 Cesar Barrabi
Cesar Barrabi Kezhong Zhang
Kezhong Zhang Ming Liu
Ming Liu Xuequn Chen
Xuequn Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 21 April 2023
Sec. Cellular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1155779
This article is part of the Research Topic Endoplasmic Reticulum Homeostasis and Metabolic Diseases View all 7 articles
In the secretory pathway of the pancreatic beta cell, proinsulin and other secretory granule proteins are first produced in the endoplasmic reticulum (ER). Beta cell ER homeostasis is vital for normal beta cell functions and is maintained by the delicate balance between protein synthesis, folding, export and degradation. Disruption of ER homeostasis leads to beta cell death and diabetes. Among the four components to maintain ER homeostasis, the role of ER export in insulin biogenesis or beta cell survival was not well-understood. COPII (coat protein complex II) dependent transport is a conserved mechanism for most cargo proteins to exit ER and transport to Golgi apparatus. Emerging evidence began to reveal a critical role of COPII-dependent ER export in beta cells. In this review, we will first discuss the basic components of the COPII transport machinery, the regulation of cargo entry and COPII coat assembly in mammalian cells, and the general concept of receptor-mediated cargo sorting in COPII vesicles. On the basis of these general discussions, the current knowledge and recent developments specific to the beta cell COPII dependent ER export are summarized under normal and diabetic conditions.
The pancreatic beta cell is the only source to synthesize and secrete insulin. In the secretory pathway of the beta cells, proinsulin and other secretory as well as membrane proteins are first produced in the endoplasmic reticulum (ER), exported to the Golgi, and then packaged into insulin secretory granules (ISG) (1, 2). Beta cell ER possesses a highly active protein synthetic, folding, and export machinery to accommodate the massive production of proinsulin and other ISG proteins. ER homeostasis is vital for normal beta cell functions and is maintained by the delicate balance between protein synthesis, folding, export and degradation (1). Disruption of ER homeostasis by genetic and environmental diabetes-causing factors leads to beta cell dysfunction or death and subsequent diabetes (3, 4). Among the four components to maintain ER homeostasis, the role of ER export in insulin biogenesis and beta cell survival was not well-understood. From yeast to mammals, COPII (coat protein complex II) dependent transport is a conserved mechanism for most cargo proteins to exit ER and transport to Golgi apparatus (5–10). Emerging evidence revealed a critical role of COPII-dependent ER export in insulin biogenesis and beta cell survival (11–13). In this review, we will discuss the basic components of the COPII transport machinery, the regulations of cargo entry and COPII coat assembly, and the current knowledge of the COPII dependent ER to Golgi transport in beta cells under healthy and diabetic conditions.
As shown in Figure 1A, according to the conventional model, the five COPII coat proteins, SAR1, SEC23, SEC24, SEC13 and SEC31, form a cage-like structure to sort cargo and bud off membrane vesicles, COPII vesicles, on specialized ribosome-free domains of the ER, known as ER exit sites (ERES) (6, 7, 14). Among them, the small GTPase, SAR1, initiates the coat assembly through GDP to GTP exchange mediated by its guanine nucleotide exchange factor SEC12. Upon activation, GTP bound SAR1 recruits SEC23.SEC24 heterodimer to form the inner coat and subsequently recruit SEC13.SEC31 heterotetramer to form the outer coat. Mammalian cells possess multiple isoforms of the COPII coat proteins, including SAR1A, SAR1B, SEC23A, SEC23B, SEC24A-D, SEC13, SEC31A, and SEC31B (6). Among the coat proteins, SEC24s directly interact and sort the cargo proteins into the COPII vesicles (15). Specific amino acid sequences are present in the cargo proteins which are recognized by the COPII coat. These sequences are called ER exit (or export) signals (or motifs) (16–18). The SEC24 isoforms have been shown to sort cargoes in an isoform- and tissue-specific manner (18–20). In addition to the five core coat proteins, other COPII associated proteins, such as SEC16, TANGO1/MIA3, and CTAGE5, also play essential roles in organizing the ERES and accommodating specific cargoes of large sizes (21–25). Several recent studies suggested that in addition to forming coated vesicles, COPII proteins can be recruited to membranes defining the boundary between the ER and ERES where they guide the cargo entry and extension of transport tubules without coating the ER export carriers (26, 27). While the basic machinery (5, 28) and structure of the COPII coat (29–32) have been well-understood, much remains to be learned regarding how COPII dependent cargo sorting and transport are regulated in a tissue-specific manner and how this regulated process may be disrupted in human diseases.
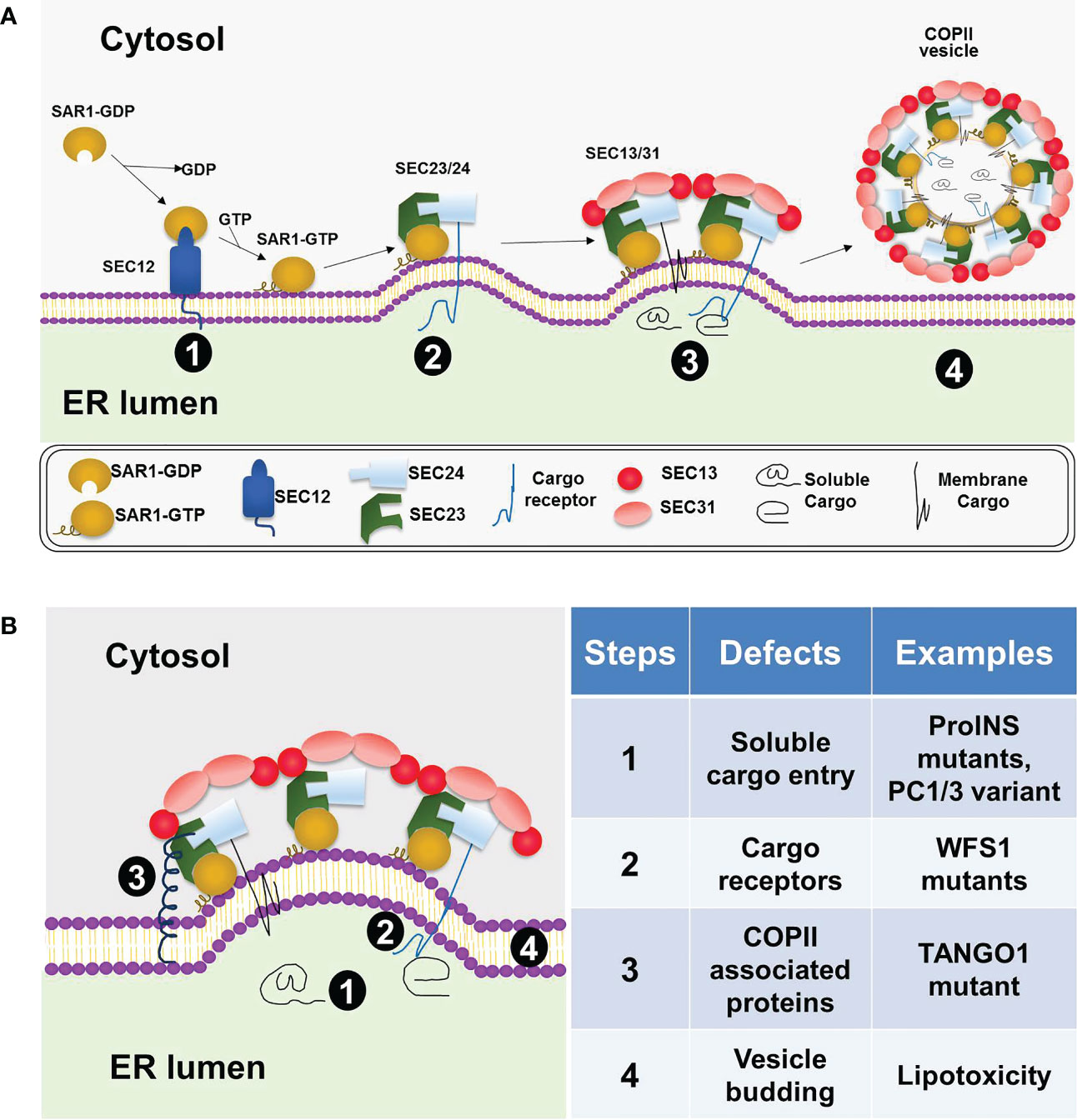
Figure 1 COPII coat assembly, cargo sorting, and vesicle formation at the ER. (A) A general model for COPII dependent ER exit in mammalian cells. COPII coat assembly on the ER membrane is initiated by SAR1 activation via SEC12 (stage 1), followed by the recruitment of the SEC23/SEC24 heterodimer and formation of the prebudding complex (stage 2). Next, the outer coat complex, SEC13/SEC31 heterotetramer, is recruited to bind the prebudding complex (stage 3). This step leads to further membrane deformation and vesicle formation. The vesicles coated with COPII proteins subsequently bud off the ER membrane (stage 4). (B) Defective ER export in beta cells under various diabetic conditions. Four different steps in the COPII-dependent ER exit process are highlighted on the left diagram and summarized with specific examples on the right table.
According to current knowledge, COPII dependent cargo export is a highly regulated process (14, 33, 34). First, cargo entry into COPII vesicles can be regulated, primarily through allowing or inhibiting the interaction between cargo and coat component, SEC24. Such examples include SREBPs, a family of ER bound transcription factors whose COPII dependent ER-Golgi translocation and activation is regulated by cellular cholesterol levels through masking the ER exit signal (35, 36). Another example is ATF6, an ER stress sensor that translocates from ER to Golgi via COPII vesicles when activated under ER stress conditions (37, 38). CRTC2, a mediator of mTOR signaling activated by insulin, was shown to competes with SEC23A to interact with SEC31A, thus disrupting SREBP1 transport (39). It was also shown that the ER/lipid droplet-associated protein Cideb selectively promoted the loading of SREBP/SCAP into COPII vesicles (40). These examples support the promise to design drugs targeting the cargo specific COPII isoforms or the interaction interfaces between specific cargo and COPII coat. The second aspect of regulation is at the COPII coat assembly on ERES. This process can be modulated by various factors such as cargo load (41–43), metabolic conditions (44, 45), or membrane lipids (46, 47). This level of regulation can be achieved through multiple mechanisms targeting different steps of coat complex assembly. For example, such regulation can be achieved at the gene expression level. It has been shown that COPII proteins were targets of unfolded protein response (UPR), and the IRE1α-mediated UPR branch regulated the expression of genes involved in ER-Golgi transport (41, 45, 48). The COPII-dependent ER export can also be regulated by post-translational modifications of the COPII coat and related proteins (33, 34). Examples of such modifications include the ubiquitylation of SEC31 (49, 50), phosphorylation of SEC24 (51), SEC16 (52), SEC12 (53), and O-GlcNAcylation of multiple COPII components (54, 55). In mammalian cells, in response to nutrient starvation, COPII proteins can be redirected to the autophagosome formation through phosphorylating SEC23B subunit (56). Interestingly, COPII-dependent ER protein transport can also be regulated by the circadian clock (57). The ER-bound stress sensor CREBH (CAMP-Responsive Element-Binding Protein, Hepatic-Specific) transits from the ER to Golgi via COPII vesicle for its activation process in response to hepatic stress, nutrient availability, or circadian cues (57–59). Under the physiological condition, the interaction between CREBH and SEC23/SEC24 and the subsequent proteolytic activation of CREBH exhibited typical circadian rhythmicity. This circadian-regulated process was controlled by the core clock oscillator BMAL1 and AKT/glycogen synthase kinase 3β (GSK3β) signaling cascade, in which the GSK3β-mediated phosphorylation of CREBH modulated the association between CREBH and SEC23/SEC24 (57). In addition to transcriptional and post-translational regulations described above, modulation of SAR1 GTPase activity has also been shown as another mechanism to regulate the COPII-dependent ER export (60, 61).
Soluble secretory proteins locate in the ER lumen and therefore cannot interact with cytoplasm-sided COPII coat proteins directly. These protein cargos enter the COPII vesicles either via bulk flow or via selective concentration through interacting with specific ER membrane proteins, called cargo receptors (8, 62). By bridging the interaction between cargos in the ER lumen and COPII proteins on the cytoplasmic side of the ER membrane, cargo receptors mediate the efficient ER export of selective proteins (63). In addition to soluble cargos, some membrane cargos have conformational constraints that prevent their direct interactions with the COPII coat. Therefore, these proteins also need cargo receptors to facilitate their entry into the COPII vesicles. After incorporation into COPII transport vesicles, cargo receptors release bound cargo in pre-Golgi or Golgi compartments, and then recycled back to the ER for additional rounds of cargo export (62). Different types of cargo receptors that recognize carbohydrate and/or polypeptide signals in secretory cargos have been characterized. Some of most extensively studied mammalian cargo receptors included LMAN1 (also known as ERGIC-53) (64) and SURF4 (65). Loss-of-function mutation in LMAN1 causes combined factor V and factor VIII deficiency in human (66) and Lman1 deficient mice exhibited reduced plasma levels of factor V and factor VIII as well as ER accumulation of α1-antitrypsin in hepatocytes (67). As the mammalian homolog of the yeast COPII cargo receptor Erv29p, SURF4 has long been hypothesized as a cargo receptor in mammalian cells. However, its putative cargos have only been identified recently and were discussed extensively in two recent well-written reviews (63, 68). Despite affecting a broad range of cargos in cell culture, SURF4 exhibited, in several recent in vivo studies, a striking priority for lipoproteins in the liver. SURF4 appears to play an important role in ER export of lipoproteins and therefore regulating lipid homeostasis in vivo (69–71).
Proinsulin is the major soluble cargo in beta cells. Its biosynthesis alone can account for up to 50% of the total protein synthesis under glucose-stimulated conditions (72, 73). However, the molecular mechanism of proinsulin ER export was poorly understood until recent years. To understand the role of the COPII machinery in proinsulin ER export and insulin biogenesis, dominant negative SAR1 mutants were over-expressed to specifically block COPII-dependent ER export in MIN6 cells (11), isolated mouse and human islets (11, 74). Results from these studies demonstrated that SAR1 mutants blocked the export of mCherry-tagged as well as the endogenous proinsulin from exiting the ER and abolished the conversion of proinsulin to insulin. These effects were confirmed by siRNA-based Sar1A and Sar1B double knockdown experiments in MIN6 cells (11, 74). Furthermore, when a well-established in vitro COPII budding assay was applied to MIN6 cells, proinsulin was shown to be incorporated into COPII vesicles with the same ATP, GTP, and SAR1 dependence as other well-characterized COPII cargo proteins including Lman1 and Sec22B (11). Collectively, results from these studies firmly established that the COPII-dependent transport machinery is required for proinsulin ER export and insulin biogenesis (11, 74).
Consistent with the key findings in the beta cell line and isolated islets (11, 74), mice with beta cell–specific knockout of Ctage5, a key player of COPII dependent ER export, also showed impaired proinsulin trafficking and reduced insulin biogenesis (12). In pancreatic sections of the wild type mice, proinsulin immunofluorescent signals were detected throughout the ER, ERGIC (ER-Golgi intermediate compartment) and Golgi, whereas they were primarily in the ER in the conditional knockout mice (12). This finding is consistent with an impaired proinsulin ER export. A similar result was found in the Sar1A/1B double knockdown MIN6 cells (74). In the knockdown cells, the impaired COPII vesicle formation resulted in ER retention of proinsulin as evidenced by the increased colocalization of proinsulin with the ER marker, protein disulfide isomerase (74). In the Ctage5 knockout mice study, it was further revealed that Ctage5 interacted with vesicle SNARE protein, Sec22b, and may coordinate with Sec22b to control the release of COPII vesicles from the ER (12). It is interesting to note that a human TANGO1 mutation was recently identified to cause insulin-dependent diabetes mellitus along with skeleton defects related to impaired collagen secretion (75). Since the diabetes is due to reduced plasma insulin (75) and TANGO1 is known to interact and cooperate with CTAGE5 (24), further investigation is warranted to understand the role of TANGO1 in beta cell and insulin secretion.
The above discussed studies revealed that the ER export of proinsulin was COPII-dependent and efficient ER export was essential for beta cell functions (11, 12, 74). However, the molecular mechanism by which proinsulin and other secretory cargos are packaged into COPII vesicles was not clearly understood. Particularly, it was unknown until recently whether their ER export is mediated by membrane cargo receptor(s). Interestingly, a recent study showed that WFS1 acted as a cargo receptor for beta cell ER export of proinsulin as well as other secretory proteins (13). In this study, an abnormal ER accumulation of proinsulin immunofluorescent signals was observed in Wfs1 knockdown but not the control INS-1 cells. This finding was confirmed in the whole-body Wfs1 knockout mice where proinsulin was mainly colocalized with the ER markers in the beta cells (13). These results together indicated that Wfs1 is required for ER export of secretory cargo proteins such as proinsulin in beta cells. Consequently, Wfs1 knockdown or knockout led to a significantly increased proinsulin to insulin ratio (13).
Consistent with being a cargo receptor for soluble cargoes, Wfs1 interacted with Sec24 isoforms via its cytosolic N-terminal domain which contains two consensus di-acidic ER export signals, 158ENE and 169ETD (13). Mutagenesis of either signal abolished the interaction between Wfs1 and Sec24. Using Bimolecular Fluorescence Complementation assay and Proximity Ligation Assay in HEK-293T cells and endogenous cargo protein immunoprecipitations in INS-1 cells, it was further shown that Wfs1 could directly interact, through its luminal C-terminal domain, with proinsulin as well as other vesicular cargo proteins such as Cpe and Scg5 (13). Interestingly, several pathogenic WFS1 mutations in Wolfram Syndrome are located within these two critical interacting domains. While the N-terminal WFS1 mutants disrupted the interaction between Wfs1 and Sec24, the C-terminal mutants failed to interact and recognize the cargo proteins (13).
In another recent study, Surf4 was shown to regulate the ER export of proinsulin in INS-1 832/13 cells (76). Under high-glucose condition, Surf4 expression was upregulated and predominantly localized to the ERES. Surf4 knockdown resulted in proinsulin retention in the ER and decreased level of mature insulin in the secretory granules. Furthermore, proinsulin could be coimmunoprecipitated with Surf4 when both proteins were overexpressed in INS-1 832/13 cells. These results supported Surf4 as a cargo receptor for proinsulin ER export in beta cell line (76). Recently, liver-specific knockout mice studies have shown that Surf4 played an important role, as a cargo receptor, in ER export of lipoproteins in vivo (69–71). In future studies, it will be interesting to examine the in vivo function of Surf4 in a beta cell specific Surf4 knockout mouse model.
ER homeostasis is vital for normal beta cell functions and is maintained by the delicate balance between protein synthesis, folding, export and degradation. Disruption of this balance by genetic and environmental diabetes-causing factors leads to ER stress, beta cell death and diabetes (4, 73). Defective UPR signaling can disrupt ER homeostasis and normal beta cell functions (77). For example, defect in the UPR transducer PERK or IRE1α function led to poor beta cell survival (78–80). Mutations in the PERK gene cause a human diabetic condition known as Wolcott-Rallison syndrome, a disease state that can be observed in PERK knockout mice (78, 81). Recent studies in MIN6 cells, isolated islets, and beta cell specific Ctage5 KO mice showed that impaired COPII-dependent ER export strongly disrupted ER homeostasis, induced ER stress, and resulted in beta cell apoptosis (11, 12). Furthermore, the ER stress induced by defective ER export was mediated via the PERK/pEIF2α and IRE1/Xbp1 pathways but not via ATF6 because its own activation was also inhibited under this condition (11). These results in beta cell line and animal model showed that defective ER export could contribute to beta cell ER stress and dysfunction. It will be important to determine the association between beta cell dysfunction in various forms of human diabetes and mutations of the COPII or the associated proteins. In this regard, the recent finding of a human TANGO1 mutation to cause insulin-dependent diabetes mellitus (75) is promising and warrant further investigation.
A follow-up study of the dominant negative SAR1 mutants in MIN6 cells and isolated islets linked defective ER export to impaired proinsulin oxidative folding in beta cell ER (74). Under this condition, misfolded proinsulin formed aberrant disulfide-linked dimers and high molecular weight proinsulin complexes. Since proinsulin is the most abundant protein in the ER and its synthesis alone accounts for up to 30-50% of total protein synthesis of beta cells stimulated by high glucose, increased misfolded proinsulin is likely an important and direct contributor to ER stress induced by defective ER export. This argument is supported by alleviation of ER stress through limiting proinsulin load in the ER using Ins1 and Ins2 knockdown (74). This study revealed the significance of efficient ER export in maintaining ER protein homeostasis and native folding of proinsulin. Given the fact that proinsulin misfolding plays an important role in diabetes (1, 73, 82–84), this study suggested that enhancing ER export may be a potential therapeutic target to prevent/delay beta-cell failure caused by proinsulin misfolding and ER stress. Additional approaches that may lessen the ER stress and beta cell failure caused by misfolded proinsulin include promoting ER associated degradation (ERAD) of proinsulin (85) and/or decreasing disulfide-linked misfolded proinsulin complexes by limiting formation of abnormal intermolecular disulfide bonds (86).
In human studies, emerging genetic evidences and functional studies in human beta cell models linked defective ER-Golgi transport to insulin deficiency and diabetes. As already discussed, a human TANGO1 truncating mutation was recently identified to cause insulin-dependent diabetes (75). In another recent human study, homozygous mutations in the YIPF5 gene were found to cause neonatal diabetes (87). YIPF5 is a multi-spanning membrane protein localized in the ER, ERGIC, as well as Golgi apparatus and plays an important role in trafficking between the ER and Golgi (88–91). Deficiency of YIPF5 in human pancreatic beta-cell models, including YIPF5 silencing in EndoC-betaH1 cells, YIPF5 knockout and mutation knockin in ESCs, and patient derived iPSCs, were used to investigate the underlying mechanisms (87). Loss of YIPF5 function in stem cell–derived islet cells resulted in proinsulin retention in the ER, marked ER stress, and beta cell failure (87). The beta cell defects found in the YIPF5 knockout are quite similar to the phenotype caused by disrupting Wfs1 (see Section above). It would be interesting to examine whether YIPF5 is required for selective cargo sorting, especially for the soluble cargoes, into COPII vesicles. While there are evidences supporting YIPF5’s role in ER membrane organization and cargo exit at ERES (88, 90, 92), there is also evidence to show that YIPF5 regulates retrograde transport from Golgi to the ER (93). Therefore, it will be worthwhile to determine the primary function of YIPF5 in beta cell ER-Golgi transport in future studies. While the above studies showed that defective ER export machinery can lead to beta cell dysfunction and death, deficiencies of individual cargo proteins in their ER export capabilities have also been linked to disease conditions. It was found that some mutations in ATP-sensitive potassium channel caused human congenital hyperinsulinism and one of these mutations disrupted the ER exit signal on the Kir6.2 channel (94). GWAS study showed prohormone convertase 1/3 (PC1/3) variants were associated with fasting proinsulin levels (95), and defective PC1/3 ER export could potentially contribute to impaired insulin biogenesis in diabetes (96).
Evidences also exist that environmental diabetogenic factors, particularly lipotoxicity, reduced ER-Golgi transport and induced ER stress in beta cells (97–99). Using GFP-tagged, temperature-sensitive vesicular stomatitis virus G protein (VSVG) to quantify the rate of ER-to-Golgi protein trafficking, it was found that pretreatment with palmitate, to mimic lipotoxic condition in vitro, significantly reduced the rate of ER-Golgi protein transport in MIN6 cells (97, 100). Using the same reporter, high-density lipoproteins were found in a later study to restore ER-Golgi trafficking in palmitate-treated MIN6 cells (101). As for the potential molecular mechanisms by which saturated fatty acids impaired ER export, a subsequent study found that chronic palmitate treatment disrupted ER lipid rafts and inhibited vesicle budding from the ER (98). Using the same temperature-sensitive VSVG-GFP reporter, another diabetes contributing factor, hypoxia, was shown to reduce ER-Golgi protein trafficking in MIN6 cells (102). These findings suggest that defective COPII dependent cargo export could be one of the underlying causes for the major beta cell defects in diabetes including impaired proinsulin maturation, loss of insulin content, abnormal insulin granule morphology, chronic ER stress and beta cell apoptosis in diabetes (1, 4, 73, 103). In fact, defective insulin maturation with increased ER localization of proinsulin was reported in the beta cells of type 2 diabetes patients (104). It will be of importance to determine if ERES organization or COPII coat assembly is altered in human diabetic islets. It is worth noting that under other circumstances, defective COPII dependent cargo export may be a consequence of chronic beta cell ER stress and dysfunction in diabetes rather than its primary cause.
Building on the well characterized COPII machinery and recent studies of COPII dependent ER export in beta cells, the essential roles of this pathway in proinsulin trafficking, insulin biogenesis and beta cell ER homeostasis have been established. Next questions to be answered will be the physiological regulation of this process in health and its dysregulation under various diabetic conditions. For example, how does the COPII-dependent ER export pathway adapt to the fluctuations of cargo (proinsulin etc.) outflow between low and high glucose? How does it prevent improperly folded cargo molecules from entering the COPII vesicles and what roles do the identified cargo receptors play in this regard? How diabetogenic factors, lipotoxicity or cytokines etc., may disrupt this process? How is ERES organization, COPII coat assembly, or COPII protein expression/PTMs altered in human diabetic islets and to what extent these changes can contribute to the defective insulin biogenesis? As we are just at the beginning of understanding these important issues, more exciting and novel scientific findings are expected and new therapeutic opportunities may unveil in the near future.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Liu M, Huang Y, Xu X, Li X, Alam M, Arunagiri A, et al. Normal and defective pathways in biogenesis and maintenance of the insulin storage pool. J Clin Invest (2021) 131:e142240. doi: 10.1172/JCI142240
2. Liu M, Wright J, Guo H, Xiong Y, Arvan P. Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. Vitamins hormones (2014) 95:35–62. doi: 10.1016/B978-0-12-800174-5.00002-8
3. Ghosh R, Colon-Negron K, Papa FR. Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol Metab (2019) 27S:S60–8. doi: 10.1016/j.molmet.2019.06.012
4. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. Therapeutic opportunities for pancreatic beta-cell ER stress in diabetes mellitus. Nat Rev Endocrinol (2021) 17:455–67. doi: 10.1038/s41574-021-00510-4
5. Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, et al. COPII: a membrane coat formed by sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell (1994) 77:895–907. doi: 10.1016/0092-8674(94)90138-4
6. Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol (2012) 14:20–8. doi: 10.1038/ncb2390
7. Miller EA, Schekman R. COPII - a flexible vesicle formation system. Curr Opin Cell Biol (2013) 25:420–7. doi: 10.1016/j.ceb.2013.04.005
8. Gomez-Navarro N, Miller E. Protein sorting at the ER-golgi interface. J Cell Biol (2016) 215:769–78. doi: 10.1083/jcb.201610031
9. Raote I, Saxena S, Malhotra V. Sorting and export of proteins at the endoplasmic reticulum. Cold Spring Harbor Perspect Biol (2022). doi: 10.1101/cshperspect.a041258 Online ahead of print
10. Aridor M. A tango for coats and membranes: new insights into ER-to-Golgi traffic. Cell Rep (2022) 38:110258. doi: 10.1016/j.celrep.2021.110258
11. Fang J, Liu M, Zhang X, Sakamoto T, Taatjes DJ, Jena BP, et al. COPII-dependent ER export: a critical component of insulin biogenesis and beta-cell ER homeostasis. Mol Endocrinol (2015) 29:1156–69. doi: 10.1210/me.2015-1012
12. Fan J, Wang Y, Liu L, Zhang H, Zhang F, Shi L, et al. cTAGE5 deletion in pancreatic beta cells impairs proinsulin trafficking and insulin biogenesis in mice. J Cell Biol (2017) 216:4153–64. doi: 10.1083/jcb.201705027
13. Wang L, Liu H, Zhang X, Song E, Wang Y, Xu T, et al. WFS1 functions in ER export of vesicular cargo proteins in pancreatic beta-cells. Nat Commun (2021) 12:6996. doi: 10.1038/s41467-021-27344-y
14. D’Arcangelo JG, Stahmer KR, Miller EA. Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim Biophys Acta (2013) 1833:2464–72. doi: 10.1016/j.bbamcr.2013.02.003
15. Gillon AD, Latham CF, Miller EA. Vesicle-mediated ER export of proteins and lipids. Biochim Biophys Acta (2012) 1821:1040–9. doi: 10.1016/j.bbalip.2012.01.005
16. Antonny B, Schekman R. ER export: public transportation by the COPII coach. Curr Opin Cell Biol (2001) 13:438–43. doi: 10.1016/S0955-0674(00)00234-9
17. Barlowe C. Signals for COPII-dependent export from the ER: what’s the ticket out? Trends Cell Biol (2003) 13:295–300. doi: 10.1016/S0962-8924(03)00082-5
18. Chatterjee S, Choi AJ, Frankel G. A systematic review of Sec24 cargo interactome. Traffic (2021) 22:412–24. doi: 10.1111/tra.12817
19. Wang B, Joo JH, Mount R, Teubner BJW, Krenzer A, Ward AL, et al. The COPII cargo adapter SEC24C is essential for neuronal homeostasis. J Clin Invest (2018) 128:3319–32. doi: 10.1172/JCI98194
20. Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. eLife (2013) 2:e00444. doi: 10.7554/eLife.00444
21. Miller EA, Barlowe C. Regulation of coat assembly–sorting things out at the ER. Curr Opin Cell Biol (2010) 22:447–53. doi: 10.1016/j.ceb.2010.04.003
22. Malhotra V, Erlmann P. Protein export at the ER: loading big collagens into COPII carriers. EMBO J (2011) 30:3475–80. doi: 10.1038/emboj.2011.255
23. Aridor M. COPII gets in shape: lessons derived from morphological aspects of early secretion. Traffic (2018) 19:823–39. doi: 10.1111/tra.12603
24. Raote I, Saxena S, Campelo F, Malhotra V. TANGO1 marshals the early secretory pathway for cargo export. Biochim Biophys Acta Biomembranes (2021) 1863:183700. doi: 10.1016/j.bbamem.2021.183700
25. Raote I, Malhotra V. Tunnels for protein export from the endoplasmic reticulum. Annu Rev Biochem (2021) 90:605–30. doi: 10.1146/annurev-biochem-080120-022017
26. Weigel AV, Chang CL, Shtengel G, Xu CS, Hoffman DP, Freeman M, et al. ER-to-Golgi protein delivery through an interwoven, tubular network extending from ER. Cell (2021) 184:2412–2429 e2416. doi: 10.1016/j.cell.2021.03.035
27. Shomron O, Nevo-Yassaf I, Aviad T, Yaffe Y, Zahavi EE, Dukhovny A, et al. COPII collar defines the boundary between ER and ER exit site and does not coat cargo containers. J Cell Biol (2021) 220:e201907224. doi: 10.1083/jcb.201907224
28. Barlowe C. Twenty-five years after coat protein complex II. Mol Biol Cell (2020) 31:3–6. doi: 10.1091/mbc.E19-11-0621
29. Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, et al. Structural basis for cargo regulation of COPII coat assembly. Cell (2008) 134:474–84. doi: 10.1016/j.cell.2008.06.024
30. Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell (2007) 129:1325–36. doi: 10.1016/j.cell.2007.05.036
31. Hutchings J, Stancheva VG, Brown NR, Cheung ACM, Miller EA, Zanetti G. Structure of the complete, membrane-assembled COPII coat reveals a complex interaction network. Nat Commun (2021) 12:2034. doi: 10.1038/s41467-021-22110-6
32. Stancheva VG, Li XH, Hutchings J, Gomez-Navarro N, Santhanam B, Babu MM, et al. Combinatorial multivalent interactions drive cooperative assembly of the COPII coat. J Cell Biol (2020) 219:e202007135. doi: 10.1083/jcb.202007135
33. McCaughey J, Stephens DJ. COPII-dependent ER export in animal cells: adaptation and control for diverse cargo. Histochem Cell Biol (2018) 150:119–31. doi: 10.1007/s00418-018-1689-2
34. Centonze FG, Farhan H. Crosstalk of endoplasmic reticulum exit sites and cellular signaling. FEBS Lett (2019) 593:2280–8. doi: 10.1002/1873-3468.13569
35. Sun LP, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to golgi: insig renders sorting signal in scap inaccessible to COPII proteins. Proc Natl Acad Sci United States America (2007) 104:6519–26. doi: 10.1073/pnas.0700907104
36. Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL. Sterol-regulated transport of SREBPs from endoplasmic reticulum to golgi: oxysterols block transport by binding to insig. Proc Natl Acad Sci United States America (2007) 104:6511–8. doi: 10.1073/pnas.0700899104
37. Schindler AJ, Schekman R. In vitro Reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc Natl Acad Sci United States America (2009) 106:17775–80. doi: 10.1073/pnas.0910342106
38. Higa A, Taouji S, Lhomond S, Jensen D, Fernandez-Zapico ME, Simpson JC, et al. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol Cell Biol (2014) 34:1839–49. doi: 10.1128/MCB.01484-13
39. Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, et al. The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature (2015) 524:243–6. doi: 10.1038/nature14557
40. Su L, Zhou L, Chen FJ, Wang H, Qian H, Sheng Y, et al. Cideb controls sterol-regulated ER export of SREBP/SCAP by promoting cargo loading at ER exit sites. EMBO J (2019) 38:e100156. doi: 10.15252/embj.2018100156
41. Farhan H, Weiss M, Tani K, Kaufman RJ, Hauri HP. Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. EMBO J (2008) 27:2043–54. doi: 10.1038/emboj.2008.136
42. Manzano-Lopez J, Perez-Linero AM, Aguilera-Romero A, Martin ME, Okano T, Silva DV, et al. COPII coat composition is actively regulated by luminal cargo maturation. Curr biology: CB (2015) 25:152–62. doi: 10.1016/j.cub.2014.11.039
43. Subramanian A, Capalbo A, Iyengar NR, Rizzo R, di Campli A, Di Martino R, et al. Auto-regulation of secretory flux by sensing and responding to the folded cargo protein load in the endoplasmic reticulum. Cell (2019) 176:1461–1476 e1423. doi: 10.1016/j.cell.2019.01.035
44. Zacharogianni M, Kondylis V, Tang Y, Farhan H, Xanthakis D, Fuchs F, et al. ERK7 is a negative regulator of protein secretion in response to amino-acid starvation by modulating Sec16 membrane association. EMBO J (2011) 30:3684–700. doi: 10.1038/emboj.2011.253
45. Liu L, Cai J, Wang H, Liang X, Zhou Q, Ding C, et al. Coupling of COPII vesicle trafficking to nutrient availability by the IRE1alpha-XBP1s axis. Proc Natl Acad Sci United States America (2019) 116:11776–85. doi: 10.1073/pnas.1814480116
46. Blumental-Perry A, Haney CJ, Weixel KM, Watkins SC, Weisz OA, Aridor M. Phosphatidylinositol 4-phosphate formation at ER exit sites regulates ER export. Dev Cell (2006) 11:671–82. doi: 10.1016/j.devcel.2006.09.001
47. Melero A, Chiaruttini N, Karashima T, Riezman I, Funato K, Barlowe C, et al. Lysophospholipids facilitate COPII vesicle formation. Curr biology: CB (2018) 28:1950–1958 e1956. doi: 10.1016/j.cub.2018.04.076
48. Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J (2011) 30:1357–75. doi: 10.1038/emboj.2011.52
49. Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, et al. Ubiquitin-dependent regulation of COPII coat size and function. Nature (2012) 482:495–500. doi: 10.1038/nature10822
50. McGourty CA, Akopian D, Walsh C, Gorur A, Werner A, Schekman R, et al. Regulation of the CUL3 ubiquitin ligase by a calcium-dependent Co-adaptor. Cell (2016) 167:525–538 e514. doi: 10.1016/j.cell.2016.09.026
51. Sharpe LJ, Luu W, Brown AJ. Akt phosphorylates Sec24: new clues into the regulation of ER-to-Golgi trafficking. Traffic (2011) 12:19–27. doi: 10.1111/j.1600-0854.2010.01133.x
52. Farhan H, Wendeler MW, Mitrovic S, Fava E, Silberberg Y, Sharan R, et al. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J Cell Biol (2010) 189:997–1011. doi: 10.1083/jcb.200912082
53. Centonze FG, Reiterer V, Nalbach K, Saito K, Pawlowski K, Behrends C, et al. LTK is an ER-resident receptor tyrosine kinase that regulates secretion. J Cell Biol (2019) 218:2470–80. doi: 10.1083/jcb.201903068
54. Dudognon P, Maeder-Garavaglia C, Carpentier JL, Paccaud JP. Regulation of a COPII component by cytosolic O-glycosylation during mitosis. FEBS Lett (2004) 561:44–50. doi: 10.1016/S0014-5793(04)00109-7
55. Cox NJ, Unlu G, Bisnett BJ, Meister TR, Condon BM, Luo PM, et al. Dynamic glycosylation governs the vertebrate COPII protein trafficking pathway. Biochemistry (2018) 57:91–107. doi: 10.1021/acs.biochem.7b00870
56. Jeong YT, Simoneschi D, Keegan S, Melville D, Adler NS, Saraf A, et al. The ULK1-FBXW5-SEC23B nexus controls autophagy. eLife (2018) 7:e42253. doi: 10.7554/eLife.42253
57. Zheng Z, Kim H, Qiu Y, Chen X, Mendez R, Dandekar A, et al. CREBH couples circadian clock with hepatic lipid metabolism. Diabetes (2016) 65:3369–83. doi: 10.2337/db16-0298
58. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell (2006) 124:587–99. doi: 10.1016/j.cell.2005.11.040
59. Zhang C, Wang G, Zheng Z, Maddipati KR, Zhang X, Dyson G, et al. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology (2012) 55:1070–82. doi: 10.1002/hep.24783
60. Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, et al. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science (2012) 337:1668–72. doi: 10.1126/science.1224947
61. Hanna MG, Mela I, Wang L, Henderson RM, Chapman ER, Edwardson JM, et al. Sar1 GTPase activity is regulated by membrane curvature. J Biol Chem (2016) 291:1014–27. doi: 10.1074/jbc.M115.672287
62. Dancourt J, Barlowe C. Protein sorting receptors in the early secretory pathway. Annu Rev Biochem (2010) 79:777–802. doi: 10.1146/annurev-biochem-061608-091319
63. Tang VT, Ginsburg D. Cargo selection in endoplasmic reticulum-to-Golgi transport and relevant diseases. J Clin Invest (2023) 133:e163838. doi: 10.1172/JCI163838
64. Appenzeller C, Andersson H, Kappeler F, Hauri HP. The lectin ERGIC-53 is a cargo transport receptor for glycoproteins. Nat Cell Biol (1999) 1:330–4. doi: 10.1038/14020
65. Emmer BT, Hesketh GG, Kotnik E, Tang VT, Lascuna PJ, Xiang J, et al. The cargo receptor SURF4 promotes the efficient cellular secretion of PCSK9. eLife (2018) 7:e38839. doi: 10.7554/eLife.38839
66. Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, et al. Mutations in the ER-golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell (1998) 93:61–70. doi: 10.1016/S0092-8674(00)81146-0
67. Zhang B, Zheng C, Zhu M, Tao J, Vasievich MP, Baines A, et al. Mice deficient in LMAN1 exhibit FV and FVIII deficiencies and liver accumulation of alpha1-antitrypsin. Blood (2011) 118:3384–91. doi: 10.1182/blood-2011-05-352815
68. Wang X, Chen XW. Cargo receptor-mediated ER export in lipoprotein secretion and lipid homeostasis. Cold Spring Harbor Perspect Biol (2022). doi: 10.1101/cshperspect.a041260 Online ahead of print
69. Wang X, Wang H, Xu B, Huang D, Nie C, Pu L, et al. Receptor-mediated ER export of lipoproteins controls lipid homeostasis in mice and humans. Cell Metab (2021) 33:350–366 e357. doi: 10.1016/j.cmet.2020.10.020
70. Tang VT, McCormick J, Xu B, Wang Y, Fang H, Wang X, et al. Hepatic inactivation of murine Surf4 results in marked reduction in plasma cholesterol. eLife (2022) 11:e82269. doi: 10.7554/eLife.82269
71. Wang B, Shen Y, Zhai L, Xia X, Gu HM, Wang M, et al. Atherosclerosis-associated hepatic secretion of VLDL but not PCSK9 is dependent on cargo receptor protein Surf4. J Lipid Res (2021) 62:100091. doi: 10.1016/j.jlr.2021.100091
72. Schuit FC, In’t Veld PA, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc Natl Acad Sci United States America (1988) 85:3865–9. doi: 10.1073/pnas.85.11.3865
73. Sun J, Cui J, He Q, Chen Z, Arvan P, Liu M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol aspects Med (2015) 42:105–18. doi: 10.1016/j.mam.2015.01.001
74. Zhu R, Li X, Xu J, Barrabi C, Kekulandara D, Woods J, et al. Defective endoplasmic reticulum export causes proinsulin misfolding in pancreatic beta cells. Mol Cell Endocrinol (2019) 493:110470. doi: 10.1016/j.mce.2019.110470
75. Lekszas C, Foresti O, Raote I, Liedtke D, Konig EM, Nanda I, et al. Biallelic TANGO1 mutations cause a novel syndromal disease due to hampered cellular collagen secretion. eLife (2020) 9:e51319. doi: 10.7554/eLife.51319
76. Saegusa K, Matsunaga K, Maeda M, Saito K, Izumi T, Sato K. Cargo receptor Surf4 regulates endoplasmic reticulum export of proinsulin in pancreatic beta-cells. Commun Biol (2022) 5:458. doi: 10.1038/s42003-022-03417-6
77. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol (2020) 21:421–38. doi: 10.1038/s41580-020-0250-z
78. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell (2001) 7:1153–63. doi: 10.1016/S1097-2765(01)00264-7
79. Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, et al. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab (2006) 4:245–54. doi: 10.1016/j.cmet.2006.07.007
80. Hassler JR, Scheuner DL, Wang S, Han J, Kodali VK, Li P, et al. The IRE1alpha/XBP1s pathway is essential for the glucose response and protection of beta cells. PloS Biol (2015) 13:e1002277. doi: 10.1371/journal.pbio.1002277
81. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-rallison syndrome. Nat Genet (2000) 25:406–9. doi: 10.1038/78085
82. Arunagiri A, Haataja L, Pottekat A, Pamenan F, Kim S, Zeltser LM, et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife (2019) 8:e44532. doi: 10.7554/eLife.44532
83. Arunagiri A, Haataja L, Cunningham CN, Shrestha N, Tsai B, Qi L, et al. Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann New York Acad Sci (2018) 1418:5–19. doi: 10.1111/nyas.13531
84. Haataja L, Arunagiri A, Hassan A, Regan K, Tsai B, Dhayalan B, et al. Distinct states of proinsulin misfolding in MIDY. Cell Mol Life sciences: CMLS (2021) 78:6017–31. doi: 10.1007/s00018-021-03871-1
85. Xu B, Allard C, Alvarez-Mercado AI, Fuselier T, Kim JH, Coons LA, et al. Estrogens promote misfolded proinsulin degradation to protect insulin production and delay diabetes. Cell Rep (2018) 24:181–96. doi: 10.1016/j.celrep.2018.06.019
86. Sun J, Xiong Y, Li X, Haataja L, Chen W, Mir SA, et al. Role of proinsulin self-association in mutant INS gene-induced diabetes of youth. Diabetes (2020) 69:954–64. doi: 10.2337/db19-1106
87. De Franco E, Lytrivi M, Ibrahim H, Montaser H, Wakeling MN, Fantuzzi F, et al. YIPF5 mutations cause neonatal diabetes and microcephaly through endoplasmic reticulum stress. J Clin Invest (2020) 130:6338–53. doi: 10.1172/JCI141455
88. Tang BL, Ong YS, Huang B, Wei S, Wong ET, Qi R, et al. A membrane protein enriched in endoplasmic reticulum exit sites interacts with COPII. J Biol Chem (2001) 276:40008–17. doi: 10.1074/jbc.M106189200
89. Kranjc T, Dempsey E, Cagney G, Nakamura N, Shields DC, Simpson JC. Functional characterisation of the YIPF protein family in mammalian cells. Histochem Cell Biol (2017) 147:439–51. doi: 10.1007/s00418-016-1527-3
90. Ran Y, Xiong MG, Xu ZS, Luo WW, Wang SY, Wang YY. YIPF5 is essential for innate immunity to DNA virus and facilitates COPII-dependent STING trafficking. J Immunol (2019) 203:1560–70. doi: 10.4049/jimmunol.1900387
91. Shaik S, Pandey H, Thirumalasetti SK, Nakamura N. Characteristics and functions of the Yip1 domain family (YIPF), multi-span transmembrane proteins mainly localized to the golgi apparatus. Front Cell Dev Biol (2019) 7:130. doi: 10.3389/fcell.2019.00130
92. Dykstra KM, Pokusa JE, Suhan J, Lee TH. Yip1A structures the mammalian endoplasmic reticulum. Mol Biol Cell (2010) 21:1556–68. doi: 10.1091/mbc.e09-12-1002
93. Kano F, Yamauchi S, Yoshida Y, Watanabe-Takahashi M, Nishikawa K, Nakamura N, et al. Yip1A regulates the COPI-independent retrograde transport from the golgi complex to the ER. J Cell Sci (2009) 122:2218–27. doi: 10.1242/jcs.043414
94. Taneja TK, Mankouri J, Karnik R, Kannan S, Smith AJ, Munsey T, et al. Sar1-GTPase-dependent ER exit of KATP channels revealed by a mutation causing congenital hyperinsulinism. Hum Mol Genet (2009) 18:2400–13. doi: 10.1093/hmg/ddp179
95. Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes (2011) 60:2624–34. doi: 10.2337/db11-0415
96. Prabhu Y, Blanco EH, Liu M, Peinado JR, Wheeler MC, Gekakis N, et al. Defective transport of the obesity mutant PC1/3 N222D contributes to loss of function. Endocrinology (2014) 155:2391–401. doi: 10.1210/en.2013-1985
97. Preston AM, Gurisik E, Bartley C, Laybutt DR, Biden TJ. Reduced endoplasmic reticulum (ER)-to-Golgi protein trafficking contributes to ER stress in lipotoxic mouse beta cells by promoting protein overload. Diabetologia (2009) 52:2369–73. doi: 10.1007/s00125-009-1506-5
98. Boslem E, Weir JM, MacIntosh G, Sue N, Cantley J, Meikle PJ, et al. Alteration of endoplasmic reticulum lipid rafts contributes to lipotoxicity in pancreatic beta-cells. J Biol Chem (2013) 288:26569–82. doi: 10.1074/jbc.M113.489310
99. Biden TJ, Boslem E, Chu KY, Sue N. Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol metabolism: TEM (2014) 25:389–98. doi: 10.1016/j.tem.2014.02.003
100. Boslem E, MacIntosh G, Preston AM, Bartley C, Busch AK, Fuller M, et al. A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem J (2011) 435:267–76. doi: 10.1042/BJ20101867
101. Petremand J, Puyal J, Chatton JY, Duprez J, Allagnat F, Frias M, et al. HDLs protect pancreatic beta-cells against ER stress by restoring protein folding and trafficking. Diabetes (2012) 61:1100–11. doi: 10.2337/db11-1221
102. Bensellam M, Maxwell EL, Chan JY, Luzuriaga J, West PK, Jonas JC, et al. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia (2016) 59:1492–502. doi: 10.1007/s00125-016-3947-y
103. Shrestha N, De Franco E, Arvan P, Cnop M. Pathological beta-cell endoplasmic reticulum stress in type 2 diabetes: current evidence. Front Endocrinol (2021) 12:650158. doi: 10.3389/fendo.2021.650158
Keywords: pancreatic beta cells, endoplasmic reticulum (ER) export, coat protein complex II (COPII), proinsulin, diabetes
Citation: Barrabi C, Zhang K, Liu M and Chen X (2023) Pancreatic beta cell ER export in health and diabetes. Front. Endocrinol. 14:1155779. doi: 10.3389/fendo.2023.1155779
Received: 31 January 2023; Accepted: 10 April 2023;
Published: 21 April 2023.
Edited by:
Guojun Shi, Sun Yat-sen University, ChinaReviewed by:
Brian Storrie, University of Arkansas for Medical Sciences, United StatesCopyright © 2023 Barrabi, Zhang, Liu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuequn Chen, eGNoZW5AbWVkLndheW5lLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.