 Yuan-Yuan Wang
Yuan-Yuan Wang Ya-Di Wang†
Ya-Di Wang† Xin-Hua Xiao
Xin-Hua Xiao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 18 February 2022
Sec. Cellular Endocrinology
Volume 13 - 2022 | https://doi.org/10.3389/fendo.2022.839849
This article is part of the Research Topic The Role of Exosomes and Organokines in Metabolic and Endocrine Disease View all 6 articles
The prevalence of obesity is escalating and has become a worldwide health challenge coinciding with the development of metabolic diseases. Emerging evidence has shown that obesity is accompanied by the infiltration of macrophages into adipose tissue, contributing to a state of low-grade chronic inflammation and dysregulated metabolism. Moreover, in the state of obesity, the phenotype of adipose tissue macrophages switches from the M2 polarized state to the M1 state, thereby contributing to chronic inflammation. Notably, multiple metabolic organs (adipose tissue, gut, skeletal muscle, and the liver) communicate with adipose tissue macrophages via secreting organokines or exosomes. In this review, we systematically summarize how the organokines (adipokines, gut microbiota and its metabolites, gut cytokines, myokines, and hepatokines) and exosomes (adipocyte-, skeletal muscle-, and hepatocyte-derived exosomes) act as important triggers for macrophage recruitment in adipose tissue and adipose tissue macrophage polarization, thus providing further insight into obesity treatment. In addition, we also highlight the complex interaction of organokines with organokines and organokines with exosomes, revealing new paths in understanding adipose tissue macrophage recruitment and polarization.
As per the World Health Organization, since the mid-1970s, the global prevalence of obesity has tripled, with an estimated 1 billion adults being overweight, as well as 650 million adults and 124 million children and adolescents being obese. Obesity escalates the risk of acquiring diverse diseases for instance diabetes mellitus (1), cardiovascular diseases (1), certain types of cancer (2), a range of musculoskeletal conditions (3), and poor mental health (4). This results in a huge financial burden on public healthcare costs worldwide. Therefore, it is imperative to improve our comprehension of the mechanisms accounting for obesity’s pathogenesis.
Obesity is defined as a chronic imbalance of caloric intake with energy expenditure. Moreover, it has been recognized as a low-grade, chronic inflammatory condition that is followed by metabolic dysfunction along with insulin resistance (IR) (5). Even though the molecular basis underlying the inflammation is not yet fully understood, researchers agree that adipose tissue macrophages (ATMs) contribute to a pro-inflammatory state in obesity and obesity-associated metabolic dysfunctions (6, 7). In an obese state, a series of chemokines, monocyte chemotactic protein-1 (MCP-1), and leukotriene B4 (LTB4), attract monocytes into the adipose tissue. These recruited monocytes are termed ATMs and are generally accompanied by a phenotypic switch (most commonly M1/M2), also known as ATM polarization (8, 9). Compared to lean mice and humans, there is an increase in macrophage infiltration into adipose tissue in obesity (6). This is because of the influx of bone marrow–originated precursors into the adipose tissue along with their subsequent differentiation to generate mature F4/80-expressing macrophages (6). More importantly, ATMs from lean mice express elevated levels of genes for instance Ym1, arginase 1 (Arg1), and IL-10, which are traditionally linked to ‘alternatively activated’ or M2-like macrophages. On the other hand, ATMs originated from obese mice express elevated levels of genes for instance tumor necrosis factor alpha (TNF-α) and inducible nitric oxide synthase (iNOS), which are characteristic of ‘classically activated’ or M1-like macrophages (9). In addition, both ATM populations can be distinguished based on the expression of surface markers, where most ATMs express CD206 in lean mice, whereas in obese conditions, CD11c is upregulated (9). Except for the classic M1 and M2 phenotypes, newly identified subpopulations of macrophages (e.g. MMe, LAMs, VAMs, and MFehi ATMs) are related to multiple metabolic regulators (10–13). However, in the coming years, it will be important to understand the precise role of these newly identified phenotypes in obesity.
Adipose tissue, the liver along with the skeletal muscle are the main metabolic organs that are involved in obesity-associated metabolic disturbances via biosynthesis coupled with secretion of adipokines, myokines, as well as hepatokines, respectively (14). It is worth noting that these metabolic organs also secrete exosomes to communicate with peripheral cells along with distant organs and modulate whole-body metabolism. In recent years, growing research evidence has demonstrated that the gut plays a crucial role in the onset and development of many metabolic diseases via secretion of gut hormones, the gut microbiome, and its metabolites (which can also be defined as an organokines) (15, 16). Here, we summarize organokines such as adipokines, gut microbiota and its metabolites, gut hormones, myokines, and hepatokines, that are responsible for recruiting macrophages and shifting the ATM phenotype, thus initiating inflammatory cascades (Figure 1). In addition, we focused on the emerging role of exosomes produced by these endocrine organs on the polarization of ATMs (Figure 1). Of note, we also emphasize these organokines and exosomes can interact and regulate each other, talking indirectly to ATMs. With increasing understanding of organokines and exosomes in regulating ATM recruitment and polarization, novel insights, as well as treatment strategies should emerge in the prevention of obesity.
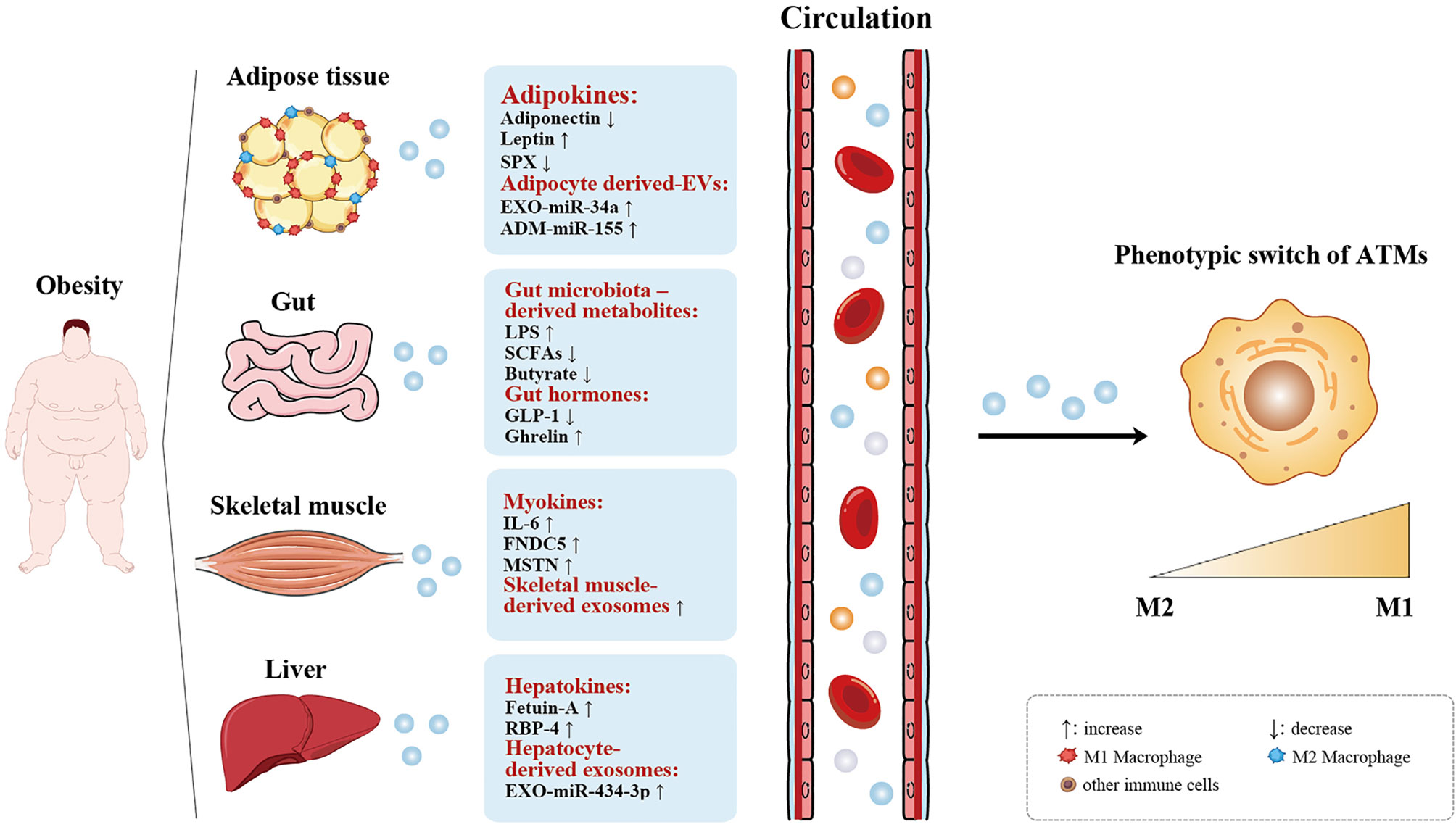
Figure 1 Organokines and exosomes-mediated ATM polarization. Adipose tissue, gut, skeletal muscle, and the liver act as endocrine organs by secreting organokines (adipokines, gut microbiota-derived metabolites, gut hormones, myokines, and hepatokines) and exoxomes (adipocyte-, skeletal muscle-, and hepatocyte-derived exosomes), which are increased or decreased in obesity, thus affecting the phenotype of ATMs switch from the anti-infammatory (M2) state to the pro-infammatory (M1) state. ↑ increase, ↓ decrease.
Adipose tissue is the primary fat storing tissue and is also recognized as the largest endocrine organ secreting adipokines systematically (17). In the setting of obesity, adipokines elicit recruitment of immune cells (mostly macrophages). Moreover, they play indispensable roles in the crosstalk of adipocytes with macrophages, thus modulating AT inflammation (17). More recently, it has been proposed that adipocyte-derived exosomes are involved in adipocyte/macrophage crosstalk and act as a remarkable mediator, whereby adipocytes modulate the polarization of ATMs in obesity (18, 19). Many research studies have aimed to elucidate the mechanisms via which adipocytes interact with macrophages and contribute to adipose tissue inflammation in obesity (20). Here, we summarize adipokines and adipocyte-derived exosomes, which are all important for macrophage recruitment and ATM activation in an obese state, thus regulating adipose tissue inflammation.
To date, many adipokines have been identified to modulate adipose tissue inflammation by switching the macrophage phenotype or accumulation of ATMs (21–23). The effect of some well-documented adipokines or lesser-known adipokines on ATMs polarization and recruitment in obesity are summarized in a chart (Supplementary Table 1). In this section, we describe in detail the role and mechanisms of three classical and newly identified adipokines (adiponectin, leptin, and SPX).
Adiponectin, the most abundant adipokine produced by adipose tissue, is downregulated in obesity and has a negative relationship with chronic inflammatory states; it is implicated in the immune response via modulating macrophage proliferation along with polarization. Cell culture investigations have documented that adiponectin influences the role of macrophages in several ways, consisting of the repression of the expressions of class A scavenger receptor (24), diminished nuclear factor-kappaB (NF-κB) activation via the Toll-like receptors (TLRs) (25), and upregulation of the anti-inflammatory cytokine IL-10 (26). Furthermore, Ohashi K et al. reported that ATMs originated from adiponectin-deficient mice display activation of M1 markers and suppression of M2 markers. In a cell culture, adding recombinant adiponectin to macrophages results in an upregulation of M2 markers, which was found to also reduce the level of M1 biologic signatures and trigger the expression of M2 biologic signatures in human monocyte-originated macrophages, as well as stromal vascular fraction (SVF) cells purified from human adipose tissue (27). As early as 2015, Hui et al. have demonstrated that adiponectin is relevant for cold-induced browning of subcutaneous white adipose tissue via promotion of M2 macrophage proliferation (28). However, other studies have demonstrated adiponectin as a negative regulatory factor of cold-induced browning/thermogenesis (29–32). One possible speculation for these conflicted results is that the effect of adiponectin on thermogenesis may be mediated simultaneously through multiple adiponectin receptors or adiponectin signaling (29). Thus, future studies need to clarify these confounding results and the underlying mechanisms of adiponectin in the regulation of energy. Overall, adiponectin directly regulates the macrophage phenotype, leading to the switch from a proinflammatory M1-like state to an anti-inflammatory M2-like state.
Obesity is linked to an elevated content of leptin in the expanding adipose tissue (33), and hyperleptinemia could result into low-grade systemic inflammation in obesity, illustrating a possible function of leptin in obesity-triggered inflammation (34). Dib et al. reported that C57BLJ mice reconstituted with db/db (leptin receptor-deficient) bone marrow, when fed on a high-fat diet (HFD), have a remarkably reduced body weight and adiposity, alleviated macrophage infiltration, and successively decreased adipose tissue inflammation, as well as a switch of ATMs from proinflammatory (M1) to anti-inflammatory (M2) in vivo (35). Importantly, two recent studies further supported that a reduction in leptin levels in an obese state serves as a novel strategy for loss of weight coupled with insulin sensitization (36, 37). Zhao et al. generated two diverse models of mouse of partial leptin insufficiency: an adipocyte-distinct congenital heterozygous leptin knockout mouse line (LepHZ) and a well-developed whole-body heterozygous leptin knockout mouse (OBHZ). In response to HFD, OBHZ and LepHZ mice were resistant to diet-triggered obesity, driven mostly via decreased fat mass expansion with no effect on lean mass. Moreover, macrophage infiltration into adipose tissue was greatly reduced in the epididymal adipose tissue depot of OBHZ and LepHZ mice on an HFD, accompanied by a reduction in adipose tissue inflammation markers (Mcp1, F4/80, and TNF-α) (36). Thus, partially leptin deficient mice under obesogenic conditions display reduced ATM recruitment, leading to a healthy adipose tissue phenotype. However, on chow fed diet, male OBHZ and LepHZ mice showed increased body weights (36), consistent with the classical physiological role attributed to leptin. The two different settings, i.e. leptin-sensitive (chow fed diet) and leptin-resistant (high fed diet) states can explain this contrary result (36, 37). In the leptin-sensitive state, increasing leptin levels did reduce food intake and body weight, while decreasing leptin levels resulted in increased food intake and body weight. In contrast, in the obese state, partial deficiency of leptin restored leptin sensitivity, resulting in a decrease in food intake and weight loss, accompanied by improved insulin sensitivity. Because secretion of catecholamines by anti-inflammatory macrophages induces adipose tissue uncoupling protein 1 (UCP1) (38), we speculated whether the reduction in proinflammatory M1 phenotypes in OBHZ and LepHZ mice on HFD plays a similar role, thus explaining the difference in body weight. Although they did not measure the expression of UCP1 in these two mouse models, we believe that this hypothesis can be explored in the future.
SPX, a 14-amino-acid peptide, constitutes a novel adipokine, which was first predicted via bioinformatics tools in 2007 (39). In diet-induced obese (DIO) mice, a low SPX plasma level, as well as low adipose tissue content were seen, and upon treatment with SPX, there was a decrease in caloric intake and body weight loss accompanied with improved glucose tolerance test (40). Evidence suggested that SPX dampens the uptake of fatty acid via adipocytes and that it represses the adipogenic process in the 3T3-L1 preadipocyte cell line (40, 41). Recently, a study evaluated the influence of SPX treatment on adipose tissue inflammation and ATM populations in a previously fructose-rich-diet (FRD) obese mouse model. This obese mouse model was characterized by an increase in body weight and epididymal adipose tissue masses, and an enhancement of inflammation in adipose tissue correlated with a decrease in M2 and an increase in M1 ATMs (42). SPX was found to reduce the percentage of M1 macrophages and increase M2 ATMs, and to improve adipose tissue inflammation, metabolic profile and adipocyte hypertrophy during FRD induced obesity (43).Furthermore, these data reported in vivo could be, at least partly, because of the direct action of SPX on macrophage activation. Thus, these data illustrate that SPX exerts a beneficial effect in inhabiting adipose tissue inflammation during obesity with a possible therapeutic potential for treating obesity.
Extracellular vesicles (EVs consisting of exosomes, microvesicles, and apoptotic bodies), a new class of microscopic endocrine hormone, mediate intercellular communication at the molecular level (44). Exosomes are nano-sized membrane-bound vesicles containing RNA (mRNA, miRNA, as well as other RNAs), protein, and lipids, many of which have gained much research attention for their roles in metabolic disorders, specifically in obesity along with its complications (44). Exosomes are generated from adipocytes and have been suggested to participate in adipocyte/macrophage crosstalk (18). Importantly, adipocyte-derived exosomes have also been found to control the ATM phenotype and play key roles in modulating adipose tissue inflammation.
Investigations have documented that adipose tissue might be a primary source of circulating exosomal miRNAs, which modulate metabolic homeostasis, as well as directly enhance IR in other organs (19). For example, a growing body of evidence has demonstrated that obese rodents and humans have greater levels of adipocyte-secreted exosomal miR-34a, and this correlates well with IR and metabolic inflammation (45–47). Importantly, Pan showed that miR-34a in adipocyte-derived exosomes suppress M2 polarization by targeting the transcription factor Kruppel-like factor 4, which in turn induces metabolic inflammation and IR. In this study, they also verified that adipose-selective ablation of miR-34a mice led to an obvious reduction in the adipose-resident M1 macrophages as well as significant upregulation of the M2 macrophages, which is accompanied by lower protein levels of the M1 macrophage marker (iNOS), coupled with an elevated abundance of the M2 macrophage marker Arg1 (47). Likewise, Zhang et al. showed that miR-155 in adipocyte-derived microvesicles could regulate macrophage polarization, hence eliciting the M2-to-M1 switch and inflammation via Janus kinase (JAK)/signal transduction and activator of transcription (STAT) signaling (48). Melatonin administration increased adipose-derived exosomal α-ketoglutarate (αKG), which was subsequently transported to macrophages; then, M2 macrophage activation was promoted, verifying the crosstalk between adipocytes and macrophages (49). Collectively, these data indicate that adipocyte-derived exosomes can regulate ATM polarization in a paracrine fashion, revealing a novel mechanism of obesity-induced chronic inflammation.
Evidence documents that the gut is a key site that is altered in obesity and obesity-linked inflammation. These alterations constitute changes in gut hormones, gut microflora, and their metabolites, which act as possible triggers of inflammation in obesity (50, 51). Among these gut-derived factors related to obesity, we will summarize existing knowledge on the potential of gut microbiota and its metabolites (including LPS, SCFAs, and butyrate), and gut hormones (GLP-1, Ghrelin) directly or indirectly modulate the activation of ATMs and mediate the pathogenesis of obesity-related inflammation.
Recently, emerging research evidences have shown that the gut microflora and its metabolites play critical roles in the onset, as well as development of obesity, and that the gut microbiota has lower bacterial richness in obese patients, along with increased IR and a markedly inflammatory phenotype (16, 52). In addition, the gut microbiota proportion was also altered in obesity, while obese people had a higher Firmicutes and lower Bacteroidetes proportion compared to lean people (50). Crosstalk between the gut microbiome and ATMs was initially observed in germ-free mice and ameliorated adiposity and resistance to obesity progression when they were fed an HFD. Caesar et al. also demonstrated that colonization of germ-free mice with Escherichia coli could result in the aggregation of macrophages in WAT and drive a shift of WAT macrophages from the anti-inflammatory M2 phenotype toward the proinflammatory M1 phenotype (53). While the role of the gut microbiome in modulating the ATM phenotype is in its early stage, more studies are needed to elaborate the complex interplay between them.
The gut microbiota produces a variety of metabolites, including lipopolysaccharide (LPS), short-chain fatty acids (SCFAs), bile acids, and a mass of amino acids, which have remarkable roles in the pathogenesis of obesity and maintenance of energy balance. Gut microbiota-derived LPS increases the plasma concentration of LPS in metabolic diseases, which is supported by the finding that obese individuals have elevated plasma contents of LPS than lean individuals (54). Mechanically, gut microbiota-derived LPS is taken up, then trafficked to adipose tissue, where it is internalized by adipocytes and ATMs, resulting in adipose tissue expansion and the transition of macrophages with the M2 phenotype to the M1 phenotype in a manner dependent on intact TLR4 signaling (55). In addition, Hersoug et al. summarized that LPS is implicated in defining the adipocyte death size and generation of crown-like structures (56–59). Based on these previous findings, microbiota-derived LPS can signal in an endocrine manner by entering the circulation to communicate with ATMs, shedding light on the potential of a microbiota-derived LPS therapeutic approach for treating obesity.
The amount of SCFA-generating bacteria and SCFAs is diminished in fecal specimens of dysmetabolic mice (57) and in humans with obesity and diabetes (58). Ni et al. indicated that SCFAs protect against LPS-triggered dysfunction of the intestinal barrier and indirectly improve LPS-triggered ATM recruitment (59). SCAFs can also cross talk with specific cell surface receptors for instance the G-protein-coupled receptor (GPR) 43 and GPR41 (60), and can also reach circulation, thus directly affecting peripheral tissue substrate metabolism and function (61). Nakajima et al. investigated the role of GPR43 in ATMs. They suggested that SCFA-activated GPR43 plays a direct role in suppressing fat accumulation and regulating inflammatory signals in adipose tissue M2-type macrophages (62). Similar to SCFAs, gut microbiota-derived succinate is akin to hormones by engaging succinate receptor 1 (SUCNR1) (63). Circulating succinate is elevated in obesity and is a promising clinical tool for early detection of metabolic dysfunction (64, 65). More recent observations point to a succinate–SUCNR1 axis acting as an integrator of macrophage immune response in metabolic disease (65). Notably, the impact of SUCNR1 insufficiency of ATMs is dependent on their location, as well as inflammatory status. SUCNR1-insufficient ATMs from subcutaneous fat show a pro-inflammatory phenotype, whereas SUCNR1-deficient ATMs from visceral fat (which display a typical pro-inflammatory phenotype) exhibit a dampened inflammatory status (66). Intriguingly, an intraperitoneal injection of butyrate in db/db mice markedly reduced the expression of inflammatory factors in subcutaneous adipose tissue and improved obesity-triggered IR in conjunction with the decreased expression of inflammatory ATM marker genes (67). Thus, gut microbiota-derived metabolites could be a possible target for furthering our current comprehension of the relationship of the gut with ATMs.
Enteroendocrine cells generate a range of gut cytokines that play key roles in metabolism, as they are associated with intestinal function, insulin secretion, and appetite (68). Among them, glucagon-like peptide-1 (GLP-1), secreted from intestinal L-cells in response to nutrient ingestion, has attracted much attention because of its mimetics for the treatment of obesity (69), lowering glucose levels, and improving insulin sensitivity in individuals with type 2 diabetes and in animal models (70). A large study population of 1462 Danish adults demonstrated that individuals who were obese or overweight had up to a 20% diminished GLP-1 response to oral glucose in contrast with normal weight individuals (71). Lee et al. initially reported that GLP-1 alleviates macrophage infiltration in the adipose tissue of leptin deficient (ob/ob) mice, which was accompanied by a reduction of M1-specific mRNAs (F4/80 and Tlr4), whereas no difference in M2 marker genes was observed through the direct inhibition of inflammatory signaling pathways (NF-κB) (72). GLP-1 receptors (GLP-1R) are abundantly expressed on the surface of numerous cell types, including pancreatic islet cells, gastrointestinal cells, neural cells, as well as mononuclear macrophages (73). It has been suggested that GLP-1/GLP-1R signaling dampens M1 activation and triggers M2 activation by cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) mediated c-Jun N-terminal kinase (JNK) downregulation in RAW264.7 cells (73). Based on these findings, we speculate that GLP-1 alleviates macrophage recruitment in adipose tissue and regulates ATMs from an M1 to M2 phenotype in an endocrine manner, thereby alleviating obesity-related inflammation.
Ghrelin, another gastrointestinal cytokines, acts via the growth hormone secretagogue receptor (GHSR) and is known to increase appetite and promote obesity (74, 75). Previous studies found that ghrelin levels in obese individuals are decreased (76, 77) and that GHSRs are expressed in ATMs (78). A previous study indicated that acyl ghrelin could enhance macrophage polarization to M1 directly in vitro, and deletion of its receptor GHSR in obese mice reduced macrophage infiltration, promoted macrophage polarization to M2 in adipose tissue, and suppressed adipose tissue inflammation (79); thus, these data suggest that Ghrelin/GHSR axis acts as an endocrine signal to induce a pro-inflammatory M1 phenotype in adipose tissue and that the deletion of GHSR promotes anti-inflammatory effects in obesity. Further investigations are needed to investigate the role of ATM-specific deletion in GHSR-induced obese mice on ATM polarization.
In recent years, considerable attention has been paid to the identification of novel functions of skeletal muscles, which release myokines and may act in an endocrine approach to facilitate tissue-to-tissue communication. Muscle contraction is a primary modulator of myokine expression and releases myokines, such as IL-6, irisin, beta-aminoisobutyric acid (BAIBA), and myonectin, exerting an effect on adipogenesis and metabolism (80–83). Importantly, skeletal muscle also releases exosomes into circulation, which can be incorporated into the distal organ and represent a new endocrine signal. Here, we summarize three candidate myokines (IL-6, FNDC5 and MSTN); we speculate that they may exert effects on the recruitment and polarization of macrophages in adipose tissue, thus regulating inflammation states in obesity.
Interleukin 6 (IL-6) was the first myokine to be discovered and is the best-studied metabolic myokine (84, 85). Intramuscular IL-6 facilitates glucose uptake and fat oxidation through PI3K and AMPK signaling pathways, respectively (86). A study showed that physical activity induces muscle IL-6 production and leads to elevated circulating IL-6 levels (87). Wedell et al. demonstrated that the exercise-induced loss of visceral adipose tissue mass was dependent on IL-6 (88). Of note, IL6 is secreted by several tissues, and the role of individual cell types can induce different inflammatory signaling responses. Han et al. demonstrated that IL-6 secreted by skeletal muscle following exercise suppresses ATM accumulation by endocrine signaling, while adipocyte IL-6 promotes HFD-induced ATM accumulation by paracrine signaling (89), thereby indicating that different sources of IL-6 production have distinct ways in which they physiologically regulate metabolism.
Fibronectin type III domain-containing protein 5 (FNDC5) is another novel myokine secreted by contracting skeletal muscles, inducing some beneficial effects of exercise via the browning of white fat (84). Recently, Xiong et al. confirmed that FNDC5 has a considerable effect on obesity-induced inflammation and IR. They found that FNDC5 overexpression attenuates adipose tissue inflammation in obesity by inhibiting macrophage recruitment and M1 phenotype polarization via AMPKα signaling in HFD-induced obese mice (90). The data highlight that the anti-inflammatory effect of FNDC5 may be involved in the beneficial roles of appropriate exercise in some chronic metabolic diseases.
Myostatin (MSTN), a myokine known to inhibit skeletal muscle growth, has been associated with the development of obesity and IR (91). The serum levels of MTSN were found to be increased in obese individuals compared to lean individuals and were not significantly upregulated in adipose tissue (91). Furthermore, it has been demonstrated that the observed differences in serum levels are likely due to the overproduction of MSTN from skeletal muscle in obesity. When MSTN was blocked in mice fed HFD by injecting a peptibody, it suppressed HFD-induced infiltration of macrophages and the expression of proinflammatory cytokines in adipose tissue (92). In addition, MSTN inhibition increased irisin production in the muscle and serum and stimulated macrophage polarization from M1 to M2 types in adipose tissue, thus suppressing inflammation. Collectively, in an obese state, MSTN functions in an endocrine manner to increase ATM infiltration and indirectly elicit an M1 phenotype in ATMs by promoting irisin expression.
Exercise-mediated contractility of skeletal muscle induces the release of exosomes into the extracellular milieu, thereby promoting metabolic systemic effects of endurance exercise (93). Compared with standard diet (SD) -fed mice, skeletal muscle releases more exosomes after HFD feeding in mice, and skeletal muscle-derived exosomes can be key communication messengers that regulate glucose uptake and metabolic homeostasis (94). Interestingly, long-term exercise-derived exosomal miR-342-5p serves as a cardio protective signal and protects against myocardial ischemia/reperfusion (MI/R) injury in rats (95). Further studies are required to demonstrate whether skeletal muscle-derived exosomes can reach the blood circulation and then regulate macrophage recruitment, the phenotypic change of ATMs, thus deciphering an exosome-based therapeutic approach for treating obesity and adipose tissue inflammation.
Like adipose tissue and skeletal muscle, the liver affects inflammation, IR, and whole-body energy homeostasis by releasing liver-derived proteins, such as hepatokines, into circulation (96). Several hepatokines may affect glucose and lipid metabolism, and thereby affect the risk of developing type 2 diabetes mellitus and metabolic disorders (97, 98). In addition, the liver modulates adipose tissue metabolic functions by directly secreting miRNA-containing hepatocyte-derived exosomes in the context of lipid overload (99). Here, we speculate that hepatokines (fetuin-A, RBP4) and hepatocyte-derived exosomes might drive ATM-mediated inflammation in obesity.
Fetuin-A, also known as alpha-2-Heremans-Schmid glycoprotein, has become one of the most important hepatokine, and has been found to act as a novel player in regulating human metabolism (98, 100, 101). A recent study showed that fetuin-A serves as an adaptor protein for saturated fatty acids and subsequently activates TLR4, triggering the production of proinflammatory cytokines (101). Human clinical data reported by Stefan and Häring also support this finding (100). In addition, some of the beneficial effects of exercise on insulin sensitivity may be explained by changes in circulating fetuin-A and free fatty acids, promoting less TLR4 signaling in adipose tissue by modulating adipose tissue macrophage-related gene expression and the M1-like phenotype (102). Interestingly, a recent study found a significant up-regulation in circulating fetuin-A after 60 days of bed rest, accompanied by deteriorative insulin sensitivity which could not be ameliorated by reactive jump training, thus indicating that fetuin-A may be a possible biological and predictive marker of metabolic diseases (103). These studies suggest that fetuin-A is secreted in adipose tissue and cultured adipocytes; fetuin-A is also considered an adipokine (104–106), since it is known primarily as a classical hepatokine. Chatterjee et al. showed that lipid-induced fetuin-A from adipocytes promoted macrophages infiltration into adipose tissue and the conversion of anti-inflammatory M2 to pro-inflammatory M1 (107). Collectively, these findings suggest that fetuin-A acts as a hepatokine or an adipokine, exerting effect on ATM in an endocrine or paracrine manner.
Retinol-binding protein-4 (RBP4), another hepatokine, is elevated in the serum of obese humans (108). Recent rodent studies have shown that elevated serum RBP4 levels cause adipose tissue inflammation, at least partly, by increasing ATMs, and the expression of both M1 and M2 macrophages was elevated in adipose tissue (109, 110). Considering that RBP4 is an adipose and liver secreted protein, it can be either a hepotokine or an adipokine (111, 112), thus regulating ATM function in an endocrine or paracrine manner. However, the studies concerning crosstalk between hepatokines and ATMs are limited, and more work is needed to integrate the hepatokines and inflammation network and to fully understand the role of cytokines in metabolic dysregulation.
Exosomes, as novel carriers in inter-tissue communication, play a role in liver-fat crosstalk. Wu et al. demonstrated that hepatic exosomes-derived miR-130a regulates energy metabolism in the adipose tissue of DIO mice (113). Mechanically, they identified that hepatic exosomes-miR-130a suppresses adipogenesis by inhibiting fatty acid synthase (FSAN) and peroxisome proliferator-activated receptor gamma (PPARγ) in 3T3-L1 cells, improving impaired glucose tolerance by suppressing pleckstrin homology domain leucine-rich repeat protein phosphatase 2 (PHLPP2) to activate the protein kinase B (AKT) – phosphorylation of Akt substrate of 160 kDa (AS160)–glucose transporter 4 (GLUT4) signaling pathway in adipocytes (113). Of note, it has been shown that the liver plays an early role in the etiology of metabolic disorders associated with lipid overload and then sends a signal to modulate the metabolic functions of adipose tissue by secreting miRNA-containing hepatocyte-derived EVs (99). Recently, it has been demonstrated that hepatocyte-derived exosomes from chronic obese mice stimulate the proinflammatory M1-like state of ATMs (114). Moreover, the proinflammation effects were due to exosomal miR-434-3p, which is enriched in these hepatocyte exosomes and this miRNA can directly promote macrophage polarization towards the M1-like state both in vitro and in vivo (114). Overall, these findings suggest that the liver plays a crucial, active metabolic regulatory role by directing crosstalk with adipose tissue via specific exosomes containing miRNAs.
As described above, we summarized the effects of various organokines and exosomes on the recruitment and polarization of ATMs in obesity. However, far beyond understanding these organokines and exosomes alone, it is crucial to emphasize that they act together in the body, forming a complex network of actions, which mediates additional favorable interactions with other organokines or exosomes and may induce indirect effects on ATMs
Obesity is accompanied by changes in the adipokine, hepatokine, myokine, gut cytokines, and gut microbiota-derived metabolites. These organokines are further participated in an intricate cross-regulation with each other and maintain a systemic inflammatory environment. For example, adiponectin (a representative adipokine) can be released by the upregulation of hormones, such as irisin, IL-15, fibroblast growth factor 21(FGF21), zinc-α2-glycoprotein, MSTN, and osteocalcin (115–118), whereas fetuin-A can inhibit the release of adiponectin. Furthermore, alteration of gut microbiota induced by antibiotic use can elevate the expression of adiponectin in HFD mice by modifying promoter DNA methylation, and supplementation of gut microbiota-derived metabolites (SCFAs, enone fatty acids) can reverse the level of adiponectin in obese mice (119–121). Thus, alterations in adiponectin levels caused by these other organokines may indirectly affect the recruitment and polarization of ATMs. Physical exercise enhances the production of myokine IL-6. In turn, it has been demonstrated to increase secretion of gut cytokines GLP-1, thus improving glucose and insulin secretion (82). Moreover, MSTN was found to promote the release of inflammatory adipokines and hepatokines while inhibiting other anti-inflammatory organokines, indicating the crosstalk among various organokines (116, 122). In a recent review, Suriano et al. suggested that the gut microbiota can modulate the profile of myokines and adipokines, and the interaction was bidirectional (123). As discussed, in obese state, the release and complex interaction of these organokines indirectly change the phenotype of ATM, further modulating chronic inflammation.
In recent years, accumulating evidence supports a role for extracellular vesicles in obesity-associated metabolic disturbance by carrying cargos (124, 125). In addition to organokines, exosomes can perform a “hormonal” function (126), and they may further interact and regulate each other and indirectly modulate the polarization of ATMs. For example, adiponectin was found to stimulate exosome biogenesis and secretion through binding to T-cadherin, leading to the reduction of cellular ceramides (127). While suppression of ceramide synthesis in whole-body and adipose tissue-specific increases anti-inflammatory M2 macrophages preferentially in subcutaneous white adipose tissue of obese mice, and promoted fibroblast growth factor 21(FGF21) and inhibited secretion of leptin (128). Considering that circulating exosome miRNA levels are substantially decreased in ADicerKO(adipose-tissue-specific knockout of the miRNA-processing enzyme Dicer) mice, it was believed that this may be due to the extreme reduction of adiponectin and adiponectin-mediated exosome synthesis in lipodystrophic mice (129, 130). Thus, adiponectin may be a crucial organokine altering the game of this small vesicle world. In addition, adipocyte-derived exosomal miR-99b could reduce FGF21 levels in the liver and impair hepatic insulin signaling pathways (129). Furthermore, it has been proved that 328 reported adipokines were identified in small extracellular vesicles derived from adipose tissue, whereas almost 75% of the known myokines were within exosomes/microvesicles (129). However, further analysis is required to explore the interplay between exosomes and organokines and their roles in ATMs.
The present review summarizes the endocrine organs release circulating triggers might crosstalk with ATMs, which are account for ATM infiltration and activation during obesity: (i) Adipose tissue, gut, skeletal muscle, and the liver can actively release secreted factors termed “organokines”, that is, adipokines, gut microbiota-derived metabolites, gut hormones, myokines, and hepatokines, respectively, which participate in macrophage recruitment and in the phenotypic change of ATMs.(ii) We address how these metabolic organs i.e. adipose tissue, skeletal muscle, and the liver secrete exosomes that may be responsible for ATM recruitment and activation. (iii) We highlight the interplay between various organokines and exosomes in obese state indirectly alter the ATMs phenotype and infiltration, further mediating chronic inflammation. Given the interaction between these triggers, it is difficult to target one organokine or exosome that regulates adipose tissue inflammation and the inflammatory phenotype of ATMs. Overall, further studies examining the relationships between various regulatory factors and ATM recruitment and polarization are likely to provide new strategies for the clinical treatment of obesity.
Y-YW: Data curation, software, visualization, writing—original draft, and writing—review and editing. Y-DW: Writing—original draft and revising manuscript. X-YQ: Data curation. Z-ZL: Data curation. Y-NM: Writing—review and editing. X-HX: Conceptualization and supervision. All authors contributed to the article and approved the submitted version.
This systematic received funding from National Natural Science Foundation of China (82070873, 82000813) and Major special projects of Hunan provincial health and family planning commission (A2017011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2022.839849/full#supplementary-material
1. Singh GM, Danaei G, Farzadfar F, Stevens GA, Woodward M, Wormser D, et al. The Age-Specific Quantitative Effects of Metabolic Risk Factors on Cardiovascular Diseases and Diabetes: A Pooled Analysis. PloS One (2013) 8(7):e65174. doi: 10.1371/journal.pone.0065174
2. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body Fatness and Cancer–Viewpoint of the IARC Working Group. N Engl J Med (2016) 375(8):794–8. doi: 10.1056/NEJMsr1606602
3. Anandacoomarasamy A, Caterson I, Sambrook P, Fransen M, March L. The Impact of Obesity on the Musculoskeletal System. Int J Obes (Lond) (2008) 32(2):211–22. doi: 10.1038/sj.ijo.0803715
4. Chu DT, Minh Nguyet NT, Nga VT, Thai Lien NV, Vo DD, Lien N, et al. An Update on Obesity: Mental Consequences and Psychological Interventions. Diabetes Metab Syndr (2019) 13(1):155–60. doi: 10.1016/j.dsx.2018.07.015
5. Hotamisligil GS. Inflammation and Metabolic Disorders. Nature (2006) 444(7121):860–7. doi: 10.1038/nature05485
6. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity Is Associated With Macrophage Accumulation in Adipose Tissue. J Clin Invest (2003) 112(12):1796–808. doi: 10.1172/JCI19246
7. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J Clin Invest (2003) 112(12):1821–30. doi: 10.1172/JCI19451
8. Bai Y, Sun Q. Macrophage Recruitment in Obese Adipose Tissue. Obes Rev (2015) 16(2):127–36. doi: 10.1111/obr.12242
9. Lumeng CN, Bodzin JL, Saltiel AR. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J Clin Invest (2007) 117(1):175–84. doi: 10.1172/JCI29881
10. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic Dysfunction Drives a Mechanistically Distinct Proinflammatory Phenotype in Adipose Tissue Macrophages. Cell Metab (2014) 20(4):614–25. doi: 10.1016/j.cmet.2014.08.010
11. Silva HM, Báfica A, Rodrigues-Luiz GF, Chi J, Santos PDA, Reis BS, et al. Vasculature-Associated Fat Macrophages Readily Adapt to Inflammatory and Metabolic Challenges. J Exp Med (2019) 216(4):786–806. doi: 10.1084/jem.20181049
12. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell (2019) 178(3):686–698.e14. doi: 10.1016/j.cell.2019.05.054
13. Orr JS, Kennedy A, Anderson-Baucum EK, Webb CD, Fordahl SC, Erikson KM, et al. Obesity Alters Adipose Tissue Macrophage Iron Content and Tissue Iron Distribution. Diabetes (2014) 63(2):421–32. doi: 10.2337/db13-0213
14. Chung HS, Choi KM. Organokines in Disease. Adv Clin Chem (2020) 94:261–321. doi: 10.1016/bs.acc.2019.07.012
15. Koliaki C, Liatis S, Dalamaga M, Kokkinos A. The Implication of Gut Hormones in the Regulation of Energy Homeostasis and Their Role in the Pathophysiology of Obesity. Curr Obes Rep (2020) 9(3):255–71. doi: 10.1007/s13679-020-00396-9
16. Wu J, Wang K, Wang X, Pang Y, Jiang C. The Role of the Gut Microbiome and Its Metabolites in Metabolic Diseases. Protein Cell (2021) 12(5):360–73. doi: 10.1007/s13238-020-00814-7
17. Kawai T, Autieri MV, Scalia R. Adipose Tissue Inflammation and Metabolic Dysfunction in Obesity. Am J Physiol Cell Physiol (2021) 320(3):C375–91. doi: 10.1152/ajpcell.00379.2020
18. Gao X, Salomon C, Freeman DJ. Extracellular Vesicles From Adipose Tissue-A Potential Role in Obesity and Type 2 Diabetes? Front Endocrinol (Lausanne) (2017) 8:202. doi: 10.3389/fendo.2017.00202
19. Zhou Y, Tan C. miRNAs in Adipocyte-Derived Extracellular Vesicles: Multiple Roles in Development of Obesity-Associated Disease. Front Mol Biosci (2020) 7:171. doi: 10.3389/fmolb.2020.00171
20. Maurizi G, Della Guardia L, Maurizi A, Poloni A. Adipocytes Properties and Crosstalk With Immune System in Obesity-Related Inflammation. J Cell Physiol (2018) 233(1):88–97. doi: 10.1002/jcp.25855
21. Martinerie C, Garcia M, Do TT, Antoine B, Moldes M, Dorothee G, et al. NOV/CCN3: A New Adipocytokine Involved in Obesity-Associated Insulin Resistance. Diabetes (2016) 65(9):2502–15. doi: 10.2337/db15-0617
22. Guo H, Jin D, Chen X. Lipocalin 2 Is a Regulator of Macrophage Polarization and NF-κb/STAT3 Pathway Activation. Mol Endocrinol (2014) 28(10):1616–28. doi: 10.1210/me.2014-1092
23. Zhu Z, Yin S, Wu K, Lee A, Liu Y, Li H, et al. Downregulation of Sfrp5 in Insulin Resistant Rats Promotes Macrophage-Mediated Pulmonary Inflammation Through Activation of Wnt5a/JNK1 Signaling. Biochem Biophys Res Commun (2018) 505(2):498–504. doi: 10.1016/j.bbrc.2018.09.070
24. Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, et al. Adipocyte-Derived Plasma Protein, Adiponectin, Suppresses Lipid Accumulation and Class A Scavenger Receptor Expression in Human Monocyte-Derived Macrophages. Circulation (2001) 103(8):1057–63. doi: 10.1161/01.cir.103.8.1057
25. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and Adiponectin Receptors in Insulin Resistance, Diabetes, and the Metabolic Syndrome. J Clin Invest (2006) 116(7):1784–92. doi: 10.1172/JCI29126
26. Kumada M, Kihara S, Ouchi N, Kobayashi H, Okamoto Y, Ohashi K, et al. Adiponectin Specifically Increased Tissue Inhibitor of Metalloproteinase-1 Through Interleukin-10 Expression in Human Macrophages. Circulation (2004) 109(17):2046–9. doi: 10.1161/01.CIR.0000127953.98131.ED
27. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin Promotes Macrophage Polarization Toward an Anti-Inflammatory Phenotype. J. Biol Chem (2010) 285(9):6153–60. doi: 10.1074/jbc.M109.088708
28. Hui X, Gu P, Zhang J, Nie T, Pan Y, Wu D, et al. Adiponectin Enhances Cold-Induced Browning of Subcutaneous Adipose Tissue via Promoting M2 Macrophage Proliferation. Cell Metab (2015) 22(2):279–90. doi: 10.1016/j.cmet.2015.06.004
29. Chen YJ, Lin CW, Peng YJ, Huang CW, Chien YS, Huang TH, et al. Overexpression of Adiponectin Receptor 1 Inhibits Brown and Beige Adipose Tissue Activity in Mice. Int J Mol Sci (2021) 22(2):906. doi: 10.3390/ijms22020906
30. Wang L, Luo Y, Luo L, Wu D, Ding X, Zheng H, et al. Adiponectin Restrains ILC2 Activation by AMPK-Mediated Feedback Inhibition of IL-33 Signaling. J Exp Med (2021) 218(2):e20191054. doi: 10.1084/jem.20191054
31. Qiao L, Hs Y, Bosco C, Lee B, GS F, Schaack J, et al. Adiponectin Reduces Thermogenesis by Inhibiting Brown Adipose Tissue Activation in Mice. Diabetologia (2014) 57(5):1027–36. doi: 10.1007/s00125-014-3180-5
32. Kajimura D, Lee HW, Riley KJ, Arteaga-Solis E, Ferron M, Zhou B, et al. Adiponectin Regulates Bone Mass via Opposite Central and Peripheral Mechanisms Through FoxO1. Cell Metab (2013) 17(6):901–15. doi: 10.1016/j.cmet.2013.04.009
33. Tilg H, Moschen AR. Adipocytokines: Mediators Linking Adipose Tissue, Inflammation and Immunity. Nat Rev Immunol (2006) 6(10):772–83. doi: 10.1038/nri1937
34. Monteiro L, Pereira JADS, Palhinha L, Moraes-Vieira PMM. Leptin in the Regulation of the Immunometabolism of Adipose Tissue-Macrophages. J Leukoc Biol (2019) 106(3):703–16. doi: 10.1002/JLB.MR1218-478R
35. Dib LH, Ortega MT, Fleming SD, Chapes SK, Melgarejo T. Bone Marrow Leptin Signaling Mediates Obesity-Associated Adipose Tissue Inflammation in Male Mice. Endocrinology (2014) 155(1):40–6. doi: 10.1210/en.2013-1607
36. Zhao S, Li N, Zhu Y, Straub L, Zhang Z, Wang MY, et al. Partial Leptin Deficiency Confers Resistance to Diet-Induced Obesity in Mice. Mol Metab (2020) 37:100995. doi: 10.1016/j.molmet.2020.100995
37. Zhao S, Zhu Y, Schultz RD, Li N, He Z, Zhang Z, et al. Partial Leptin Reduction as an Insulin Sensitization and Weight Loss Strategy. Cell Metab (2019) 30(4):706–719.e6. doi: 10.1016/j.cmet.2019.08.005
38. Nguyen KD, Qiu Y, Cui X, Goh YP, Mwangi J, David T, et al. Alternatively Activated Macrophages Produce Catecholamines to Sustain Adaptive Thermogenesis. Nature (2011) 480(7375):104–8. doi: 10.1038/nature10653
39. Mirabeau O, Perlas E, Severini C, Audero E, Gascuel O, Possenti R, et al. Identification of Novel Peptide Hormones in the Human Proteome by Hidden Markov Model Screening. Genome Res (2007) 17(3):320–7. doi: 10.1101/gr.5755407
40. Walewski JL, Ge F, Lobdell H4, Levin N, Schwartz GJ, Vasselli JR, et al. Spexin Is a Novel Human Peptide That Reduces Adipocyte Uptake of Long Chain Fatty Acids and Causes Weight Loss in Rodents With Diet-Induced Obesity. Obes (Silver Spring) (2014) 22(7):1643–52. doi: 10.1002/oby.20725
41. Kolodziejski PA, Pruszynska-Oszmalek E, Micker M, Skrzypski M, Wojciechowicz T, Szwarckopf P, et al. Spexin: A Novel Regulator of Adipogenesis and Fat Tissue Metabolism. Biochim Biophys Acta Mol Cell Biol Lipids (2018) 1863(10):1228–36. doi: 10.1016/j.bbalip.2018.08.001
42. Gambaro SE, Zubiría MG, Portales AE, Rey MA, Rumbo M, Giovambattista A. M1 Macrophage Subtypes Activation and Adipocyte Dysfunction Worsen During Prolonged Consumption of a Fructose-Rich Diet. J Nutr Biochem (2018) 61:173–82. doi: 10.1016/j.jnutbio.2018.08.004
43. Gambaro SE, Zubiría MG, Giordano AP, Portales AE, Alzamendi A, Rumbo M, et al. “Spexin Improves Adipose Tissue Inflammation and Macrophage Recruitment in Obese Mice”. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(7):158700. doi: 10.1016/j.bbalip.2020.158700
44. Latifkar A, Hur YH, Sanchez JC, Cerione RA, Antonyak MA. New Insights Into Extracellular Vesicle Biogenesis and Function. J Cell Sci (2019) 132(13):jcs222406. doi: 10.1242/jcs.222406
45. Ortega FJ, Moreno-Navarrete JM, Pardo G, Sabater M, Hummel M, Ferrer A, et al. MiRNA Expression Profile of Human Subcutaneous Adipose and During Adipocyte Differentiation. PloS One (2010) 5(2):e9022. doi: 10.1371/journl.0009022
46. Fu T, Seok S, Choi S, Huang Z, Suino-Powell K, Xu HE, et al. MicroRNA 34a Inhibits Beige and Brown Fat Formation in Obesity in Part by Suppressing Adipocyte Fibroblast Growth Factor 21 Signaling and SIRT1 Function. Mol Cell Biol (2014) 34(22):4130–42. doi: 10.1128/MCB.00596-14
47. Pan Y, Hui X, Hoo RLC, Ye D, Chan CYC, Feng T, et al. Adipocyte-Secreted Exosomal microRNA-34a Inhibits M2 Macrophage Polarization to Promote Obesity-Induced Adipose Inflammation. J Clin Invest (2019) 129(2):834–49. doi: 10.1172/JCI123069
48. Zhang Y, Mei H, Chang X, Chen F, Zhu Y, Han X. Adipocyte-Derived Microvesicles From Obese Mice Induce M1 Macrophage Phenotype Through Secreted miR-155. J Mol Cell Biol (2016) 8(6):505–17. doi: 10.1093/jmcb/mjw040
49. Liu Z, Gan L, Zhang T, Ren Q, Sun C. Melatonin Alleviates Adipose Inflammation Through Elevating α-Ketoglutarate and Diverting Adipose-Derived Exosomes to Macrophages in Mice. J Pineal Res (2018) 64(1):e12455. doi: 10.1111/jpi.12455
50. Tseng CH, Wu CY. The Gut Microbiome in Obesity. J Formos Med Assoc (2019) 118 Suppl 1:S3–9. doi: 10.1016/j.jfma.2018.07.009
51. Mishra AK, Dubey V, Ghosh AR. Obesity: An Overview of Possible Role(s) of Gut Hormones, Lipid Sensing and Gut Microbiota. Metabolism (2016) 65(1):48–65. doi: 10.1016/j.metabol.2015.10.008
52. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of Human Gut Microbiome Correlates With Metabolic Markers. Nature (2013) 500(7464):541–6. doi: 10.1038/nature12506
53. Caesar R, Reigstad CS, Bäckhed HK, Reinhardt C, Ketonen M, Lundén GÖ, et al. Gut-Derived Lipopolysaccharide Augments Adipose Macrophage Accumulation But Is Not Essential for Impaired Glucose or Insulin Tolerance in Mice. Gut (2012) 61(12):1701–7. doi: 10.1136/gutjnl-2011-301689
54. Trøseid M, Nestvold TK, Rudi K, Thoresen H, Nielsen EW, Lappegård KT. Plasma Lipopolysaccharide Is Closely Associated With Glycemic Control and Abdominal Obesity: Evidence From Bariatric Surgery. Diabetes Care (2013) 36(11):3627–32. doi: 10.2337/dc13-0451
55. Hersoug LG, Møller P, Loft S. Gut Microbiota-Derived Lipopolysaccharide Uptake and Trafficking to Adipose Tissue: Implications for Inflammation and Obesity. Obes Rev (2016) 17(4):297–312. doi: 10.1111/obr.12370
56. Hersoug LG, Møller P, Loft S. Role of Microbiota-Derived Lipopolysaccharide in Adipose Tissue Inflammation, Adipocyte Size and Pyroptosis During Obesity. Nutr Res Rev (2018) 31(2):153–63. doi: 10.1017/S0954422417000269
57. Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut Bacteria Selectively Promoted by Dietary Fibers Alleviate Type 2 Diabetes. Science (2018) 359(6380):1151–6. doi: 10.1126/science.aao5774
58. Makki K, Deehan EC, Walter J, Bäckhed F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe (2018) 23(6):705–15. doi: 10.1016/j.chom.2018.05.01
59. Ni Y, Ni L, Zhuge F, Xu L, Fu Z, Ota T. Adipose Tissue Macrophage Phenotypes and Characteristics: The Key to Insulin Resistance in Obesity and Metabolic Disorders. Obes (Silver Spring) (2020) 28(2):225–34. doi: 10.1002/oby.22674
60. Clark T. G-Protein Coupled Receptors: Answers From Simulations. Beilstein J Org Chem (2017) 13:1071–8. doi: 10.3762/bjoc.13.106
61. Blaak EE, Canfora EE, Theis S, Frost G, Groen AK, Mithieux G, et al. Short Chain Fatty Acids in Human Gut and Metabolic Health. Benef Microbes (2020) 11(5):411–55. doi: 10.3920/BM2020.0057
62. Nakajima A, Nakatani A, Hasegawa S, Irie J, Ozawa K, Tsujimoto G, et al. The Short Chain Fatty Acid Receptor GPR43 Regulates Inflammatory Signals in Adipose Tissue M2-Type Macrophages. PloS One (2017) 12(7):e0179696. doi: 10.1371/journal.pone.0179696
63. He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric Acid Cycle Intermediates as Ligands for Orphan G-Protein-Coupled Receptors. Nature (2004) 429(6988):188–93. doi: 10.1038/nature02488
64. Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño MI, Bernal R, Gomez-Huelgas R, et al. Elevated Circulating Levels of Succinate in Human Obesity Are Linked to Specific Gut Microbiota. ISME J (2018) 12(7):1642–57. doi: 10.1038/s41396-018-0068-2
65. Fernández-Veledo S, Ceperuelo-Mallafré V, Vendrell J. Rethinking Succinate: An Unexpected Hormone-Like Metabolite in Energy Homeostasis. Trends Endocrinol Metab (2021) 32(9):680–92. doi: 10.1016/j.tem.2021.06.003
66. Keiran N, Ceperuelo-Mallafré V, Calvo E, Hernández-Alvarez MI, Ejarque M, Núñez-Roa C, et al. SUCNR1 Controls an Anti-Inflammatory Program in Macrophages to Regulate the Metabolic Response to Obesity. Nat Immunol (2019) 20(5):581–92. doi: 10.1038/s41590-019-0372-7
67. Wang X, He G, Peng Y, Zhong W, Wang Y, Zhang B. Sodium Butyrate Alleviates Adipocyte Inflammation by Inhibiting NLRP3 Pathway. Sci Rep (2015) 5:12676. doi: 10.1038/srep12676
68. Martin AM, Sun EW, Keating DJ. Mechanisms Controlling Hormone Secretion in Human Gut and Its Relevance to Metabolism. J Endocrinol (2019) 244(1):R1–R15. doi: 10.1530/JOE-19-0399
69. O’Neil PM, Birkenfeld AL, McGowan B, Mosenzon O, Pedersen SD, Wharton S, et al. Efficacy and Safety of Semaglutide Compared With Liraglutide and Placebo for Weight Loss in Patients With Obesity: A Randomised, Double-Blind, Placebo and Active Controlled, Dose-Ranging, Phase 2 Trial. Lancet (2018) 392(10148):637–49. doi: 10.1016/S0140-6736(18)31773-2
70. Arakawa M, Mita T, Azuma K, Ebato C, Goto H, Nomiyama T, et al. Inhibition of Monocyte Adhesion to Endothelial Cells and Attenuation of Atherosclerotic Lesion by a Glucagon-Like Peptide-1 Receptor Agonist, Exendin-4. Diabetes (2010) 59(4):1030–7. doi: 10.2337/db09-1694
71. Færch K, Torekov SS, Vistisen D, Johansen NB, Witte DR, Jonsson A, et al. GLP-1 Response to Oral Glucose Is Reduced in Prediabetes, Screen-Detected Type 2 Diabetes, and Obesity and Influenced by Sex: The ADDITION-PRO Study. Diabetes (2015) 64(7):2513–25. doi: 10.2337/db14-1751
72. Lee YS, Park MS, Choung JS, Kim SS, Oh HH, Choi CS, et al. Glucagon-Like Peptide-1 Inhibits Adipose Tissue Macrophage Infiltration and Inflammation in an Obese Mouse Model of Diabetes. Diabetologia (2012) 55(9):2456–68. doi: 10.1007/s00125-012-2592-3
73. Wan S, Sun H. Glucagon-Like Peptide-1 Modulates RAW264.7 Macrophage Polarization by Interfering With the JNK/STAT3 Signaling Pathway. Exp Ther Med (2019) 17(5):3573–9. doi: 10.3892/etm.2019.7347
74. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide From Stomach. Nature (1999) 402(6762):656–60. doi: 10.1038/45230
75. Tschöp M, Smiley DL, Heiman ML. Ghrelin Induces Adiposity in Rodents. Nature (2000) 407(6806):908–13. doi: 10.1038/35038090
76. Shiiya T, Nakazato M, Mizuta M, Date Y, Mondal MS, Tanaka M, et al. Plasma Ghrelin Levels in Lean and Obese Humans and the Effect of Glucose on Ghrelin Secretion. J Clin Endocrinol Metab (2002) 87(1):240–4. doi: 10.1210/jcem.87.1.8129
77. Tschöp M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating Ghrelin Levels Are Decreased in Human Obesity. Diabetes (2001) 50(4):707–9. doi: 10.2337/diabetes.50.4.707
78. Ma X, Lin L, Yue J, Pradhan G, Qin G, Minze LJ, et al. Ghrelin Receptor Regulates HFCS-Induced Adipose Inflammation and Insulin Resistance. Nutr Diabetes (2013) 3(12):e99. doi: 10.1038/nutd.2013.41
79. Yuan F, Ma J, Xiang X, Lan H, Xu Y, Zhao J, et al. Improvement of Adipose Macrophage Polarization in High Fat Diet-Induced Obese GHSR Knockout Mice. BioMed Res Int (2018) 2018:4924325. doi: 10.1155/2018/4924325
80. Seldin MM, Peterson JM, Byerly MS, Wei Z, Wong GW. Myonectin (CTRP15), a Novel Myokine That Links Skeletal Muscle to Systemic Lipid Homeostasis. J Biol Chem (2012) 287(15):11968–80. doi: 10.1074/jbc.M111.336834
81. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-Like Development of White Fat and Thermogenesis. Nature (2012) 481(7382):463–8. doi: 10.1038/nature10777
82. Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, et al. Interleukin-6 Enhances Insulin Secretion by Increasing Glucagon-Like Peptide-1 Secretion From L Cells and Alpha Cells. Nat Med (2011) 17(11):1481–9. doi: 10.1038/nm.2513
83. Pedersen BK. Physical Activity and Muscle-Brain Crosstalk. Nat Rev Endocrinol (2019) 15(7):383–92. doi: 10.1038/s41574-019-0174-x
84. Ostrowski K, Rohde T, Zacho M, Asp S, Pedersen BK. Evidence That Interleukin-6 Is Produced in Human Skeletal Muscle During Prolonged Running. J Physiol (1998) 508(Pt3):949–53. doi: 10.1111/j.1469-7793.1998.949bp.x
85. Steensberg A, van Hall G, Osada T, Sacchetti M, Saltin B, Klarlund Pedersen B. Production of Interleukin-6 in Contracting Human Skeletal Muscles can Account for the Exercise-Induced Increase in Plasma Interleukin-6. J Physiol (2000) 529 Pt 1(Pt 1):237–42. doi: 10.1111/j.1469-7793.2000.00237.x
86. Giudice J, Taylor JM. Muscle as a Paracrine and Endocrine Organ. Curr Opin Pharmacol (2017) 34:49–55. doi: 10.1016/j.coph.2017.05.005
87. Pedersen BK, Febbraio MA. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol Rev (2008) 88(4):1379–406. doi: 10.1152/physrev.90100.2007
88. Wedell-Neergaard AS, Lang Lehrskov L, Christensen RH, Legaard GE, Dorph E, Larsen MK, et al. Exercise-Induced Changes in Visceral Adipose Tissue Mass Are Regulated by IL-6 Signaling: A Randomized Controlled Trial. Cell Metab (2019) 29(4):844–855.e3. doi: 10.1016/j.cmet.2018.12.007
89. Han MS, White A, Perry RJ, Camporez JP, Hidalgo J, Shulman GI, et al. Regulation of Adipose Tissue Inflammation by Interleukin 6. Proc Natl Acad Sci U S A (2020) 117(6):2751–60. doi: 10.1073/pnas.1920004117
90. Xiong XQ, Geng Z, Zhou B, Zhang F, Han Y, Zhou YB, et al. FNDC5 Attenuates Adipose Tissue Inflammation and Insulin Resistance via AMPK-Mediated Macrophage Polarization in Obesity. Metabolism (2018) 83:31–41. doi: 10.1016/j.metabol.2018.01.013
91. Amor M, Itariu BK, Moreno-Viedma V, Keindl M, Jürets A, Prager G, et al. Serum Myostatin Is Upregulated in Obesity and Correlates With Insulin Resistance in Humans. Exp Clin Endocrinol Diabetes (2019) 127(8):550–6. doi: 10.1055/a-0641-5546
92. Dong J, Dong Y, Dong Y, Chen F, Mitch WE, Zhang L. Inhibition of Myostatin in Mice Improves Insulin Sensitivity via Irisin-Mediated Cross Talk Between Muscle and Adipose Tissues. Int J Obes (Lond) (2016) 40(3):434–42. doi: 10.1038/ijo.2015.200
93. Safdar A, Saleem A, Tarnopolsky MA. The Potential of Endurance Exercise-Derived Exosomes to Treat Metabolic Diseases. Nat Rev Endocrinol (2016) 12(9):504–17. doi: 10.1038/nrendo.2016.76
94. Aswad H, Forterre A, Wiklander OP, Vial G, Danty-Berger E, Jalabert A, et al. Exosomes Participate in the Alteration of Muscle Homeostasis During Lipid-Induced Insulin Resistance in Mice. Diabetologia (2014) 57(10):2155–64. doi: 10.1007/s00125-014-3337-2
95. Hou Z, Qin X, Hu Y, Zhang X, Li G, Wu J, et al. Longterm Exercise-Derived Exosomal miR-342-5p: A Novel Exerkine for Cardioprotection. Circ Res (2019) 124(9):1386–400. doi: 10.1161/CIRCRESAHA.118.314635
96. Iroz A, Couty JP, Postic C. Hepatokines: Unlocking the Multi-Organ Network in Metabolic Diseases. Diabetologia (2015) 58(8):1699–703. doi: 10.1007/s00125-015-3634-4
97. Jung TW, Yoo HJ, Choi KM. Implication of Hepatokines in Metabolic Disorders and Cardiovascular Diseases. BBA Clin. BBA Clin (2016) 5:108–13. doi: 10.1016/j.bbacli.2016.03.002
98. Stefan N, Häring HU. The Role of Hepatokines in Metabolism. Nat Rev Endocrinol (2013) 9(3):144–52. doi: 10.1038/nrendo.2012.258
99. Zhao Y, Zhao MF, Jiang S, Wu J, Liu J, Yuan XW, et al. Liver Governs Adipose Remodelling via Extracellular Vesicles in Response to Lipid Overload. Nat Commun (2020) 11(1):719. doi: 10.1038/s41467-020-14450-6
100. Stefan N, Häring HU. Circulating Fetuin-A and Free Fatty Acids Interact to Predict Insulin Resistance in Humans. Nat Med (2013) 19(4):394–5. doi: 10.1038/nm.3116
101. Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A Acts as an Endogenous Ligand of TLR4 to Promote Lipid-Induced Insulin Resistance. Nat Med (2012) 18(8):1279–85. doi: 10.1038/nm.2851
102. Lee S, Norheim F, Gulseth HL, Langleite TM, Kolnes KJ, Tangen DS, et al. Interaction Between Plasma Fetuin-A and Free Fatty Acids Predicts Changes in Insulin Sensitivity in Response to Long-Term Exercise. Physiol Rep (2017) 5(5):e13183. doi: 10.14814/phy2.13183
103. Ward K, Mulder E, Frings-Meuthen P, O’Gorman DJ, Cooper D. Fetuin-A as a Potential Biomarker of Metabolic Variability Following 60 Days of Bed Rest. Front Physiol (2020) 11:573581. doi: 10.3389/fphys.2020.573581
104. Pérez-Sotelo D, Roca-Rivada A, Larrosa-García M, Castelao C, Baamonde I, Baltar J, et al. Visceral and Subcutaneous Adipose Tissue Express and Secrete Functional Alpha2hsglycoprotein (Fetuin a) Especially in Obesity. Endocrine (2017) 55(2):435–46. doi: 10.1007/s12020-016-1132-1
105. Jialal I, Devaraj S, Bettaieb A, Haj F, Adams-Huet B. Increased Adipose Tissue Secretion of Fetuin-A, Lipopolysaccharide-Binding Protein and High-Mobility Group Box Protein 1 in Metabolic Syndrome. Atherosclerosis (2015) 241(1):130–7. doi: 10.1016/j.atherosclerosis.2015.04.814
106. Jialal I, Pahwa R. Fetuin-A Is Also an Adipokine. Lipids Health Dis (2019) 18(1):73. doi: 10.1186/s12944-019-1021-8
107. Chatterjee P, Seal S, Mukherjee S, Kundu R, Mukherjee S, Ray S, et al. Adipocyte Fetuin-A Contributes to Macrophage Migration Into Adipose Tissue and Polarization of Macrophages. J Biol Chem (2013) 288(39):28324–30. doi: 10.1074/jbc.C113.495473
108. Moraes-Vieira PM, Yore MM, Dwyer PM, Syed I, Aryal P, Kahn BB. RBP4 Activates Antigen-Presenting Cells, Leading to Adipose Tissue Inflammation and Systemic Insulin Resistance. Cell Metab (2014) 19(3):512–26. doi: 10.1016/j.cmet.2014.01.018
109. Graham TE, Yang Q, Blüher M, Hammarstedt A, Ciaraldi TP, Henry RR, et al. Retinol-Binding Protein 4 and Insulin Resistance in Lean, Obese, and Diabetic Subjects. N Engl J Med (2006) 354(24):2552–63. doi: 10.1056/NEJMoa054862
110. Moraes-Vieira PM, Yore MM, Sontheimer-Phelps A, Castoldi A, Norseen J, Aryal P, et al. Retinol Binding Protein 4 Primes the NLRP3 Inflammasome by Signaling Through Toll-Like Receptors 2 and 4. Proc Natl Acad Sci U.S.A. (2020) 117(49):31309–18. doi: 10.1073/pnas.2013877117
111. Ma X, Zhou Z, Chen Y, Wu Y, Liu Y. RBP4 Functions as a Hepatokine in the Regulation of Glucose Metabolism by the Circadian Clock in Mice. Diabetologia (2016) 59(2):354–62. doi: 10.1007/s00125-015-3807-1
112. Kotnik P, Fischer-Posovszky P, Wabitsch M. RBP4: A Controversial Adipokine. Eur J Endocrinol (2011) 165(5):703–11. doi: 10.1530/EJE-11-0431
113. Wu J, Dong T, Chen T, Sun J, Luo J, He J, et al. Hepatic Exosome-Derived miR-130a-3p Attenuates Glucose Intolerance via Suppressing PHLPP2 Gene in Adipocyte. Metabolism (2020) 103:154006. doi: 10.1016/j.metabol.2019.154006
114. Ji Y, Luo Z, Gao H, Dos Reis FCG, Bandyopadhyay G, Jin Z, et al. Hepatocyte-Derived Exosomes From Early Onset Obese Mice Promote Insulin Sensitivity Through miR-3075. Nat Metab (2021) 3(9):1163–74. doi: 10.1038/s42255-021-00444-1
115. Gonzalez-Gil AM, Elizondo-Montemayor L. The Role of Exercise in the Interplay Between Myokines, Hepatokines, Osteokines, Adipokines, and Modulation of Inflammation for Energy Substrate Redistribution and Fat Mass Loss: A Review. Nutrients (2020) 12(6):1899. doi: 10.3390/nu12061899
116. de Oliveira Dos Santos AR, de Oliveira Zanuso B, Miola VFB, Barbalho SM, Santos Bueno PC, Flato UAP, et al. Adipokines, Myokines, and Hepatokines: Crosstalk and Metabolic Repercussions. Int J Mol Sci (2021) 22(5):2639. doi: 10.3390/ijms22052639
117. Wang L, Liu M, Ning D, Zhu H, Shan G, Wang D, et al. Low Serum ZAG Levels Correlate With Determinants of the Metabolic Syndrome in Chinese Subjects. Front Endocrinol (Lausanne) (2020) 11:154. doi: 10.3389/fendo.2020.00154
118. Suzuki ST, Zhao B, Yang J. Enhanced Muscle by Myostatin Propeptide Increases Adipose Tissue Adiponectin, PPAR-Alpha, and PPAR-Gamma Expressions. Biochem Biophys Res Commun (2008) 369(2):767–73. doi: 10.1016/j.bbrc.2008.02.092
119. Yao H, Fan C, Lu Y, Fan X, Xia L, Li P, et al. Alteration of Gut Microbiota Affects Expression of Adiponectin and Resistin Through Modifying DNA Methylation in High-Fat Diet-Induced Obese Mice. Genes Nutr (2020) 15(1):12. doi: 10.1186/s12263-020-00671-3
120. Lu Y, Fan C, Liang A, Fan X, Wang R, Li P, et al. Effects of SCFA on the DNA Methylation Pattern of Adiponectin and Resistin in High-Fat-Diet-Induced Obese Male Mice. Br J Nutr (2018) 120(4):385–92. doi: 10.1017/S0007114518001526
121. Yang HE, Li Y, Nishimura A, Jheng HF, Yuliana A, Kitano-Ohue R, et al. Synthesized Enone Fatty Acids Resembling Metabolites From Gut Microbiota Suppress Macrophage-Mediated Inflammation in Adipocytes. Mol Nutr Food Res (2017) 61(10):1700064. doi: 10.1002/mnfr.201700064
122. Oh KJ, Lee DS, Kim WK, Han BS, Lee SC, Bae KH. Metabolic Adaptation in Obesity and Type II Diabetes: Myokines, Adipokines and Hepatokines. Int J Mol Sci (2016) 18(1):8. doi: 10.3390/ijms18010008
123. Suriano F, Van Hul M, Cani PD. Gut Microbiota and Regulation of Myokine-Adipokine Function. Curr Opin Pharmacol (2020) 52:9–17. doi: 10.1016/j.coph.2020.03.006
124. Huang-Doran I, Zhang CY, Vidal-Puig A. Extracellular Vesicles: Novel Mediators of Cell Communication In Metabolic Disease. Trends Endocrinol Metab (2017) 28(1):3–18. doi: 10.1016/j.tem.2016.10.003
125. Akbar N, Azzimato V, Choudhury RP, Aouadi M. Extracellular Vesicles in Metabolic Disease. Diabetologia (2019) 62(12):2179–87. doi: 10.1007/s00125-019-05014-5
126. Vlachakis D, Mitsis τ, Nicolaides N, Efthimiadou A, Giannakakis A, Bacopoulou F, et al. Functions, Pathophysiology and Current Insights of Exosomal Endocrinology (Review). Mol Med Rep (2021) 23(1):26. doi: 10.3892/mmr.2020.11664
127. Obata Y, Kita S, Koyama Y, Fukuda S, Takeda H, Takahashi M, et al. Adiponectin/T-Cadherin System Enhances Exosome Biogenesis and Decreases Cellular Ceramides by Exosomal Release. JCI Insight (2018) 3(8):e99680. doi: 10.1172/jci.insight.99680
128. Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DL, et al. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metab (2016) 24(6):820–34. doi: 10.1016/j.cmet.2016.10.002
129. Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, et al. Adipose-Derived Circulating miRNAs Regulate Gene Expression in Other Tissues. Nature (2017) 542(7642):450–5. doi: 10.1038/nature21365
Keywords: obesity, adipose tissue inflammation, adipose tissue macrophages, organokines, exosomes
Citation: Wang Y-Y, Wang Y-D, Qi X-Y, Liao Z-Z, Mai Y-N and Xiao X-H (2022) Organokines and Exosomes: Integrators of Adipose Tissue Macrophage Polarization and Recruitment in Obesity. Front. Endocrinol. 13:839849. doi: 10.3389/fendo.2022.839849
Received: 20 December 2021; Accepted: 24 January 2022;
Published: 18 February 2022.
Edited by:
Jae B. Kim, Seoul National University, South KoreaReviewed by:
Shunbun Kita, Osaka University, JapanCopyright © 2022 Wang, Wang, Qi, Liao, Mai and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin-Hua Xiao, eGluaHVhMDEwMkAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.