 Pui Li1Kuraym Khalid Kuraym Alenazi1
Pui Li1Kuraym Khalid Kuraym Alenazi1 Jordanna Dally1Emma Louise Woods1
Jordanna Dally1Emma Louise Woods1 Rachel Jane Waddington2
Rachel Jane Waddington2 Ryan Moseley1*
Ryan Moseley1*
- 1Disease Mechanisms Group, School of Dentistry, College of Biomedical and Life Sciences, Cardiff University, Cardiff, United Kingdom
- 2Biomaterials Group, School of Dentistry, College of Biomedical and Life Sciences, Cardiff University, Cardiff, United Kingdom
Impaired bone healing is a significant complication observed in individuals with type 2 diabetes mellitus (T2DM), leading to prolonged recovery, increased risk of complications, impaired quality of life, and increased risk of patient morbidity. Oxidative stress, resulting from an imbalance between the generation of reactive oxygen species (ROS) and cellular/tissue antioxidant defence mechanisms, has been identified as a critical contributor to the pathogenesis of impaired bone healing in T2DM. Antioxidants have shown promise in mitigating oxidative stress and promoting bone repair, particularly non-enzymic antioxidant entities. This comprehensive narrative review aims to explore the underlying mechanisms and intricate relationship between oxidative stress, impaired bone healing and T2DM, with a specific focus on the current preclinical and clinical evidence advocating the potential of antioxidant therapeutic interventions in improving bone healing outcomes in individuals with T2DM. From the ever-emerging evidence available, it is apparent that exogenously supplemented antioxidants, especially non-enzymic antioxidants, can ameliorate the detrimental effects of oxidative stress, inflammation, and impaired cellular function on bone healing processes during uncontrolled hyperglycaemia; and therefore, hold considerable promise as novel efficacious therapeutic entities. However, despite such conclusions, several important gaps in our knowledge remain to be addressed, including studies involving more sophisticated enzymic antioxidant-based delivery systems, further mechanistic studies into how these antioxidants exert their desirable reparative effects; and more extensive clinical trial studies into the optimisation of antioxidant therapy dosing, frequency, duration and their subsequent biodistribution and bioavailability. By enhancing our understanding of such crucial issues, we can fully exploit the oxidative stress-neutralising properties of these antioxidants to develop effective antioxidant interventions to mitigate impaired bone healing and reduce the associated complications in such T2DM patient populations.
1 Introduction
Bone repair is the physiological process that occurs at sites of lost tissue, with the ultimate aim of re-establishing normal bone structure and function (1). The various stages of bone healing are clinically important in the fields of dentistry and orthopaedics, as these are responsible for achieving the successful healing of bone defects caused by trauma, bone diseases (such as osteonecrosis and tumours), or surgical procedures that involve bone manipulation, including dental extractions, implant placement or bone augmentation techniques (2–7). Therefore, it is important to create and maintain a favourable environment to allow optimal healing at the area of bone injury or defect.
However, despite being a highly organised process, it is recognised that mechanisms underlying bone repair can be significantly disrupted or impaired by the local tissue microenvironment, including via metabolic, cellular and molecular changes induced through the uncontrolled glycaemic control and hyperglycaemia associated with type 2 diabetes mellitus (T2DM). T2DM is a chronic metabolic disorder characterised by insulin resistance and elevated blood glucose levels (8). T2DM and its associated patient co-morbidities represent major medical and public health concerns, due to their ever-increasing global prevalence. Indeed, T2DM is estimated to affect approximately 451 million people worldwide, with projections expecting rises to 693 million by 2045 (9). Consequently, such clinical situations provide significant economic burdens to healthcare providers. In this regard, uncontrolled T2DM is recognised as a mediator of disordered bone metabolism and homeostasis, being associated with various complications including delayed or compromised bone formation following trauma or surgical intervention, resulting in prolonged recovery time, non-union, heightened risk of other post-injury complications and reduced functional outcomes (10–12). Thus, T2DM is now an established risk factor for the development or exacerbation of dental/orthopaedic fractures, periodontal disease and implant failure (13–15), whilst normal glycaemic control is imperative to the success of these reparative processes.
Concurent with impaired bone healing in T2DM patients are several mechanisms implicated in causing dysfunctional cellular behaviour and osteogenic activities, with one of the most well-established mediators of these disrupted processes being oxidative stress (16–21). However, despite oxidative stress being widely acknowledged as a key influential factor on the various cell types involved in normal bone repair responses, our current knowledge and understanding of the roles and therapeutic potentials of the antioxidants responsible for counteracting the elevated levels and deleterious effects of oxidative stress in biological systems, including bone, remains much less in comparison. Therefore, this review article aims to explore the existing scientific preclinical and clinical evidence to provide a much needed and detailed overview of our current understanding of the mechanistic roles which oxidative stress plays in mediating altered cell signalling pathways leading to dysfunctional cellular repair responses in T2DM bone, coupled with the evidence available to support or discount the potential use of antioxidants as novel therapeutics for the alleviation of such impaired T2DM-associated healing outcomes in future.
2 Cellular and molecular mechanisms of normal bone repair
Bone healing is regarded as a complex, but tightly organised process, consisting of several highly coordinated overlapping phases, involving inflammation, repair and remodelling; mediated through cooperation between various cell types and intracellular/extracellular signalling molecules to re-establish normal bone architecture and function [(1, 22–25); summarised in Figure 1]. Bone repair commences when a bone is injured, leading to vascular disruption. Platelet-mediated fibrin clot formation occurs during haemostasis and acts as a provisional matrix, releasing chemotactic growth factors and pro-inflammatory cytokines, such as interleukin-1 (IL-1), IL-6 and tumour necrosis factor-α (TNF-α) (23). Subsequently, inflammation occurs, leading to the recruitment of neutrophils, classical pro-inflammatory M1 subtype macrophages and lymphocytes to the wound site (24–27). These inflammatory cells are primarily responsible for the removal of bacteria, minimising the risk of wound site infection.
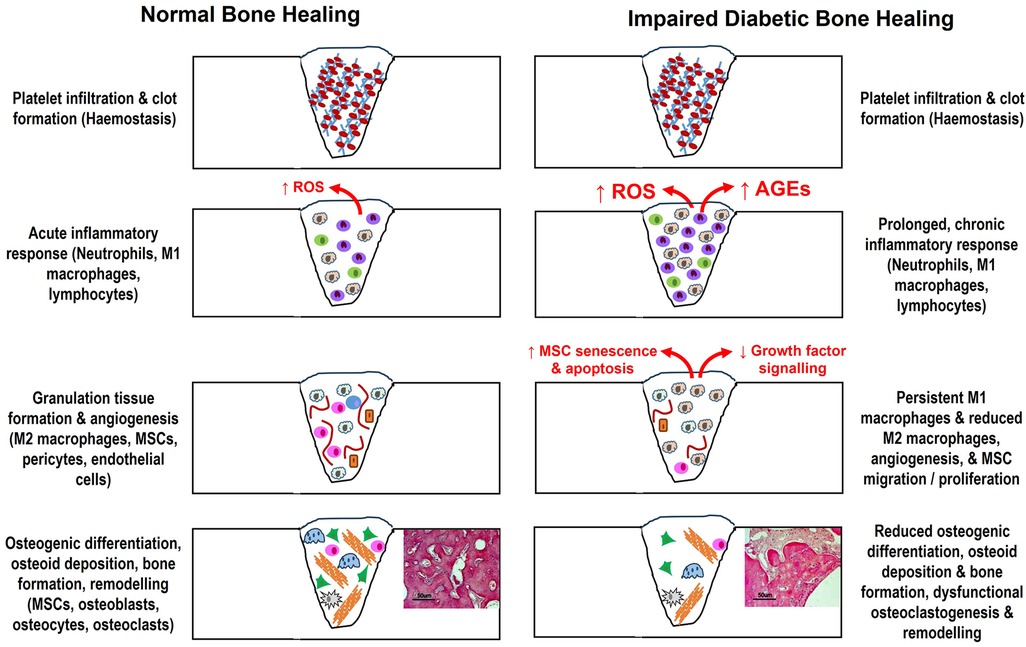
Figure 1. Summary of the various overlapping phases and cell types associated with bone repair to re-establish normal bone architecture and function, and how these cellular and molecular events are disrupted by the uncontrolled hyperglycaemia associated with T2DM to cause impaired healing. AGEs, advanced glycation end products; MSCs, mesenchymal stromal cells; ROS, reactive oxygen species.
Following inflammation, the blood clot transitions into granulation tissue, where mesenchymal stromal cells (MSCs) are recruited to the wound site from the bone marrow and periosteum; along with alternative anti-inflammatory M2 subtype macrophages, responsible for removing cellular debris and the promotion of angiogenesis, facilitated via production of a wide range of anti-inflammatory mediators as healing progresses (24–26). Such important roles for MSCs in bone healing are attributed to their self-renewal properties and responses to growth factors, which initiate migratory and proliferative responses, in addition to their osteogenic lineage differentiation commitment to form mature osteoblasts via upregulation of transcription factors, such as Runx2 and Osterix. Osteoblasts are responsible for initiation of extracellular matrix (ECM) synthesis, deposition, remodelling and subsequent mineralisation (22, 23). Pericyte recruitment is also essential to stimulate endothelial cell formation; whilst neo-angiogenesis and vascular ingrowth support tissue repair via supplementation of nutrients and additional MSCs to wound sites, both of which are necessary for osteogenesis to occur (28, 29).
Central to these osteogenic and angiogenic responses at wound sites are the upstream activation of cell signalling pathways, principally orchestrated through the stimulation of endogenous connective tissue cells by a plethora of growth factors, including transforming growth factor-β1 (TGF-β1), bone morphogenetic proteins (BMPs) and vascular endothelial growth factor (VEGF) (30–32). Such growth factors possess osteoinductive, osteoconductive and osteoadaptive properties that instigate ossification. Maintenance of MSC populations is achieved through the presence of niches, located within various bone regions (33, 34).
During bone repair, MSCs are primarily sourced from two distinct niches within the bone marrow cavity (35). These include the highly vascularised perivascular/sub-endosteal niche, which contains endothelial cells, hematopoietic stem cells (HSCs) and uncommitted MSCs (36–38); whereas the endosteal niche, located at the interface between trabecular bone and bone marrow, comprises uncommitted MSCs, pre-osteoblasts and osteoblasts lining compact bone (39–41). It was originally proposed that bone marrow-derived MSCs are the main source responsible for bone repair. However, it has since been shown that MSCs within both the perivascular and endosteal niches possess important roles in facilitating bone repair processes overall (34, 35). Indeed, committed lineage restricted MSCs lining the endosteum also play key roles in mediating bone repair responses, acting as “first responders” during mineralised tissue repair (41–43).
MSC osteogenic differentiation into mature osteoblasts is followed by the synthesis and secretion of bone osteoid, an immature bone ECM consisting of type I collagen, proteoglycans, such as the small leucine-rich proteoglycans (SLRPs), decorin and biglycan; and various bone glycoproteins (such as osteocalcin, osteonectin, osteopontin and bone sialoprotein), which possess distinct roles in regulating normal matrix-mediated mineralisation (1, 37–39). Herein, some osteoblasts become embedded within the mineralised ECM to form osteocytes, with roles in regulating bone homeostasis and osteoblast/osteoclast formation via hormonal and mechanical cues; whilst others undergo apoptosis to arrest further bone synthesis (44, 45).
The latter phase is tissue remodelling involves replacement of immature woven bone with mature lamellar bone over time, driven by mechanical loading and mediated via the coordinated action of osteoblasts and bone resorbing osteoclasts (1, 37, 39, 46–50). Osteoblasts regulate osteoclastic differentiation from HSCs, via secretion of receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin (OPG), which control RANKL interaction with receptor activator of nuclear factor-kappa B (RANK) on HSC surfaces and osteoclast formation overall. Hence, the type and quality of bone formed not only relies on the tissue and anatomical location of the wound, but also the mechanical conditions in the wound site (38, 47, 50).
3 Type 2 diabetes mellitus and impaired bone healing
Due to the varied mineralised tissue perturbations observed in T2DM patients, extensive preclinical studies have been performed to enhance our understanding of the cellular, molecular and metabolic events that support delayed/dysfunctional bone formation and the macro/microscopic changes in bone architecture associated with T2DM, such as deteriorations in composition, volume, quality and biomechanical properties. Indeed, it is known that T2DM influences bone quantity properties, such as the relative mineral density and porosity, which significantly affect overall bone quality. Consequently, various biomechanical analyses have shown that diabetic bone derived from humans and rodent model studies are accompanied by increased fragility and fatigue, with reductions in crucial parameters, such as mechanical load, elasticity, energy absorption and stiffness (51–55).
Impaired bone healing observed in individuals with T2DM is a multifactorial phenomenon, affecting all stages of bone repair and mediated by numerous initiators, including hyperglycaemia, chronic inflammation, oxidative stress, advanced glycation end products (AGEs), polyol pathway, high protein kinase C (PKC) activity and hexosamine biosynthesis pathways; which abrogate bone healing and angiogenic processes [(8, 10–15); summarised in Figure 1]. Chronic inflammation, which is commonly observed in T2DM, further exacerbates the impairment of bone healing processes, although it has been proposed that the chemotactic ability to recruit inflammatory cells to wound healing sites declines, compared to normo-glycaemic environments. The diabetic bone environment is further associated with a delayed, but sustained increase in the secretion of pro-inflammatory cytokines, such as IL-1β and TNF-α, capable of inhibiting osteoblast differentiation and activity, but activating osteoclast formation and bone resorption (46, 49). Furthermore, dysregulated immune responses and the prolonged presence of pro-inflammatory M1 macrophages during T2DM, at the expense of reparative M2 macrophages, may lead to delayed resolution of inflammation and compromised tissue repair (56–63).
Additional studies have suggested that impaired diabetic bone healing is due to the delayed onset of osteogenic responses during T2DM, culminating in histopathological features such as osteopenia and decreased bone formation in vivo (56, 63–67). Hyperglycaemia is well-established to significantly impact normal bone marrow-derived MSC and osteoblast responses, which exhibit reduced proliferative capabilities due to shortened telomeres and early-onset replicative senescence, negatively influencing MSC viability and apoptosis, colony-forming efficiency, multi-potency and osteogenic differentiation capabilities overall (68–74). Impaired osteogenesis under hyperglycaemic conditions has been reported to occur due to preferential MSC adipogenic differentiation, driven by peroxisome proliferator-activated receptor-γ (PPAR-γ) (69, 71, 75).
Despite many studies supporting the detrimental effects of hyperglycaemia on MSC and osteoblast responses in bone, other studies suggest limited effects of hyperglycaemia on MSC and osteoblast functions (76–79). These inter-study variations are a proposed consequence of factors, such as the varied MSC isolation procedures implemented, the source, purity and heterogeneity of isolated MSC populations, and the contrasting glucose concentrations/exposure periods used during in vitro studies (41). Indeed, despite most in vitro hyperglycaemia studies demonstrating significant deleterious effects of hyperglycaemic conditions on the proliferation and osteogenic differentiation of human or rodent MSC responses derived from the perivascular niche (68–74), recent evidence suggests that hyperglycaemia has limited impact on the proliferative and stem cell characteristics of MSC populations derived from the endosteal niche of compact bone (39–43, 80). However, endosteal niche-derived MSCs are susceptible to reduced osteogenic and adipogenic differentiation capabilities under such hyperglycaemic conditions.
In addition to direct effects of uncontrolled glucose levels on MSC and osteoblast wound healing responses, the diabetic bone microenvironment can further disrupt cellular reparative functions via dysregulation of growth factor signalling and alterations in bone ECM composition, both of which have further repercussions for bone repair overall. Despite various growth factors exhibiting essential roles in the regulation of normal bone repair (30–32), T2DM induces an imbalance in the expression and signalling of many growth factors, including TGF-β1, BMPs, VEGF, fibroblast growth factor-2 (FGF-2) and insulin-like growth factor-1 (IGF-1), which disrupts their bioavailability and cell signalling mechanisms, contributing to dysregulated angiogenesis and bone healing overall.
The delayed, but sustained secretion of TGF-β1 have been reported, associated with high glucose-treated MSCs and other in vivo models of diabetic bone repair, in addition to sera derived from T2DM patients (56, 63, 73, 81–84). Such elevated TGF-β1 levels have been shown to be predominantly derived from MSCs following short-term exposure to hyperglycaemia, whilst prolonged high glucose exposure significantly retarded TGF-β1 expression and secretion by MSCs (63). Both scenarios could have severe consequences for normal bone repair, as high TGF-β1 levels are known to be inhibitory towards osteoblast differentiation and ECM deposition during the latter stages of osteogenesis (85–88); whilst reduced TGF-β1 levels with long-term glucose exposure could result in complete attenuation of osteogenic differentiation by MSCs and subsequent bone ECM deposition and mineralisation (31, 32). Similar profiles have also been shown with BMP-2, BMP-4 and BMP-6, where delayed expression, followed by subsequent elevated levels and sustained disruption to the normal secretion and cell signalling profiles have been reported (83, 89–93). Decreased VEGF, FGF-2 and IGF-1 levels have further been associated with diabetic bone healing, which impedes both osteogenic and angiogenic responses within the healing tissue (55, 92–96).
Such direct effects on growth factor signalling and MSC osteogenic differentiation further impact on the deposition and remodelling of the bone ECM, with significant perturbations in composition and subsequent mineralisation reported. A prominent feature of impaired diabetic bone healing is decreased type I collagen synthesis. In combination with the established structural changes to collagen molecules, due to AGEs forming non-enzymatic irreversible crosslinks within the collagen triple helix and between adjacent fibres, normal collagen enzymatic crosslinking processes are compromised, resulting in impaired bone stability, strength and quality (97–101). As these AGE-based crosslinks impede the proteolytic remodelling of type I collagen by osteoclasts, their reduced turnover leads to an accumulation of abnormal type 1 collagen that contribute to diminishing biomechanical properties in diabetic bone (102).
In addition to type I collagen, the expression, protein levels and structural properties of various non-collagenous ECM components are further altered during T2DM. Regarding the prominent proteoglycans within bone, decorin and biglycan, recent studies have demonstrated that MSC expression and secretion are enhanced following short-term hyperglycaemic exposure (63, 102). However, prolonged exposure to high glucose conditions significantly arrested decorin and biglycan expression and protein levels. Numerous glycoproteins commonly localised within bone are also influenced by hyperglycaemic conditions, largely exhibiting altered expression of osteocalcin, osteopontin and bone sialoprotein in vitro, with delayed and/or sustained levels identified in vivo (56, 68, 81, 85, 103–107). Such ECM component dysregulation has collectively been proposed to perturb the sequence and orchestration of events that occur during normal bone mineralisation and repair, such as collagen fibrillogenesis, mineral deposition, crystal growth, TGF-β1 bioavailability and osteogenic cell signalling; further contributing to delayed or impaired bone healing overall (108–110).
There is also substantial evidence confirming that the uncontrolled glycaemic control associated with T2DM disrupts normal endothelial cells functions and neo-angiogenesis, essential for oxygen and nutrient provision and successful bone repair (28, 29). Specifically, preclinical and clinical studies have demonstrated that hyperglycaemia induces alterations in hypoxia-inducible factor 1 (HIF-1) levels. Consequently, pro-angiogenic signalling pathways are abrogated, resulting in decreased VEGF expression, as well as that of other markers, such as PECAM-1; whilst promoting the activation of anti-angiogenic signals (55, 93, 97, 107, 111, 112). Additionally, endothelial progenitor cells (EPCs) within the bone marrow vascular niches are significantly reduced in number by hyperglycaemia (113, 114). Such cellular losses lead to impaired EPC and microvascular endothelial cell functions, via reduced proliferation, migration and tube formation, together with increased autophagy and apoptosis, in part mediated via transcription factor, FOXO1; and culminating in defective revascularisation (61, 112, 114–118).
Due to such hyperglycaemic and pro-inflammatory environments, osteoblast differentiation and functions are significantly disrupted during T2DM. Such events may be abrogated further due to increased RANKL and reduced OPG expression, in favour of enhanced HSC differentiation, osteoclast formation and bone resorption (104, 119, 120). That said, the role of osteoclastogenesis in T2DM is quite contentious, as there is conflicting evidence to suggest opposing low RANKL and high OPG expression profiles in osteoblasts under high glucose conditions, potentially limiting bone resorptive activities (74, 121). Similarly, despite hyperglycaemia leading to aberrant osteoclast differentiation and increased bone resorption, in vitro experiments suggest that changes in bone architecture typical for T2DM are not a direct consequence of excessive bone resorption (74). As it has also been proposed that osteoclasts possess decreased bone resorption activity under hyperglycaemic conditions (107, 122), it is plausible that both osteogenesis and osteoclastogenesis are impaired during T2DM.
Hence, the impairment of normal osteoblast and osteoclast reparative responses, coupled with dysfunctional inflammation and attenuated angiogenic responses that compromise blood supply to the wound site, collectively lead to delayed healing and reduced bone formation during T2DM. Consequently, understanding the intricate relationship between oxidative stress, impaired bone healing and T2DM, is crucial for developing targeted therapeutic strategies to improve bone repair outcomes in diabetic individuals. Therefore, the remainder of this narrative review focusses on providing a comprehensive analysis of the mechanisms underlying impaired bone healing associated with T2DM, with a specific emphasis on the role of oxidative stress and potential interventions involving antioxidants. By targeting these underlying oxidative mechanisms and restoring the redox balance between reactive oxygen species (ROS) production and antioxidant defences, it may be possible to develop novel therapeutic strategies that address the altered inflammatory, angiogenic and osteogenic responses, to restore bone healing and mitigate the clinical complications associated with T2DM.
4 Oxidative stress and antioxidants
Oxidative stress, referring to an imbalance between ROS production and cellular/tissue antioxidant defence mechanisms, plays a pivotal role in the pathogenesis of numerous diseases, including T2DM and impaired bone healing (16–21). ROS, such as superoxide radicals (O2·−), hydrogen peroxide (H2O2), and hydroxyl radicals (·OH), are generated as by-products of normal cellular metabolism and can be further produced by various enzymatic sources, including NADPH oxidases, xanthine oxidase, and the mitochondrial electron transport chain (16–18, 21). Additionally, in uncontrolled chronic diabetic environments, AGEs can be produced. AGEs are a diverse group of compounds spontaneously produced by non-enzymatic glycation or oxidation of various proteins, including type I collagen (97–102), and increased in T2DM patients due to hyperglycaemia and the accompanying elevations in oxidative stress (123). AGEs also promote inflammation and the production of pro-inflammatory cytokines, perpetuating impaired healing (124). Consequently, low ROS levels are purported to play important roles in regulating cell signalling and functions (125). However, excessive production of highly reactive ROS and AGE molecules can cause indiscriminate modifications and damage to cellular and ECM components, including DNA, proteins, lipids and carbohydrates, which negatively impacts normal biomolecular and cellular functions (16, 17). Furthermore, interactions between AGEs and their cell surface receptor, receptor for AGEs (RAGE), can enhance further pro-inflammatory cytokine and ROS production (123, 126), resulting in a continual cycle of chronic inflammation and bone resorption.
ROS levels are tightly regulated by enzymic and non-enzymic antioxidant defence mechanisms, which act by neutralising and scavenging ROS, maintaining redox homeostasis, and preventing cellular damage (16, 17, 19, 21, 127). Antioxidants are compounds that can neutralise or reduce the harmful effects of ROS, which can be divided into endogenous and exogenous antioxidants that help mitigate oxidative stress. Endogenous antioxidant mechanisms include enzymes, such as superoxide dismutases (SODs), catalase, and glutathione peroxidases (GPx), as well as non-enzymatic antioxidants such as glutathione.
Three distinct SOD isoforms have been identified in mammalian cells, with the copper-zinc containing SOD (SOD1) and manganese containing SOD (SOD2) being the most significant. SOD1 and SOD2 are both ubiquitously expressed in aerobic cells, being localised intracellularly within the cytosol and mitochondria, respectively (128, 129). In contrast, the copper-zinc containing SOD isoform (SOD3) possesses more restricted expression and is particularly localised within pericellular and ECM environments (128, 130). Nonetheless, wherever located, SOD isoforms have key roles in alleviating oxidative stress by catalysing the dismutation reaction to convert O2·− to H2O2 (16, 17). Most aerobic cells also contain the haem containing enzyme, catalase, particularly localised in the cytosol within the peroxisomes (131), which detoxifies H2O2 into H2O (16, 17). In addition to catalase, H2O2 decomposition is aided by glutathione-metabolising enzymes, including glutathione peroxidases (GPXs), S-transferases (GSTs), reductases (GSRs) and synthetases (GSSs) (132, 133). GPXs catalyse the oxidation of reduced glutathione (GSH) to oxidised glutathione (GSSG) by H2O2 within the cytosol, with GSH synthesis being regulated and restored through the actions of GSRs and GSSs.
Enzymic antioxidants are themselves regulated at a gene level by nuclear factor E2-related actor 2 (Nrf2), a transcription factor normally located in the cytosol under basal oxidative conditions, being tightly regulated by Kelch-like ECH-associated protein 1 (Keap1), a redox-sensitive ubiquitin ligase which tethers Nrf2 within the cytosolic compartment (19, 134, 135). Under conditions of oxidative stress, Keap1 is oxidised at reactive cysteine residues, resulting in Keap1 inactivation, stabilisation of Nrf2 and subsequent translocation to the nucleus where antioxidant response element (ARE) binding and gene transcription is initiated. Notable gene targets of Nrf2 regulation include glutathione regulatory genes, such as GPx, glutathione disulfide reductase 1 (GSR1) and glutathione synthetase (GSS) (134, 135). In contrast, exogenous antioxidants are primarily comprised of dietary nutrients, including ascorbic acid, α-tocopherol, selenium and various phytochemicals, which provide additional protection against oxidative damage (16, 17).
4.1 Oxidative stress and impaired bone healing during T2DM
During normal bone healing, a delicate balance exists between oxidative stress and antioxidant defence mechanisms, which permit tissue repair to proceed. Normal glucose metabolism occurs through the tricarboxylic acid cycle, involving the activation of electron pump channels and electron movement across mitochondrial membranes to generate energy. However, in individuals with T2DM, high glucose levels increase the voltage across the membranes to significantly elevate O2·− production, disrupting the ROS/antioxidant balance (18, 21, 126, 127). This imbalance induces detrimental effects in bone cells, ECM and tissues, via disruption of cell signalling pathways involved in inflammation, angiogenesis and bone formation, exacerbating impaired healing (16–21, 123–127). In addition to elevated ROS production by neutrophils and M1 macrophages during T2DM-associated chronic inflammation (56–63), hyperglycaemia contributes to ROS overproduction through multiple pathways, including increased glucose auto-oxidation, AGE formation, PKC activation and various other signalling pathways involved in inflammation, angiogenesis and osteoblast differentiation [(16–21, 123–127); summarised in Figure 2].
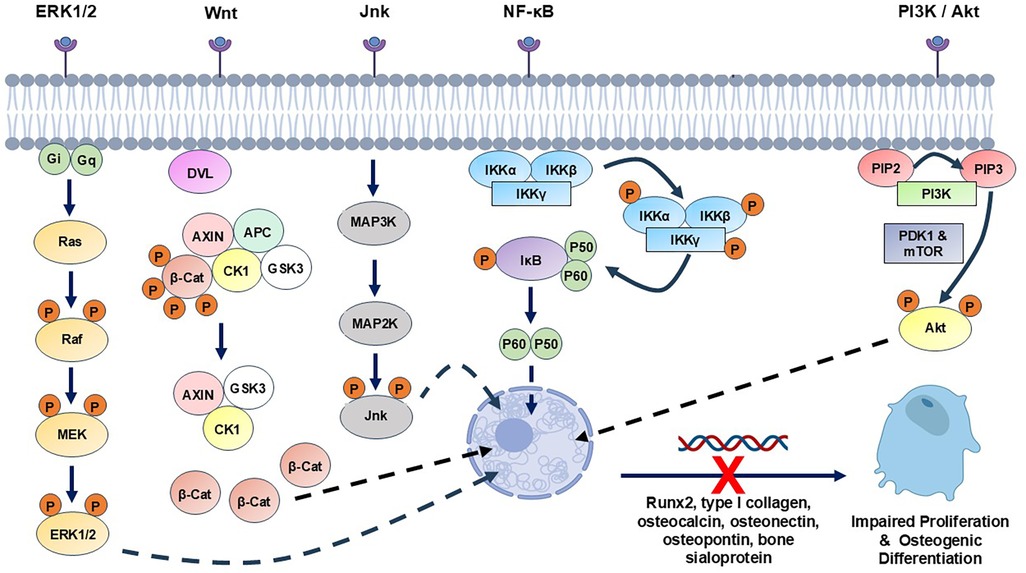
Figure 2. Summary of the main cell signalling pathway mechanisms reportedly disrupted by hyperglycaemia-associated oxidative stress and AGEs in MSCs, osteoblasts and osteocytes, potentially contributing to impaired bone healing in T2DM patients. ERK1/2, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor κB; PI3K, phosphoinositide 3-kinase.
Several studies have demonstrated the association between oxidative stress and impaired bone healing with T2DM. The excessive ROS generated under such diabetic states can induce direct deleterious effects in resident bone cells, including osteoblasts, osteoclasts, and osteocytes, disrupting their functions and survival. MSCs and osteoblasts are particularly susceptible to oxidative stress-induced damage and dysfunction, leading to reduced proliferation and migration, increased senescence and apoptosis, in addition to impaired MSC osteogenic differentiation and ECM synthesis (64, 136–142); thereby promoting osteopenia and compromised mineralisation overall (64, 65, 143–145).
Excessive ROS exposure is also capable of inducing comparable impairment in osteocyte activities, culminating in disrupted bone homeostasis (44, 45, 146–151). Furthermore, analogous studies involving MSC, osteoblast and osteocyte interaction with AGEs have reported similar findings (69, 73, 152–162), with responses orchestrated by altered mitochondrial function, enhanced ROS production and the induction of endoplasmic reticulum (ER) stress (69, 152, 153, 161, 162). ROS- and AGE-induced alterations in extracellular signal-regulated kinase (ERK1/2), c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK), nuclear factor κB (NF-κB), Wnt and phosphoinositide 3-kinases (PI3K)/Akt signalling in MSCs, osteoblasts or osteocytes, have each been implicated in the dysregulation of normal osteogenic repair responses, bone formation and function [(136, 140, 141, 148, 154, 163, 164); Figure 2].
Although ROS and AGE effects on osteogenic differentiation would impede ECM component expression by mature osteoblasts (68, 70, 85, 97, 136, 154, 157, 158), ROS and AGEs are also capable of disrupting bone tissue architecture and mineralisation through the direct modification and degradation of ECM components commonly localised within bone. As detailed above, a large body of evidence exists to demonstrate that AGE reactions with type I collagen fibres negatively affects the biomechanical properties of bone, such as bone stability, strength and quality (97–101). However, ROS are also well-established mediators of type I collagen modification and degradation, especially the ·OH species (16). ROS exposure initially promotes a reduction in collagen gelation, increased cross-linking, aggregation, and collagen insolubility, followed by extensive degradation to low molecular weight peptides, and an increased susceptibility to proteolysis (165–168). The basis of these alterations in collagen structure is the modification and loss of functional groups of certain amino acids, such as methionine, histidine and tyrosine residues (16, 168). Considering the essential requirement to maintain the correct composition, architecture and orientation of type I collagen fibres to facilitate the initiation and progression of normal bone mineralisation within the gap zones between tropocollagen molecules (1, 37, 110), such type I collagen structural modifications would have severe implications to the propagation of bone mineralisation in T2DM patients.
Similar structural changes have also been identified in bone proteoglycans following ROS exposure. Bone proteoglycans, predominantly the SLRPs decorin and biglycan, have been shown to be susceptible to ROS-induced degradation, with degradative effects particularly manifested as amino acid modification within the core protein structure, such as leucine, proline, tyrosine and phenylalanine residues, leading to protein cleavage (16, 169). In contrast, the proteoglycan chondroitin 4-sulphate glycosaminoglycan (GAG) chains remained relatively intact, unless exposed to ·OH species (16, 169, 170). As these proteoglycans possess pivotal roles in the initiation and progression of bone mineralisation, through the regulation of collagen fibrillogenesis, mineral deposition, crystal growth, TGF-β1 bioavailability and osteogenic cell signalling; such manifestations could additionally impact on normal bone mineralisation events in T2DM bone (108–110, 171).
Oxidative stress has also been suggested to promote osteoclast differentiation and activity, leading to excessive bone resorption and impaired bone. Although osteoclastogenesis may be activated via the elevated levels of pro-inflammatory cytokines, such as IL-1β, IL-6 and TNF-α, associated with chronic diabetic inflammation (7, 46, 49, 56–63); most preclinical studies propose that ROS and AGEs can exert direct stimulatory effects on HSCs and osteoclasts to enhance bone resorption. Increased H2O2, ER stress, autophagy, inactivation of Nrf2 and elevated RANKL/OPG ratios, are all implicated in mediating these responses (104, 121, 151, 172–180). Such events are activated via several signalling pathways, including p38 MAPK, JNK, ERK1/2 and NF-κB, which further exacerbates bone repair (148, 177, 179). However, conflicting preclinical studies have suggested that hyperglycaemic conditions actually disrupts normal bone resorptive mechanisms in osteoclasts, via reductions in RANKL/OPG ratios, leading to dysfunctional bone turnover (74, 181, 182).
In addition to direct influences on bone cells and the ECM, there are numerous reports on the effects ROS and AGEs on immature and mature endothelial cells, and the bone vasculature, which could further induce contributory factors to delayed bone healing. Indeed, ROS and AGEs have been shown to impaired endothelial progenitor and endothelial cell responses, such as proliferation and migration, with increased apoptosis, leading to abrogated neo-vascularisation via p38 and p44/42 MAPK activation and attenuated angiopoietin-1 (Ang-1) signalling (164, 183–189).
Elevated ROS generation and oxidative stress during T2DM induce the increased detection of DNA, protein and lipid oxidative stress biomarkers associated with diabetic bone pathology and/or diminished healing (143–145, 190–192). These events are enhanced by compromised antioxidant defences, due to reduced enzymic antioxidant expression/activities and decreased levels of endogenous antioxidant capacity overall (16, 17, 19, 127–135). Indeed, the expression/activities of Nrf2, SODs, catalase and GPx have been observed to diminish in diabetic animal models and T2DM patients with impaired bone healing (143–145, 192, 193). Thus, such an antioxidant imbalance in hyperglycaemic bone would promote the uncontrolled accumulation of O2·−, H2O2 and ·OH, capable of altering normal bone healing responses by affecting cell functions and viability, the ECM and the various cell signalling pathways involved in the healing processes.
5 Antioxidants as potential therapies for impaired bone healing during T2DM
As it is established that antioxidant defences can be compromised in T2DM patients (143–145, 192, 193), numerous studies have investigated the potential therapeutic benefits of exogenous antioxidant supplementation, to address cellular ROS/antioxidant imbalances and improve bone healing outcomes associated with T2DM. By neutralising ROS, enhancing antioxidant defence mechanisms and/or inhibiting their damaging effects on bone and vascular cell responses, antioxidants can aid the restoration of redox balance and promote favourable conditions for bone healing overall (16, 17, 19, 127–135).
5.1 Enzymic antioxidants and diabetic bone repair
Despite the considerable reductions in enzymic antioxidant expression/activities accompanying impaired bone healing with T2DM (16, 17, 19, 127–135), few studies have evaluated the potential delivery of exogeneous enzymic antioxidants to hyperglycaemic bone defects, via strategies such as gene therapy, SOD/catalase and GPx mimetics (Salens and Ebselen, respectively) and nanozymes, or recombinant protein approaches, in order to alleviate oxidative stress-induced inflammation, cell/tissue damage and impaired bone healing; unlike other T2DM-related complications (194–202). Instead, as both enzymic and non-enzymic antioxidants would be expected to play essential roles in mitigating oxidative damage and promoting bone healing in individuals with T2DM, most reported antioxidant interventional studies have relied upon the application of non-enzymic antioxidants to achieve oxidative stress-counteracting outcomes.
5.2 Non-enzymic antioxidants and diabetic bone repair
In contrast to the status with exogenous enzymic antioxidants, considerable evidence exists supporting the promise of numerous non-enzymic antioxidant entities in improving bone healing outcomes associated with T2DM, in line with the findings of the considerable randomised controlled trials (RCTs) performed to establish the therapeutic effects of systemic antioxidant supplementation in improving insulin sensitivity, promoting glycaemic control and alleviating complications in T2DM patients, through improvements in oxidant/antioxidant status (203–205). Although not universally validated in all RCTs performed, largely due to factors associated with trial design, such as experimental group sample sizes, mono-antioxidant entity assessments, and a limited understanding of the optimal antioxidant dosing regimen (206–208), these findings still provide some support to the concept that non-enzymic antioxidant supplementation could possess therapeutic potential in diabetes management and treatment overall. Thus, it is reasonable to speculate that such systemic protective benefits would be further evident with more localised diabetic complications, including impaired diabetic bone healing. However, in contrast to the extensive number of RCTs that have previously been performed, which have shown beneficial effects of exogenous non-enzymic antioxidant supplementation on systemic complications associated with T2DM (203–205), the number of RCTs which have specifically examined the efficacies of supplementation with various non-enzymic antioxidants on bone health and healing outcomes in T2DM patients, are far fewer in comparison. Nonetheless, the findings and conclusions of these limited number of reported RCTs to date (4 in total), are summarised in Table 1.
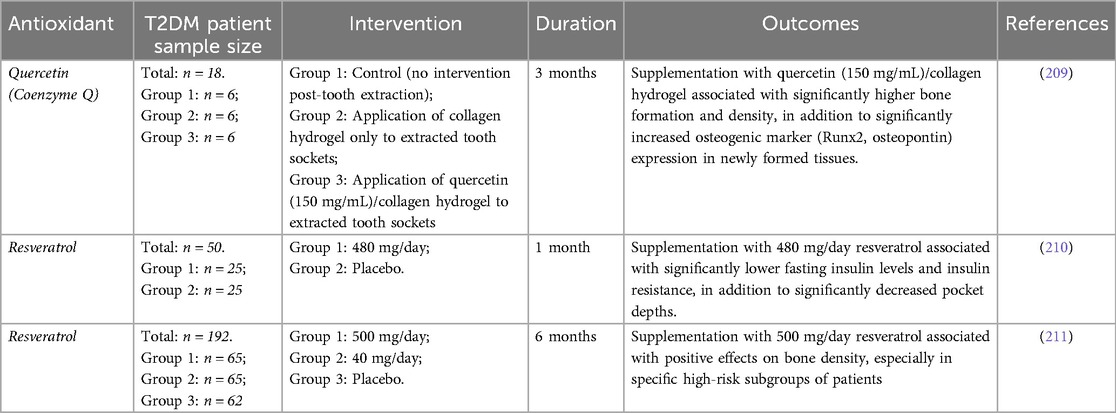
Table 1. Summary of the findings of reported randomized controlled trial (RCT) studies, evaluating the efficacies of various non-enzymic antioxidants as potential therapeutics for the maintenance of bone health and healing in T2DM patients.
As the non-enzymic antioxidants suggested to exhibit bone healing efficacies under hyperglycaemic conditions are commonly derived from dietary sources, it would be rational to assume that the healthy diet and lifestyle changes advocated by clinicians for diagnosed T2DM patients would begin to address any pre-existing deficiencies in a patient's non-enzymic antioxidant profiles and total antioxidant capabilities (204, 205, 207). The principal non-enzymic antioxidants evaluated as therapeutics against impaired bone healing associated with uncontrolled hyperglycaemia and T2DM, are summarised below. Table 2 also highlights these various therapeutic interventions and key mechanistic findings from in vitro and in vivo animal model and human clinical trials.
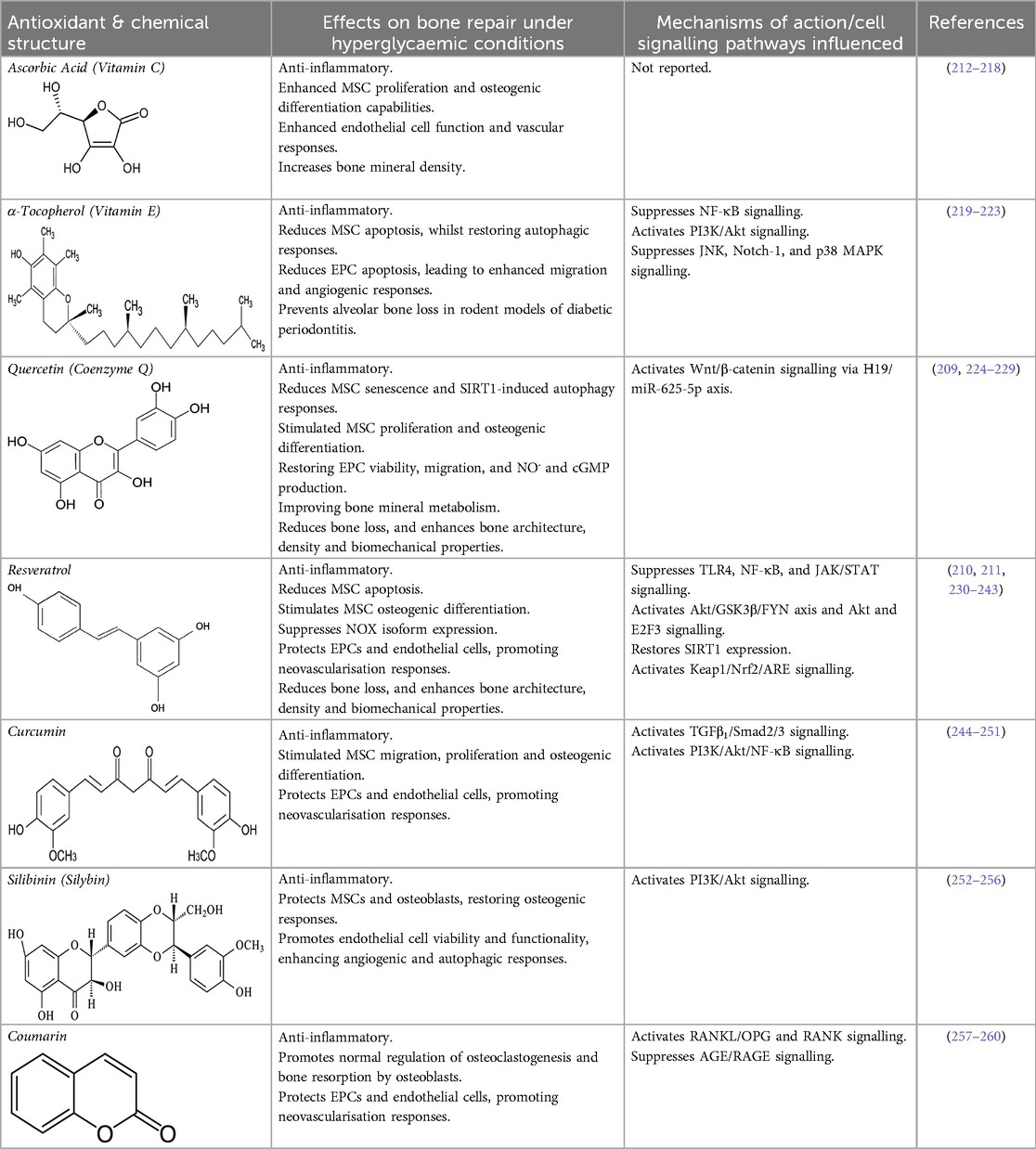
Table 2. Non-enzymic antioxidants evaluated as potential therapeutics against impaired bone healing associated with uncontrolled hyperglycaemia and T2DM, and their proposed mechanisms of action based on preclinical and clinical findings.
5.2.1 Ascorbic acid (Vitamin C)
Ascorbic acid is an essential micronutrient and potent hydrophilic antioxidant, which plays a crucial role in collagen synthesis and bone formation (207, 208). Ascorbate directly scavenges ROS and subsequently protects against lipid peroxidation and protein glycation. Studies have also revealed that diabetics have lowered ascorbic acid levels, compared to their non-diabetic counterparts (207). To address such inadequacies, numerous studies have shown that ascorbate supplementation can counteract the cytotoxic effects of oxidative stress and AGEs on MSC proliferation, osteogenic differentiation, and the restoration of their paracrine signalling mechanism capabilities during hyperglycaemic conditions (212, 213); in addition to aiding the resolution of immuno-inflammatory responses in diabetic bone (214–216) and restoring endothelial cell function and other vascular responses (217). Such beneficial effects on bone reparative mechanisms are reflected in T2DM patients by the positive associations evident between high levels of circulating ascorbate and the possession of significantly greater bone mineral densities (218).
5.2.2 α-Tocopherol (Vitamin E)
α-Tocopherol is another powerful lipid-soluble antioxidant, highly effective in protecting lipids from peroxidation (261, 262). α-Tocopherol has also been shown to reduce oxidative stress and inflammation via scavenging ROS and suppressing NF-κB signalling pathways (207). In contrast, reduced α-tocopherol levels are associated with the onset and development of T2DM. α-Tocopherol is well-known for influencing bone metabolism (219), demonstrating positive antioxidant and anti-inflammatory effects on MSCs derived from rodent T2DM models, especially at lower concentrations (220). Furthermore, α-tocopherol treatment reduces MSC apoptosis, whilst repairing autophagy and restoring PI3K/Akt (protein kinase B) signalling (221). Similarly, α-tocopherol reduces high glucose/hypoxia-induced cell apoptosis in EPCs, by promoting B-cell lymphoma 2 (Bcl-2) and Akt expression, and by inhibiting NF-κB p65, JNK, neurogenic locus notch homolog protein 1 (Notch-1), and p38 MAPK expression, subsequently resulting in enhanced EPC migration and increased capillary density in vivo (222). The elevated presence of serum α-tocopherol levels has further been shown to correlate with reductions in alveolar bone loss in rodent models of diabetic periodontitis (223).
5.2.3 Quercetin (Coenzyme Q)
Quercetin is a flavonoid compound abundantly found in various fruit and vegetables, and particularly shown to be beneficial for T2DM due to its anti-hyperglycaemic and antioxidant properties (263–265). The direct ROS scavenging activity of quercetin is due to its β-ring catechol arrangement, in addition to the -OH group at position 3 of the adjoining AC rings. It is also capable of attenuating oxidative stress via the Nrf2/ARE pathway (266). Recent in-silico studies have further suggested that quercetin acts via the PI3K/Akt, MAPK, PKC, and JNK signalling pathways to elicit ROS-protective effects (267, 268).
Consistent with reports that quercetin possesses both anti-hyperglycaemic and antioxidant capabilities (263–265), several studies have proposed that quercetin decreases blood glucose levels and oxidative stress in rodent animal models of experimental periodontitis diabetic bone healing; improving bone mineral metabolism, serum antioxidant levels and diminishing bone loss, and thereby preventing disease progression and leading to enhanced bone architecture and biomechanical properties (224–228). At a cellular level, quercetin may facilitate bone haemostasis by limiting inflammation and by reducing senescent cells and sirtuin 1 (SIRT1)-induced autophagy. Quercetin has been shown to stimulate MSC proliferation and osteogenic differentiation, partly regulated through the H19/miR-625-5p axis to activate the Wnt/β-catenin signalling pathway (229). Furthermore, quercetin can protect EPCs from hyperglycaemia and ROS, lowering oxidative stress biomarker levels and restoring cell viability, migration responsiveness, nitric oxide (NO.) production, and cyclic guanosine 3′, 5′-cyclic monophosphate (cGMP) levels (269).
Due to these properties identified during preclinical studies, a RCT was performed involving quercetin delivery into tooth extraction sockets via a collagen hydrogel to promote mandibular alveolar socket augmentation post-tooth extraction, particularly prevalent with T2DM patients (209). Quercetin was shown to significantly increase bone density vs. controls, due to enhanced MSC osteogenic differentiation.
5.2.4 Resveratrol
Resveratrol is a natural polyphenolic compound found in red grapes and other plant sources, which possesses both antioxidant and anti-inflammatory properties (270, 271). Resveratrol exhibits antioxidant effects by directly scavenging ROS, inhibition of NF-κB, and enhancing the antioxidant enzymes, such as SODs. These responses are largely mediated through the presence of 3′, 4′, and 5′-hydroxyl groups in its phenolic rings (272, 273). Furthermore, studies have identified that sequential proton loss electron transfer (SPLET), and hydrogen atom transfer (HAT) are the two major mechanisms underlying the direct ROS scavenging activity of resveratrol (274). Intriguingly, resveratrol has also been shown to exhibit pro-oxidant effects by activating various redox-associated signalling pathways. For instance, resveratrol promotes the production of antioxidant enzymes by activating the AMPK-FOXO1 pathway, which promotes the expression of catalase and SOD2 (275). Both Nrf2 and SIRT1 plays important roles in the protective effects of resveratrol, modulating the expression of antioxidant enzymes as well as promotes mitochondrial biogenesis, through the targeting of downstream transcription factors, including the FOXO family.
Based on these properties, there has been much interest in resveratrol from a therapeutic viewpoint in relation to the pathologies associated with T2DM, including impaired bone healing. Resveratrol has been shown to possess protective roles in ameliorating bone loss during various stages of the repair process under hyperglycaemic conditions, exhibiting anti-inflammatory effects through the suppression of pro-inflammatory cytokine levels and Toll-like receptor 4 (TLR4) and NF-κB signalling (230–232). Furthermore, resveratrol reduces MSC apoptosis and promotes osteogenic differentiation, resulting in accelerated bone healing with improved glycaemic control, bone density and trabecular architecture, and enhanced biomechanical properties (210, 211, 232–239). Amelioration of the detrimental impact of hyperglycaemia on osteogenic dysfunction has been reported to be mediated through Nrf2 activation via the Akt/glycogen synthase kinase 3β (GSK3β)/FYN axis and the restoration of SIRT1 expression in MSCs and osteoblasts (236, 237).
In addition to direct effects on osteogenesis, studies have confirmed similar protective resveratrol responses in vascular cells, with the suppression of NADPH oxidase (NOX) isoform expression coupled with the up-regulation of Keap1/Nrf2/ARE signalling and the antioxidant cascade, leading to reduced oxidative stress (240). Such antioxidant capabilities induce anti-inflammatory marker expression in endothelial cells via the down-regulation of NF-κB and JAK/STAT signalling (241), whilst further protecting and promoting neovascularisation by EPCs and endothelial cells via regulation of Akt and E2F3 signalling (242, 243).
5.2.5 Other non-enzymic antioxidants with proposed diabetic bone repair capabilities
Curcumin, derived from the spice turmeric, possesses potent antioxidant, anti-inflammatory, and anti-diabetic properties (244, 245). Subsequently, studies have highlighted its beneficial effects of exogenously administered curcumin on bone healing under diabetic conditions by reducing oxidative stress, inflammation, and stimulating high quality bone formation via enhancement of MSC proliferation, migration and osteogenic differentiation, orchestrated via TGFβ1/Smad2/3 pathway activation (246, 247). Similarly, curcumin has been demonstrated to further bestow protective and pro-angiogenic properties to EPCs and endothelial cells, mediated through PI3K/Akt/NF-κB signalling (248–251).
Silibinin (also referred to as silybin) is a polyphenolic flavonoid present in foods, such as artichokes, which has been shown to possess potent antioxidant capabilities in various clinical situations, including those associated with T2DM (252, 253). Indeed, silibinin alleviates MSC and osteoblast dysfunction associated with oxidative stress and hyperglycaemia, by modulating PI3K/Akt signalling (254, 255). Moreover, it promotes endothelial cell viability and functionality under hyperglycaemic conditions, through its antioxidant properties enhancing angiogenic and autophagic responses (256, 276).
Coumarin is a heterocyclic compound belonging to the class of benzopyrone enriched in numerous edible plants, which possess a wide array of bioactive properties, including anti-inflammatory and antioxidant capabilities (257, 258). Hence, coumarin have been shown to ameliorate impaired bone turnover and remodelling under diabetic conditions, by promoting the expression of OPG and RANK in osteoblasts and osteoclasts, respectively, thereby restoring the normal balance between RANKL/OPG and RANK regulating osteoblast function, osteoclastogenesis and osteoclast resorptive activities (259). Coumarin has been reported to induce such effects by suppressing the interaction between AGEs and its receptor, RAGE. Such anti-AGE effects have also been identified in endothelial cells, reducing oxidative stress and inflammation (260).
Therefore, from the comprehensive range of studies performed, it is evident that such preclinical studies have largely confirmed the beneficial effects of various non-enzymic antioxidants, such as quercetin, resveratrol and curcumin, in promoting bone healing outcomes under hyperglycaemic conditions. Such studies complement recent systematic review findings of their efficacies in promoting repair during various other non-diabetic and diabetic situations associated with impaired osteogenesis (18, 228, 277). These antioxidants influence multiple signalling pathways at a cellular level to exert such diverse effects, ultimately modulating oxidative stress, inflammation, and the promotion of osteoblastic activities, leading to improved bone repair overall.
6 Future perspectives and implications for clinical practice
From the information above, it is irrefutable that impaired bone healing is a significant complication associated with T2DM, posing challenges for patients and healthcare providers worldwide. Recognition of the significant roles that oxidative stress and antioxidants play in impaired bone healing associated with T2DM has important clinical implications. The accumulating preclinical evidence suggesting that antioxidants, especially non-enzymic antioxidant entities, can mitigate the detrimental effects of oxidative stress, inflammation, and impaired cellular function on bone healing processes has received much attention; and holds much promise.
Despite such advances, several important considerations and challenges remain to be addressed. For instance, further studies are needed to unravel the precise cellular and molecular mechanisms underlying the effects of oxidative stress on bone cells and the specific mechanisms through which antioxidants exert their protective effects. Additionally, as inflammation, angiogenesis and new bone formation are inextricably linked during bone healing, investigating the crosstalk between oxidative stress and the other cell signalling pathways involved in the various stages of bone repair would provide a comprehensive understanding of these complex interactions. The performance of additional mechanistic studies would further help elucidate how antioxidants exert their desirable reparative effects, which would provide valuable insights into the identification of specific cellular and molecular targets for more effective therapeutic intervention development; and justification for the development of antioxidant-based therapies to improve bone healing outcomes in T2DM patients.
Although much of the preclinical evidence supporting antioxidant supplementation as a therapeutic strategy to enhance bone repair in T2DM patients has focussed upon non-enzymic antioxidants, including quercetin, resveratrol and curcumin, further research into the potential benefits of more sophisticated enzymic antioxidant-based delivery strategies should be explored. Gene therapy, SOD/catalase and GPx mimetics, nanozymes, and recombinant protein approaches show therapeutic potential in their abilities to counteract excessive oxidative stress (188–196), although the evidence supporting their effectiveness in alleviating inflammation and impaired bone healing responses associated with uncontrolled hyperglycaemia is much less in comparison. Thus, such novel interventional approaches to enhance depleted endogenous antioxidant defences accompanying impaired bone healing with T2DM (16, 17, 19, 127–135), could hold promise as future therapeutic strategies.
One significant challenge to this concept that requires major consideration for both enzymic and non-enzymic antioxidants are their pharmacological properties, including their respective mechanisms of delivery, half-life, biodistribution and bioavailability of individual antioxidant entities (199, 278, 279). Indeed, their poor bioavailability remains a significant constraint to antioxidant therapy development and efficacy, be they directly delivered locally to bone defect sites within the oral cavity, or following oral intake, gastrointestinal absorption and systemic circulation to the bone tissues, as conventionally achieved with dietary antioxidants. Thus, various antioxidant administration approaches, including both conventional and novel drug delivery systems, have been explored in attempts to enhance their pharmacological action via drug targeting and increased bioavailability. These have included numerous biomaterial- and nanomedicine-based approaches, such as 3D scaffolds, hydrogels, nanoparticles, micelles and liposomes (199, 278, 280).
From a clinical perspective, RCTs evaluating the use of non-enzymic antioxidants in impaired bone healing associated with T2DM are severely limited [(209–211), Table 1]; but show some promising results. However, in comparison, no RCTs have, to date, been performed to assess the efficacies of enzymic antioxidant-based delivery systems as novel therapeutics to address impaired healing in T2DM patients. Consequently, due to the overall lack of RCTs evaluating the potential beneficial effects of the exogenously supplemented enzymic and non-enzymic antioxidants on bone health and repair overall, there is a considerable need to address this situation in future. If successful, these studies should be subsequently progressed towards evaluations of antioxidant dosing, frequency and treatment duration, and subsequent biodistribution and bioavailability. Further comparisons of monotherapy vs. combinational antioxidant therapies, and their respective efficacies and potential synergistic effects in improving bone healing outcomes, whilst reducing the risk of complications in T2DM patients should also be prioritised. Healthcare providers involved in the care of individuals with T2DM, and impaired bone healing should consider the assessment of oxidative stress markers as part of the diagnostic and treatment process. Monitoring markers, such as ROS, oxidative damage biomarker and antioxidant enzyme levels, may help identify patients at higher risk for complications and guide the selection of appropriate interventions.
Lifestyle modifications aimed at reducing oxidative stress, such as maintaining tight glycaemic control, adopting a healthy diet rich in antioxidants, regular physical activity, and smoking cessation, could also be recommended to support bone healing processes. Furthermore, patient supplementation with exogenous antioxidants, such as ascorbic acid, tocopherol, resveratrol or quercetin, could be considered as adjunctive therapies to enhance bone healing outcomes in individuals with T2DM. However, it is important to note that the optimal dosage, treatment duration, and specific patient populations that would benefit the most from antioxidant supplementation require further investigation. Personalised treatment plans should further be developed based on individual patient characteristics, genetic factors, oxidative stress biomarker levels, response to antioxidants and T2DM severity; thereby tailoring antioxidant therapies based on patient characteristics and disease profiles that may lead to improved treatment outcomes.
While significant progress has been made in understanding the role of oxidative stress and antioxidants in impaired bone healing associated with T2DM, further studies are warranted to optimise antioxidant dosages, treatment durations, formulations and delivery methods to promote maximum efficacy and to better understand the specific mechanisms through which antioxidants modulate bone healing in the context of T2DM. Furthermore, the undertaking of comparative studies into the use of antioxidant monotherapies vs. the potential synergistic effects of combining different antioxidants as therapies or with other interventions, such as pharmacological agents or regenerative therapies should be explored, to potentially enhance bioavailability, efficacy and/or reduce side-effects. Addressing these future perspectives and challenges will enhance our understanding of the therapeutic potential of antioxidants in treating impaired bone healing associated with T2DM; and facilitate the development of safe, efficacious and accessible, antioxidant-based therapeutic interventions to improve bone healing outcomes in individuals with T2DM.
7 Conclusions
In summary, it is acknowledged that oxidative stress and antioxidants play critical roles in impaired bone healing associated with T2DM. Although mainly ascertained through the various preclinical studies performed, these have provided valuable insights into the potential mechanisms and therapeutic applications of antioxidants in counteracting the deleterious effects of T2DM on bone repair processes, particularly where non-enzymic antioxidants are concerned. Through a greater understanding of these mechanisms and harnessing of the potential that antioxidants can offer, we can undoubtedly aid the development of novel treatment strategies to attenuate oxidative stress and enhance bone healing outcomes in individuals with T2DM.
Exogenous antioxidant supplementation already shows promise as a therapeutic approach to mitigate oxidative stress and improve bone healing outcomes in experimental studies. However, although their ability to counteract oxidative stress, inflammation and cellular dysfunction provides a rationale for further exploration in clinical settings, additional translational studies and more comprehensive, well-designed RCTs are needed, to confirm the potential of targeted exogenous antioxidant interventions, whilst optimising dosages, treatment durations, biodistribution, bioavailability, and delivery methods to maximise their pharmacokinetic, pharmacological and therapeutic benefits; ultimately alleviating the burden of impaired bone healing and improving the quality of life in T2DM patient cohorts.
Author contributions
PL: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. KA: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. JD: Conceptualization, Supervision, Writing – review & editing. EW: Conceptualization, Supervision, Writing – review & editing. RW: Conceptualization, Supervision, Writing – review & editing. RM: Conceptualization, Data curation, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dimitriou R, Tsiridis E, Giannoudis PV. Current concepts of molecular aspects of bone healing. Injury. (2005) 36:1392–404. doi: 10.1016/j.injury.2005.07.019
2. Marco F, Milena F, Gianluca G, Vittoria O. Peri-implant osteogenesis in health and osteoporosis. Micron. (2005) 36:630–44. doi: 10.1016/j.micron.2005.07.008
3. McAllister BS, Haghighat K. Bone augmentation techniques. J Periodontol. (2007) 78:377–96. doi: 10.1902/jop.2007.060048
4. Pagni G, Pellegrini G, Giannobile WV, Rasperini G. Postextraction alveolar ridge preservation: biological basis and treatments. Int J Dent. (2012) 2012:151030. doi: 10.1155/2012/151030
5. Araújo MG, Silva CO, Misawa M, Sukekava F. Alveolar socket healing: what can we learn? Periodontol 2000. (2015) 68:122–34. doi: 10.1111/prd.12082
6. Wang W, Yeung KWK. Bone grafts and biomaterials substitutes for bone defect repair: a review. Bioactive Mater. (2017) 2:224–47. doi: 10.1016/j.bioactmat.2017.05.007
7. Irandoust S, Müftü S. The interplay between bone healing and remodeling around dental implants. Sci Rep. (2020) 10:4335. doi: 10.1038/s41598-020-60735-7
8. American Diabetes Association. Standards of medical care in diabetes—2021. Diabetes Care. (2020) 44:S1–232. doi: 10.2337/dc21-Sint
9. Cho NH, Shaw JE, Karuranga S, Huang Y, Da RFJ, Ohlrogge AW, et al. IDF Diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
10. Jiao H, Xiao E, Graves DT. Diabetes and its effect on bone and fracture healing. Curr Osteoporos Rep. (2015) 13:327–35. doi: 10.1007/s11914-015-0286-8
11. Marin C, Luyten FP, Van der Schueren B, Kerckhofs G, Vandamme K. The impact of type 2 diabetes on bone fracture healing. Front Endocrinol (Lausanne). (2018) 9:6. doi: 10.3389/fendo.2018.00006
12. Ko KI, Sculean A, Graves DT. Diabetic wound healing in soft and hard oral tissues. Transl Res. (2021) 236:72–86. doi: 10.1016/j.trsl.2021.05.001
13. Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. (2011) 7:738–48. doi: 10.1038/nrendo.2011.106
14. Moseley KF. Type 2 diabetes and bone fractures. Curr Opin Endocrinol Diabetes Obes. (2012) 19:128–35. doi: 10.1097/MED.0b013e328350a6e1
15. Monje A, Catena A, Borgnakke WS. Association between diabetes mellitus/hyperglycaemia and peri-implant diseases: systematic review and meta-analysis. J Clin Periodontol. (2017) 44:636–48. doi: 10.1111/jcpe.12724
16. Waddington RJ, Moseley R, Embery G. Reactive oxygen species: a potential role in the pathogenesis of periodontal diseases. Oral Dis. (2000) 6:138–51. doi: 10.1111/j.1601-0825.2000.tb00325.x
17. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. (2015) 4:180–83. doi: 10.1016/j.redox.2015.01.002
18. Bacevic M, Brkovic B, Albert A, Rompen E, Radermecker RP, Lambert F. Does oxidative stress play a role in altered characteristics of diabetic bone? A systematic review. Calcif Tissue Int. (2017) 101:553–63. doi: 10.1007/s00223-017-0327-7
19. Kubo Y, Wruck CJ, Fragoulis A, Drescher W, Pape HC, Lichte P, et al. Role of Nrf2 in fracture healing: clinical aspects of oxidative stress. Calcif Tissue Int. (2019) 105:341–52. doi: 10.1007/s00223-019-00576-3
20. Schröder K. NADPH Oxidases in bone homeostasis and osteoporosis. Free Radic Biol Med. (2019) 132:67–72. doi: 10.1016/j.freeradbiomed.2018.08.036
21. Marques-Carvalho A, Kim HN, Almeida M. The role of reactive oxygen species in bone cell physiology and pathophysiology. Bone Rep. (2024) 19:101664. doi: 10.1016/j.bonr.2023.101664
22. McKee MD, Addison WN, Kaartinen MT. Hierarchies of extracellular matrix and mineral organization in bone of the craniofacial complex and skeleton. Cells Tissues Organs. (2005) 181:176–88. doi: 10.1159/000091379
23. Claes L, Recknagel S, Ignatius A. Fracture healing under healthy and inflammatory conditions. Nat Rev Rheumatol. (2012) 8:133–43. doi: 10.1038/nrrheum.2012.1
24. Hankenson KD, Gagne K, Shaughnessy M. Extracellular signaling molecules to promote fracture healing and bone regeneration. Adv Drug Deliv Revs. (2015) 94:3–12. doi: 10.1016/j.addr.2015.09.008
25. Loi F, Córdova LA, Pajarinen J, Lin TH, Yao Z, Goodman SB. Inflammation, fracture and bone repair. Bone. (2016) 86:119–30. doi: 10.1016/j.bone.2016.02.020
26. Schlundt C, Fischer H, Bucher CH, Rendenbach C, Duda GN, Schmidt-Bleek K. The multifaceted roles of macrophages in bone regeneration: a story of polarization, activation and time. Acta Biomater. (2021) 133:46–57. doi: 10.1016/j.actbio.2021.04.052
27. Chapple ILC, Hirschfeld J, Kantarci A, Wilensky A, Shapira L. The role of the host-neutrophil biology. Periodontol 2000. (in press). doi: 10.1111/prd.12490
28. Sivaraj KK, Adams RH. Blood vessel formation and function in bone. Development. (2016) 143:2706–15. doi: 10.1242/dev.136861
29. Diomede F, Marconi GD, Fonticoli L, Pizzicanella J, Merciaro I, Bramanti P, et al. Functional relationship between osteogenesis and angiogenesis in tissue regeneration. Int J Mol Sci. (2020) 21:3242. doi: 10.3390/ijms21093242
30. Hu K, Olsen BR. The roles of vascular endothelial growth factor in bone repair and regeneration. Bone. (2016) 91:30–8. doi: 10.1016/j.bone.2016.06.013
31. Wu M, Chen G, Li Y-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. (2016) 4:16009. doi: 10.1038/boneres.2016.9
32. Jann J, Gascon S, Roux S, Faucheux N. Influence of the TGF-β superfamily on osteoclasts/osteoblasts balance in physiological and pathological bone conditions. Int J Mol Sci. (2020) 21:7597. doi: 10.3390/ijms21207597
33. Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Revs Mol Cell Biol. (2008) 9:11–21. doi: 10.1038/nrm2319
34. Herrmann M, Jakob F. Bone marrow niches for skeletal progenitor cells and their inhabitants in health and disease. Curr Stem Cell Res Ther. (2019) 14:305–19. doi: 10.2174/1574888X14666190123161447
35. Knight MN, Hankenson KD. Mesenchymal stem cells in bone regeneration. Adv Wound Care. (2013) 2:306–16. doi: 10.1089/wound.2012.0420
36. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. (2006) 25:977–88. doi: 10.1016/j.immuni.2006.10.016
37. Lévesque JP, Winkler IG. Hierarchy of immature hematopoietic cells related to blood flow and niche. Curr Op Hematol. (2011) 18:220–5. doi: 10.1097/MOH.0b013e3283475fe7
38. Balduino A, Mello-Coelho V, Wang Z, Taichman RS, Krebsbach PH, Weeraratna AT, et al. Molecular signature and in vivo behavior of bone marrow endosteal and subendosteal stromal cell populations and their relevance to hematopoiesis. Exp Cell Res. (2012) 318:2427–37. doi: 10.1016/j.yexcr.2012.07.009
39. Köhler A, Schmithorst V, Filippi MD, Ryan MA, Daria D, Gunzer M, et al. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones. Blood. (2009) 114:290–8. doi: 10.1182/blood-2008-12-195644
40. Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. (2009) 457:97–101. doi: 10.1038/nature07639
41. Yusop N, Battersby P, Alraies A, Sloan AJ, Moseley R, Waddington RJ. Isolation and characterisation of mesenchymal stem cells from rat bone marrow and the endosteal niche: a comparative study. Stem Cells Int. (2018) 2018:6869128. doi: 10.1155/2018/6869128
42. Russell KC, Phinney DG, Lacey MR, Barrilleaux BL, Meyertholen KE, O'Connor KC. In vitro high-capacity assay to quantify the clonal heterogeneity in tri-lineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells. (2010) 28:788–98. doi: 10.1002/stem.312
43. James S, Fox J, Afsari F, Lee J, Clough S, Knight C, et al. Multiparameter analysis of human bone marrow stromal cells identifies distinct immunomodulatory and differentiation-competent subtypes. Stem Cell Rep. (2015) 4:1004–15. doi: 10.1016/j.stemcr.2015.05.005
44. Mackie EJ. Osteoblasts: novel roles in orchestration of skeletal architecture. Int J Biochem Cell Biol. (2003) 35:1301–5. doi: 10.1016/s1357-2725(03)00107-9
45. Delgado-Calle J, Bellido T. The osteocyte as a signaling cell. Physiol Rev. (2022) 102:379–410. doi: 10.1152/physrev.00043.2020
46. Boyce BF. Advances in the regulation of osteoclasts and osteoclast functions. J Dent Res. (2013) 92:860–7. doi: 10.1177/0022034513500306
47. Ghiasi MS, Chen J, Vaziri A, Rodriguez EK, Nazarian A. Bone fracture healing in mechanobiological modeling: a review of principles and methods. Bone Rep. (2017) 6:87–100. doi: 10.1016/j.bonr.2017.03.002
48. Kim JM, Lin C, Stavre Z, Greenblatt MB, Shim JH. Osteoblast-osteoclast communication and bone homeostasis. Cells. (2020) 9:2073. doi: 10.3390/cells9092073
49. McDonald MM, Kim AS, Mulholland BS, Rauner M. New insights into osteoclast biology. JBMR Plus. (2021) 5:e10539. doi: 10.1002/jbm4.10539
50. Ma Q, Miri Z, Haugen HJ, Moghanian A, Loca D. Significance of mechanical loading in bone fracture healing, bone regeneration, and vascularization. J Tiss Eng. (2023) 14:20417314231172573. doi: 10.1177/20417314231172573
51. Kerckhofs G, Durand M, Vangoitsenhoven R, Marin C, Van der Schueren B, Carmeliet G, et al. Changes in bone macro- and microstructure in diabetic obese mice revealed by high resolution microfocus x-ray computed tomography. Sci Rep. (2016) 6:35517. doi: 10.1038/srep35517
52. Karim L, Moulton J, Van Vliet M, Velie K, Robbins A, Malekipour F, et al. Bone microarchitecture, biomechanical properties, and advanced glycation end-products in the proximal femur of adults with type 2 diabetes. Bone. (2018) 114:32–9. doi: 10.1016/j.bone.2018.05.030
53. Morgan EF, Unnikrisnan GU, Hussein AI. Bone mechanical properties in healthy and diseased states. Ann Rev Biomed Eng. (2018) 20:119–43. doi: 10.1146/annurev-bioeng-062117-121139
54. Karim L, Rezaee T, Vaidya R. The effect of type 2 diabetes on bone biomechanics. Curr Osteoporos Rep. (2019) 17:291–300. doi: 10.1007/s11914-019-00526-w
55. Cai F, Liu Y, Liu K, Zhao R, Chen W, Yusufu A, et al. Diabetes mellitus impairs bone regeneration and biomechanics. J Orthop Surg Res. (2023) 18:169. doi: 10.1186/s13018-023-03644-5
56. Colombo JS, Balani D, Sloan AJ, Crean S, Okazaki J, Waddington RJ. Delayed osteoblast differentiation and altered inflammatory response around implants placed inincisor sockets of type 2 diabetic rats. Clin Oral Impl Res. (2011) 22:578–86. doi: 10.1111/j.1600-0501.2010.01992.x
57. Loi F, Córdova LA, Zhang R, Pajarinen J, Lin TH, Goodman SB, et al. The effects of immunomodulation by macrophage subsets on osteogenesis in vitro. Stem Cell Res Ther. (2016) 7:15. doi: 10.1186/s13287-016-0276-5
58. He S, Hu Q, Xu X, Niu Y, Chen Y, Lu Y, et al. Advanced glycation end products enhance M1 macrophage polarization by activation the MAPK pathway. Biochem Biophys Res Commun. (2020) 525:334–40. doi: 10.1016/j.bbrc.2020.02.053
59. Shahen VA, Gerbaix M, Koeppenkastrop S, Lim SF, McFarlane KE, Nguyen ANL, et al. Multifactorial effects of hyperglycaemia, hyperinsulinemia and inflammation on bone remodelling in type 2 diabetes mellitus. Cytokine Growth Fact Rev. (2020) 55:109–18. doi: 10.1016/j.cytogfr.2020.04.001
60. Shen X, Shen X, Li B, Zhu W, Fu Y, Xu R, et al. Abnormal macrophage polarization impedes the healing of diabetes-associated tooth sockets. Bone. (2020) 143:115618. doi: 10.1016/j.bone.2020.115618
61. Xiang G, Huang X, Wang T, Wang J, Zhao G, Wang H, et al. The impact of sitagliptin on macrophage polarity and angiogenesis in the osteointegration of titanium implants in type 2 diabetes. Biomed Pharmacother. (2020) 126:110078. doi: 10.1016/j.biopha.2020.110078
62. Lee RSB, Hamlet SM, Moon HJ, Ivanovski S. Re-establishment of macrophage homeostasis by titanium surface modification in type II diabetes promotes osseous healing. Biomaterials. (2021) 267:120464. doi: 10.1016/j.biomaterials.2020.120464
63. Yusop N, Moseley R, Waddington RJ. Hyperglycemia exerts disruptive effects on the secretion by TGF-β1 and it’s matrix ligands, decorin and biglycan, by mesenchymal sub-populations and macrophages during bone repair. Front Dent Med. (2023) 4:1200122. doi: 10.3389/fdmed.2023.1200122
64. Hamada Y, Kitazawa S, Kitazawa R, Fujii H, Kasuga M, Fukagawa M. Histomorphometric analysis of diabetic osteopenia in streptozotocin-induced diabetic mice: a possible role of oxidative stress. Bone. (2007) 40:1408–14. doi: 10.1016/j.bone.2006.12.057
65. Fujii H, Hamada Y, Fukagawa M. Bone formation in spontaneously diabetic torii-newly established model of non-obese type 2 diabetes rats. Bone. (2008) 42:372–9. doi: 10.1016/j.bone.2007.10.007
66. Sakai D, Okazaki J, Komasa Y. Bone healing of tooth extraction socket in type 2 diabetes. J Oral Tiss Eng. (2008) 5:134–44. doi: 10.11223/jarde.5.134
67. Wang F, Song Y-L, Li D-H, Li C-X, Wang Y, Zhang N, et al. Type 2 diabetes mellitus impairs bone healing of dental implants in GK rats. Diabetes Res Clin Pract. (2010) 88:e7–9. doi: 10.1016/j.diabres.2010.01.017
68. Lu H, Kraut D, Gerstenfeld LC, Graves DT. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology. (2003) 144:346–52. doi: 10.1210/en.2002-220072
69. Kume S, Kato S, Yamagishi S, Inagaki Y, Ueda S, Arima N, et al. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage and bone. J Bone Miner Res. (2005) 20:1647–58. doi: 10.1359/JBMR.050514
70. Wang W, Zhang X, Zheng J, Yang J. High glucose stimulates adipogenic and inhibits osteogenic differentiation in MG-63 cells through cAMP/protein kinase A/extracellular signal-regulated kinase pathway. Mol Cell Biochem. (2010) 338:115–22. doi: 10.1007/s11010-009-0344-6
71. Zhen D, Chen Y, Tang X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J Diabetes Compl. (2010) 24:334–44. doi: 10.1016/j.jdiacomp.2009.05.002
72. Zhao YF, Zeng DL, Xia LG, Zhang SM, Xu LY, Jiang XQ, et al. Osteogenic potential of bone marrow stromal cells derived from streptozotocin-induced diabetic rats. Int J Mol Med. (2013) 31:614–20. doi: 10.3892/ijmm.2013.1227
73. Notsu M, Yamaguchi T, Okazaki K, Tanaka K, Ogawa N, Kanazawa I, et al. Advanced glycation end product 3 (AGE3) suppresses the mineralization of mouse stromal ST2 cells and human mesenchymal stem cells by increasing TGF-β expression and secretion. Endocrinology. (2014) 155:2402–10. doi: 10.1210/en.2013-1818
74. Cunha JS, Ferreira VM, Maquigussa E, Naves MA, Boim MA. Effects of high glucose and high insulin concentrations on osteoblast function in vitro. Cell Tiss Res. (2014) 358:249–56. doi: 10.1007/s00441-014-1913-x
75. Cramer C, Freisinger E, Jones RK, Slakey DP, Dupin CL, Newsome ER, et al. Persistent high glucose concentrations alter the regenerative potential of mesenchymal stem cells. Stem Cells Dev. (2010) 19:1875–84. doi: 10.1089/scd.2010.0009
76. Stolzing A, Coleman N, Scutt A. Glucose-induced replicative senescence in mesenchymal stem cells. Rejuvenation Res. (2006) 9:31–5. doi: 10.1089/rej.2006.9.31
77. Li YM, Schilling T, Benisch P, Zeck S, Meissner-Weigl J, Schneider D, et al. Effects of high glucose on mesenchymal stem cell proliferation and differentiation. Biochem Biophys Res Commun. (2007) 363:209–15. doi: 10.1016/j.bbrc.2007.08.161
78. Weil BR, Abarbanell AM, Herrmann JL, Wang Y, Meldrum DR. High glucose concentration in cell culture medium does not acutely affect human mesenchymal stem cell growth factor production or proliferation. Am J Physiol Reg Integr Comp Physiol. (2009) 296:R1735–43. doi: 10.1152/ajpregu.90876.2008
79. Stolzing A, Sellers D, Llewelyn O, Scutt A. Diabetes induced changes in rat mesenchymal stem cells. Cells Tiss Organs. (2010) 191:453–65. doi: 10.1159/000281826
80. Al-Qarakhli AMA, Yusop Y, Moseley R, Waddington RJ. Effects of high glucose conditions on the expansion and differentiation capabilities of mesenchymal stromal cells derived from rat endosteal niche. BMC Mol Cell Biol. (2019) 20:51. doi: 10.1186/s12860-019-0235-y
81. Freude T, Braun KF, Haug A, Pscherer S, Stöckle U, Nussler AK, et al. Hyperinsulinemia reduces osteoblast activity in vitro via upregulation of TGF-β. J Mol Med (Berlin). (2012) 90:1257–66. doi: 10.1007/s00109-012-0948-2
82. Ehnert S, Freude T, Ihle C, Mayer L, Braun B, Graeser J. Factors circulating in the blood of type 2 diabetes mellitus patients affect osteoblast maturation—description of a novel in vitro model. Exp Cell Res. (2015) 332:247–58. doi: 10.1016/j.yexcr.2014.12.011,
83. Xu MT, Sun S, Zhang L, Xu F, Du SL, Zhang XD, et al. Diabetes mellitus affects the biomechanical function of the callus and the expression of TGF-β1 and BMP2 in an early stage of fracture healing. Braz J Med Biol Res. (2016) 49:e4736. doi: 10.1590/1414-431X20154736
84. Zhang P, Zhang H, Lin J, Xiao T, Xu R, Fu Y, et al. Insulin impedes osteogenesis of BMSCs by inhibiting autophagy and promoting premature senescence via the TGF-β1 pathway. Aging (Albany NY). (2020) 12:2084–100. doi: 10.18632/aging.102723
85. Alliston T, Choy L, Ducy P, Karsenty G, Derynck R. TGF-β-induced repression of CBFA1 by Smad3 decreases cbfa1 and osteocalcin expression and inhibits osteoblast differentiation. EMBO J. (2001) 20:2254–72. doi: 10.1093/emboj/20.9.2254
86. Maeda S, Hayashi M, Komiya S, Imamura T, Miyazono K. Endogenous TGF-β signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. (2004) 23:552–63. doi: 10.1038/sj.emboj.7600067
87. Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfect. Nat Med. (2014) 20:670–5. doi: 10.1038/nm.3544
88. Nam B, Park H, Lee YL, Oh Y, Park J, Kim SY, et al. TGFβ1 suppressed matrix mineralization of osteoblasts differentiation by regulating SMURF1-C/EBPβ-DKK1 axis. Int J Mol Sci. (2020) 21:9771. doi: 10.3390/ijms21249771
89. Bostrom KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. (2011) 108:446–57. doi: 10.1161/CIRCRESAHA.110.236596
90. Schall N, Garcia JJ, Kalyanaraman H, China SP, Lee JJ, Sah RL, et al. Protein kinase G1 regulates bone regeneration and rescues diabetic fracture healing. JCI Insight. (2020) 5:e135355. doi: 10.1172/jci.insight.135355
91. Bhatti FUR, Dadwal UC, Valuch CR, Tewari NP, Awosanya OD, de Andrade Staut C, et al. The effects of high fat diet, bone healing, and BMP-2 treatment on endothelial cell growth and function. Bone. (2021) 146:115883. doi: 10.1016/j.bone.2021.115883
92. Li J, Wei J, Li A, Liu H, Sun J, Qiao H. A dual peptide sustained-release system based on nanohydroxyapatite/polyamide 66 scaffold for synergistic-enhancing diabetic rats’ fracture healing in osteogenesis and angiogenesis. Front Bioeng Biotechnol. (2021) 9:657699. doi: 10.3389/fbioe.2021.657699
93. Wang M, Qiu Y, Gao L, Qi F, Bi L. The impact of IGF-1 on alveolar bone remodeling and BMP-2 expression in orthodontic tooth movement in diabetic rats. Adv Clin Exp Med. (2023) 32:349–56. doi: 10.17219/acem/153956
94. Fowlkes JL, Bunn RC, Thrailkill KM. Contributions of the insulin/insulin-like growth factor-1 axis to diabetic osteopathy. J Diabetes Metab. (2011) 1:S1-003. doi: 10.4172/2155-6156.S1-003
95. Chen QQ, Wang WM. Expression of FGF-2 and IGF-1 in diabetic rats with fracture. Asian Pac J Trop Med. (2014) 7:71–5. doi: 10.1016/S1995-7645(13)60195-9
96. Lim JC, Ko KI, Mattos M, Fang M, Zhang C, Feinberg D, et al. TNFα contributes to diabetes impaired angiogenesis in fracture healing. Bone. (2017) 99:26–38. doi: 10.1016/j.bone.2017.02.014
97. Entz L, Falgayrac G, Chauveau C, Pasquier G, Lucas S. The extracellular matrix of human bone marrow adipocytes and glucose concentration differentially alter mineralization quality without impairing osteoblastogenesis. Bone Rep. (2022) 17:101622. doi: 10.1016/j.bonr.2022.101622
98. Khosravi R, Sodek KL, Faibish M, Trackman PC. Collagen advanced glycation inhibits its discoidin domain receptor 2 (DDR2)-mediated induction of lysyl oxidase in osteoblasts. Bone. (2014) 58:33–41. doi: 10.1016/j.bone.2013.10.001
99. Poundarik AA, Wu PC, Evis Z, Sroga GE, Ural A, Rubin M, et al. A direct role of collagen glycation in bone fracture. J Mech Behav Biomed Mater. (2015) 52:120–30. doi: 10.1016/j.jmbbm.2015.08.012
100. Willett TL, Voziyan P, Nyman JS. Causative or associative: a critical review of the role of advanced glycation end-products in bone fragility. Bone. (2022) 163:116485. doi: 10.1016/j.bone.2022.116485
101. Voziyan P, Uppuganti S, Leser M, Rose KL, Nyman JS. Mapping glycation and glycoxidation sites in collagen I of human cortical bone. BBA Adv. (2023) 3:100079. doi: 10.1016/j.bbadva.2023.100079
102. Valcourt U, Merle B, Gineyts E, Viguet-Carrin S, Delmas PD, Garnero P. Non-enzymatic glycation of bone collagen modifies osteoclastic activity and differentiation. J Biol Chem. (2007) 282:5691–703. doi: 10.1074/jbc.M610536200
103. Haug AT, Braun KF, Ehnert S, Mayer L, Stöckle U, Nüssler AK, et al. Gene expression changes in cancellous bone of type 2 diabetics: a biomolecular basis for diabetic bone disease. Langenbecks Arch Surg. (2014) 399:639–47. doi: 10.1007/s00423-014-1188-4
104. García-Hernández A, Arzate H, Gil-Chavarría I, Rojo R, Moreno-Fierros L. High glucose concentrations alter the biomineralization process in human osteoblastic cells. Bone. (2012) 50:276–88. doi: 10.1016/j.bone.2011.10.032
105. Paglia DN, Wey A, Breitbart EA, Faiwiszewski J, Mehta SK, Al-Zube L, et al. Effects of local insulin delivery on subperiosteal angiogenesis and mineralized tissue formation during fracture healing. J Orthop Res. (2013) 31:783–91. doi: 10.1002/jor.22288
106. Fu C, Zhang X, Ye F, Yang J. High insulin levels in KK-ay diabetic mice cause increased cortical bone mass and impaired trabecular micro-structure. Int J Mol Sci. (2015) 16:8213–26. doi: 10.3390/ijms16048213
107. Wallner C, Schira J, Wagner JM, Schulte M, Fischer S, Hirsch T, et al. Application of VEGFA and FGF-9 enhances angiogenesis, osteogenesis and bone remodeling in type 2 diabetic long bone regeneration. PLoS One. (2015) 10:e0118823. doi: 10.1371/journal.pone.0118823
108. Waddington RJ, Roberts HC, Sugars RV, Schönherr E. Differential roles for small leucine-rich proteoglycans in bone formation. Eur Cell Mater. (2003) 6:12–21. doi: 10.22203/ecm.v006a02
109. Nikitovic D, Aggelidakis J, Young MF, Iozzo RV, Karamanos NK, Tzanakakis GN. The biology of small leucine-rich proteoglycans in bone pathophysiology. J. Biol. Chem. (2012) 287:33926–33. doi: 10.1074/jbc.R112.379602
110. Lin X, Patil S, Gao YG, Qian A. The bone extracellular matrix in bone formation and regeneration. Front Pharmacol. (2020) 11:757. doi: 10.3389/fphar.2020.00757
111. Howangyin KY, Silvestre JS. Diabetes mellitus and ischemic diseases: molecular mechanisms of vascular repair dysfunction. Arterioscler Thromb Vasc Biol. (2014) 34:1126–35. doi: 10.1161/ATVBAHA.114.303090
112. Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. (2002) 106:2781–6. doi: 10.1161/01.cir.0000039526.42991.93
113. Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. (2005) 45:1449–57. doi: 10.1016/j.jacc.2004.11.067
114. Saito H, Yamamoto Y, Yamamoto H. Diabetes alters subsets of endothelial progenitor cells that reside in blood, bone marrow, and spleen. Am J Physiol Cell Physiol. (2012) 302:C892–901. doi: 10.1152/ajpcell.00380.2011
115. D’Souza DR, Salib MM, Bennett J, Mochin-Peters M, Asrani K, Goldblum SE, et al. Hyperglycemia regulates RUNX2 activation and cellular wound healing through the aldose reductase polyol pathway. J Biol Chem. (2009) 284:17947–55. doi: 10.1074/jbc.M109.002378
116. Kim KA, Shin YJ, Akram M, Kim ES, Choi KW, Suh H, et al. High glucose condition induces autophagy in endothelial progenitor cells contributing to angiogenic impairment. Biol Pharm Bull. (2014) 37:1248–52. doi: 10.1248/bpb.b14-00172
117. Tie L, Chen LY, Chen DD, Xie HH, Channon KM, Chen AF. GTP Cyclohydrolase I prevents diabetic-impaired endothelial progenitor cells and wound healing by suppressing oxidative stress/thrombospondin-1. Am J Physiol Endocrinol Metab. (2014) 306:E1120–31. doi: 10.1152/ajpendo.00696.2013
118. Rezabakhsh A, Cheraghi O, Nourazarian A, Hassanpour M, Kazemi M, Ghaderi S. Type 2 diabetes inhibited human mesenchymal stem cells angiogenic response by over-activity of the autophagic pathway. J Cell Biochem. (2017) 118:1518–30. doi: 10.1002/jcb.25814
119. Plut A, Sprogar Š, Drevenšek G, Hudoklin S, Zupan J, Marc J, et al. Bone remodeling during orthodontic tooth movement in rats with type 2 diabetes. Am J Orthod Dentofacial Orthop. (2015) 148:1017–25. doi: 10.1016/j.ajodo.2015.05.031
120. Catalfamo DL, Britten TM, Storch DL, Calderon NL, Sorenson HL, Wallet SM. Hyperglycemia induced and intrinsic alterations in type 2 diabetes-derived osteoclast function. Oral Dis. (2013) 19:303–12. doi: 10.1111/odi.12002
121. Sassi F, Buondonno I, Luppi C, Spertino E, Stratta E, Di Stefano M, et al. Type 2 diabetes affects bone cells precursors and bone turnover. BMC Endocr Disord. (2018) 18:55. doi: 10.1186/s12902-018-0283-x
122. Hu Z, Ma C, Liang Y, Zou S, Liu X. Osteoclasts in bone regeneration under type 2 diabetes mellitus. Acta Biomater. (2019) 84:402–13. doi: 10.1016/j.actbio.2018.11.052
123. Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced glycation end-products (AGEs): formation, chemistry, classification, receptors, and diseases related to AGEs. Cells. (2022) 11:1312. doi: 10.3390/cells11081312
124. Yan SF, Yan SD, Ramasamy R, Schmidt AM. Tempering the wrath of RAGE: an emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann Med. (2009) 41:408–22. doi: 10.1080/07853890902806576
125. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS And ROS-mediated cellular signaling. Oxid Med Cell Longev. (2016) 2016:4350965. doi: 10.1155/2016/4350965
126. González P, Lozano P, Ros G, Solano F. Hyperglycemia and oxidative stress: an integral, updated and critical overview of their metabolic interconnections. Int J Mol Sci. (2023) 24:9352. doi: 10.3390/ijms24119352
127. Domazetovic V, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Oxidative stress in bone remodeling: role of antioxidants. Clin Cases Miner Bone Metab. (2017) 14:209–16. doi: 10.11138/ccmbm/2017.14.1.209
128. Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. (2002) 33:337–49. doi: 10.1016/s0891-5849(02)00905-x
129. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. (2011) 15:1583–606. doi: 10.1089/ars.2011.3999
130. Nozik-Grayck E, Suliman HB, Piantadosi CA. Extracellular superoxide dismutase. Int J Biochem Cell Biol. (2005) 37:2466–71. doi: 10.1016/j.biocel.2005.06.012
131. Kirkman HN, Gaetani GF. Mammalian catalase: a venerable enzyme with new mysteries. Trends Biochem Sci. (2007) 32:44–50. doi: 10.1016/j.tibs.2006.11.003
132. Board PG, Menon D. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim Biophys Acta. (2013) 1830:3267–88. doi: 10.1016/j.bbagen.2012.11.019
133. Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim Biophys Acta. (2013) 1830:3217–66. doi: 10.1016/j.bbagen.2012.09.018
134. Tonelli C, Chio IIC, Tuveson DA. Transcriptional regulation by Nrf2. Antioxid Red Signal. (2018) 29:1727–45. doi: 10.1089/ars.2017.7342
135. Suzuki T, Takahashi J, Yamamoto M. Molecular basis of the KEAP1-NRF2 signaling pathway. Mol Cells. (2023) 46:133–41. doi: 10.14348/molcells.2023.0028
136. Bai X, Lu D, Bai J, Zheng KZ, Li X, Luo S. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-κB. Biochem Biophys Res Commun. (2004) 314:197–207. doi: 10.1016/j.bbrc.2003.12.073
137. Chen RM, Wu GJ, Chang HC, Chen JT, Chen TF, Lin YL, et al. 2,6-Diisopropylphenol Protects osteoblasts from oxidative stress-induced apoptosis through suppression of caspase-3 activation. Ann N Y Acad Sci. (2005) 1042:448–59. doi: 10.1196/annals.1338.038
138. Fatokun AA, Stone TW, Smith RA. Hydrogen peroxide-induced oxidative stress in MC3T3-E1 cells: the effects of glutamate and protection by purines. Bone. (2006) 39:542–51. doi: 10.1016/j.bone.2006.02.062
139. Vanella L, Jr SC, Kim DH, Abraham NG, Ebraheim N. Oxidative stress and heme oxygenase-1 regulated human mesenchymal stem cells differentiation. Int J Hypertens. (2012) 2012:890671. doi: 10.1155/2012/890671
140. Zhang Y, Yang JH. Activation of the PI3K/akt pathway by oxidative stress mediates high glucose-induced increase of adipogenic differentiation in primary rat osteoblasts. J Cell Biochem. (2013) 114:2595–602. doi: 10.1002/jcb.24607
141. Zhao X, Zhang G, Wu L, Tang Y, Guo C. Inhibition of ER stress-activated JNK pathway attenuates TNF-α-induced inflammatory response in bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. (2021) 541:8–14. doi: 10.1016/j.bbrc.2020.12.101
142. Lin C, Yang Q, Guo D, Xie J, Yang YS, Chaugule S, et al. Impaired mitochondrial oxidative metabolism in skeletal progenitor cells leads to musculoskeletal disintegration. Nat Commun. (2022) 13:6869. doi: 10.1038/s41467-022-34694-8
143. Waddington RJ, Alraies A, Colombo JS, Sloan AJ, Okazaki J, Moseley R. Characterization of oxidative stress status during diabetic bone healing. Cells Tiss Organs. (2011) 194:307–12. doi: 10.1159/000324251
144. Feng YF, Wang L, Zhang Y, Li X, Ma ZS, Zou JW, et al. Effect of reactive oxygen species overproduction on osteogenesis of porous titanium implant in the present of diabetes mellitus. Biomaterials. (2013) 34:2234–43. doi: 10.1016/j.biomaterials.2012.12.023
145. Saito N, Mikami R, Mizutani K, Takeda K, Kominato H, Kido D, et al. Impaired dental implant osseointegration in rat with streptozotocin-induced diabetes. J Periodontal Res. (2022) 57:412–24. doi: 10.1111/jre.12972
146. Kar R, Riquelme MA, Werner S, Jiang JX. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J Bone Miner Res. (2013) 28:1611–21. doi: 10.1002/jbmr.1917
147. Yang Y, Zheng X, Li B, Jiang S, Jiang L. Increased activity of osteocyte autophagy in ovariectomized rats and its correlation with oxidative stress status and bone loss. Biochem Biophys Res Commun. (2014) 451:86–92. doi: 10.1016/j.bbrc.2014.07.069
148. Fontani F, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Glutathione, N-acetylcysteine and lipoic acid down-regulate starvation-induced apoptosis, RANKL/OPG ratio and sclerostin in osteocytes: involvement of JNK and ERK1/2 signalling. Calcif Tissue Int. (2015) 96:335–46. doi: 10.1007/s00223-015-9961-0
149. Sánchez-de-Diego C, Pedrazza L, Pimenta-Lopes C, Martinez-Martinez A, Dahdah N, Valer JA, et al. NRF2 function in osteocytes is required for bone homeostasis and drives osteocytic gene expression. Redox Biol. (2021) 40:101845. doi: 10.1016/j.redox.2020.101845
150. Kaur J, Khosla S, Farr JN. Effects of diabetes on osteocytes. Curr Opin Endocrinol Diabetes Obes. (2022) 29:310–7. doi: 10.1097/MED.0000000000000733
151. Zhang J, Riquelme MA, Hua R, Acosta FM, Gu S, Jiang JX. Connexin 43 hemichannels regulate mitochondrial ATP generation, mobilization, and mitochondrial homeostasis against oxidative stress. Elife. (2022) 11:e82206. doi: 10.7554/eLife.82206
152. Zhang C, Wei W, Chi M, Wan Y, Li X, Qi M, et al. FOXO1 mediates advanced glycation end products induced mouse osteocyte-like MLO-Y4 cell apoptosis and dysfunctions. J Diabetes Res. (2019) 2019:6757428. doi: 10.1155/2019/6757428
153. Eckhardt BA, Rowsey JL, Thicke BS, Fraser DG, O'Grady KL, Bondar OP, et al. Accelerated osteocyte senescence and skeletal fragility in mice with type 2 diabetes. JCI Insight. (2020) 5:e135236. doi: 10.1172/jci.insight.135236
154. Park SY, Choi KH, Jun JE, Chung HY. Effects of advanced glycation end products on differentiation and function of osteoblasts and osteoclasts. J Korean Med Sci. (2021) 36:e239. doi: 10.3346/jkms.2021.36.e239
155. Phimphilai M, Pothacharoen P, Chattipakorn N, Kongtawelert P. Receptors of advanced glycation end product (RAGE) suppression associated with a preserved osteogenic differentiation in patients with prediabetes. Front Endocrinol (Lausanne). (2022) 13:799872. doi: 10.3389/fendo.2022.799872
156. Sakasai-Sakai A, Takata T, Takeuchi M. The association between accumulation of toxic advanced glycation end-products and cytotoxic effect in MC3T3-E1 cells. Nutrients. (2022) 14(5):990. doi: 10.3390/nu14050990
157. Waqas K, Muller M, Koedam M, El Kadi Y, Zillikens MC, van der Eerden BCJ. Methylglyoxal—an advanced glycation end products (AGEs) precursor—inhibits differentiation of human MSC-derived osteoblasts in vitro independently of receptor for AGEs (RAGE). Bone. (2022) 164:116526. doi: 10.1016/j.bone.2022.116526
158. Tanaka K, Yamaguchi T, Kaji H, Kanazawa I, Sugimoto T. Advanced glycation end products suppress osteoblastic differentiation of stromal cells by activating endoplasmic reticulum stress. Biochem Biophys Res Commun. (2013) 438:463–7. doi: 10.1016/j.bbrc.2013.07.126
159. Suh KS, Choi EM, Rhee SY, Kim YS. Methylglyoxal induces oxidative stress and mitochondrial dysfunction in osteoblastic MC3T3-E1 cells. Free Radic Res. (2014) 48:206–17. doi: 10.3109/10715762.2013.859387
160. Suzuki R, Fujiwara Y, Saito M, Arakawa S, Shirakawa JI, Yamanaka M, et al. Intracellular accumulation of advanced glycation end products induces osteoblast apoptosis via endoplasmic reticulum stress. J Bone Miner Res. (2020) 35:1992–2003. doi: 10.1002/jbmr.4053
161. Aikawa T, Matsubara H, Ugaji S, Shirakawa J, Nagai R, Munesue S, et al. Contribution of methylglyoxal to delayed healing of bone injury in diabetes. Mol Med Rep. (2017) 16:403–9. doi: 10.3892/mmr.2017.6589
162. Phimphilai M, Pothacharoen P, Kongtawelert P, Chattipakorn N. Impaired osteogenic differentiation and enhanced cellular receptor of advanced glycation end products sensitivity in patients with type 2 diabetes. J Bone Miner Metab. (2017) 35:631–41. doi: 10.1007/s00774-016-0800-9
163. Li G, Xu J, Li Z. Receptor for advanced glycation end products inhibits proliferation in osteoblast through suppression of wnt, PI3K and ERK signaling. Biochem Biophys Res Commun. (2012) 423:684–9. doi: 10.1016/j.bbrc.2012.06.015
164. Wang Y, Jiang C, Shang Z, Qiu G, Yuan G, Xu K, et al. AGEs/RAGE promote osteogenic differentiation in rat bone marrow-derived endothelial progenitor cells via MAPK signaling. J Diabetes Res. (2022) 2022:4067812. doi: 10.1155/2022/4067812
165. Greenwald RA, Moy WW. Inhibition of collagen gelation by action of the superoxide radical. Arthritis Rheum. (1979) 22:251–9. doi: 10.1002/art.1780220307
166. Chace KV, Carubelli R, Nordquist RE. The role of nonenzymatic glycosylation, transition metals, and free radicals in the formation of collagen aggregates. Arch Biochem Biophys. (1991) 288:473–80. doi: 10.1016/0003-9861(91)90223-6
167. Mukhopadhyay CK, Chatterjee IB. Oxidative fragmentation of collagen and prolyl peptide by cu(II)/H2O2. Conversion of proline residue to 2-pyrrolidone. J Biol Chem. (1992) 267:23646–51. doi: 10.1016/S0021-9258(18)43797-0
168. Kato Y, Uchida K, Kawakishi S. Free metal ion-independent oxidative damage of collagen. Protection by ascorbic acid. J Biol Chem. (1994) 269:30200–5. doi: 10.1016/S0021-9258(18)35887-3
169. Moseley R, Waddington RJ, Embery G, Rees SG. The modification of alveolar bone proteoglycans by reactive oxygen species in vitro. Conn Tiss Res. (1998) 37:13–28. doi: 10.3109/03008209809028897
170. Moseley R, Waddington R, Evans P, Halliwell B, Embery G. The chemical modification of glycosaminoglycan structure by oxygen-derived species in vitro. Biochim Biophys Acta. (1995) 1244:245–52. doi: 10.1016/0304-4165(95)00010-9
171. Hiebert LM. Proteoglycans and diabetes. Curr Pharm Des. (2017) 23:1500–9. doi: 10.2174/1381612823666170125154915
172. Wang K, Niu J, Kim H, Kolattukudy PE. Osteoclast precursor differentiation by MCPIP via oxidative stress, endoplasmic reticulum stress, and autophagy. J Mol Cell Biol. (2011) 3:360–8. doi: 10.1093/jmcb/mjr021
173. Hyeon S, Lee H, Yang Y, Jeong W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic Biol Med. (2013) 65:789–99. doi: 10.1016/j.freeradbiomed.2013.08.005
174. Kanzaki H, Shinohara F, Kajiya M, Kodama T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J Biol Chem. (2013) 288:23009–20. doi: 10.1074/jbc.M113.478545
175. Callaway DA, Jiang JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Miner Metab. (2015) 33:359–70. doi: 10.1007/s00774-015-0656-4
176. Li Z, Li C, Zhou Y, Chen W, Luo G, Zhang Z, et al. Advanced glycation end products biphasically modulate bone resorption in osteoclast-like cells. Am J Physiol Endocrinol Metab. (2016) 310:E355–66. doi: 10.1152/ajpendo.00309.2015
177. Wang X, Chen B, Sun J, Jiang Y, Zhang H, Zhang P, et al. Iron-induced oxidative stress stimulates osteoclast differentiation via NF-κB signaling pathway in mouse model. Metab Clin Exp. (2018) 83:167–76. doi: 10.1016/j.metabol.2018.01.005
178. Agidigbi TS, Kim C. Reactive oxygen species in osteoclast differentiation and possible pharmaceutical targets of ROS-mediated osteoclast diseases. Int J Mol Sci. (2019) 20:3576. doi: 10.3390/ijms20143576
179. Moon JS, Lee SY, Kim JH, Choi YH, Yang DW, Kang JH, et al. Synergistic alveolar bone resorption by diabetic advanced glycation end products and mechanical forces. J Periodontol. (2019) 90(12):1457–69. doi: 10.1002/JPER.18-0453
180. Wang L, Liang Y, Zhou X, Tian Y, Miao Z, Ko CC, et al. Nrf2 differentially regulates osteoclast and osteoblast differentiation for bone homeostasis. Biochem Biophys Res Commun. (2023) 674:19–26. doi: 10.1016/j.bbrc.2023.06.080
181. Miranda C, Giner M, Montoya MJ, Vázquez MA, Miranda MJ, Pérez-Cano R. Influence of high glucose and advanced glycation end-products (ages) levels in human osteoblast-like cells gene expression. BMC Musculoskelet Disord. (2016) 17:377. doi: 10.1186/s12891-016-1228-z
182. Tanaka K, Yamagata K, Kubo S, Nakayamada S, Sakata K, Matsui T, et al. Glycolaldehyde-modified advanced glycation end-products inhibit differentiation of human monocytes into osteoclasts via upregulation of IL-10. Bone. (2019) 128:115034. doi: 10.1016/j.bone.2019.115034
183. Scheubel RJ, Kahrstedt S, Weber H, Holtz J, Friedrich I, Borgermann J, et al. Depression of progenitor cell function by advanced glycation end products (AGEs): potential relevance for impaired angiogenesis in advanced age and diabetes. Exp Gerontol. (2006) 41:540–8. doi: 10.1016/j.exger.2006.01.002
184. Liu ZJ, Velazquez OC. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid Redox Signal. (2008) 10:1869–82. doi: 10.1089/ars.2008.2121
185. Di Stefano V, Cencioni C, Zaccagnini G, Magenta A, Capogrossi MC, Martelli F. P66shca modulates oxidative stress and survival of endothelial progenitor cells in response to high glucose. Cardiovasc Res. (2009) 82:421–9. doi: 10.1093/cvr/cvp082
186. Sliman SM, Eubank TD, Kotha SR, Kuppusamy ML, Sherwani SI, Butler ES, et al. Hyperglycemic oxoaldehyde, glyoxal, causes barrier dysfunction, cytoskeletal alterations, and inhibition of angiogenesis in vascular endothelial cells: aminoguanidine protection. Mol Cell Biochem. (2010) 333:9–26. doi: 10.1007/s11010-009-0199-x
187. Devi MS, Sudhakaran PR. Differential modulation of angiogenesis by advanced glycation end products. Exp Biol Med (Maywood). (2011) 236:52–61. doi: 10.1258/ebm.2010.010087
188. Xie J, Li R, Wu H, Chen J, Li G, Chen Q, et al. Advanced glycation end products impair endothelial progenitor cell migration and homing via syndecan 4 shedding. Stem Cells. (2017) 35:522–31. doi: 10.1002/stem.2506
189. Zhou H, Chen T, Li Y, You J, Deng X, Chen N, et al. Glycation of tie-2 inhibits angiopoietin-1 signaling activation and angiopoietin-1-induced angiogenesis. Int J Mol Sci. (2022) 23:7137. doi: 10.3390/ijms23137137
190. Hamada Y, Fujii H, Kitazawa R, Yodoi J, Kitazawa S, Fukagawa M. Thioredoxin-1 overexpression in transgenic mice attenuates streptozotocin-induced diabetic osteopenia: a novel role of oxidative stress and therapeutic implications. Bone. (2009) 44:936–41. doi: 10.1016/j.bone.2008
191. Bastos AS, Graves DT, Loureiro AP, Rossa Júnior C, Abdalla DS, Faulin Tdo E, et al. Lipid peroxidation is associated with the severity of periodontal disease and local inflammatory markers in patients with type 2 diabetes. J Clin Endocrinol Metab. (2012) 97:E1353–62. doi: 10.1210/jc.2011-3397
192. Li X, Sun X, Zhang X, Mao Y, Ji Y, Shi L, et al. Enhanced oxidative damage and Nrf2 downregulation contribute to the aggravation of periodontitis by diabetes mellitus. Oxid Med Cell Longev. (2018) 2018:9421019. doi: 10.1155/2018/9421019
193. Chen J, Song M, Yu S, Gao P, Yu Y, Wang H, et al. Advanced glycation end products alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation end products mediate overexpression of cell oxidant stress. Mol Cell Biochem. (2010) 335:137–46. doi: 10.1007/s11010-009-0250-y
194. Zhou J, Xu H, Huang K. Organoselenium small molecules and chromium (III) complexes for intervention in chronic low-grade inflammation and type 2 diabetes. Curr Top Med Chem. (2016) 16:823–34. doi: 10.2174/1568026615666150827094815
195. Srivastava S, Singh D, Patel S, Singh MR. Role of enzymatic free radical scavengers in management of oxidative stress in autoimmune disorders. Int J Biol Macromol. (2017) 101:502–17. doi: 10.1016/j.ijbiomac.2017.03.100
196. Bonetta R. Potential therapeutic applications of MnSODs and SOD-mimetics. Chemistry. (2018) 24:5032–41. doi: 10.1002/chem.201704561
197. Zhu MR, Zhou J, Jin Y, Gao LH, Li L, Yang JR, et al. A manganese-salen complex as dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Int J Biol Macromol. (2018) 120:1232–9. doi: 10.1016/j.ijbiomac.2018.08.089
198. Greenberger JS, Mukherjee A, Epperly MW. Gene therapy for systemic or organ specific delivery of manganese superoxide dismutase. Antioxidants (Basel). (2021) 10:1057. doi: 10.3390/antiox10071057
199. Rosa AC, Bruni N, Meineri G, Corsi D, Cavi N, Gastaldi D, et al. Strategies to expand the therapeutic potential of superoxide dismutase by exploiting delivery approaches. Int J Biol Macromol. (2021) 168:846–65. doi: 10.1016/j.ijbiomac.2020.11.149
200. Polianskyte-Prause Z, Tolvanen TA, Lindfors S, Kon K, Hautala LC, Wang H, et al. Ebselen enhances insulin sensitivity and decreases oxidative stress by inhibiting SHIP2 and protects from inflammation in diabetic mice. Int J Biol Sci. (2022) 18:1852–64. doi: 10.7150/ijbs.66314
201. Singh N, Sherin GR, Mugesh G. Antioxidant and prooxidant nanozymes: from cellular redox regulation to next-generation therapeutics. Angew Chem Int Ed Engl. (2023) 62:e202301232. doi: 10.1002/anie.202301232
202. Singh S. Antioxidant nanozymes as next-generation therapeutics to free radical-mediated inflammatory diseases: a comprehensive review. Int J Biol Macromol. (2024) 260:129374. doi: 10.1016/j.ijbiomac.2024.129374
203. Ashor AW, Werner AD, Lara J, Willis ND, Mathers JC, Siervo M. Effects of vitamin C supplementation on glycaemic control: a systematic review and meta-analysis of randomised controlled trials. Eur J Clin Nutr. (2017) 71:1371–80. doi: 10.1038/ejcn.2017.24
204. Grabež M, Škrbić R, Stojiljković MP, Vučić V, Rudić Grujić V, Jakovljević V, et al. A prospective, randomized, double-blind, placebo-controlled trial of polyphenols on the outcomes of inflammatory factors and oxidative stress in patients with type 2 diabetes mellitus. Rev Cardiovasc Med. (2022) 23:57. doi: 10.31083/j.rcm2302057
205. Shrivastav D, Dabla PK, Sharma J, Viswas A, Mir R. Insights on antioxidant therapeutic strategies in type 2 diabetes mellitus: a narrative review of randomized control trials. World J Diabetes. (2023) 14:919–29. doi: 10.4239/wjd.v14.i6.919
206. Thakur P, Kumar A, Kumar A. Targeting oxidative stress through antioxidants in diabetes mellitus. J Drug Target. (2018) 26:766–76. doi: 10.1080/1061186X.2017.1419478
207. Bhatti JS, Sehrawat A, Mishra J, Sidhu IS, Navik U, Khullar N, et al. Oxidative stress in the pathophysiology of type 2 diabetes and related complications: current therapeutics strategies and future perspectives. Free Radic Biol Med. (2022) 184:114–34. doi: 10.1016/j.freeradbiomed.2022.03.019
208. Aghajanian P, Hall S, Wongworawat MD, Mohan S. The roles and mechanisms of actions of vitamin C in bone: new developments. J Bone Miner Res. (2015) 30:1945–55. doi: 10.1002/jbmr.2709
209. Ghanem M, Heikal L, Abdel Fattah H, El Ashwah A, Fliefel R. The effect of coenzyme Q10/collagen hydrogel on bone regeneration in extraction socket prior to implant placement in type II diabetic patients: a randomized controlled clinical trial. J Clin Med. (2022) 11:3059. doi: 10.3390/jcm11113059
210. Javid AZ, Hormoznejad R, Yousefimanesh HA, Zakerkish M, Haghighi-zadeh MH, Dehghan P, et al. The impact of resveratrol supplementation on blood glucose, insulin, insulin resistance, triglyceride, and periodontal markers in type 2 diabetic patients with chronic periodontitis. Phytother Res. (2017) 31:108–14. doi: 10.1002/ptr.5737
211. Bo S, Gambino R, Ponzo V, Cioffi I, Goitre I, Evangelista A, et al. Effects of resveratrol on bone health in type 2 diabetic patients. A double-blind randomized-controlled trial. Nutr Diabetes. (2018) 8:51. doi: 10.1038/s41387-018-0059-4
212. Maartens M, Kruger MJ, van de Vyver M. The effect of N-acetylcysteine and ascorbic acid-2-phosphate supplementation on mesenchymal stem cell function in B6.C-lep(ob)/J type 2 diabetic mice. Stem Cells Dev. (2021) 30:1179–89. doi: 10.1089/scd.2021.0139
213. Bo T, Nohara H, Yamada KI, Miyata S, Fujii J. Ascorbic acid protects bone marrow from oxidative stress and transient elevation of corticosterone caused by x-ray exposure in Akr1a-knockout mice. Antioxidants (Basel). (2024) 13:152. doi: 10.3390/antiox13020152
214. Ellulu MS, Rahmat A, Patimah I, Khaza'ai H, Abed Y. Effect of vitamin C on inflammation and metabolic markers in hypertensive and/or diabetic obese adults: a randomized controlled trial. Drug Des Devel Ther. (2015) 9:3405–12. doi: 10.2147/DDDT.S83144
215. Ellulu MS. Obesity, cardiovascular disease, and role of vitamin C on inflammation: a review of facts and underlying mechanisms. Inflammopharmacology. (2017) 25:313–28. doi: 10.1007/s10787-017-0314-7
216. Toraman A, Arabaci T, Aytekin Z, Albayrak M, Bayir Y. Effects of vitamin C local application on ligature-induced periodontitis in diabetic rats. J Appl Oral Sci. (2020) 28:e20200444. doi: 10.1590/1678-7757-2020-0444
217. Ashor AW, Lara J, Mathers JC, Siervo M. Effect of vitamin C on endothelial function in health and disease: a systematic review and meta-analysis of randomised controlled trials. Atherosclerosis. (2014) 235:9–20. doi: 10.1016/j.atherosclerosis.2014.04.004
218. Jain SK, McLean WE, Stevens CM, Dhawan R. The positive association of plasma levels of vitamin C and inverse association of VCAM-1 and total adiponectin with bone mineral density in subjects with diabetes. Nutrients. (2022) 14:3893. doi: 10.3390/nu14193893
219. Norazlina M, Ima-Nirwana S, Gapor MT, Khalid BA. Palm vitamin E is comparable to α-tocopherol in maintaining bone mineral density in ovariectomised female rats. Exp Clin Endocrinol Diabetes. (2000) 108:305–10. doi: 10.1055/s-2000-7758
220. Noguchi M, Yamawaki I, Takahashi S, Taguchi Y, Umeda M. Effects of α-tocopherol on bone marrow mesenchymal cells derived from type II diabetes mellitus rats. J Oral Sci. (2018) 60:579–57. doi: 10.2334/josnusd.17-0422
221. Mubarak HA, Kamal MM, Mahmoud Y, Abd-Elsamea FS, Abdelbary E, Gamea MG, et al. The ameliorating effects of mesenchymal stem cells compared to α-tocopherol on apoptosis and autophagy in streptozotocin-induced diabetic rats: implication of PI3K/akt signaling pathway and entero-insular axis. J Cell Biochem. (2023) 124:1705–19. doi: 10.1002/jcb.30482
222. Wu Z, Zheng X, Meng L, Fang X, He Y, Li D, et al. α-Tocopherol, especially α-tocopherol phosphate, exerts antiapoptotic and angiogenic effects on rat bone marrow-derived endothelial progenitor cells under high-glucose and hypoxia conditions. J Vasc Surg. (2018) 67:1263–73. doi: 10.1016/j.jvs.2017.02.051
223. Lu M, Zhang Y, Yuan X, Zhang Y, Zhou M, Zhang T, et al. Increased serum α-tocopherol acetate mediated by gut microbiota ameliorates alveolar bone loss through the STAT3 signalling pathway in diabetic periodontitis. J Clin Periodontol. (2023) 50:1539–52. doi: 10.1111/jcpe.13862
224. Kanter M, Altan MF, Donmez S, Ocakci A, Kartal ME. The effects of quercetin on bone minerals, biomechanical behavior, and structure in streptozotocin-induced diabetic rats. Cell Biochem Funct. (2007) 25:747–52. doi: 10.1002/cbf.1397
225. Liang W, Luo Z, Ge S, Li M, Du J, Yang M, et al. Oral administration of quercetin inhibits bone loss in rat model of diabetic osteopenia. Eur J Pharmacol. (2011) 670:317–24. doi: 10.1016/j.ejphar.2011.08.014
226. Baş A, Albeniz I. Investigation of the effects of eugenol and quercetin on bone loss in STZ-NA induced diabetic rats utilizing micro CT. J Diabetes Metab Disord. (2022) 21:637–46. doi: 10.1007/s40200-022-01026-y
227. Yurteri A, Yildirim A, Çelik ZE, Vatansev H, Durmaz MS. The effect of quercetin on bone healing in an experimental rat model. Jt Dis Relat Surg. (2023) 34:365–73. doi: 10.52312/jdrs.2023.870
228. Laky M, Arslan M, Zhu X, Rausch-Fan X, Moritz A, Sculean A, et al. Quercetin in the prevention of induced periodontal disease in animal models: a systematic review and meta-analysis. Nutrients. (2024) 16:735. doi: 10.3390/nu16050735
229. Bian W, Xiao S, Yang L, Chen J, Deng S. Quercetin promotes bone marrow mesenchymal stem cell proliferation and osteogenic differentiation through the H19/miR-625-5p axis to activate the wnt/beta-catenin pathway. BMC Complement Med Ther. (2021) 21:243. doi: 10.1186/s12906-021-03418-8
230. Guo R, Liu B, Wang K, Zhou S, Li W, Xu Y. Resveratrol ameliorates diabetic vascular inflammation and macrophage infiltration in db/db mice by inhibiting the NF-κB pathway. Diab Vasc Dis Res. (2014) 11:92–102. doi: 10.1177/1479164113520332
231. Zhen L, Fan DS, Zhang Y, Cao XM, Wang LM. Resveratrol ameliorates experimental periodontitis in diabetic mice through negative regulation of TLR4 signaling. Acta Pharmacol Sin. (2015) 36:221–8. doi: 10.1038/aps.2014.131
232. Cirano FR, Molez AM, Ribeiro FV, Tenenbaum HC, Casati MZ, Corrêa MG, et al. Resveratrol and insulin association reduced alveolar bone loss and produced an antioxidant effect in diabetic rats. J Periodontol. (2021) 92:748–59. doi: 10.1002/JPER.19-0718
233. Pino DS, Casarin RC, Pimentel SP, Cirano FR, Corrêa MG, Ribeiro FV. Effect of resveratrol on critical-sized calvarial defects of diabetic rats: histometric and gene expression analysis. J Oral Maxillofac Surg. (2017) 75:2561.e1–10. doi: 10.1016/j.joms.2017.07.167
234. Hua Y, Bi R, Li Z, Li Y. Resveratrol treatment promotes titanium implant osseointegration in diabetes mellitus rats. J Orthop Res. (2020) 38:2113–9. doi: 10.1002/jor.24651
235. Corrêa MG, Ribeiro FV, Pimentel SP, Benatti BB, Felix Silva PH, Casati MZ, et al. Impact of resveratrol in the reduction of the harmful effect of diabetes on peri-implant bone repair: bone-related gene expression, counter-torque and micro-CT analysis in rats. Acta Odontol Scand. (2021) 79:174–81. doi: 10.1080/00016357.2020.1797159
236. Xuan Y, Wang J, Zhang X, Wang J, Li J, Liu Q, et al. Resveratrol attenuates high glucose-induced osteoblast dysfunction via AKT/GSK3β/FYN-mediated NRF2 activation. Front Pharmacol. (2022) 13:862618. doi: 10.3389/fphar.2022.862618
237. Deng X, Deng L, Xu M, Sun Y, Yang M. Effects of SIRT1 on osteogenic differentiation of bone marrow mesenchymal stem cells in type 2 diabetic patients. Endocr Metab Immune Disord Drug Targets. (2023) 23:1077–86. doi: 10.2174/1871530323666230109124631
238. Wajima CS, Pitol-Palin L, de Souza Batista FR, Dos Santos PH, Matsushita DH, Okamoto R. Morphological and biomechanical characterization of long bones and peri-implant bone repair in type 2 diabetic rats treated with resveratrol. Sci Rep. (2024) 14:2860. doi: 10.1038/s41598-024-53260-4
239. Hwang SM, Kim TY, Kim A, Kim YG, Park JW, Lee JM, et al. Resveratrol facilitates bone formation in high-glucose conditions. Front Physiol. (2024) 15:1347756. doi: 10.3389/fphys.2024.1347756
240. Ungvari Z, Bagi Z, Feher A, Recchia FA, Sonntag WE, Pearson K, et al. Resveratrol confers endothelial protection via activation of the antioxidant transcription factor Nrf2. Am J Physiol Heart Circ Physiol. (2010) 299:H18–24. doi: 10.1152/ajpheart.00260.2010
241. Zheng X, Zhu S, Chang S, Cao Y, Dong J, Li J, et al. Protective effects of chronic resveratrol treatment on vascular inflammatory injury in steptozotocin-induced type 2 diabetic rats: role of NF-kappa B signaling. Eur J Pharmacol. (2013) 720:147–57. doi: 10.1016/j.ejphar.2013.10.034
242. Wu H, Chen Z, Chen JZ, Xie J, Xu B. Resveratrol improves tube formation in AGE-induced late endothelial progenitor cells by suppressing syndecan-4 shedding. Oxid Med Cell Longev. (2018) 2018:9045976. doi: 10.1155/2018/9045976
243. Ding X, Yao W, Zhu J, Mu K, Zhang J, Zhang JA. Resveratrol attenuates high glucose-induced vascular endothelial cell injury by activating the E2F3 pathway. Biomed Res Int. (2020) 2020:6173618. doi: 10.1155/2020/6173618
244. Abrahams S, Haylett WL, Johnson G, Carr JA, Bardien S. Antioxidant effects of curcumin in models of neurodegeneration, aging, oxidative and nitrosative stress: a review. Neuroscience. (2019) 406:1–21. doi: 10.1016/j.neuroscience.2019.02.020
245. Marton LT, Pescinini-E-Salzedas LM, Camargo MEC, Barbalho SM, Haber JFDS, Sinatora RV, et al. The effects of curcumin on diabetes mellitus: a systematic review. Front Endocrinol (Lausanne). (2021) 12:669448. doi: 10.3389/fendo.2021.669448
246. Li Y, Zhang ZZ. Sustained curcumin release from PLGA microspheres improves bone formation under diabetic conditions by inhibiting the reactive oxygen species production. Drug Des Devel Ther. (2018) 12:1453–66. doi: 10.2147/DDDT.S154334
247. Liang Y, Zhu B, Li S, Zhai Y, Yang Y, Bai Z, et al. Curcumin protects bone biomechanical properties and microarchitecture in type 2 diabetic rats with osteoporosis via the TGFβ/Smad2/3 pathway. Exp Ther Med. (2020) 20:2200–08. doi: 10.3892/etm.2020.8943
248. You J, Sun J, Ma T, Yang Z, Wang X, Zhang Z, et al. Curcumin induces therapeutic angiogenesis in a diabetic mouse hindlimb ischemia model via modulating the function of endothelial progenitor cells. Stem Cell Res Ther. (2017) 8:182. doi: 10.1186/s13287-017-0636-9
249. Zhang Z, Li K. Curcumin attenuates high glucose-induced inflammatory injury through the reactive oxygen species-phosphoinositide 3-kinase/protein kinase B-nuclear factor-κB signaling pathway in rat thoracic aorta endothelial cells. J Diabetes Investig. (2018) 9:731–40. doi: 10.1111/jdi.12767
250. Kadam S, Kanitkar M, Dixit K, Deshpande R, Seshadri V, Kale V. Curcumin reverses diabetes-induced endothelial progenitor cell dysfunction by enhancing MnSOD expression and activity in vitro and in vivo. J Tissue Eng Regen Med. (2018) 12:1594–607. doi: 10.1002/term.2684
251. Jin QH, Hu XJ, Zhao HY. Curcumin activates autophagy and attenuates high glucose-induced apoptosis in HUVECs through the ROS/NF-κB signaling pathway. Exp Ther Med. (2022) 24:596. doi: 10.3892/etm.2022.11533
252. Surai PF. Silymarin as a natural antioxidant: an overview of the current evidence and perspectives. Antioxidants (Basel). (2015) 4:204–47. doi: 10.3390/antiox4010204
253. Chu C, Li D, Zhang S, Ikejima T, Jia Y, Wang D, et al. Role of silibinin in the management of diabetes mellitus and its complications. Arch Pharm Res. (2018) 41:785–96. doi: 10.1007/s12272-018-1047-x
254. Ying X, Chen X, Liu H, Nie P, Shui X, Shen Y, et al. Silibinin alleviates high glucose-suppressed osteogenic differentiation of human bone marrow stromal cells via antioxidant effect and PI3K/akt signaling. Eur J Pharmacol. (2015) 765:394–401. doi: 10.1016/j.ejphar.2015.09.005
255. Wang T, Cai L, Wang Y, Wang Q, Lu D, Chen H, et al. The protective effects of silibinin in the treatment of streptozotocin-induced diabetic osteoporosis in rats. Biomed Pharmacother. (2017) 89:681–8. doi: 10.1016/j.biopha.2017.02.018
256. Palomino OM, Gouveia NM, Ramos S, Martín MA, Goya L. Protective effect of Silybum marianum and silibinin on endothelial cells submitted to high glucose concentration. Planta Med. (2017) 83:97–103. doi: 10.1055/s-0042-113135
257. Garg SS, Gupta J, Sharma S, Sahu D. An insight into the therapeutic applications of coumarin compounds and their mechanisms of action. Eur J Pharm Sci. (2020) 152:105424. doi: 10.1016/j.ejps.2020.105424
258. Ranđelović S, Bipat R. A review of coumarins and coumarin-related compounds for their potential antidiabetic effect. Clin Med Insights Endocrinol Diabetes. (2021) 14:11795514211042023. doi: 10.1177/11795514211042023
259. Lee EJ, Kang MK, Kim YH, Kim DY, Oh H, Kim SI, et al. Coumarin ameliorates impaired bone turnover by inhibiting the formation of advanced glycation end products in diabetic osteoblasts and osteoclasts. Biomolecules. (2020) 10:1052. doi: 10.3390/biom10071052
260. Dang BT, Gény C, Blanchard P, Rouger C, Tonnerre P, Charreau B, et al. Advanced glycation inhibition and protection against endothelial dysfunction induced by coumarins and procyanidins from Mammea neurophylla. Fitoterapia. (2014) 96:65–75. doi: 10.1016/j.fitote.2014.04.005
261. Tucker JM, Townsend DM. α-Tocopherol: roles in prevention and therapy of human disease. Biomed Pharmacother. (2005) 59:380–7. doi: 10.1016/j.biopha.2005.06.005
262. Meulmeester FL, Luo J, Martens LG, Mills K, van Heemst D, Noordam R. Antioxidant supplementation in oxidative stress-related diseases: what have we learned from studies on α-tocopherol? Antioxidants (Basel). (2022) 11:2322. doi: 10.3390/antiox11122322
263. Xu D, Hu MJ, Wang YQ, Cui YL. Antioxidant activities of quercetin and its complexes for medicinal application. Molecules. (2019) 24:1123. doi: 10.3390/molecules24061123
264. Dhanya R. Quercetin for managing type 2 diabetes and its complications, an insight into multitarget therapy. Biomed Pharmacother. (2022) 146:112560. doi: 10.1016/j.biopha.2021.112560
265. Yan L, Vaghari-Tabari M, Malakoti F, Moein S, Qujeq D, Yousefi B, et al. Quercetin: an effective polyphenol in alleviating diabetes and diabetic complications. Crit Rev Food Sci Nutr. (2023) 63:9163–86. doi: 10.1080/10408398.2022.2067825
266. Arredondo F, Echeverry C, Abin-Carriquiry JA, Blasina F, Antúnez K, Jones DP, et al. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free Radic Biol Med. (2010) 49:738–47. doi: 10.1016/j.freeradbiomed.2010.05.020
267. Zaplatic E, Bule M, Shah SZA, Uddin MS, Niaz K. Molecular mechanisms underlying protective role of quercetin in attenuating Alzheimer’s disease. Life Sci. (2019) 224:109–19. doi: 10.1016/j.lfs.2019.03.055
268. Zu G, Sun K, Li L, Zu X, Han T, Huang H. Mechanism of quercetin therapeutic targets for Alzheimer disease and type 2 diabetes mellitus. Sci Rep. (2021) 11:22959. doi: 10.1038/s41598-021-02248-5
269. Zhao LR, Du YJ, Chen L, Liu ZG, Pan YH, Liu JF, et al. Quercetin protects against high glucose-induced damage in bone marrow-derived endothelial progenitor cells. Int J Mol Med. (2014) 34:1025–31. doi: 10.3892/ijmm.2014.1852
270. Meng X, Zhou J, Zhao CN, Gan RY, Li HB. Health benefits and molecular mechanisms of resveratrol: a narrative review. Foods. (2020) 9:340. doi: 10.3390/foods9030340
271. Meng T, Xiao D, Muhammed A, Deng J, Chen L, He J. Anti-inflammatory action and mechanisms of resveratrol. Molecules. (2021) 26:229. doi: 10.3390/molecules26010229
272. Li Y, Cao Z, Zhu H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol Res. (2006) 53:6–15. doi: 10.1016/j.phrs.2005.08.002
273. Prabhakar O. Cerebroprotective effect of resveratrol through antioxidant and anti-inflammatory effects in diabetic rats. Naunyn Schmiedebergs Arch Pharmacol. (2013) 386:705–10. doi: 10.1007/s00210-013-0871-2
274. Shang YJ, Qian YP, Liu XD, Dai F, Shang XL, Jia WQ, et al. Radical-scavenging activity and mechanism of resveratrol-oriented analogues: influence of the solvent, radical, and substitution. J Org Chem. (2009) 74:5025–31. doi: 10.1021/jo9007095
275. Yun H, Park S, Kim MJ, Yang WK, Im DU, Yang KR, et al. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. FEBS J. (2014) 281:4421–38. doi: 10.1111/febs.12949
276. Rezabakhsh A, Fathi F, Bagheri HS, Malekinejad H, Montaseri A, Rahbarghazi R, et al. Silibinin protects human endothelial cells from high glucose-induced injury by enhancing autophagic response. J Cell Biochem. (2018) 119:8084–94. doi: 10.1002/jcb.26735
277. Inchingolo AD, Inchingolo AM, Malcangi G, Avantario P, Azzollini D, Buongiorno S, et al. Effects of resveratrol, curcumin and quercetin supplementation on bone metabolism—a systematic review. Nutrients. (2022) 14:3519. doi: 10.3390/nu14173519
278. Losada-Barreiro S, Sezgin-Bayindir Z, Paiva-Martins F, Bravo-Díaz C. Biochemistry of antioxidants: mechanisms and pharmaceutical applications. Biomedicines. (2022) 10:3051. doi: 10.3390/biomedicines10123051
279. Żyżelewicz D, Oracz J. Bioavailability and bioactivity of plant antioxidants. Antioxidants (Basel). (2022) 11:2336. doi: 10.3390/antiox11122336
Keywords: bone repair, type II diabetes mellitus, hyperglycaemia, mesenchymal stromal cell, osteoblast, angiogenesis, oxidative stress, antioxidant
Citation: Li P, Alenazi KKK, Dally J, Woods EL, Waddington RJ and Moseley R (2024) Role of oxidative stress in impaired type II diabetic bone repair: scope for antioxidant therapy intervention? Front. Dent. Med 5:1464009. doi: 10.3389/fdmed.2024.1464009
Received: 12 July 2024; Accepted: 2 October 2024;
Published: 14 October 2024.
Edited by:
Roberta Gasparro, University of Naples Federico II, ItalyReviewed by:
Gennaro Musella, University of Foggia, ItalyGiuseppe Sangiovanni, University of Salerno, Italy
Copyright: © 2024 Li, Alenazi, Dally, Woods, Waddington and Moseley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan Moseley, TW9zZWxleVJAY2FyZGlmZi5hYy51aw==