 Xuefei Zhao
Xuefei Zhao Xuedong An
Xuedong An Cunqing Yang
Cunqing Yang Wenjie Sun
Wenjie Sun Fengmei Lian
Fengmei Lian- Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Insulin resistance (IR) plays a crucial role in the development and progression of metabolism-related diseases such as diabetes, hypertension, tumors, and nonalcoholic fatty liver disease, and provides the basis for a common understanding of these chronic diseases. In this study, we provide a systematic review of the causes, mechanisms, and treatments of IR. The pathogenesis of IR depends on genetics, obesity, age, disease, and drug effects. Mechanistically, any factor leading to abnormalities in the insulin signaling pathway leads to the development of IR in the host, including insulin receptor abnormalities, disturbances in the internal environment (regarding inflammation, hypoxia, lipotoxicity, and immunity), metabolic function of the liver and organelles, and other abnormalities. The available therapeutic strategies for IR are mainly exercise and dietary habit improvement, and chemotherapy based on biguanides and glucagon-like peptide-1, and traditional Chinese medicine treatments (e.g., herbs and acupuncture) can also be helpful. Based on the current understanding of IR mechanisms, there are still some vacancies to follow up and consider, and there is also a need to define more precise biomarkers for different chronic diseases and lifestyle interventions, and to explore natural or synthetic drugs targeting IR treatment. This could enable the treatment of patients with multiple combined metabolic diseases, with the aim of treating the disease holistically to reduce healthcare expenditures and to improve the quality of life of patients to some extent.
1 Introduction
More than 100 years have passed since the discovery of insulin, an important regulator of blood sugar, vasodilation, cell growth and protein metabolism. Decreased peripheral target tissue responsiveness to insulin action leads to insulin resistance (IR), a complex pathophysiological condition with reduced sensitivity, impaired ability to inhibit glucose production and stimulate peripheral glucose elimination, and often accompanied with hyperinsulinemia to maintain blood sugar stability (1). IR is characterized by insulin-mediated blood glucose management disorders, blood glucose utilization disorders, abnormal lipid accumulation, and increased lipid decomposition activities in adipocytes, which can be called insulin resistance syndrome or metabolic syndrome. As a hotbed, IR breeds obesity, type 2 diabetes and its complications, non-alcoholic fatty liver disease (NAFLD), tumor, cardiovascular disease and other metabolic diseases. Any disease or disorder that leads to an abnormal metabolic process can be defined as metabolic disease, which poses a major threat to human health and affects the quality of life. Therefore, it is of great necessity to understand IR clearly and explore innovative therapeutic approaches to reduce the burden of disease.
For the diagnosis of IR, the hyperinsulinemic-positive glucose clamp test (HEGC) is considered as the gold standard, but its clinical universality is poor due to its complexity and limitations. There are some less invasive approximations can be used to measure IR, including quantitative insulin sensitivity test index, homeostatic model assessment (HOMA), fasting insulin test, insulin release test, and oral glucose tolerance test (2). Moreover, in clinical and epidemiological studies, the measurement of blood glucose, insulin, and adipokine levels has replaced HEGC for the evaluation of IR (3); for example, elevated levels of branched-chain amino acids and reduced levels of glycine are currently more reliable amino acid markers for IR (4). And some other biomarkers closely related to obesity and metabolic syndrome components, such as adiponectin, fetuin-A and Peptidase M20 domain containing 1 (PM20D1) measurement of their serum concentrations may be valuable for clinical diagnosis of IR-related metabolic and cardiovascular diseases (5–7).
The prevalence of IR and metabolic syndrome is commonly thought to be associated with obesity and T2DM, which inflicts nearly one-third of the world’s population (8, 9). Since IR plays a crucial role in many serious chronic diseases such as type 2 diabetes, cardiovascular and cerebrovascular diseases, the subsequent rise in the incidence of these diseases has made them a major cause of mortality and morbidity worldwide. These metabolism-related diseases not only cause psychological and physical distress to patients, but also place a tremendous burden on health systems, with the total cost, including medical costs and potential loss of economic activity, running into trillions of dollars (8). And patients with metabolic underlying diseases are more vulnerable than healthy individuals in the face of epidemic disease; for example, up to 50% of those who die from COVID-19 have metabolic and vascular disease (10). The increasing incidence of IR and metabolic diseases and the toll they take has prompted an in-depth study of the mechanisms involved. Furthermore, because all these metabolic diseases, as well as IR and obesity, are interrelated through complex molecular-biochemical and immune-related mechanisms, it has been found that many patients encountered in clinical practice today have a combination of multiple metabolic diseases. They interact in a causal manner, with the onset or exacerbation of one disease also affecting other metabolic diseases. For example, among patients with T2DM, the global prevalence of NAFLD is 55.5% (95% CI 47.3-63.7) (11) and up to 50% of hypertensive patients have NAFLD (12). Therefore, if the mechanisms by which IR and metabolic diseases act together can be recognized, so that metabolic diseases with the same mechanisms can be treated with drugs that target IR, this could improve multiple problems for patients and reduce the economic burden. This is crucial for understanding and developing new therapies for many chronic diseases, such as tapping into drugs with multi-target therapeutic effects like metformin, which has good clinical guidance.
The aim of this review is to focus on the key role of IR in a variety of metabolic diseases at multiple levels, including etiology, mechanisms and therapeutic approaches. Moreover, we summarize some of the most recent advances on the pathogenesis and mechanisms of IR. In addition, we outline the available methods for the treatment of IR in terms of non-pharmacological treatment and chemotherapy.
2 IR and metabolic disease
As mentioned before, IR is a powerful risk factor for the occurrence and development of a bunch of serious chronic diseases. The relationship between IR and these diseases will be described in turn, based on the clinical researches. Chronic metabolic diseases that may be induced by IR are demonstrated in Figure 1.
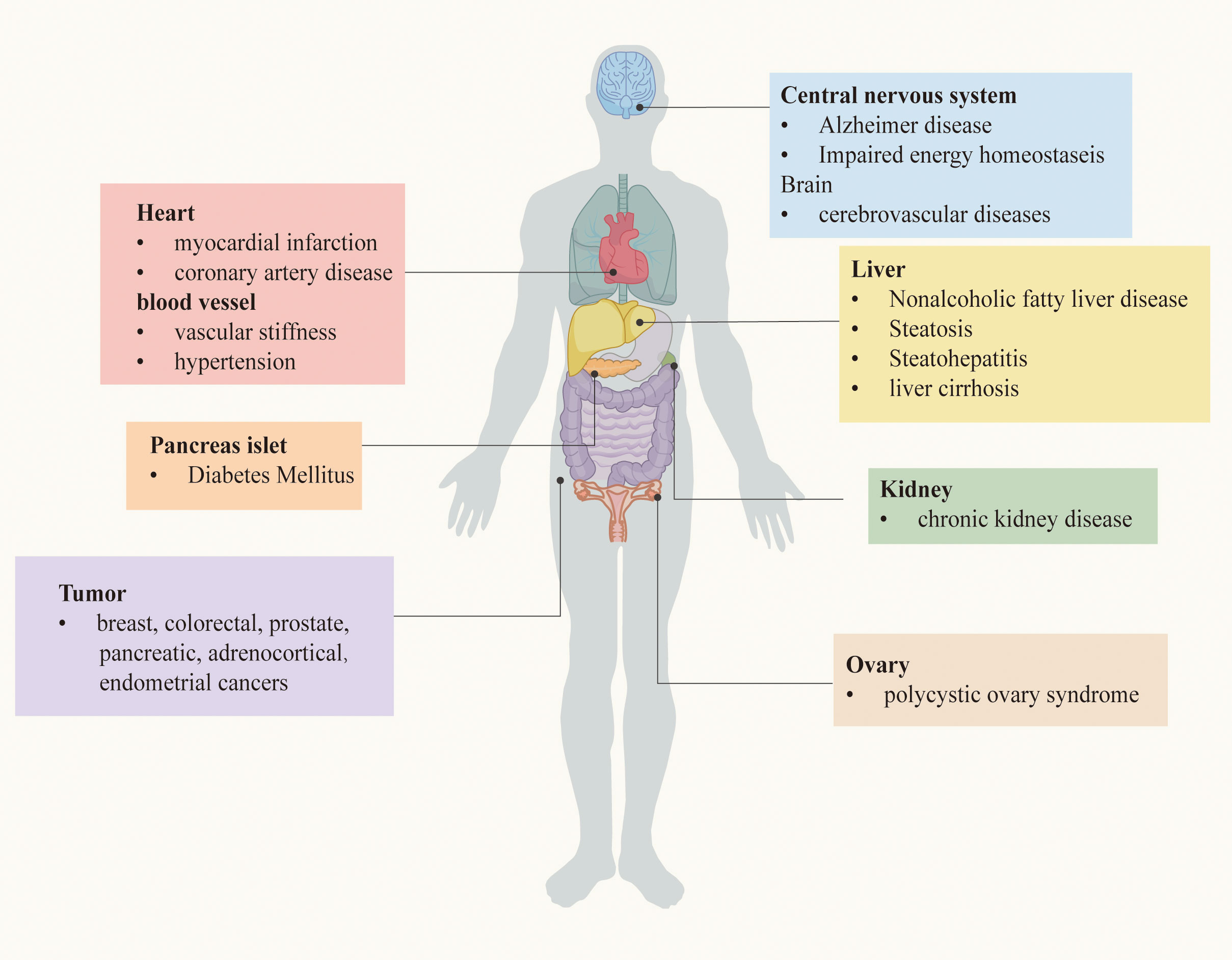
Figure 1 Chronic metabolic diseases may be induced by IR.
2.1 IR and diabetes mellitus
According to the 10th edition of the IDF Diabetes Atlas, 536.6 million people worldwide have diabetes, which means that more than 10.5% of the world’s adult population has the disease, and this number continues to grow, predicted to rise to 12.2% (783.2 million people) by 2045 (13). Since insulin is a pivotal hormone that regulates blood sugar, IR is closely associated with all stages of DM, including prediabetes, diabetes, and its complications. Impaired β-cell compensation in response to increased IR is a pathophysiological factor associated with poor glucose tolerance, which contributes to the development of DM.
Type 1 DM (T1DM) is caused by the primary loss of β-cells — the cells that release insulin — and the complex autoimmune process of continuous insulin deficiency. Nevertheless, clinical and experimental evidence have shown that patients with T1DM exhibit IR (14), which is a prominent feature in adolescents and adults (15–17), mainly involving the liver, peripheral, and adipose tissue (18). Insulin injections are currently the conventional treatment for T1DM, and prolonged overexposure to insulin itself is a trigger for insulin resistance. patients with T1DM eventually also develop insulin resistance and other features of T2DM, such as cardiovascular disease (19).
Type 2 DM (T2DM) is characterized by defective insulin secretion from pancreatic beta cells. Under normal conditions, increased insulin release by pancreatic β-cells is sufficient of insulin action and maintain normal glucose tolerance (20). However, under the circumstances of IR combined with environmental factors and genetic factors related to T2D, persistent overnutrition sets up a vicious spiral of hyperinsulinemia and insulin resistance, ultimately leading to beta cell failure, possibly due to glucose and lipid toxicity and other factors leading to significant T2D (21). There is a lot of evidence suggesting that both IR and T2D are associated with obesity, especially with high proportion of intra-abdominal and intra-hepatic fat, which is the most crucial factor contributes to the emergence of metabolic disease (22, 23). IR at the beta-cell level may play a role in the pathogenesis of insulin release defects. Reduced insulin release may impair adipocyte metabolism, leading to increased lipolysis and elevated levels of non-esterified fatty acid (NEFA). Elevation of NEFA and glucose can work together to impair islet health and insulin action. Therefore, this process may slowly progress forward to develop T2D (22).
In addition, IR was independently associated with each of the chronic macrovascular and microvascular complications from diabetes (24). Triglyceride-glucose index (TyG index) is a convenient measure of IR. In a large Chinese inpatient cohort study, inpatients with elevated TyG index were shown to be at higher risk for lower extremity macrovascular stenosis, arterial stiffness and renal microvascular injury (25, 26). In particular, IR or hyperinsulinemia is responsible for the development of diabetic cardiomyopathy by pathophysiological mechanisms including impaired insulin signaling, cardiac mitochondrial dysfunction, endoplasmic reticulum stress, impaired autophagy, impaired myocardial calcium handling, abnormal coronary microcirculation, inappropriate neurohumoral activation and maladaptive immune responses (27, 28). Regarding chronic kidney disease, although this remains to be proven, IR is considered to be a factor contributing to the development and progression of diabetic nephropathy (DN), as well as a consequence of DN. IR is exacerbated during the development of DN, possibly due to some potentially modifiable changes in circulating hormones, neuroendocrine pathways, and chronic inflammation (29).
2.2 IR and tumor
In recent years, a wealth of experimental, epidemiological and clinical evidence has suggested that IR and its compensatory hyperinsulinemia have a synergistic relationship with the development and progression of certain types of cancer, including breast, colorectal, prostate, pancreatic, adrenocortical and endometrial cancers (30–32). To put it in perspective, IR and hyperinsulinemia, even in individuals without diabetes, are independently and positively associated with increased mortality from pancreatic cancer (33). Besides, according to a large observational study, breast cancer incidence in women with high HOMA-IR is associated with all-cause mortality, especially in postmenopausal women (34). Although the underlying mechanisms of the association between IR and tumor remain unclear, it may rely on several mechanisms and is not necessarily the same for different types of cancers. It is clear that IR-related factors, including chronic persistent hyperinsulinemia, INSRs, IGF1Rs and INSR/IGF1R hybrids, as well as chronic inflammation, ncRNAs and microbiota, have been suggested as factors that may play a role in all tumor stages (35). In addition, the mitogen-activated protein kinase (MAPK) insulin pathway is the basis of many obesity-related malignancies that control cell growth and mitosis (36), whereas insulin can directly promote cell proliferation and survival via the phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) and Ras/MAPK pathways (37). On the other hand, IR is closely associated with visceral adipose dysfunction and systemic inflammation, both of which favor creating an environment conducive to tumorigenesis (38, 39). Additionally, epigenetic modifications which are triggered by IR and other environmental factors and chronic disease often involve in oncogenesis, such as DNA methylation, histone modifications, and non-coding RNA (35, 40, 41). In addition to the mechanisms described above, recent studies indicate that gut microbiota may be a contributing factor in the relationship between IR and cancer, due to gut dysbiosis (42). Therefore, increasing knowledge about the role of IR in cancer has important implications for cancer prevention and tumor growth inhibition.
2.3 IR and cardiovascular and cerebrovascular diseases
IR is thought to be a key risk factor leading to cardiovascular and cerebrovascular diseases in different populations, whether normal or diabetic (43–46). The results of a mathematic analysis indicate that IR, which is responsible for approximately 42% of myocardial infarctions, is probably the most important single cause of coronary artery disease (CAD) (47). Another research showed that compared with patients with lower value of HOMA-IR, those with high value HOMA-IR (≥4.14) had significantly lower global longitudinal strain (GLS), vascular stiffness, and increased pulse wave velocity (PWV) measured in the carotid artery, which have implications for myocardial and vascular function (48). Increased plasma levels of fatty acids in patients with IR and dyslipidemia, with or without diabetes, may lead to the development of metabolism-related cardiomyopathy (49). An example is diabetic cardiomyopathy, which is characterized by diastolic dysfunction and left ventricular hypertrophy in the absence of vascular defects. Diabetic dyslipidemia and lipid accumulation in the myocardium are key pathologic features (50). In animal experiments, mice have shown that when IR develops, insulin receptor substrate-1 (IRS1) and insulin receptor substrate-2 (IRS2) signaling will be impaired, resulting in impaired expression of cardiac energy metabolism genes and activation of p38α mitogen-activated protein kinase (p38), ultimately leading to abnormal cardiac function (51). The strong association between IR and CVD may be due to the fact that the heart is a target organ for insulin, which requires greater energy consumption, yet when IR occurs, it impedes the normal function of the heart and increases the incidence of CVD (52, 53). What’s more, the higher relative risk of cardiovascular events in male with IR compared to female, especially younger women, can be explained by the attenuated relationship between IR and CVD risk (54, 55). Therefore, improving insulin sensitivity not only reduces plasma glucose concentrations in patients with T2DM, but also reduces the risk of cerebrovascular disease independent of the control of blood glucose levels (43, 56).
2.4 IR and nonalcoholic fatty liver disease
The liver is one of the main organs controlling the metabolic balance and there is a close relationship between IR and NAFLD, which could be described as a two-way street (57, 58). NAFLD is characterized by excessive accumulation of lipids in hepatocytes. Lipids and metabolites secreted by the liver, including lipoproteins, ketones, acylcarnitine and bile acids, may act as signaling molecules and regulate insulin action (59, 60). Hyperinsulinemia can drive hepatic lipogenesis and lipid accumulation directly as well as through indirect mechanisms, including excess circulating FFA, that impede the ability of insulin to inhibit hepatic glucose production (61). High IR was found to be the most important predictor of NAFLD in both obese and lean subjects (62), and studies have shown that serum insulin levels are strongly associated with hepatic lobular inflammation and histological progression such as ballooning (63). Similarly, in patients with NAFLD, glycerol appearance and lipid oxidation were markedly increased, and IR also increased with the degree of steatosis (64, 65). A meta-analysis showed that compared with those without NAFLD, the risk of T2DM was more than two times higher in patients with NAFLD, with the highest risk particularly in patients with nonalcoholic steatohepatitis (NASH) (66). In the condition of mildly active hepatic steatosis, IR is associated with hepatocellular injury and atherosclerotic dyslipidemia. While in steatohepatitis, IR is combined with cytokine pro-inflammatory status and fibrosis indicators (67).
2.5 IR and polycystic ovary syndrome
PCOS is a complex gynecologic endocrine disease, which is characterized by hyperandrogenism, menoxenia, ovulatory dysfunction and infertility. A study of obese adolescent girls indicates that the PCOS phenotype with high androgen levels has the greatest degree of insulin resistance and inflammation (68). Although the etiology and pathogenesis behind PCOS remain to be determined, IR and its compensatory hyperinsulinemia is considered to be an important pathological change that led to progression of PCOS and the main pathological basis for its reproductive dysfunction (69–71). Excessive insulin secretion triggers insulin receptors in the pituitary gland, promoting androgen secretion from the ovaries and adrenal glands through the pituitary-ovary and adrenal axes, and increases free testosterone levels by inhibiting hepatic sex binding globulin (SHBG) synthesis (72, 73). Moreover, insulin, as a reproductive as well as metabolic hormone, has direct effect of stimulating ovarian androgen production by stimulating 17α-hydroxylase activity in the ovarian theca cells and enhance the activity of insulin-like growth factor-1 (IGF-1) receptor in the ovary, thus increasing its free IGF level and promoting androgen production (74, 75). Also, IR has long-term and deleterious effects on the metabolism of women with polycystic ovary syndrome. Irrespective of obesity, 50% of patients with PCOS develop IR (76, 77).
2.6 IR and other diseases
In addition to the diseases described above, IR is also associated with many other diseases of various systems throughout the body. This includes liver cirrhosis, which is associated with changes in glucose homeostasis, even in intact liver function. Essential features of the association between cirrhosis and IR include endocrine dysregulation, liver inflammation, changes in muscle mass and composition, changes in the gut microbiota, and permeability (78). IR may also affect the association between insulinemia and bone mass, and Yi-Hsiu Fu et al. found an increased risk of osteoporosis when HOMA-β≥100 and HOMA-IR≥2 in a diverse population (79). Additionally, IR is a crucial risk factor for deterioration of renal function in non-diabetic chronic kidney disease (CKD) and hypertension (80). We also noted the effect of IR in the studies related to postburn trauma (81), postadolescent acne (82), gastro-esophageal reflux disease (GERD) (83) and other diseases.
3 Pathogeny of IR
The pathogenesis of IR is the result of the interaction of environmental and genetic factors. Its mechanism of development mainly includes abnormalities in the internal environment, such as inflammation, hypoxia, lipotoxicity, immune environment abnormalities, and abnormal metabolic functions, including metabolic tissues and metabolites.
3.1 Heredity
IR and metabolic disorders are commonly clustered in families, which is thought to be the result of an interaction of environmental and genetic factors, although the full genetic background of these conditions remains incomplete (84, 85). Genetic factors associated with IR can be classified as abnormal structure of insulin, genetic defects in the insulin signaling system, genetic defects related to substance metabolism, and other related genetic defects. Mutations in certain insulin-related genes produce mutant human insulins, including Chicago insulin (F25BL*), Los Angeles insulin (F25BS) and Wakayama insulin (V3AL), which have been shown to have significantly reduced insulin bioactivity and decreased binding affinity to the insulin receptor, with consequent effects on insulin sensitivity (86, 87). There are also rare mutations in insulin receptor genes leading to reduced number of cell surface receptors and defective insulin receptor pathways causing hereditary IR, which are found in patients with genetic syndromes of severe IR, such as type A syndrome of extreme IR, leprechaunism, Rabson-Mendenhall syndrome and Donohue syndrome (88, 89). More importantly, since many molecular pathways are involved in energy homeostasis and metabolism, IR is the result of a certain number of mutations in multiple genes, such as those related to type 4 glucose transporter (GLUT4), glucokinase, and Peroxisome proliferator-activated receptor (PPAR) nuclear receptor family, among others (90, 91). Mutations in lipid metabolic pathways, such as mutations in adipocyte-derived hormones such as leptin, adiponectin, resistin or their receptors, mutations in peroxisome proliferator-activated receptors α, γ, and δ, mutations in the lipoprotein lipase gene, and other mutations in genes related to adipose tissue formation can affect the development of glycolipid metabolism and IR (92). For example, the mutation of AKT2/PKBβ in cultured cells may disrupt insulin signaling and inhibit AKT/PKB co-expression (93). The latest advances in high-throughput genetics have revealed the relationship between protein tyrosine phosphatase N1 (PTPN1) and IR, and that the association is mediated by differences in DNA sequences outside the coding region of PTPN1 (94). Healthy carriers of the T allele of TCF7L2 rs7903146, may increase insulin secretion and lead to impaired β-cell function, which is associated with an increased risk of T2DM (95).
3.2 Environment
3.2.1 Obesity
Obesity-induced IR is characterized by impaired insulin function that inhibits hepatic glucose output and promotes glucose uptake in adipose tissue and muscle (96). It has been found that weight loss/gain could increase/decrease insulin sensitivity, and obesity and insulin resistance are causally related (97). Using the insulin clamp test, the sensitivity of tissues to insulin decreased by 30% to 40% when body weight exceeded 35% to 40% of ideal body weight. It has been found that waist circumference is closely related to IR, and an increase in waist circumference corresponds to a decrease in glucose consumption or an increase in IR. Conversely, a 10% reduction in BMI improved IR in patients with obesity and T2DM (98). Hence, obesity, especially central obesity, may induce the development of IR due to the massive accumulation of adipose tissue inducing systemic insulin resistance, including endocrine dysregulation and inflammation (99). In obese individuals, especially in those with abdominal obesity, the increase in adipose tissue tends to be more lipolytic, resulting in higher plasma free fatty acid (FFA) levels and intracellular lipid accumulation. Elevated FFA can enhance the phosphorylation of serine residues of insulin receptor substrate (IRS) by activating a series of protein kinases such as c-Jun N-terminal kinase (JNK), whose activity is abnormally increased in obese patients (100, 101). Another mechanism linking obesity and IR is chronic inflammatory responses, including increased production and release of pro-inflammatory factors such as TNF-α, IL-6, and C-reactive protein, which cause insulin resistance in liver, skeletal muscle, and adipose tissue through insulin-interfering signaling pathways (102).
3.2.2 The effect of diseases and drugs
Several physiopathological factors and therapeutic causes, such as chronic hyperglycemia, high free fatty acidemia, certain drugs, such as glucocorticoids, pregnancy, and increased insulin-antagonistic hormones in the body all contribute to the occurrence of IR. There is a pathophysiological relationship between chronic obstructive pulmonary disease (COPD) and IR, partly because the two conditions share common risk factors, such as smoking and lack of physical activity. In addition, systemic effects (deterioration of physical inactivity and sedentary behavior, inflammation) and corticosteroid therapy in patients with COPD may also play a role (103). Also, IR is a common condition after organ transplantation, which leads to new-onset diabetes and metabolic syndrome after transplantation, and subsequent hyperglycemia may significantly increase the morbidity and mortality of cardiovascular disease after kidney transplantation (104, 105). This is due to post-transplant treatment with immunosuppressive agents such as sirolimus, cyclosporine, steroids, etc., leading to IR and metabolic complications (106, 107). In both rodents and humans, exogenous synthetic glucocorticoids such as prednisolone and dexamethasone may induce a number of adverse effects when administered in excess or for prolonged periods, including the development of glucose intolerance, islet-cell dysfunction, IR, hyperglycemia, and dyslipidemia (108, 109). In contrast, almost all morphophysiological changes induced by dexamethasone in the endocrine pancreas are reversed after cessation of treatment (110). Statins may increase IR in peripheral tissues by impairing insulin sensitivity and islet β-cell secretion after long-term use, as it impairs Ca2+ signaling in pancreatic β-cells and downregulates GLUT4 in adipocytes (111).
3.2.3 Aging
Advanced age is an important factor in increasing susceptibility to IR. With increasing age, there is insufficient insulin secretion and a progressive decrease in glucose tolerance, as well as increasing IR due to sarcopenia, excess adiposity and osteoporosis (112, 113). According to epidemiology, the prevalence of IR and T2DM is high in the elderly population (112, 114). This is associated with an increased prevalence of central obesity and increased visceral fat in the aging population (99, 115). In addition to this, factors that increase the risk of IR in the elderly are free radicals that contribute to oxidative stress in old age, and mitochondrial dysfunction (115–117). The paper by Petersen et al. published in the journal Science mentions that older subjects clearly showed reduced insulin-stimulated muscle glucose metabolism compared to younger subjects. To explore the reasons for this, the investigators found increased fat accumulation in muscle and liver tissue as assessed by 1H NMR spectroscopy and an approximately 40% reduction in mitochondrial oxidative and phosphorylation activity as assessed by in vivo 13C/31P NMR spectroscopy (118). According to the result of an animal experiment, compared with young mice, aged mice are more susceptible to IR, due to reduced levels of glycolytic proteins and reduced flexible to diet, caused by reduced mitochondrial β-oxidation capacity (119). However, these hypotheses still need to be further tested and further understanding of the metabolic changes associated with aging.
4 The mechanism of IR
The balance of insulin action involves multiple processes in several glucose-utilizing organs or organs, including the liver, adipose tissue, skeletal muscle and kidneys. These metabolic processes receive complex signal regulation. The etiology and pathogenesis of IR are complicated, and the main pathological mechanisms include abnormalities in receptor binding, environment inside the host, intracellular factors, autophagy and intestinal microecology. It is noteworthy that the mechanisms of IR occur somewhat differently in different insulin receptor tissues, and IR appears in a different order, where the initial appearance of IR is in adipose tissue. However, they interact with each other and may eventually develop into systemic IR, a phenomenon verified in observational studies in humans (120–122). In-depth study of the pathogenesis of IR and multiple research directions have become the key to solving the challenges of IR and its related metabolic diseases today. The effects of insulin signaling pathways and the effects of inflammatory cytokines and FFA on them are shown in Figure 2.
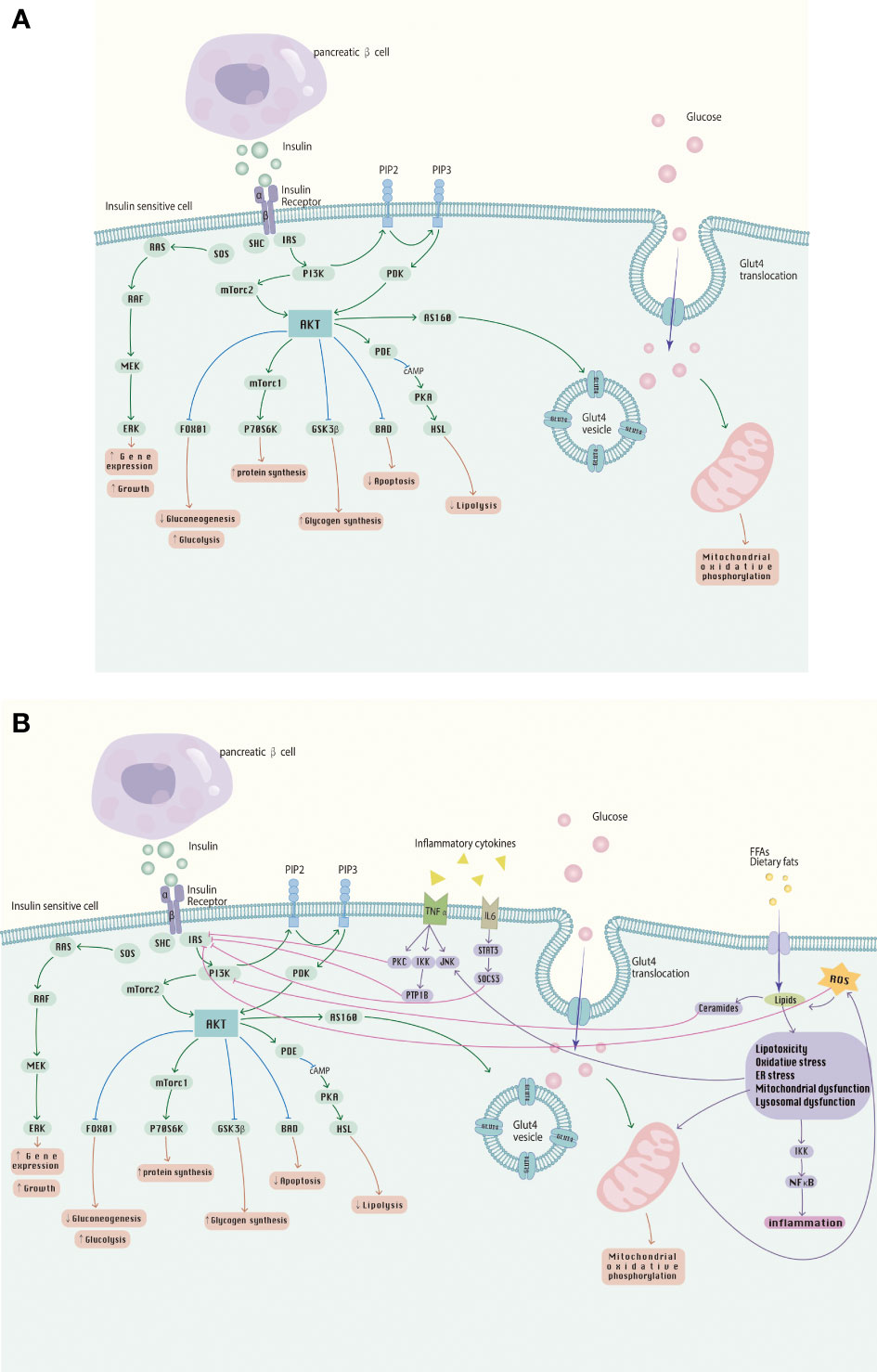
Figure 2 (A) The insulin signaling pathway; (B) Abnormalities in the insulin signaling pathway caused by inflammatory cytokines, FFA, etc., involved in the development of IR.
4.1 Insulin receptor defects
Insulin receptors (INSR) which is a tyrosine kinase, bind specifically to insulin and play a key role in insulin-mediated glucose homeostasis and cell growth (123, 124). Impaired INSR binding mainly refers to a decrease in the affinity and number of target receptors on the cell membrane or structural abnormalities of the target receptors that affect insulin binding to the receptor (125). There are several, albeit rare, severe diseases of insulin resistance, including leprechaun’s disease, Rabson-Mendenhall syndrome, or type A insulin resistance syndrome, where insulin binding is severely reduced due to mutations in the insulin receptor gene (88). The insulin receptor substrate protein is generally considered a node in the insulin signaling system, which is closely related to the development of insulin insensitivity. At the molecular level, the crosstalk between the downstream nucleotide-binding oligomerization domain (NOD) 1 effector and the insulin receptor pathway may inhibit insulin signaling by reducing the action of insulin receptor substrates (126). Insulin activates insulin receptor tyrosine kinases, which are capable of aggregating and phosphorylating various substrate docking proteins, such as the insulin receptor substrate (IRS) protein family. Of the four mammalian IRS proteins (IRS-1, IRS-2, IRS-3, IRS-4), IRS1 and IRS2 play key roles in regulating growth and survival, metabolism and aging. They are key substrates of insulin signaling and play an important role in insulin signaling by binding to PI3K and inducing downstream pathways. At the molecular level, dysregulation of the signaling pathway by insulin receptor substrates (IRS) is one of the most common causes of this disease. The current data suggest that insulin-stimulated kinases mediate feedback serine/threonine residues phosphorylation in the IRS and desensitization of proximal insulin signaling plays an important role in the pathogenesis of IR (127). For example the double-stranded RNA-dependent protein kinase (PKR) has also been shown to upregulate the inhibitory phosphorylation of IRS1 and the expression of IRS2 in liver and muscle cells, thereby regulating the insulin signaling pathway. Mediated by two other protein kinases, JNK and IKK, PKR upregulated the phosphorylation of IRS1 at Ser312 and inhibited the tyrosine phosphorylation of IRS1 (128, 129). IRS1 has also been shown to be a target of ceramide-induced Pbx regulating protein 1 (Prep1) and p160 in muscle cells, and the Prep1-p160 axis also affects IRS-1 stability (130). In addition, protein tyrosine phosphatase 1B (PTP-1B), protein kinase C (PKC) and tyrosine residue receptor phosphorylation levels are involved in the regulation of receptor-insulin binding in target tissues. It has been shown that inhibition of PTP1B, a main negative regulator of insulin receptor signaling, can improve glucose homeostasis and insulin signaling (131). In the insulin receptor signaling cascade, protein tyrosine kinase amplifies the insulin signaling response, and phosphatase is necessary to regulate the rate and duration of the reaction (132).
IR occurs in a variety of tissues, including skeletal muscle, liver, kidney and adipose tissue, and its mechanisms are specific. Among the target organs of insulin, bone, as an endocrine organ, can regulate energy homeostasis by altering insulin sensitivity, dietary behavior, and adipocytes (133). There seems to be a bilateral relationship between bone and IR that binds them together in a biological partnership (134). Among them, skeletal muscle estrogen receptor α plays a crucial role in maintaining systemic glucose homeostasis and insulin sensitivity (135). Beyond that, skeletal muscle is an important insulin-sensitive tissue, accounting for approximately 80% of insulin-dependent glucose uptake. It has been repeatedly demonstrated that skeletal muscle tissue plays an important role in the maintenance of systemic glucose homeostasis and overall metabolic health. In addition, the crosstalk between muscle factors and adipokines leads to negative feedback, which in turn aggravates muscle reduction obesity and IR (136). In the kidney, the effector cells of insulin are podocytes in which nucleotide-binding oligomerization domain 2 (NOD2) is highly expressed. NOD2 is a major member of the NOD receptor family and is involved in the innate immune response. It induces podocyte IR by activating the inflammatory response (137). In terms of hepatic IR, IRA, one of the isoforms of the insulin receptor, whose expression in the liver of mice on a high-fat diet increase hepatic glucose uptake, decrease lipid accumulation, and reduce or at least delay the development of fatty liver and NASH. This suggests that a gene therapy approach to hepatic IRA expression could act as a facilitator of glucose uptake in IR states (138–140).
4.2 Abnormal insulin signaling
Insulin acts by binding to the INSR and activating downstream signaling pathways which have been extensively studied. After binding to INSR, insulin acts mainly through two major signaling pathways, the phosphatidylinositol 3-kinase (PI3K)–serine-threonine kinase (AKT)/protein kinase B (PKB) pathway, which plays a part in regulating metabolism, and the Ras–mitogen-activated protein kinase (MAPK) pathway, which is mainly responsible for controlling cell growth and differentiation (141). Although where the defect occurs in the insulin signaling pathway remains a matter of doubt, many key insulin signaling pathway components have been identified. These components can be divided into proximal components, including insulin receptors, insulin receptor substrates, PI3K, and AKT/PKB, and distal components, representing various components downstream of AKT/PKB, including TBC1D4, GSK3, and PDE3B. IR is caused by defects in one or more of these signaling components (142).
4.3 Abnormal internal environment
Environment, such as diet and exercise, and genetics, as well as the interaction between the two, play a major role in the development of IR and metabolic disease. Exercise and dietary habits may directly or indirectly drive changes in the host internal microenvironment. Current research suggests that extracellular influences such as inflammation, hypoxic environments, lipotoxicity or immune abnormalities can trigger intracellular stress in key metabolic target tissues, which impairs the normal metabolic function of insulin in these cells thereby causing the progression of whole-body IR (143).
4.3.1 Inflammation
Obesity characterized by a chronic, low-grade inflammatory state is closely associated with IR. In the obese state induced by diet, there is a significant increase in lipid accumulation and increased secretion of pro-inflammatory cytokines by adipocytes and macrophages, including pro-inflammatory response factors such as tumor necrosis factor‐α (TNF-α), monocyte chemotactic protein‐1 (MCP‐1) and interleukins (IL), as well as increased production and release of C-reactive protein (CRP), which induce insulin resistance through multiple mechanisms, including activation of Ser/Thr kinases, decreasing IRS-1, GLUT4, and PPARγ expression, or activation of SOCS3 in adipocytes (99, 144–146).
4.3.1.1 Inflammatory factors
The mechanisms of inflammation leading to IR mainly include inflammatory factors acting on the insulin signaling system to interfere with INSR signal transduction. TNF-α and IL-1β are additional macrophage-derived pro-inflammatory mediators that directly affect insulin sensitivity (147, 148). TNF-α stimulates insulin-resistant adipose tissue through IRS protein interference by abnormal signals on phosphorylated serine residues of IRS1 (149). In addition, TNF-α could affect insulin signaling through serine phosphorylation and kinase pathway defects (99, 150). CRP is another marker of inflammation associated with IR and metabolic diseases and is a widely used clinical biomarker. CRP binds to leptin, blocks leptin signaling and modulates its central action and hypothalamic signaling, thereby directly interfering with energy homeostasis, insulin sensitivity and glucose homeostasis (151, 152).
4.3.1.2 Inflammatory pathway
Furthermore, the activity of signaling molecules in inflammatory pathways such as IκBα kinase β (IKKβ)/nuclear factor-kappaB (NF-κB) and JNK1 was found to be activated in adipose tissue and liver (100, 153). The above pro-inflammatory cytokines exert their effects by stimulating major intracellular inflammatory pathways, and the activation of these pathways also promotes increased expression of the inflammatory factors involved in IR. Toll-like receptor (TLR), especially TLR4, participates in IR-related inflammation by increasing the gene expression of IKKβ, NF-κB transcription factors, and pro-inflammatory mediators in adipose tissue macrophages (154–157). IKK is an enzyme complex that activates the NF-κB transcription factor (144). It has also been shown that NF-κB receptor activator (RANKL) is a potent stimulator of NF-κB and that systemic or hepatic blockade of RANKL signaling leads to significant improvements in hepatic insulin sensitivity and prevents the development of diabetes (158). On the other hand, metabolic stress activates the JNK signaling pathway, which can inhibit the tyrosine phosphorylation level of IRS in target tissue cell membranes, which in turn affects downstream insulin-related signaling such as PI3K and Akt/PKB, resulting in IR (99, 159). And JNK signaling in adipocytes leads to an increase in circulating concentrations of hepatic factor fibroblast growth factor 21 (FGF21), which regulates systemic metabolism (160).
4.3.1.3 Immunocytes
In the pathogenesis of IR and metabolic diseases, immune cells play a crucial role. Adipose tissue contains most types of immune cells, which under conditions of obesity contribute to a complex network of inflammation and IR with activation and infiltration of pro-inflammatory immune cells in adipose tissue, including macrophages, neutrophils, eosinophils, mast cells, NK cells, MAIT cells, CD4 T cells, CD8 T cells, regulatory T cells and B cells, as well as high levels of pro-inflammatory molecules (161). Among them, adipose tissue macrophages can be divided into M1 phenotype (pro-inflammatory macrophages) and M2 phenotype (anti-inflammatory macrophages), representing the two extremes of macrophage polarization. M1 macrophages are highly antimicrobial and antigen-presenting, producing pro-inflammatory cytokines, such as TNF-α, and reactive oxygen species (ROS) that worsen inflammation, mast cells, neutrophils and dendritic cells directly or indirectly exacerbate IR (162). In contrast, M2 macrophages help maintain insulin sensitivity in lean adipose tissue, as well as eosinophils and innate lymphocytes appear to have a protective effect on glucose homeostasis and insulin sensitivity (163–168). Crosstalk between M1-M2 macrophage polarization plays an important role in IR through the shift from M1 to M2 phenotype and activation of transcription factors (162, 169). Dysregulation of visceral adipose tissue macrophage (ATM) response to microenvironmental changes underlies the development of abnormal local and systemic inflammation and IR (170). In the obese state, enhanced macrophage infiltration and secretion of various inflammatory cytokines in white adipose tissue activate JNK and NF-κB, causing local and systemic IR (171, 172). Macrophages can alter their phenotype in response to changes in the microenvironment and macrophage differentiation.
In the past, more attention has been paid to the regulation of insulin sensitivity by innate immune cells, particularly macrophage mediated, which have been mentioned before. Cells of the adaptive immune system, B lymphocytes and T lymphocytes, and their respective subsets, are also thought to be important regulators of glucose homeostasis and play an important role in the immunopathogenesis of autoimmune diabetes (168, 173, 174). Studies have shown that CD4(+) T lymphocytes in visceral adipose tissue (VAT) control the progression of metabolic abnormalities associated with obesity, including expansion of adiposity and pathogenic VAT T cells, which can be successfully reversed by immunotherapy (175, 176). Impaired through an adaptive immune response, IR can also be driven by inflammation and dysregulation of the gut microbiota, as in pathogen-induced periodontitis (177). In addition, the intestinal immune system is an important regulator of glucose homeostasis and obesity-related IR in turn affects intestinal permeability and thus systemic IR (178). Another essential part of the immune defense system is the complement system. It plays an important role in activating innate and adaptive immune responses, promoting apoptosis, and eliminating damaged endogenous cells. Patients with obesity exhibit activation of the complement system in their adipose tissue, which is connected to changes in glucose metabolism and subclinical inflammation (179).
4.3.2 Hypoxia
Adipose tissue hypoxia is causally related to obesity-induced IR, especially in high-fat diet (HFD) fed and early obese patients, as adipocyte respiration becomes uncoupled, resulting in a state of increased oxygen consumption and relative adipocyte hypoxia (180). Clinically, obstructive sleep apnea (OSA), characterized by intermittent hypoxia (IH), is a widely prevalent respiratory disorder with a particularly high prevalence in obese patients and is associated with IR and metabolic diseases such as hypertension, cardiovascular risk and NAFLD (181, 182). Not only in obese individuals, but an animal study found that IH cause acute IR in lean or healthy mice, which is related to reduced glucose utilization in oxidized muscle fibers. As the glucose infusion rate decreased, hypoxia induced systemic IRA (183). The key regulators of oxygen homeostasis in response to hypoxia are the hypoxia-inducible factors (HIFs), a family of transcription factors activated by hypoxia. Adipocyte hypoxia could trigger HIF-1α induction causing adipose tissue inflammation and IR (180, 184). HIF-1-mediated activation of NOX4 transcription and the consequent increase in H2O2 led to intermittent hypoxia-induced pancreatic β-cell dysfunction (185). In hypoxic adipocytes, HIF-1α activates the NLRP3 inflammasome pathway and stimulates IR by upregulating the expression of pla2g16. In obesity-induced intestinal hypoxia, HIF-2α increases the production of ceramide, to promote the expression of the key enzyme sialidase 3 encoding Neu3, which leads to the development of IR in obese mice induced by a high-fat diet (186). While in skeletal muscle, hypoxia is a stimulus stimulating GLUT4 translocation via activation of AMPK, causing defects of glucose transport and this may counteract IR (187).
4.3.3 Lipotoxicity
Insulin regulates lipid metabolism through the typical insulin signaling cascade, while metabolites can also directly regulate insulin sensitivity by modulating components of the insulin signaling pathway (188). Lipids have multiple roles as signaling molecules, metabolic substrates and cell membrane components, and can also alter proteins that affect insulin sensitivity (189). Lipotoxicity is when the storage capacity of adipose tissue is overloaded due to obesity, overnutrition, etc., leading to uncontrolled accumulation of lipids in ectopic non-adipose tissues (e.g., liver, heart, pancreas, and muscle). High concentrations of lipids and lipid derivatives cause deleterious effects on cells through mechanisms including oxidative stress, endoplasmic reticulum (ER) stress, c-Jun NH2-terminal kinase (JNK)-induced toxicity, and BH3-pure protein-induced mitochondrial and lysosomal dysfunction (190, 191). Numerous studies have reported that Adipose tissue dysfunction and lipotoxicity play a role in metabolic disorders and IR (192, 193). This is associated with a chronic elevation of free fatty acids (FFA, also called non-esterified fatty acids) in plasma due to adipose tissue dysfunction (99). Adipose malnutrition or adipose tissue dysfunction can lead to pathologically elevated FFAs. Chronically elevated FFAs appear to cause adipocyte production of inflammatory factors, decreased insulin biosynthesis, glucose-stimulated insulin secretion, and glucose sensitivity in β-cells. The ER stress pathway is a key mediator of inflammation induced by serum excess FFA and IR in various cell types, and PERK and IKKβ are key signaling components (194). The obesity-induced increase in adipocyte volume and tissue mass will lead to inflammation, additional disturbances in adipose tissue function, and ultimately adipose tissue fibrosis (195). Adipose tissue macrophages are an abundant immune component of hypertrophy, which plays a key role in diet-induced T2DM and IR (196).
In renal ectopic lipid accumulation, lipotoxicity promotes podocyte injury, tubular injury, thylakoid proliferation, endothelial cell activation and macrophage-derived foam cell formation, which contribute to the development of renal IR and other renal diseases, especially diabetic nephropathy (197). In skeletal muscle, sustained nutrient overload of L6 myotubes leads to lipotoxicity that promotes activation of the IKKβ-NFkB pathway in muscle cells, inducing increased cellular ROS and impaired insulin action in the myotubes (198). Saturated fatty acids are known to increase the production of lipotoxic products such as ceramide and diacylglycerol, which disrupt islet beta-cell function, vascular reactivity and mitochondrial metabolism, and also play a key role in the induction of muscle IR (199–201). Similarly, defective fatty acid oxidation (FAO) and consequent lipotoxicity in cardiac cells induce a range of pathological responses, including oxidative stress, DNA damage, inflammation and insulin resistance. The obesity-mediated atrial fibrillation and structural remodeling can be attenuated by promoting FAO, activating AMPK signaling and attenuating atrial lipotoxicity through levocarnitine (LCA) (202). Lysophosphatidic acid (LPA) is an effective, biologically active lipid. After binding to G protein-coupled receptors, it can profoundly affect cell signal transduction and function. Metabolic and inflammatory disorders, including obesity and IR, are associated with modifications in LPA signaling as well as the production and function of autocrine motility factors (203). Additionally, it has been discovered that the anti-adipogenic transcription factor GATA-3 is a possible molecular target that affects adipogenesis. Those with obesity and IR exhibit increased GATA-3 expression when compared to insulin-sensitive individuals with BMI matches (204). While lifestyle interventions such as physical activity have been confirmed to have a positive effect on insulin sensitivity in skeletal muscle, affecting lipid metabolism (205).
4.3.3.1 Ceramide accumulation
Ceramides are a family of lipid molecules consisting of sphingosine and a fatty acid. The synthesis of de novo ceramides depends on the availability of free fatty acids, especially palmitate, whose over-intake may lead to an excessive accumulation of ceramides (206). In addition to their function in lipid bilayers, these molecules are also thought to be biologically active agents involved in a variety of intracellular pathways, such as free radical production, release of inflammatory cytokines, apoptotic processes, and regulation of gene expression. Ceramides are metabolic products that accumulate in individuals suffering from obesity or dyslipidemia and alter cellular processes in response to fuel overload (207). ceramides accumulation over time modulates signaling and metabolic pathways that drive lipotoxicity and IR, causing tissue dysfunction (208). Numerous studies have been conducted in recent years to confirm the critical role played by ceramides in glucose homeostasis and insulin signaling (199). These evidence are particularly strong in skeletal muscle, while the data in liver and WA are somewhat more equivocal (209, 210). Ceramides are synthesized by ceramide synthase (CerS) through N-acylation. To date, six mammalian CerS have been identified (CerS1-6) that show different affinities for the fatty acid acyl-CoA chain length used for sphingomyelin N-acylation. Among them, CerS1 is most abundant in skeletal muscle and is responsible for the synthesis of C18:0-Cer, which negatively regulates insulin sensitivity in obese and/or T2D subjects (211). CerS2 is the major isomer in the liver that preferentially makes extra-long-chain (C22/C24/C24:1) ceramides, which inhibits β-oxidation, leads to a compensatory increase in long-chain C16-ceramides, and makes one susceptible to diet-induced fatty liver and IR (212). CerS6 is specific for C14 and C16 acyl chain lengths, and CerS6 levels are significantly increased in obese adipose tissue (212, 213). The main mechanism by which ceramides promote IR is through inhibition of proximal insulin signaling components, such as Akt/PKB activity. Ceramide can inhibit Akt/PKB activity by increasing protein phosphatase 2A (PP2A) activity to stimulate Akt/PKB dephosphorylation and blocking Akt/PKB translocation through PKCζ (214). Activation of PP2A inhibits Akt/PKB by impairing serine phosphorylation of Akt/PKB, thereby reducing the transfer of GLUT4 to the plasma membrane and thus reducing glucose uptake (215, 216). In addition, ceramide may cause IR by accumulating in mitochondria and causing mitochondrial reactive oxygen species (ROS) or by promoting the secretion of pro-inflammatory factors (217).
4.3.3.2 Diacylglycerols accumulation
Another lipid metabolite closely associated with IR is DAG, whose accumulation in skeletal muscle, adipocytes and liver is thought to promote IR by altering cellular signaling at its specific location, due to increased serum FFA levels (218). The DAG hypothesis of IR is that the interference of activated PKC, especially the novel PKC isoforms including δ, ϵ, ν, and θ, with insulin signaling is due to the accumulation of DAG in insulin-sensitive tissues (219, 220). In particular, 1,2-DAG, which derives from esterification and accumulates mainly in the membranes, is clearly associated with PKC activation, and these isoforms then phosphorylate IRS1 serine with the result that decrease PI3K activation (211, 221).
It is worth noting that the role of intracellular ceramide and DAG in IR is controversial and that defects in these components are unlikely to be the sole cause of IR. It is true that not all studies have confirmed a role for the DAG-PKC-insulin receptor pathway in IR; for example, some studies have shown that PKCϵ deficiency in the liver has no effect on systemic insulin sensitivity in mice (222), and there are also experiments in which acute knockout of PKCϵ in the liver protects rats from IR (218). Therefore, more in-depth studies on proximal insulin signaling with DAG and ceramide are still needed.
4.4 Organelle interaction
Organelles, including the endoplasmic reticulum (ER), mitochondria and endoplasmata, contribute to a range of cellular functions through their unique local environment and molecular composition. Organelles can actively communicate and cooperate with each other through vesicle trafficking pathways and membrane contact points (MCSs) to maintain cellular homeostasis, which facilitates the exchange of metabolites and other information required for normal cellular physiology (223). Imbalances in organelle interactions may lead to various pathological processes, such as imbalances in cellular energy metabolism (224). Recent studies have shown that mitochondria could interact with various organelles (225), which are essential for energy metabolism and cell survival, and increasing evidence shows that mitochondrial dysfunction in skeletal muscle and mitochondrial overactivation may induce IR (226).
The production of mitochondrial ROS is thought to adjust skeletal muscle insulin sensitivity. Mitochondrial quality control mechanisms are regulated by PGC-1α, which may affect age-related mitochondrial dysfunction and insulin sensitivity (227). The continuous processes that occur in the skeletal muscle after excessive intake of a high-fat diet include the accumulation of cytosolic fatty acids, increased production of ROS, mutation, and aging. The ensuing mitochondrial dysfunction could lead to decreased β-oxidation, respiratory function, and increased glycolipid toxicity. Together, these events induce IR in the skeletal muscle (228). The physical contact site between the mitochondria and endoplasmic reticulum (ER) is called the mitochondrial-associated membrane (MAM). The imbalance of MAMs significantly leads to IR. ER stress may be the main mechanism by which MAM induces IR in the brain, especially in the hypothalamus (229, 230). Exosome-like vesicles (ELVs) are the smallest type of extracellular vesicles released from cells that play a role in cell crosstalk because they regulate insulin signaling and β-cell quality, and released ELVs leading to IR or β-cell apoptosis (231).
4.5 Other influence elements
4.5.1 Phosphatase and tensin homolog
PTEN is not only a tumor suppressor gene but also a metabolic regulator. Under physiological and T2D conditions, PTEN also has a negative regulatory function in insulin signaling through its inhibition in the PI3K pathway (232, 233). PTEN reduces the level of phosphatidylinositol-3, 4, 5-phosphate (PIP3). This leads to impaired insulin signaling and promotion of IR in the pathogenesis of T2D. The function of PTEN in regulating insulin signaling in different organs has been identified. The role of PTEN in the regulation of insulin action in many cell types has been elucidated through mouse models of lacking PTEN in metabolic organs and in vitro cell culture (234, 235). Interventions targeting PTEN regulatory signaling may therefore be a promising target aimed at reversing insulin resistance. Interventions targeting PTEN regulatory signaling may therefore be a promising target aimed at reversing insulin resistance.
4.5.2 Vitamin D
In addition to its effects on skeleton, Vit D has significant effects on pancreatic β-cells function and metabolic syndrome including blood pressure, abdominal obesity, glucose metabolism associated with it, as calcitriol functions as a chemical messenger by interacting with calcium flux-regulating receptors on beta cells (236). As the results of a meta-analysis showed, there was an inverse relationship between serum Vit D concentration and metabolic syndrome risk in the general adult population in cross-sectional studies (237). The molecular mechanisms of Vit D deficiency involved in the development of IR may be because it maintains normal resting levels of ROS and Ca2+ in pancreatic β-cells and also reduces the degree of IR-related pathological degrees, such as oxidative stress and inflammation (238). Vitro studies showed that Vit D could regulate lipid and glucose metabolism in adipose tissue, skeletal muscle and liver, and pancreatic insulin secretion (239).
4.5.3 Minerals
Minerals are essential micronutrients for the human body. Deficiencies in certain micronutrients due to differences in diet composition may lead to imbalances in glucose homeostasis and IR (240).
Magnesium is a cofactor required for glucose access to cells and carbohydrate metabolism, and it has the function of regulating the electrical activity of pancreatic beta cells and insulin secretion (240). Mechanistically explained, magnesium is a cofactor in the downstream action of the insulin cascade. Low magnesium ion levels lead to defective tyrosine kinase activity, blocking intracellular insulin action and altered cellular glucose transport, thus promoting IR (241). On the other hand, magnesium deficiency inhibits cellular defenses against oxidative damage and triggers chronic systemic inflammation that enhances IR. As demonstrated in a longitudinal study, magnesium intake was also inversely associated with high-sensitivity CRP, IL-6 and fibrinogen levels, as well as HOMA-IR (242). There is evidence suggesting that magnesium supplementation attenuates IR in patients with hypomagnesemia-associated IR (243). Also, animal studies have shown that dietary magnesium supplementation to increase plasma magnesium concentrations reduces blood glucose levels, improves mitochondrial function, and reduces oxidative stress in diabetic mice (244). However, new intervention studies are still needed to clarify the role of nutrients in the prevention of this metabolic disorder, as well as to standardize the type, dose, and timing of magnesium supplementation.
Zinc is an essential micronutrient for metabolism, which plays a particularly critical role in the islets. Diabetes affects zinc homeostasis, and disturbances in zinc homeostasis have been associated with diabetes and IR (245). Because zinc is an essential component of insulin, it regulates islet cell secretion and promotes its binding to hepatocyte membranes while maintaining phosphorylation and dephosphorylation levels of the receptor. Zinc influx mediated by Slc39a5, a zinc exporter in pancreatic β-cells, plays a role in insulin processing and secretion by inducing Glut2 expression through Sirt1-mediated activation of Pgc-1α (246). In addition, zinc acts as a pro-antioxidant to reduce the formation of ROS, which is particularly beneficial in aging and IR (247). Mineral deficiencies are directly or indirectly associated with oxidative stress, which ultimately leads to IR or diabetes (240).
4.6 Nervous system effects
The brain is also an insulin-sensitive organ with a large number of insulin receptors distributed (248, 249). The action of insulin in the brain produces a variety of behavioral and metabolic effects that influence eating behavior, peripheral metabolism, and cognitive performance (250). Disturbances in the role of insulin in the brain reveal a possible link between metabolism and cognitive health. The hypothalamus plays a fundamental role in the survival and control of physiological processes necessary for vital physical functions, including various endocrine functions. Injecting insulin via intranasal administration leads to an increase and subsequent decrease in plasma insulin, affecting peripheral metabolism, and a decrease in BOLD signaling and cerebral blood flow in the hypothalamus is observed (250, 251). It appears that the effects of central insulin may have a biphasic effect on peripheral insulin sensitivity (251). Insulin signaling has been shown to affect the molecular cascade of hippocampal plasticity, learning, and memory (252). Furthermore, the insulin-responsive glucose transporter GluT4 has a key part in hippocampal memory processes, and reduced activation of this transporter may underlie IR-induced cognitive deficits (253).
4.7 Autophagy
Autophagy is a self-degrading process that is conserved in all eukaryotic cells and plays a crucial role in balancing energy sources during critical periods of development and in response to nutritional stress. Autophagy also promotes cellular senescence and cell surface antigen presentation, prevents genomic instability and necrosis, and it is an important mechanism for a variety of physiological processes, such as cellular homeostasis, senescence, immunity, oxidation, differentiation, and cell death and survival (254). Recent studies have shown that autophagy is an important regulator of organelle function and insulin signaling, and that loss of autophagy is a key component of defective insulin action in obesity, which may be specifically related to ER function (255). It has been found that autophagy deficiency and its resulting mitochondrial dysfunction increase fibroblast growth factor 21 (Fgf21) expression through the induction of Atf4. The induction of Fgf21 promotes protection against diet-induced obesity and IR (256). In addition, exercise induces autophagy through the regulator BCL2, which may contribute to beneficial metabolic effects and improve IR in muscle (257).
4.8 Intestinal microecology
In addition to the aforementioned influences such as metabolites and cytokines, the 100 trillion bacterial colonized gut microbiota can also contribute to IR (220, 258). Patients with metabolic syndrome showed increased insulin sensitivity after six weeks of infusion of gut microbiota from lean individuals. Levels of gut microbiota producing butyrate, which has been shown to prevent and treat diet-induced insulin resistance in mice by promoting energy expenditure and inducing mitochondrial function, were also increased (259, 260). Dietary reasons for obesity may promote IR both through mechanisms independent of the gut microbiota and through mechanisms dependent on the bacterial community (261). Intestinal dysbiosis is associated with the transfer of bacterial lipopolysaccharide (LPS) into the systemic circulation and its induction of metabolic endotoxemia, leading to a chronic subclinical inflammatory process and the development of IR through activation of toll-like receptor 4 (TLR4) (261–263). In addition to the LPA mentioned above, branched-chain amino acids (BCAAs) are another harmful gut microbially regulated metabolite whose levels are increased in the serum metabolome of IR individuals. Prevotella copri has been shown in mice experiments to induce IR, exacerbate glucose intolerance and increase circulating levels of BCAAs (264). Moreover, gut microbiota-derived short-chain fatty acids (SCFA) may improve IR and prevent T2DM by reducing the secretion of pro-inflammatory cytokines and chemokines and decreasing local macrophage infiltration, as well as increasing the lipid storage capacity of white adipose tissue (121, 265, 266). Taken together, targeting gut microbes may have the potential to reduce IR and decrease the incidence of related metabolic diseases.
5 Treatment of IR
5.1 Nondrug treatment
Today’s modern lifestyle is characterized by reduced energy expenditure, consumption of high-calorie junk food and fast food, sedentary lifestyle, irregular eating occasions and eating times especially for late night snacking, and chronic psychological stress. This lifestyle triggers several mechanisms such as the development of IR that aggravate metabolic stress. Studies have shown that lifestyle interventions through small weight loss (7-10%), 150 minutes of moderate intensity exercise per week and behavioral therapy approaches are very effective in preventing and treating IR and T2DM (267). According to the ADA/EASD consensus guidelines, lifestyle recommendations are the first-line therapy, followed by metformin for T2D. Next, the contribution of non-pharmacological therapies, including exercise and diet, to the alleviation of IR will be elaborated.
5.1.1 Physical exercises
Exercise is well known to improve metabolic disease by improving obesity and enhancing insulin sensitivity. A meta-analysis determined the effectiveness of a structured exercise intervention program for IR in T2DM, and the evidence highlights that regular exercise improves glycemic control and therefore can be recommended for reducing IR with a moderate level of evidence (268). As we know, physical exercise increases the oxidative capacity and biogenesis of mitochondrial substrates in skeletal muscle. It was shown that treadmill training modulates the increase in mitochondrial substrate oxidation in liver and skeletal muscle induced by a high-energy diet in mice, disconnecting it from pyruvate and acetyl CoA-driven lipid synthesis. This may help prevent the long-term deleterious effects of excessive nutritional intake on liver mitochondrial function and insulin sensitivity, thereby preventing the development of metabolic diseases such as fatty liver and NAFLD (269). As described in the mechanism section, intermittent hypoxia leads to disturbances in the gut microbiota-circulating exosome pathway, disrupting adipocyte homeostasis and leading to metabolic dysfunction manifested as IR, whereas experiments have shown that such changes can be attenuated by physical activity, as regular non-strenuous activity will lead to substantial improvements in the gut microbiota-exosome pathway (270). In addition, available data suggest that aerobic exercise can lead to increased insulin sensitivity and enhanced glucose metabolism through a variety of different molecular mechanisms, including upregulation of insulin transporters on cell membranes of insulin-dependent cells, reduction of adipokines, normalization of redox status, improvement of β-cell function, regulation of IRS-1 phosphorylation, reduction of ceramide plasma levels, and induction of angiogenesis, which may lead to a reduced incidence of diabetic complications, as well as other metabolic effects (271, 272). Other forms of exercise, such as yoga, have also been shown to improve IR. Several meta-analyses have shown that yoga is a safe and effective intervention to reduce waist circumference and systolic blood pressure in patients with metabolic syndrome, particularly in improving cardio-metabolic health (273, 274). Some traditional Chinese health exercises, such as qigong and tai chi, have also been shown to have a measurable effect on weight, waist circumference, leg strength, increase HDL cholesterol, and result in significant improvements in IR (275, 276).
5.1.2 Diet and nutrition therapy
As mentioned above, high-fat diets and the obesity they induce are a major cause of IR. Conversely, weight loss, when necessary, and dietary interventions such as intermittent fasting programs that reduce carbohydrates in the diet can significantly improve glycemic and insulin responses. From the available scientific data, reducing total daytime carbohydrate intake to 40-50% of daily energy intake, such as a Mediterranean-style diet and high protein diet, is one of the key dietary habits for improving IR (267). The Mediterranean diet is characterized by a wide range of cardio-protective nutrients, with beneficial effects on several outcomes related to metabolic health, and significant beneficial changes in metabolic risk factors, including HOMA-IR index (277–279). There are also RCT studies reporting that a high-protein diet is more effective in controlling IR and glycemic variability compared to a Mediterranean diet, which may be related to the satiety and increased metabolic rate associated with a high-protein, low-sugar diet (280).
In terms of dietary composition, a key dietary strategy for treating IR and improving glycemic control is to consume foods and meals that reduce the glucose fluctuations known to induce oxidative stress and beta cell damage (281). The contribution of high-fat diets to obesity and IR is well known. However, a single-minded approach to weight loss by replacing fat intake with carbohydrates is counterproductive because it could exacerbate IR. Researchers suggest that calorie restriction for weight loss and rationing of the macronutrient composition of the diet is important. What’s more, according to the suggestion from a recent meta-analysis, low-fat dairy intake has beneficial effects on abdominal adiposity and body weight, which may be associated with a reduced risk of IR and metabolic diseases (282, 283). The possible mechanism for this is that calcium and vitamin D in supplemental dairy products may facilitate lipolysis and optimize glucose metabolism (284). Carbohydrates are the main macro-nutrient influencing the glycemic response, especially after a meal. In recent years, some researchers have proposed that consumption of carbohydrates rich in dietary fiber and low glycemic index, such as whole grains, is beneficial in improving insulin sensitivity and metabolic flexibility, independent of gut hormones (285, 286). A recent meta-analysis reported that increasing daily fiber intake by 15 or 35 grams compared to a low-fiber diet reduced homeostatic model assessment of insulin resistance (HOMA-IR), leading to improvements in glycemic control, lipids, weight, and inflammation, as well as a reduction in premature mortality (287). Not only is the amount of carbohydrate intake important, but the timing of major carbohydrate intake during the day is also a determining factor in the increase in glucose and insulin after meals and the improvement or otherwise of IR (267). The results of some randomized controlled trial (RCT) studies suggest that it is advisable to consume at least half of the carbohydrates at lunch and to avoid consuming large amounts of carbohydrates at breakfast or dinner in order to control blood glucose spikes, which may be related to diurnal variations in insulin sensitivity (288–290). Results of another study showed that 10 hours of restrictive eating improved quality of life by reducing body weight and improving blood glucose, insulin sensitivity and related metabolic disorders (291). Other dietary strategies have been shown to prevent high-fat diet-induced IR, such as the intake of flavonoid-rich natural products, like flavonoids, which upregulate the expression of related genes through cell surface G protein-coupled estrogen receptors (292).
5.2 Pharmacological treatments
Although lifestyle modification and weight loss are highly recommended to improve IR and its associated metabolic disorders, they have limited effectiveness, slow onset of action, and low feasibility. Pharmacological treatments to increase insulin sensitivity will be described next.
5.2.1 Chemotherapy
Currently, the main drugs that can effectively improve IR are anti-hyperglycemic drugs, including metformin, thiazolidinediones (TZD), sodium glucose cotransporter (SGLT)-2 inhibitors (SGLT2i), etc., which are listed in Table 1 and will be described sequentially below.

Table 1 Clinically used antihyperglycemic drugs to improve IR.
Metformin, the most commonly used insulin-sensitizing agent, has been a guideline-recommended first-line treatment for T2DM for decades and has recently found new applications in the prevention and treatment of various diseases, including metabolic disorders and cardiovascular diseases (298). A meta-analysis summarizing 31 RCT trials confirmed that treatment with metformin in populations at high risk for diabetes improved weight, lipid profile, and IR and resulted in a 40% reduction in new-onset diabetes (299). Metformin improves IR by modulating metabolic mechanisms and mitochondrial biogenesis through altering microRNAs levels by AMPK-dependent or AMPK-independent mechanisms (300). TZDs, such as pioglitazone, are potent insulin sensitizers targeting PPARγ and PI3K, regulating the transcription of nuclear transcription factors, stimulating mainly white adipose tissue remodeling, and regulating lipid flux for insulin sensitization and beta cell protection (294, 301). SGLT2i is a relatively new class of glucose-lowering drug that not only lowers blood glucose by inhibiting renal glucose reuptake, leading to increased urinary glucose excretion and lower blood glucose, but also improves insulin sensitivity in patients with T2DM by reducing body weight or glucose toxicity (302, 303). And in a randomized, double-blind, placebo-controlled clinical trial, it was shown that 8 weeks of treatment with SGLT2i empagliflozin restored insulin sensitivity in the hypothalamus of patients with prediabetes (304). Glucose-lowering drugs have also shown good, stacked effects in patients who do not have good response with one drug alone. For example, the addition of rosiglitazone to metformin can be clinically important in improving glycemic control, insulin sensitivity and beta-cell function (305). The addition of sitagliptin or metformin to pioglitazone monotherapy also leads to faster and better improvement in IR and inflammatory status parameters (306).
Other therapies, as well as some new drugs in clinical trials, such as anti-inflammatory drugs, drugs that target hepatic lipid and energy metabolism, renin-angiotensin-aldosterone system blockers, vitamin D, antioxidants, probiotics and fecal transplants, have also shown improvement in IR (220). Among them, selected clinical trials in the last decade have been listed in Table 2. As mentioned previously, low-grade chronic inflammation is associated with IR and metabolic disturbances. For example, in in vitro and in vivo mouse models of diet-induced hyperinsulinemia, low-dose naltrexone attenuates hyperinsulinemia-induced proinflammatory cytokine release and restores insulin sensitivity (307). However, it is worth noting that corticosteroids can cause IR and hyperglycemia due to their metabolic effects, and statins also increase the risk of IR, although they can reduce circulating inflammatory markers (220).
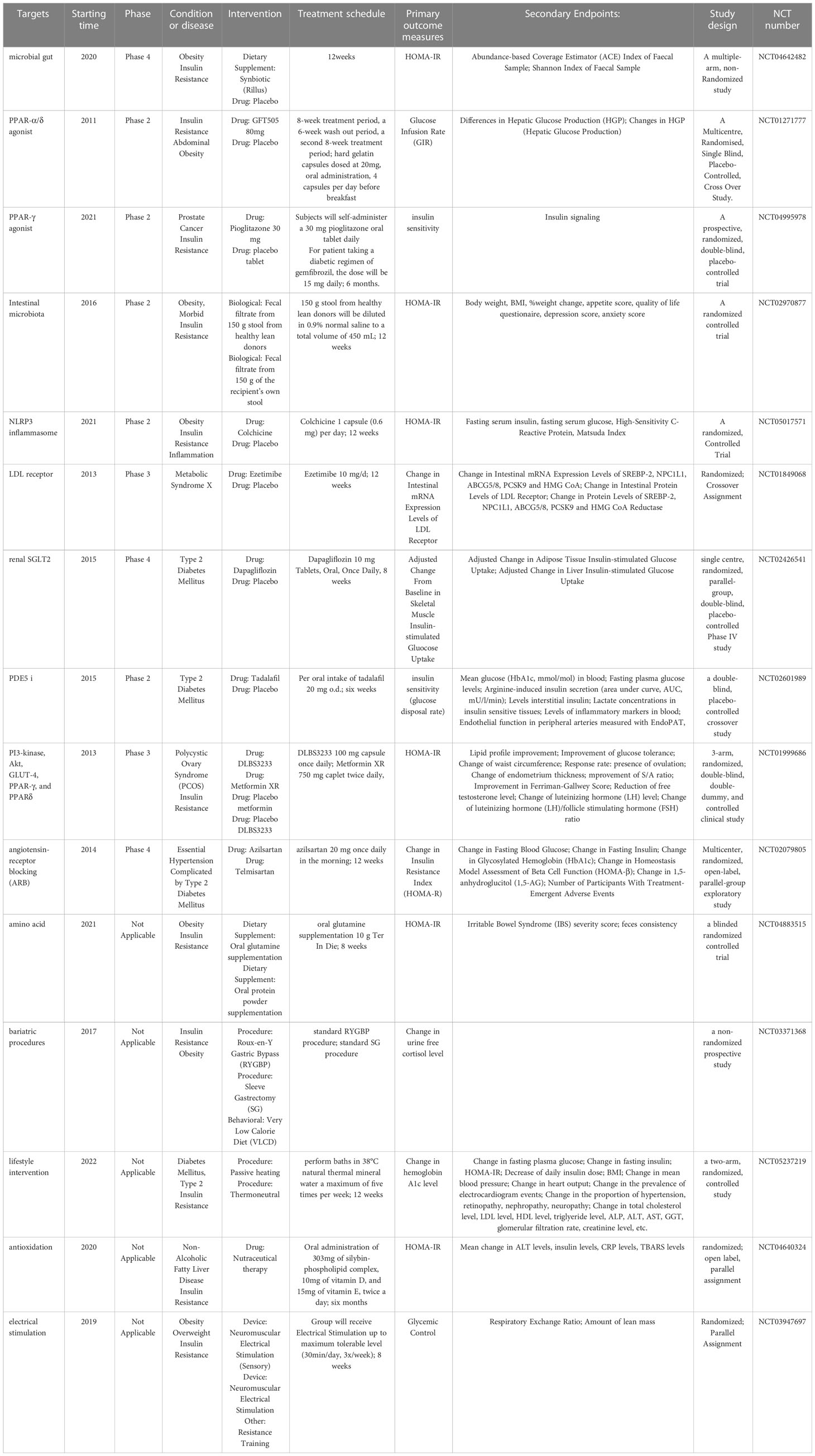
Table 2 Currently developing drugs targeting insulin resistance.
5.2.2 Traditional Chinese medicine treatment
TCM plays an equally critical role in the treatment of many acute and chronic diseases, especially its adeptness in restoring the dynamic balance of the body in systemic diseases. Its main therapeutic measures include herbal medicine, acupuncture and Tui Na. Several classical herbal formulations have been widely used in the clinical treatment of T2DM and various other metabolic disorders. For example, GegenQinlian decoction improves IR in fat, liver and muscle tissue through a variety of compounds, targets, pathways and mechanisms (308). Yi Qi Zeng Min Tang has been shown to improve IR in high-fat fed Sprague-Dawley rats without increasing body weight (309). Because it reduced the expression of PI3K p85 mRNA and IRS1 protein, Fu Fang Zhen Zhu Tiao Zhi formula similarly improved IR in vitro and in rats with metabolic syndrome (310).Gui Zhi Fu Ling Wan, Dingkun Pill and Liuwei Dihuang Pills are herbal formulas widely used in the treatment of gynecological disorders and have the effect of harmonizing Qi and blood or dispelling blood stasis in Chinese medical theory. In modern clinical and animal studies, they have been found to be very effective in the treatment of PCOS and also slightly improve IR by alleviating inflammation, remodeling intestinal homeostasis, or by regulating the PI3K/Akt signaling pathway, among other mechanisms (311–314). In addition, the efficacy of acupuncture in improving IR is equally impressive, as a recent meta-analysis showed that acupuncture improved HOMA-IR and ISI as well as fasting blood glucose (FBG), 2h postprandial blood glucose (2hPG) and fasting insulin (FINS) levels, with fewer adverse events (315).
6 Conclusion and perspective
The increased incidence of IR and its vital role as a major and common cause of numerous metabolic diseases have created an urgent need to gain insight into the etiology and pathogenesis of IR, as well as to explore better early diagnostic methods and therapeutic strategies for it. The diagnosis of insulin resistance is currently inconclusive, while it is important to detect IR early and predict individual response to treatment. In addition to the few simple indices of IR calculated from biochemical or anthropometric variables currently in use, emerging biomarkers may now be the way forward, but this still needs to be supported by clinical data. Different ranges and criteria are also needed for the diagnosis and monitoring of different metabolic diseases. As mentioned above, IR is a central mechanism in many metabolic diseases. Since this is the case, IR should be considered as a therapeutic target for patients with a combination of multiple metabolic diseases so that multiple diseases can be treated simultaneously with the same treatment approach, thereby reducing healthcare expenditures. Although there is no universally accepted theory to explain the mechanisms that cause IR. Nevertheless, there is growing evidence linking ectopic lipid accumulation, ER stress, plasma concentration of inflammatory cytokines, oxidative stress, abnormalities in insulin signaling, and other factors to IR. In recent years, the exploration of the molecular mechanisms of IR has also led to the emergence of new therapeutic concepts beyond metformin and TZD. Regardless lifestyle modification remains the most basic and least costly intervention. Normative criteria need to be developed for different metabolic diseases considering IR as a focus.
Author contributions
FL and HJ provided the idea of the manuscript. XZ, XA, and CY contributed equally to this manuscript. XZ, XA, WS, and CY drafted the manuscript and searched the relevant literature. XZ and XA drafted the figures, and all authors approved the final version of the manuscript. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine. (No: ZYYCXTD-D-202001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology (2005) 42(5):987–1000. doi: 10.1002/hep.20920
2. Sharma VR, Matta ST, Haymond MW, Chung ST. Measuring insulin resistance in humans. Horm Res Paediatr (2020) 93(11-12):577–88. doi: 10.1159/000515462
3. Muniyappa R, Madan R, Varghese RT. Assessing insulin sensitivity and resistance in humans. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, editors. Endotext. South Dartmouth (MA: MDText.com; Inc. Copyright © 2000-2021; MDText.com; Inc.; 2000. eng.
4. Gar C, Rottenkolber M, Prehn C, Adamski J, Seissler J, Lechner A. Serum and plasma amino acids as markers of prediabetes; insulin resistance; and incident diabetes. Crit Rev Clin Lab Sci (2018) 55(1):21–32. doi: 10.1080/10408363.2017.1414143
5. Park SE, Park CY, Sweeney G. Biomarkers of insulin sensitivity and insulin resistance: Past; present and future. Crit Rev Clin Lab Sci (2015) 52(4):180–90. doi: 10.3109/10408363.2015.1023429
6. Milburn MV, Lawton KA. Application of metabolomics to diagnosis of insulin resistance. Annu Rev Med (2013) 64:291–305. doi: 10.1146/annurev-med-061511-134747
7. Yang R, Hu Y, Lee CH, Liu Y, Diaz-Canestro C, Fong CHY, et al. PM20D1 is a circulating biomarker closely associated with obesity; insulin resistance and metabolic syndrome. Eur J Endocrinol (2021) 186(2):151–61. doi: 10.1530/eje-21-0847
8. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep (2018) 20(2):12. doi: 10.1007/s11906-018-0812-z
9. Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism (2019) 92:6–10. doi: 10.1016/j.metabol.2018.09.005
10. Steenblock C, Schwarz PEH, Ludwig B, Linkermann A, Zimmet P, Kulebyakin K, et al. COVID-19 and metabolic disease: mechanisms and clinical management. Lancet Diabetes Endocrinol (2021) 9(11):786–98. doi: 10.1016/s2213-8587(21)00244-8
11. Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, et al. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J Hepatol (2019) 71(4):793–801. doi: 10.1016/j.jhep.2019.06.021
12. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension; diabetes; atherosclerosis and NASH: Cause or consequence? (2018) 68(2):335–52. doi: 10.1016/j.jhep.2017.09.021
13. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes atlas: Global; regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
14. Kaul K, Apostolopoulou M, Roden M. Insulin resistance in type 1 diabetes mellitus. Metabolism (2015) 64(12):1629–39. doi: 10.1016/j.metabol.2015.09.002
15. Nadeau KJ, Regensteiner JG, Bauer TA, Brown MS, Dorosz JL, Hull A, et al. Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab (2010) 95(2):513–21. doi: 10.1210/jc.2009-1756
16. Cree-Green M, Newcomer BR, Brown MS, Baumgartner AD, Bergman B, Drew B, et al. Delayed skeletal muscle mitochondrial ADP recovery in youth with type 1 diabetes relates to muscle insulin resistance. Diabetes (2015) 64(2):383–92. doi: 10.2337/db14-0765
17. Schauer IE, Snell-Bergeon JK, Bergman BC, Maahs DM, Kretowski A, Eckel RH, et al. Insulin resistance; defective insulin-mediated fatty acid suppression; and coronary artery calcification in subjects with and without type 1 diabetes: The CACTI study. Diabetes (2011) 60(1):306–14. doi: 10.2337/db10-0328
18. Donga E, Dekkers OM, Corssmit EP, Romijn JA. Insulin resistance in patients with type 1 diabetes assessed by glucose clamp studies: systematic review and meta-analysis. Eur J Endocrinol (2015) 173(1):101–9. doi: 10.1530/eje-14-0911
19. Liu HY, Cao SY, Hong T, Han J, Liu Z, Cao W. Insulin is a stronger inducer of insulin resistance than hyperglycemia in mice with type 1 diabetes mellitus (T1DM). J Biol Chem (2009) 284(40):27090–100. doi: 10.1074/jbc.M109.016675
20. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab (2019) 29(5):1028–44. doi: 10.1016/j.cmet.2019.03.009
21. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia (2003) 46(1):3–19. doi: 10.1007/s00125-002-1009-0
22. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature (2006) 444(7121):840–6. doi: 10.1038/nature05482
23. Rattarasarn C. Dysregulated lipid storage and its relationship with insulin resistance and cardiovascular risk factors in non-obese asian patients with type 2 diabetes. Adipocyte (2018) 7(2):71–80. doi: 10.1080/21623945.2018.1429784
24. Pop A, Clenciu D, Anghel M, Radu S, Socea B, Mota E, et al. Insulin resistance is associated with all chronic complications in type 1 diabetes. J Diabetes. (2016) 8(2):220–8. doi: 10.1111/1753-0407.12283
25. Pan Y, Zhong S, Zhou K, Tian Z, Chen F, Liu Z, et al. Association between diabetes complications and the triglyceride-glucose index in hospitalized patients with type 2 diabetes. J Diabetes Res (2021) 2021:8757996. doi: 10.1155/2021/8757996
26. Wang S, Shi J, Peng Y, Fang Q, Mu Q, Gu W, et al. Stronger association of triglyceride glucose index than the HOMA-IR with arterial stiffness in patients with type 2 diabetes: a real-world single-centre study. Cardiovasc Diabetol (2021) 20(1):82. doi: 10.1186/s12933-021-01274-x
27. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia (2018) 61(1):21–8. doi: 10.1007/s00125-017-4390-4
28. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol (2016) 12(3):144–53. doi: 10.1038/nrendo.2015.216
29. Svensson M, Eriksson JW. Insulin resistance in diabetic nephropathy–cause or consequence? Diabetes Metab Res Rev (2006) 22(5):401–10. doi: 10.1002/dmrr.648
30. Godsland IF. Insulin resistance and hyperinsulinaemia in the development and progression of cancer. Clin Sci (Lond). (2009) 118(5):315–32. doi: 10.1042/cs20090399
31. Hernandez AV, Pasupuleti V, Benites-Zapata VA, Thota P, Deshpande A, Perez-Lopez FR. Insulin resistance and endometrial cancer risk: A systematic review and meta-analysis. Eur J Cancer. (2015) 51(18):2747–58. doi: 10.1016/j.ejca.2015.08.031
32. Yin DT, He H, Yu K, Xie J, Lei M, Ma R, et al. The association between thyroid cancer and insulin resistance; metabolic syndrome and its components: A systematic review and meta-analysis. Int J Surg (2018), 57:66–75. doi: 10.1016/j.ijsu.2018.07.013
33. Kim NH, Chang Y, Lee SR, Ryu S, Kim HJ. Glycemic status; insulin resistance; and risk of pancreatic cancer mortality in individuals with and without diabetes. Am J Gastroenterol (2020) 115(11):1840–8. doi: 10.14309/ajg.0000000000000956
34. Pan K, Chlebowski RT, Mortimer JE, Gunter MJ, Rohan T, Vitolins MZ, et al. Insulin resistance and breast cancer incidence and mortality in postmenopausal women in the women's health initiative. Cancer (2020) 126(16):3638–47. doi: 10.1002/cncr.33002
35. Chiefari E, Mirabelli M, La Vignera S, Tanyolaç S, Foti DP, Aversa A, et al. Insulin resistance and cancer: In search for a causal link. Int J Mol Sci (2021) 22(20). doi: 10.3390/ijms222011137
36. Barber TM, Kyrou I, Randeva HS, Weickert MO. Mechanisms of insulin resistance at the crossroad of obesity with associated metabolic abnormalities and cognitive dysfunction. Int J Mol Sci (2021) 22(2). doi: 10.3390/ijms22020546
37. Mu N, Zhu Y, Wang Y, Zhang H, Xue F. Insulin resistance: a significant risk factor of endometrial cancer. Gynecol Oncol (2012) 125(3):751–7. doi: 10.1016/j.ygyno.2012.03.032
38. Arcidiacono B, Iiritano S, Nocera A, Possidente K, Nevolo MT, Ventura V, et al. Insulin resistance and cancer risk: an overview of the pathogenetic mechanisms. Exp Diabetes Res (2012) 2012:789174. doi: 10.1155/2012/789174
39. Inoue M, Tsugane S. Insulin resistance and cancer: epidemiological evidence. Endocr Relat Cancer (2012) 19(5):F1–8. doi: 10.1530/erc-12-0142
40. Kong Y, Hsieh CH, Alonso LC. ANRIL: A lncRNA at the CDKN2A/B locus with roles in cancer and metabolic disease. Front Endocrinol (Lausanne) (2018) 9:405. doi: 10.3389/fendo.2018.00405
41. Ramos-Lopez O, Riezu-Boj JI, Milagro FI, Martinez JA. DNA methylation signatures at endoplasmic reticulum stress genes are associated with adiposity and insulin resistance. Mol Genet Metab (2018) 123(1):50–8. doi: 10.1016/j.ymgme.2017.11.011
42. Kwa M, Plottel CS, Blaser MJ, Adams S. The intestinal microbiome and estrogen receptor-positive female breast cancer. J Natl Cancer Inst (2016) 108(8). doi: 10.1093/jnci/djw029
43. Adeva-Andany MM, Martínez-Rodríguez J, González-Lucán M, Fernández-Fernández C, Castro-Quintela E. Insulin resistance is a cardiovascular risk factor in humans. Diabetes Metab Syndr (2019) 13(2):1449–55. doi: 10.1016/j.dsx.2019.02.023
44. Adeva-Andany MM, Fernández-Fernández C, Carneiro-Freire N, Castro-Quintela E, Pedre-Piñeiro A, Seco-Filgueira M. Insulin resistance underlies the elevated cardiovascular risk associated with kidney disease and glomerular hyperfiltration. Rev Cardiovasc Med (2020) 21(1):41–56. doi: 10.31083/j.rcm.2020.01.5102
45. Saely CH, Aczel S, Marte T, Langer P, Hoefle G, Drexel H. The metabolic syndrome; insulin resistance; and cardiovascular risk in diabetic and nondiabetic patients. J Clin Endocrinol Metab (2005) 90(10):5698–703. doi: 10.1210/jc.2005-0799
46. Zhang X, Li J, Zheng S, Luo Q, Zhou C, Wang C. Fasting insulin; insulin resistance; and risk of cardiovascular or all-cause mortality in non-diabetic adults: a meta-analysis. Biosci Rep (2017) 37(5). doi: 10.1042/bsr20170947
47. Eddy D, Schlessinger L, Kahn R, Peskin B, Schiebinger R. Relationship of insulin resistance and related metabolic variables to coronary artery disease: a mathematical analysis. Diabetes Care (2009) 32(2):361–6. doi: 10.2337/dc08-0854
48. Novo G, Manno G, Russo R, Buccheri D, Dell'Oglio S, Morreale P, et al. Impact of insulin resistance on cardiac and vascular function. Int J Cardiol (2016) 221:1095–9. doi: 10.1016/j.ijcard.2016.07.087
49. Wang M, Li Y, Li S, Lv J. Endothelial dysfunction and diabetic cardiomyopathy. Front Endocrinol (2022) 13:851941. doi: 10.3389/fendo.2022.851941
50. Nakamura M, Sadoshima J. Cardiomyopathy in obesity, insulin resistance and diabetes. J Physiol (2020) 598(14):2977–93. doi: 10.1113/jp276747
51. Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, et al. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes (2013) 62(11):3887–900. doi: 10.2337/db13-0095
52. Razani B, Chakravarthy MV, Semenkovich CF. Insulin resistance and atherosclerosis. Endocrinol Metab Clin North Am (2008) 37(3):603–21. doi: 10.1016/j.ecl.2008.05.001
53. Fernández-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr Rev (2003) 24(3):278–301. doi: 10.1210/er.2002-0010
54. Kim SH, Reaven G. Sex differences in insulin resistance and cardiovascular disease risk. J Clin Endocrinol Metab (2013) 98(11):E1716–21. doi: 10.1210/jc.2013-1166
55. Robins SJ, Rubins HB, Faas FH, Schaefer EJ, Elam MB, Anderson JW, et al. Insulin resistance and cardiovascular events with low HDL cholesterol: the veterans affairs HDL intervention trial (VA-HIT). Diabetes Care (2003) 26(5):1513–7. doi: 10.2337/diacare.26.5.1513
56. Abdul-Ghani MA, Jayyousi A, DeFronzo RA, Asaad N, Al-Suwaidi J. Insulin resistance the link between T2DM and CVD: Basic mechanisms and clinical implications. Curr Vasc Pharmacol (2019) 17(2):153–63. doi: 10.2174/1570161115666171010115119
57. Muzurović E, Mikhailidis DP, Mantzoros C. Non-alcoholic fatty liver disease; insulin resistance; metabolic syndrome and their association with vascular risk. Metabolism (2021) 119:154770. doi: 10.1016/j.metabol.2021.154770
58. Valenti L, Bugianesi E, Pajvani U, Targher G. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int (2016) 36(11):1563–79. doi: 10.1111/liv.13185
59. Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med (2018) 283(4):356–70. doi:10.1111/joim.12719
60. Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr Rev (2019) Oct 1;40(5):1367-1393. eng. doi:10.1210/er.2019-00034
61. Titchenell PMLazar MABirnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol Metab (2017) 28(7):497–505. doi: 10.1016/j.tem.2017.03.003
62. Huang JF, Tsai PC, Yeh ML, Huang CF, Huang CI, Hsieh MH, et al. Risk stratification of non-alcoholic fatty liver disease across body mass index in a community basis. J Formos Med Assoc (2020) 119(1 Pt 1):89–96. doi: 10.1016/j.jfma.2019.03.014
63. Enooku K, Kondo M, Fujiwara N, Sasako T, Shibahara J, Kado A, et al. Hepatic IRS1 and ß-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. J Gastroenterol (2018) 53(12):1261–75. doi: 10.1007/s00535-018-1472-0
64. Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia (2005) 48(4):634–42. doi: 10.1007/s00125-005-1682-x
65. Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology (2001) 120(5):1183–92. doi: 10.1053/gast.2001.23256
66. Mantovani A, Byrne CD, Bonora E, Targher G. Nonalcoholic fatty liver disease and risk of incident type 2 diabetes: A meta-analysis. Diabetes Care (2018) 41(2):372–82. doi: 10.2337/dc17-1902
67. Shipovskaya AA, Dudanova OP, Kurbatova IV. The clinical significance of insulin resistance in non-diabetic patients with early forms of non-alcoholic fatty liver disease. Ter Arkh. (2018) 90(8):63–8. doi: 10.26442/terarkh201890863-68
68. Alemzadeh R, Kichler J, Calhoun M. Spectrum of metabolic dysfunction in relationship with hyperandrogenemia in obese adolescent girls with polycystic ovary syndrome. Eur J Endocrinol (2010) 162(6):1093–9. doi: 10.1530/eje-10-0205
69. Macut D, Bjekić-Macut J, Rahelić D, Doknić M. Insulin and the polycystic ovary syndrome. Diabetes Res Clin Pract (2017) 130:163–70. doi: 10.1016/j.diabres.2017.06.011
70. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev (2012) 33(6):981–1030. doi: 10.1210/er.2011-1034
71. Legro RS, Arslanian SA, Ehrmann DA, Hoeger KM, Murad MH, Pasquali R, et al. Diagnosis and treatment of polycystic ovary syndrome: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2013) 98(12):4565–92. doi: 10.1210/jc.2013-2350
72. Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, Legro RS. Scientific statement on the diagnostic criteria; epidemiology; pathophysiology; and molecular genetics of polycystic ovary syndrome. Endocr Rev (2015) 36(5):487–525. doi: 10.1210/er.2015-1018
73. He FF, Li YM. Role of gut microbiota in the development of insulin resistance and the mechanism underlying polycystic ovary syndrome: a review. J Ovarian Res (2020) 13(1):73. doi: 10.1186/s13048-020-00670-3
74. Falcone T, Finegood DT, Fantus IG, Morris D. Androgen response to endogenous insulin secretion during the frequently sampled intravenous glucose tolerance test in normal and hyperandrogenic women. J Clin Endocrinol Metab (1990) 71(6):1653–7. doi: 10.1210/jcem-71-6-1653
75. Vrbikova J, Hill M, Bendlova B, Grimmichova T, Dvorakova K, Vondra K, et al. Incretin levels in polycystic ovary syndrome. Eur J Endocrinol (2008) 159(2):121–7. doi: 10.1530/eje-08-0097
76. Stepto NK, Cassar S, Joham AE, Hutchison SK, Harrison CL, Goldstein RF, et al. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum Reprod (2013) 28(3):777–84. doi: 10.1093/humrep/des463
77. Rosenfield RL, Ehrmann DA. The pathogenesis of polycystic ovary syndrome (PCOS): The hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr Rev (2016) 37(5):467–520. doi: 10.1210/er.2015-1104
78. Clarembeau F, Bale G, Lanthier N. Cirrhosis and insulin resistance: current knowledge; pathophysiological mechanisms; complications and potential treatments. Clin Sci (Lond). (2020) 134(16):2117–35. doi: 10.1042/cs20200022
79. Fu YH, Liu WJ, Lee CL, Wang JS. Associations of insulin resistance and insulin secretion with bone mineral density and osteoporosis in a general population. Front Endocrinol (Lausanne) (2022) 13:971960. doi: 10.3389/fendo.2022.971960
80. Kobayashi H, Tokudome G, Hara Y, Sugano N, Endo S, Suetsugu Y, et al. Insulin resistance is a risk factor for the progression of chronic kidney disease. Clin Nephrol. (2009) 71(6):643–51. doi: 10.5414/cnp71643
81. Cree MG, Wolfe RR. Postburn trauma insulin resistance and fat metabolism. Am J Physiol Endocrinol Metab (2008) 294(1):E1–9. doi: 10.1152/ajpendo.00562.2007
82. Nagpal M, De D, Handa S, Pal A, Sachdeva N. Insulin resistance and metabolic syndrome in young men with acne. JAMA Dermatol (2016) 152(4):399–404. doi: 10.1001/jamadermatol.2015.4499
83. Hsu CS, Wang PC, Chen JH, Su WC, Tseng TC, Chen HD, et al. Increasing insulin resistance is associated with increased severity and prevalence of gastro-oesophageal reflux disease. Aliment Pharmacol Ther (2011) 34(8):994–1004. doi: 10.1111/j.1365-2036.2011.04817.x
84. Carmelli D, Cardon LR, Fabsitz R. Clustering of hypertension; diabetes; and obesity in adult male twins: same genes or same environments? Am J Hum Genet (1994) 55(3):566–73.
85. Lin HF, Boden-Albala B, Juo SH, Park N, Rundek T, Sacco RL. Heritabilities of the metabolic syndrome and its components in the northern manhattan family study. Diabetologia (2005) 48(10):2006–12. doi: 10.1007/s00125-005-1892-2
86. Wan ZL, Huang K, Xu B, Hu SQ, Wang S, Chu YC, et al. Diabetes-associated mutations in human insulin: crystal structure and photo-cross-linking studies of a-chain variant insulin wakayama. Biochemistry (2005) 44(13):5000–16. doi: 10.1021/bi047585k
87. Tager H, Given B, Baldwin D, Mako M, Markese J, Rubenstein A, et al. A structurally abnormal insulin causing human diabetes. Nature (1979) 281(5727):122–5. doi: 10.1038/281122a0
88. Taylor SI, Kadowaki T, Kadowaki H, Accili D, Cama A, McKeon C. Mutations in insulin-receptor gene in insulin-resistant patients. Diabetes Care (1990) 13(3):257–79. doi: 10.2337/diacare.13.3.257
89. Verdecchia F, Akcan N, Dastamani A, Morgan K, Semple RK, Shah P. Unusual glycemic presentations in a child with a novel heterozygous intragenic INSR deletion. Horm Res Paediatr (2020) 93(6):396–401. doi: 10.1159/000510462
90. Brown AE, Walker M. Genetics of insulin resistance and the metabolic syndrome. Curr Cardiol Rep (2016) 18(8):75. doi: 10.1007/s11886-016-0755-4
91. Mercado MM, McLenithan JC, Silver KD, Shuldiner AR. Genetics of insulin resistance. Curr Diabetes Rep (2002) 2(1):83–95. doi: 10.1007/s11892-002-0063-9
92. Al-Beltagi M, Bediwy AS, Saeed NK. Insulin-resistance in paediatric age: Its magnitude and implications. World J Diabetes. (2022) 13(4):282–307. doi: 10.4239/wjd.v13.i4.282
93. Kuglin B, Kolb H, Greenbaum C, Maclaren NK, Lernmark A, Palmer JP. The fourth international workshop on the standardisation of insulin autoantibody workshop. Diabetologia (1990) 33(10):638–9. doi: 10.1007/bf00400213
94. Bowden DW. Association of the PTPN1 gene with type 2 diabetes and insulin resistance. Discovery Med (2004) 4(24):427–32.
95. Alibegovic AC, Sonne MP, Højbjerre L, Hansen T, Pedersen O, van Hall G, et al. The t-allele of TCF7L2 rs7903146 associates with a reduced compensation of insulin secretion for insulin resistance induced by 9 days of bed rest. Diabetes (2010) 59(4):836–43. doi: 10.2337/db09-0918
96. Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature (2001) 414(6865):799–806. doi: 10.1038/414799a
97. Freidenberg GR, Reichart D, Olefsky JM, Henry RR. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin-dependent diabetes mellitus. Effect weight loss. J Clin Invest. (1988) 82(4):1398–406. doi: 10.1172/jci113744
98. Abd El-Kader SM, Al-Jiffri OH. Impact of weight reduction on insulin resistance; adhesive molecules and adipokines dysregulation among obese type 2 diabetic patients. Afr Health Sci (2018) 18(4):873–83. doi: 10.4314/ahs.v18i4.5
99. Ye J. Mechanisms of insulin resistance in obesity. Front Med (2013) 7(1):14–24. doi: 10.1007/s11684-013-0262-6
100. Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature (2002) 420(6913):333–6. doi: 10.1038/nature01137
101. Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med (2006) 119(5 Suppl 1):S10–6. doi: 10.1016/j.amjmed.2006.01.009
102. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. (2008) 118(9):2992–3002. doi: 10.1172/jci34260
103. Machado FVC, Pitta F, Hernandes NA, Bertolini GL. Physiopathological relationship between chronic obstructive pulmonary disease and insulin resistance. Endocrine (2018) 61(1):17–22. doi: 10.1007/s12020-018-1554-z
104. Cosio FG, Kudva Y, van der Velde M, Larson TS, Textor SC, Griffin MD, et al. New onset hyperglycemia and diabetes are associated with increased cardiovascular risk after kidney transplantation. Kidney Int (2005) 67(6):2415–21. doi: 10.1111/j.1523-1755.2005.00349.x
105. Porrini E, Delgado P, Bigo C, Alvarez A, Cobo M, Checa MD, et al. Impact of metabolic syndrome on graft function and survival after cadaveric renal transplantation. Am J Kidney Dis (2006) 48(1):134–42. doi: 10.1053/j.ajkd.2006.04.078
106. Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab (1982) 54(1):131–8. doi: 10.1210/jcem-54-1-131
107. Lopes PC, Fuhrmann A, Carvalho F, Sereno J, Santos MR, Pereira MJ, et al. Cyclosporine a enhances gluconeogenesis while sirolimus impairs insulin signaling in peripheral tissues after 3 weeks of treatment. Biochem Pharmacol (2014) 91(1):61–73. doi: 10.1016/j.bcp.2014.06.014
108. Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther (2002) 96(1):23–43. doi: 10.1016/s0163-7258(02)00297-8
109. Rafacho A, Ortsäter H, Nadal A, Quesada I. Glucocorticoid treatment and endocrine pancreas function: implications for glucose homeostasis; insulin resistance and diabetes. J Endocrinol (2014) 223(3):R49–62. doi: 10.1530/joe-14-0373
110. Rafacho A, Quallio S, Ribeiro DL, Taboga SR, Paula FM, Boschero AC, et al. The adaptive compensations in endocrine pancreas from glucocorticoid-treated rats are reversible after the interruption of treatment. Acta Physiol (Oxf). (2010) 200(3):223–35. doi: 10.1111/j.1748-1716.2010.02146.x
111. Galicia-Garcia U, Jebari S, Larrea-Sebal A, Uribe KB, Siddiqi H, Ostolaza H, et al. Statin treatment-induced development of type 2 diabetes: From clinical evidence to mechanistic insights. Int J Mol Sci (2020) 21(13). doi: 10.3390/ijms21134725
112. Chang AM, Halter JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab (2003) 284(1):E7–12. doi: 10.1152/ajpendo.00366.2002
113. Krentz AJ, Viljoen A, Sinclair A. Insulin resistance: a risk marker for disease and disability in the older person. Diabetes Med (2013) 30(5):535–48. doi: 10.1111/dme.12063
114. Resnick HE, Harris MI, Brock DB, Harris TB. American diabetes association diabetes diagnostic criteria; advancing age; and cardiovascular disease risk profiles: results from the third national health and nutrition examination survey. Diabetes Care (2000) 23(2):176–80. doi: 10.2337/diacare.23.2.176
115. Gabriely I, Ma XH, Yang XM, Atzmon G, Rajala MW, Berg AH, et al. Removal of visceral fat prevents insulin resistance and glucose intolerance of aging: an adipokine-mediated process? Diabetes (2002) 51(10):2951–8. doi: 10.2337/diabetes.51.10.2951
116. Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab (2007) 5(2):151–6. doi: 10.1016/j.cmet.2007.01.008
117. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science (2005) 307(5708):384–7. doi: 10.1126/science.1104343
118. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science (2003) 300(5622):1140–2. doi: 10.1126/science.1082889
119. Vieira-Lara MA, Dommerholt MB, Zhang W, Blankestijn M, Wolters JC, Abegaz F, et al. Age-related susceptibility to insulin resistance arises from a combination of CPT1B decline and lipid overload. BMC Biol (2021) 19(1):154. doi: 10.1186/s12915-021-01082-5
120. Minokoshi Y, Kahn CR, Kahn BB. Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem (2003) 278(36):33609–12. doi: 10.1074/jbc.R300019200
121. Gancheva S, Jelenik T, Álvarez-Hernández E, Roden M. Interorgan metabolic crosstalk in human insulin resistance. Physiol Rev (2018) 98(3):1371–415. doi: 10.1152/physrev.00015.2017
122. Severinsen MCK, Pedersen BK. Muscle-organ crosstalk: The emerging roles of myokines. Endocr Rev (2020) 41(4):594–609. doi: 10.1210/endrev/bnaa016
123. Horita S, Nakamura M, Suzuki M, Satoh N, Suzuki A, Seki G. Selective insulin resistance in the kidney. BioMed Res Int (2016) 2016:5825170. doi: 10.1155/2016/5825170
124. Ashraf A, Palakkott A, Ayoub MA. Anti-insulin receptor antibodies in the pathology and therapy of diabetes mellitus. Curr Diabetes Rev (2021) 17(2):198–206. doi: 10.2174/1573399816666200604122345
125. Hall C, Yu H, Choi E. Insulin receptor endocytosis in the pathophysiology of insulin resistance. Exp Mol Med (2020) 52(6):911–20. doi: 10.1038/s12276-020-0456-3
126. Rivers SL, Klip A, Giacca A. NOD1: An interface between innate immunity and insulin resistance. Endocrinology (2019) 160(5):1021–30. doi: 10.1210/en.2018-01061
127. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia (2012) 55(10):2565–82. doi: 10.1007/s00125-012-2644-8
128. Carvalho-Filho MA, Carvalho BM, Oliveira AG, Guadagnini D, Ueno M, Dias MM, et al. Double-stranded RNA-activated protein kinase is a key modulator of insulin sensitivity in physiological conditions and in obesity in mice. Endocrinology (2012) 153(11):5261–74. doi: 10.1210/en.2012-1400
129. Hage Hassan R, Pacheco de Sousa AC, Mahfouz R, Hainault I, Blachnio-Zabielska A, Bourron O, et al. Sustained Action of Ceramide on the Insulin Signaling Pathway in Muscle Cells: IMPLICATION OF THE DOUBLE-STRANDED RNA-ACTIVATED PROTEIN KINASE. J Biol Chem (2016) 291(6):3019–29. doi: 10.1074/jbc.M115.686949
130. Cimmino I, Lorenzo V, Fiory F, Doti N, Ricci S, Cabaro S, et al. A peptide antagonist of Prep1-p160 interaction improves ceramide-induced insulin resistance in skeletal muscle cells. Oncotarget (2017) 8(42):71845–58. doi: 10.18632/oncotarget.18286
131. Abdelsalam SS, Korashy HM, Zeidan A, Agouni A. The role of protein tyrosine phosphatase (PTP)-1B in cardiovascular disease and its interplay with insulin resistance. Biomolecules (2019) 9(7). doi: 10.3390/biom9070286
132. Sevillano J, Sánchez-Alonso MG, Pizarro-Delgado J, Ramos-Álvarez MDP. Role of receptor protein tyrosine phosphatases (RPTPs) in insulin signaling and secretion. Int J Mol Sci (2021) 22(11). doi: 10.3390/ijms22115812
133. Zhou R, Guo Q, Xiao Y, Guo Q, Huang Y, Li C, et al. Endocrine role of bone in the regulation of energy metabolism. Bone Res (2021) 9(1):25. doi: 10.1038/s41413-021-00142-4
134. Conte C, Epstein S, Napoli N. Insulin resistance and bone: a biological partnership. Acta Diabetol (2018) 55(4):305–14. doi: 10.1007/s00592-018-1101-7
135. Hevener AL, Zhou Z, Drew BG, Ribas V. The role of skeletal muscle estrogen receptors in metabolic homeostasis and insulin sensitivity. Adv Exp Med Biol (2017) 1043:257–84. doi: 10.1007/978-3-319-70178-3_13
136. Hong SH, Choi KM. Sarcopenic obesity; insulin resistance; and their implications in cardiovascular and metabolic consequences. Int J Mol Sci (2020) 21(2). doi: 10.3390/ijms21020494
137. Du P, Fan B, Han H, Zhen J, Shang J, Wang X, et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int (2013) 84(2):265–76. doi: 10.1038/ki.2013.113
138. Lopez-Pastor AR, Gomez-Hernandez A, Diaz-Castroverde S, Gonzalez-Aseguinolaza G, Gonzalez-Rodriguez A, Garcia G, et al. Liver-specific insulin receptor isoform a expression enhances hepatic glucose uptake and ameliorates liver steatosis in a mouse model of diet-induced obesity. Dis Model Mech (2019) 12(2). doi: 10.1242/dmm.036186
139. Diaz-Castroverde S, Baos S, Luque M, Di Scala M, González-Aseguinolaza G, Gómez-Hernández A, et al. Prevalent role of the insulin receptor isoform a in the regulation of hepatic glycogen metabolism in hepatocytes and in mice. Diabetologia (2016) 59(12):2702–10. doi: 10.1007/s00125-016-4088-z
140. Diaz-Castroverde S, Gómez-Hernández A, Fernández S, García-Gómez G, Di Scala M, González-Aseguinolaza G, et al. Insulin receptor isoform a ameliorates long-term glucose intolerance in diabetic mice. Dis Model Mech (2016) 9(11):1271–81. doi: 10.1242/dmm.025288
141. Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol (2006) 7(2):85–96. doi: 10.1038/nrm1837
142. James DE, Stöckli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol (2021) 22(11):751–71. doi: 10.1038/s41580-021-00390-6
143. Fazakerley DJ, Krycer JR, Kearney AL, Hocking SL, James DE. Muscle and adipose tissue insulin resistance: malady without mechanism? J Lipid Res (2019) 60(10):1720–32. doi: 10.1194/jlr.R087510
144. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol (Lausanne). (2013) 4:52. doi: 10.3389/fendo.2013.00052
145. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med (2012) 18(3):363–74. doi: 10.1038/nm.2627
146. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol (2014) 6(1). doi: 10.1101/cshperspect.a009191
147. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science (1996) 271(5249):665–8. doi: 10.1126/science.271.5249.665
148. Donath MY, Dalmas É, Sauter NS, Böni-Schnetzler M. Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab (2013) 17(6):860–72. doi: 10.1016/j.cmet.2013.05.001
149. Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem (2008) 114(3):183–94. doi: 10.1080/13813450802181047
150. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell (2012) 148(5):852–71. doi: 10.1016/j.cell.2012.02.017
151. Chen K, Li F, Li J, Cai H, Strom S, Bisello A, et al. Induction of leptin resistance through direct interaction of c-reactive protein with leptin. Nat Med (2006) 12(4):425–32. doi: 10.1038/nm1372
152. Yang M, Qiu S, He Y, Li L, Wu T, Ding N, et al. Genetic ablation of c-reactive protein gene confers resistance to obesity and insulin resistance in rats. Diabetologia (2021) 64(5):1169–83. doi: 10.1007/s00125-021-05384-9
153. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of ikkbeta. Science (2001) 293(5535):1673–7. doi: 10.1126/science.1061620
154. Yin J, Peng Y, Wu J, Wang Y, Yao L. Toll-like receptor 2/4 links to free fatty acid-induced inflammation and β-cell dysfunction. J Leukoc Biol (2014) 95(1):47–52. doi: 10.1189/jlb.0313143
155. Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab (2014) 99(1):39–48. doi: 10.1210/jc.2013-3092
156. Kim JJ, Sears DD. TLR4 and insulin resistance. Gastroenterol Res Pract (2010) 2010. doi: 10.1155/2010/212563
157. Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, et al. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab (2009) 10(5):419–29. doi: 10.1016/j.cmet.2009.09.006
158. Kiechl S, Wittmann J, Giaccari A, Knoflach M, Willeit P, Bozec A, et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med (2013) 19(3):358–63. doi: 10.1038/nm.3084
159. Kaneto H, Matsuoka TA, Nakatani Y, Kawamori D, Miyatsuka T, Matsuhisa M, et al. Oxidative stress; ER stress; and the JNK pathway in type 2 diabetes. J Mol Med (Berl). (2005) 83(6):429–39. doi: 10.1007/s00109-005-0640-x
160. Han MS, Perry RJ, Camporez JP, Scherer PE, Shulman GI, Gao G, et al. A feed-forward regulatory loop in adipose tissue promotes signaling by the hepatokine FGF21. Genes Dev (2021) 35(1-2):133–46. doi: 10.1101/gad.344556.120
161. Apostolopoulos V, de Courten MP, Stojanovska L, Blatch GL, Tangalakis K, de Courten B. The complex immunological and inflammatory network of adipose tissue in obesity. Mol Nutr Food Res (2016) 60(1):43–57. doi: 10.1002/mnfr.201500272
162. Orliaguet L, Dalmas E, Drareni K, Venteclef N, Alzaid F. Mechanisms of macrophage polarization in insulin signaling and sensitivity. Front Endocrinol (Lausanne). (2020) 11:62. doi: 10.3389/fendo.2020.00062
163. McNelis JC, Olefsky JM. Macrophages; immunity; and metabolic disease. Immunity (2014) 41(1):36–48. doi: 10.1016/j.immuni.2014.05.010
164. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes (2008) 57(12):3239–46. doi: 10.2337/db08-0872
165. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med (2012) 18(9):1407–12. doi: 10.1038/nm.2885
166. Stefanovic-Racic M, Yang X, Turner MS, Mantell BS, Stolz DB, Sumpter TL, et al. Dendritic cells promote macrophage infiltration and comprise a substantial proportion of obesity-associated increases in CD11c+ cells in adipose tissue and liver. Diabetes (2012) 61(9):2330–9. doi: 10.2337/db11-1523
167. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332(6026):243–7. doi: 10.1126/science.1201475
168. Chng MH, Alonso MN, Barnes SE, Nguyen KD, Engleman EG. Adaptive immunity and antigen-specific activation in obesity-associated insulin resistance. Mediators Inflamm (2015) 2015:593075. doi: 10.1155/2015/593075
169. Zand H, Morshedzadeh N, Naghashian F. Signaling pathways linking inflammation to insulin resistance. Diabetes Metab Syndr (2017), S307–s309. doi: 10.1016/j.dsx.2017.03.006
170. Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia (2018) 61(4):942–53. doi: 10.1007/s00125-017-4526-6
171. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. central role of tumor necrosis factor-alpha. J Clin Invest. (1994) 94(4):1543–9. doi: 10.1172/jci117495
172. Olefsky JM, Glass CK. Macrophages; inflammation; and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi: 10.1146/annurev-physiol-021909-135846
173. Tan T, Xiang Y, Deng C, Cao C, Ren Z, Huang G, et al. Variable frequencies of peripheral t-lymphocyte subsets in the diabetes spectrum from type 1 diabetes through latent autoimmune diabetes in adults (LADA) to type 2 diabetes. Front Immunol (2022) 13:974864. doi: 10.3389/fimmu.2022.974864
174. Deng C, Xiang Y, Tan T, Ren Z, Cao C, Huang G, et al. Altered peripheral b-lymphocyte subsets in type 1 diabetes and latent autoimmune diabetes in adults. Diabetes Care (2016) 39(3):434–40. doi: 10.2337/dc15-1765
175. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med (2009) 15(8):921–9. doi: 10.1038/nm.2001
176. Revelo XS, Tsai S, Lei H, Luck H, Ghazarian M, Tsui H, et al. Perforin is a novel immune regulator of obesity-related insulin resistance. Diabetes (2015) 64(1):90–103. doi: 10.2337/db13-1524
177. Blasco-Baque V, Garidou L, Pomié C, Escoula Q, Loubieres P, Le Gall-David S, et al. Periodontitis induced by porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut (2017) 66(5):872–85. doi: 10.1136/gutjnl-2015-309897
178. Khan S, Luck H, Winer S, Winer DA. Emerging concepts in intestinal immune control of obesity-related metabolic disease. Nat Commun (2021) 12(1):2598. doi: 10.1038/s41467-021-22727-7
179. Moreno-Navarrete JM, Fernández-Real JM. The complement system is dysfunctional in metabolic disease: Evidences in plasma and adipose tissue from obese and insulin resistant subjects. Semin Cell Dev Biol (2019) 85:164–72. doi: 10.1016/j.semcdb.2017.10.025
180. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1α; causing inflammation and insulin resistance in obesity. Cell (2014) 157(6):1339–52. doi: 10.1016/j.cell.2014.05.012
181. Malhotra A, White DP. Obstructive sleep apnoea. Lancet (2002) 360(9328):237–45. doi: 10.1016/s0140-6736(02)09464-3
182. Mesarwi OA, Loomba R, Malhotra A. Obstructive sleep apnea; hypoxia; and nonalcoholic fatty liver disease. Am J Respir Crit Care Med (2019) 199(7):830–41. doi: 10.1164/rccm.201806-1109TR
183. Iiyori N, Alonso LC, Li J, Sanders MH, Garcia-Ocana A, O'Doherty RM, et al. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med (2007) 175(8):851–7. doi: 10.1164/rccm.200610-1527OC
184. Catrina SB, Zheng X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia (2021) 64(4):709–16. doi: 10.1007/s00125-021-05380-z
185. Wang N, Shi XF, Khan SA, Wang B, Semenza GL, Prabhakar NR, et al. Hypoxia-inducible factor-1 mediates pancreatic β-cell dysfunction by intermittent hypoxia. Am J Physiol Cell Physiol (2020) 319(5):C922–c932. doi: 10.1152/ajpcell.00309.2020
186. Xia QS, Lu FE, Wu F, Huang ZY, Dong H, Xu LJ, et al. New role for ceramide in hypoxia and insulin resistance. World J Gastroenterol (2020) 26(18):2177–86. doi: 10.3748/wjg.v26.i18.2177
187. O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. (2011) 108(38):16092–7. doi: 10.1073/pnas.1105062108
188. Yang Q, Vijayakumar A, Kahn BB. Metabolites as regulators of insulin sensitivity and metabolism. Nat Rev Mol Cell Biol (2018) 19(10):654–72. doi: 10.1038/s41580-018-0044-8
189. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol (2008) 9(2):162–76. doi: 10.1038/nrm2335
190. Lipke K, Kubis-Kubiak A, Piwowar A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States-Current View of Knowledge. Cells (2022) 11(5). doi: 10.3390/cells11050844
191. Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism (2016) 65(8):1049–61. doi: 10.1016/j.metabol.2016.02.014
192. Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci (2019) 20(9). doi: 10.3390/ijms20092358
193. Ahmed B, Sultana R, Greene MW. Adipose tissue and insulin resistance in obese. BioMed Pharmacother. (2021) 137:111315. doi: 10.1016/j.biopha.2021.111315
194. Jiao P, Ma J, Feng B, Zhang H, Diehl JA, Chin YE, et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obes (Silver Spring). (2011) 19(3):483–91. doi: 10.1038/oby.2010.200
195. DeBari MK, Abbott RD. Adipose tissue fibrosis: Mechanisms; models; and importance. Int J Mol Sci (2020) 21(17). doi: 10.3390/ijms21176030
196. Dahik VD, Frisdal E, Le Goff W. Rewiring of lipid metabolism in adipose tissue macrophages in obesity: Impact on insulin resistance and type 2 diabetes. Int J Mol Sci (2020) 21(15). doi: 10.3390/ijms21155505
197. Opazo-Ríos L, Mas S, Marín-Royo G, Mezzano S, Gómez-Guerrero C, Moreno JA, et al. Lipotoxicity and diabetic nephropathy: Novel mechanistic insights and therapeutic opportunities. Int J Mol Sci (2020) 21(7). doi: 10.3390/ijms21072632
198. Nisr RB, Shah DS, Ganley IG, Hundal HS. Proinflammatory NFkB signalling promotes mitochondrial dysfunction in skeletal muscle in response to cellular fuel overloading. Cell Mol Life Sci (2019) 76(24):4887–904. doi: 10.1007/s00018-019-03148-8
199. Chaurasia B, Summers SA. Ceramides - lipotoxic inducers of metabolic disorders: (Trends in endocrinology and metabolism 26; 538-550; 2015). Trends Endocrinol Metab (2018) 29(1):66–7. doi: 10.1016/j.tem.2017.09.005
200. Chaurasia B, Summers SA. Ceramides in Metabolism: Key Lipotoxic Players. Annu Rev Physiol (2021) 83:303–30. doi: 10.1146/annurev-physiol-031620-093815
201. Bandet CL, Tan-Chen S, Bourron O, Le Stunff H, Hajduch E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int J Mol Sci (2019) 20(3). doi: 10.3390/ijms20030479
202. Zhang Y, Fu Y, Jiang T, Liu B, Sun H, Zhang Y, et al. Enhancing fatty acids oxidation via l-carnitine attenuates obesity-related atrial fibrillation and structural remodeling by activating AMPK signaling and alleviating cardiac lipotoxicity. Front Pharmacol (2021) 12:771940. doi: 10.3389/fphar.2021.771940
203. D'Souza K, Paramel GV, Kienesberger PC. Lysophosphatidic acid signaling in obesity and insulin resistance. Nutrients (2018) 10(4). doi: 10.3390/nu10040399
204. Al-Jaber H, Al-Mansoori L, Elrayess MA. GATA-3 as a potential therapeutic target for insulin resistance and type 2 diabetes mellitus. Curr Diabetes Rev (2021) 17(2):169–79. doi: 10.2174/1573399816666200705210417
205. Imierska M, Kurianiuk A, Błachnio-Zabielska A. The influence of physical activity on the bioactive lipids metabolism in obesity-induced muscle insulin resistance. Biomolecules (2020) 10(12). doi: 10.3390/biom10121665
206. Drazba MA, Holásková I, Sahyoun NR, Ventura Marra M. Associations of Adiposity and Diet Quality with Serum Ceramides in Middle-Aged Adults with Cardiovascular Risk Factors. J Clin Med (2019) 8(4). doi: 10.3390/jcm8040527
207. Summers SA, Chaurasia B, Holland WL. Metabolic messengers: ceramides. Nat Metab (2019) 1(11):1051–8. doi: 10.1038/s42255-019-0134-8
208. Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science (2019) 365(6451):386–92. doi: 10.1126/science.aav3722
209. Petersen MC, Jurczak MJ. CrossTalk opposing view: Intramyocellular ceramide accumulation does not modulate insulin resistance. J Physiol (2016) 594(12):3171–4. doi: 10.1113/jp271677
210. Summers SA, Goodpaster BH. CrossTalk proposal: Intramyocellular ceramide accumulation does modulate insulin resistance. J Physiol (2016) 594(12):3167–70. doi: 10.1113/jp271676
211. Perreault L, Newsom SA, Strauss A, Kerege A, Kahn DE, Harrison KA, et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight (2018) 3(3). doi: 10.1172/jci.insight.96805
212. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab (2014) 20(4):687–95. doi: 10.1016/j.cmet.2014.09.015
213. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab (2014) 20(4):678–86. doi: 10.1016/j.cmet.2014.08.002
214. Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of akt/protein kinase b. J Biol Chem (2004) 279(35):36608–15. doi: 10.1074/jbc.M406499200
215. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev (2018) 98(4):2133–223. doi: 10.1152/physrev.00063.2017
216. Reali F, Morine MJ, Kahramanoğulları O, Raichur S, Schneider HC, Crowther D, et al. Mechanistic interplay between ceramide and insulin resistance. Sci Rep (2017) 7:41231. doi: 10.1038/srep41231
217. Chaurasia B, Talbot CL, Summers SA. Adipocyte ceramides-the nexus of inflammation and metabolic disease. Front Immunol (2020) 11:576347. doi: 10.3389/fimmu.2020.576347
218. Lyu K, Zhang Y, Zhang D, Kahn M, Ter Horst KW, Rodrigues MRS, et al. A membrane-bound diacylglycerol species induces PKC-mediated hepatic insulin resistance. Cell Metab (2020) 32(4):654–664.e5. doi: 10.1016/j.cmet.2020.08.001
219. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature (2019) 576(7785):51–60. doi: 10.1038/s41586-019-1797-8
220. Mastrototaro L, Roden M. Insulin resistance and insulin sensitizing agents. Metabolism (2021) 125:154892. doi: 10.1016/j.metabol.2021.154892
221. Schmitz-Peiffer C, Biden TJ. Protein kinase c function in muscle; liver; and beta-cells and its therapeutic implications for type 2 diabetes. Diabetes (2008) 57(7):1774–83. doi: 10.2337/db07-1769
222. Brandon AE, Liao BM, Diakanastasis B, Parker BL, Raddatz K, McManus SA, et al. Protein kinase c epsilon deletion in adipose tissue; but not in liver; improves glucose tolerance. Cell Metab (2019) 29(1):183–191.e7. doi: 10.1016/j.cmet.2018.09.013
223. Cohen S, Valm AM, Lippincott-Schwartz J. Interacting organelles. Curr Opin Cell Biol (2018) 53:84–91. doi: 10.1016/j.ceb.2018.06.003
224. Huang J, Meng P, Wang C, Zhang Y, Zhou L. The relevance of organelle interactions in cellular senescence. Theranostics (2022) 12(5):2445–64. doi: 10.7150/thno.70588
225. Keenan SN, Watt MJ, Montgomery MK. Inter-organelle communication in the pathogenesis of mitochondrial dysfunction and insulin resistance. Curr Diabetes Rep (2020) 20(6):20. doi: 10.1007/s11892-020-01300-4
226. Kruse R, Sahebekhtiari N, Højlund K. The mitochondrial proteomic signatures of human skeletal muscle linked to insulin resistance. Int J Mol Sci (2020) 21(15). doi: 10.3390/ijms21155374
227. Halling JF, Pilegaard H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl Physiol Nutr Metab (2020) 45(9):927–36. doi: 10.1139/apnm-2020-0005
228. Jana BA, Chintamaneni PK, Krishnamurthy PT, Wadhwani A, Mohankumar SK. Cytosolic lipid excess-induced mitochondrial dysfunction is the cause or effect of high fat diet-induced skeletal muscle insulin resistance: a molecular insight. Mol Biol Rep (2019) 46(1):957–63. doi: 10.1007/s11033-018-4551-7
229. Sun Y, Ding S. ER-mitochondria contacts and insulin resistance modulation through exercise intervention. Int J Mol Sci (2020) 21(24). doi: 10.3390/ijms21249587
230. Cheng H, Gang X, He G, Liu Y, Wang Y, Zhao X, et al. The molecular mechanisms underlying mitochondria-associated endoplasmic reticulum membrane-induced insulin resistance. Front Endocrinol (Lausanne). (2020) 11:592129. doi: 10.3389/fendo.2020.592129
231. Ge Q, Xie XX, Xiao X, Li X. Exosome-like vesicles as new mediators and therapeutic targets for treating insulin resistance and β-cell mass failure in type 2 diabetes mellitus. J Diabetes Res (2019) 2019:3256060. doi: 10.1155/2019/3256060
232. Nakashima N, Sharma PM, Imamura T, Bookstein R, Olefsky JM. The tumor suppressor PTEN negatively regulates insulin signaling in 3T3-L1 adipocytes. J Biol Chem (2000) 275(17):12889–95. doi: 10.1074/jbc.275.17.12889
233. Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature (2018) 560(7719):499–503. doi: 10.1038/s41586-018-0343-4
234. Li YZ, Di Cristofano A, Woo M. Metabolic role of PTEN in insulin signaling and resistance. Cold Spring Harb Perspect Med (2020) 10(8). doi: 10.1101/cshperspect.a036137
235. Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H. Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol (2005) 25(6):2498–510. doi: 10.1128/mcb.25.6.2498-2510.2005
236. Contreras-Bolívar V, García-Fontana B, García-Fontana C, Muñoz-Torres M. Mechanisms involved in the relationship between vitamin d and insulin resistance: Impact on clinical practice. Nutrients (2021) 13(10). doi: 10.3390/nu13103491
237. Hajhashemy Z, Shahdadian F, Moslemi E, Mirenayat FS, Saneei P. Serum vitamin d levels in relation to metabolic syndrome: A systematic review and dose-response meta-analysis of epidemiologic studies. Obes Rev (2021) 22(7):e13223. doi: 10.1111/obr.13223
238. Szymczak-Pajor I, Śliwińska A. Analysis of association between vitamin d deficiency and insulin resistance. Nutrients (2019) 11(4). doi: 10.3390/nu11040794
239. Pramono A, Jocken JWE, Blaak EE. Vitamin d deficiency in the aetiology of obesity-related insulin resistance. Diabetes Metab Res Rev (2019) 35(5):e3146. doi: 10.1002/dmrr.3146
240. Dubey P, Thakur V, Chattopadhyay M. Role of minerals and trace elements in diabetes and insulin resistance. Nutrients (2020) 12(6). doi: 10.3390/nu12061864
241. Kostov K. Effects of magnesium deficiency on mechanisms of insulin resistance in type 2 diabetes: Focusing on the processes of insulin secretion and signaling. Int J Mol Sci (2019) 20(6). doi: 10.3390/ijms20061351
242. Kim DJ, Xun P, Liu K, Loria C, Yokota K, Jacobs DR Jr, et al. Magnesium intake in relation to systemic inflammation; insulin resistance; and the incidence of diabetes. Diabetes Care (2010) 33(12):2604–10. doi: 10.2337/dc10-0994
243. Morais JBS, Severo JS, de Alencar GRR, de Oliveira ARS, Cruz KJC, Marreiro DDN, et al. Effect of magnesium supplementation on insulin resistance in humans: A systematic review. Nutrition (2017) 38:54–60. doi: 10.1016/j.nut.2017.01.009
244. Liu M, Jeong EM, Liu H, Xie A, So EY, Shi G, et al. Magnesium supplementation improves diabetic mitochondrial and cardiac diastolic function. JCI Insight (2019) 4(1). doi: 10.1172/jci.insight.123182
245. Wijesekara N, Chimienti F, Wheeler MB. Zinc; a regulator of islet function and glucose homeostasis. Diabetes Obes Metab (2009) 11:202–14. doi: 10.1111/j.1463-1326.2009.01110.x
246. Wang X, Gao H, Wu W, Xie E, Yu Y, He X, et al. The zinc transporter Slc39a5 controls glucose sensing and insulin secretion in pancreatic β-cells via Sirt1- and pgc-1α-mediated regulation of Glut2. Protein Cell (2019) 10(6):436–49. doi: 10.1007/s13238-018-0580-1
247. Kloubert V, Rink L. Zinc as a micronutrient and its preventive role of oxidative damage in cells. Food Funct (2015) 6(10):3195–204. doi: 10.1039/c5fo00630a
248. Baskin DG, Brewitt B, Davidson DA, Corp E, Paquette T, Figlewicz DP, et al. Quantitative autoradiographic evidence for insulin receptors in the choroid plexus of the rat brain. Diabetes (1986) 35(2):246–9. doi: 10.2337/diab.35.2.246
249. Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature (1978) 272(5656):827–9. doi: 10.1038/272827a0
250. Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Häring HU. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev (2016) 96(4):1169–209. doi: 10.1152/physrev.00032.2015
251. Heni M, Kullmann S, Ketterer C, Guthoff M, Linder K, Wagner R, et al. Nasal insulin changes peripheral insulin sensitivity simultaneously with altered activity in homeostatic and reward-related human brain regions. Diabetologia (2012) 55(6):1773–82. doi: 10.1007/s00125-012-2528-y
252. Spinelli M, Fusco S, Grassi C. Brain insulin resistance and hippocampal plasticity: Mechanisms and biomarkers of cognitive decline. Front Neurosci (2019) 13:788. doi: 10.3389/fnins.2019.00788
253. McNay EC, Pearson-Leary J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp Neurol (2020) 323:113076. doi: 10.1016/j.expneurol.2019.113076
254. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
255. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab (2010) 11(6):467–78. doi: 10.1016/j.cmet.2010.04.005
256. Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med (2013) 19(1):83–92. doi: 10.1038/nm.3014
257. He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature (2012) 481(7382):511–5. doi: 10.1038/nature10758
258. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev (2010) 90(3):859–904. doi: 10.1152/physrev.00045.2009
259. Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology (2012) 143(4):913–6.e7. doi: 10.1053/j.gastro.2012.06.031
260. Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes (2009) 58(7):1509–17. doi: 10.2337/db08-1637
261. Saad MJ, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiol (Bethesda). (2016) 31(4):283–93. doi: 10.1152/physiol.00041.2015
262. Velloso LA, Folli F, Saad MJ. TLR4 at the crossroads of nutrients; gut microbiota; and metabolic inflammation. Endocr Rev (2015) 36(3):245–71. doi: 10.1210/er.2014-1100
263. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes (2007) 56(7):1761–72. doi: 10.2337/db06-1491
264. Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature (2016) 535(7612):376–81. doi: 10.1038/nature18646
265. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean; but not obese; fat is enriched for a unique population of regulatory t cells that affect metabolic parameters. Nat Med (2009) 15(8):930–9. doi: 10.1038/nm.2002
266. Kim YA, Keogh JB, Clifton PM. Probiotics; prebiotics; synbiotics and insulin sensitivity. Nutr Res Rev (2018) 31(1):35–51. doi: 10.1017/s095442241700018x
267. Papakonstantinou E, Oikonomou C, Nychas G, Dimitriadis GD. Effects of diet; lifestyle; chrononutrition and alternative dietary interventions on postprandial glycemia and insulin resistance. Nutrients (2022) 14(4). doi: 10.3390/nu14040823
268. Sampath Kumar A, Maiya AG, Shastry BA, Vaishali K, Ravishankar N, Hazari A, et al. Exercise and insulin resistance in type 2 diabetes mellitus: A systematic review and meta-analysis. Ann Phys Rehabil Med (2019) 62(2):98–103. doi: 10.1016/j.rehab.2018.11.001
269. Hoene M, Kappler L, Kollipara L, Hu C, Irmler M, Bleher D, et al. Exercise prevents fatty liver by modifying the compensatory response of mitochondrial metabolism to excess substrate availability. Mol Metab (2021) 22:101359. doi: 10.1016/j.molmet.2021.101359
270. Khalyfa A, Ericsson A, Qiao Z, Almendros I, Farré R, Gozal D. Circulating exosomes and gut microbiome induced insulin resistance in mice exposed to intermittent hypoxia: Effects of physical activity. EBioMedicine (2021) 64:103208. doi: 10.1016/j.ebiom.2021.103208
271. Yaribeygi H, Atkin SL, Simental-Mendía LE, Sahebkar A. Molecular mechanisms by which aerobic exercise induces insulin sensitivity. J Cell Physiol (2019) 234(8):12385–92. doi: 10.1002/jcp.28066
272. Marson EC, Delevatti RS, Prado AK, Netto N, Kruel LF. Effects of aerobic; resistance; and combined exercise training on insulin resistance markers in overweight or obese children and adolescents: A systematic review and meta-analysis. Prev Med (2016) 93:211–8. doi: 10.1016/j.ypmed.2016.10.020
273. Cramer H, Langhorst J, Dobos G, Lauche R. Yoga for metabolic syndrome: A systematic review and meta-analysis. Eur J Prev Cardiol (2016) 23(18):1982–93. doi: 10.1177/2047487316665729
274. Chu P, Gotink RA, Yeh GY, Goldie SJ, Hunink MG. The effectiveness of yoga in modifying risk factors for cardiovascular disease and metabolic syndrome: A systematic review and meta-analysis of randomized controlled trials. Eur J Prev Cardiol (2016) 23(3):291–307. doi: 10.1177/2047487314562741
275. Liu X, Miller YD, Burton NW, Chang JH, Brown WJ. Qi-gong mind-body therapy and diabetes control. A randomized Controlled trial. Am J Prev Med (2011) 41(2):152–8. doi: 10.1016/j.amepre.2011.04.007
276. Chau JPC, Leung LYL, Liu X, Lo SHS, Choi KC, Zhao J, et al. Effects of tai chi on health outcomes among community-dwelling adults with or at risk of metabolic syndrome: A systematic review. Complement Ther Clin Pract (2021) 44:101445. doi: 10.1016/j.ctcp.2021.101445
277. Papadaki A, Nolen-Doerr E, Mantzoros CS. The effect of the mediterranean diet on metabolic health: A systematic review and meta-analysis of controlled trials in adults. Nutrients (2020) 12(11). doi: 10.3390/nu12113342
278. Widmer RJ, Flammer AJ, Lerman LO, Lerman A. The mediterranean diet; its components; and cardiovascular disease. Am J Med (2015) 128(3):229–38. doi: 10.1016/j.amjmed.2014.10.014
279. Galié S, García-Gavilán J, Papandreou C, Camacho-Barcía L, Arcelin P, Palau-Galindo A, et al. Effects of mediterranean diet on plasma metabolites and their relationship with insulin resistance and gut microbiota composition in a crossover randomized clinical trial. Clin Nutr (2021) 40(6):3798–806. doi: 10.1016/j.clnu.2021.04.028
280. Tettamanzi F, Bagnardi V, Louca P, Nogal A, Monti GS, Mambrini SP, et al. A high protein diet is more effective in improving insulin resistance and glycemic variability compared to a mediterranean diet-a cross-over controlled inpatient dietary study. Nutrients (2021) 13(12). doi: 10.3390/nu13124380
281. Ceriello A, Esposito K, Piconi L, Ihnat MA, Thorpe JE, Testa R, et al. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes (2008) 57(5):1349–54. doi: 10.2337/db08-0063
282. Sochol KM, Johns TS, Buttar RS, Randhawa L, Sanchez E, Gal M, et al. The effects of dairy intake on insulin resistance: A systematic review and meta-analysis of randomized clinical trials. Nutrients (2019) 11(9). doi: 10.3390/nu11092237
283. O'Connor S, Turcotte AF, Gagnon C, Rudkowska I. Increased dairy product intake modifies plasma glucose concentrations and glycated hemoglobin: A systematic review and meta-analysis of randomized controlled trials. Adv Nutr (2019) 10(2):262–79. doi: 10.1093/advances/nmy074
284. Seida JC, Mitri J, Colmers IN, Majumdar SR, Davidson MB, Edwards AL, et al. Clinical review: Effect of vitamin D3 supplementation on improving glucose homeostasis and preventing diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab (2014) 99(10):3551–60. doi: 10.1210/jc.2014-2136
285. Malin SK, Kullman EL, Scelsi AR, Haus JM, Filion J, Pagadala MR, et al. A whole-grain diet reduces peripheral insulin resistance and improves glucose kinetics in obese adults: A randomized-controlled trial. Metabolism (2018) 82:111–7. doi: 10.1016/j.metabol.2017.12.011
286. Malin SK, Kullman EL, Scelsi AR, Godin JP, Ross AB, Kirwan JP. A whole-grain diet increases glucose-stimulated insulin secretion independent of gut hormones in adults at risk for type 2 diabetes. Mol Nutr Food Res (2019) 63(7):e1800967. doi: 10.1002/mnfr.201800967
287. Reynolds AN, Akerman AP, Mann J. Dietary fibre and whole grains in diabetes management: Systematic review and meta-analyses. PloS Med (2020) 17(3):e1003053. doi: 10.1371/journal.pmed.1003053
288. Pedersen E, Lange K, Clifton P. Effect of carbohydrate restriction in the first meal after an overnight fast on glycemic control in people with type 2 diabetes: a randomized trial. Am J Clin Nutr (2016) 104(5):1285–91. doi: 10.3945/ajcn.116.135343
289. Pearce KL, Noakes M, Keogh J, Clifton PM. Effect of carbohydrate distribution on postprandial glucose peaks with the use of continuous glucose monitoring in type 2 diabetes. Am J Clin Nutr (2008) 87(3):638–44. doi: 10.1093/ajcn/87.3.638
290. Chang CR, Francois ME, Little JP. Restricting carbohydrates at breakfast is sufficient to reduce 24-hour exposure to postprandial hyperglycemia and improve glycemic variability. Am J Clin Nutr (2019) 109(5):1302–9. doi: 10.1093/ajcn/nqy261
291. Che T, Yan C, Tian D, Zhang X, Liu X, Wu Z. Time-restricted feeding improves blood glucose and insulin sensitivity in overweight patients with type 2 diabetes: a randomised controlled trial. Nutr Metab (Lond). (2021) 18(1):88. doi: 10.1186/s12986-021-00613-9
292. Engin AB, Tsatsakis AM, Tsoukalas D, Engin A. Do flavanols-rich natural products relieve obesity-related insulin resistance? Food Chem Toxicol (2018) 112:157–67. doi: 10.1016/j.fct.2017.12.055
293. Herman R, Kravos NA, Jensterle M, Janež A, Dolžan V. Metformin and insulin resistance: A review of the underlying mechanisms behind changes in GLUT4-mediated glucose transport. Int J Mol Sci (2022) 23(3). doi: 10.3390/ijms23031264
294. Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends Pharmacol Sci (2011) 32(10):607–16. doi: 10.1016/j.tips.2011.06.006
295. Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia (2017) 60(2):215–25. doi: 10.1007/s00125-016-4157-3
296. Beck-Nielsen H, Hjøllund E, Pedersen O, Richelsen B, Sørensen NS. Sulfonylureas improve insulin binding and insulin action in non-insulin-dependent diabetes mellitus. Diabetes Care (1984) 7:100–5.
297. Yaribeygi H, Sathyapalan T, Sahebkar A. Molecular mechanisms by which GLP-1 RA and DPP-4i induce insulin sensitivity. Life Sci (2019) 234:116776. doi: 10.1016/j.lfs.2019.116776
298. Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab (2003) 29(4 Pt 2):6s28–35. doi: 10.1016/s1262-3636(03)72785-2
299. Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med (2008) 121(2):149–157.e2. doi: 10.1016/j.amjmed.2007.09.016
300. Alimoradi N, Firouzabadi N, Fatehi R. Metformin and insulin-resistant related diseases: Emphasis on the role of microRNAs. BioMed Pharmacother. (2021) 139:111662. doi: 10.1016/j.biopha.2021.111662
301. Biondo LA, Teixeira AAS, de OSFKC, Neto JCR. Pharmacological strategies for insulin sensitivity in obesity and cancer: Thiazolidinediones and metformin. Curr Pharm Des (2020) 26(9):932–45. doi: 10.2174/1381612826666200122124116
302. Ferrannini G, Hach T, Crowe S, Sanghvi A, Hall KD, Ferrannini E. Energy balance after sodium-glucose cotransporter 2 inhibition. Diabetes Care (2015) 38(9):1730–5. doi: 10.2337/dc15-0355
303. Yaribeygi H, Sathyapalan T, Maleki M, Jamialahmadi T, Sahebkar A. Molecular mechanisms by which SGLT2 inhibitors can induce insulin sensitivity in diabetic milieu: A mechanistic review. Life Sci (2020) 240:117090. doi: 10.1016/j.lfs.2019.117090
304. Kullmann S, Hummel J, Wagner R, Dannecker C, Vosseler A, Fritsche L, et al. Empagliflozin improves insulin sensitivity of the hypothalamus in humans with prediabetes: A randomized; double-blind; placebo-controlled; phase 2 trial. Diabetes Care (2021). doi: 10.2337/dc21-1136
305. Jones TA, Sautter M, Van Gaal LF, Jones NP. Addition of rosiglitazone to metformin is most effective in obese; insulin-resistant patients with type 2 diabetes. Diabetes Obes Metab (2003) 5(3):163–70. doi: 10.1046/j.1463-1326.2003.00258.x
306. Derosa G, Maffioli P, Salvadeo SA, Ferrari I, Ragonesi PD, Querci F, et al. Effects of sitagliptin or metformin added to pioglitazone monotherapy in poorly controlled type 2 diabetes mellitus patients. Metabolism (2010) 59(6):887–95. doi: 10.1016/j.metabol.2009.10.007
307. Choubey A, Girdhar K, Kar AK, Kushwaha S, Yadav MK, Ghosh D, et al. Low-dose naltrexone rescues inflammation and insulin resistance associated with hyperinsulinemia. J Biol Chem (2020) 295(48):16359–69. doi: 10.1074/jbc.RA120.013484
308. Cao Z, Zeng Z, Wang B, Liu C, Liu C, Wang Z, et al. Identification of potential bioactive compounds and mechanisms of GegenQinlian decoction on improving insulin resistance in adipose; liver; and muscle tissue by integrating system pharmacology and bioinformatics analysis. J Ethnopharmacol (2021) 264:113289. doi: 10.1016/j.jep.2020.113289
309. Zhang Z, Xue HL, Liu Y, Wang WJ. Yi-Qi-Zeng-Min-Tang; a chinese medicine; ameliorates insulin resistance in type 2 diabetic rats. World J Gastroenterol (2011) 17(8):987–95. doi: 10.3748/wjg.v17.i8.987
310. Hu X, Wang M, Bei W, Han Z, Guo J. The chinese herbal medicine FTZ attenuates insulin resistance via IRS1 and PI3K in vitro and in rats with metabolic syndrome. J Transl Med (2014) 12:47. doi: 10.1186/1479-5876-12-47
311. Deng Y, Xue W, Wang YF, Liu XH, Zhu SY, Ma X, et al. Insulin resistance in polycystic ovary syndrome improved by chinese medicine dingkun pill (): A randomized controlled clinical trial. Chin J Integr Med (2019) 25(4):246–51. doi: 10.1007/s11655-018-2947-1
312. Zhu Y, Li Y, Liu M, Hu X, Zhu H. Guizhi fuling wan; chinese herbal medicine; ameliorates insulin sensitivity in PCOS model rats with insulin resistance via remodeling intestinal homeostasis. Front Endocrinol (Lausanne). (2020) 11:575. doi: 10.3389/fendo.2020.00575
313. Qiu Z, Dong J, Xue C, Li X, Liu K, Liu B, et al. Liuwei dihuang pills alleviate the polycystic ovary syndrome with improved insulin sensitivity through PI3K/Akt signaling pathway. J Ethnopharmacol (2020) 250:111965. doi: 10.1016/j.jep.2019.111965
314. Liu M, Zhu H, Zhu Y, Hu X. Guizhi fuling wan reduces autophagy of granulosa cell in rats with polycystic ovary syndrome via restoring the PI3K/AKT/mTOR signaling pathway. J Ethnopharmacol (2021) 270:113821. doi: 10.1016/j.jep.2021.113821
Keywords: insulin resistance, metabolic disease, obesity, T2DM, NAFLD
Citation: Zhao X, An X, Yang C, Sun W, Ji H and Lian F (2023) The crucial role and mechanism of insulin resistance in metabolic disease. Front. Endocrinol. 14:1149239. doi: 10.3389/fendo.2023.1149239
Received: 21 January 2023; Accepted: 07 March 2023;
Published: 28 March 2023.
Edited by:
Jean-François Tanti, INSERM U1065 Centre Méditerranéen de Médecine Moléculaire, FranceReviewed by:
Eric Hajduch, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceXavier Prieur, INSERM U1087 Institut du Thorax, France
Copyright © 2023 Zhao, An, Yang, Sun, Ji and Lian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengmei Lian, bGZtNTY1QHNvaHUuY29t; Hangyu Ji, amloYW5neXVlY2hvQDE2My5jb20=
†These authors share first authorship