
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 17 August 2023
Sec. Diabetes: Molecular Mechanisms
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1226655
This article is part of the Research Topic Mechanistic and Physiological Implications of Insulin Resistance in Metabolic Diseases View all 9 articles
 Thi Kim Chung Le1*
Thi Kim Chung Le1* Xuan Dat Dao1
Xuan Dat Dao1 Dang Vung Nguyen1Duc Huy Luu2
Dang Vung Nguyen1Duc Huy Luu2 Thi Minh Hanh Bui1
Thi Minh Hanh Bui1 Thi Huong Le1
Thi Huong Le1 Huu Thang Nguyen1Tran Ngoan Le1Toshio Hosaka3
Huu Thang Nguyen1Tran Ngoan Le1Toshio Hosaka3 Thi Thu Thao Nguyen1*
Thi Thu Thao Nguyen1*The discovery of insulin in 1921 introduced a new branch of research into insulin activity and insulin resistance. Many discoveries in this field have been applied to diagnosing and treating diseases related to insulin resistance. In this mini-review, the authors attempt to synthesize the updated discoveries to unravel the related mechanisms and inform the development of novel applications. Firstly, we depict the insulin signaling pathway to explain the physiology of insulin action starting at the receptor sites of insulin and downstream the signaling of the insulin signaling pathway. Based on this, the next part will analyze the mechanisms of insulin resistance with two major provenances: the defects caused by receptors and the defects due to extra-receptor causes, but in this study, we focus on post-receptor causes. Finally, we discuss the recent applications including the diseases related to insulin resistance (obesity, cardiovascular disease, Alzheimer’s disease, and cancer) and the potential treatment of those based on insulin resistance mechanisms.
Insulin signaling beginning at its receptors produces multidirectional impacts on the metabolism, survival, and proliferation of targeted cells. Insulin transduces its effects by insulin receptors that signal through many pathways including participation in protein and lipid phosphorylation, management of trafficking procedures, management of a system of enzymes, and control of a system of transcriptional factors. Numerous studies have indicated that insulin resistance is related to many diseases: type 2 diabetes, obesity, cardiovascular disease, Alzheimer’s disease, and cancer, which are the medical challenges in the first century of the new millennium (1–3). Therefore, it is necessary to update the physiology of insulin signaling and the disordered physiological processes associated with insulin resistance, henceforth proposing more effective therapies for treating diseases related to insulin resistance.
The insulin signaling pathway is an intracellular signaling pathway that is responsible for the metabolism of the body, especially in metabolism, growth, and survival. The process of the insulin signaling pathway comprises several steps, wherein the initial stage is the participation of insulin and insulin-like growth factors (IGF) which bind to insulin and IGF receptors. The second step is binding an insulin receptor (IR) to its direct substrates including Growth factor receptor-bound protein 2 (GRB2), Src homology 2 domain-containing (SHC), insulin receptor substrate (IRS), SH2B adapter protein 2/adapter protein with a PH and SH2 domain (SH2B2/APS), and Growth factor receptor-bound protein 10 (GRB10). This binding could exert several cellular signaling pathways for mitogenesis and metabolism. Our review will briefly unravel every step of the insulin signaling pathway (2) (Figure 1).
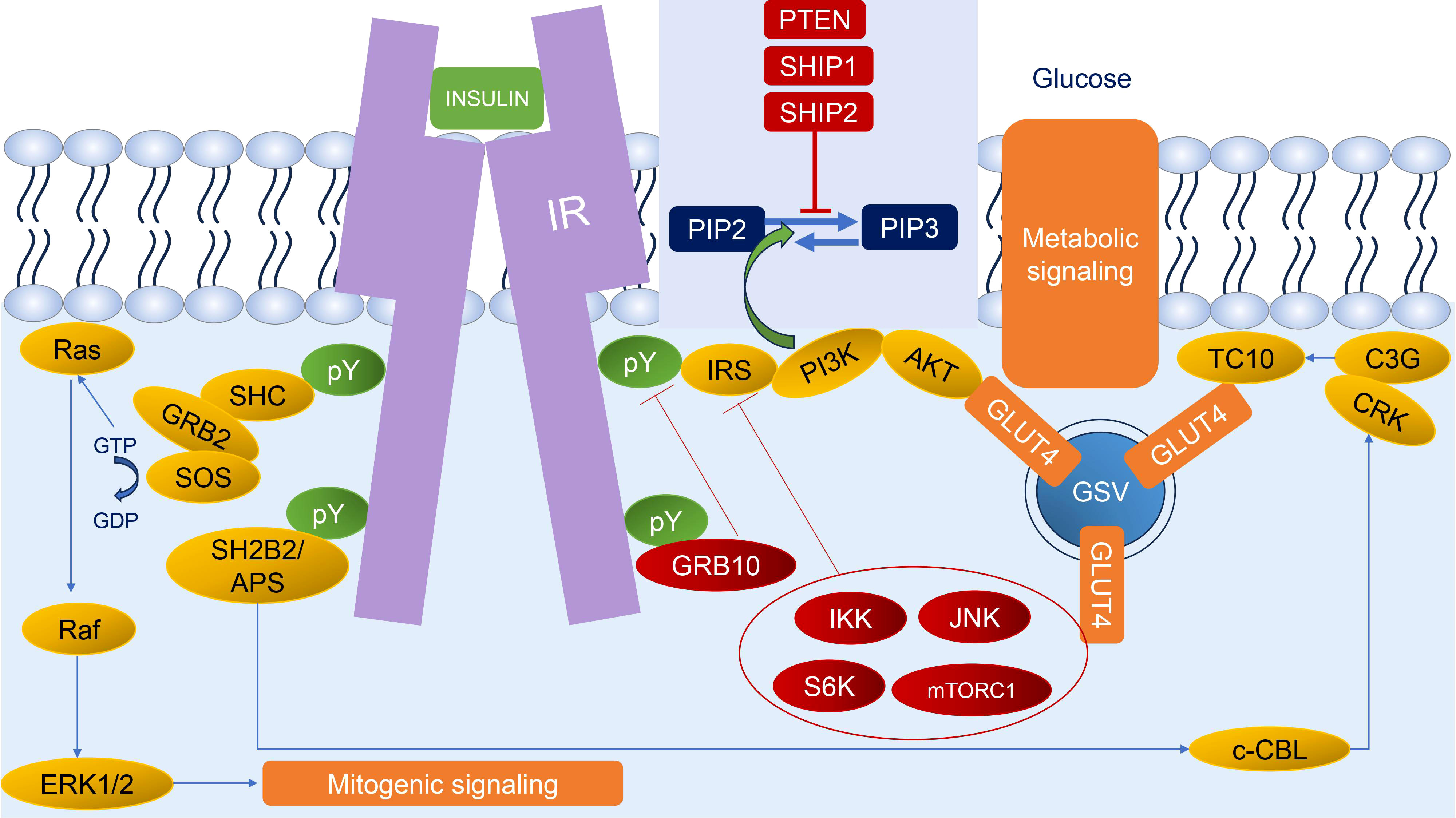
Figure 1 Insulin signaling. Insulin binds to IR leading to autophosphorylation of IR; thus, IR could recruit diverse substrates. The two main responses of insulin signaling are mitogenic signaling (begin with SHC and GRB2 through the ERK1/2 pathway) and metabolic signaling (begin with IRS through the AKT pathway and SH2B2/APS through CRK/TC10 pathway). The regulation of insulin signaling could be characterized by negative feedback mechanisms, such as stabilization and recruitment of GRB10 to the IR; activation of lipid phosphatases (PTEN, SHIP1, and SHIP2) that dephosphorylate PIP3; and activation of several stress kinases (IKK, JNK, S6K, and mTORC1) to phosphorylate and inhibit IR and IRS. Green circles and arrows represent activating events; red circles and lines represent inhibitory events. pY: phosphorylated tyrosine residue.
Insulin is classified in the peptide hormone group that is secreted from beta cells in the islet of Langerhans of the pancreas. In mammals, apart from insulin, IGF-1 and IGF-2 are also synthesized by the insulin gene family. Insulin could bind to the specific receptors on target cells and exert the metabolic response. Similarly, IGF-1 and IGF-2 would trigger the mitogenic response, and promote proliferation and differentiation of cells (4, 5). Nevertheless, these functional distinctions are blurry due to the high similarity between the insulin and IGF receptors and are common in many downstream effectors (3, 6). Therefore, there is a relationship between hyperinsulinemia and neurodegeneration, cardiovascular disease, and several cancers (7–9).
IR is a heterotetrameric receptor tyrosine kinase that was formed by four subunits composed of two extracellular α subunits and two transmembrane β subunits. Owing to this structure, IR has two isoforms, A and B, and there are several differences between them. IR-A, generated by the splicing out of exon 11 that leads to the absence of 12 amino acids at the carboxyl terminus of the α subunit, is highly expressed during fetal life and has more potency in metabolism, promoting glycogenesis compared to IR-B. IR-B is differentiated by alternative splicing and includes an additional 12-amino acid sequence encoded by exon 11 at the C-terminal of the α subunit; the corresponding sequence in type 1 IGF is vacant. The presence of IR-B in the liver, muscle, and white adipocyte tissue is more predominant than IR-A due to the high affinity of IR-B compared to IR-A (6, 10, 11).
Insulin binding to IR creates trans-autophosphorylation into IR, which is an essential event for the recruitment of IR’s direct substrates. These substrates bind to IR by the bindings between the phosphotyrosine-binding (PTB) domain of the substrates and the tyrosine residues of IR. The downstream signaling of IR activation comprises two parts: metabolic and mitogenic signals. The metabolic signals need a lower insulin concentration to be triggered compared to the mitogenic signals (2, 3).
The metabolic arm of insulin signaling is initiated by IRS and SH2B2/APS (2). Together, these pathways warrant the effective translocation of glucose transporter type 4 (GLUT4) to the membrane by adapting signaling platforms at the cell surface which are composed of lipids, protein kinases, small GTPases, and adaptor proteins. To be specific, GLUT4 is in the GLUT family of transmembrane hexose transporter, which has a high attractiveness to glucose, and the majority of it is expressed in muscle cells and adipocytes (12). The translocation of GLUT4 in response to insulin or exercise results in a ten-fold increase in glucose uptake (13); when insulin is absent, only approximately 5% of the total GLUT4 is found on the membrane (14, 15). GLUT4 trafficking has a key role in the maintenance of glucose homeostasis by preventing gluconeogenesis in the liver and promoting glucose uptake into the muscle and adipose tissues, but it depends on the response to insulin and coordination of insulin signaling (PI3K signaling pathway and APS signaling pathway) in every step of GLUT4 trafficking, including GLUT4 endocytosis, GLUT4 sorting and retention, and exocytosis of GLUT4 storage vesicles (GSVs) (12).
The best-unraveled substrate for the role of IR scaffolds is IRS. The IRS family has six members, wherein IRS1 and IRS2 are claimed to mediate virtually the metabolic responses of IR activation. IRS has NH2-terminal pleckstrin homology (PH) and phosphotyrosine-binding domains (PTB domains) which are responsible for guiding them to activate IR. After binding to IR, the IRS tyrosine residues are phosphorylated which enlist downstream effectors to enhance the insulin response (16). Particularly, phosphorylated IRS attract phosphoinositide-3-kinase (PI3K) heterodimers containing a regulatory subunit with SH2 domain (p85) and a catalytic subunit (p110) that stimulate glucose transport (17, 18). Besides, PI3K catalyzes the reaction, converting phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). On the contrary, phosphatase and tensin homolog deleted on chromosome 10 (PTEN) catalyze the reverse reaction. Interestingly, insulin could block the activity of PTEN by obscure mechanisms, which may be explained by the interactions with phosphatidylinositol 3,4,5-trisphosphate-dependent Rac exchanger 2 (P-REX2) through two joints, i.e., PH domain of P-REX2 binding to the catalytic region of PTEN and the inositol polyphosphate 4-phosphatase domain of P-REX2 interacting with the postsynaptic density-95/Discs large/zona occludens-1-binding domain of PTEN. These interactions request the phosphorylation of the C-terminal of PTEN to discharge the PH domain of P-REX2 from its adjoining diffuse B-cell lymphoma homology domain (19). Therefore, activating PI3K as well as inhibiting PTEN by insulin leads to the accumulation of PIP3. PIP3 then pulls in proteins with PH domains to the plasma membrane including phosphoinositide-dependent kinase 1 (PDK1) and Protein kinase B/AKT. PDK1 phosphorylates AKT in its activation loop after binding to PIP3, and the mechanistic target of rapamycin complex 2 (mTORC2) phosphorylates its hydrophobic motif (20–22). AKT is a bridge protein that connects insulin signaling to downstream effectors of GLUT4 trafficking. It is involved in glucose absorption in the muscle and adipose tissue and increases intracellular glucose production (12, 23). AKT promotes GLUT4 storage vesicle (GSV) exocytosis by phosphorylating and dephosphorylating RAB GAP AS160 (also known as TBC1 domain family member 4, TBC1D4) and the RAL-GTPase-activating protein complex (RAL-GAP complex, RGC), which control small GTPases involved in GSV retention and localization to the cell surface. Particularly, RAB GAP AS160 is necessary for insulin activation of GLUT4 exocytosis. The evidence supporting this process is that the inhibitory-induced mutant of AS160 inhibits insulin activation of exocytosis at a prior step of the fusion of GSVs with the cell membrane. Nevertheless, this mutant does not constrain insulin-induced inhibition of GLUT4 endocytosis. RGC is a complex, including an RGC1 regulatory subunit and an RGC2 catalytic subunit, which directly activates the guanosine triphosphate hydrolysis of RalA, whereas the small GTPase RalA downstream of PI3K plays the role as an essential part of redistribution of GLUT4 by marshalling the exocyst complex for GLUT4 vesicle targeting in adipocytes. Thus, RGC complex regulates the transport machineries responsible for GLUT4 translocation, connecting with PI3K signal (12, 24, 25).
Another pathway of metabolic insulin response is the APS-insulin signaling pathway. Activated IR recruits APS, which contain PH and Src homology 2 (SH2) domains and is so also known as SH2B2, with high affinity. Following that, APS pulls in a complex that includes the proto-oncogene c-CBL and c-CBL-associated protein (CAP) and phosphorylate c-CBL (12, 26). Activated c-CBL recruits CRK, which is a complex with the guanine nucleotide exchange factor (GEF) C3G to the plasma membrane, then C3G activates TC10, a member of the RHO-family of small GTPases (12, 27, 28). GTP-bound TC10 interacts with the exocyst tethering complex, resulting in GSV sites docking at the cell surface. Apart from that, TC10 also binds to CDC42-interacting protein 4 (CIP4), forming a stable complex with Rab GEF GAPex5 and regulating the activity of Rab5 family GTPases involved in GSV retention and delivery (12).
The second major response of insulin signaling is mitogenic action. In this pathway, insulin acts as a growth factor although it is a considerably weaker mitogen than its cousins, the IGF, and other relatives such as platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and EGF. It has been considered to be the most anabolic hormone that governs proliferation and migration, and it also inhibits apoptosis (3, 29–31). The mitogenic action is triggered by the binding between IR and SHC protein, which recruits the GRB2-SOS (son of sevenless) complex. SOS regulates the exchange of GTP for GDP only when Ras is attached to the plasma membrane; consequently, SOS GTP loading activates the Ras protein. Farnesylation of Ras facilitates its translocation to the cell surface, particularly when farnesyltransferase (FTase) catalyzes the attachment of a farnesyl moiety to a cysteine residue of Ras. Simultaneously, with the phosphorylating IR, insulin could activate FTase, thus increasing the farnesylation of Ras. Furthermore, insulin-activating FTase requires an intact IR but not an IGF receptor, implying that this action is driven solely by IR and does not include the interaction of IR and the IGF receptor. Ras protein then activates the cascade from Raf protein to the mitogen-activated protein kinase (MAPK). The activated MAPK, extracellular signal-regulated kinase (ERK1/2, also referred to as p44/p42 MAPK), is a key effector of the insulin mitogenic signal that promotes cell growth, cell division, migration, and apoptosis (3, 30). ERK1/2 activates a pleiotropy of substrates (approximately 200 different substrates have been identified), including cytosolic targets (PLA2, Raf-1, Caspase-9, Bcl-2, etc.) and cytoskeletal targets (MAP2, MAP4, Tau, etc.) which regulate specific activities within certain organelles, such as transcription in the nucleus and mitochondria, and bring components of the ERK1/2 cascade into proper localizations where it phosphorylates specific proteins without remarkable nuclear translocation. Half of the currently identified ERK1/2 substrates are nuclear targets including Elk-1, c-Myc, c-Fos, and HIF-1α, which regulate many stimulated nuclear processes, such as transcription, chromatin remodeling, and nuclear translocation (32, 33). Evidence of these effects of the ERK1/2 pathway was found in a variety of tissues, including the adipose tissue, muscle, and pancreatic beta-cells. ERK-1 signaling has been shown to promote adipogenesis, and ERK-1 knockdown animals had decreased adipose mass (3). Pre-adipocytes have very little IR, but as they move through adipogenesis, their IR levels rise to levels that are greater than those of IGF-1R. In studies using cell culture, high insulin dosages stimulate ERK activation and support the early proliferative stage of adipogenesis. ERK-1 activity decreases when adipogenesis increases and IR expression rises, and metabolic insulin signaling takes over. The IGF-1R stimulation is necessary to activate ERK and causes adipogenesis that is provided in vivo by high levels of IGF ligands during embryonic and postnatal growth and development in FIRKO mice. As adipogenesis expands and IR expression rises, ERK-1 activation declines, and metabolic insulin signaling takes over. In vivo, high levels of IGF ligands throughout embryonic and postnatal growth and development provide the IGF-1R stimulation required to activate ERK and drive adipogenesis, and FIRKO mice with IR missing in adipose tissue, nonetheless, form fat pads (3).
Insulin signaling is always under tight control due to the fact that any abnormal signal could exert severe perturbations in metabolism and tumorigenesis. Hence, rapidly turning off the insulin signal at various levels is critical. Signaling is attenuated by the activity of several phosphatases, stress kinases, and adaptor proteins (34).
Tyrosine phosphatases comprise transmembrane phosphatases, such as Leukocyte common antigen-related (LAR) protein and cytoplasmic protein tyrosine phosphatases, especially PTP1B, that could dephosphorylate the tyrosine residues on IR and IRS, thus reducing their actions (2, 34).
The serine/threonine phosphatases comprise protein phosphatase 1 (PP1) and protein phosphatase 2 (PP2, including PP2A, PP2B, and PP2C). PP1 controls several types of enzymes involved in the metabolism of glucose, including glycogen synthase. In the PP2 family, multiple protein kinases linked to insulin function are regulated by PP2A, i.e., AKT, ERK, PP2B, and PP2C (in particular, the PH domain leucine-rich repeat protein phosphatases PHLPP-1 and -2) could dephosphorylate AKT. Thereby, they could induce insulin resistance (35–38).
Lipid phosphatases, such as PTEN and SH2 domain-containing inositol 5-phosphatases (SHIP) 1 and 2, could dephosphorylate PIP3, hence antagonizing PI3K signaling in cells (34, 37, 39).
The increased serine/threonine phosphorylation of IR or IRS that has an inhibitor effect on tyrosine kinase activity has been witnessed in IR in humans, thus inhibiting insulin signaling (34, 40). Multiple kinases including c-Jun amino-terminal kinase (JNK), inhibitor of κB kinase (IKK), traditional and innovative PKCs but also mTORC1, and S6 kinase (S6K) could inhibit insulin signaling by increasing serine/threonine phosphorylation and reducing IRS tyrosine phosphorylation (2, 34).
Grb10 and Grb14, as well as proteins of the suppressor of cytokine signaling (SOCS) family (especially SOCS1 and SOCS3), are negative regulators of the tyrosine kinase activity of the IR, proving that it has a role in insulating the contact of IR substrates to the activated receptors. (34)
Tribbles homolog 3 (Trb3) pertains to a family of pseudokinases that could bind to AKT and block its activation, hence inhibiting insulin signaling. Besides, inositol phosphate IP7 could inhibit AKT translocation to the cell surface, thus attenuating insulin signaling (34).
Insulin resistance is typical with a state when plasma insulin is at a normal range; however, target tissues cannot exert a physiological response of glucose-lowering, implicating the inhibition of gluconeogenesis, lipolysis, cellular glucose uptake, and glyconeogenesis (2, 41). The state of insulin resistance commands enhanced insulin secretion to recompense; hence, fasting plasma insulin is in high concentrations (42). In this review, insulin resistance is defined as a curve of responding to insulin dose with increased EC50 (half maximal effective concentration), with or without declined maximal response (43).
The physiological mechanism of insulin resistance owes to deficient insulin action at target cells. To pinpoint the deficiency of cellular insulin action, a lot of effort has been induced and two major origins have been indicated: receptor defects (decreased IR expression at the plasma membrane) and post-receptor defects (impaired signal transduction) (44). In the case of decreased IR expression (receptor defects), the dose response increased but the maximal biological response remained the same at the normal value unless the IR fell to 5-10% of the normal value. Distinctly, not only was there an increase in dose response but the maximal response also decreased in the case of post-receptor defects (2, 44, 45).
Receptor defects lead to insulin resistance by mutations of IR in the majority. Mutations of IR exert the type A insulin resistance syndrome, which occurs in at least 0.05% of the general Japanese population (46, 47). Apart from that, type B insulin resistance syndrome causes insulin resistance by autoantibodies blocking the binding sites of insulin on IR. This syndrome is frequently accompanied by autoimmune conditions (48). Besides, Rabson–Mendenhall syndrome and Donohue syndrome are also brought about by mutations of IR, most of which are in both alleles but are more serious compared to type A insulin resistance syndrome, which is usually associated with intractable diabetes (47, 49).
Post-receptor defects comprise abnormalities in metabolic signals and mitogenic signals; it includes the effects on substrates and negative modulators of insulin signaling.
Many single nucleotide polymorphisms (SNPs) and gene mutations of IRS have been identified that are associated with decreased insulin signaling. The SNPs G972R, Gly972Arg, and rs1801278 of IRS-1 have shown a high prevalence in patients with type 2 diabetes and are caused by disrupted insulin signaling (50–52). It showed that the T608R mutation was located in the highly conversed region of IRS-1 that impairs PI 3-kinase activation and binding, as well as GLUT4 translocation in adipose cells. On the other hand, the I65S, R66S, and G86R mutations reflexed amino acids in the phosphotyrosine binding site of the IRS-1 protein. These changes in amino acids could lead to a shift in binding energies and trigger conformational variations in the L1 domain of the IR structure. Besides, in the ligand-binding area of the IR structure, I65S, R66S, and G86R mutations of IRS1 structures’ positional displacement were caused by the observation of variable binding mode orientations. All of these factors could be reasons for the varied interactions of IRS1 and IR, which lead to aberrant insulin transduction, followed by insulin resistance (53, 54).
The activity of PI3K depends on an equivalence between the activities of two subunits: the regulatory and catalytic subunits (55). In the regulatory subunit, several mutations have shown their association with signal transduction. The mutations of PIK3R1, encoding the p85α regulatory subunit of PI3K, including the most frequent mutation Arg649Trp and 11 other PIK3R1 mutations have been recognized in patients with SHORT syndrome and genetic insulin resistance. The majority of the mutations in this syndrome are in the area encoding the C-terminal SH2 domain of p85, which is required for PI3K binding to tyrosine-phosphorylated proteins including insulin receptor and (IR) substrate (IRS-1). The 649 amino acid position on the SH2 domain of the p85 regulatory subunit is arginine (wild-type) and highly conserved, and it participates in a bivalent interaction between PI3K and IR or IRS with the linkage of oxygen atoms of the phosphotyrosine. The improved link to the phosphopeptide was focused within the binding pocket. The mutation Arg649Trp at this location changes the amino acid sequence from Arginine to Tryptophan, which decreases affinity to the phosphopeptide (49). Several works of research have indicated that PIK3R1 mutations are the second common origin in the rank of single-gene insulin resistance (56–58). Mutations of PIK3R2, encoding the p85β regulatory subunit of PI3K, have shown the ability to augment PI3K signaling, which has been perceived in patients with hypoglycemia and either segmental overgrowth or megalencephaly (59). Somatic mutations of PIK3CA, encoding the p110α catalytic subunit of PI3K have been seen generally in tumor cells and segmental overgrowth tissues less frequently, while germline mutations of PIK3CA have been observed in patients with segmental overgrowth or megalencephaly (59, 60).
AKT substrate includes three isoforms: AKT1, AKT2, and AKT3; wherein AKT1 and AKT2 are universally occurring, AKT3 is mainly expressed in the central nervous system (2). The mutations of AKT1 and AKT3 have not shown an association with insulin sensitivity; however, in contrast, mutations of AKT2 are an identified origin of the monogenic disorder of glucose metabolism (61). A heterozygous Arg274His mutation of AKT2 gives rise to a blocked impact on insulin activity, which was observed in patients with severe postprandial hyperinsulinemia (62). A lower function AKT2 coding variant, p.Pro50Thr, occurs with high frequency (1.1%) in the Finnish population, and influences glucose uptake negotiated by insulin in target tissues, thus escalating the risk of type 2 diabetes in carriers (23, 63).
TBC1D4 is involved in GSV translocation, and its mutations may affect insulin action. A truncated Arg363Ter mutation was identified in a family who suffered from postprandial hyperinsulinemia (64). A homozygous Arg684Ter variant of TBC1D4, which appeared with a great allele prevalence (17%) in the Greenlandic population, has shown a remarkable increase in the risk of the progress of type 2 diabetes (65). Deletion of TBC1D4 generating abolished glucose uptake negotiated by insulin is related to impaired glycemic control (66).
The variants in PTPN11 are possibly responsible for the blockage of tyrosine kinase signaling and downstream Ras-MAPK signaling, hence exerting Noonan syndrome. The individuals harboring mutations of PTPN11 manifest insulin resistance with impairment in glucose uptake and glycogen synthesis (67, 68).
Patients with Cowden syndrome are heterozygous mutations in PTEN for diminished function and also have elevated insulin sensitivity. PTEN haploinsufficiency and PTEN deletion ameliorate insulin sensitivity and defend against systemic insulin resistance associated with obesity. Besides, the inactivation of PTEN gives rise to the decreased hyperproliferation of cells (69, 70).
Several factors could exert increased activity of stress kinases including JNK, IKK, and PKC, thus increasing insulin resistance state. Some of these factors include free fatty acids (FFA), diacylglycerol (DAG), sphingolipid ceramide, hyperglycemia, reactive oxygen species (ROS), and endoplasmic reticulum (ER) stress (34).
Obesity has an association with insulin resistance, which is characterized by the progress of chronic inflammation at a low level (71). Adipocytes secreting the chemokine MCP-1 promote macrophage agglomeration into adipose tissues and exert insulin resistance (72). Several cytokines, such as TNF-α, IL1β, and IL-6, are secreted by immune cells and adipocytes that give rise to insulin resistance by increased serine/threonine phosphorylation or activation of SOCS3 (an adaptor protein) in adipocytes (73, 74).
Interestingly, in the context of insulin resistance, when the metabolic signals such as PI3K signaling is inhibited, the mitogenic signals like the Ras-Raf-MAPK pathway of insulin are not interrupted and are possibly upregulated (75).
Insulin resistance leads to increased insulin secretion to compensate; this state is known as hyperinsulinemia (42). Insulin resistance and/or hyperinsulinemia could exert many complications, such as obesity, cardiovascular disease, neurodegeneration especially Alzheimer’s disease, and cancer (3).
Indeed, insulin resistance could result in lipoproteins’ profile alterations. ApoB is a protein that could enhance the assemblage and excretion of VLDL. Normally, through the PI3K pathway, insulin controls the deterioration of apoB; however, under insulin resistance, this process is inadequate. Besides, insulin resistance also decreases the activity of lipoprotein lipase, which is a dominant factor in VLDL clearance. Thereby, insulin resistance gives rise to an escalation in VLDL formation, which accounts for hypertriglyceridemia under the insulin resistance state (76–79). Insulin resistance also exerts the synthesis of small dense LDL (sdLDL) and decreases HDL levels, which are created by the transferring of VLDL’s triglycerides to LDL and HDL under the catalysis of cholesteryl ester transfer protein (CETP), provoking triglyceride-rich LDL and decreasing HDL-C. Triglyceride-rich LDL is lipolysis by hepatic lipase, leading to the formation of sdLDL. sdLDL has several properties including lessened affinity for the LDL receptor, accelerated entry into the arterial wall, dominant arterial detention, major sensitivity to oxidation, and greater half-life that promotes the atherogenic activity of sdLDL. Strikingly, increased sdLDL levels could not be measured by the LDL levels, due to the lower level of sdLDL than LDL. These variations of lipoproteins profile are called dyslipidemia, which is characterized by the lipid trinity: hypertriglyceridemia, low levels of HDL, and the occurrence of sdLDL, which leads to obesity. Dyslipidemia is a sturdy predictor of the development of type 2 diabetes, which is owed to insulin resistance and cardiovascular disease (9, 80–82). A strong association between insulin resistance and the risk of cardiovascular disease (CVD) has been indicated (83). Several molecular mechanisms contributing to this correlation include atherosclerosis development and vascular function (9, 84). Dyslipidemia is the main reason for atheroma plaque formation. Besides, insulin resistance could exert vascular abnormality by activating the MAPK pathway generating vasoconstriction improvement and inhibiting the NO synthesis which has an essential role in vascular strength through the ability of vasodilation (85).
Several studies have revealed that brain insulin resistance, which means dysregulated brain insulin signaling, is one of the factors of cognitive disorders and Alzheimer’s disease (AD) and is also considered a risk factor for sporadic AD development (86, 87). Indeed, the major pathology of AD including neurofibrillary tangles (NFT) and amyloid-β plaques could owe to insulin resistance (8). Firstly, insulin, IGF-1, and IGF-2 have been indicated to have inhibited activity on apoptosis in brain neurons through IR and regulate many neurobiological processes, inhibiting the degradation of amyloid-β (88–90). Moreover, amyloid-β could compete with insulin for binding to IR, leading to impairment of insulin-binding affinity to IR, and thus exerting insulin resistance. This state has been proven to aggravate the already present AD pathology (91). The increased serine phosphorylation of IRS-1, PI3K pathway dysfunction, and inhibition of AKT causing stimulation of Glycogen synthase kinase 3β (GSK-3β) and mTOR phosphorylation lead to hyperphosphorylation of tau protein, which is the main component of NFT. Besides, activated GSK-3β could enhance the amyloid-β plaque deposition. Both of them may give rise to microglia-mediated neuroinflammation that produces AD pathogenesis (92–96). Apart from that, the activation of the Ras-MAPK pathway enhances the transcription of several particular genes related to the role of the survival of neurons which are associated with synaptic plasticity and tau phosphorylation and are connected with amyloid-β plaques accumulation and NFT formation, which are major features of AD (97, 98).
Several investigations have indicated the association between hyperinsulinemia with the risk of certain types of cancers. This phenomenon could be analyzed by direct or indirect impacts of insulin resistance on the development of cancer (3). The direct impacts due to the overexpression of insulin resistance in specific tumor cell lines enhance the proliferative response to insulin, and ERK has been pointed out to be responsible for activating the cell cycle process in cancer cells (99, 100). The indirect effects account for the ability of insulin to stimulate increased levels of many modulators of proliferation and differentiation including IGF-1, cytokines, and growth factors, such as leptin, VEGF, and IL-6 (101–103). These effects may accelerate the growth of neoplasms, angiogenic process, and metastasis, and could be the mechanisms by which hyperinsulinemia and/or insulin resistance increases the carcinogenesis risk of many kinds of cancer, such as cancer of the breast, colon, liver, pancreas, and endometrium (104–107).
Based on these observations, it would seem that IR modulator therapies are effective in the treatment of diseases related to IR, especially for those substances that more preferentially stimulate metabolic signaling (AKT), minimally activate mitogenic responses (ERK), and limit insulin-like growth factor receptor (IGFR) activation (3, 108). Several reports have declared some IR agonist molecules that would be satisfied to become a safe and efficient IR agonist (selectively activating the AKT pathway over the ERK signaling): the peptide S597 and the human monoclonal antibody XMetA (3, 109, 110).
The complications of insulin signaling do not only owe to the requirement for maintaining the plasma glucose within a narrow range and the regulation of other metabolic substances but also control the growth and survival of multiple cells. The ability to design IR agonists provides variable potencies for treatment. While extensive investigations have been made, a lot of gaps persist in our knowledge of the molecular mechanisms involved in insulin signaling and will stand as the highlight of years of studies. It is hoped that this review would serve as a resource for assistance in the progress of more efficient next-generation therapies for diseases related to insulin resistance.
CL and TN have equally contributed as the main authors to the supervision of the final manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Saltiel AR. Insulin signaling in health and disease. J Clin Invest (2021) 131(1):142241. doi: 10.1172/JCI142241
2. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev (2018) 98(4):2133–223. doi: 10.1152/physrev.00063.2017
3. Bedinger DH, Adams SH. Metabolic, anabolic, and mitogenic insulin responses: A tissue-specific perspective for insulin receptor activators. Mol Cell Endocrinol (2015) 415:143–56. doi: 10.1016/j.mce.2015.08.013
4. Jin Chan S, Steiner DF. Insulin through the ages: phylogeny of a growth promoting and metabolic regulatory hormone1. Am Zool (2000) 40(2):213–22. doi: 10.1093/icb/40.2.213
5. Wu Q, Brown MR. Signaling and function of insulin-like peptides in insects. Annu Rev Entomol (2006) 51:1–24. doi: 10.1146/annurev.ento.51.110104.151011
6. Siddle K. Signalling by insulin and IGF receptors: supporting acts and new players. J Mol Endocrinol (2011) 47(1):R1–10. doi: 10.1530/JME-11-0022
7. Perseghin G, Calori G, Lattuada G, Ragogna F, Dugnani E, Garancini MP, et al. Insulin resistance/hyperinsulinemia and cancer mortality: the Cremona study at the 15th year of follow-up. Acta Diabetol (2012) 49(6):421–8. doi: 10.1007/s00592-011-0361-2
8. Akhtar A, Sah SP. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem Int (2020) 135:104707. doi: 10.1016/j.neuint.2020.104707
9. Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol (2018) 17(1):122. doi: 10.1186/s12933-018-0762-4
10. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol (2018) 19(1):31–44. doi: 10.1038/nrm.2017.89
11. Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin receptor isoforms in physiology and disease: an updated view. Endocr Rev (2017) 38(5):379–431. doi: 10.1210/er.2017-00073
12. Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol (2012) 13(6):383–96. doi: 10.1038/nrm3351
13. Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol (2002) 3(4):267–77. doi: 10.1038/nrm782
14. Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol (1991) 113(1):123–35. doi: 10.1083/jcb.113.1.123
15. Martin S, Millar CA, Lyttle CT, Meerloo T, Marsh BJ, Gould GW, et al. Effects of insulin on intracellular GLUT4 vesicles in adipocytes: evidence for a secretory mode of regulation. J Cell Sci (2000) 113 Pt 19:3427–38. doi: 10.1242/jcs.113.19.3427
16. Hubbard SR. The insulin receptor: both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb Perspect Biol (2013) 5(3):a008946. doi: 10.1101/cshperspect.a008946
17. Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol (2005) 25(5):1596–607. doi: 10.1128/MCB.25.5.1596-1607.2005
18. Clarke JF, Young PW, Yonezawa K, Kasuga M, Holman GD. Inhibition of the translocation of GLUT1 and GLUT4 in 3T3-L1 cells by the phosphatidylinositol 3-kinase inhibitor, wortmannin. Biochem J (1994) 300(Pt 3):631–5. doi: 10.1042/bj3000631
19. Hodakoski C, Hopkins BD, Barrows D, Mense SM, Keniry M, Anderson KE, et al. Regulation of PTEN inhibition by the pleckstrin homology domain of P-REX2 during insulin signaling and glucose homeostasis. Proc Natl Acad Sci USA (2014) 111(1):155–60. doi: 10.1073/pnas.1213773111
20. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J (1996) 15(23):6541–51. doi: 10.1002/j.1460-2075.1996.tb01045.x
21. Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science (1998) 279(5351):710–4. doi: 10.1126/science.279.5351.710
22. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science (2005) 307(5712):1098–101. doi: 10.1126/science.1106148
23. Latva-Rasku A, Miikka-Juhani H, Stancakova A, Koistinen H, Kuusisto J, Guan L, et al. A partial loss-of-function variant in AKT2 is associated with reduced insulin-mediated glucose uptake in multiple insulin-sensitive tissues: A genotype-based callback positron emission tomography study. Diabetes (2017) 67(2):334–42. doi: 10.2337/db17-1142
24. Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on rabGAP AS160. Mol Biol Cell (2004) 15(10):4406–15. doi: 10.1091/mbc.e04-04-0333
25. Chen XW, Leto D, Xiong T, Yu G, Cheng A, Decker S, et al. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell (2011) 22(1):141–52. doi: 10.1091/mbc.e10-08-0665
26. Liu J, Kimura A, Baumann CA, Saltiel AR. APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol Cell Biol (2002) 22(11):3599–609. doi: 10.1128/MCB.22.11.3599-3609.2002
27. Knudsen BS, Feller SM, Hanafusa H. Four proline-rich sequences of the guanine-nucleotide exchange factor C3G bind with unique specificity to the first Src homology 3 domain of Crk. J Biol Chem (1994) 269(52):32781–7. doi: 10.1016/S0021-9258(20)30059-4
28. Chiang SH, Baumann CA, Kanzaki M, Thurmond DC, Watson RT, Neudauer CL, et al. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature (2001) 410(6831):944–8. doi: 10.1038/35073608
29. Chakrabarti P, Kim JY, Singh M, Shin YK, Kim J, Kumbrink J, et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol Cell Biol (2013) 33(18):3659–66. doi: 10.1128/MCB.01584-12
30. Draznin B. Mitogenic action of insulin: friend, foe or ‘frenemy’? Diabetologia (2010) 53(2):229–33. doi: 10.1007/s00125-009-1558-6
31. Sasaoka T, Rose DW, Jhun BH, Saltiel AR, Draznin B, Olefsky JM. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem (1994) 269(18):13689–94. doi: 10.1016/S0021-9258(17)36885-0
32. Wortzel I, Seger R. The ERK cascade. Genes Cancer (2011) 2(3):195–209. doi: 10.1177/1947601911407328
33. Kohno M, Pouyssegur J. Targeting the ERK signaling pathway in cancer therapy. Ann Med (2006) 38(3):200–11. doi: 10.1080/07853890600551037
34. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol (2014) 6(1):a009191. doi: 10.1101/cshperspect.a009191
35. Kowluru A, Matti A. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem Pharmacol (2012) 84(5):591–7. doi: 10.1016/j.bcp.2012.05.003
36. Brady MJ, Saltiel AR. The role of protein phosphatase-1 in insulin action. Recent Prog Horm Res (2001) 56:157–73. doi: 10.1210/rp.56.1.157
37. Ni YG, Wang N, Cao DJ, Sachan N, Morris DJ, Gerard RD, et al. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci USA (2007) 104(51):20517–22. doi: 10.1073/pnas.0610290104
38. Brognard J, Newton AC. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab (2008) 19(6):223–30. doi: 10.1016/j.tem.2008.04.001
39. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene (2008) 27(41):5527–41. doi: 10.1038/onc.2008.247
40. Shao J, Catalano PM, Yamashita H, Ruyter I, Smith S, Youngren J, et al. Decreased insulin receptor tyrosine kinase activity and plasma cell membrane glycoprotein-1 overexpression in skeletal muscle from obese women with gestational diabetes mellitus (GDM): evidence for increased serine/threonine phosphorylation in pregnancy and GDM. Diabetes (2000) 49(4):603–10. doi: 10.2337/diabetes.49.4.603
41. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature (2006) 444(7121):840–6. doi: 10.1038/nature05482
42. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med (2017) 23(7):804–14. doi: 10.1038/nm.4350
43. Kahn CR. Insulin resistance, insulin insensitivity, and insulin unresponsiveness: a necessary distinction. Metabolism (1978) 27(12 Suppl 2):1893–902. doi: 10.1016/s0026-0495(78)80007-9
44. Kolterman OG, Gray RS, Griffin J, Burstein P, Insel J, Scarlett JA, et al. Receptor and postreceptor defects contribute to the insulin resistance in noninsulin-dependent diabetes mellitus. J Clin Invest (1981) 68(4):957–69. doi: 10.1172/JCI110350
45. Olefsky JM, Kolterman OG, Scarlett JA. Insulin action and resistance in obesity and noninsulin-dependent type II diabetes mellitus. Am J Physiol (1982) 243(1):E15–30. doi: 10.1152/ajpendo.1982.243.1.E15
46. Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM, et al. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N Engl J Med (1976) 294(14):739–45. doi: 10.1056/NEJM197604012941401
47. Takeuchi T, Ishigaki Y, Hirota Y, Hasegawa Y, Yorifuji T, Kadowaki H, et al. Clinical characteristics of insulin resistance syndromes: A nationwide survey in Japan. J Diabetes Investig (2020) 11(3):603–16. doi: 10.1111/jdi.13171
48. Flier JS, Bar RS, Muggeo M, Kahn CR, Roth J, Gorden P. The evolving clinical course of patients with insulin receptor autoantibodies: spontaneous remission or receptor proliferation with hypoglycemia. J Clin Endocrinol Metab (1978) 47(5):985–95. doi: 10.1210/jcem-47-5-985
49. Taylor SI, Cama A, Accili D, Barbetti F, Quon MJ, de la Luz Sierra M, et al. Mutations in the insulin receptor gene. Endocr Rev (1992) 13(3):566–95. doi: 10.1210/edrv-13-3-566
50. Hribal ML, Tornei F, Pujol A, Menghini R, Barcaroli D, Lauro D, et al. Transgenic mice overexpressing human G972R IRS-1 show impaired insulin action and insulin secretion. J Cell Mol Med (2008) 12(5B):2096–106. doi: 10.1111/j.1582-4934.2008.00246.x
51. Burguete-Garcia AI, Cruz-Lopez M, Madrid-Marina V, Lopez-Ridaura R, Hernández-Avila M, Cortina B, et al. Association of Gly972Arg polymorphism of IRS1 gene with type 2 diabetes mellitus in lean participants of a national health survey in Mexico: a candidate gene study. Metabolism (2010) 59(1):38–45. doi: 10.1016/j.metabol.2009.07.007
52. Martínez-Gómez LE, Cruz M, Martínez-Nava GA, Madrid-Marina V, Parra E, García-Mena J, et al. A replication study of the IRS1, CAPN10, TCF7L2, and PPARG gene polymorphisms associated with type 2 diabetes in two different populations of Mexico. Ann Hum Genet (2011) 75(5):612–20. doi: 10.1111/j.1469-1809.2011.00668.x
53. Esposito DL, Li Y, Vanni C, Mammarella S, Veschi S, Della Loggia F, et al. A novel T608R missense mutation in insulin receptor substrate-1 identified in a subject with type 2 diabetes impairs metabolic insulin signaling. J Clin Endocrinol Metab (2003) 88(4):1468–75. doi: 10.1210/jc.2002-020933
54. Veeraragavulu PC, Yellapu NK, Yerrathota S, Adi PJ, Matcha B. Three novel mutations I65S, R66S, and G86R divulge significant conformational variations in the PTB domain of the IRS1 gene. ACS Omega (2019) 4(1):2217–24. doi: 10.1021/acsomega.8b01712
55. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170(4):605–35. doi: 10.1016/j.cell.2017.07.029
56. Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet (2013) 93(1):141–9. doi: 10.1016/j.ajhg.2013.05.019
57. Bárcena C, Quesada V, De Sandre-Giovannoli A, Puente DA, Fernández-Toral J, Sigaudy S, et al. Exome sequencing identifies a novel mutation in PIK3R1 as the cause of SHORT syndrome. BMC Med Genet (2014) 15(1):51. doi: 10.1186/1471-2350-15-51
58. Avila M, Dyment DA, Sagen JV, St-Onge J, Moog U, Chung BHY, et al. Clinical reappraisal of SHORT syndrome with PIK3R1 mutations: toward recommendation for molecular testing and management. Clin Genet (2016) 89(4):501–6. doi: 10.1111/cge.12688
59. Rivière JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet (2012) 44(8):934–40. doi: 10.1038/ng.2331
60. Lindhurst MJ, Parker VER, Payne F, Sapp JC, Rudge S, Harris J, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet (2012) 44(8):928–33. doi: 10.1038/ng.2332
61. Kushi R, Hirota Y, Ogawa W. Insulin resistance and exaggerated insulin sensitivity triggered by single-gene mutations in the insulin signaling pathway. Diabetol Int (2021) 12(1):62–7. doi: 10.1007/s13340-020-00455-5
62. George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science (2004) 304(5675):1325–8. doi: 10.1126/science.1096706
63. Manning A, Highland HM, Gasser J, Sim X, Tukiainen T, Fontanillas P, et al. A low-frequency inactivating AKT2 variant enriched in the finnish population is associated with fasting insulin levels and type 2 diabetes risk. Diabetes (2017) 66(7):2019–32. doi: 10.2337/db16-1329
64. Dash S, Sano H, Rochford JJ, Semple RK, Yeo G, Hyden CSS, et al. A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc Natl Acad Sci USA (2009) 106(23):9350–5. doi: 10.1073/pnas.0900909106
65. Moltke I, Grarup N, Jørgensen ME, Bjerregaard P, Treebak JT, Fumagalli M, et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature (2014) 512(7513):190–3. doi: 10.1038/nature13425
66. Binsch C, Barbosa DM, Hansen-Dille G, Hubert M, Hodge SM, Kolasa M, et al. Deletion of Tbc1d4/As160 abrogates cardiac glucose uptake and increases myocardial damage after ischemia/reperfusion. Cardiovasc Diabetol (2023) 22(1):17. doi: 10.1186/s12933-023-01746-2
67. El Bouchikhi I, Belhassan K, Moufid FZ, Iraqui Houssaini M, Bouguenouch L, Samri I, et al. Noonan syndrome-causing genes: Molecular update and an assessment of the mutation rate. Int J Pediatr Adolesc Med (2016) 3(4):133–42. doi: 10.1016/j.ijpam.2016.06.003
68. Ranza E, Guimier A, Verloes A, Capri Y, Marques C, Auclair M, et al. Overlapping phenotypes between SHORT and Noonan syndromes in patients with PTPN11 pathogenic variants. Clin Genet (2020) 98(1):10–8. doi: 10.1111/cge.13746
69. Pal A, Barber TM, Van de Bunt M, Rudge SA, Zhang Q, Lachlan KL, et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med (2012) 367(11):1002–11. doi: 10.1056/NEJMoa1113966
70. Li A, Qiu M, Zhou H, Wang T, Guo W. PTEN, insulin resistance and cancer. Curr Pharm Des (2017) 23(25):3667–76. doi: 10.2174/1381612823666170704124611
71. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med (2012) 18(3):363–74. doi: 10.1038/nm.2627
72. Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem (2006) 281(36):26602–14. doi: 10.1074/jbc.M601284200
73. Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem (2010) 285(8):5347–60. doi: 10.1074/jbc.M109.076976
74. Steppan CM, Wang J, Whiteman EL, Birnbaum MJ, Lazar MA. Activation of SOCS-3 by resistin. Mol Cell Biol (2005) 25(4):1569–75. doi: 10.1128/MCB.25.4.1569-1575.2005
75. Goalstone M, Carel K, Leitner JW, Draznin B. Insulin stimulates the phosphorylation and activity of farnesyltransferase via the Ras-mitogen-activated protein kinase pathway. Endocrinology (1997) 138(12):5119–24. doi: 10.1210/endo.138.12.5621
76. Semenkovich CF. Insulin resistance and atherosclerosis. J Clin Invest (2006) 116(7):1813–22. doi: 10.1172/JCI29024
77. Haas ME, Attie AD, Biddinger SB. The regulation of ApoB metabolism by insulin. Trends Endocrinol Metab (2013) 24(8):391–7. doi: 10.1016/j.tem.2013.04.001
78. Vergès B. Pathophysiology of diabetic dyslipidaemia: where are we? Diabetologia (2015) 58(5):886–99. doi: 10.1007/s00125-015-3525-8
79. Pont F, Duvillard L, Florentin E, Gambert P, Vergès B. Early kinetic abnorMalities of apoB-containing lipoproteins in insulin-resistant women with abdominal obesity. Arteriosclerosis Thrombosis Vasc Biol (2002) 22(10):1726–32. doi: 10.1161/01.ATV.0000032134.92180.41
80. Goldberg IJ. Clinical review 124: Diabetic dyslipidemia: causes and consequences. J Clin Endocrinol Metab (2001) 86(3):965–71. doi: 10.1210/jcem.86.3.7304
81. Sung KC, Park HY, Kim MJ, Reaven G. Metabolic markers associated with insulin resistance predict type 2 diabetes in Koreans with normal blood pressure or prehypertension. Cardiovasc Diabetol (2016) 15:47. doi: 10.1186/s12933-016-0368-7
82. Ginsberg HN, Zhang YL, Hernandez-Ono A. Metabolic syndrome: focus on dyslipidemia. Obes (Silver Spring) (2006) 14 Suppl 1:41S–9S. doi: 10.1038/oby.2006.281
83. Gast KB, Tjeerdema N, Stijnen T, Smit JWA, Dekkers OM. Insulin resistance and risk of incident cardiovascular events in adults without diabetes: meta-analysis. PloS One (2012) 7(12):e52036. doi: 10.1371/journal.pone.0052036
84. Laakso M, Kuusisto J. Insulin resistance and hyperglycaemia in cardiovascular disease development. Nat Rev Endocrinol (2014) 10(5):293–302. doi: 10.1038/nrendo.2014.29
85. Zhou MS, Schulman IH, Raij L. Vascular inflammation, insulin resistance, and endothelial dysfunction in salt-sensitive hypertension: role of nuclear factor kappa B activation. J Hypertens (2010) 28(3):527–35. doi: 10.1097/HJH.0b013e3283340da8
86. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest (2012) 122(4):1316–38. doi: 10.1172/JCI59903
87. Bhat NR. Linking cardiometabolic disorders to sporadic Alzheimer’s disease: a perspective on potential mechanisms and mediators. J Neurochem (2010) 115(3):551–62. doi: 10.1111/j.1471-4159.2010.06978.x
88. Steen E, Terry BM J, Rivera E, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? J Alzheimer’s Dis (2005) 7(1):63–80. doi: 10.3233/JAD-2005-7107
89. Logan S, Pharaoh GA, Marlin MC, Masser DR, Matsuzaki S, Wronowski B, et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol Metab (2018) 9:141–55. doi: 10.1016/j.molmet.2018.01.013
90. Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, et al. Stimulation of β-amyloid precursor protein trafficking by insulin reduces intraneuronal β-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci (2001) 21(8):2561–70. doi: 10.1523/JNEUROSCI.21-08-02561.2001
91. Xie L, Helmerhorst E, Taddei K, Plewright B, van Bronswijk W, Martins R. Alzheimer’s β-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci (2002) 22(10):RC221–1. doi: 10.1523/JNEUROSCI.22-10-j0001.2002
92. Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging (2010) 31(2):224–43. doi: 10.1016/j.neurobiolaging.2008.04.002
93. Lee CWC, Lau KF, Miller CCJ, Shaw PC. Glycogen synthase kinase-3β-mediated tau phosphorylation in cultured cell lines. NeuroReport (2003) 14(2):257. doi: 10.1097/00001756-200302100-00020
94. Kyoung Pyo H, Lovati E, Pasinetti GM, Ksiezak-Reding H. Phosphorylation of tau at THR212 and SER214 in human neuronal and glial cultures: The role of AKT. Neuroscience (2004) 127(3):649–58. doi: 10.1016/j.neuroscience.2004.05.036
95. Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J Neurochem (2008) 104(6):1433–9. doi: 10.1111/j.1471-4159.2007.05194.x
96. Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J (2005) 272(16):4211–20. doi: 10.1111/j.1742-4658.2005.04833.x
97. Hu Y, Russek SJ. BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem (2008) 105(1):1–17. doi: 10.1111/j.1471-4159.2008.05237.x
98. Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology (2010) 58(3):561–8. doi: 10.1016/j.neuropharm.2009.11.010
99. Djiogue S, Nwabo Kamdje AH, Vecchio L, Kipanyula MJ, Farahna M, Aldebasi Y, et al. Insulin resistance and cancer: the role of insulin and IGFs. Endocr Relat Cancer (2013) 20(1):R1–17. doi: 10.1530/ERC-12-0324
100. Wang CF, Zhang G, Zhao LJ, Qi WJ, Li XP, Wang JL, et al. Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth. PloS One (2013) 8(8):e69001. doi: 10.1371/journal.pone.0069001
101. Baxter RC. IGF binding proteins in cancer: mechanistic and clinical insights. Nat Rev Cancer (2014) 14(5):329–41. doi: 10.1038/nrc3720
102. Gong Y, Dou LJ, Liang J. Link between obesity and cancer: role of triglyceride/free fatty acid cycling. Eur Rev Med Pharmacol Sci (2014) 18(19):2808–20.
103. Rose DP, Vona-Davis L. The cellular and molecular mechanisms by which insulin influences breast cancer risk and progression. Endocr Relat Cancer (2012) 19(6):R225–241. doi: 10.1530/ERC-12-0203
104. Espelund U, Cold S, Frystyk J, Ørskov H, Flyvbjerg A. Elevated free IGF2 levels in localized, early-stage breast cancer in women. Eur J Endocrinol (2008) 159(5):595–601. doi: 10.1530/EJE-08-0154
105. Erarslan E, Coşkun Y, Türkay C, Köktener A, Aydoğan T. IGF-I levels and visceral fat accumulation in colonic neoplasia. Clin Res Hepatol Gastroenterol (2014) 38(1):99–105. doi: 10.1016/j.clinre.2013.06.004
106. Arcidiacono B, Iiritano S, Nocera A, Possidente K, Nevolo MT, Ventura V, et al. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res (2012) 2012:789174. doi: 10.1155/2012/789174
107. Yee LD, Mortimer JE, Natarajan R, Dietze EC, Seewaldt VL. Metabolic health, insulin, and breast cancer: why oncologists should care about insulin. Front Endocrinol (Lausanne) (2020) 11:58. doi: 10.3389/fendo.2020.00058
108. Hansen BF, Kurtzhals P, Jensen AB, Dejgaard A, Russell-Jones D. Insulin X10 revisited: a super-mitogenic insulin analogue. Diabetologia (2011) 54(9):2226–31. doi: 10.1007/s00125-011-2203-8
109. Jensen M, Hansen B, De Meyts P, Schäffer L, Ursø B. Activation of the insulin receptor by insulin and a synthetic peptide leads to divergent metabolic and mitogenic signaling and responses. J Biol Chem (2007) 282(48):35179–86. doi: 10.1074/jbc.M704599200
Keywords: insulin signaling, insulin resistance, application, insulin signal pathway, receptor, post-receptor signal transduction
Citation: Le TKC, Dao XD, Nguyen DV, Luu DH, Bui TMH, Le TH, Nguyen HT, Le TN, Hosaka T and Nguyen TTT (2023) Insulin signaling and its application. Front. Endocrinol. 14:1226655. doi: 10.3389/fendo.2023.1226655
Received: 22 May 2023; Accepted: 19 July 2023;
Published: 17 August 2023.
Edited by:
Hilda Ghadieh, University of Balamand, LebanonReviewed by:
Muhammad Sajid Hamid Akash, Government College University, Faisalabad, PakistanCopyright © 2023 Le, Dao, Nguyen, Luu, Bui, Le, Nguyen, Le, Hosaka and Nguyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thi Thu Thao Nguyen, dGhhb25ndXllbjk0Lm50M0BnbWFpbC5jb20=; Thi Kim Chung Le, bGVraW1jaHVuZ0BobXUuZWR1LnZu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.