 Sitara Niranjan
Sitara Niranjan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 15 June 2023
Sec. Clinical Diabetes
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1193373
This article is part of the Research TopicDiabetes and Non-Alcoholic Fatty Liver Disease: Points of Physiological and Mechanistic Intersection and Current Co-Therapeutic ApproachesView all 17 articles
Diabetes mellitus is a metabolic disease clinically-characterized as acute and chronic hyperglycemia. It is emerging as one of the common conditions associated with incident liver disease in the US. The mechanism by which diabetes drives liver disease has become an intense topic of discussion and a highly sought-after therapeutic target. Insulin resistance (IR) appears early in the progression of type 2 diabetes (T2D), particularly in obese individuals. One of the co-morbid conditions of obesity-associated diabetes that is on the rise globally is referred to as non-alcoholic fatty liver disease (NAFLD). IR is one of a number of known and suspected mechanism that underlie the progression of NAFLD which concurrently exhibits hepatic inflammation, particularly enriched in cells of the innate arm of the immune system. In this review we focus on the known mechanisms that are suspected to play a role in the cause-effect relationship between hepatic IR and hepatic inflammation and its role in the progression of T2D-associated NAFLD. Uncoupling hepatic IR/hepatic inflammation may break an intra-hepatic vicious cycle, facilitating the attenuation or prevention of NAFLD with a concurrent restoration of physiologic glycemic control. As part of this review, we therefore also assess the potential of a number of existing and emerging therapeutic interventions that can target both conditions simultaneously as treatment options to break this cycle.
Diabetes mellitus is a metabolic disease clinically-characterized as acute and chronic hyperglycemia (1). It is emerging as one of the common conditions associated with incident liver disease in the US. The spectrum of liver disease ranges from mild transaminitis to non-alcoholic fatty liver disease (NAFLD). NAFLD encompasses non-alcoholic steatosis (fatty liver) without inflammation (normal transaminases), non-alcoholic steatohepatitis (NASH) without fibrosis, NASH with fibrosis eventually progressing to cirrhosis, hepatocellular carcinoma and liver failure culminating in death (1, 2). In clinical practice, most patients with NAFLD are asymptomatic with possible hepatomegaly. They are diagnosed when liver enzymes ALT and/or AST are elevated, or steatosis is detected on abdominal imaging. It is a diagnosis of exclusion, and normal liver enzymes do not eliminate a diagnosis of NAFLD (3–7). Worldwide, the pooled prevalence of NAFLD (umbrella term of macrovesicular fat deposition) is 25.24% (8). In the US, a comparison of 3 cycles of the National Health and Nutrition Examination Survey (NHANES) based on transaminitis alone, demonstrated a steady increase in the prevalence of NAFLD from 5.5% in 1988 to 11% in 2008. The inclusion of steatosis with normal transaminases may account for an even higher prevalence (9). The prevalence of NAFLD’s closely associated metabolic counterparts such as essential hypertension, obesity and diabetes has trended up as well (10). Studies in multiple countries have demonstrated that NAFLD has a higher prevalence in men. Prevalence in women increases with age, while it remains stable in men. Sex hormones, menopausal status and obesity are major contributing factors to this disparity (11).
The mechanisms by which diabetes drives liver disease have become a topic of intense discussion and highly sought-after therapeutic targets. Traditionally, diabetes has been classified into type 1 (T1D) and type 2 (T2D). T1D begins as an autoimmune process culminating in an autoimmune inflammation-mediated, selective impairment of the pancreatic beta cells and overt hyperglycemia. T2D, instead, is characterized by peripheral insulin resistance (IR) compensated for by the production of more insulin culminating in overt hyperglycemia. Accumulating evidence suggests that these seemingly divergent conditions share many etiopathogenetic and clinical features other than just hyperglycemia. Thus, latent autoimmune diabetes of adults (LADA) presents features of both T1D and T2D and IR is seen in overweight T1D patients (12). On the other hand, some T2D patients exhibit pancreatic autoimmunity (13).
Broadly-understood, IR is coupled to impaired insulin action at multiple points in the signaling cascade in the main glucose-utilizing, insulin-responsive tissues, particularly skeletal muscle, adipose, and the liver. These as well as possible pressure points of therapeutic interest are illustrated in Figure 1. These include the action of lipid mediators, cellular stress, mitochondrial abnormalities, and leukocyte-derived soluble molecules (14). Lipid-induced IR has been observed in the liver (15) as the consequence of high fat diet (HFD) or lipolysis, where the concentration of FFA exceeds that of the intracellular fatty acid oxidation and storage rate, as demonstrated in humans and rodent models (14). Increased concentrations of diacylglycerol (DAG) also lead to IR by impairing insulin signaling (14). For example, plasmalemmal accumulation of intrahepatic DAG stimulates protein kinase Cϵ and inhibitory insulin receptor kinase phosphorylation on threonine (16, 17) resulting in IR. These results were consistent in rodent models and humans. In addition to protein kinase Cϵ, increased activity of the δ enzymatic isoform in livers of obese humans has been observed to cause hepatic IR (18). Human study outcomes and rodent models have shown that activation of other protein kinase C isoforms (δ, ϵ, ν, θ) have been implicated in DAG release and IR onset or progression (14). Non-FFA-derived lipids are another species implicated in the onset of hepatic IR in humans exhibiting NASH. A number of studies in humans revealed elevated intra-hepatic FFA concentrations concurrent with hepatic oxidative stress and inflammation (19). While ceramides have also been implicated in hepatic IR under obese conditions and T2D evolution, this has been well-reviewed elsewhere (20) and remains outside the topic of the current review. While HFD-facilitated elevations in circulating FFAs and lipids as a basis of IR is strongly-supported by many lines of animal and human investigation (21), not all situations of IR are a consequence of this. Cellular stress, instead, is a better predictor of IR in the obese state. Endoplasmic reticulum (ER) stress, particularly, is a common finding in the liver among obese men and women (22, 23). Nevertheless, exposure to HFD in rodents leads to an expansion of lipid deposition inside the liver followed by hepatic IR even in the absence of peripheral fat accumulation and peripheral IR. Under such diet conditions, insulin signaling has been shown to be impaired, partly due to activation of PKCε and JNK1 (24). Estrogen has a protective role against hepatic steatosis and insulin resistance by decreasing triglyceride synthesis and increasing hepatic FFA oxidation (25). Circulating 17-beta estradiol also suppresses hepatic gluconeogenesis via FoxO1 signaling, independent of IRS-1 and IRS- 2 (26). In mice IRS-2 is transcriptionally-attenuated as a function of sterol-regulatory element binding protein (SREBP) activation and FoxO suppression (27–31). This is possibly a consequence of hyperinsulinemia-induced downregulation of IRS-2 facilitating hepatic IR (32, 33). Further, growing evidence indicates that hepatic DAG accumulation potentiates hepatic IR (34) and DAG levels inside hepatocyte lipid droplets were particularly-informative predictors of IR in humans (35).
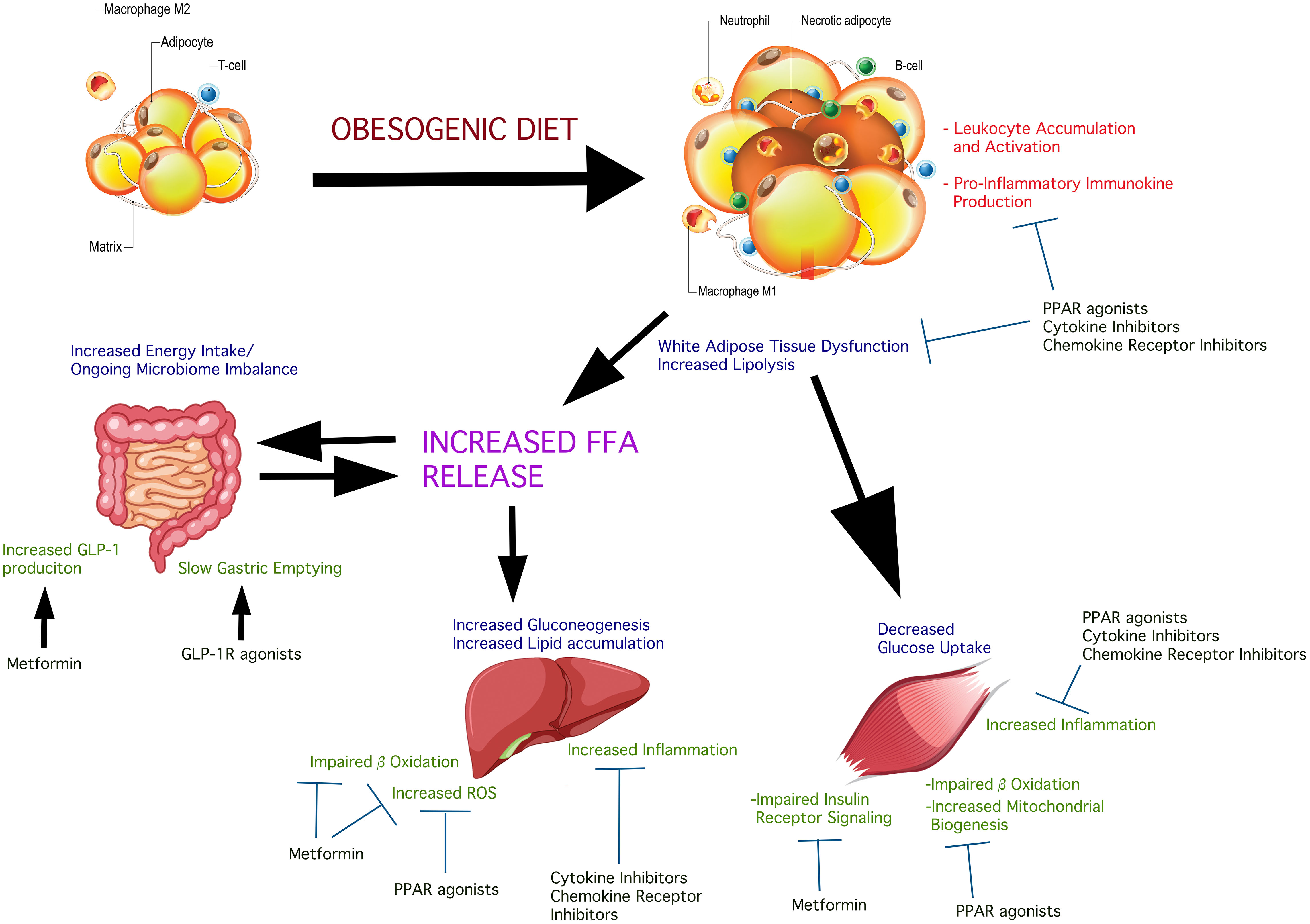
Figure 1 Insulin-sensitive inter-organ effects of obesogenic diets in the deterioration of insulin sensitivity and possible treatment pressure points. Obesogenic diets promote a state of systemic low-grade inflammation which contributes to, and is response to pathologic changes in glucolipometabolism in the main insulin-sensitive tissues and organs. The intestinal microbiome is altered causing changes in the complement of short-chain fatty acids produced. These are released into the circulation affecting insulin sensitivity and potentiate systemic and insulin-sensitive tissue inflammation. Obesogenic diet causes adipose hypertrophy and expansion, resulting in the conversion of resident M2 macrophages into pro-inflammatory M1 macrophage. Concurrent core adipose hypoxia creates an environment that signals “danger”. This initiates the accumulation of neutrophils and other leukocytes which become activated, further potentiating local inflammation. The net result is peripheral insulin resistance, consequent to insulin receptor signaling impairment due to the action of immunokines produced by the accumulating pro-inflammatory leukocytes. IL-6, for example, acting via the IL-6 receptor on adipocytes, impairs insulin-stimulated phosphorylation of signaling components downstream of the insulin receptor tyrosine kinase. Skeletal muscle is susceptible to expanding fat and accumulation of lipid droplets, as well as the effects of circulating FFA on hepatocytes cause the accumulation of leukocytes and their activation, with a net effect of insulin signaling impairment. The different classes of agents shown in the Figure have shown variable beneficial effects on insulin sensitivity. Combination approaches could simultaneously act systemically and on the key insulin-sensitive tissues, attenuating inflammation thus facilitating better insulin sensitivity.
One of the molecular pathways of insulin signaling is the activation of Akt which, as it suppresses hepatic gluconeogenesis, in parallel causes activation of sterol regulatory element binding protein 1c (SREBP1c). As demonstrated in transgenic rat hepatocytes, this is a consequence of Akt-stimulated mammalian target of rapamycin complex-1 (mTORC1) activity which regulates the transcription and stability of SREBP1c (36). Activated SREBP1c stimulates increased expression of genes encoding key enzymes in FA biosynthesis including those of the fatty acid elongase complex, fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), and ATP citrate lyase (37). A seeming paradox is observed inside the liver with developing obesity and progression towards T2D-associated NAFLD. Impairment of gluconeogenesis suppression occurs concomitant with de novo lipogenesis (DNL) and IR. This can be partially explained as a function of liver insulin signaling stimulating hepatic DNL whose biochemical pathway products predispose and drive the impairment of gluconeogenesis suppression. These biochemical pathway products and their concentrations, evidence suggests, determine the onset and rate of hepatic structural and cellular damage observed in the onset of NAFLD in mice (38). The question that remains to be better understood is, what is the point in hepatic insulin signaling where its effects on glucose and lipid metabolism diverge?
Some evidence suggests that mTORC1 may be one such point of divergence at the level of hepatic hyperinsulinemia and resistance. Studies in rodents have shown that the blockade of Akt and PI3K activity prevents insulin-mediated expression of genes of enzymes involved in gluconeogenesis while mTORC1 prevented insulin-dependent induction of SREBP1c without any effect on suppression of expression of gluconeogenetic genes (39). mTORC1 is a nutrient-sensing biochemical control point promoting its re-distribution to the lysosome (40–42). However, as demonstrated in transgenic mouse models, mTORC1, on its own, is insufficient to cause de novo lipogenesis and NASH, at least in the absence of Akt2 (43). The nuance in these observations is best evaluated noting the role of tuberous sclerosis complex (TSC) proteins (44, 45). A number of mouse models have shown that Akt stimulation of mTORC1 is conditioned on TSC2 inhibition. Hepatic deletion of TSC1 results in an insulin-depended mTORC1 activation and protects from steatosis and de novo lipogenesis (44, 46). Additional studies in mice exhibiting hepatocyte-targeted inactivating genetic modifications of Akt, FoxO1, and TSC1, insulin-dependent co-ordinate activation of mTORC1 and FoxO1 inhibition were considered to be sufficient and possibly-necessary for insulin-dependent de novo lipogenesis (47, 48).
Co-incident with the onset of NAFLD, are a series of changes inside hepatocytes indicating an acute stress response; changes concomitant with intra-hepatic inflammation (49). Central to this stress response is the unfolded protein response (UPR) with its fulcrum point the endoplasmic reticulum (ER). Hepatic ER stress has been observed in NAFLD (50) and related to its progression, including its mechanistic relationship with hepatic insulin resistance (51). ER stress has been coupled to steatohepatitis-associated insulin resistance (52). Moreover, de novo lipogenesis in the liver has also been linked to hepatocyte ER stress (53). Pharmacologic suppression of Caspase-2 as well as Caspase-2 disruption, observed in hepatocyte ER stress-associated NASH prevented fibrosis and inflammation by preventing SREBP1 and SREBP2 activation. These observations suggested that ER stress could participate in the early onset of hepatic insulin resistance, de novo lipogenesis and the progression towards NAFLD.
It stands to reason that, especially under HFD conditions, lipids and FFAs are widely-viewed as the basis of IR, systemic or hepatic, however, other metabolites, especially in high fat “Western diets” have been implicated. Several amino acids (AA) have been shown to contribute to IR (15). In humans, AA elevation in plasma impairs insulin-stimulated glucose disposal in skeletal muscle. The mechanism appears to be through the mammalian target of rapamycin (mTOR)/S6 kinase pathway and phosphorylation of IRS-1 (54). Branched-chain (BC) AA are constituents of liver gluconeogenesis and their levels in the circulation have been found to be correlated with IR in humans (55). In skeletal muscle under hypersinulinemic conditions, BCAA impair glucose disposal and augment ATP synthesis without any effect on mitochondrial abundance of DNA (56, 57). In contrast, transient dietary reduction of BCAA reduces post-prandial insulin secretion and improves adipose metabolism (58).
Macrophages are possibly the first leukocytes to accumulate inside the liver of obese individuals concomitant to IR onset (peripheral and/or hepatic) (59). These cells impair insulin signaling mainly via secreted immunokines (60). Liver-resident macrophages have been implicated in the onset and progression of hepatic IR and a number of overlapping mechanisms have been identified in their activation. While the following observations have been made mainly in skeletal muscle, and muscle-associated adipose, one can anticipate similar mechanisms to participate in hepatic IR: Accumulation of lipids inside myotubes in humans and rodent models, stimulates NF-κB nuclear translocation, attenuated mitochondrial respiration, fragmentation and mitophagy and elevated production of reactive oxygen species (ROS) (61). Systemic IR is widely-reported to co-incide with macrophage accumulation and activation inside adipose (62), however, adipose IR can manifest adipose macrophage accumulation and activation (63), suggesting that, at least in some instances, IR can precede an inflammatory state and may in fact represent a “danger” signal causing the eventual activation of Kupffer cells and liver macrophages. Potential mechanisms underlying an IR-first cause could involve local hyperinsulinemia-stimulated activation of these leukocytes and/or hyperinsulinemia-stimulated increase in microvascular blood flow, hyperoxygenation and hepatic cell stress. Hyperinsulinemia would then be a consequence of pancreatic β cell impairment. A number of known mechanisms of peripheral IR could cause beta cell impairment via stress induction, UPR, and failure to sense glucose/produce insulin (64, 65).
Overnutrition and obesity lead to a systemic low grade chronic inflammatory state referred to as meta-inflammation, characterized by adipocyte necrosis and altered secretory phenotype in adipocytes (66–68). This results in the recruitment and release of proinflammatory cells and cytokines, such as TNFα expressed by macrophages and monocytes infiltrating obese adipocytes. Adipose tissue contains predominantly M2 macrophages, with a phenotypic switch to M1 in obese persons. M1 macrophages produce chemokines such as MCP-1 which recruit circulating monocytes to the liver and adipose tissue where they can undergo maturation into the pro-inflammatory M1 phenotype. Adipocytes also produce low levels of TNFα, leading to MCP-1 production and macrophage infiltration in adipocytes, triggering release of pro-inflammatory cytokines, such as IL-6 and IL-1β (69). The level of pro inflammatory cytokines in subcutaneous abdominal adipose tissue, inversely correlates with hepatic and systemic insulin sensitivity. Obese individuals with NAFLD have shown a decrease in hepatocyte insulin signaling compared to obese individuals with normal intrahepatic triglycerides (70). This low grade chronic inflammatory state in adipose tissue further contributes to IR via TNFα mediated serine/threonine phosphorylation of IRS-1, leading to enhanced lipolysis and increased exposure of hepatocytes to lipids (71, 72), fueling the progression of NASH.
More recent human and rodent studies, however, show that macrophages alone may not be sufficient to be involved in hepatic pathology concomitant to obesity-driven IR. Accumulation of neutrophils occurs very close to, or concurrent with that of macrophages (73). Indeed, more recent data demonstrate a prominent role of neutrophils over macrophages as being pivotal leukocytes that license and co-operate with macrophages in the onset of IR and T2D (74, 75). Neutrophil migration to sites of “danger” and their activation is a function of the balance of the CXCR2/CXCR4 chemokine receptor density on their surface (76). Neutrophil-attracting CXCR2 ligands are expressed in the pancreas, adipose and liver (77), suggesting that under potentially-stressful states, their secretion can be expected to recruit and activate neutrophils, which in turn would exacerbate and amplify a low grade inflammatory condition (78).
With the activation of leukocytes inside the liver, such as macrophages, growing intra-hepatic lipid deposition results in immunokine release [reviewed in (79) and (80)] which potentiates adipocyte lipolysis (81) concomitant to inhibition of hepatic insulin signaling (81, 82). Immunokines promote not only hepatic, but also systemic IR (83, 84), and cytokines like TNFα are detectable and upregulated in concentration inside the liver and adipose tissue of NASH patients (85), suggesting that upregulated TNFα in adipose might potentiate the progression of NAFLD in two ways: systemic IR and activation of a peripheral inflammation of insulin-responsive tissues (86). For example, adipose-produced IL-6 in liver stimulates hepatic SOCS3, suppressing insulin signaling, resulting in hepatic IR (87). Serum IL-6 concentrations are elevated in NAFLD and NASH (88).
The most obvious approaches to improving insulin sensitivity are diet changes and exercise that result in weight loss. However, work-life balance, in many instances, can impede commitment to defined diet and even low-level exercise activity. The distinct sex related disparities in the prevalence of NAFLD due to an interplay of sex hormones, age related hormonal changes as well as diseases such as polycystic ovarian syndrome and Turner’s Syndrome may warrant exploration into sex-specific therapeutic strategies that have been presented and/or reviewed elsewhere (89–93).
An array of different medicinals has been developed specifically to lower glucose concentrations, improve insulin production and/or correct weight and attenuate inflammation. Table 1 presents the clinical studies where insulin sensitivity, and hepatic insulin sensitivity in particular, was one of the outcome measures. Other classes of drugs have been repurposed for these indications. Their effects on IR have been mild to variable. A single class of agent to improve insulin sensitivity together with prevention of IR-associated liver pathology remains to be discovered, although we have shown that a neutrophil-targeting CXCR2 antagonist could offer such a solution [see below, (105)].
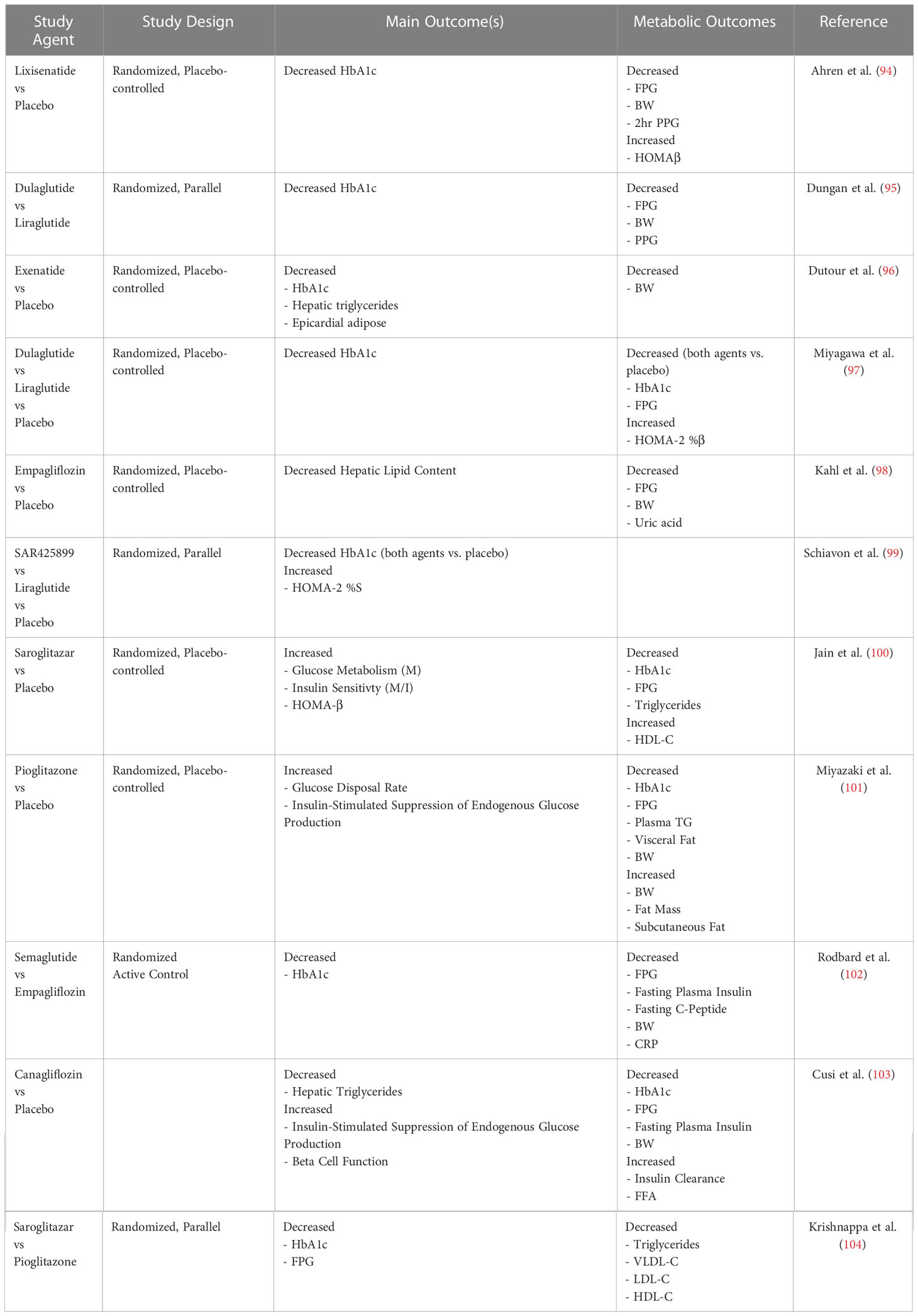
Table 1 Clinical trials assessing the 3-month (and greater) outcomes on insulin sensitivity in overweight/obese individuals with or without type 2 diabetes.
Sulfonylureas lower blood sugar concentrations by stimulating insulin secretion independent of food intake, however, they are associated with hypoglycemia. While some studies demonstrated beneficial effects on IR, others could not (106, 107). Sulfonylurea use is slowly being replaced by newer agent classes to treat hyperglycemia.
Metformin remains a first-line glucose lowering agent. Although the underlying mechanism of action remains incompletely understood, it appears that it inhibits the hepatic glycerol-3-phosphate dehydrogenase activity, resulting in suppression of glycerol-induced gluconeogenesis and increased cytosolic redox state. Together, these actions lead to a reduction in lactate dehydrogenase and lactate-induced endogenous glucose production (108). Other possible mechanisms of action include the inhibition of complex I followed by increased AMP, activating AMP kinase and facilitating fatty acid oxidation in liver and reduced expression of genes encoding enzymes involved in gluconeogenesis. Additionally, AMP interferes with glucagon signaling and gluconeogenesis (108). In non-hepatic tissues, metformin increases insulin stimulated glucose utilization (108) and AMP kinase activity (109). A meta-analytic inspection of 11 randomized controlled trials (RCT) in obese and overweight adolescents, revealed that metformin reduced fasting plasma glucose (FPG) at less than 6 months, without impacting insulin sensitivity (110). Another meta-analysis of 31 RCT using metformin for more than 8 weeks in individuals at high risk for T2D revealed that it improved insulin sensitivity concurrent with a reduced incidence T2D (111). An additional meta-analysis in patients with NAFLD revealed benefit in insulin sensitivity without, however, any improvement in NAFLD liver histology (112).
PPAR agonists, particularly those for PPARγ, have shown promising efficacy in improving IR and liver histology in T2D-associated NAFLD. As a class, they also suppress the production of pro-inflammatory immunokines concurrent with stimulation of adiponectin production (113, 114). Pioglitazone treatment of T2D patients has resulted in beneficial outcomes in NAFLD (62) resulting in improved liver and peripheral insulin sensitivity (101). While its use has been somewhat questioned due to adverse event concerns (115), a more recently-developed agent, lobeglitazone, exhibits improved safety with improvements in insulin sensitivity and liver steatosis in T2D-associated NAFLD (116). Another PPARγ-sparing agent, MSDC-0602K, also achieves insulin-sensitizing peripheral effects safely (117). More recently, CHS-131 demonstrated significant dual-target outcomes, improving fasting insulin levels and insulin sensitivity, total plasma cholesterol, triglycerides, liver enzymes, and increased plasma adiponectin levels. Most importantly, CHS‐131 improved liver histology and markers of hepatic fibrosis (118). Fibrates, ligands of PPARα, reduce fasting plasma glucose, insulin, and improve insulin sensitivity (119) although some questions remain about their true efficacy (120). Seladelpar and GW501516 are PPARδ agonists shown to improve insulin sensitivity in obese individuals (120, 121) with mechanisms of action that include increased fatty acid oxidation in skeletal muscle and attenuation of macrophage pro-inflammatory state (122). Another PPAR agent is Elafibranor, a PPARα/δ agonist, which reduces inflammation and enhances both peripheral and liver insulin sensitivity under obese conditions (123, 124), although the latter findings remain to be validated (125). Saroglitazar is a dual PPARα/γ agonist with whole body insulin sensitivity improvement without adverse events noted with the use of other PPARα/γ agonists (100, 104). A pan-PPAR agonist, lanifibranor, is currently being tested in phase II studies, with enabling data showing improved insulin sensitivity in T2D and improved intra-hepatic lipid content in T2D-associated NAFLD (clinicaltrials.gov #NCT03459079).
A randomized single blinded phase 2a clinical trial evaluated the efficacy of a fatty acid synthetase inhibitor TVB-2640 on de novo lipogenesis in a population of NASH patients (126). Fatty acid synthetases convert metabolites of simple sugars to palmitate (126). The rationale behind this was to reduce de novo lipogenesis in patients with NASH. The outcome demonstrated decreased liver fat by 9.6% in a population with fatty liver and fibrosis that included subjects with diabetes.
GLP-1 agonists like exenatide, liraglutide, semaglutide, and lixisenide can improve insulin sensitivity, although it is not clear if this effect is in the periphery or in the liver as well (94, 96, 102, 127, 128). Glucose-dependent insulinotropic polypeptide (GIP; tirzepatide) use also achieved some insulin sensitivity improvement in T2D, although again it is unknown if this acted at the level of the liver (127). Reduced hepatic inflammation and lipid deposition was demonstrated in T2D-associated liver pathology following a tri-pathway-targeting approach using HM1521, an agent that targets glucagon/GIP/GLP-1Ra in mice and in humans (117, 127).
While α-Glucosidase inhibitors (AGI) are not a priori thought of as agents that could affect IR, clinical studies have shown that they can, following establishment of a steady dose level (129, 130). These effects are expected to be extra-hepatic and a consequence of attenuation of hyperglycemia. In a similar manner, Sodium Glucose Co-transporter-2 Inhibitors (SGLT2I) have also demonstrated some insulin sensitivity enhancing effect (103, 131, 132) including a positive effect on liver IR (103) with neutral outcomes on non-hepatic IR (133).
It stands to reason that the accumulation of pro-inflammatory leukocytes and elevation of the concentration of their pro-inflammatory soluble mediators inside insulin-sensitive tissues is a high-priority target of therapy aimed to restore normal insulin-sensitivity in T2D as well as prevent any T2D-associated liver impairment that can be a consequence of, or drive hepatic IR. Salicylates were among the earliest agents tested for this objective and demonstrated mild improvement in peripheral glucose disposal (134, 135).
Inhibition of TNFα action with a variety of antibodies (etanercept, infliximab, adalimumab) improved insulin sensitivity in some patients, however, the heterogeneity of the study populations requires validation of those outcomes (135, 136). Targeting the IL-1β system (using IL-1 receptor antagonist protein, or antibodies like canakinumab and gevokizumab) improves glucoregulation overall, absent of any discernible effects on insulin resistance in T2D (135). In contrast, using the IL-6-targeting antibody tocilizumab, which aims to break the IL-6-mediated interference of insulin signaling, achieved statistically-relevant improvement of insulin sensitivity in obese patients (137).
Some excitement was generated when initial results from pre-clinical and early-clinical outcomes were reported showing improved hepatic function with the use of cenicriviroc, a dual CCR2/CCR5 chemokine receptor antagonist in hepatic pathology, however these reactions were tempered when the agent was unable to improve insulin sensitivity in patients with NASH (138).
As neutrophil accumulation into areas characterized by molecular and microenvironmental structural anomaly is a function mainly of the balance of CXCR2 and CXCR4 ligands and the neutrophil cell surface ratio of CXCR2:CXCR4 chemokine receptors (76), modulation of signaling via these receptors was proposed to be potentially therapeutic for T2D progression, IR, and possibly NAFLD. CXCR2-deficient mice are resistant from high fat diet-induced IR and T2D and are characterized by reduced macrophage accumulation in adipose (139). We recently demonstrated that a selective CXCR2 antagonist, AZD5069 (140) treatment of high fat diet-fed mice, improved insulin sensitivity and insulin-induced suppression of hepatic glucose production, decreased hepatic lipid storage, and significantly-prevented the progression towards liver pathology reminiscent of NAFLD.
Myeloperoxidase (MPO) is a key enzyme in neutrophil respiratory burst, that generates reactive oxidation species. Studies have shown an increase in the prevalence of MPO-positive Kupffer cells and neutrophils in the liver during NASH. The free radicals produced by MPO could participate in liver damage, directly (on hepatocytes) and/or on the stroma. MPO-deficient mice fed a high fat diet were protected against NASH-related liver injury. Additionally, mice fed with an oral MPO inhibitor exhibited reduced transaminitis and fibrosis (141). Thus, this enzyme, targeted alone or together with CXCR2 inhibitors/antagonists could represent a novel therapeutic approach in liver IR-related NASH (142, 143).
Currently there are no FDA-approved single agent treatments for the concurrent management of insulin sensitivity and the prevention (or at least the attenuation of progression to) to NAFLD/NASH in individuals with metabolic syndrome and T2D. The closest drug to market is obeticholic acid which recently completed a phase 3 clinical trial, but has yet to be approved by the FDA due to safety concerns in long term adverse effects (144). Our outcomes with AZD5069, as a single agent, showing benefits in the prevention of progression of insulin resistance and liver pathology reminiscent of NASH/NAFLD, as well as clinical trials in humans showing that AZD5069 was very well-tolerated with few side effects (145), offer an opportunity for this and possibly other similar drugs (e.g. ladarixin (146),) to enter clinical consideration as adjunctive treatments to standard of care of obesity and T2D to prevent and/or attenuate insulin resistance and liver pathology.
AZD5069 and similar agents may be found to exert their overall effects in a wider-ranging manner. For example, by preventing CXCR2-stimulated inhibition of insulin-induced glucose transport in muscle cells (147). Additionally, by preventing the effects of IL-8 (produced by growing adipose) on insulin-induced Akt phosphorylation in adipocytes (148, 149). This furthers strengthens the rationale that these agents can be potentially helpful treatments in insulin resistance-incident obesity and T2D. Finally, ongoing studies in our laboratory will soon determine if neutrophil antagonism impacts macrophage accumulation and function and thus, in an indirect manner, AZD5069 and similar agents, such as ladarixin (146), could prevent accumulation and further activation of liver-resident macrophages.
Improvement in insulin sensitivity in obesity and T2D-associated NAFLD have been achieved using lipid metabolism-modifying agents like ketohexokinase inhibitor, a protein tyrosine phosphatase-1B inhibitor, or an ω3-fatty acid [reviewed in (117)]. Liver-targeted dinitriophenyl (DNP)-methyl ether (DNPME) and mitochondrial protonophore (CRMP) aiming to motivate hepatic fatty acid oxidation while reducing lipid accumulation improved systemic IR in rodent and non-human primate models of obesity-associated NAFLD (150). Another mitochondrion-acting agent, BAM15, also showed evidence of improving systemic IR and liver inflammation as well as pathology in mouse models of obesity (150). Precise targeting of sensitive points inside these pathways without systemic adverse events or toxicities remains a largely-unexplored area of T2D pharmaceutical research, especially for the objective of improving IR concurrent with delaying or obviating liver pathology.
It is now evident that inflammation dependent pathways have a clear pathological role in the propagation of NAFLD. Initially, IR and hepatic lipid accumulation result in oxidative stress and activation of inflammatory pathways in the liver. In fact, inflammation plays a key role in IR as well. Overnutrition and increased caloric intake, set the stage for IR via multiple mechanisms. IR and hepatic lipid accumulation result in oxidative stress and activation of inflammatory pathways in the liver. Additionally, ER stress culminates in the UPR aimed at reducing ER burden while simultaneously increasing the translation of pro-apoptotic proteins. Finally, obesity-mediated adipocyte inflammation and necrosis results in a systemic meta-inflammation mediated by macrophages and cytokines such as TNFα and IL-8. IR contributes to hepatic steatosis through an increase in the circulating FFA, further leading to inflammation dependent liver injury resulting in NASH. This happens through liver macrophages in combination with, as emerging evidence indicates, the increased recruitment of neutrophils through CXCR2 signals. This recruitment of inflammatory cells to the liver plays a key role in the pathogenesis of NASH. Functionally, peripheral IR, especially in the liver further impairs systemic glucoregulation. The liver is a key site of gluconeogenesis, typically down regulated by insulin via the interference in transcription of gluconeogenic genes. Insulin physiologically favors lipogenesis and inhibits gluconeogenesis. Paradoxically, during IR states in the liver, there continues to be an increase in lipogenesis and gluconeogenesis referred to as selective IR. This culminates in NASH and systemic hyperglycemia, contributing to the diabetic phenotype.
With respect to therapeutics, a novel approach is to target IR and interfere with the natural disease progression of NASH. Bearing in mind that IR often precedes NASH and has an overlapping pathogenesis in the form of systemic meta-inflammation, combination therapy targeting at least two distinct inflammation networks would have maximum synergistic value. CXCR2 antagonists are a novel approach that have demonstrated both an improvement in insulin sensitivity and interference in the natural disease progression of NASH, through an interference in recruitment of inflammatory cells. CXCR2 antagonists in combination with PPARγ agonists may have a synergistic role considering the latter’s proven efficacy in improving insulin sensitivity and potential in NASH treatment. PPARγ agonists improve insulin sensitivity by increasing adiponectin and GLUT-4 translocation. Though limited by their side effects such as pulmonary edema in clinical practices new alternatives like CHS-131 show promise in this aspect, alone or in combination.
SN and BP wrote the original draft of the manuscript with significant intellectual guidance by NG. SN and NG edited all versions of the manuscript. NG assumes responsibility of the final submitted draft. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tolman KG, Fonseca V, Dalpiaz A, Tan MH. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care (2007) 30(3):734–43. doi: 10.2337/dc06-153910.2337/dc06-1539
2. Flisiak-Jackiewicz M, Bobrus-Chociej A, Wasilewska N, Lebensztejn DM. From nonalcoholic fatty liver disease (NAFLD) to metabolic dysfunction-associated fatty liver disease (MAFLD)-new terminology in pediatric patients as a step in good scientific direction? J Clin Med (2021) 10(5). doi: 10.3390/jcm1005092410.3390/jcm10050924
3. Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology (1994) 107(4):1103–9. doi: 10.1016/0016-5085(94)90235-610.1016/0016-5085(94)90235-6
4. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc (1980) 55(7):434–8.
5. Lee RG. Nonalcoholic steatohepatitis: a study of 49 patients. Hum Pathol (1989) 20(6):594–8. doi: 10.1016/0046-8177(89)90249-910.1016/0046-8177(89)90249-9
6. Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology (1999) 116(6):1413–9. doi: 10.1016/s0016-5085(99)70506-810.1016/s0016-5085(99)70506-8
7. Amarapurkar D, Kamani P, Patel N, Gupte P, Kumar P, Agal S, et al. Prevalence of non-alcoholic fatty liver disease: population based study. Ann Hepatol (2007) 6(3):161–3. doi: 10.1016/S1665-2681(19)31922-2
8. Mitra S, De A, Chowdhury A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl Gastroenterol Hepatol (2020) 5:16. doi: 10.21037/tgh.2019.09.0810.21037/tgh.2019.09.08
9. Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, et al. Prevalence of nonalcoholic fatty liver disease in the united states: the third national health and nutrition examination survey, 1988-1994. Am J Epidemiol (2013) 178(1):38–45. doi: 10.1093/aje/kws44810.1093/aje/kws448
10. Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the united states from 1988 to 2008. Clin Gastroenterol Hepatol (2011) 9(6):524–530.e1. doi: 10.1016/j.cgh.2011.03.02010.1016/j.cgh.2011.03.020
11. Nagral A, Bangar M, Menezes S, Bhatia S, Butt N, Ghosh J, et al. Gender differences in nonalcoholic fatty liver disease. Euroasian J Hepatogastroenterol (2022) 12(Suppl 1):S19–s25. doi: 10.5005/jp-journals-10018-1370
12. Zaharieva ET, Velikova TV, Tsakova AD, Kamenov ZA. Prevalence of positive diabetes-associated autoantibodies among type 2 diabetes and related metabolic and inflammatory differences in a sample of the Bulgarian population. J Diabetes Res (2017) 2017:9016148. doi: 10.1155/2017/901614810.1155/2017/9016148
13. de Marco R, Locatelli F, Zoppini G, Verlato G, Bonora E, Muggeo M. Cause-specific mortality in type 2 diabetes. Verona Diabetes Study. Diabetes Care (1999) 22(5):756–61. doi: 10.2337/diacare.22.5.75610.2337/diacare.22.5.756
14. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature (2019) 576(7785):51–60. doi: 10.1038/s41586-019-1797-8
15. Gancheva S, Jelenik T, Alvarez-Hernandez E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev (2018) 98(3):1371–415. doi: 10.1152/physrev.00015.2017
16. Lyu K, Zhang Y, Zhang D, Kahn M, Ter Horst KW, Rodrigues MRS, et al. A membrane-bound diacylglycerol species induces PKC-mediated hepatic insulin resistance. Cell Metab (2020) 32(4):654–664.e5. doi: 10.1016/j.cmet.2020.08.001
17. Lyu K, Zhang D, Song J, Li X, Perry RJ, Samuel VT, et al. Short-term overnutrition induces white adipose tissue insulin resistance through sn-1,2-diacylglycerol/PKCepsilon/insulin receptor Thr1160 phosphorylation. JCI Insight (2021) 6(4). doi: 10.1172/jci.insight.139946
18. Bezy O, Tran TT, Pihlajamaki J, Suzuki R, Emanuelli B, Winnay J, et al. PKCdelta regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J Clin Invest (2011) 121(6):2504–17. doi: 10.1172/JCI46045
19. Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E, et al. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care (2018) 41(6):1235–43. doi: 10.2337/dc17-1318
20. Sokolowska E, Blachnio-Zabielska A. The role of ceramides in insulin resistance. Front Endocrinol (Lausanne) (2019) 10:577. doi: 10.3389/fendo.2019.00577
21. Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, et al. Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest (1996) 97(12):2859–65. doi: 10.1172/JCI118742
22. Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, et al. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes (2009) 58(3):693–700. doi: 10.2337/db08-1220
23. Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab (2008) 93(11):4532–41. doi: 10.1210/jc.2008-1001
24. Samuel VT, Liu ZX, Qu X, Elder BD, Bilz S, Befroy D, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem (2004) 279(31):32345–53. doi: 10.1074/jbc.M313478200
25. Varlamov O, Bethea CL, Roberts CT Jr.Sex-specific differences in lipid and glucose metabolism. Front Endocrinol (Lausanne) (2014) 5:241. doi: 10.3389/fendo.2014.00241
26. Yan H, Yang W, Zhou F, Li X, Pan Q, Shen Z, et al. Estrogen improves insulin sensitivity and suppresses gluconeogenesis via the transcription factor Foxo1. Diabetes (2019) 68(2):291–304. doi: 10.2337/db18-0638
27. Zhang J, Ou J, Bashmakov Y, Horton JD, Brown MS, Goldstein JL. Insulin inhibits transcription of IRS-2 gene in rat liver through an insulin response element (IRE) that resembles IREs of other insulin-repressed genes. Proc Natl Acad Sci U.S.A. (2001) 98(7):3756–61. doi: 10.1073/pnas.071054598
28. Hirashima Y, Tsuruzoe K, Kodama S, Igata M, Toyonaga T, Ueki K, et al. Insulin down-regulates insulin receptor substrate-2 expression through the phosphatidylinositol 3-kinase/Akt pathway. J Endocrinol (2003) 179(2):253–66. doi: 10.1677/joe.0.1790253
29. Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell (2000) 6(1):77–86. doi: 10.1016/S1097-2765(05)00010-9
30. Ide T, Shimano H, Yahagi N, Matsuzaka T, Nakakuki M, Yamamoto T, et al. SREBPs suppress IRS-2-mediated insulin signalling in the liver. Nat Cell Biol (2004) 6(4):351–7. doi: 10.1038/ncb1111
31. Tsunekawa S, Demozay D, Briaud I, McCuaig J, Accili D, Stein R, et al. FoxO feedback control of basal IRS-2 expression in pancreatic beta-cells is distinct from that in hepatocytes. Diabetes (2011) 60(11):2883–91. doi: 10.2337/db11-0340
32. Krause C, Geissler C, Tackenberg H, El Gammal AT, Wolter S, Spranger J, et al. Multi-layered epigenetic regulation of IRS2 expression in the liver of obese individuals with type 2 diabetes. Diabetologia (2020) 63(10):2182–93. doi: 10.1007/s00125-020-05212-6
33. Honma M, Sawada S, Ueno Y, Murakami K, Yamada T, Gao J, et al. Selective insulin resistance with differential expressions of IRS-1 and IRS-2 in human NAFLD livers. Int J Obes (Lond) (2018) 42(9):1544–55. doi: 10.1038/s41366-018-0062-9
34. Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci (2017) 38(7):649–65. doi: 10.1016/j.tips.2017.04.004
35. Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U.S.A. (2011) 108(39):16381–5. doi: 10.1073/pnas.1113359108
36. Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE, et al. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc Natl Acad Sci U.S.A. (2012) 109(40):16184–9. doi: 10.1073/pnas.1213343109
37. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest (2002) 109(9):1125–31. doi: 10.1172/JCI15593
38. Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab (2008) 7(2):95–6. doi: 10.1016/j.cmet.2007.12.009
39. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U.S.A. (2010) 107(8):3441–6. doi: 10.1073/pnas.0914798107
40. Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol (2014) 24(7):400–6. doi: 10.1016/j.tcb.2014.03.003
41. Bond P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J Int Soc Sports Nutr (2016) 13:8. doi: 10.1186/s12970-016-0118-y
42. Zhang S, Lin X, Hou Q, Hu Z, Wang Y, Wang Z. Regulation of mTORC1 by amino acids in mammalian cells: a general picture of recent advances. Anim Nutr (2021) 7(4):1009–23. doi: 10.1016/j.aninu.2021.05.003
43. Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR, et al. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab (2011) 14(4):516–27. doi: 10.1016/j.cmet.2011.09.001
44. Kenerson HL, Yeh MM, Yeung RS. Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PloS One (2011) 6(3):e18075. doi: 10.1371/journal.pone.0018075
45. Xiang X, Lan H, Tang H, Yuan F, Xu Y, Zhao J, et al. Tuberous sclerosis complex 1-mechanistic target of rapamycin complex 1 signaling determines brown-to-white adipocyte phenotypic switch. Diabetes (2015) 64(2):519–28. doi: 10.2337/db14-0427
46. Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab (2011) 14(1):21–32. doi: 10.1016/j.cmet.2011.06.002
47. Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C, et al. Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab (2016) 23(6):1154–66. doi: 10.1016/j.cmet.2016.04.022
48. Uehara K, Sostre-Colon J, Gavin M, Santoleri D, Leonard KA, Jacobs RL, et al. via dual regulation of VLDL-TAG secretion and De novo lipogenesis. Cell Mol Gastroenterol Hepatol (2022) 13(6):1625–47. doi: 10.1016/j.jcmgh.2022.02.015
49. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology (2010) 52(5):1836–46. doi: 10.1002/hep.24001
50. Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology (2008) 134(2):568–76. doi: 10.1053/j.gastro.2007.10.039
51. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol (2011) 54(4):795–809. doi: 10.1016/j.jhep.2010.11.005
52. Sasako T, Ohsugi M, Kubota N, Itoh S, Okazaki Y, Terai A, et al. Hepatic Sdf2l1 controls feeding-induced ER stress and regulates metabolism. Nat Commun (2019) 10(1):947. doi: 10.1038/s41467-019-08591-6
53. Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, et al. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell (2018) 175(1):133–145 e15. doi: 10.1016/j.cell.2018.08.020
54. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, et al. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes (2005) 54(9):2674–84. doi: 10.2337/diabetes.54.9.2674
55. Wurtz P, Soininen P, Kangas AJ, Ronnemaa T, Lehtimaki T, Kahonen M, et al. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care (2013) 36(3):648–55. doi: 10.2337/dc12-0895
56. Tatpati LL, Irving BA, Tom A, Bigelow ML, Klaus K, Short KR, et al. The effect of branched chain amino acids on skeletal muscle mitochondrial function in young and elderly adults. J Clin Endocrinol Metab (2010) 95(2):894–902. doi: 10.1210/jc.2009-1822
57. Smith GI, Yoshino J, Stromsdorfer KL, Klein SJ, Magkos F, Reeds DN, et al. Protein ingestion induces muscle insulin resistance independent of leucine-mediated mTOR activation. Diabetes (2015) 64(5):1555–63. doi: 10.2337/db14-1279
58. Karusheva Y, Koessler T, Strassburger K, Markgraf D, Mastrototaro L, Jelenik T, et al. Short-term dietary reduction of branched-chain amino acids reduces meal-induced insulin secretion and modifies microbiome composition in type 2 diabetes: a randomized controlled crossover trial. Am J Clin Nutr (2019) 110(5):1098–107. doi: 10.1093/ajcn/nqz191
59. Weinstock A, Moura Silva H, Moore KJ, Schmidt AM, Fisher EA. Leukocyte heterogeneity in adipose tissue, including in obesity. Circ Res (2020) 126(11):1590–612. doi: 10.1161/CIRCRESAHA.120.316203
60. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest (2011) 121(6):2111–7. doi: 10.1172/JCI57132
61. Nisr RB, Shah DS, Ganley IG, Hundal HS. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell Mol Life Sci (2019) 76(24):4887–904. doi: 10.1007/s00018-019-03148-8
62. Dewidar B, Kahl S, Pafili K, Roden M. Metabolic liver disease in diabetes - from mechanisms to clinical trials. Metabolism (2020) 111S:154299. doi: 10.1016/j.metabol.2020.154299
63. Hernandez EA, Kahl S, Seelig A, Begovatz P, Irmler M, Kupriyanova Y, et al. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J Clin Invest (2017) 127(2):695–708. doi: 10.1172/JCI89444
64. Hudish LI, Reusch JE, Sussel L. Beta cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest (2019) 129(10):4001–8. doi: 10.1172/JCI129188
65. Wysham C, Shubrook J. Beta-cell failure in type 2 diabetes: mechanisms, markers, and clinical implications. Postgrad Med (2020) 132(8):676–86. doi: 10.1080/00325481.2020.1771047
66. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, DeFuria 2J, Jick Z, et al. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes (2007) 56(12):2910–8. doi: 10.2337/db07-0767
67. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature (2017) 542(7640):177–85. doi: 10.1038/nature21363
68. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
69. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112(12):1821–30. doi: 10.1172/jci1945110.1172/JCI19451
70. Fuchs A, Samovski D, Smith GI, Cifarelli V, Farabi SS, Yoshino J, et al. Associations among adipose tissue immunology, inflammation, exosomes and insulin sensitivity in people with obesity and nonalcoholic fatty liver disease. Gastroenterology (2021) 161(3):968–981.e12. doi: 10.1053/j.gastro.2021.05.008
71. Devisscher L, Verhelst X, Colle I, Van Vlierberghe H, Geerts A. The role of macrophages in obesity-driven chronic liver disease. J Leukoc Biol (2016) 99(5):693–8. doi: 10.1189/jlb.5RU0116-016R
72. Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology (2009) 50(1):185–97. doi: 10.1002/hep.22952
73. Michailidou Z, Gomez-Salazar M, Alexaki VI. Innate immune cells in the adipose tissue in health and metabolic disease. J Innate Immun 1-27 (2021) 14(1):4–30. doi: 10.1159/000515117
74. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta (2014) 1842(3):446–62. doi: 10.1016/j.bbadis.2013.05.017
75. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18(9):1407–12. doi: 10.1038/nm.2885
76. Strydom N, Rankin SM. Regulation of circulating neutrophil numbers under homeostasis and in disease. J Innate Immun (2013) 5(4):304–14. doi: 10.1159/00035028210.1159/000350282
77. Wilson GC, Kuboki S, Freeman CM, Nojima H, Schuster RM, Edwards MJ, et al. CXC chemokines function as a rheostat for hepatocyte proliferation and liver regeneration. PloS One (2015) 10(3):e0120092. doi: 10.1371/journal.pone.0120092
78. Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, et al. Chemokine receptor redundancy and specificity are context dependent. Immunity (2019) 50(2):378–389.e5. doi: 10.1016/j.immuni.2019.01.009
79. Herder C, Carstensen M, Ouwens DM. Anti-inflammatory cytokines and risk of type 2 diabetes. Diabetes Obes Metab (2013) 15 Suppl:3, 39–50. doi: 10.1111/dom.12155
80. Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol (2017) 13(9):509–20. doi: 10.1038/nrendo.2017.56
81. Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, et al. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest (2001) 107(2):181–9. doi: 10.1172/JCI10934
82. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of ikkbeta. Science (2001) 293(5535):1673–7. doi: 10.1126/science.1061620
83. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest (1995) 95(5):2409–15. doi: 10.1172/JCI117936
84. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi: 10.1126/science.7678183
85. Crespo J, Cayon A, Fernandez-Gil P, Hernandez-Guerra M, Mayorga M, Dominguez-Diez A, et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology (2001) 34(6):1158–63. doi: 10.1053/jhep.2001.29628
86. Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes (2007) 56(4):1010–3. doi: 10.2337/db06-1656
87. Sabio G, Das M, Mora A, Zhang Z, Jun JY, Ko HJ, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science (2008) 322(5907):1539–43. doi: 10.1126/science.1160794
88. Coulon S, Francque S, Colle I, Verrijken A, Blomme B, Heindryckx F, et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine (2012) 59(2):442–9. doi: 10.1016/j.cyto.2012.05.001
89. Kardashian A, Serper M, Terrault N, Nephew LD. Health disparities in chronic liver disease. Hepatology (2023) 77(4):1382–403. doi: 10.1002/hep.32743
90. Della Torre S. Beyond the X factor: relevance of sex hormones in NAFLD pathophysiology. Cells 10(9) (2021) 10(9):2502–2546. doi: 10.3390/cells10092502
91. Beaudry KM, Devries MC. Sex-based differences in hepatic and skeletal muscle triglyceride storage and metabolism (1). Appl Physiol Nutr Metab (2019) 44(8):805–13. doi: 10.1139/apnm-2018-0635
92. Della Torre S. Non-alcoholic fatty liver disease as a canonical example of metabolic inflammatory-based liver disease showing a sex-specific prevalence: relevance of estrogen signaling. Front Endocrinol (Lausanne) (2020) 11:572490. doi: 10.3389/fendo.2020.572490
93. Tobari M, Hashimoto E. Characteristic features of nonalcoholic fatty liver disease in Japan with a focus on the roles of age, sex and body mass index. Gut Liver (2020) 14(5):537–45. doi: 10.5009/gnl19236
94. Ahren B, Leguizamo Dimas A, Miossec P, Saubadu S, Aronson R. Efficacy and safety of lixisenatide once-daily morning or evening injections in type 2 diabetes inadequately controlled on metformin (GetGoal-m). Diabetes Care (2013) 36(9):2543–50. doi: 10.2337/dc12-2006
95. Dungan KM, Povedano ST, Forst T, Gonzalez JG, Atisso C, Sealls W, et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet (2014) 384(9951):1349–57. doi: 10.1016/S0140-6736(14)60976-4
96. Dutour A, Abdesselam I, Ancel P, Kober F, Mrad G, Darmon P, et al. Exenatide decreases liver fat content and epicardial adipose tissue in patients with obesity and type 2 diabetes: a prospective randomized clinical trial using magnetic resonance imaging and spectroscopy. Diabetes Obes Metab (2016) 18(9):882–91. doi: 10.1111/dom.12680
97. Miyagawa J, Odawara M, Takamura T, Iwamoto N, Takita Y, Imaoka T. Once-weekly glucagon-like peptide-1 receptor agonist dulaglutide is non-inferior to once-daily liraglutide and superior to placebo in Japanese patients with type 2 diabetes: a 26-week randomized phase III study. Diabetes Obes Metab (2015) 17(10):974–83. doi: 10.1111/dom.12534
98. Kahl S, Gancheva S, Strassburger K, Herder C, Machann J, Katsuyama H, et al. Empagliflozin effectively lowers liver fat content in well-controlled type 2 diabetes: a randomized, double-blind, phase 4, placebo-controlled trial. Diabetes Care (2020) 43(2):298–305. doi: 10.2337/dc19-0641
99. Schiavon M, Visentin R, Gobel B, Riz M, Cobelli C, Klabunde T, et al. Improved postprandial glucose metabolism in type 2 diabetes by the dual glucagon-like peptide-1/glucagon receptor agonist SAR425899 in comparison with liraglutide. Diabetes Obes Metab (2021) 23(8):1795–805. doi: 10.1111/dom.14394
100. Jain N, Bhansali S, Kurpad AV, Hawkins M, Sharma A, Kaur S, et al. Effect of a dual PPAR alpha/gamma agonist on insulin sensitivity in patients of type 2 diabetes with hypertriglyceridemia- randomized double-blind placebo-controlled trial. Sci Rep (2019) 9(1):19017. doi: 10.1038/s41598-019-55466-3
101. Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab (2002) 87(6):2784–91. doi: 10.1210/jcem.87.6.8567
102. Rodbard HW, Rosenstock J, Canani LH, Deerochanawong C, Gumprecht J, Lindberg SO, et al. Oral semaglutide versus empagliflozin in patients with type 2 diabetes uncontrolled on metformin: the PIONEER 2 trial. Diabetes Care (2019) 42(12):2272–81. doi: 10.2337/dc19-0883
103. Cusi K, Bril F, Barb D, Polidori D, Sha S, Ghosh A, et al. Effect of canagliflozin treatment on hepatic triglyceride content and glucose metabolism in patients with type 2 diabetes. Diabetes Obes Metab (2019) 21(4):812–21. doi: 10.1111/dom.13584
104. Krishnappa M, Patil K, Parmar K, Trivedi P, Mody N, Shah C, et al. Effect of saroglitazar 2 mg and 4 mg on glycemic control, lipid profile and cardiovascular disease risk in patients with type 2 diabetes mellitus: a 56-week, randomized, double blind, phase 3 study (PRESS XII study). Cardiovasc Diabetol (2020) 19(1):93. doi: 10.1186/s12933-020-01073-w
105. Phillips BE, Lantier L, Engman C, Garciafigueroa Y, Singhi A, Trucco M, et al. Improvement in insulin sensitivity and prevention of high fat diet-induced liver pathology using a CXCR2 antagonist. Cardiovasc Diabetol (2022) 21(1):130. doi: 10.1186/s12933-022-01564-y
106. Koshiba K, Nomura M, Nakaya Y, Ito S. Efficacy of glimepiride on insulin resistance, adipocytokines, and atherosclerosis. J Med Invest (2006) 53(1-2):87–94. doi: 10.2152/jmi.53.87
107. Korytkowski M, Thomas A, Reid L, Tedesco MB, Gooding WE, Gerich J. Glimepiride improves both first and second phases of insulin secretion in type 2 diabetes. Diabetes Care (2002) 25(9):1607–11. doi: 10.2337/diacare.25.9.1607
108. LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocr Rev (2021) 42(1):77–96. doi: 10.1210/endrev/bnaa023
109. Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes (2002) 51(7):2074–81. doi: 10.2337/diabetes.51.7.2074
110. Sun J, Wang Y, Zhang X, He H. The effects of metformin on insulin resistance in overweight or obese children and adolescents: a PRISMA-compliant systematic review and meta-analysis of randomized controlled trials. Med (Baltimore) (2019) 98(4):e14249. doi: 10.1097/MD.0000000000014249
111. Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med (2008) 121(2):149–157 e2. doi: 10.1016/j.amjmed.2007.09.016
112. Li Y, Liu L, Wang B, Wang J, Chen D. Metformin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. BioMed Rep (2013) 1(1):57–64. doi: 10.3892/br.2012.18
113. Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends Pharmacol Sci (2011) 32(10):607–16. doi: 10.1016/j.tips.2011.06.006
114. Natali A, Ferrannini E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: a systematic review. Diabetologia (2006) 49(3):434–41. doi: 10.1007/s00125-006-0141-7
115. Pafili K, Roden M. Nonalcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol Metab (2021) 50:101122. doi: 10.1016/j.molmet.2020.101122
116. Lee YH, Kim JH, Kim SR, Jin HY, Rhee EJ, Cho YM, et al. Lobeglitazone, a novel thiazolidinedione, improves non-alcoholic fatty liver disease in type 2 diabetes: its efficacy and predictive factors related to responsiveness. J Korean Med Sci (2017) 32(1):60–9. doi: 10.3346/jkms.2017.32.1.60
117. Gastaldelli A, Stefan N, Haring HU. Liver-targeting drugs and their effect on blood glucose and hepatic lipids. Diabetologia (2021) 64(7):1461–79. doi: 10.1007/s00125-021-05442-2
118. Perakakis N, Joshi A, Peradze N, Stefanakis K, Li G, Feigh M, et al. The selective peroxisome proliferator-activated receptor gamma modulator CHS-131 improves liver histopathology and metabolism in a mouse model of obesity and nonalcoholic steatohepatitis. Hepatol Commun (2020) 4(9):1302–15. doi: 10.1002/hep4.1558
119. Simental-Mendia LE, Simental-Mendia M, Sanchez-Garcia A, Banach M, Atkin SL, Gotto AM Jr., et al. Effect of fibrates on glycemic parameters: a systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol Res (2018) 132:232–41. doi: 10.1016/j.phrs.2017.12.030
120. Riserus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes (2008) 57(2):332–9. doi: 10.2337/db07-1318
121. Bays HE, Schwartz S, Littlejohn T, Kerzner B, Krauss RM, Karpf DB, et al. Roberts: MBX-8025, a novel peroxisome proliferator receptor-delta agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab (2011) 96(9):2889–97. doi: 10.1210/jc.2011-1061
122. Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes (2005) 54(8):2460–70. doi: 10.2337/diabetes.54.8.2460
123. Cariou B, Hanf R, Lambert-Porcheron S, Zair Y, Sauvinet V, Noel B, et al. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care (2013) 36(10):2923–30. doi: 10.2337/dc12-2012
124. Hanf R, Millatt LJ, Cariou B, Noel B, Rigou G, Delataille P, et al. The dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 exerts anti-diabetic effects in db/db mice without peroxisome proliferator-activated receptor gamma-associated adverse cardiac effects. Diabetes Vasc Dis Res (2014) 11(6):440–7. doi: 10.1177/1479164114548027
125. Westerouen Van Meeteren MJ, Drenth JPH, Tjwa E. Elafibranor: a potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs (2020) 29(2):117–23. doi: 10.1080/13543784.2020.1668375
126. Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled phase 2a trial. Gastroenterology (2021) 161(5):1475–86. doi: 10.1053/j.gastro.2021.07.02510.1053/j.gastro.2021.07.025
127. Baggio LL, Drucker DJ. Glucagon-like peptide-1 receptor co-agonists for treating metabolic disease. Mol Metab (2021) 46:101090. doi: 10.1016/j.molmet.2020.101090
128. Jinnouchi H, Sugiyama S, Yoshida A, Hieshima K, Kurinami N, Suzuki T, et al. Liraglutide, a glucagon-like peptide-1 analog, increased insulin sensitivity assessed by hyperinsulinemic-euglycemic clamp examination in patients with uncontrolled type 2 diabetes mellitus. J Diabetes Res (2015) 2015:706416. doi: 10.1155/2015/706416
129. Delgado H, Lehmann T, Bobbioni-Harsch E, Ybarra J, Golay A. Acarbose improves indirectly both insulin resistance and secretion in obese type 2 diabetic patients. Diabetes Metab (2002) 28(3):195–200.
130. Yokoyama H, Kannno S, Ishimura I, Node K. Miglitol increases the adiponectin level and decreases urinary albumin excretion in patients with type 2 diabetes mellitus. Metabolism (2007) 56(11):1458–63. doi: 10.1016/j.metabol.2007.06.010
131. Ferrannini E. Sodium-glucose Co-transporters and their inhibition: clinical physiology. Cell Metab (2017) 26(1):27–38. doi: 10.1016/j.cmet.2017.04.011
132. Ferrannini G, Hach T, Crowe S, Sanghvi A, Hall KD, Ferrannini E. Energy balance after sodium-glucose cotransporter 2 inhibition. Diabetes Care (2015) 38(9):1730–5. doi: 10.2337/dc15-0355
133. Merovci A, Solis-Herrera C, Daniele G, Eldor R, Fiorentino TV, Tripathy D, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest (2014) 124(2):509–14. doi: 10.1172/JCI70704
134. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, et al. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest (2002) 109(10):1321–6. doi: 10.1172/JCI14955
135. Goldfine AB, Shoelson SE. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J Clin Invest (2017) 127(1):83–93. doi: 10.1172/JCI88884
136. Wascher TC, Lindeman JH, Sourij H, Kooistra T, Pacini G, Roden M. Chronic TNF-alpha neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol Med (2011) 17(3-4):189–93. doi: 10.2119/molmed.2010.00221
137. Wueest S, Seelig E, Timper K, Lyngbaek MP, Karstoft K, Donath MY, et al. IL-6 receptor blockade increases circulating adiponectin levels in people with obesity: an explanatory analysis. Metabolites (2021) 11(2). doi: 10.3390/metabo11020079
138. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (2018) 67(5):1754–67. doi: 10.1002/hep.29477
139. Neels JG, Badeanlou L, Hester KD, Samad F. Keratinocyte-derived chemokine in obesity: expression, regulation, and role in adipose macrophage infiltration and glucose homeostasis. J Biol Chem (2009) 284(31):20692–8. doi: 10.1074/jbc.M109.01855610.1074/jbc.M109.018556
140. O’Byrne PM, Metev H, Puu M, Richter K, Keen C, Uddin M, et al. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med (2016) 4(10):797–806. doi: 10.1016/S2213-2600(16)30227-2
141. Koop AC, Thiele ND, Steins D, Michaëlsson E, Wehmeyer M, Scheja L, et al. Therapeutic targeting of myeloperoxidase attenuates NASH in mice. Hepatol Commun (2020) 4(10):1441–58. doi: 10.1002/hep4.156610.1002/hep4.1566
142. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol (2009) 175(4):1473–82. doi: 10.2353/ajpath.2009.08099910.2353/ajpath.2009.080999
143. Malle E, Furtmüller PG, Sattler W, Obinger C. Myeloperoxidase: a target for new drug development? Br J Pharmacol (2007) 152(6):838–54. doi: 10.1038/sj.bjp.070735810.1038/sj.bjp.0707358
144. Sharma M, Premkumar M, Kulkarni AV, Kumar P, Reddy DN, Rao NP. Drugs for non-alcoholic steatohepatitis (NASH): quest for the holy grail. J Clin Transl Hepatol (2021) 9(1):40–50. doi: 10.14218/JCTH.2020.00055
145. Cullberg M, Arfvidsson C, Larsson B, Malmgren A, Mitchell P, Wahlby Hamren U, et al. Pharmacokinetics of the oral selective CXCR2 antagonist AZD5069: a summary of eight phase I studies in healthy volunteers. Drugs R D (2018) 18(2):149–59. doi: 10.1007/s40268-018-0236-x
146. Castelli V, Brandolini L, d’Angelo M, Giorgio C, Alfonsetti M, Cocchiaro P, et al. CXCR1/2 inhibitor ladarixin ameliorates the insulin resistance of 3T3-L1 adipocytes by inhibiting inflammation and improving insulin signaling. Cells (2021) 10(9). doi: 10.3390/cells10092324
147. Chavey C, Lazennec G, Lagarrigue S, Clape C, Iankova I, Teyssier J, et al. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metab (2009) 9(4):339–49. doi: 10.1016/j.cmet.2009.03.002
148. Henkel J, Klauder J, Statz M, Wohlenberg AS, Kuipers S, Vahrenbrink M, et al. Enhanced palmitate-induced interleukin-8 formation in human macrophages by insulin or prostaglandin E2. Biomedicines (2021) 9(5). doi: 10.3390/biomedicines9050449
149. Kobashi C, Asamizu S, Ishiki M, Iwata M, Usui I, Yamazaki K, et al. Inhibitory effect of IL-8 on insulin action in human adipocytes via MAP kinase pathway. J Inflammation (Lond) (2009) 6:25. doi: 10.1186/1476-9255-6-25
Keywords: insulin resistance, type 2 diabetes, NAFLD, NASH, hepatic insulin resistance
Citation: Niranjan S, Phillips BE and Giannoukakis N (2023) Uncoupling hepatic insulin resistance – hepatic inflammation to improve insulin sensitivity and to prevent impaired metabolism-associated fatty liver disease in type 2 diabetes. Front. Endocrinol. 14:1193373. doi: 10.3389/fendo.2023.1193373
Received: 24 March 2023; Accepted: 01 June 2023;
Published: 15 June 2023.
Edited by:
Christian Göbl, Medical University of Vienna, AustriaReviewed by:
Vincenza Cifarelli, Saint Louis University, United StatesCopyright © 2023 Niranjan, Phillips and Giannoukakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick Giannoukakis, ngn1@andrew.cmu.edu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.