 treatment in mdx mice or patients, possibly due to the short duration of these trials. GH secretagogues have been explored in mice, including ghrelin and the synthetic secretagogues EP80317 and JMV2894. Both studies demonstrated decreased muscle fibrosis and inflammation, and increased muscle strength (206, 207), but neither study evaluated the bone phenotype in these mice. Conversely, the idea that GH inhibition might be beneficial stemmed from the observation that a patient with DMD and GH deficiency had no clinical evidence of muscular weakness before initiation of GH replacement therapy; and treatment with human GH resulted in appearance of symptoms of easy fatigability and proximal muscle weakness, which suggest that increased growth may worsen muscle function in DMD (208). However, clinical trials investigating mazindol, a postulated inhibitor of growth hormone release, have shown inconsistent effects on growth suppression and no benefits on muscle strength (209, 210). In light of limited data and ongoing controversy, routine use of rhGH or secretagogues in DMD population is not recommended. Any decision to use rhGH should be made on a case-by-case assessment with biochemical evidence of growth hormone deficiency, weighing the risks and benefits.
Delayed or absent pubertal development in DMD patients on glucocorticoids is common due to hypogonadotropic hypogonadism (211). Testosterone therapy is recommended for pubertal induction in these individuals, typically initiated by age 14 (22, 212–214). Clinical trials have shown that testosterone therapy not only induces puberty but also preserves muscle mass, improves growth velocity, and prevents bone deterioration (212–216). However, these benefits are observed only during treatment. A recent follow up study on patients that were treated with testosterone for 2 years demonstrated persistent lower testosterone levels and testicular volumes than adult reference values; muscle functional volume increased during the intervention but declined in the years after cessation of supplementation (214). Given the known additional advantages of testosterone on bone health, muscle, and well-being (217), the benefits of restoring testosterone to normal physiologic levels are deemed to outweigh potential risks. Nevertheless, further research is warranted to elucidate the optimal timing, duration, and regimen for testosterone supplementation in DMD patients and its potential impact on bone health.
PTH administration in DMD mouse models improved or maintained BMD in several studies (218–220). Research on corticosteroid-induced bone loss suggests that PTH may be more effective than bisphosphonates (221). Although its safety and efficacy in improving bone health in DMD still need to be validated in clinical trials, a small cohort treated with teriparatide, a PTH analogue, demonstrated decreased fracture risk (218, 222).
As previously mentioned, multiple hormonal pathways are affected in DMD. This complexity prompted an investigation to evaluate the efficacy of hGH, testosterone, and zolendronic acid (ZA, a bisphosphonate) in various combinations after vertebral fractures (15). The study revealed that hGH and testosterone, either alone or in conjunction with ZA, significantly delayed the occurrence of subsequent vertebral fractures compared to ZA treatment alone. Moreover, multivariate analysis demonstrated that hGH was the greatest contributor in enhancing this protective effect (15). This study highlights the intricate hormonal imbalances contributing to osteoporosis in DMD patients, which point out the need for a holistic management strategy that targets these hormonal disruptions.
3.6 Novel anti-inflammatory and immunomodulatory agents
In addition to corticosteroids, monoclonal antibodies targeting specific immune modulators are increasingly used to treat inflammatory conditions. Tocilizumab, an anti-IL-6 receptor monoclonal antibody, is used to treat certain autoimmune conditions and may attenuate bone loss (223–225). Ex vivo and in vitro experiments found that tocilizumab rescued the anti-osteogenic/pro-osteoclastogenic effect induced by DMD patient serum (7). In mdx mice, tocilizumab treatment improved skeletal muscle phenotype by improving muscle diameter, reducing fibrosis, and decreasing serum CK, suggesting it has promise for altering disease progression (104). Although no studies have evaluated its effect on bone in vivo, this may be a promising avenue to treat muscle and bone degeneration. Anti-IL-1 receptor antagonist anakinra and anti-TNF-α monoclonal antibody adalimumab are also used to treat autoimmune diseases and may also attenuate bone loss (63, 226). Neither has been tested for effects on bone in DMD models, but they have been studied for their effects on muscle. Anti-TNF-α treatment decreased muscle damage, degeneration, and fibrosis in mdx mice, but appeared detrimental to cardiac function (107, 109). Anti-IL-1R treatment in mdx mice improved forelimb strength (111). The effects of these treatments on disease progression and osteoporosis in DMD requires more investigation, but thus far tocilizumab appears to be most promising.
3.7 Novel therapies targeting myokines
Myokines secreted from dystrophic muscle significantly contribute to the development of bone abnormalities in DMD. Targeting these myokines holds considerable promise for mitigating dystrophic bone loss. Of the myokines discussed, RANKL, FGF21, myostatin, irisin, and lipocalin 2 have been studied in mouse models. Only myostatin inhibition has been translated to the clinic.
In preclinical studies, inhibition of FGF21 or lipocalin 2, and supplementation of irisin or LIF have been studied with some promising results. Inhibition of FGF21 via neutralizing antibody increased BMD in a severe DMD mouse model by decreasing osteoclastogenesis (77). The effect of FGF21 inhibition on dystrophic muscle has yet to be determined. Lipocalin 2 has been studied in dystrophic mice by neutralizing antibody and global knockout of lcn2, which resulted in increased trabecular volume, grip strength, and reduced diaphragm fibrosis (100). A study by Reza et al. (130) revealed that recombinant irisin treatment increased muscle mass and strength in addition to reducing muscle fibrosis and necrosis, although its effects on bone are unknown. Several studies have also evaluated LIF supplementation, which improved dystrophic phenotype by improving repair and preventing fibrosis (142–144), but given its role in promoting solid tumor progression, further research should be cautious of potential oncogenic effects (227). With the popularity of monoclonal antibody treatments, inhibition of FGF21 and lipocalin 2 could be promising therapeutic targets. Irisin could be supplemented by recombinant protein or mimetics (97, 228). While the effects of FGF21 on muscle and irisin and LIF on bone are unstudied, their known roles in muscle and bone pathology warrant further investigation in pre-clinical models.
In preclinical studies involving DMD animal models, myostatin inhibition led to increased muscle mass and strength, and decreased fibrosis (125–128, 229), as well as improved bone mass and strength (88). However, these positive results have not translated to positive outcomes in clinical trials (230). Three distinct myostatin inhibitors have been developed and all have failed in clinical trials [as recently reviewed in (94, 230)]. The discrepancy of efficacy between animal models and humans is likely due to the differing expression levels of myostatin between mdx mice and human patients. Myostatin levels in mice are approximately 5 to 9-fold higher than in humans (124). Thus, further exploring direct inhibition of myostatin is unlikely to be fruitful. However, increased understanding of myostatin action, such as downstream signaling pathways, may provide alternative druggable targets.
4 Conclusions
The pathogenesis of bone abnormalities in DMD is multifactorial, extending beyond corticosteroid usage and loss of ambulation. While factors such as mechanical loading, corticosteroid usage, nutritional deficiencies, hormonal imbalances, systemic inflammation, and myokine dysregulation contribute to bone health deterioration in DMD patients, the precise interplay among these factors requires further elucidation. Moreover, the management of bone health in DMD necessitates a multidisciplinary approach that includes PT, nutritional supplementation, hormonal therapy, anti-inflammatory interventions, and potential myokine-targeted therapies. It is evident that a more comprehensive clinical management plan is imperative to address the complex pathophysiology of bone abnormalities in DMD and must take into account patient age. Future research efforts should focus on unraveling the underlying mechanisms driving bone pathology in DMD and developing tailored therapeutic strategies to mitigate bone loss and improve skeletal health outcomes.
Author contributions
AH-N: Writing – review & editing, Writing – original draft. DC: Writing – review & editing, Writing – original draft. KM: Writing – review & editing, Writing – original draft. LW: Writing – review & editing, Writing – original draft. HL: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Investigation, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. HL, LW, and AH-N receive research support from NIH grants 1R01AR076357 and 1R01AR083398. LW also receives research support from NIH grant 5R01HL098032. AH-N and DC are also supported by the University of Iowa MSTP training grant T32 GM139776.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ward LM, Hadjayannakis S, McMillan HJ, Noritz G, Weber DR. Bone health and osteoporosis management of the patient with duchenne muscular dystrophy. Pediatrics. (2018) 142:S34–42. doi: 10.1542/peds.2018-0333E
2. Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) 17:347–61. doi: 10.1016/S1474-4422(18)30025-5
3. Specht S, Zhukova I, Westhoff JH, Erb L, Ziegler A, Kölker S, et al. Fat embolism syndrome in Duchenne muscular dystrophy: Report on a novel case and systematic literature review. Eur J Paediatr Neurol. (2023) 48:91–100. doi: 10.1016/j.ejpn.2023.11.012
4. Bianchi ML, Mazzanti A, Galbiati E, Saraifoger S, Dubini A, Cornelio F, et al. Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos Int. (2003) 14:761–7. doi: 10.1007/s00198-003-1443-y
5. Perera N, Sampaio H, Woodhead H, Farrar M. Fracture in duchenne muscular dystrophy: natural history and vitamin D deficiency. J Child Neurol. (2016) 31:1181–7. doi: 10.1177/0883073816650034
6. Buckner JL, Bowden SA, Mahan JD. Optimizing bone health in duchenne muscular dystrophy. Int J Endocrinol. (2015) 2015:928385. doi: 10.1155/2015/928385
7. Rufo A, Del Fattore A, Capulli M, Carvello F, De Pasquale L, Ferrari S, et al. Mechanisms inducing low bone density in Duchenne muscular dystrophy in mice and humans. J Bone Miner Res. (2011) 26:1891–903. doi: 10.1002/jbmr.410
8. Ward LM, Weber DR. Growth, pubertal development, and skeletal health in boys with Duchenne Muscular Dystrophy. Curr Opin Endocrinol Diabetes Obes. (2019) 26:39–48. doi: 10.1097/MED.0000000000000456
9. Feder D, Koch ME, Palmieri B, Fonseca FLA, Carvalho AAds. Fat embolism after fractures in Duchenne muscular dystrophy: an underdiagnosed complication? A systematic review. Ther Clin Risk Manag. (2017) 13:1357–61. doi: 10.2147/TCRM.S143317
10. Eiholzer U, Awano H, Lee T, Takeshima Y, Matsuo M, Iijima K, et al. Short stature: a common feature in Duchenne muscular dystrophy. Eur J Pediatr. (1988) 147:602–5. doi: 10.1007/BF00442472
11. Matsumoto M, Awano H, Lee T, Takeshima Y, Matsuo M, Iijima K, et al. Patients with Duchenne muscular dystrophy are significantly shorter than those with Becker muscular dystrophy, with the higher incidence of short stature in Dp71 mutated subgroup. Neuromuscul Disord. (2017) 27:1023–8. doi: 10.1016/j.nmd.2017.06.007
12. Crabtree NJ, Roper H, Shaw NJ. Cessation of ambulation results in a dramatic loss of trabecular bone density in boys with Duchenne muscular dystrophy (DMD). Bone. (2022) 154:116248. doi: 10.1016/j.bone.2021.116248
13. Bergmann P, Body JJ, Boonen S, Boutsen Y, Devogelaer JP, Goemaere S, et al. Loading and skeletal development and maintenance. J Osteoporos. (2010) 2011:786752. doi: 10.4061/2011/786752
14. Barzegar M, Niknam E, Habibi P, Shiva S, Tahmasebi S. Bone mineral density and bone metabolism in patients with duchenne muscular dystrophy. Iran J Child Neurol. (2018) 12:77–83.
15. Loscalzo E, See J, Bharill S, Yousefzadeh N, Gough E, Wu M, et al. Growth hormone and testosterone delay vertebral fractures in boys with muscular dystrophy on chronic glucocorticoids. Osteoporos Int. (2023) 35(2):327–38. doi: 10.1007/s00198-023-06951-z
16. Bianchi ML, Morandi L, Andreucci E, Vai S, Frasunkieqwicz J, Cottafava R, et al. Low bone density and bone metabolism alterations in Duchenne muscular dystrophy: response to calcium and vitamin D treatment. Osteoporos Int. (2011) 22:529–39. doi: 10.1007/s00198-010-1275-5
17. Boulanger Piette A, Hamoudi D, Marcadet L, Morin F, Argaw A, Ward L, et al. Targeting the muscle-bone unit: filling two needs with one deed in the treatment of duchenne muscular dystrophy. Curr Osteoporos Rep. (2018) 16:541–53. doi: 10.1007/s11914-018-0468-2
18. Abou-Khalil R, Yang F, Mortreux M, Lieu S, Yu YY, Wurmser M, et al. Delayed bone regeneration is linked to chronic inflammation in murine muscular dystrophy. J Bone Miner Res. (2014) 29:304–15. doi: 10.1002/jbmr.2038
19. Tung JY, Lam TP, Chan SH. Bone microarchitectural alterations in boys with Duchenne muscular dystrophy on long-term glucocorticoid treatment. J Bone Miner Metab. (2021) 39:606–11. doi: 10.1007/s00774-020-01196-w
20. Yoon SH, Grynpas MD, Mitchell J. Growth hormone increases bone toughness and decreases muscle inflammation in glucocorticoid-treated mdx mice, model of duchenne muscular dystrophy. J Bone Miner Res. (2019) 34:1473–86. doi: 10.1002/jbmr.3718
21. Hamoudi D, Marcadet L, Piette Boulanger A, Yagita H, Bouredji Z, Argaw A, et al. An anti-RANKL treatment reduces muscle inflammation and dysfunction and strengthens bone in dystrophic mice. Hum Mol Genet. (2019) 28:3101–12. doi: 10.1093/hmg/ddz124
22. Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) 17:251–67. doi: 10.1016/S1474-4422(18)30024-3
23. Annexstad EJ, Bollerslev J, Westvik J, Myhre AG, Godang K, Holm I, et al. The role of delayed bone age in the evaluation of stature and bone health in glucocorticoid treated patients with Duchenne muscular dystrophy. Int J Pediatr Endocrinol. (2019) 2019:4. doi: 10.1186/s13633-019-0070-0
24. Shimonty A, Bonewald LF, Pin F. Role of the osteocyte in musculoskeletal disease. Curr Osteoporos Rep. (2023) 21:303–10. doi: 10.1007/s11914-023-00788-5
25. King WM, Ruttencutter R, Nagaraja HN, Matkovic V, Landoll J, Hoyle C, et al. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology. (2007) 68:1607–13. doi: 10.1212/01.wnl.0000260974.41514.83
26. Alman BA, Raza SN, Biggar WD. Steroid treatment and the development of scoliosis in males with duchenne muscular dystrophy. J Bone Joint Surg Am. (2004) 86:519–24. doi: 10.2106/00004623-200403000-00009
27. Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. (2006) 16:249–55. doi: 10.1016/j.nmd.2006.01.010
28. Quattrocelli M, Barefield DY, Warner JL, Vo AH, Hadhazy M, Earley JU, et al. Intermittent glucocorticoid steroid dosing enhances muscle repair without eliciting muscle atrophy. J Clin Invest. (2017) 127:2418–32. doi: 10.1172/JCI91445
29. Quattrocelli M, Zelikovich AS, Salamone IM, Fischer JA, McNally EM. Mechanisms and clinical applications of glucocorticoid steroids in muscular dystrophy. J Neuromuscul Dis. (2021) 8:39–52. doi: 10.3233/JND-200556
30. Crabtree NJ, Roper H, McMurchie H, Shaw NJ. Regional changes in bone area and bone mineral content in boys with duchenne muscular dystrophy receiving corticosteroid therapy. J Pediatr. (2010) 156:450–5. doi: 10.1016/j.jpeds.2009.09.010
31. Guglieri M, Bushby K, McDermott MP, Hart KA, Tawil R, Martens WB, et al. Effect of different corticosteroid dosing regimens on clinical outcomes in boys with duchenne muscular dystrophy: A randomized clinical trial. JAMA. (2022) 327:1456–68. doi: 10.1001/jama.2022.4315
32. James KA, Cunniff C, Apkon SD, Mathews K, Lu Z, Holtzer C, et al. Risk factors for first fractures among males with duchenne or becker muscular dystrophy. J Pediatr Orthop. (2015) 35:640–4. doi: 10.1097/BPO.0000000000000348
33. Weber FJ, Latshang TD, Blum MR, Kohler M, Wertli MM. Prognostic factors, disease course, and treatment efficacy in Duchenne muscular dystrophy: A systematic review and meta-analysis. Muscle Nerve. (2022) 66:462–70. doi: 10.1002/mus.27682
34. Joseph S, Wang C, Di Marco M, Horrocks I, Abu-Arafeh I, Baxter A, et al. Fractures and linear growth in a nationwide cohort of boys with duchenne muscular dystrophy with and without glucocorticoid treatment: results from the UK northStar database. JAMA Neurol. (2019) 76:701–9. doi: 10.1001/jamaneurol.2019.0242
35. Bell JM, Shields MD, Watters J, Hamilton A, Beringer T, Elliott M, et al. Interventions to prevent and treat corticosteroid-induced osteoporosis and prevent osteoporotic fractures in Duchenne muscular dystrophy. Cochrane Database Syst Rev. (2017) 1:CD010899. doi: 10.1002/14651858.CD010899.pub2
36. Kim S, Zhu Y, Romitti PA, Fox DJ, Sheehan DW, Valdez R, et al. Associations between timing of corticosteroid treatment initiation and clinical outcomes in Duchenne muscular dystrophy. Neuromuscul Disord. (2017) 27:730–7. doi: 10.1016/j.nmd.2017.05.019
37. Singh A, Schaeffer EK, Reilly CW. Vertebral fractures in duchenne muscular dystrophy patients managed with deflazacort. J Pediatr Orthop. (2018) 38:320–4. doi: 10.1097/BPO.0000000000000817
38. Suthar R, Chaithanya Reddy BV, Malviya M, Sirari T, Attri SV, Patial A, et al. Bone density and bone health alteration in boys with Duchenne Muscular Dystrophy: a prospective observational study. J Pediatr Endocrinol Metab. (2021) 34:573–81. doi: 10.1515/jpem-2020-0680
39. Joseph S, Wang C, Di Marco M, Horrocks I, Abu-Arafeh I, Baxter A, et al. Fractures and bone health monitoring in boys with Duchenne muscular dystrophy managed within the Scottish Muscle Network. Neuromuscul Disord. (2019) 29:59–66. doi: 10.1016/j.nmd.2018.09.005
40. Compston J. Glucocorticoid-induced osteoporosis: an update. Endocrine. (2018) 61:7–16. doi: 10.1007/s12020-018-1588-2
41. Wang L, Heckmann BL, Yang X, Long H. Osteoblast autophagy in glucocorticoid-induced osteoporosis. J Cell Physiol. (2019) 234:3207–15. doi: 10.1002/jcp.v234.4
42. Hathout Y, Conklin LS, Seol H, Gordish-Dressman H, Brown KJ, Morgenroth LP, et al. Serum pharmacodynamic biomarkers for chronic corticosteroid treatment of children. Sci Rep. (2016) 6:31727. doi: 10.1038/srep31727
43. Dhawan P, Christakos S. Novel regulation of 25-hydroxyvitamin D3 24-hydroxylase (24(OH)ase) transcription by glucocorticoids: cooperative effects of the glucocorticoid receptor, C/EBP beta, and the Vitamin D receptor in 24(OH)ase transcription. J Cell Biochem. (2010) 110:1314–23. doi: 10.1002/jcb.v110:6
44. Nagel BH, Mortier W, Elmlinger M, Wollmann HA, Schmitt K, Ranke MB, et al. Short stature in Duchenne muscular dystrophy: a study of 34 patients. Acta Paediatr. (1999) 88:62–5. doi: 10.1111/j.1651-2227.1999.tb01270.x
45. Kao KT, Joseph S, Capaldi N, Brown S, Di Marco M, Dunne J, et al. Skeletal disproportion in glucocorticoid-treated boys with Duchenne muscular dystrophy. Eur J Pediatr. (2019) 178:633–40. doi: 10.1007/s00431-019-03336-5
46. Lavi E, Cohen A, Dor T, Tsabari R, Zangen D. Growth hormone therapy for children with Duchenne muscular dystrophy and glucocorticoid induced short stature. Growth Horm IGF Res. (2023) 72-73:101558. doi: 10.1016/j.ghir.2023.101558
47. Wood CL, Straub V, Guglieri M, Bushby K, Cheetham T. Short stature and pubertal delay in Duchenne muscular dystrophy. Arch Dis Child. (2016) 101:101–6. doi: 10.1136/archdischild-2015-308654
48. Peng G, Sun H, Jiang H, Wang Q, Gan L, Tian Y, et al. Exogenous growth hormone functionally alleviates glucocorticoid-induced longitudinal bone growth retardation in male rats by activating the Ihh/PTHrP signaling pathway. Mol Cell Endocrinol. (2022) 545:111571. doi: 10.1016/j.mce.2022.111571
49. Lui JC, Baron J. Effects of glucocorticoids on the growth plate. Endocr Dev. (2011) 20:187–93. doi: 10.1159/000321244
50. Mazziotti G, Giustina A. Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol. (2013) 9:265–76. doi: 10.1038/nrendo.2013.5
51. Mehls O, Himmele R, Hömme M, Kiepe D, Klaus G. The interaction of glucocorticoids with the growth hormone-insulin-like growth factor axis and its effects on growth plate chondrocytes and bone cells. J Pediatr Endocrinol Metab. (2001) 14 Suppl 6:1475–82.
52. Callewaert F, Sinnesael M, Gielen E, Boonen S, Vanderschueren D. Skeletal sexual dimorphism: relative contribution of sex steroids, GH-IGF1, and mechanical loading. J Endocrinol. (2010) 207:127–34. doi: 10.1677/JOE-10-0209
53. Bian Q, McAdam L, Grynpas M, Mitchell J, Harrington J. Increased rates of vitamin D insufficiency in boys with duchenne muscular dystrophy despite higher vitamin D(3) supplementation. Glob Pediatr Health. (2019) 6:2333794X19835661. doi: 10.1177/2333794X19835661
54. Forrest KY, Stuhldreher WL. Prevalence and correlates of vitamin D deficiency in US adults. Nutr Res. (2011) 31:48–54. doi: 10.1016/j.nutres.2010.12.001
55. Vranic L, Mikolasevic I, Milic S. Vitamin D deficiency: consequence or cause of obesity? Medicina (Kaunas). (2019) 55:541. doi: 10.3390/medicina55090541
56. Oonk S, Spitali P, Hiller M, Sqitzar L, Dalebout H, Calissano M, et al. Comparative mass spectrometric and immunoassay-based proteome analysis in serum of Duchenne muscular dystrophy patients. Proteomics Clin Appl. (2016) 10:290–9. doi: 10.1002/prca.201500044
57. Bouillon R, Bouillon R, Schuit F, Antonio L, Rastinejad F. Vitamin D binding protein: A historic overview. Front Endocrinol (Lausanne). (2019) 10:910. doi: 10.3389/fendo.2019.00910
58. Bez Batti Angulski A, Hosny N, Cohen H, Martin AA, Hahn D, Bauer J, et al. Duchenne muscular dystrophy: disease mechanism and therapeutic strategies. Front Physiol. (2023) 14:1183101. doi: 10.3389/fphys.2023.1183101
59. Cruz-Guzman Odel R, Rodriguez-Cruz M, Escobar Cedillo RE. Systemic inflammation in duchenne muscular dystrophy: association with muscle function and nutritional status. BioMed Res Int. (2015) 2015:891972. doi: 10.1155/2015/891972
60. Maurel DB, Jahn K, Lara-Castillo N. Muscle-bone crosstalk: emerging opportunities for novel therapeutic approaches to treat musculoskeletal pathologies. Biomedicines. (2017) 5:62. doi: 10.3390/biomedicines5040062
61. Ogundele M, Zhang JS, Goswami MV, Barbieri ML, Dang UJ, Novak JS, et al. Validation of chemokine biomarkers in duchenne muscular dystrophy. Life (Basel). (2021) 11:827. doi: 10.3390/life11080827
62. Zhou S, Qian B, Wang L, Zhang C, Hogan MV, Li H, et al. Altered bone-regulating myokine expression in skeletal muscle Of Duchenne muscular dystrophy mouse models. Muscle Nerve. (2018) 58:573–82. doi: 10.1002/mus.26195
63. Kwan Tat S, Padrines M, Théoleyre S, Heymann D, Fortun Y, et al. IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. (2004) 15:49–60. doi: 10.1016/j.cytogfr.2003.10.005
64. Siddiqui JA, Partridge NC. CCL2/monocyte chemoattractant protein 1 and parathyroid hormone action on bone. Front Endocrinol (Lausanne). (2017) 8:49. doi: 10.3389/fendo.2017.00049
65. Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev. (2009) 8:379–83. doi: 10.1016/j.autrev.2008.12.002
66. Lee JH, Kim HN, Kim KO, Jin WJ, Lee S, Kim HH, et al. CXCL10 promotes osteolytic bone metastasis by enhancing cancer outgrowth and osteoclastogenesis. Cancer Res. (2012) 72:3175–86. doi: 10.1158/0008-5472.CAN-12-0481
67. Li G, Zhang L, Wang D, AlQudsy L, Jiang JX, Xu H, et al. Muscle-bone crosstalk and potential therapies for sarco-osteoporosis. J Cell Biochem. (2019) 120:14262–73. doi: 10.1002/jcb.v120.9
68. Severinsen MCK, Pedersen BK. Muscle-organ crosstalk: the emerging roles of myokines. Endocr Rev. (2020) 41:594–609. doi: 10.1210/endrev/bnaa016
69. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. (2008) 473:139–46. doi: 10.1016/j.abb.2008.03.018
70. Dufresne SS, Dumont NA, Couchard P, Lavergne E, Penninger JM, Frenette J, et al. Osteoprotegerin protects against muscular dystrophy. Am J Pathol. (2015) 185:920–6. doi: 10.1016/j.ajpath.2015.01.006
71. Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest. (2019) 129:3214–23. doi: 10.1172/JCI125915
72. Jayash SN, Hamoudi D, Stephen L, Argaw A, Huesa C, Joseph S, et al. Anti-RANKL therapy prevents glucocorticoid-induced bone loss and promotes muscle function in a mouse model of duchenne muscular dystrophy. Calcif Tissue Int. (2023) 113:449–68. doi: 10.1007/s00223-023-01116-w
73. Akhtar Ali S, Kang H, Olney R, Ramos-Platt L, Ryabets-Lienhard A, Cheung C, et al. Evaluating RANKL and OPG levels in patients with Duchenne muscular dystrophy. Osteoporos Int. (2019) 30:2283–8. doi: 10.1007/s00198-019-05077-5
74. Dolegowska K, Marchelek-Mysliwiec M, Nowosiad-Magda M, Slawinski M, Dolegowska B. FGF19 subfamily members: FGF19 and FGF21. J Physiol Biochem. (2019) 75:229–40. doi: 10.1007/s13105-019-00675-7
75. Sun H, Sherrier M, Li H. Skeletal muscle and bone - emerging targets of fibroblast growth factor-21. Front Physiol. (2021) 12:625287. doi: 10.3389/fphys.2021.625287
76. Flippo KH, Potthoff MJ. Metabolic messengers: FGF21. Nat Metab. (2021) 3:309–17. doi: 10.1038/s42255-021-00354-2
77. Li H, Sun H, Qian B, Feng W, Carney D, Miller J, et al. Increased expression of FGF-21 negatively affects bone homeostasis in dystrophin/utrophin double knockout mice. J Bone Miner Res. (2020) 35:738–52. doi: 10.1002/jbmr.3932
78. Lovadi E, Csereklyei M, Markli H, FüLöp K, SeboK Á, Karcagi V, et al. Elevated FGF 21 in myotonic dystrophy type 1 and mitochondrial diseases. Muscle Nerve. (2017) 55:564–9. doi: 10.1002/mus.25364
79. Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proc Natl Acad Sci U.S.A. (2012) 109:3143–8. doi: 10.1073/pnas.1200797109
80. Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdort DJ, Kliewer SA, et al. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. (2008) 8:77–83. doi: 10.1016/j.cmet.2008.05.006
81. Milman S, Huffman DM, Barzilai N. The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab. (2016) 23:980–9. doi: 10.1016/j.cmet.2016.05.014
82. Wu S, Levenson A, Kharitonenkov A, De Luca F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J Biol Chem. (2012) 287:26060–7. doi: 10.1074/jbc.M112.343707
83. Wu S, Grunwald T, Kharitonenkov A, Dam J, Jockers R, De Luca F, et al. Increased expression of fibroblast growth factor 21 (FGF21) during chronic undernutrition causes growth hormone insensitivity in chondrocytes by inducing leptin receptor overlapping transcript (LEPROT) and leptin receptor overlapping transcript-like 1 (LEPROTL1) expression. J Biol Chem. (2013) 288:27375–83. doi: 10.1074/jbc.M113.462218
84. Carnac G, Vernus B, Bonnieu A. Myostatin in the pathophysiology of skeletal muscle. Curr Genomics. (2007) 8:415–22. doi: 10.2174/138920207783591672
85. Chen YS, Guo Q, Guo LJ, Liu T, Wu XP, Lin ZY, et al. GDF8 inhibits bone formation and promotes bone resorption in mice. Clin Exp Pharmacol Physiol. (2017) 44:500–8. doi: 10.1111/cep.2017.44.issue-4
86. Cui Y, Ti Q, Sun W, Huang D, Zhang H, Duan L, et al. Molecular basis and therapeutic potential of myostatin on bone formation and metabolism in orthopedic disease. Biofactors. (2023) 49:21–31. doi: 10.1002/biof.v49.1
87. Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Med. (2015) 21:1085–90. doi: 10.1038/nm.3917
88. Puolakkainen T, Ma H, Kainulainen H, Pasternack A, Rantalainen T, Ritvos O, et al. Treatment with soluble activin type IIB-receptor improves bone mass and strength in a mouse model of Duchenne muscular dystrophy. BMC Musculoskelet Disord. (2017) 18:20. doi: 10.1186/s12891-016-1366-3
89. Hamrick MW, Shi X, Zhang W, Pennington C, Thakore H, Haque M, et al. Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bone. (2007) 40:1544–53. doi: 10.1016/j.bone.2007.02.012
90. Lee SJ, Lehar A, Meir JU, Koch C, Morgan A, Warren LE, et al. Targeting myostatin/activin A protects against skeletal muscle and bone loss during spaceflight. Proc Natl Acad Sci U.S.A. (2020) 117:23942–51. doi: 10.1073/pnas.2014716117
91. Anaya-Segura MA, Garcia-Martinez FA, Montes-Almanza LA, Diaz BG, Avila-Ramirez G, Alvarez-Maya I, et al. Non-invasive biomarkers for duchenne muscular dystrophy and carrier detection. Molecules. (2015) 20:11154–72. doi: 10.3390/molecules200611154
92. Awano H, Takeshima Y, Okizuka Y, Saiki K, Yagi M, Matsuo M, et al. Wide ranges of serum myostatin concentrations in Duchenne muscular dystrophy patients. Clin Chim Acta. (2008) 391:115–7. doi: 10.1016/j.cca.2008.01.024
93. Wojciechowski J, Purohit VS, Harnisch LO, Dua P, Tan B, Nicholas T, et al. Population PK and PD analysis of domagrozumab in pediatric patients with duchenne muscular dystrophy. Clin Pharmacol Ther. (2022) 112:1291–302. doi: 10.1002/cpt.v112.6
94. Markati T, De Waele L, Schara-Schmidt U, Servias L. Lessons learned from discontinued clinical developments in duchenne muscular dystrophy. Front Pharmacol. (2021) 12:735912. doi: 10.3389/fphar.2021.735912
95. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. (2012) 481:463–8. doi: 10.1038/nature10777
96. Buccoliero C, Oranger A, Colaianni G, Pignataro P, Zerlotin R, Lovero R, et al. The effect of Irisin on bone cells in vivo and in vitro. Biochem Soc Trans. (2021) 49:477–84. doi: 10.1042/BST20200978
97. Reza MM, Subramaniyam N, Sim CM, Ge X, Sathiakumar D, McFarlane C, et al. Irisin is a pro-myogenic factor that induces skeletal muscle hypertrophy and rescues denervation-induced atrophy. Nat Commun. (2017) 8:1104. doi: 10.1038/s41467-017-01131-0
98. Barp A, Carraro E, Goggi G, Lizio A, Zanolini A, Messina C, et al. Body composition and myokines in a cohort of patients with Becker muscular dystrophy. Muscle Nerve. (2022) 66:63–70. doi: 10.1002/mus.27565
99. Arikan S, Alaca N, Ozbeyli D, Elmas MA, Arbak S, Suyen G, et al. Effects of moderate aerobic exercise, low-level laser therapy, or their combination on muscles pathology, oxidative stress and irisin levels in the mdx mouse model of Duchenne muscular dystrophy. Lasers Med Sci. (2022) 37:2925–36. doi: 10.1007/s10103-022-03562-8
100. Ponzetti M, Ucci A, Maurizi A, Giacchi L, Teti A, Rucci N, et al. Lipocalin 2 influences bone and muscle phenotype in the MDX mouse model of duchenne muscular dystrophy. Int J Mol Sci. (2022) 23:958. doi: 10.3390/ijms23020958
101. Costa D, Lazzarini E, Canciani B, Giuliani A, Spano R, Marozzi K, et al. Altered bone development and turnover in transgenic mice over-expressing lipocalin-2 in bone. J Cell Physiol. (2013) 228:2210–21. doi: 10.1002/jcp.v228.11
102. Wang T, He C. TNF-alpha and IL-6: the link between immune and bone system. Curr Drug Targets. (2020) 21:213–27. doi: 10.2174/1389450120666190821161259
103. Miyatake S, Shimizu-Motohashi Y, Takeda S, Aoki Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: focus on skeletal muscle-releasing factors. Drug Des Devel Ther. (2016) 10:2745–58. doi: 10.2147/DDDT.S110163
104. Wada E, Tanihata J, Iwamura A, Takeda S, Hayashi YK, Matsuda R, et al. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet Muscle. (2017) 7:23. doi: 10.1186/s13395-017-0140-z
105. Riddell DO, Hildyard JCW, Harron RCM, Hornby NL, Wells DJ, Piercy RJ, et al. Serum inflammatory cytokines as disease biomarkers in the DE50-MD dog model of Duchenne muscular dystrophy. Dis Model Mech. (2022) 15:dmm049394. doi: 10.1242/dmm.049394
106. Zhou J, Liu B, Liang C, Li Y, Song YH. Cytokine signaling in skeletal muscle wasting. Trends Endocrinol Metab. (2016) 27:335–47. doi: 10.1016/j.tem.2016.03.002
107. Piers AT, Lavin T, Radley-Crabb HG, Bakker AJ, Grounds MD, Pinniger GJ, et al. Blockade of TNF in vivo using cV1q antibody reduces contractile dysfunction of skeletal muscle in response to eccentric exercise in dystrophic mdx and normal mice. Neuromuscul Disord. (2011) 21:132–41. doi: 10.1016/j.nmd.2010.09.013
108. Pierno S, Nico B, Burdi R, Liantonio A, Didonna MP, Cippone V, et al. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol. (2007) 33:344–59. doi: 10.1111/j.1365-2990.2007.00798.x
109. Ermolova NV, Martinez L, Vetrone SA, Jordan MC, Roos KP, Sweeny HL, et al. Long-term administration of the TNF blocking drug Remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul Disord. (2014) 24:583–95. doi: 10.1016/j.nmd.2014.04.006
110. Li W, Moylan JS, Chambers MA, Smith J, Reid MB, et al. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am J Physiol Cell Physiol. (2009) 297:C706–14. doi: 10.1152/ajpcell.00626.2008
111. Benny Klimek ME, Sali A, Parayvarapu S, Van der Meulen JH, Nagaraju K. Effect of the IL-1 receptor antagonist kineret(R) on disease phenotype in mdx mice. PloS One. (2016) 11:e0155944. doi: 10.1371/journal.pone.0155944
112. Blanc RS, Kallenbach JG, Bachman JF, Mitchell A, Paris ND, Chakkalakal JV, et al. Inhibition of inflammatory CCR2 signaling promotes aged muscle regeneration and strength recovery after injury. Nat Commun. (2020) 11:4167. doi: 10.1038/s41467-020-17620-8
113. Lu H, Huang D, Ransohoff RM, Zhou L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. (2011) 25:3344–55. doi: 10.1096/fj.10-178939
114. Deyhle MR, Hafen PS, Parmley J, Preece CN, Robison M, Sorensen JR, et al. CXCL10 increases in human skeletal muscle following damage but is not necessary for muscle regeneration. Physiol Rep. (2018) 6:e13689. doi: 10.14814/phy2.13689
115. Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T, et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol Med. (2014) 6:1476–92. doi: 10.15252/emmm.201403967
116. Atilano-Miguel S, Barbosa-Cortes L, Ortiz-Muniz R. Duchenne muscular dystrophy: RANK/RANKL/OPG (receptor activator of nuclear factor-kB/RANK ligand/osteoprotegerin) system and glucocorticoids. Bol Med Hosp Infant Mex. (2022) 79:275–83. doi: 10.24875/BMHIM.21000171
117. Marcadet L, Bouredji Z, Argaw A, Frenette J. The roles of RANK/RANKL/OPG in cardiac, skeletal, and smooth muscles in health and disease. Front Cell Dev Biol. (2022) 10:903657. doi: 10.3389/fcell.2022.903657
118. Gao X, Tang Y, Amra S, Sun X, Cui Y, Cheng H, et al. Systemic investigation of bone and muscle abnormalities in dystrophin/utrophin double knockout mice during postnatal development and the mechanisms. Hum Mol Genet. (2019) 28:1738–51. doi: 10.1093/hmg/ddz012
119. Marcadet L, Juracic ES, Khan N, Bouredji Z, Yagita H, Ward LM, et al. RANKL inhibition reduces cardiac hypertrophy in mdx mice and possibly in children with duchenne muscular dystrophy. Cells. (2023) 12:1538. doi: 10.3390/cells12111538
120. Hung C, Mathews KD, Shibli-Rahhal A. Effect of denosumab on bone health in adult patients with duchenne/becker muscular dystrophy: A report of 2 cases. JBJS Case Connect. (2022) 12. doi: 10.2106/JBJS.CC.21.00664
121. Kumaki D, Nahamura Y, Sakai N, Kosho T, Nakamura A, Hirabayashi S, et al. Efficacy of denosumab for glucocorticoid-induced osteoporosis in an adolescent patient with duchenne muscular dystrophy: A case report. JBJS Case Connect. (2018) 8:e22. doi: 10.2106/JBJS.CC.17.00190
122. Kubicky RA, Wu S, Kharitonenkov A, De Luca F. Role of fibroblast growth factor 21 (FGF21) in undernutrition-related attenuation of growth in mice. Endocrinology. (2012) 153:2287–95. doi: 10.1210/en.2011-1909
123. Tang Y, Zhang M. Fibroblast growth factor 21 and bone homeostasis. BioMed J. (2023) 46:100548. doi: 10.1016/j.bj.2022.07.002
124. Omosule CL, Phillips CL. Deciphering myostatin’s regulatory, metabolic, and developmental influence in skeletal diseases. Front Genet. (2021) 12:662908. doi: 10.3389/fgene.2021.662908
125. Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. (2002) 52:832–6. doi: 10.1002/ana.10385
126. Bogdanovich S, Krag TOB, Barton ER, Morris LD, Whittemore LA, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. (2002) 420:418–21. doi: 10.1038/nature01154
127. Bogdanovich S, Perkins KJ, Krag TOB, Whittemore LA, Khurana TS. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. (2005) 19:543–9. doi: 10.1096/fj.04-2796com
128. Qiao C, Li J, Jiang J, Zhu X, Wang B, Li J, Xiao X, et al. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther. (2008) 19:241–54. doi: 10.1089/hum.2007.159
129. Campbell C, McMillan HJ, Mah JK, Tarnopolsky M, Selby K, McClure T, et al. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve. (2017) 55:458–64. doi: 10.1002/mus.25268
130. Reza MM, Sim CM, Subramaniyam N, Ge X, Sharma M, Kambadur R, et al. Irisin treatment improves healing of dystrophic skeletal muscle. Oncotarget. (2017) 8:98553–66. doi: 10.18632/oncotarget.21636
131. Ponzetti M, Aielli F, Ucci A, Cappariello A, Lombardi G, Teti A, et al. Lipocalin 2 increases after high-intensity exercise in humans and influences muscle gene expression and differentiation in mice. J Cell Physiol. (2022) 237:551–65. doi: 10.1002/jcp.v237.1
132. Wang Z, An J, Zhu D, Chen H, Lin A, Kang J, et al. Periostin: an emerging activator of multiple signaling pathways. J Cell Commun Signal. (2022) 16:515–30. doi: 10.1007/s12079-022-00674-2
133. Delany AM, Hankenson KD. Thrombospondin-2 and SPARC/osteonectin are critical regulators of bone remodeling. J Cell Commun Signal. (2009) 3:227–38. doi: 10.1007/s12079-009-0076-0
134. Liu S, Jin Z, Cao M, Hao D, Li C, Li D, et al. Periostin regulates osteogenesis of mesenchymal stem cells from ovariectomized rats through actions on the ILK/Akt/GSK-3beta Axis. Genet Mol Biol. (2021) 44:e20200461. doi: 10.1590/1678-4685-GMB-2020-0461
135. Ghanemi A, Melouane A, Yoshioka M, St-Amand J. Secreted protein acidic and rich in cysteine and bioenergetics: Extracellular matrix, adipocytes remodeling and skeletal muscle metabolism. Int J Biochem Cell Biol. (2019) 117:105627. doi: 10.1016/j.biocel.2019.105627
136. Jorgensen LH, Petersson SJ, Sellathurai J, Andersen DC, Thayssen S, Sant DJ, et al. Secreted protein acidic and rich in cysteine (SPARC) in human skeletal muscle. J Histochem Cytochem. (2009) 57:29–39. doi: 10.1369/jhc.2008.951954
137. Kos K, Wong S, Tan B, Gummesson A, Jernas M, Franck N, et al. Regulation of the fibrosis and angiogenesis promoter SPARC/osteonectin in human adipose tissue by weight change, leptin, insulin, and glucose. Diabetes. (2009) 58:1780–8. doi: 10.2337/db09-0211
138. Hara M, Yokota K, Saito T, Kobayakawa K, Kijima K, Yoshizaki S, et al. Periostin promotes fibroblast migration and inhibits muscle repair after skeletal muscle injury. J Bone Joint Surg Am. (2018) 100:e108. doi: 10.2106/JBJS.17.01230
139. Ito N, Miyagoe-Suzuki Y, Takeda S, Kudo A. Periostin is required for the maintenance of muscle fibers during muscle regeneration. Int J Mol Sci. (2021) 22:3627. doi: 10.3390/ijms22073627
140. Heymann D, Rousselle AV. gp130 Cytokine family and bone cells. Cytokine. (2000) 12:1455–68. doi: 10.1006/cyto.2000.0747
141. Hunt LC, White J. The role of leukemia inhibitory factor receptor signaling in skeletal muscle growth, injury and disease. Adv Exp Med Biol. (2016) 900:45–59. doi: 10.1007/978-3-319-27511-6_3
142. Austin L, Bower JJ, Bennett TM, Lynch GS, Kapsa R, White JD, et al. Leukemia inhibitory factor ameliorates muscle fiber degeneration in the mdx mouse. Muscle Nerve. (2000) 23:1700–5. doi: 10.1002/1097-4598(200011)23:11<1700::AID-MUS5>3.0.CO;2-W
143. Welc SS, Flores I, Wehling-Henricks M, Ramos J, Wang Y, Bertoni C, et al. Targeting a therapeutic LIF transgene to muscle via the immune system ameliorates muscular dystrophy. Nat Commun. (2019) 10:2788. doi: 10.1038/s41467-019-10614-1
144. White JD, Davies M, McGeachie J, Grounds MD. An evaluation of leukaemia inhibitory factor as a potential therapeutic agent in the treatment of muscle disease. Neuromuscul Disord. (2002) 12:909–16. doi: 10.1016/S0960-8966(02)00117-7
145. Di Maggio N, Banfi A. The osteo-angiogenic signaling crosstalk for bone regeneration: harmony out of complexity. Curr Opin Biotechnol. (2022) 76:102750. doi: 10.1016/j.copbio.2022.102750
146. Tidball JG, Wehling-Henricks M. Nitric oxide synthase deficiency and the pathophysiology of muscular dystrophy. J Physiol. (2014) 592:4627–38. doi: 10.1113/jphysiol.2014.592.issue-21
147. Thomas GD. Functional muscle ischemia in Duchenne and Becker muscular dystrophy. Front Physiol. (2013) 4:381. doi: 10.3389/fphys.2013.00381
148. Kodippili K, Throne PK, Laughlin MH, Duan D. Dystrophin deficiency impairs vascular structure and function in the canine model of Duchenne muscular dystrophy. J Pathol. (2021) 254:589–605. doi: 10.1002/path.v254.5
149. Podkalicka P, Mucha O, Dulak J, Loboda A. Targeting angiogenesis in Duchenne muscular dystrophy. Cell Mol Life Sci. (2019) 76:1507–28. doi: 10.1007/s00018-019-03006-7
150. Palladino M, Gatto I, Neri V, Straino S, Smith RC, Silver M, et al. Angiogenic impairment of the vascular endothelium: a novel mechanism and potential therapeutic target in muscular dystrophy. Arterioscler Thromb Vasc Biol. (2013) 33:2867–76. doi: 10.1161/ATVBAHA.112.301172
151. Miike T, Sugino S, Ohtani Y, Taku K, Yoshioka K, et al. Vascular endothelial cell injury and platelet embolism in Duchenne muscular dystrophy at the preclinical stage. J Neurol Sci. (1987) 82:67–80. doi: 10.1016/0022-510X(87)90007-4
152. Loufrani L, Dubroca C, You D, Li Z, Levy B, Paulin D, et al. Absence of dystrophin in mice reduces NO-dependent vascular function and vascular density: total recovery after a treatment with the aminoglycoside gentamicin. Arterioscler Thromb Vasc Biol. (2004) 24:671–6. doi: 10.1161/01.ATV.0000118683.99628.42
153. Lees D, Fabbrizio E, Mornet D, Harricane MC, Travo P. Dystrophin (Xp21), a new phenotype marker of cultured rat aortic myocytes. Exp Cell Res. (1994) 210:230–5. doi: 10.1006/excr.1994.1034
154. Rivier F, Robert A, Hugon G, Mornet D. Different utrophin and dystrophin properties related to their vascular smooth muscle distributions. FEBS Lett. (1997) 408:94–8. doi: 10.1016/S0014-5793(97)00398-0
155. Kaplan KM, Morgan KG. The importance of dystrophin and the dystrophin associated proteins in vascular smooth muscle. Front Physiol. (2022) 13:1059021. doi: 10.3389/fphys.2022.1059021
156. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. (2010) 9:177–89. doi: 10.1016/S1474-4422(09)70272-8
157. Spaulding HR, Selsby JT. Is exercise the right medicine for dystrophic muscle? Med Sci Sports Exerc. (2018) 50:1723–32. doi: 10.1249/MSS.0000000000001639
158. Ratel S, Duche P, Williams CA. Muscle fatigue during high-intensity exercise in children. Sports Med. (2006) 36:1031–65. doi: 10.2165/00007256-200636120-00004
159. Bianchi ML, Vai S, Baranello G, Broggi F, Judex S, Hangartner T, et al. Low-intensity vibration protects the weight-bearing skeleton and suppresses fracture incidence in boys with duchenne muscular dystrophy: A prospective, randomized, double-blind, placebo-controlled clinical trial. JBMR Plus. (2022) 6:e10685. doi: 10.1002/jbm4.10685
160. Townsend EL, Bibeau C, Holmes TM. Supported standing in boys with duchenne muscular dystrophy. Pediatr Phys Ther. (2016) 28:320–9. doi: 10.1097/PEP.0000000000000251
161. Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Mol Cell Biochem. (1998) 179:111–23. doi: 10.1023/A:1006812004945
162. Baumann CW, Warren GL, Lowe DA. Plasmalemma function is rapidly restored in mdx muscle after eccentric contractions. Med Sci Sports Exerc. (2020) 52:354–61. doi: 10.1249/MSS.0000000000002126
163. Childers MK, Okamura CS, Bogan DJ, Bogan JR, Petroski GF, McDonald K, et al. Eccentric contraction injury in dystrophic canine muscle. Arch Phys Med Rehabil. (2002) 83:1572–8. doi: 10.1053/apmr.2002.35109
164. Motohashi N, Asakura A. Muscle satellite cell heterogeneity and self-renewal. Front Cell Dev Biol. (2014) 2:1. doi: 10.3389/fcell.2014.00001
165. Filippelli RL, Chang NC. Empowering muscle stem cells for the treatment of duchenne muscular dystrophy. Cells Tissues Organs. (2022) 211:641–54. doi: 10.1159/000514305
166. Jiang C, Wen Y, Kuroda K, Hannon K, Rudnicki MA, Kuang S, et al. Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis Model Mech. (2014) 7:997–1004. doi: 10.1242/dmm.015917
167. Lindsay A, Larson AA, Verma M, Ervasti JM, Lowe DA, et al. Isometric resistance training increases strength and alters histopathology of dystrophin-deficient mouse skeletal muscle. J Appl Physiol (1985). (2019) 126:363–75. doi: 10.1152/japplphysiol.00948.2018
168. Dudley-Javoroski S, Petrie MA, McHenry CL, Amelon RE, Saha PK, Shields RK, et al. Bone architecture adaptations after spinal cord injury: impact of long-term vibration of a constrained lower limb. Osteoporos Int. (2016) 27:1149–60. doi: 10.1007/s00198-015-3326-4
169. Dudley-Javoroski S, Saha PK, Li C, Gao Z, Shields RK. High dose compressive loads attenuate bone mineral loss in humans with spinal cord injury. Osteoporos Int. (2012) 23:2335–46. doi: 10.1007/s00198-011-1879-4
170. Cheung WH, Wong RMY, Choy VMH, Li MCM, Cheng KYK, Chow SKH, et al. Enhancement of osteoporotic fracture healing by vibration treatment: The role of osteocytes. Injury. (2021) 52 Suppl 2:S97–S100. doi: 10.1016/j.injury.2020.05.020
171. Thompson WR, Yen SS, Rubin J. Vibration therapy: clinical applications in bone. Curr Opin Endocrinol Diabetes Obes. (2014) 21:447–53. doi: 10.1097/MED.0000000000000111
172. Adams V. Electromyostimulation to fight atrophy and to build muscle: facts and numbers. J Cachexia Sarcopenia Muscle. (2018) 9:631–4. doi: 10.1002/jcsm.12332
173. Chan AS, Hardee JP, Blank M, Cho EHJ, McGregor NE, Sims NA, et al. Increasing muscle contractility through low-frequency stimulation alters tibial bone geometry and reduces bone strength in mdx and dko dystrophic mice. J Appl Physiol (1985). (2023) 135:77–87. doi: 10.1152/japplphysiol.00651.2022
174. Swolin-Eide D, Magnusson P. Does whole-body vibration treatment make children’s bones stronger? Curr Osteoporos Rep. (2020) 18:471–9. doi: 10.1007/s11914-020-00608-0
175. Biggar WD, Skalsky A, McDonald CM. Comparing deflazacort and prednisone in duchenne muscular dystrophy. J Neuromuscul Dis. (2022) 9:463–76. doi: 10.3233/JND-210776
176. Connolly AM, Schierbecker J, Renna R, Florence J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord. (2002) 12:917–25. doi: 10.1016/S0960-8966(02)00180-3
177. Connolly AM, Zaidman CM, Golumbek PT, Cradock MM, Flanigan KM, Kuntz NL, et al. Twice-weekly glucocorticosteroids in infants and young boys with Duchenne muscular dystrophy. Muscle Nerve. (2019) 59:650–7. doi: 10.1002/mus.26441
178. Wintzinger M, Miz K, York A, Demonbreun AR, Molkentin JD, McNally EM, et al. Effects of glucocorticoids in murine models of duchenne and limb-girdle muscular dystrophy. Methods Mol Biol. (2023) 2587:467–78. doi: 10.1007/978-1-0716-2772-3_24
179. Escolar DM, Hache LP, Clemens PR, Cnaan A, McDonald CM, Viswanathan V, et al. Randomized, blinded trial of weekend vs daily prednisone in Duchenne muscular dystrophy. Neurology. (2011) 77:444–52. doi: 10.1212/WNL.0b013e318227b164
180. Fenichel GM, Mendell JR, Moxley RT 3rd, Griggs RC, Brooke MH, Miller JP, et al. A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch Neurol. (1991) 48:575–9. doi: 10.1001/archneur.1991.00530180027012
181. Heier CR, Damsker JM, Yu Q, Dillingham C, Huynh T, Van der Meulen JH, et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. (2013) 5:1569–85. doi: 10.1002/emmm.201302621
182. Heier CR, Yu Q, Fiorillo AA, Tully CB, Tucker A, Mazala DA, et al. Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci Alliance. (2019) 2(1):e201800186. doi: 10.26508/lsa.201800186
183. Liu X, Wang Y, Gutierrez JS, Damsker JM, Nagaraju K, Hoffman EP, et al. Disruption of a key ligand-H-bond network drives dissociative properties in vamorolone for Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U.S.A. (2020) 117:24285–93. doi: 10.1073/pnas.2006890117
184. Conklin LS, Damsker JM, Hoffman EP, Jusko WJ, Mavroudis PD, Schwartz BD, et al. Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacol Res. (2018) 136:140–50. doi: 10.1016/j.phrs.2018.09.007
185. Mah JK, Clemens PR, Guglieri M, Smith EC, Finkel RS, Tulinius M, et al. Efficacy and safety of vamorolone in duchenne muscular dystrophy: A 30-month nonrandomized controlled open-label extension trial. JAMA Netw Open. (2022) 5:e2144178. doi: 10.1001/jamanetworkopen.2021.44178
186. Guglieri M, Clemens PR, Perlman SJ, Smith EC, Horrocks I, Finkel RS, et al. Efficacy and safety of vamorolone vs placebo and prednisone among boys with duchenne muscular dystrophy: A randomized clinical trial. JAMA Neurol. (2022) 79:1005–14. doi: 10.1001/jamaneurol.2022.2480
187. Damsker JM, Dillingham BC, Rose MC, Balsley MA, Heier CR, Watson AM, et al. VBP15, a glucocorticoid analogue, is effective at reducing allergic lung inflammation in mice. PloS One. (2013) 8:e63871. doi: 10.1371/journal.pone.0063871
188. Dang UJ, Damsker JM, Guglieri M, Clemens PR, Perlman SJ, Smith EC, et al. Efficacy and safety of vamorolone over 48 weeks in boys with duchenne muscular dystrophy: A randomized controlled trial. Neurology. (2024) 102:e208112. doi: 10.1212/WNL.0000000000208112
189. Wang D, Tang X, Shi Q, Wang R, Ji T, Tang X, et al. Denosumab in pediatric bone disorders and the role of RANKL blockade: a narrative review. Transl Pediatr. (2023) 12:470–86. doi: 10.21037/tp-22-276
190. Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. (2008) 83:1032–45. doi: 10.4065/83.9.1032
191. Landfeldt E, Phung K, Zaman F, Astrom E, Abner S, Lochmuller H, et al. Bisphosphonates in glucocorticoid-treated patients with duchenne muscular dystrophy: A systematic review and grading of the evidence. Neurology. (2024) 102:e207948. doi: 10.1212/WNL.0000000000207948
192. Hanley DA, Adachi JC, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract. (2012) 66:1139–46. doi: 10.1111/ijcp.2012.66.issue-12
193. Srinivasan R, Rawlings D, Wood CL, Cheetham T, Jimenez Moreno AC, Mayhew A, et al. Prophylactic oral bisphosphonate therapy in duchenne muscular dystrophy. Muscle Nerve. (2016) 54:79–85. doi: 10.1002/mus.24991
194. Tian C, Wong BL, Hornung L, Khoury JC, Rybalsky I, Shellenbarger KC, et al. Oral bisphosphonate treatment in patients with Duchenne muscular dystrophy on long term glucocorticoid therapy. Neuromuscul Disord. (2020) 30:599–610. doi: 10.1016/j.nmd.2020.06.005
195. Eghbali-Fatourechi G. Bisphosphonate therapy in pediatric patients. J Diabetes Metab Disord. (2014) 13:109. doi: 10.1186/s40200-014-0109-y
196. Misof BM, Roschger P, McMillan HJ, Ma J, Klaushofer K, Rauch F, et al. Histomorphometry and bone matrix mineralization before and after bisphosphonate treatment in boys with duchenne muscular dystrophy: A paired transiliac biopsy study. J Bone Miner Res. (2016) 31:1060–9. doi: 10.1002/jbmr.2756
197. Jensen PR, Andersen TL, Chavassieux P, Roux JP, Delaisse JM. Bisphosphonates impair the onset of bone formation at remodeling sites. Bone. (2021) 145:115850. doi: 10.1016/j.bone.2021.115850
198. Wawrzyniak A, Balawender K. Structural and metabolic changes in bone. Anim (Basel). (2022) 12:1946. doi: 10.3390/ani12151946
199. Dufresne SS, Boulanger-Piette A, Bosse S, Argaw A, Hamoudi D, Marcadet L, et al. Genetic deletion of muscle RANK or selective inhibition of RANKL is not as effective as full-length OPG-fc in mitigating muscular dystrophy. Acta Neuropathol Commun. (2018) 6:31. doi: 10.1186/s40478-018-0533-1
200. Grassi G, Chiodini I, Palmieri S, Cairoli E, Arosio M, Eller-Vainicher C, et al. Bisphosphonates after denosumab withdrawal reduce the vertebral fractures incidence. Eur J Endocrinol. (2021) 185:387–96. doi: 10.1530/EJE-21-0157
201. Cummings SR, Ferrari S, Eastell R, Gilchrist N, Jensen JEB, McClung M, et al. Vertebral fractures after discontinuation of denosumab: A post hoc analysis of the randomized placebo-controlled FREEDOM trial and its extension. J Bone Miner Res. (2018) 33:190–8. doi: 10.1002/jbmr.3337
202. Tsaknakis K, Jäckle K, Lüders KA, Lorenz HM, Braunschweig L, Hell AK, et al. Reduced bone mineral density in adolescents with Duchenne Muscular Dystrophy (DMD) and scoliosis. Osteoporos Int. (2022) 33:2011–8. doi: 10.1007/s00198-022-06416-9
203. Cittadini A, Comi LI, Longobardi S, Petretta VR, Casaburi C, Passamano L, et al. A preliminary randomized study of growth hormone administration in Becker and Duchenne muscular dystrophies. Eur Heart J. (2003) 24:664–72. doi: 10.1016/S0195-668X(02)00740-6
204. Rutter MM, Collins J, Rose SR, Woo JG, Sucharew H, Sawnani H, et al. Growth hormone treatment in boys with Duchenne muscular dystrophy and glucocorticoid-induced growth failure. Neuromuscul Disord. (2012) 22:1046–56. doi: 10.1016/j.nmd.2012.07.009
205. Rutter MM, Wong BL, Collins JJ, Sawnani H, Taylor MD, Horn PS, et al. Recombinant human insulin-like growth factor-1 therapy for 6 months improves growth but not motor function in boys with Duchenne muscular dystrophy. Muscle Nerve. (2020) 61:623–31. doi: 10.1002/mus.26846
206. Boccanegra B, Cappellari O, Mantuano P, Trisciuzzi D, Mele A, Tulimiero L, et al. Growth hormone secretagogues modulate inflammation and fibrosis in mdx mouse model of Duchenne muscular dystrophy. Front Immunol. (2023) 14:1119888. doi: 10.3389/fimmu.2023.1119888
207. Chang L, Niu F, Chen J, Cao X, Liu Z, Bao X, et al. Ghrelin improves muscle function in dystrophin-deficient mdx mice by inhibiting NLRP3 inflammasome activation. Life Sci. (2019) 232:116654. doi: 10.1016/j.lfs.2019.116654
208. Zatz M, Betti RT. Benign Duchenne muscular dystrophy in a patient with growth hormone deficiency: a five years follow-up. Am J Med Genet. (1986) 24:567–72. doi: 10.1002/ajmg.1320240323
209. Zatz M, Rapaport D, Vainzof M, Pavanello Rde, Rocha JM, Betti RT, et al. Effect of mazindol on growth hormone levels in patients with Duchenne muscular dystrophy. Am J Med Genet. (1988) 31:821–33. doi: 10.1002/ajmg.1320310415
210. Griggs RC, Moxley RT 3rd, Mendell JR, Fenichel GM, Brooke MH, Miller PJ, et al. Randomized, double-blind trial of mazindol in Duchenne dystrophy. Muscle Nerve. (1990) 13:1169–73. doi: 10.1002/mus.880131212
211. Dooley JM, Bobbitt SA, Cummings EA. The impact of deflazacort on puberty in Duchenne muscular dystrophy. Pediatr Neurol. (2013) 49:292–3. doi: 10.1016/j.pediatrneurol.2013.05.004
212. Lee SL, Lim A, Munns C, Simm PJ, Zacharin M. Effect of testosterone treatment for delayed puberty in duchenne muscular dystrophy. Horm Res Paediatr. (2020) 93:108–18. doi: 10.1159/000508290
213. Wood CL, Cheetham TD, Guglieri M, Bushby K, Owen C, Johnstone H, et al. Testosterone treatment of pubertal delay in duchenne muscular dystrophy. Neuropediatrics. (2015) 46:371–6. doi: 10.1055/s-0035-1563696
214. Wood CL, Hollingsworth KG, Bokaie E, Hughes E, Muni-Lofra R, Mayhew A, et al. Is ongoing testosterone required after pubertal induction in Duchenne muscular dystrophy? Endocr Connect. (2023) 12(12):e230245. doi: 10.1530/EC-23-0245
215. Welle S, Jozefowicz R, Forbes G, Griggs RC. Effect of testosterone on metabolic rate and body composition in normal men and men with muscular dystrophy. J Clin Endocrinol Metab. (1992) 74:332–5. doi: 10.1210/jcem.74.2.1730811
216. Wood CL, Hollingsworth KG, Hughes E, Punniyakodi S, Muni-Lofra R, Mayhew A, et al. Pubertal induction in adolescents with DMD is associated with high satisfaction, gonadotropin release and increased muscle contractile surface area. Eur J Endocrinol. (2021) 184:67–79. doi: 10.1530/EJE-20-0709
217. Hartgens F, Kuipers H. Effects of androgenic-anabolic steroids in athletes. Sports Med. (2004) 34:513–54. doi: 10.2165/00007256-200434080-00003
218. Catalano A, Vita GL, Russo M, Vita G, Lasco A, Morabito N, Messina S, et al. Effects of teriparatide on bone mineral density and quality of life in Duchenne muscular dystrophy related osteoporosis: a case report. Osteoporos Int. (2016) 27:3655–9. doi: 10.1007/s00198-016-3761-x
219. Gray SK, McGee-Lawrence ME, Sanders JL, Condon KW, Tsai CJ, Donahue SW, et al. Black bear parathyroid hormone has greater anabolic effects on trabecular bone in dystrophin-deficient mice than in wild type mice. Bone. (2012) 51:578–85. doi: 10.1016/j.bone.2012.05.003
220. Yoon SH, Grynpas M, Mitchell J. Intermittent PTH treatment improves bone and muscle in glucocorticoid treated Mdx mice: A model of Duchenne Muscular Dystrophy. Bone. (2019) 121:232–42. doi: 10.1016/j.bone.2019.01.028
221. Saag KG, Shane E, Boonen S, Marín F, Donley DW, Taylor KA, et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. (2007) 357:2028–39. doi: 10.1056/NEJMoa071408
222. Nasomyont N, Keefe C, Tian C, Hornung L, Khoury J, Tilden JC, et al. Safety and efficacy of teriparatide treatment for severe osteoporosis in patients with Duchenne muscular dystrophy. Osteoporos Int. (2020) 31:2449–59. doi: 10.1007/s00198-020-05549-z
223. Poutoglidou F, Pourzitaki C, Manthou ME, Samoladas E, Saitis A, Malliou F, et al. The inhibitory effect of tocilizumab on systemic bone loss and tendon inflammation in a juvenile Collagen-Induced arthritis rat model. Connect Tissue Res. (2022) 63:577–89. doi: 10.1080/03008207.2022.2042275
224. Tanaka K, Hashizume M, Mihara M, Yoshida H, Suzuki M, Matsumoto Y, et al. Anti-interleukin-6 receptor antibody prevents systemic bone mass loss via reducing the number of osteoclast precursors in bone marrow in a collagen-induced arthritis model. Clin Exp Immunol. (2014) 175:172–80. doi: 10.1111/cei.12201
225. Chen YM, Chen HH, Huang WN, Liao TL, Chen JP, Chao WC, et al. Tocilizumab potentially prevents bone loss in patients with anticitrullinated protein antibody-positive rheumatoid arthritis. PloS One. (2017) 12:e0188454. doi: 10.1371/journal.pone.0188454
226. Confavreux CB, Chapurlat RD. Systemic bone effects of biologic therapies in rheumatoid arthritis and ankylosing spondylitis. Osteoporos Int. (2011) 22:1023–36. doi: 10.1007/s00198-010-1462-4
227. Wang J, Chang CY, Yang X, Zhou F, Liu J, Feng Z, et al. Leukemia inhibitory factor, a double-edged sword with therapeutic implications in human diseases. Mol Ther. (2023) 31:331–43. doi: 10.1016/j.ymthe.2022.12.016
228. Kan T, He Z, Du J, Xu M, Cui J, Han X, et al. Irisin promotes fracture healing by improving osteogenesis and angiogenesis. J Orthop Translat. (2022) 37:37–45. doi: 10.1016/j.jot.2022.07.006
229. Morine KJ, Bish LT, Selsby JT, Gazzara JA, Pendrak K, Sleeper MM, et al. Activin IIB receptor blockade attenuates dystrophic pathology in a mouse model of Duchenne muscular dystrophy. Muscle Nerve. (2010) 42:722–30. doi: 10.1002/mus.21743
230. Wagner KR. The elusive promise of myostatin inhibition for muscular dystrophy. Curr Opin Neurol. (2020) 33:621–8. doi: 10.1097/WCO.0000000000000853
231. Hourde C, Joanne P, Medja F, Mougenot N, Jacquet A, Mouisel E, et al. Voluntary physical activity protects from susceptibility to skeletal muscle contraction-induced injury but worsens heart function in mdx mice. Am J Pathol. (2013) 182:1509–18. doi: 10.1016/j.ajpath.2013.01.020
232. Barbin IC, Pereira JA, Bersan Rovere M, de Oliveira Moreira D, Marques MJ, Santo Neto H, et al. Diaphragm degeneration and cardiac structure in mdx mouse: potential clinical implications for Duchenne muscular dystrophy. J Anat. (2016) 228:784–91. doi: 10.1111/joa.2016.228.issue-5
Keywords: DMD, skeletal abnormality, osteoporosis, myokines, muscle and bone crosstalk
Citation: Hurley-Novatny A, Chang D, Murakami K, Wang L and Li H (2024) Poor bone health in Duchenne muscular dystrophy: a multifactorial problem beyond corticosteroids and loss of ambulation. Front. Endocrinol. 15:1398050. doi: 10.3389/fendo.2024.1398050
Received: 08 March 2024; Accepted: 31 October 2024;
Published: 28 November 2024.
Edited by:
Jiajia Xu, Southern Medical University, ChinaReviewed by:
Ramona Meanti, University of Milano Bicocca, ItalyKhashayar Sakhaee, University of Texas Southwestern Medical Center, United States
Chun Ming Wu, Johns Hopkins University, United States
Copyright © 2024 Hurley-Novatny, Chang, Murakami, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongshuai Li, aG9uZ3NodWFpLWxpQHVpb3dhLmVkdQ==