
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 10 October 2022
Sec. Mucosal Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.954869
This article is part of the Research TopicInterplay Between Diets, Microbiota, Bacterial Metabolites and Host for Intestinal Health and DiseaseView all 17 articles
 Nicolás Ortiz-López1,2†
Nicolás Ortiz-López1,2† Catalina Fuenzalida1,2†
Catalina Fuenzalida1,2† María Soledad Dufeu1,2†
María Soledad Dufeu1,2† Araceli Pinto-León1
Araceli Pinto-León1 Alejandro Escobar3Jaime Poniachik4
Alejandro Escobar3Jaime Poniachik4 Juan Pablo Roblero4
Juan Pablo Roblero4 Lucía Valenzuela-Pérez1,2*
Lucía Valenzuela-Pérez1,2* Caroll J. Beltrán1,2*
Caroll J. Beltrán1,2*Non-alcoholic fatty liver disease (NAFLD) is a complex and heterogeneous disorder considered a liver-damaging manifestation of metabolic syndrome. Its prevalence has increased in the last decades due to modern-day lifestyle factors associated with overweight and obesity, making it a relevant public health problem worldwide. The clinical progression of NAFLD is associated with advanced forms of liver injury such as fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). As such, diverse pharmacological strategies have been implemented over the last few years, principally focused on metabolic pathways involved in NAFLD progression. However, a variable response rate has been observed in NAFLD patients, which is explained by the interindividual heterogeneity of susceptibility to liver damage. In this scenario, it is necessary to search for different therapeutic approaches. It is worth noting that chronic low-grade inflammation constitutes a central mechanism in the pathogenesis and progression of NAFLD, associated with abnormal composition of the intestinal microbiota, increased lymphocyte activation in the intestine and immune effector mechanisms in liver. This review aims to discuss the current knowledge about the role of the immune response in NAFLD development. We have focused mainly on the impact of altered gut-liver-microbiota axis communication on immune cell activation in the intestinal mucosa and the role of subsequent lymphocyte homing to the liver in NAFLD development. We further discuss novel clinical trials that addressed the control of the liver and intestinal immune response to complement current NAFLD therapies.
Non-alcoholic fatty liver disease (NAFLD) is a liver disorder characterized by fat accumulation in at least 5% of the liver cells in individuals without significant alcohol consumption (1) and no secondary causes of chronic liver disease such as hepatitis, medications, toxicants in the environment, parenteral nutrition, Wilson’s disease, and chronic liver disease (hemochromatosis, autoimmune liver disease, chronic viral hepatitis, fatty liver of pregnancy, and tyrosinemia) (2). NAFLD has become a term encompassing a clinicopathological spectrum ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), the more severe form of NAFLD that can lead to advanced fibrosis and cirrhosis. The global prevalence of NAFLD has increased over the last decades; a recent meta-analysis that included 363 studies from 40 countries or regions worldwide reported a pooled estimated prevalence of 29.38% (3). A population-based observational study including 21 regions and 195 countries reported a rise in prevalence from 8.2% in 1990 to 10.9% in 2017, demonstrating a global public health problem (4). NAFLD is commonly related to metabolic syndrome, which in turn is characterized by an increase in the risk of cardiovascular disease (CVD) (5). Interestingly, CVD is the leading cause of NAFLD-related deaths after cirrhosis (6). This evidence emphasizes the need for broad clinical management of the disease to reduce the associated cardiovascular risk.
Despite increasing advances in the understanding of the pathophysiology of NAFLD, the exact mechanisms involved in the progression towards liver damage remain unknown. The “two-hit” hypothesis has been postulated to explain the pathogenesis of NAFLD. Here, the first event or “hit” denotes an increase in lipolysis and the consequent accumulation of triglycerides in the liver (7). The second “hit” is subsequently generated by an imbalance between reactive nitrogen and reactive oxygen species, leading to increased inflammatory injury that is accompanied by the release of various cytokines that contribute to hepatocellular injury and fibrosis (8). In addition to this hypothesis, a decade ago, an alternative model of multiple parallel hits emerged that includes various factors, such as the interaction between the gut microbiota and immune system, that promote the progression from simple steatosis to NASH; these factors have not been wholly described thus far (9).
Elevated immune cell activation has been widely described in the pathophysiology of NAFLD (10). However, the main studies have been centered on evaluating the role of these inflammatory processes in the liver; the activation of the immune response in the gut and its impact on liver function are poorly described. The intestine comprises one of the most oversized compartments of the immune system that is continually exposed to antigens from the diet and the microbiota (11). The balance in the adequate activation of the immune response, either to pathogenic luminal antigens or to commensal tolerogenic stimuli, determines the type of activation of a chronic inflammatory response that could promote liver damage.
This review aims to provide an integrated overview of the current state of knowledge on the role of the immune response in NAFLD. We mainly focused on the innate and adaptive mucosal activation of the intestinal immune response in this disease as a part of the altered communication of the microbiota-gut-liver axis. In addition, based on this knowledge, we expose possible therapeutic targets directed at controlling hepatic and intestinal inflammation and adaptive immune responses.
The pathogenesis of NAFLD is complex and multifactorial, including genetic predisposition, obesity, insulin resistance, increased immune response, altered gut microbiota, and environmental factors such as diet. These factors configure metabolic syndrome, which is characterized by increased serum-free fatty acid, triglycerides, LDL, and total cholesterol and decreased HDL levels, as well as adipocyte dysfunction. The excess of free fatty acid in the liver leads to steatosis and lipotoxicity that induce mitochondrial dysfunction, oxidative stress, and endoplasmic reticulum stress (12). Accumulated evidence suggests that this oxidative stress process and consequent liver damage through activation of the liver immune response plays an important role in the pathogenesis of NAFLD (13). As result, the activation of these processes leads to a systemic low-grade inflammation with increased levels of cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, proposed as inflammatory markers of NAFLD (14).
In 1982, a positive correlation between intestinal bacterial overgrowth and hepatic steatosis was observed in patients, being the first evidence of the role of the intestinal microbiota in hepatic steatosis (15). Since then, the influence of the intestinal microbiota on the development of liver disease has been highlighted. Many research works have focused on studying NAFLD pathogenesis regarding the interaction between the gut microbiota and liver function (Figure 1). These studies even investigated the intermediary role of the gut immune system in the interplay between the microbiota and liver function. In this regard, it has been observed that microbiota composition is significantly influenced by genetic and environmental factors, such as diet, which induce metabolic changes in the activity of the gut microorganisms. The metabolites released by the microbiota promote inflammatory processes that contribute to the pathogenesis of the disease (16). Therefore, the interaction among the components of the microbiota-gut-liver axis defines the behavior of diverse dependent mechanisms, such as intestinal barrier function, systemic immune responses, and hepatic inflammation, all of which are seriously altered in NAFLD.
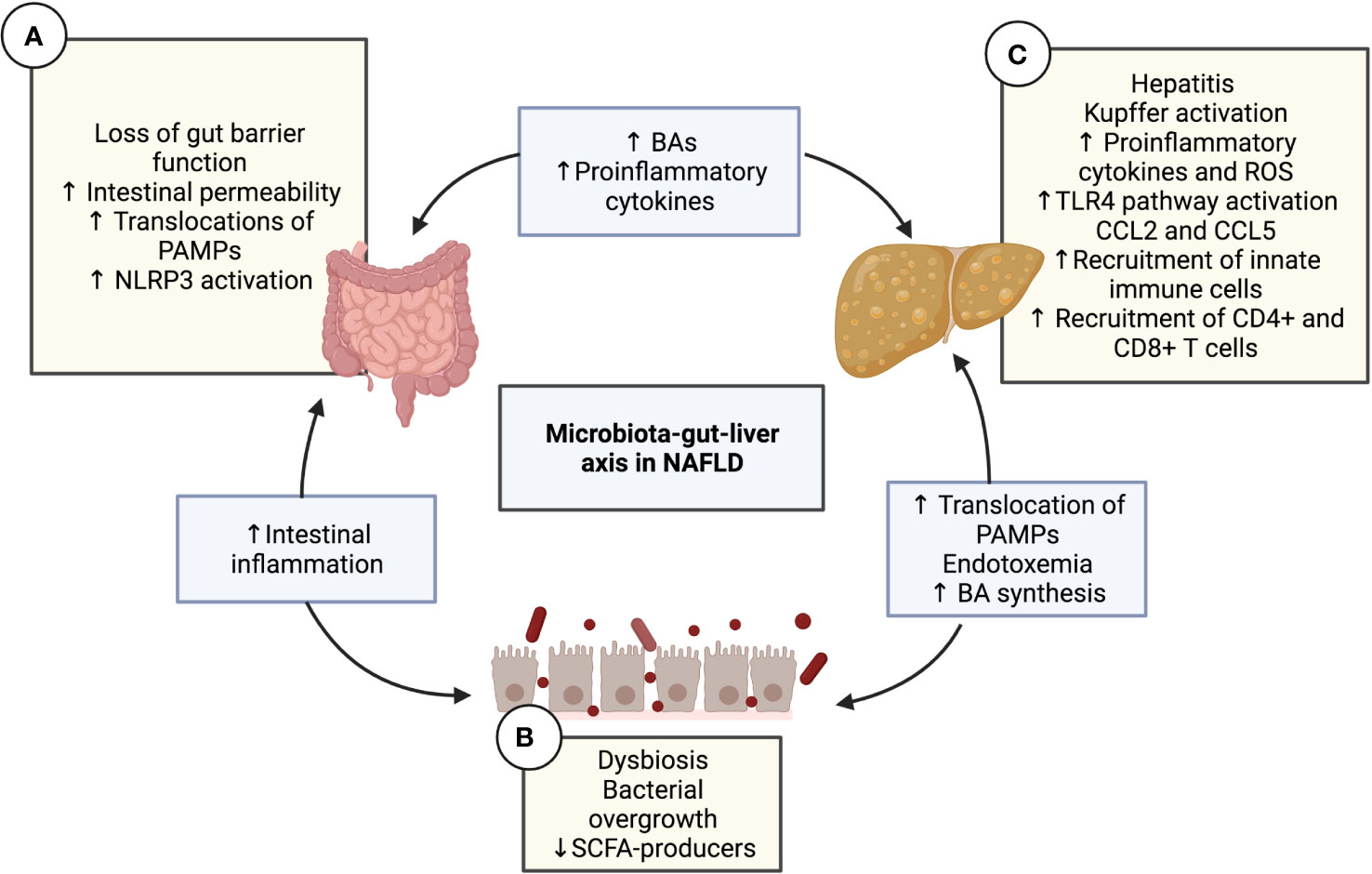
Figure 1 The microbiota-gut-liver axis in NAFLD. Interaction diagram of the different mechanisms of the microbiota-gut-liver axis participating in the pathogenesis of NAFLD. (A) Intestinal gut barrier disruption and increased permeability have been demonstrated in patients with NAFLD along with the decreased expression of junctional adhesion molecule A, zonula occludens-1, and occludin. This alteration causes the transfer of pro-inflammatory products and PAMPs (such as LPS or PGN) to the liver circulation, configuring intestinal inflammation and endotoxemia. The translation of PAMPs causes TLR signaling in the mucosa, which leads to the activation of NLRP3. (B) Diet nutrient composition can affect the quantitative and qualitative composition of the gut microbiota, leading to intestinal dysbiosis and bacterial overgrowth, which impacts the immune response, favoring NAFLD progression. Dysbiosis contributes to the disruption of the intestinal barrier, increasing mucosal permeability, which produces more dysbiosis, thereby creating a vicious cycle. Another consequence of dysbiosis is the alteration in the homeostasis of microbe-derived metabolites, such as a decrease in SCFAs and an increase in BAs. (C) The liver is a vital organ in fat metabolization and undergoes many changes in patients with metabolic syndrome, including the over-accumulation of free fatty acid, activation of KCs due to and the TLR4 pathway, lipotoxicity, increased reactive oxygen species and cytokines, and finally, steatosis. Hepatic CCL5 expression levels have been shown to increase in NAFLD patients. The release of the chemokines CCL2 and CCL5 is crucial in the recruitment of lymphocytes to the liver. The migration of mesenteric lymph node cells into the liver is mediated by CCL5, which induces hepatic CD4+ T and CD8+ T cell activation, subsequently leading to liver injury and the progression of NAFLD. BA, Bile acid; CD, Cluster of differentiation; CCL, C-C motif chemokine ligand; KC, Kupffer cell; LPS, Lipopolysaccharide; NAFLD, Non-alcoholic fatty liver disease; NLRP3, NLR family pyrin domain containing 3; PAMP, pathogen-associated molecular pattern; PGN, Peptidoglycan; ROS, Reactive oxygen species; SCFA, Short-chain fatty acid; TLR, Toll-like receptor.
A loss of intestinal homeostasis induces changes in the diversity and composition of the gut microbiota that impact the mucosal immune response and promote NAFLD progression. Likewise, bacterial overgrowth in the small and large intestine has been observed in patients with NAFLD (17, 18), which has been associated with impairment of the intestinal barrier functions and an activated intestinal immune response in an NAFLD/NASH mouse model (19). In this regard, it is important to consider that the intestinal barrier controls the transport of substances from the gut to the enterohepatic circulation by preventing the translocation of pathogens and molecules, such as pathogen- and damage-associated molecular patterns (PAMPs and DAMPs). Its function involves diverse components, among them the mucus lining and an epithelial monolayer of specialized cells that are bound by junctional complexes. These complexes include tight junctions (TJs), which play a sealing role in the intercellular space to control paracellular passage (20). Increased intestinal permeability and elevated levels of inflammation are positively correlated with the onset and progression of NAFLD (21). Altered gut barrier function in this disorder is related to decreased expression of the TJ proteins zonula occludens-1 (ZO-1) and occludin (22). Under normal conditions, the liver exerts an immune vigilance role by supporting the clearance of bacterial products from the portal circuit. However, when intestinal barrier function is altered, LPS and other bacteria-derived compounds rise in the circulation, increasing the activation of toll-like receptors (TLRs) and other pathogen recognition receptors (PRRs) in the liver, thereby triggering inflammatory responses in this organ (23). Several studies have shown higher LPS content in the serum as well as hepatocytes in NAFLD patients (24, 25). In the liver, through its interaction with TLR4 expressed on resident hepatic macrophages known as Kupffer cells (KCs), LPS triggers a signaling pathway toward activating nuclear factor kappa B (NF-κB), which promotes the expression of pro-inflammatory cytokine genes such as IL-6. A higher number of TLR4-positive macrophages were observed in NASH patients in comparison to patients with simple steatosis and controls, where TLR4 expression was positively correlated with serum LPS levels (24). These data support that the restoration of intestinal permeability via microbiota modulation can be an attractive therapeutic target for NAFLD (26).
Intestinal dysbiosis induces alterations in the produced microbe-derived metabolites including short-chain fatty acids (SCFAs) and secondary bile acids (BAs). The role of each in NAFLD is described below.
SCFAs are produced by anaerobic bacterial fermentation that influences the intestinal epithelial barrier function and immune response. It has been shown that SCFAs modulate the differentiation of several immune cells, such as macrophages, dendritic cells (DCs), and T regulatory cells (Tregs), and contribute to the control of some immune cell functions, like the phagocytic activity of macrophages (27). In this regard, murine colonic macrophages treated with oral butyrate, one of the most common SCFAs in the human intestine, enhanced their antimicrobial activity without an increased inflammatory cytokine response, suggesting that increased intestinal butyrate might represent a strategy to bolster host defenses without damaging tissue inflammation (28). Additionally, it has been demonstrated that butyrate, as well as the other common SCFAs acetate and propionate, can directly promote T cell differentiation into CD4+ T cells producing IL-17, interferon-γ, or IL-10, depending on the cytokine milieu (29). This evidence suggests that the intestinal microbiota, through the production of SCFAs, exerts a regulatory role on the immune response. In vitro studies in several cell types, including monocytes, macrophages, and KCs, have demonstrated that SCFAs suppress the LPS and cytokine-stimulated production of proinflammatory mediators, such as TNF-α, IL-6, and NO (30). Currently, only evidence based on animal models is available; a high-fat diet (HFD) mouse model study showed that the intragastric administration of sodium butyrate ameliorated HFD-induced hepatic steatosis, inflammation, and gut microbiota imbalance in the mice (31). In summary, these findings suggest that SCFAs regulate the development of inflammatory responses in NAFLD, have important anti-inflammatory activity, and may have a beneficial effect. Further research is necessary to determine the specific mechanism by which SCFAs affect the occurrence and development of NAFLD.
The microbiota-gut-liver axis is essential to regulating systemic metabolism (32). BAs are steroid-derivative components of bile that participate in communication along this axis. They have a significant role in many physiological processes, such as the digestion and solubilization of lipids, the regulation of hepatic glucose levels, and inflammation (33). Under diverse pathological conditions, such as NAFLD, the size of the BA pool and its composition are altered. In patients with NAFLD, the serum’s total, primary, and conjugated BAs are significantly increased, with slight changes in unconjugated BAs. However, secondary BAs were observed to considerably increase in some studies and decrease in others (34). Thus, there is no clear consensus among the studies of hepatic BAs in patients with NAFLD. Despite these contradictory findings, limited clinical studies concluded that hepatic BA homeostasis is dysregulated in this pathology (34), which can be associated with alterations in the regulation of BA homeostasis by the dysbiotic intestinal microbiota. Increased intestinal permeability is associated with alterations in BA composition as well as metabolic endotoxemia and inflammation, which are common findings in patients with NAFLD (35). A study by Gupta et al. in a murine model of NAFLD demostrated that the use of sevelamer hydrochloride to sequester intestinal BAs decreased mucosal inflammation and improved intestinal barrier function. This was correlated with reduced liver injury and reduced hepatosteatosis, demonstrating the therapeutic potential of targeting BAs in NAFLD (36). This evidence suggests that the modulation of BAs and the microbiota can be a good therapeutic target in NAFLD.
In addition, impaired BA signaling has been shown to be an essential mechanism for NAFLD development (37). Through interaction with one of the BA receptors, the farsenoid X receptor (FXR), BA can increase insulin sensitivity and decrease hepatic gluconeogenesis and circulating triglycerides (38). In this regard, Yang et al. observed lower levels of hepatic FXR and elevated triglyceride levels in patients with NAFLD compared to normal controls (39).
Despite evidence that BA can directly modulate both innate and adaptive immune cell responses (40, 41), its role in NAFLD immunity is not well-explored. Diverse BA receptor agonists have attracted the attention as drug candidates for intestinal inflammation (42, 43); however, there are no clinical trials evaluating the targeting of this mechanism in NAFLD.
The mucosa of the gastrointestinal tract (GIT) is the most extensive area of the immune system in the human body. Microbial colonization of the GIT in early life is crucial to the proper education and maturation of immune cells since it will determine an effective response against pathogens and tolerance to commensal antigens. The gut mucosal immune response occurs in two different compartments, namely the inductive and effector sites. The first site, in which the adaptive immune response occurs with the priming and differentiation of lymphocytes, includes the mesenteric lymph nodes (MLNs) and the gut-associated lymphoid tissues (GALT) consisting of Peyer’s patches (44) and isolated lymphoid follicles (ILFs), distributed along the small and large intestines (45). The second site comprises the epithelium and intestinal lamina propria, in which immune cells are located and activated to promote intestinal barrier functions (44). In a coordinated manner, both innate and adaptive responses are exerted at the two mucosal sites to respond against pathogenic insult and commensal stimuli.
Oral substances absorbed in the intestine reach the liver via the portal circulation, continuously exposing this organ to potential antigens. Liver damage can be initiated and enhanced by a local intestinal immune response whose activation promotes inflammation and the migration of immune cells to the liver. The leaky gut processes contribute to intestinal inflammation (Figure 2). The loss of epithelial barrier integrity increases the translocation of microbial components to the lamina propria and liver, activating receptors that initiate signaling conducted by the gene expression of diverse elements of innate immunity. Systemic inflammation contributes to the pathogenesis of NAFLD, characterized by a high number of neutrophils and macrophages in the liver (46). This response leads to liver cell death that supports the disease’s progression (47). Moreover, diverse resident liver cells, like parenchymal hepatocytes and lymphoid and non-lymphoid cells play an essential role in the homeostasis of the liver immune response, apart from being involved in modulating NAFLD progression (48).
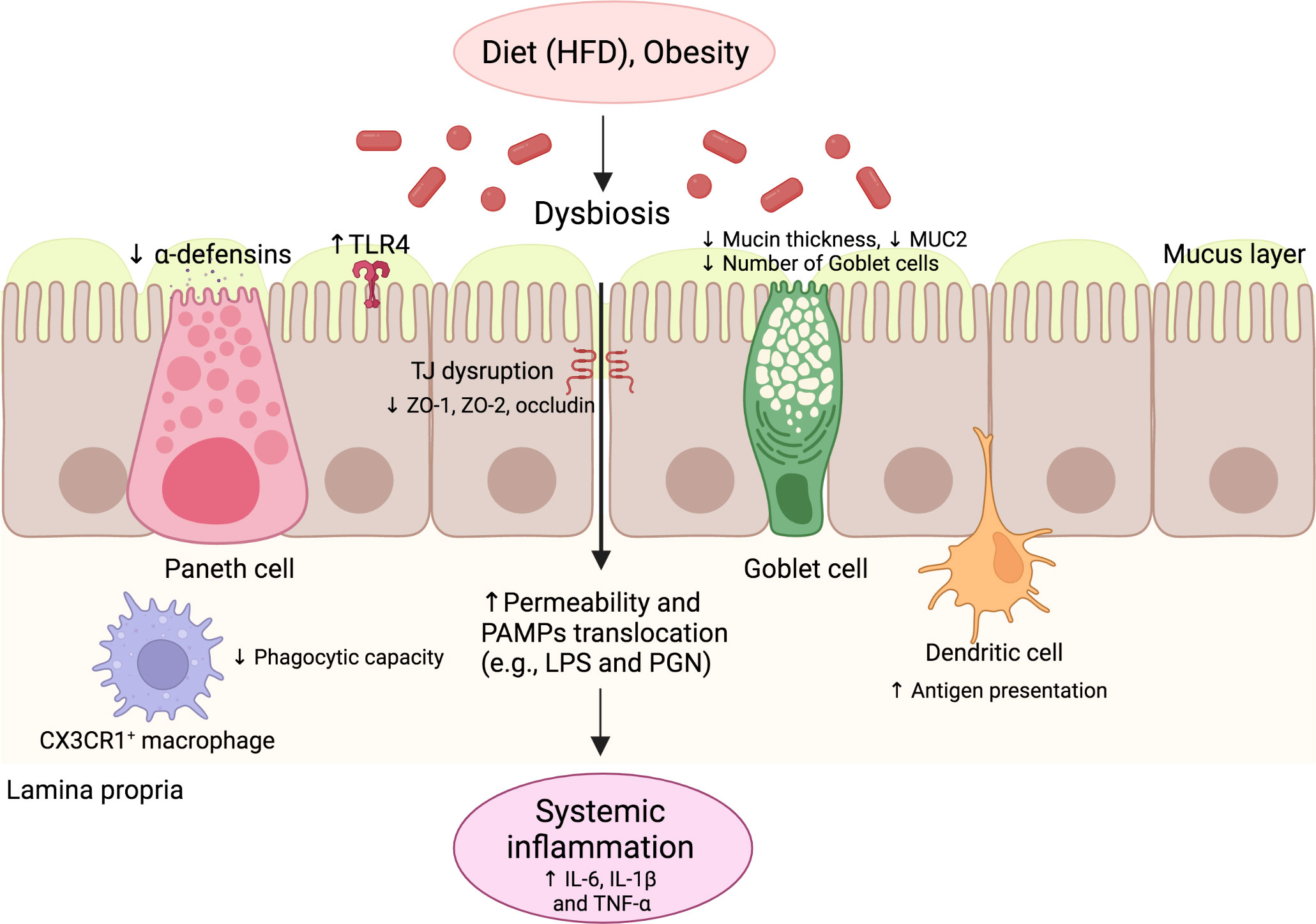
Figure 2 The innate immune response of the gut in NAFLD. Environmental factors such as diet and obesity promote dysbiosis in the gut microbiota, leading to increased levels of PAMPs and impaired intestinal barrier function. The intestinal barrier dysfunction is characterized by the impaired function of several cells, such as goblet cells and Paneth cells, with decreased production of mucin and antimicrobial peptides, respectively. TJ structure and composition disruption also occurs, characterized by reduced ZO-1, ZO-2, and occludin expression. Additionally, alterations of antigen-presenting cell function occur, including decreased phagocytic capacity and increased antigen presentation. These alterations promote increased intestinal permeability and the translocation of PAMPs, leading to increased serum levels of pro-inflammatory cytokines and systemic inflammation. HFD, High-fat diet; IL, Interleukin; LPS, Lipopolysaccharide; NAFLD, Nonalcoholic fatty liver disease; PAMP, pathogen-associated molecular pattern; PGN, Peptidoglycan; TLR, Toll-like receptor; TNF, Tumor necrosis factor.
The intestinal epithelium is a single layer of luminal lining cells in the gut, considered the most significant communication barrier between the internal and external environments (49). The intercellular surface in the epithelium includes diverse junctional complexes, among them desmosomes, adherens junctions (AJs), and TJs. While AJs and desmosomes are essential for the mechanical linkage of adjacent cells (49), TJs play a sealing role that controls the paracellular transport of luminal agents towards the lamina propia (49). It has been observed that the composition of TJ proteins in the small intestine is altered in NAFLD, with decreased expression of ZO-1 (21). As already mentioned, the intestinal microbiota influences intestinal epithelial integrity. In the HFD mouse model, oral treatment with Faecalibacterium prausnitzii, considered a bacterial indicator of a healthy gut, significantly increased the expression of the intestinal TJ protein-encoding Tjp1 gene that encodes the ZO-1 protein (50). Similarly, Briskey et al. showed significant liver steatosis and reduced expression of TJ proteins such as ZO-1 and ZO-2 in an HFD mouse model (51). Interestingly, in this study, probiotic supplementation mitigated the severity of steatosis by partially preventing TJ expression, suggesting that the modulation of TJs is a good strategy to reverse the progression of steatosis in NAFLD.
Other components of the intestinal barrier include the goblet cells and Paneth cells, which can secrete mucins that are part of the intestinal mucus (52) and antimicrobial peptides (53) that control bacterial load at the lumen (54, 55), respectively. These cells are in close interaction with a sizeable population of immune cells immersed in the epithelial layer, such as intraepithelial lymphocytes, which contribute to the first line of defense in the gastrointestinal mucosa (56). An alteration in the number and function of these cell components has been described in animal models of obesity and NAFLD patients. HFD-fed mice showed a decreased number of goblet cells in the ileal crypts and intestinal MUC2 expression (57, 58). These alterations were reversed through the administration of nuciferine, a bioactive component derived from the lotus leaf that could have a protective role in the epithelial layer (57). Additionally, resistin-like molecule β (RELMβ), expressed in the secretory granules of intestinal goblet cells, has been described to regulate gut microbiota composition, contributing to the maintenance of immune response the gut homeostasis. Increased intestinal expression and serum concentrations of RELMβ, which promote insulin resistance, have been observed in HFD-fed mice (59). Furthermore, RELMβ knockout mice were resistant to a methionine- and choline-deficient (MCD) diet, suggesting the contribution of increases in RELMβ to NASH development and raising the possibility that RELMβ is a novel therapeutic target for this pathology (60).
Paneth cells produce defensins (α and β), cathelicidins (LL-37/CRAMP), and C-type lectins (RegIII α/γ/β), which constitute the AMP gut repertoire. These AMPs control the interaction of the gut microbiota with the intestinal mucosa. Mice lacking RegIIIγ have been shown to display an altered mucus distribution that increases the proximity of microbiota to the intestinal epithelium, inducing inflammation in the ileal mucosa. As AMP promotes barrier integrity, a low number of Paneth cells impairs a proper bacterial defense and favors NAFLD progression (61). Moreover, the depletion of Paneth cells’ granules by intravenous dithizone, a zinc chelating agent, has been observed to ameliorate the severity of NAFLD in HFD-fed mice; this effect was associated with changes in gut microbiota composition (62). Furthermore, insufficiency of vitamin D is considered one of the risk factors for metabolic syndrome and NAFLD, which is associated with the decreased production of Paneth cell defensins. In this regard, HFD-fed mice with vitamin D deficiency (HFD+VDD) showed a decrease in the ileum-specific α-defensins of Paneth cells, associated with increased intestinal permeability and gut dysbiosis (58). In the same study, the oral administration of α-defensin-5 in the HFD+VDD model restored the eubiotic state, decreasing Helicobacter hepaticus, a bacteria that causes hepatitis and liver tumors in mouse models, and increasing Akkermansia muciniphila, a symbiotic bacteria, that restored metabolic disturbances (i.e., glucose levels, body mass, and liver fat content) (58). This evidence suggests a protective role for Paneth cell defensins in NAFLD development, mediated by the modulation of gut microbiota composition.
A delicate interplay between the gut microbiota, epithelium, and immune cells in the mucosa allows for the maintenance of selective permeability in the intestine (Kolodziejczyk, 2019). PRRs, such as TLRs and NLRs, are responsible for recognizing molecular patterns inducing inflammatory responses that are crucial in the pathogenesis of NAFLD. The intestinal epithelial cells express several TLRs, including TLR1, TLR2, TLR4, TLR5, and TLR9 (63). Regarding the role of intestinal PRRs in NAFLD, upregulated signal activity of TLR4 has been observed in HFD-fed mice, which was associated with the elevated transcription of inflammatory cytokines such as IL-6 and IL-1β (64). TLR4 is expressed in immune cells, mainly of myeloid origin, including monocytes, macrophages, and DCs (65). TLR4 activation in these cells induces the release of inflammatory cytokines and chemokines that promote the further recruitment of innate immune cells to the intestinal mucosa (66). Among the receptors involved in the recruitment of immune cells, the chemokine receptor CX3CR1 is responsible for the maintenance of mononuclear cell populations in the lamina propria (67). It has been seen that CX3CR1 deficiency is associated with a reduced number of resident intestinal macrophages in HFD-fed mice. This finding was associated with an elevation in the translocation of bacterial components to the liver (68), demonstrating that innate cell turnover is essential for intestinal homeostasis and lessening the inflammatory damage in NAFLD.
Due to the absence of pharmacological treatments for NAFLD, diverse receptors involved in controlling metabolic disturbances have been studied, among them the PPAR. Three isoforms of PPAR have been described: PPARα, located mainly in the liver; PPARδ (also known as PPARβ), in the skeletal muscle, adipose tissue, and skin; and PPARγ in adipose tissue (69). Free fatty acids, eicosanoids, and other complex lipids are endogenous ligands for PPAR. Once the ligand is bound, a heterodimeric complex with the nuclear retinoid X receptor (RXR) is formed.
Consequently, the expression of several genes and proteins involved in beta-oxidation, fatty acid absorption, adipogenesis, and adipocyte differentiation is upregulated to control the metabolism of lipids and glucose (70). The role of PPARs in innate and adaptive immunity has been widely described, which suggests that their modulation can be considered a target for NAFLD treatment. Activated PPARs regulate the expression of several inflammatory genes expressed in a wide variety of tissues and immune cells, such as macrophages, DCs, T cells, and B cells (71). Indeed, PPARα signal activation suppresses the inflammatory gene expression mediated by the NF-κB pathway, decreasing inflammatory cytokine secretion by diverse cell types. In addition, PPARα regulates the absorption of fatty acids, beta-oxidation, ketogenesis, and bile acid secretion (72). Based on these mechanisms, PPARα regulates the hepatic metabolism of fats (73) and glucose (74). A protective role of these receptors has been demonstrated for hepatic steatosis in the context of HFD-fed mice. Thus, PPARα KO mice developed more severe steatohepatitis with MCD diet compared to wild-type mice. Even treatment with a potent agonist for PPARα, wy-14643, prevented the accumulation of hepatic triglycerides as well as liver damage in wild-type mice but not the KO group (75). In this regard, PPARα activation prevents the accumulation of triglycerides by increasing the catabolism of fatty acids.
Moreover, regarding the the anti-inflammatory role of PPAR, studies have evidenced the inhibitory effect of the agonist of this receptor on the NF-κB signaling pathway (76). In this regard, Delerive et al. observed in primary human hepatocytes that synthetic PPARα activators, such as wy-14643 and fibrates, upregulate the expression of inhibitor of nuclear factor-kappa B alpha (IκBα) and reduce the binding activity of NF-κB to DNA; however, these effects not were observed in PPARα-null mice (77), supporting the use of PPARα agonists as a potential treatment for inflammatory diseases. Further studies are needed to evaluate the role of PPARα agonists in preventing and reversing the inflammatory damage in NAFLD progression.
Regarding the role of other PPAR isoforms, PPARδ is expressed mainly in the skeletal muscle, which regulates mitochondrial metabolism and beta-oxidation. It is localized in hepatocytes, KCs, and hepatic stellate cells in the liver, preventing inflammation and fibrosis (78). A study evaluating the role of PPARδ using the synthetic agonist GW501516 showed reductions in obesity development in the HFD-fed mouse model caused by improved insulin resistance and the prevention of lipid accumulation in the liver (79).
Furthermore, PPARγ ligands can inhibit the activation of macrophages and the production of inflammatory cytokines such as TNF-α, IL-6, and IL-1β (80). PPARγ is mainly expressed in adipose tissue, regulating adipocyte differentiation, adipogenesis, and lipid metabolism (81). Regarding innate immunity, its activation is involved in macrophage phenotype polarization, from M1 (inflammatory) to M2 (anti-inflammatory) (82). Additionally, PPARγ has also been implicated in colonic inflammation, having been identified as a target of mesalazine, a 5-aminosalicylate (5-ASA) (83). As previously reported, the proinflammatory activation of KCs contributes to the progression of NAFLD. In this regard, Lumeng et al. demonstrated that diet-induced obesity leads to a shift in the phenotype activation of macrophages from adipose tissue from M2 to M1, which contributes to insulin resistance (84) in an HFD-fed mouse model. Indeed, another study showed that HFD induced hepatic steatosis and the local proinflammatory response associated with the M1-predominant profiling of KCs. Macrophages and M1 KC activation are regulated by several transcription factors, among them NF-κB. Transcriptional regulation conduced by increased NF-κB signaling in the liver has been observed in HFD-fed mice, resulting in an M1 proinflammatory predominance. Thus, by PPARγ activation, M1 phenotype activation can be diverted to the M2 phenotype. In this regard, Luo et al. (2017) demonstrated a shift in lipid-induced macrophage polarization through the use of PPARγ agonists from M1 to the M2 phenotype mediated by a direct interaction between PPARγ and NF-κBp65. These findindings allow the conclusion that the PPARγ agonist could improve hepatic steatosis by M1 KC polarization in HFD-fed mice (85). In conclusion, diverse strategies focused on modulating PPARγ activity can be effective approaches to control the innate profiling that conduces the progression of inflammatory liver damage.
Unlike innate immunity, the adaptive response induces highly specific responses against harmful antigens. The adaptative immune response starts with the antigen presentation process. The professional antigen-presenting cells (APCs), such as DCs, macrophages, and B cells, induce the activation and differentiation of mucosal T cells in the GALT (86). Consequently, the effector and memory CD4+ helper T cells and CD8+ cytotoxic T cells are generated (87). CD4+ T cells differentiate into Th1, Th2, Th17, Th9, Th22, follicular helper T (Tfh), and peripheral (p) Treg cells, while CD8+ T cells differentiate into Tc1 and Tc2, Tc17, Tc9, and CD8+ reg T cells as major subtypes in response to the cytokine profile that are received during priming (88, 89).
Increased activation of the adaptive immune response has been shown in NAFLD in both the intestinal and liver compartments. In this regard, intestinal DCs have emerged as essential mediators of immune responses in non-infectious chronic fibro-inflammatory conditions, such as pulmonary fibrosis, inflammatory bowel disease, and acute pancreatitis (90). Indeed, more DCs have been observed in the small intestine in HFD-induced NAFLD mice compared to normal diet-fed mice (91). Additionally, an MCD diet-induced NASH model in rats characterized by induced liver inflammation showed an increased number of macrophages and DCs in the ileal tissue (92). Specific DC subsets that are altered in NAFLD remain to be identified; therefore, further research is needed to distinguish their phenotype, location, and functional properties to identify specific new therapeutic targets. Altogether, this evidence suggests an increased number of APCs in NAFLD, which may correlate to increased lymphocyte activation.
Th1 cells primarily produce IFN-γ and TNF-α, which activate macrophages and conduce cytotoxic CD8+ T cell responses, respectively, promoting the elimination of intracellular pathogens such as viruses and bacteria (93). In contrast, Th2 cells produce cytokines such as IL-4, IL-5, IL-9, IL-10, and IL-13, which promote humoral immune responses. A predominance of Th2 responses is observed in pathogenic processes such as allergy (94) and gastrointestinal helminth infections (95). Th17 cells produce IL-17 and IL-22, crucial for host protection against several extracellular pathogens (96). Additionally, Th17 cells secrete IL-22 and granulocyte-macrophage colony-stimulating factor (GM-CSF) to induce neutrophil recruitment (97). Despite Th17 cells playing a relevant physiological role in maintaining populations of commensal bacteria at the gut barrier, they are involved in the progression of many autoimmune diseases and inflammatory disorders (93), among them NASH (98). Th22 cells produce IL-22, IL-13, IL-26, TNF-α, and granzyme B and regulate different antimicrobial proteins produced by intestinal epithelial cells, such as β-defensin 2 (99, 100). It has been shown that Th22 cells may be involved in allergies, autoimmune diseases, intestinal diseases, and tumors (100). Additionally, Th9 cells predominantly secrete the pro-inflammatory cytokine IL-9, which provides immunity against helminths and antitumor immunity (101). pTreg lymphocytes are generated in response to antigen exposure by APCs at the site of inflammation, and their primary function is to promote mucosal tolerance (102). The numerous functions performed by each T cell population highlights its critical role in the intestine based on its specific abilities in controlling intestinal homeostasis, which requires a delicate balance between effector and regulatory responses (103). Furthermore, B cells differentiate into plasma cells that produce immunoglobulins (Igs) in response to direct antigen recognition by surface Igs. The principal Ig secreted by plasma cells is the IgA class, which contributes to intestinal mucosal immunity and barrier function (104).
In addition to the altered number of APCs, an abnormal T cell effector response has been described in animal models and NAFLD patients. In animal models, an HFD has been demonstrated to induce a change in the percentage of intestinal immune T cell populations in diverse murine models. A study by Su et al. showed an increased CD4+/CD8+ ratio in the PP of rats fed an HFD for 12 weeks (105). Concerning CD4+ profile development in NAFLD, an increase in the level of IFNγ-producing Th1 cells has been reported in an HFD-fed mouse model compared to mice fed a normal diet (106). Similarly, Su et al. observed an increase in the Th1 cell proportion of CD4+ T cells and a reduction in the Th2 cell proportion of CD4+ T cells in the MLNs in the same HFD-fed mouse model (107). Additionally, the number of CD4+ and CD8+ T cells in the duodenal lamina propria was observed to be lower in NAFLD patients than in healthy subjects, indicating that intestinal immune function is impaired in NAFLD (108). Furthermore, the number of CD4+ and CD8+ T cells in the duodenal lamina propria was found to be lower in in NAFLD patients than in healthy subjects, indicating that intestinal immune function is impaired in NAFLD (108). In parallel, an increased Th1/Th2 ratio was consistently observed in liver samples, suggesting a relationship between the immune response in the intestine and the liver in NAFLD (107).
The pathogenic role of the Th17 profile response in NAFLD is controversial. An obesity-driven activation of the IL-17 axis is associated with the development and progression of NAFLD (109). Along with it, an increased proportion of Th17 cells in the full CD4+ T cell population has been reported in the MLNs in mice fed an HFD for 12 weeks (107). Conversely, a reduced proportion of Th17 cells within CD4+ T cells was detected in the small intestinal lamina propria of mice fed an HFD for 10 weeks compared to those fed a normal diet (110). Similarly, in the MLNs, a lower Th17 cell proportion of CD4+ T cells was found in the MCD diet-induced NASH mouse model compared to mice fed a normal diet (111). To these controverted findings, we can add that HFD-fed mice present increased IL-17-producing γδ T cells in the small and large intestinal lamina propria (106). Considering that γδ T cells participate in maintaining the integrity of the intestinal barrier by an interaction with enterocytes and other immune cells (112), the inflammatory role of the Th17 profile remains to be elucidated in this pathology. It is noteworthy that a similar dual role of γδ T cells in inflammation has also been reported in some murine models of colitis (113).
Intestinal Treg cells regulate mucosal immune responses through diverse mechanisms, including cell-to-cell contact suppression and the secretion of soluble suppressive factors, maintaining immune tolerance to dietary and microbiota components (114, 115). In HFD-fed mice, decreased levels of FOXP3+ Treg cells in the intestinal lamina propria (106) and a reduced proportion of CD4+/FOXP3+ cells in the MLNs compared to mice fed a normal diet (107) have been reported. In contrast, the MCD diet-induced NASH mouse model presented an increased FOXP3+ Treg cell proportion of CD4+ T cells in the MLNs compared to mice fed a normal diet (111). A therapeutic strategy based on oral anti-CD3 mAbs to elicit Treg induction has been explored in NASH treatment (116). This approach is based on the binding properties of the mAbs to the CD3/T cell receptor (TCR complex) of lamina propria T cells that trigger the upregulation of membrane-bound TGF-β and the conversion to the Th3 reg phenotype. Through the release of TGF-β and IL-10, Th3 cells contribute to the tolerogenic intestinal microenvironment (117).
Regarding the role of the gut humoral response, an altered density of IgA+ cells and full IgA content in the intestine have been observed in NAFLD. In particular, Su et al. observed increased levels of intestinal IgA in the small intestinal fractions of the 12-week scheme HFD rat model, which were associated with an impairment of gut barrier function demonstrated by a high serum level of endotoxin and D-xylose (105). In contrast, Matsumoto et al. demonstrated that MCD diet-fed mice had lower IgA+ cell numbers in the ileal and colonic tissues and decreased IgA content in the feces in comparison to a normal diet. Interestingly, these alterations were prevented by adding fructooligosaccharides to the diet, suggesting a feasible prebiotic role that modifies the gastrointestinal microbiota (118). In line with these results, another study reported a decrease in IgA content in the small intestine mucus in HFD-induced NAFLD rats (119). Considering the role of IgA as a soluble factor that controls the load and composition of the microbiota in the lumen, these results suggest that IgA deficiency contributes to the pathogenesis of liver diseases associated with an altered gut microbiota composition (120).
Intestinal lymphocyte recruitment from the bloodstream depends on sequential events of lymphocyte-endothelial cell adhesion molecule (CAM) interactions (121). Lymphocyte location in various intestinal compartments, such as the lamina propria and epithelia, is determined by specific homing pathways that are conducted by chemokines released by inflamed tissue. Cytokines and co-stimulatory molecules from mature DCs mediate the specific expression of CAMs in lymphocytes during the antigen presentation process. DCs loaded with specific antigens migrate to the secondary lymphoid tissue located proximal to the site of antigen entry, such as Peyer’s patches, MLNs, and ILFs. In the steady state, the intestinal T cells can be activated by retinoic acid-primed intestinal mucosa DCs (CD103+) to maintain a permanent lymphocyte pool population, principally in the small intestine. This activation induces the upregulation of α4β7 integrin and CCR9 receptors that are crucial for the migration of the lymphocytes toward the small intestine. Further, CCR9 upregulates α4β7 integrin expression via interaction with C-C motif chemokine ligand 25 (CCL25), which is selectively and constitutively expressed by intestinal epithelial cells. α4β7 integrin can bind the mucosal addressin cell adhesion molecule 1 (MAdCAM-1), which is strongly expressed on Peyer’s patches’ high endothelial venules, allowing the entry of lymphocytes from the bloodstream into the lamina propria (122). Despite the scarcity of information about the mechanism that controls the homing to diverse areas of the intestine, the anatomical distribution of lymphocytes is known to depend on the differential expression of CCR9 and retinoic acid availability. In contrast to the small intestine, CD8+ T cell homing to the large intestine involves the chemokine receptors CXCR3 and GPR15 α4β7 but not CCR9 (98, 123). It is worth noting that CXCR3, CXCR6, and CCR5 induce T cell trafficking to the liver to maintain lymphocyte permanency as a liver-resident cell phenotype (109).
The gut-primed lymphocytes can migrate to extra-intestinal tissues, such as the liver, lung, and skin. This process is highlighted in certain pathological conditions, such as inflammatory bowel disease (IBD) and NAFLD. In this regard, a “gut-lymphocyte homing” hypothesis has been proposed to explain the pathophysiology of NAFLD (Figure 3). This concept was initially proposed by Grant et al., who observed a strong association between primary sclerosing cholangitis (PSC) and IBD (124). In this regard, intestinal mucosal T cells were observed in the pool of liver-infiltrating lymphocytes of PSC patients, which were recruited by the aberrant expression of gut-specific CCL25 on the hepatic endothelium. These findings were in agreement with abnormal MAdCAM-1 expression in the hepatic endothelium of IBD patients, especially those with PSC comorbidity (125).
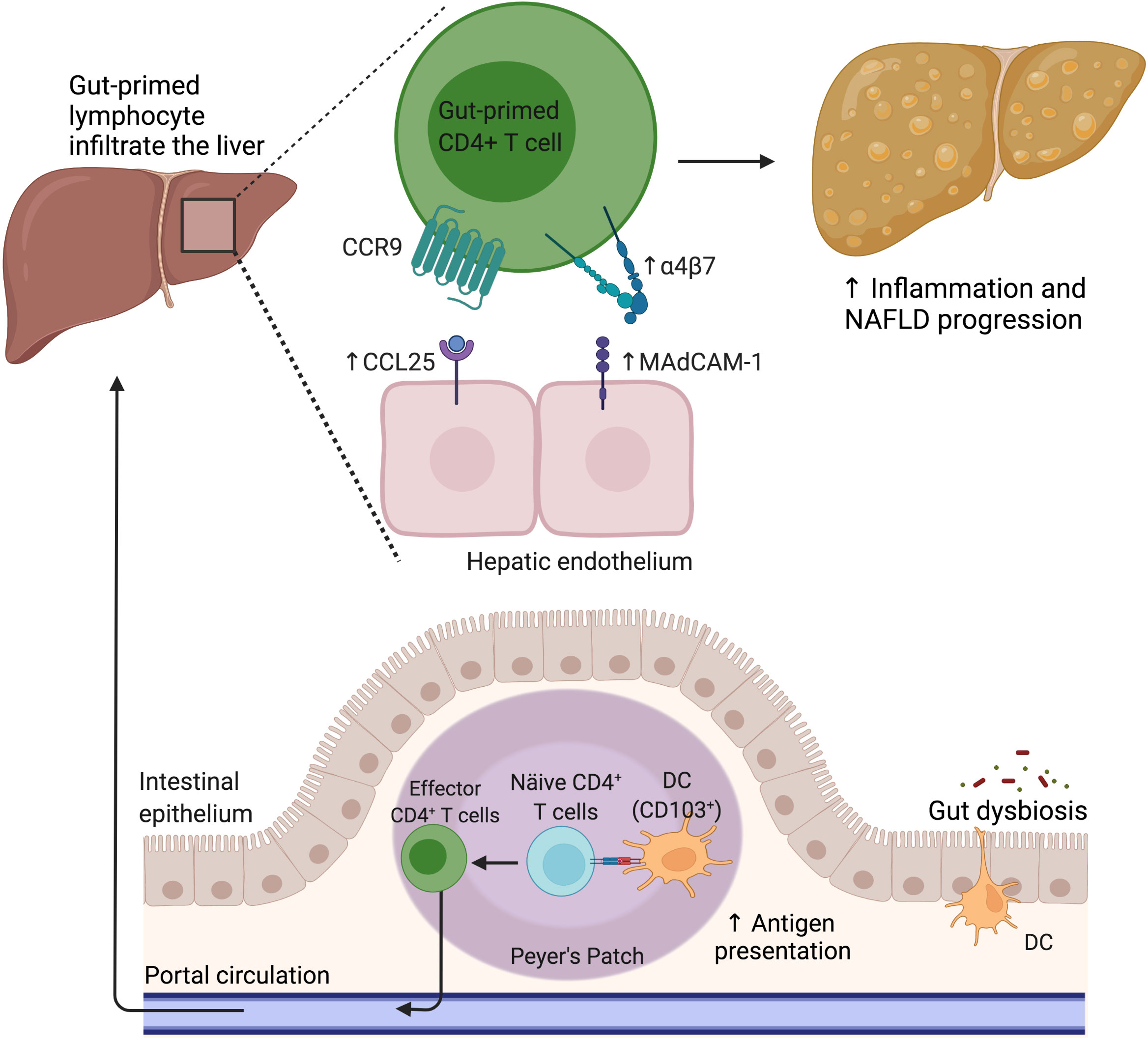
Figure 3 The gut lymphocyte is homing in the NAFLD liver. Under physiological conditions, dendritic cells imprint gut-homing specificity on T cells in the Peyer’s patches or mesenteric lymph nodes by inducing the upregulation of α4β7 integrin and CCR9. Nevertheless, during NAFLD, the hepatic endothelium aberrantly expresses CCL25 and MAdCAM-1, allowing the pathologic recruitment of gut-primed lymphocytes into the liver. CCL25, C-C motif chemokine ligand 25; CCR9, C-C motif chemokine receptor 9; DC, Dendritic cell; MAdCAM-1, Mucosal addressin cell adhesion molecule 1; NAFLD, Nonalcoholic fatty liver disease.
In addition, recently, the MLN has been considered a potential source of liver lymphocytes in NAFLD (107, 126, 127), supporting the hypothesis of gut-lymphocyte homing in this pathology. In this regard, an increased number of CCR9+ cells in the liver and an elevated serum CCL25 level were observed in NASH patients in comparison to healthy controls, as well as an increased number of CCR9+ macrophages and CCR9+ hepatic stellate cells (HSCs) in a high-fat/high-cholesterol (HFHC)-diet mouse model. These findings were associated with liver disease severity and prevented by CCR9 antagonist treatment, suggesting that approaches orientated at blocking the CCR9/CCL25 axis can effectively prevent liver fibrosis progression (128). Furthermore, an increased proportion of Th1 and Th17 in MNL CD4+ T cells was observed in HFD-fed NAFLD mice, with increased in vitro chemotaxis of MNL CD4+ T cells to the liver extract from HFD-fed NAFLD mice (107).
Similarly, a high level of migration to the liver of adoptive-transferred, gut-derived lymphocytes from HFD-induced NAFLD donor mice to NAFLD recipient mice was observed compared to that in control recipient mice. This migration bias was associated with exacerbated liver damage. Upregulated CCL5 expression was observed in the liver of NAFLD recipient mice, along with CCL5 receptor CCR3 in transferred MLN immune cells. The MLN liver migration was inhibited by using a CCL5-blocking antibody in vitro (129). Additionally, the increased microbiota-driven intestinal and hepatic expression of MAdCAM-1 contributes to α4β7+ CD4+ T cell recruitment to the intestine and liver in murine steatohepatitis models (130, 131).
Interestingly, MAdCAM-1 and β7 have been reported to have opposing roles in liver-damaging progression, wherein the upregulation of MAdCAM-1 has been described as being pathogenic in contrast to the expression of β7, which appears to be protective in hepatic inflammation and the liver oxidative response (130). These shreds of evidence suggest that the increased propensity of gut lymphocytes to migrate to the liver in NAFLD can reinforce the inflammatory environment of liver injury. Thus, diverse strategies targeting the molecular mechanisms involved in the migration of activated immune cells in the intestinal environment to the liver, like MAdCAM-1, promise to reduce liver damage progression in this pathology.
The contribution of the liver immune response to NAFLD progression has been widely described. In this regard, high activation of resident immune cells, hepatocytes, and sinusoidal endothelial cells plays a significant role in NAFLD pathogenesis by contributing to the chronic inflammatory status. Both innate and adaptive immune responses are involved in the onset of liver damage, reinforcing the recruitment of immune cells, such as monocytes, macrophages, neutrophils, innate lymphoid cells, and CD8+ and CD4+ T cells, to the site of inflammation (132, 133).
The liver tissue is composed of diverse types of cells that are involved in liver immune responses. Hepatocytes constitute 60% of the liver mass and have a metabolic function and depurative role regarding waste products and substances harmful to the organism. In response to diverse stimuli, hepatocytes contribute to innate immune responses through their ability to produce and secrete various inflammatory proteins. Several innate receptors are expressed in hepatocytes. TLRs have been described to have significant roles in NAFLD pathogenesis (134). Elevated circulating LPS levels are observed in diverse NAFLD mouse models, associated with increased intestinal permeability and altered microbiota.
As resident macrophages in the liver, KCs are located in the hepatic sinusoids close to parenchymal and nonparenchymal cells. KCs act as sentinels in the sinusoidal barrier and prevent the spread of filtered products from the intestinal wall to the systemic circulation. KCs exert tolerogenic responses to gut-derived substances, commensal antigens, and death cell products under healthy conditions. Indeed, these cells are permanently exposed to low LPS levels from the gut microbiota that trigger the activation of inflammatory control mechanisms mediated by the release of IL-10 (135). However, under pathogenic conditions, KCs can switch to an inflammatory phenotype, acting as APCs and releasing pro-inflammatory mediators (cytokines, prostanoids, nitric oxide, and oxidizing agents) that contribute to liver inflammation (136–138). Increased activation of TLR4 signaling has also been observed for KCs during NAFLD development, with TLR4-mutant mice being resistant to this damaging process (139). In this regard, massive TLR4 stimulation in response to increased LPS levels from the portal circulation leads to KCs producing several chemokines, such as CCL2 (10). Likewise, a significantly increased expression of CCL5 was detected in the liver of HFD-fed mice, associated with significant hepatic steatosis (140). These findings agree with the increased CCL5 mRNA expression observed in liver samples from patients with fibrotic NASH compared with subjects with simple steatosis, suggesting that CCL5 expression is a marker of fibrotic liver disease (141). Both CCL2 and CCL5 are involved in the recruitment of lymphocytes to the liver (10). It has been shown that CD4+ T and CD8+ T cells activated in the MLNs can migrate to the liver, leading to liver injury, while CCL5 blockade prevents the recruitment of T cells. These findings suggest a crucial role for CCL5 in the migration of gut-derived lymphocytes in the NAFLD mouse model (129).
Additionally, augmented infiltration of LY6C2+ monocytes into the liver has been described, mediated by the CCR2–CCL2 interaction. This event is considered critical in steatohepatitis development and subsequent fibrosis progression (10, 142). A pro-inflammatory M1 phenotype can be induced in KCs by liver metabolic abnormalities. Under this condition, KCs perpetuate the effects of a high-fat diet by increasing triglyceride accumulation in hepatocytes and decreasing fatty acid oxidation and insulin responsiveness, which is attenuated by the neutralization of TNFα in vitro. Considering that the depletion of KCs limits the development of liver inflammation, insulin resistance, and alterations in hepatic lipid metabolism and fibrosis, this evidence suggests that KCs are a crucial intermediary in the cross-talk of immune-metabolic liver functions (143).
Natural killer (43) cells and natural killer T (NKT) cells are relevant components of the innate immune response in NAFLD. Both cells have a significant role in the progression of liver damage due to their cytotoxic impact and the promotion of pro-inflammatory responses, demonstrated in viral hepatitis and chronic liver inflammation (144). Liver-resident NKs possess immunophenotypic and functional characteristics that differ from peripheral NKs, sharing functional properties with the innate lymphoid cells of mucosal tissues (145). NKT cells reside in the sinusoids, playing an immune surveillance role, acting as sentinels, and eliminating pre-malignant senescent hepatocytes. Two subsets of NKTs have been described: a proinflammatory type I phenotype, principally activated by lipids, and type II, abundant in the livers of both mice and humans compared to the type I subtype. NK T cell types I and II have opposing immune functions, with a protective role described for type II cells in liver inflammation (146). Interestingly, HFD-fed NKT cell-deficient mice, lacking the NKT type I and II phenotypes, are prone to developing diet-induced obesity and metabolic perturbations due to increased inflammatory responses and steatosis in the liver (147). Unlike these effects, the systemic depletion of NK cells in an HFD mouse model of obesity decreased macrophage infiltration into the adipose tissue, reducing systemic inflammation and insulin resistance (148). This evidence suggests that NKT cells may be a therapeutic target in modulating metabolic disorders in the liver, among them NAFLD. Unlike NKT cells, NK cells’ role in metabolic disturbances, liver inflammation, and damaging progression remains controversial.
Lipotoxic lipid species induce hepatocyte damage and the release of neutrophil-recruiting chemokines, such as CXCL1 and IL-8 (149). The increased liver infiltration of neutrophils in NAFLD initiates and enhances inflammation, reinforcing the recruitment of macrophages and the interaction with APCs. Changes in neutrophil granular content and composition have been associated with diverse inflammatory processes in several NASH models (149). Myeloperoxidase (MPO), a pro-oxidant enzyme released by neutrophils, enhances macrophage cytotoxicity and promotes inflammation and fibrosis in HFD-fed mice. In contrast, MPO-deficient mice attenuate NASH development (150). Additionally, neutrophils promote insulin resistance and inflammation by releasing neutrophil elastase levels in HFD-fed mice (151). This evidence shows that the increased infiltration and activity of neutrophils contribute to the inflammatory damage in and progression of NAFLD. As for human models, increased neutrophils and elevated MPO plasma levels have been observed in patients with NASH compared with those with fatty liver. Thus, MPO activity in the liver is associated with increased inflammation in these patients (152).
Mast cells (MCs) have been highlighted as important regulators of pathogenic processes in liver disease progression (153). In NASH patients, the number of hepatic MCs positively correlates with the stage of fibrosis (154). This evidence suggests that the modulation of MCs may be another attractive therapeutic target for treating NASH.
Regarding adaptive immune responses, it has been demonstrated that DC activation and its immune phenotype commitment are essential for the perpetuation of liver damage (127). Among the adaptive immune cells, DCs are responsible for initiating and limiting liver inflammation through their properties of presenting antigens to lymphocytes in the neighboring lymphoid organs as well as eliminating apoptotic and necrotic waste. The liver contains several types of DCs that are usually located surrounding the central veins and the portal system. In the normal liver, resident DCs exhibit an immature phenotype imprinted by a tolerogenic IL-10-enriched microenvironment (155). Under inflammatory conditions, DCs are recruited from the hepatic sinusoids to periportal areas. LPS and peptidoglycan induce the upregulation of costimulatory molecules, such as CD40, CD80, and CD86, and the release of inflammatory cytokines by hepatic DCs through TLR4/MD2 complex activation (156). The role of hepatic DCs in NAFLD pathogenesis is controversial due to liver population heterogeneity and differences in the mouse models of disease used in various studies (157). Although murine CD103+ DC subtype (classical type-1 DCs and cDC1s) influences the pro-and anti-inflammatory balance and protects the liver from metabolic and inflammatory damage (158), increased activation and abundance of these cell types have been observed in NAFLD patients, promoting inflammatory T cell reprogramming in NASH (159).
Regarding the role of liver B and T lymphocytes in NAFLD, increased infiltration of B2 cells and CD4+ and CD8+ T cells as well as elevated circulating antibodies have been observed in NASH (133). In terms of T cell subsets, an increase in Th1 and Th17 cells and a reduction in Treg levels in the liver have been observed in NAFLD patients compared to healthy controls, principally in patients with steatohepatitis compared with those with simple steatosis (133). Also, Th1, Th17, and CD8+ lymphocytes contribute to hepatic macrophage activation and NKT cell recruitment in NASH murine models (133). Additionally, increased γδT cell recruitment to the liver has been associated with the progression of steatohepatitis, mainly due to the IL-17-secreting subset (160). Considering that CD4, CD8, and γδ T cells can recognize microbial peptides and lipid antigens, this suggests that the overall stimulation of the intrahepatic T cell subsets can be directly influenced by the homeostasis of the gut microbiota (161).
Moreover, recent evidence supports that gut microbial factors drive the pathogenic function of B cells during NASH development. Fecal microbiota transplantation from humans with NAFLD into recipient mice induces the increased accumulation and activation of intrahepatic B cells, predominantly pro-inflammatory IgM+ IgD+ B2 cells (162). These findings suggest that the adaptive immune response is highly activated in NAFLD, associated with a decreased tolerogenic response and disease progression.
The current treatment for NAFLD focuses on improving patients’ lifestyles by promoting a healthy diet, physical activity, and weight loss. Despite compelling evidence of the effectiveness of these recommendations in reducing liver damage and even reversing liver fibrosis (163), low treatment adherence leads groups of patients to search for therapeutics directed at complementing current clinical protocols.
The first two drugs evaluated for NAFLD treatment were vitamin E and pioglitazone, an antidiabetic agent. Although there is diverse evidence of their beneficial effect on liver function and NASH resolution (164–166), the use of these drugs is limited due to the high risk of side effects under prolonged administration. Recently, two not yet fully approved hypoglycemic drugs, glucagon-like peptide-1 (GLP-1) agonists and sodium-glucose cotransporter-2 (SGLT-2), showed effectiveness in reducing liver inflammation and fibrosis, mainly in a subgroup of diabetic patients in phase II and III trials (167, 168). Moreover, different therapeutic approaches for liver diseases are focused on modulating the gut microbiota, including prebiotics and probiotics (169). The results of their evaluations support the benefit of this complementary strategy in NAFLD pharmacological therapy (170, 171).
There are several potential strategies to normalize altered hepatic metabolism in NAFLD. These include therapies aimed at reducing hepatic steatosis by modulating the lipid metabolism. BAs have been described as promising alternatives in NAFLD treatment. Since BAs regulate carbohydrate and lipid metabolism in the liver, approaches directed at increasing their function have resulted in an interesting strategy to reduce the liver metabolic overload and liver damage in NAFLD. By interacting with their nuclear receptor FXR, BAs can activate the gene expression of diverse components involved in the entero-hepatic metabolic pathways (172). Obeticholic acid (OCA), an analog of chenodeoxycholic acid, is a semi-synthetic BA with high affinity and selectivity for FXR that has been trialed in NASH. Along with FXR activation in the ileum, which induces the secretion of fibroblast growth factor 19 (FGF-19) and its subsequent transport to the portal system, OCA decreases the production of BAs, stimulates beta-oxidation, and decreases lipogenesis and gluconeogenesis in the liver (173). Randomized studies in patients with NASH have shown that OCA significantly improves steatosis, inflammation, ballooning, and liver fibrosis compared to placebo, with a low rate of side effects such as pruritus and an increase in LDL being the most frequent (174). These studies are consistent with previous multicenter, randomized, placebo-controlled trials in NASH patients (FLINT and REGENERATE) in which improvements in liver histology, including a decrease in the fibrosis stage, were achieved by the use of OCA (53, 175). The therapies described above are among those most frequently addressed in NASH. However, the modulation of the immune response has been little discussed in these studies.
The modulation of the immune response has been a secondary aim in NAFLD therapy since the main approaches are focused on reducing liver fat loading to lessen inflammation. However, few works focus on controlling the immune response at the intestinal level because most studies have been directed at the immune response in the liver. Thus, possible targets in the intestinal response would be interesting to study. To classify the immune targets explored in NAFLD, we have divided the strategies into treatments focused on controlling inflammation, including modulating agents of the innate immune response and receptors associated with inflammatory pathways, and those that control the adaptive immune response (Table 1).
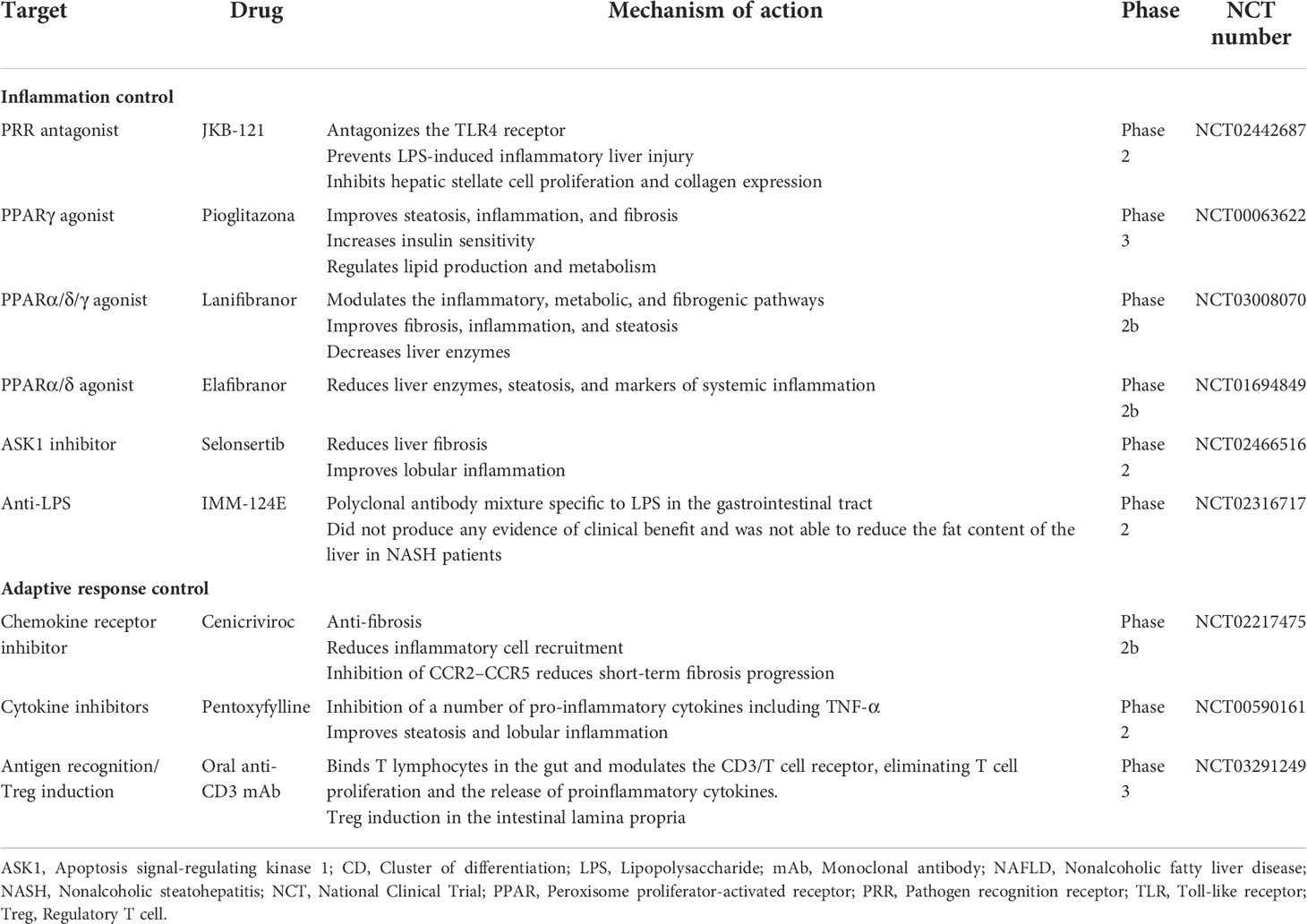
Table 1 Clinical trials of pharmacological approaches targeting the immune response in NAFLD.
The liver is exposed permanently to gut-derived endotoxins, such as LPS, that enter the enterohepatic circuit after passing across the intestinal epithelial barrier. LPS in the liver activates KCs by TLR4 signaling, provoking pro-inflammatory gene expression and the subsequent release of mediators that induce hepatic injury and fibrosis. Thus, strategies that block the TLR4 pathway appear promising to prevent the progression of liver inflammatory damage. A preclinical study in mice showed that KC depletion by the intravenous injection of clodronate liposomes reduced histological evidence of steatohepatitis and prevented the increase of TLR4 expression in the liver, which demonstrates that the link between KCs and TLR4 signaling plays a central role in the pathogenesis of steatohepatitis (176). Other studies also assessed this approach and focused on evaluating the impact of TLR4 signaling antagonists on liver damage. JKB-121, a non-selective opioid TLR4 antagonist, has been shown to prevent LPS-induced inflammatory liver injury in an MCD diet-fed rat model of NAFLD (177). Additionally, in vitro experiments have shown that JKB-121 could reduce the release of LPS-induced inflammatory cytokines and inhibit hepatic stellate cell activation (178). However, a randomized, double-blind, placebo-controlled phase II trial of JKB-121 showed unsatisfactory results, wherein JKB-121 did not improve the liver fat content and liver fibrosis biomarkers in patients with NASH compared to placebo (NCT02442687) (179). Given the multiple relevant biological pathways of TLR4 in the pathogenesis of NAFLD, further investigation of TLR4 inhibition is necessary (NCT02442687). In line with this evidence, a phase II study in NASH patients using a polyclonal antibody mixture specific to LPS and other pathogenic bacterial components (IMM-124E) did not produce any evidence of clinical benefit. The intervention could not reduce the liver fat content but decreased serum LPS levels and AST and ALT biomarkers associated with liver function (180).
Another therapeutic approach studied for NASH treatment is the inhibition of the activation of apoptosis signal-regulating kinase 1 (ASK1). Under pathological conditions, an increase in oxidative stress in hepatocytes induces ASK1 autocleavage, leading to increased p38/JNK pathway activation that worsens hepatic inflammation, apoptosis, and fibrosis (181). Selonsertib, a selective inhibitor of ASK1, was evaluated in a phase II clinical trial for NASH (NCT02466516) and improved liver fibrosis and decreased fibrosis progression rates over a 24-week treatment period, indicating its potential as an anti-fibrotic therapy (181). Specifically, 18 mg of selonsertib was shown to lead to improvements in 43% of patients in at least one stage of fibrosis, compared to 30% for selonsertib 6 mg. However, other phase III studies using selonsertib in NAFLD did not achieve the expected results at week 48 of treatment and were terminated (NCT03053050 and NCT03053063).
As discussed above, the role of PPARs in regulating the liver immune response has been well described. Multiple studies have been conducted to modulate PPAR nuclear activity by using receptor agonists. In a phase IIb study, elafibranor, a PPARα/δ dual agonist, resolved NASH after 52 weeks of treatment by reducing liver enzymes, steatosis, and systemic levels of inflammatory markers (NCT01694849) (182). Another phase III study (NCT02704403) using elafibranor showed reduced liver fibrosis stages in the subgroup of patients that reached NASH resolution compared to the group without NASH resolution; however, this study was suspended because it did not meet the primary endpoint. The drug did not worsen the fibrosis, but only 24.5% of patients who received elafibranor 120 mg achieved fibrosis improvement of at least one stage compared to 22.4% in the placebo group. In this regard, in a phase IIb trial, the pan-PPAR agonist lanifibranol demonstrated effectiveness in modulating inflammatory, metabolic, and fibrogenic pathways in NAFLD pathogenesis in patients with non-cirrhotic NASH who were treated for 24 weeks (183).
Since obesity is associated with the induction of pro-inflammatory profiling in gut-immune populations, anti-inflammatory therapies targeting the gut, such as mesalazine (5-aminosalicylate, 5-ASA), have been investigated in HFD-fed mouse models. Treatment with mesalazine reduced liver steatosis compared to the non-treated group (106). This finding suggests that controlling the inflammatory response in the intestine contributes to the lessening of the liver damage induced via the accumulation of fatty acids in the liver.
The adaptive immune response plays an essential role in liver damage due to the recruitment and migration of cells to the liver, leading to the generation of proinflammatory immune profiles responsible for the increased oxidative and inflammatory damage in NASH. Therapeutic strategies focused on modulating the activation of the adaptive immune response in the gut have been explored (116). An oral anti-CD3 monoclonal antibody treatment induced intestinal regulatory T cells that suppress the chronic inflammatory state associated with NASH. The anti-CD3 monoclonal antibody foralumab is currently in phase II of development (NCT03291249).
In NASH, several studies have focused on chemokine blockage to prevent the elevation of immune cell recruitment in the liver. Cenicriviroc is a dual antagonist that inhibits the CCR2/CCR5b chemokine receptors. This experimental drug can potently block the infiltration of pro-inflammatory monocytes and macrophages via the antagonism of CCR2. It also has antifibrotic activity in the liver due to the modulation of immune cells and hepatic stellate cells via CCR5 inhibition. A phase IIb multinational, randomized, double-blinded, placebo-controlled study has shown that patients with NASH treated with oral cenicriviroc had reduced circulating biomarkers of systemic inflammation, such as high-sensitivity C-reactive protein, IL-6, fibrinogen, and IL-1ß, as well as reduced monocyte activation, compared with the placebo group. These results suggest that cenicriviroc exhibited anti-inflammatory and anti-fibrotic effects at Year 1; specifically, twice as many subjects on cenicriviroc achieved improvement in fibrosis of ≥1 stage and no worsening of steatohepatitis compared to those on placebo (184). Despite the promising results obtained in the phase II trial, the phase III clinical study was interrupted early due to lack of efficacy (NCT03028740).
The pathogenesis of NAFLD is complex and directly related to metabolic syndrome. Environmental factors including diet, lifestyle, obesity, insulin resistance, and psychosocial stress determine its development based on the individual characteristics of susceptibility, genetics, gut microbiota, and immune response of the patients. Metabolic syndrome establishment has several consequences in the tissues, such as increasing serum free fatty acid and cholesterol levels, leading to adipocyte dysfunction. This syndrome has hepatic implications due to the over-accumulation of free fatty acids, leading to lipotoxicity, the release of pro-inflammatory cytokines, and steatosis.
Gut-derived metabolites, whether dietary or microbial, are transported via the portal vein to the liver, continuously exposing this organ to potential antigens. Conversely, liver-derived factors, such as BAs, are transported to the gut, influencing gut microbiota composition and function. Liver diseases, including NAFLD, are associated with compositional and functional alterations of the gut microbiota, known as dysbiosis. Dysbiosis impacts the host’s immune and metabolic systems and intestinal barrier integrity. Metabolic mechanisms include effects on glucose and lipid metabolism, mainly mediated by changes in BA composition and alterations in the production of SCFAs. Immune mechanisms include delicate crosstalk between the gut microbiota, intestinal epithelial cells, and gut mucosal system. The disruption of this crosstalk leads to alterations in the modulation of inflammasome signaling through microbial metabolites, activation of TLRs and NLRs, and the shifting of the balance between regulatory and pro-inflammatory T cells. Concerning the intestinal barrier, dysbiosis disrupts its integrity, causing a leaky gut and the increased translocation of microbial components to the MNLs and the GALT. Additionally, when the intestinal barrier is compromised, the liver becomes overloaded with metabolites from the gut, leading to a loss of liver tolerance. Antigens such as LPS derived from the microbiota induce inflammation by binding to the TLRs of KCs. Signaling via TLRs leads to pro-inflammatory changes in the liver, and failure to regulate gut microbiota results in further disease progression.
The elevation in gut inflammation and the adaptive immune cell priming at the lymphoid tissue causes aberrant homing of gut lymphocytes to the liver, reinforcing the inflammatory damage of hepatocytes. Gut lymphocyte homing was initially described in NAFLD as involving the aberrant expression of homing receptors in the liver and increased hepatic oxidative stress and inflammation. Nevertheless, the mechanisms of gut immune cell homing to the liver, the immune profiles, and the specific composition of the subsets of these populations have not been fully described. In addition, the characterization of this mechanism should consider the different stages of NAFLD and their association with changes in the gut microbiota composition. It is worth noting that although these mechanisms reinforce and promote damage to the liver (from gut to the liver), whether the deterioration of liver functions implies a metabolic alteration that may affect the synthesis of proteins necessary for the structure of the intestinal epithelial TJs and the mechanisms of regulation of the systemic inflammatory response has not been studied. We propose that the study of gut mucosal immunity in the context of NAFLD could provide novel insights into the development and progression of this disease. In this context, the modulation of intestinal mucosal immunity stands out due to its direct relationship with the liver.
Despite several advances in the understanding of the pathophysiology underlying NAFLD, no drugs have been approved by the Food and Drug Administration (FDA) to treat either simple steatosis or NASH. Thus, no specific therapy can be firmly recommended, and any drug treatment would be off-label (185). Clinical trials have focused mainly on the metabolism as a target, while studies that focus on drugs whose primary target is the modulation of the immune response are scarce. Interestingly, gut lymphocyte homing is a process involved in NAFLD pathogenesis, as has been shown in the experimental NASH model where MAdCAM-1 deficiency improved the disease (130); therefore, further research aimed at evaluating the pharmacological modulation of gut lymphocyte homing as a therapeutic strategy is needed.
Based on this revised information, we propose that modulating intestinal inflammatory events at the onset of the disease could be a possible therapeutic target that has remained unexplored thus far. Further research is needed to fill the gaps in knowledge of the immunological mechanisms altered in NAFLD to identify specific new potential therapeutic targets.
To date, the primary treatment for NAFLD is based on metabolic control measures, such as weight loss or exercise. However, these medical indications require high patient adherence, which is not always reached. Current research in this area has allowed a greater understanding of the pathogenesis of the disease, giving rise to new treatment options. As we described, the altered immune response in NAFLD is associated with the dysfunction of the gut-liver axis. Indeed, increased immune cell activation plays a crucial role in the onset of the disease and the course of chronic liver damage. Based on this, determining inflammatory mediators and identifying activated immune cell profiles could significantly contribute to the determination of predictive biomarkers of NAFLD progression toward NASH or cirrhosis. On the other hand, new research focused on describing immune response dynamics in this pathology will allow us to propose new therapeutic targets for the modulation of adaptive immunity. In this regard, it is essential to highlight the role of increased gut-to-liver lymphocyte homing in the pathogenesis of NAFLD. In this regard, therapies based on the blockage of activated lymphocyte migration, such as CCR9 antagonists, could reduce lymphocytic infiltration in the liver and prevent tissue damage that leads to fibrosis progression.
NO-L, CF, MD and AP-L: drafting the original manuscript. NO-L, CF, MD, LV-P, and CB conceived the idea and the aim of the review. CB, LV-P, AP-L, AE, JR and JP reviewing. CB, AE and LV-P: editing. NO-L, CF, MD, AP, AE, JR, JP, LV-P, and CB: validation. CB and LV-P: supervision. CB: funding acquisition. All authors contributed to the article and approved the submitted version.
Financed by FONDECYT 1181699, ANID.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ando Y, Jou JH. Nonalcoholic fatty liver disease and recent guideline updates. Clin Liver Dis (2021) 17(1):23–8. doi: 10.1002/cld.1045
2. Dumitrascu DL, Neuman MG. Non-alcoholic fatty liver disease: An update on diagnosis. Clujul Med (2018) 91(2):147–50. doi: 10.15386/cjmed-993
3. Liu J, Tian Y, Fu X, Mu C, Yao M, Ni Y, et al. Estimating global prevalence, incidence, and outcomes of non-alcoholic fatty liver disease from 2000 to 2021: Systematic review and meta-analysis. Chin Med J (Engl) (2022) 135(14):1682–91. doi: 10.1097/cm9.0000000000002277
4. Ge X, Zheng L, Wang M, Du Y, Jiang J. Prevalence trends in non-alcoholic fatty liver disease at the global, regional and national levels, 1990-2017: A population-based observational study. BMJ Open (2020) 10(8):e036663. doi: 10.1136/bmjopen-2019-036663
5. Käräjämäki AJ, Hukkanen J, Kauma H, Kesäniemi YA, Ukkola O. Metabolic syndrome but not genetic polymorphisms known to induce nafld predicts increased total mortality in subjects with nafld (Opera study). Scand J Clin Lab Invest (2020) 80(2):106–13. doi: 10.1080/00365513.2019.1700428
6. Paik JM, Henry L, De Avila L, Younossi E, Racila A, Younossi ZM. Mortality related to nonalcoholic fatty liver disease is increasing in the united states. Hepatol Commun (2019) 3(11):1459–71. doi: 10.1002/hep4.1419
7. Day CP, James OF. Steatohepatitis: A tale of two “Hits”? Gastroenterology (1998) 114(4):842–5. doi: 10.1016/s0016-5085(98)70599-2
8. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radical Biol Med (2020) 152:116–41. doi: 10.1016/j.freeradbiomed.2020.02.025
9. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology (2010) 52(5):1836–46. doi: 10.1002/hep.24001
10. Wang H, Mehal W, Nagy LE, Rotman Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell Mol Immunol (2021) 18(1):73–91. doi: 10.1038/s41423-020-00579-3
11. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol (2014) 14(10):667–85. doi: 10.1038/nri3738
12. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (Nafld). Metabolism (2016) 65(8):1038–48. doi: 10.1016/j.metabol.2015.12.012
13. Poeta M, Pierri L, Vajro P. Gut–liver axis derangement in non-alcoholic fatty liver disease. Children (2017) 4(8):66. doi: 10.3390/children4080066
14. Jamali R, Arj A, Razavizade M, Aarabi MH. Prediction of nonalcoholic fatty liver disease via a novel panel of serum adipokines. Medicine (Baltimore) (2016) 95(5):2630. doi: 10.1097/md.0000000000002630
15. Drenick EJ, Fisler J, Johnson D. Hepatic steatosis after intestinal bypass–prevention and reversal by metronidazole, irrespective of protein-calorie malnutrition. Gastroenterology (1982) 82(3):535–48. doi: 10.1016/S0016-5085(82)80403-4
16. Tumani MF, Tapia G, Aguirre C, Obregón AM, Pettinelli P. Rol de la microbiota intestinal en El desarrollo del hígado graso no alcohólico. Rev médica Chile (2021) 149:570–9. doi: 10.4067/s0034-98872021000400570
17. Wieland A, Frank DN, Harnke B, Bambha K. Systematic review: Microbial dysbiosis and nonalcoholic fatty liver disease. Alimentary Pharmacol Ther (2015) 42(9):1051–63. doi: 10.1111/apt.13376
18. Wijarnpreecha K, Lou S, Watthanasuntorn K, Kroner PT, Cheungpasitporn W, Lukens FJ, et al. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease: A systematic review and meta-analysis. Eur J Gastroenterol Hepatol (2020) 32(5):601–8. doi: 10.1097/meg.0000000000001541
19. Mao JW, Tang HY, Zhao T, Tan XY, Bi J, Wang BY, et al. Intestinal mucosal barrier dysfunction participates in the progress of nonalcoholic fatty liver disease. Int J Clin Exp Pathol (2015) 8(4):3648–58.
20. Martín-Mateos R, Albillos A. The role of the gut-liver axis in metabolic dysfunction-associated fatty liver disease. Front Immunol (2021) 12:660179. doi: 10.3389/fimmu.2021.660179
21. Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology (2009) 49(6):1877–87. doi: 10.1002/hep.22848
22. Xin D, Zong-Shun L, Bang-Mao W, Lu Z. Expression of intestinal tight junction proteins in patients with non-alcoholic fatty liver disease. Hepatogastroenterology (2014) 61(129):136–40. doi: 10.5754/hge12760
23. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in Nafld/Nash. Dig Dis Sci (2016) 61(5):1294–303. doi: 10.1007/s10620-016-4049-x
24. Carpino G, Del Ben M, Pastori D, Carnevale R, Baratta F, Overi D, et al. Increased liver localization of lipopolysaccharides in human and experimental nafld. Hepatology (2020) 72(2):470–85. doi: 10.1002/hep.31056
25. Hegazy MA, Mogawer SM, Alnaggar A, Ghoniem OA, Abdel Samie RM. Serum lps and Cd163 biomarkers confirming the role of gut dysbiosis in overweight patients with Nash. Diabetes Metab Syndr Obes (2020) 13:3861–72. doi: 10.2147/dmso.S249949
26. Milosevic I, Vujovic A, Barac A, Djelic M, Korac M, Radovanovic Spurnic A, et al. Gut-liver axis, gut microbiota, and its modulation in the management of liver diseases: A review of the literature. Int J Mol Sci (2019) 20(2):395. doi: 10.3390/ijms20020395
27. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72(3):558–77. doi: 10.1016/j.jhep.2019.10.003
28. Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity (2019) 50(2):432–45.e7. doi: 10.1016/j.immuni.2018.12.018
29. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mtor-S6k pathway. Mucosal Immunol (2015) 8(1):80–93. doi: 10.1038/mi.2014.44
30. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients (2011) 3(10):858–76. doi: 10.3390/nu3100858
31. Zhou D, Pan Q, Xin FZ, Zhang RN, He CX, Chen GY, et al. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J Gastroenterol (2017) 23(1):60–75. doi: 10.3748/wjg.v23.i1.60
32. Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of aldafermin, an engineered Fgf19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology (2021) 160(1):219–31.e1. doi: 10.1053/j.gastro.2020.08.004
33. Auguet T, Bertran Ramos L, Binetti J. Intestinal dysbiosis and non-alcoholic fatty liver disease. Beloborodova NV, Grechko AV , editors. Human Microbiome (London: IntechOpen) (2020). doi: 10.5772/intechopen.92972
34. Zhang X, Deng R. Dysregulation of bile acids in patients with nafld. In: Gad EH, editor. Nonalcoholic Fatty Liver Disease - An Update. (London: IntechOpen) (2018). doi: 10.5772/intechopen.81474
35. Aragonès G, González-García S, Aguilar C, Richart C, Auguet T. Gut microbiota-derived mediators as potential markers in nonalcoholic fatty liver disease. BioMed Res Int (2019) 2019:8507583. doi: 10.1155/2019/8507583
36. Gupta B, Rai R, Oertel M, Raeman R. Intestinal barrier dysfunction in fatty liver disease: Roles of microbiota, mucosal immune system, and bile acids. Semin Liver Dis (2022) 42(2):122–37. doi: 10.1055/s-0042-1748037
37. Wang C, Zhu C, Shao L, Ye J, Shen Y, Ren Y. Role of bile acids in dysbiosis and treatment of nonalcoholic fatty liver disease. Med Inflamm (2019) 2019:7659509. doi: 10.1155/2019/7659509
38. Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res (2012) 53(9):1723–37. doi: 10.1194/jlr.R024794
39. Yang Z-X, Shen W, Sun H. Effects of nuclear receptor fxr on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol Int (2010) 4(4):741–8. doi: 10.1007/s12072-010-9202-6
40. Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature (2020) 581(7809):475–9. doi: 10.1038/s41586-020-2193-0
41. Godlewska U, Bulanda E, Wypych TP. Bile acids in immunity: Bidirectional mediators between the host and the microbiota. Front Immunol (2022) 13:949033. doi: 10.3389/fimmu.2022.949033
42. Sakanaka T, Inoue T, Yorifuji N, Iguchi M, Fujiwara K, Narabayashi K, et al. The effects of a Tgr5 agonist and a dipeptidyl peptidase iv inhibitor on dextran sulfate sodium-induced colitis in mice. J Gastroenterol Hepatol (2015) 30(S1):60–5. doi: 10.1111/jgh.12740
43. Stojancevic M, Stankov K, Mikov M. The impact of farnesoid X receptor activation on intestinal permeability in inflammatory bowel disease. Can J Gastroenterol (2012) 26(9):631–7. doi: 10.1155/2012/538452
44. Mörbe UM, Jørgensen PB, Fenton TM, von Burg N, Riis LB, Spencer J, et al. Human gut-associated lymphoid tissues (Galt); diversity, structure, and function. Mucosal Immunol (2021) 14(4):793–802. doi: 10.1038/s41385-021-00389-4
45. Fenton TM, Jørgensen PB, Niss K, Rubin SJS, Mörbe UM, Riis LB, et al. Immune profiling of human gut-associated lymphoid tissue identifies a role for isolated lymphoid follicles in priming of region-specific immunity. Immunity (2020) 52(3):557–70.e6. doi: 10.1016/j.immuni.2020.02.001
46. Nati M, Chung KJ, Chavakis T. The role of innate immune cells in nonalcoholic fatty liver disease. J Innate Immun (2022) 14(1):31–41. doi: 10.1159/000518407
47. Gao B, Tsukamoto H. Inflammation in alcoholic and nonalcoholic fatty liver disease: Friend or foe? Gastroenterology (2016) 150(8):1704–9. doi: 10.1053/j.gastro.2016.01.025
48. Meli R, Mattace Raso G, Calignano A. Role of innate immune response in non-alcoholic fatty liver disease: Metabolic complications and therapeutic tools. Front Immunol (2014) 5:177. doi: 10.3389/fimmu.2014.00177
49. Groschwitz KR, Hogan SP. Intestinal barrier function: Molecular regulation and disease pathogenesis. J Allergy Clin Immunol (2009) 124(1):3–22. doi: 10.1016/j.jaci.2009.05.038
50. Munukka E, Rintala A, Toivonen R, Nylund M, Yang B, Takanen A, et al. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J (2017) 11(7):1667–79. doi: 10.1038/ismej.2017.24
51. Briskey D, Heritage M, Jaskowski LA, Peake J, Gobe G, Subramaniam VN, et al. Probiotics modify tight-junction proteins in an animal model of nonalcoholic fatty liver disease. Therap Adv Gastroenterol (2016) 9(4):463–72. doi: 10.1177/1756283x16645055
52. Nicoletti A, Ponziani FR, Biolato M, Valenza V, Marrone G, Sganga G, et al. Intestinal permeability in the pathogenesis of liver damage: From non-alcoholic fatty liver disease to liver transplantation. World J Gastroenterol (2019) 25(33):4814–34. doi: 10.3748/wjg.v25.i33.4814
53. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (2019) 394(10215):2184–96. doi: 10.1016/s0140-6736(19)33041-7
54. Elphick DA, Mahida YR. Paneth cells: Their role in innate immunity and inflammatory disease. Gut (2005) 54(12):1802–9. doi: 10.1136/gut.2005.068601
55. Lueschow SR, McElroy SJ. The paneth cell: The curator and defender of the immature small intestine. Front Immunol (2020) 11:587. doi: 10.3389/fimmu.2020.00587
56. Kolodziejczyk AA, Zheng D, Shibolet O, Elinav E. The role of the microbiome in nafld and Nash. EMBO Mol Med (2019) 11(2):e9302. doi: 10.15252/emmm.201809302
57. Fan J, Sun J, Li T, Yan X, Jiang Y. Nuciferine prevents hepatic steatosis associated with improving intestinal mucosal integrity, mucus-related microbiota and inhibiting Tlr4/Myd88/Nf-Kb pathway in high-fat induced rats. J Funct Foods (2022) 88:104859. doi: 10.1016/j.jff.2021.104859
58. Su D, Nie Y, Zhu A, Chen Z, Wu P, Zhang L, et al. Vitamin d signaling through induction of paneth cell defensins maintains gut microbiota and improves metabolic disorders and hepatic steatosis in animal models. Front Physiol (2016) 7:498. doi: 10.3389/fphys.2016.00498
59. Okubo H, Kushiyama A, Nakatsu Y, Yamamotoya T, Matsunaga Y, Fujishiro M, et al. Roles of gut-derived secretory factors in the pathogenesis of non-alcoholic fatty liver disease and their possible clinical applications. Int J Mol Sci (2018) 19(10):3064. doi: 10.3390/ijms19103064
60. Okubo H, Kushiyama A, Sakoda H, Nakatsu Y, Iizuka M, Taki N, et al. Involvement of resistin-like molecule B in the development of methionine-choline deficient diet-induced non-alcoholic steatohepatitis in mice. Sci Rep (2016) 6(1):20157. doi: 10.1038/srep20157
61. Fernandez-Cantos MV, Garcia-Morena D, Iannone V, El-Nezami H, Kolehmainen M, Kuipers OP. Role of microbiota and related metabolites in gastrointestinal tract barrier function in nafld. Tissue Barriers (2021) 9(3):1879719. doi: 10.1080/21688370.2021.1879719
62. Zhang S, Tun HM, Zhang D, Chau H-T, Huang F-Y, Kwok H, et al. Alleviation of hepatic steatosis: Dithizone-related gut microbiome restoration during paneth cell dysfunction. Front Microbiol (2022) 13:813783. doi: 10.3389/fmicb.2022.813783
63. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat Rev Immunol (2010) 10(2):131–44. doi: 10.1038/nri2707
64. Chandrashekaran V, Seth RK, Dattaroy D, Alhasson F, Ziolenka J, Carson J, et al. Hmgb1-rage pathway drives peroxynitrite signaling-induced ibd-like inflammation in murine nonalcoholic fatty liver disease. Redox Biol (2017) 13:8–19. doi: 10.1016/j.redox.2017.05.005
65. Ciesielska A, Matyjek M, Kwiatkowska K. Tlr4 and Cd14 trafficking and its influence on lps-induced pro-inflammatory signaling. Cell Mol Life Sci (2021) 78(4):1233–61. doi: 10.1007/s00018-020-03656-y
66. Kamdar K, Nguyen V, DePaolo RW. Toll-like receptor signaling and regulation of intestinal immunity. Virulence (2013) 4(3):207–12. doi: 10.4161/viru.23354
67. Medina-Contreras O, Geem D, Laur O, Williams IR, Lira SA, Nusrat A, et al. Cx3cr1 regulates intestinal macrophage homeostasis, bacterial translocation, and colitogenic Th17 responses in mice. J Clin Invest (2011) 121(12):4787–95. doi: 10.1172/JCI59150
68. Schneider KM, Bieghs V, Heymann F, Hu W, Dreymueller D, Liao L, et al. Cx3cr1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology (2015) 62(5):1405–16. doi: 10.1002/hep.27982
69. Tailleux A, Wouters K, Staels B. Roles of ppars in nafld: Potential therapeutic targets. Biochim Biophys Acta (2012) 1821(5):809–18. doi: 10.1016/j.bbalip.2011.10.016
70. Cave MC, Clair HB, Hardesty JE, Falkner KC, Feng W, Clark BJ, et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim Biophys Acta (2016) 1859(9):1083–99. doi: 10.1016/j.bbagrm.2016.03.002
71. Daynes RA, Jones DC. Emerging roles of ppars in inflammation and immunity. Nat Rev Immunol (2002) 2(10):748–59. doi: 10.1038/nri912
72. Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr Rev (1999) 20(5):649–88. doi: 10.1210/edrv.20.5.0380
73. Yoon M. The role of pparalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on pparalpha actions. Pharmacol Res (2009) 60(3):151–9. doi: 10.1016/j.phrs.2009.02.004
74. Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, et al. Pgc-1 promotes insulin resistance in liver through ppar-Alpha-Dependent induction of trb-3. Nat Med (2004) 10(5):530–4. doi: 10.1038/nm1044
75. Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of pparalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology (2003) 38(1):123–32. doi: 10.1053/jhep.2003.50307
76. Vanden Berghe W, Vermeulen L, Delerive P, De Bosscher K, Staels B, Haegeman G. A paradigm for gene regulation: Inflammation, nf-kappab and ppar. Adv Exp Med Biol (2003) 544:181–96. doi: 10.1007/978-1-4419-9072-3_22
77. Delerive P, Gervois P, Fruchart JC, Staels B. Induction of ikappabalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem (2000) 275(47):36703–7. doi: 10.1074/jbc.M004045200
78. Monroy-Ramirez HC, Galicia-Moreno M, Sandoval-Rodriguez A, Meza-Rios A, Santos A, Armendariz-Borunda J. Ppars as metabolic sensors and therapeutic targets in liver diseases. Int J Mol Sci (2021) 22(15):8298. doi: 10.3390/ijms22158298
79. Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci U.S.A. (2003) 100(26):15924–9. doi: 10.1073/pnas.0306981100
80. Decara J, Rivera P, López-Gambero AJ, Serrano A, Pavón FJ, Baixeras E, et al. Peroxisome proliferator-activated receptors: Experimental targeting for the treatment of inflammatory bowel diseases. Front Pharmacol (2020) 11:730. doi: 10.3389/fphar.2020.00730
81. Wu H, Li X, Shen C. Peroxisome proliferator-activated receptor gamma in white and brown adipocyte regulation and differentiation. Physiol Res (2020) 69(5):759–73. doi: 10.33549/physiolres.934411
82. Abdalla HB, Napimoga MH, Lopes AH, de Macedo Maganin AG, Cunha TM, Van Dyke TE, et al. Activation of ppar-Γ induces macrophage polarization and reduces neutrophil migration mediated by heme oxygenase 1. Int Immunopharmacol (2020) 84:106565. doi: 10.1016/j.intimp.2020.106565
83. Ye B, van Langenberg DR. Mesalazine preparations for the treatment of ulcerative colitis: Are all created equal? World J Gastrointest Pharmacol Ther (2015) 6(4):137–44. doi: 10.4292/wjgpt.v6.i4.137
84. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117(1):175–84. doi: 10.1172/JCI29881
85. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of ppar-Γ activity on kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep (2017) 7(1):44612. doi: 10.1038/srep44612
86. Choy MC, Visvanathan K, De Cruz P. An overview of the innate and adaptive immune system in inflammatory bowel disease. Inflamm Bowel Dis (2017) 23(1):2–13. doi: 10.1097/mib.0000000000000955
87. Kelley N, Jeltema D, Duan Y, He Y. The Nlrp3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
88. DiToro D, Basu R. Emerging complexity in Cd4(+)T lineage programming and its implications in colorectal cancer. Front Immunol (2021) 12:694833. doi: 10.3389/fimmu.2021.694833
89. Mittrücker H-W, Visekruna A, Huber M. Heterogeneity in the differentiation and function of Cd8+ T cells. Archivum Immunologiae Therapiae Experimentalis (2014) 62(6):449–58. doi: 10.1007/s00005-014-0293-y
90. Henning JR, Graffeo CS, Rehman A, Fallon NC, Zambirinis CP, Ochi A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatol (Baltimore Md) (2013) 58(2):589–602. doi: 10.1002/hep.26267
91. Fang JH, Yu W, Zhou G, Shi JP, Yang WJ, Shen XY, et al. [Study on the correlation between small intestinal dendritic cells and non-alcoholic fatty liver disease in mice]. Zhonghua Gan Zang Bing Za Zhi (2019) 27(9):698–703. doi: 10.3760/cma.j.issn.1007-3418.2019.09.008
92. Tsujimoto T, Kawaratani H, Kitazawa T, Uemura M, Fukui H. Innate immune reactivity of the ileum–liver axis in nonalcoholic steatohepatitis. Digestive Dis Sci (2012) 57(5):1144–51. doi: 10.1007/s10620-012-2073-z
93. Imam T, Park S, Kaplan MH, Olson MR. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol (2018) 9:1212. doi: 10.3389/fimmu.2018.01212
94. Weigmann B, Neurath MF. T-Bet and mucosal Th1 responses in the gastrointestinal tract. Gut (2002) 51(3):301–3. doi: 10.1136/gut.51.3.301
95. Bamias G, Cominelli F. Role of type 2 immunity in intestinal inflammation. Curr Opin Gastroenterol (2015) 31(6):471–6. doi: 10.1097/MOG.0000000000000212
96. Mucida D, Salek-Ardakani S. Regulation of Th17 cells in the mucosal surfaces. J Allergy Clin Immunol (2009) 123(5):997–1003. doi: 10.1016/j.jaci.2009.03.016
97. Koo S-Y, Park E-J, Lee C-W. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp Mol Med (2020) 52(8):1209–19. doi: 10.1038/s12276-020-0480-3
98. Taniki N, Nakamoto N, Chu PS, Ichikawa M, Teratani T, Kanai T. Th17 cells in the liver: Balancing autoimmunity and pathogen defense. Semin Immunopathol (2022) 503–26. doi: 10.1007/s00281-022-00917-9
99. Jia L, Wu C. The biology and functions of Th22 cells. Adv Exp Med Biol (2014) 841:209–30. doi: 10.1007/978-94-017-9487-9_8
100. Jiang Q, Yang G, Xiao F, Xie J, Wang S, Lu L, et al. Role of Th22 cells in the pathogenesis of autoimmune diseases. Front Immunol (2021) 12:688066. doi: 10.3389/fimmu.2021.688066
101. Vyas SP, Goswami R. A decade of Th9 cells: Role of Th9 cells in inflammatory bowel disease. Front Immunol (2018) 9:1139. doi: 10.3389/fimmu.2018.01139
102. Yadav M, Bluestone J, Stephan S. Peripherally induced tregs – role in immune homeostasis and autoimmunity. Front Immunol (2013) 4:232. doi: 10.3389/fimmu.2013.00232
103. van Wijk F, Cheroutre H. Mucosal T cells in gut homeostasis and inflammation. Expert Rev Clin Immunol (2010) 6(4):559–66. doi: 10.1586/eci.10.34
104. Wang L, Zhu L, Qin S. Gut microbiota modulation on intestinal mucosal adaptive immunity. J Immunol Res (2019) 2019:4735040–. doi: 10.1155/2019/4735040
105. Su L, Wang JH, Cong X, Wang LH, Liu F, Xie XW, et al. Intestinal immune barrier integrity in rats with nonalcoholic hepatic steatosis and steatohepatitis. Chin Med J (Engl) (2012) 125(2):306–11. doi: 10.3760/cma.j.issn.0366-6999.2012.02.026
106. Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab (2015) 21(4):527–42. doi: 10.1016/j.cmet.2015.03.001
107. Su L, Wu Z, Chi Y, Song Y, Xu J, Tan J, et al. Mesenteric lymph node Cd4+ T lymphocytes migrate to liver and contribute to non-alcoholic fatty liver disease. Cell Immunol (2019) 337:33–41. doi: 10.1016/j.cellimm.2019.01.005
108. Jiang W, Wu N, Wang X, Chi Y, Zhang Y, Qiu X, et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci Rep (2015) 5:8096–. doi: 10.1038/srep08096
109. Wang Y, Zhang C. The roles of liver-resident lymphocytes in liver diseases. Front Immunol (2019) 10:1582. doi: 10.3389/fimmu.2019.01582
110. Hong CP, Park A, Yang BG, Yun CH, Kwak MJ, Lee GW, et al. Gut-specific delivery of T-helper 17 cells reduces obesity and insulin resistance in mice. Gastroenterology (2017) 152(8):1998–2010. doi: 10.1053/j.gastro.2017.02.016
111. Gao W, Wang Y, Bi J, Chen X, Li N, Wang Y, et al. Impaired Ccr9/Ccl25 signalling induced by inefficient dendritic cells contributes to intestinal immune imbalance in nonalcoholic steatohepatitis. Biochem Biophys Res Commun (2021) 534:34–40. doi: 10.1016/j.bbrc.2020.12.007
112. Suzuki T, Hayman L, Kilbey A, Edwards J, Coffelt SB. Gut Γδ T cells as guardians, disruptors, and instigators of cancer. Immunol Rev (2020) 298(1):198–217. doi: 10.1111/imr.12916
113. Catalan-Serra I, Sandvik AK, Bruland T, Andreu-Ballester JC. Gammadelta T cells in crohn’s disease: A new player in the disease pathogenesis? J Crohn’s Colitis (2017) 11(9):1135–45. doi: 10.1093/ecco-jcc/jjx039
114. Sojka DK, Huang Y-H, Fowell DJ. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology (2008) 124(1):13–22. doi: 10.1111/j.1365-2567.2008.02813.x
115. Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nat Rev Immunol (2016) 16(5):295–309. doi: 10.1038/nri.2016.36
116. Ilan Y, Shailubhai K, Sanyal A. Immunotherapy with oral administration of humanized anti-Cd3 monoclonal antibody: A novel gut-immune system-based therapy for metaflammation and Nash. Clin Exp Immunol (2018) 193(3):275–83. doi: 10.1111/cei.13159
117. Kuhn C, Weiner HL. Therapeutic anti-Cd3 monoclonal antibodies: From bench to bedside. Immunotherapy (2016) 8(8):889–906. doi: 10.2217/imt-2016-0049
118. Matsumoto K, Ichimura M, Tsuneyama K, Moritoki Y, Tsunashima H, Omagari K, et al. Fructo-oligosaccharides and intestinal barrier function in a methionine-Choline-Deficient mouse model of nonalcoholic steatohepatitis. PloS One (2017) 12(6):e0175406–e. doi: 10.1371/journal.pone.0175406
119. Li S, Wu W-C, He C-Y, Han Z, Jin D-Y, Wang L. Change of intestinal mucosa barrier function in the progress of non-alcoholic steatohepatitis in rats. World J Gastroenterol (2008) 14(20):3254–8. doi: 10.3748/wjg.14.3254
120. Inamine T, Schnabl B. Immunoglobulin a and liver diseases. J Gastroenterol (2018) 53(6):691–700. doi: 10.1007/s00535-017-1400-8
121. Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol (2006) 6(3):244–51. doi: 10.1038/nri1784
122. de Chaisemartin L. Lymphocyte homing and trafficking. In: Parnham MJ, editor. Compendium of inflammatory diseases. Basel: Springer Basel (2016). p. 857–64.
123. Dzutsev A, Hogg A, Sui Y, Solaymani-Mohammadi S, Yu H, Frey B, et al. Differential T cell homing to colon vs. small intestine is imprinted by local Cd11c+ apcs that determine homing receptors. J Leukocyte Biol (2017) 102(6):1381–8. doi: 10.1189/jlb.1A1116-463RR
124. Grant AJ, Lalor PF, Salmi M, Jalkanen S, Adams DH. Homing of mucosal lymphocytes to the liver in the pathogenesis of hepatic complications of inflammatory bowel disease. Lancet (2002) 359(9301):150–7. doi: 10.1016/s0140-6736(02)07374-9
125. Eksteen B, Grant AJ, Miles A, Curbishley SM, Lalor PF, Hübscher SG, et al. Hepatic endothelial Ccl25 mediates the recruitment of Ccr9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med (2004) 200(11):1511–7. doi: 10.1084/jem.20041035
126. Hu Y, Zhang H, Li J, Cong X, Chen Y, He G., et al Gut-derived lymphocyte recruitment to liver and induce liver injury in non-alcoholic fatty liver disease mouse model. J Gastroenterol Hepatol (2016) 31(3):676–84. doi: 10.1111/jgh.13183
127. Seike T, Mizukoshi E, Kaneko S. Role of Cd4<Sup>+ </Sup>T-cells in the pathology of non-alcoholic fatty liver disease and related diseases. Hepatoma Res (2021) 7:46. doi: 10.20517/2394-5079.2021.46
128. Morikawa R, Nakamoto N, Amiya T, Chu PS, Koda Y, Teratani T, et al. Role of cc chemokine receptor 9 in the progression of murine and human non-alcoholic steatohepatitis. J Hepatol (2021) 74(3):511–21. doi: 10.1016/j.jhep.2020.09.033
129. Hu Y, Zhang H, Li J, Cong X, Chen Y, He G, et al. Gut-derived lymphocyte recruitment to liver and induce liver injury in non-alcoholic fatty liver disease mouse model. J Gastroenterol Hepatol (2016) 31(3):676–84. doi: 10.1111/jgh.13183
130. Drescher HK, Schippers A, Clahsen T, Sahin H, Noels H, Hornef M, et al. B (7)-integrin and madcam-1 play opposing roles during the development of non-alcoholic steatohepatitis. J Hepatol (2017) 66(6):1251–64. doi: 10.1016/j.jhep.2017.02.001
131. Rai RP, Liu Y, Iyer SS, Liu S, Gupta B, Desai C, et al. Blocking integrin# ± 4²7-mediated Cd4 T cell recruitment to the intestine and liver protects mice from Western diet-induced non-alcoholic steatohepatitis. J Hepatol (2020):1013–22. doi: 10.1016/j.jhep.2020.05.047
132. Luci C, Bourinet M, Leclère PS, Anty R, Gual P. Chronic inflammation in non-alcoholic steatohepatitis: Molecular mechanisms and therapeutic strategies. Front Endocrinol (2020) 11:597648. doi: 10.3389/fendo.2020.597648
133. Sutti S, Albano E. Adaptive immunity: An emerging player in the progression of nafld. Nat Rev Gastroenterol Hepatol (2020) 17(2):81–92. doi: 10.1038/s41575-019-0210-2
134. Miura K, Seki E, Ohnishi H, Brenner DA. Role of toll-like receptors and their downstream molecules in the development of nonalcoholic fatty liver disease. Gastroenterol Res Pract (2010) 2010:362847. doi: 10.1155/2010/362847
135. Knolle P, Schlaak J, Uhrig A, Kempf P, Meyer zum Büschenfelde KH, Gerken G. Human kupffer cells secrete il-10 in response to lipopolysaccharide (Lps) challenge. J Hepatol (1995) 22(2):226–9. doi: 10.1016/0168-8278(95)80433-1
136. Chen L, Zhang Q, Yu C, Wang F, Kong X. Functional roles of Ccl5/Rantes in liver disease. Liver Res (2020) 4(1):28–34. doi: 10.1016/j.livres.2020.01.002
137. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3(2):785–97. doi: 10.1002/cphy.c120026
138. Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to Cd14 and enhances lipopolysaccharide-mediated tnf-alpha production in human monocytes. J Immunol (2008) 180(7):5067–74. doi: 10.4049/jimmunol.180.7.5067
139. Miura K, Ohnishi H. Role of gut microbiota and toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol (2014) 20(23):7381–91. doi: 10.3748/wjg.v20.i23.7381
140. Kirovski G, Gäbele E, Dorn C, Moleda L, Niessen C, Weiss TS, et al. Hepatic steatosis causes induction of the chemokine rantes in the absence of significant hepatic inflammation. Int J Clin Exp Pathol (2010) 3(7):675–80.
141. Berres M-L, Koenen RR, Rueland A, Zaldivar MM, Heinrichs D, Sahin H, et al. Antagonism of the chemokine Ccl5 ameliorates experimental liver fibrosis in mice. J Clin Invest (2010) 120(11):4129–40. doi: 10.1172/JCI41732
142. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine Ccl2 (Mcp-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61(3):416–26. doi: 10.1136/gutjnl-2011-300304
143. Huang W, Metlakunta A, Dedousis N, Zhang P, Sipula I, Dube JJ, et al. Depletion of liver kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes (2010) 59(2):347–57. doi: 10.2337/db09-0016
144. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol (2016) 13(2):88–110. doi: 10.1038/nrgastro.2015.200
145. Giraud J, Chalopin D, Blanc J-F, Saleh M. Hepatocellular carcinoma immune landscape and the potential of immunotherapies. Front Immunol (2021) 12:655697. doi: 10.3389/fimmu.2021.655697
146. Singh AK, Tripathi P, Cardell SL. Type ii nkt cells: An elusive population with immunoregulatory properties. Front Immunol (2018) 9:1969. doi: 10.3389/fimmu.2018.01969
147. Martin-Murphy BV, You Q, Wang H, de la Houssaye BA, Reilly TP, Friedman JE, et al. Mice lacking natural killer T cells are more susceptible to metabolic alterations following high fat diet feeding. PloS One (2014) 9(1):e80949–e. doi: 10.1371/journal.pone.0080949
148. O’Rourke RW, Meyer KA, Neeley CK, Gaston GD, Sekhri P, Szumowski M, et al. Systemic nk cell ablation attenuates intra-abdominal adipose tissue macrophage infiltration in murine obesity. Obes (Silver Spring) (2014) 22(10):2109–14. doi: 10.1002/oby.20823
149. Hwang S, Yun H, Moon S, Cho YE, Gao B. Role of neutrophils in the pathogenesis of nonalcoholic steatohepatitis. Front Endocrinol (2021) 12:751802. doi: 10.3389/fendo.2021.751802
150. Rensen SS, Bieghs V, Xanthoulea S, Arfianti E, Bakker JA, Shiri-Sverdlov R, et al. Neutrophil-derived myeloperoxidase aggravates non-alcoholic steatohepatitis in low-density lipoprotein receptor-deficient mice. PloS One (2012) 7(12):e52411–e. doi: 10.1371/journal.pone.0052411
151. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18(9):1407–12. doi: 10.1038/nm.2885
152. Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol (2009) 175(4):1473–82. doi: 10.2353/ajpath.2009.080999
153. Jarido V, Kennedy L, Hargrove L, Demieville J, Thomson J, Stephenson K, et al. The emerging role of mast cells in liver disease. Am J Physiology-Gastrointestinal Liver Physiol (2017) 313(2):G89–G101. doi: 10.1152/ajpgi.00333.2016
154. Lombardo J, Broadwater D, Collins R, Cebe K, Brady R, Harrison S. Hepatic mast cell concentration directly correlates to stage of fibrosis in Nash. Hum Pathol (2019) 86:129–35. doi: 10.1016/j.humpath.2018.11.029
155. Lau AH, Thomson AW. Dendritic cells and immune regulation in the liver. Gut (2003) 52(2):307. doi: 10.1136/gut.52.2.307
156. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology (2008) 48(1):322–35. doi: 10.1002/hep.22306
157. Hirsova P, Bamidele AO, Wang H, Povero D, Revelo XS. Emerging roles of T cells in the pathogenesis of nonalcoholic steatohepatitis and hepatocellular carcinoma. Front Endocrinol (2021) 12:760860. doi: 10.3389/fendo.2021.760860
158. Heier EC, Meier A, Julich-Haertel H, Djudjaj S, Rau M, Tschernig T, et al. Murine Cd103(+) dendritic cells protect against steatosis progression towards steatohepatitis. J Hepatol (2017) 66(6):1241–50. doi: 10.1016/j.jhep.2017.01.008
159. Deczkowska A, David E, Ramadori P, Pfister D, Safran M, Li B, et al. Xcr1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med (2021) 27(6):1043–54. doi: 10.1038/s41591-021-01344-3
160. Zhou QH, Wu FT, Pang LT, Zhang TB, Chen Z. Role of Γδt cells in liver diseases and its relationship with intestinal microbiota. World J Gastroenterol (2020) 26(20):2559–69. doi: 10.3748/wjg.v26.i20.2559
161. Gebru YA, Gupta H, Kim HS, Eom JA, Kwon GH, Park E, et al. T Cell subsets and natural killer cells in the pathogenesis of nonalcoholic fatty liver disease. Int J Mol Sci (2021) 22(22):12190. doi: 10.3390/ijms222212190
162. Barrow F, Khan S, Fredrickson G, Wang H, Dietsche K, Parthiban P, et al. Microbiota-driven activation of intrahepatic b cells aggravates Nash through innate and adaptive signaling. Hepatology (2021) 74(2):704–22. doi: 10.1002/hep.31755
163. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology (2015) 149(2):367–78.e5. doi: 10.1053/j.gastro.2015.04.005
164. Lange NF, Graf V, Caussy C, Dufour J-F. Ppar-targeted therapies in the treatment of non-alcoholic fatty liver disease in diabetic patients. Int J Mol Sci (2022) 23(8):4305. doi: 10.3390/ijms23084305
165. Luo Q, Wei R, Cai Y, Zhao Q, Liu Y, Liu WJ. Efficacy of off-label therapy for non-alcoholic fatty liver disease in improving non-invasive and invasive biomarkers: A systematic review and network meta-analysis of randomized controlled trials. Front Med (2022) 9:793203. doi: 10.3389/fmed.2022.793203
166. Perumpail BJ, Li AA, John N, Sallam S, Shah ND, Kwong W, et al. The role of vitamin e in the treatment of nafld. Diseases (2018) 6(4):86. doi: 10.3390/diseases6040086
167. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med (2021) 384(12):1113–24. doi: 10.1056/NEJMoa2028395
168. Rezaei S, Tabrizi R, Nowrouzi-Sohrabi P, Jalali M, Atkin SL, Al-Rasadi K, et al. Glp-1 receptor agonist effects on lipid and liver profiles in patients with nonalcoholic fatty liver disease: Systematic review and meta-analysis. Can J Gastroenterol Hepatol (2021) 2021:8936865–. doi: 10.1155/2021/8936865
169. Fuenzalida C, Dufeu MS, Poniachik J, Roblero JP, Valenzuela-Pérez L, Beltrán CJ. Probiotics-based treatment as an integral approach for alcohol use disorder in alcoholic liver disease. Front Pharmacol (2021) 12:729950. doi: 10.3389/fphar.2021.729950
170. Yang R, Shang J, Zhou Y, Liu W, Tian Y, Shang H. Effects of probiotics on nonalcoholic fatty liver disease: A systematic review and meta-analysis. Expert Rev Gastroenterol Hepatol (2021) 15(12):1401–9. doi: 10.1080/17474124.2022.2016391
171. Yao M, Qv L, Lu Y, Wang B, Berglund B, Li L. An update on the efficacy and functionality of probiotics for the treatment of non-alcoholic fatty liver disease. Engineering (2021) 7(5):679–86. doi: 10.1016/j.eng.2020.01.017
172. Matsubara T, Li F, Gonzalez FJ. Fxr signaling in the enterohepatic system. Mol Cell Endocrinol (2013) 368(1-2):17–29. doi: 10.1016/j.mce.2012.05.004
173. Sawangjit R, Chongmelaxme B, Phisalprapa P, Saokaew S, Thakkinstian A, Kowdley KV, et al. Comparative efficacy of interventions on nonalcoholic fatty liver disease (Nafld): A prisma-compliant systematic review and network meta-analysis. Med (Baltimore) (2016) 95(32):e4529. doi: 10.1097/md.0000000000004529
174. Xi Y, Li H. Role of farnesoid X receptor in hepatic steatosis in nonalcoholic fatty liver disease. Biomed Pharmacother (2020) 121:109609. doi: 10.1016/j.biopha.2019.109609
175. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (Flint): A multicentre, randomised, placebo-controlled trial. Lancet (2015) 385(9972):956–65. doi: 10.1016/s0140-6736(14)61933-4
176. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47(4):571–9. doi: 10.1016/j.jhep.2007.04.019
177. Vonghia L, Van Herck MA, Weyler J, Francque S. Targeting myeloid-derived cells: New frontiers in the treatment of non-alcoholic and alcoholic liver disease. Front Immunol (2019) 10:563. doi: 10.3389/fimmu.2019.00563
178. Sumida Y, Yoneda M. Current and future pharmacological therapies for Nafld/Nash. J Gastroenterol (2018) 53(3):362–76. doi: 10.1007/s00535-017-1415-1
179. Diehl AM, Harrison S, Caldwell S, Rinella M, Paredes A, Moylan C, et al. Jkb-121 in patients with nonalcoholic steatohepatitis: A phase 2 double blind randomized placebo control study. J Hepatol (2018) 68:103. doi: 10.1016/S0168-8278(18)30425-2
180. Fraile JM, Palliyil S, Barelle C, Porter AJ, Kovaleva M. Non-alcoholic steatohepatitis (Nash) - a review of a crowded clinical landscape, driven by a complex disease. Drug Des Devel Ther (2021) 15:3997–4009. doi: 10.2147/DDDT.S315724
181. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The Ask1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology (2018) 67(2):549–59. doi: 10.1002/hep.29514
182. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-a and -Δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology (2016) 150(5):1147–59.e5. doi: 10.1053/j.gastro.2016.01.038
183. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-ppar agonist lanifibranor in Nash. New Engl J Med (2021) 385(17):1547–58. doi: 10.1056/NEJMoa2036205
184. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology (2018) 67(5):1754–67. doi: 10.1002/hep.29477
Keywords: non-alcoholic fatty liver disease (NAFLD), microbiota-gut-liver axis, low-grade inflammation, liver lymphocyte homing, steatohepatitis (NASH), microbiota, liver diseases, liver fibrosis
Citation: Ortiz-López N, Fuenzalida C, Dufeu MS, Pinto-León A, Escobar A, Poniachik J, Roblero JP, Valenzuela-Pérez L and Beltrán CJ (2022) The immune response as a therapeutic target in non-alcoholic fatty liver disease. Front. Immunol. 13:954869. doi: 10.3389/fimmu.2022.954869
Received: 27 May 2022; Accepted: 21 September 2022;
Published: 10 October 2022.
Edited by:
Robert-J Brummer, Örebro University, SwedenReviewed by:
Houkai Li, Shanghai University of Traditional Chinese Medicine, ChinaCopyright © 2022 Ortiz-López, Fuenzalida, Dufeu, Pinto-León, Escobar, Poniachik, Roblero, Valenzuela-Pérez and Beltrán. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroll J. Beltrán, Y2Fyb2xsYmVsdHJhbm1AdWNoaWxlLmNs; Y2Fyb2xsYmVsdHJhbm1AZ21haWwuY29t; Lucía Valenzuela-Pérez, bHVjaWEudmFsZW56dWVsYUB1Y2hpbGUuY2w=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.