 Su Sheng Quach
Su Sheng Quach Alan Zhu
Alan Zhu- School of Dentistry, The University of Queensland, Herston, QLD, Australia
Despite over 50 years of research into the immunology of periodontal disease, the precise mechanisms and the role of many cell types remains an enigma. Progress has been limited by the inability to determine disease activity clinically. Understanding the immunopathogenesis of periodontal disease, however, is fundamental if immunomodulation is to be used as a therapeutic strategy. It is important for the clinician to understand what could be modulated and why. In this context, potential targets include different immune cell populations and their subsets, as well as various cytokines. The aim of this review is to examine the role of the principal immune cell populations and their cytokines in the pathogenesis of periodontal disease and their potential as possible therapeutic targets.
Introduction
Periodontitis is defined as a microbially-associated, host-mediated inflammation that results in loss of periodontal attachment and apical migration of the junctional epithelium (1). Analysis of the Global Burden of Disease Study 2019 showed that there were 1.1 billion cases of severe periodontitis globally and that the age-standardized prevalence rate of severe periodontitis had increased 8.44% worldwide from 1990 to 2019 (2, 3). In conjunction with growing evidence of periodontal-systemic associations (4, 5), periodontitis is responsible for widespread and significant morbidity.
Although periodontal bacteria are the etiological agents in periodontitis, the host immune response to these bacteria is of fundamental importance (6, 7). Over the past few decades different models have been proposed to explain the pathogenesis of periodontitis, examining the dynamics between the host-immune responses and bacteria (8–10). Both innate and adaptive immune responses have been extensively studied (Figure 1), however, despite over 50 years of research into the immunology of periodontal disease, the precise mechanisms and the roles of many different immune cells are still not well understood.
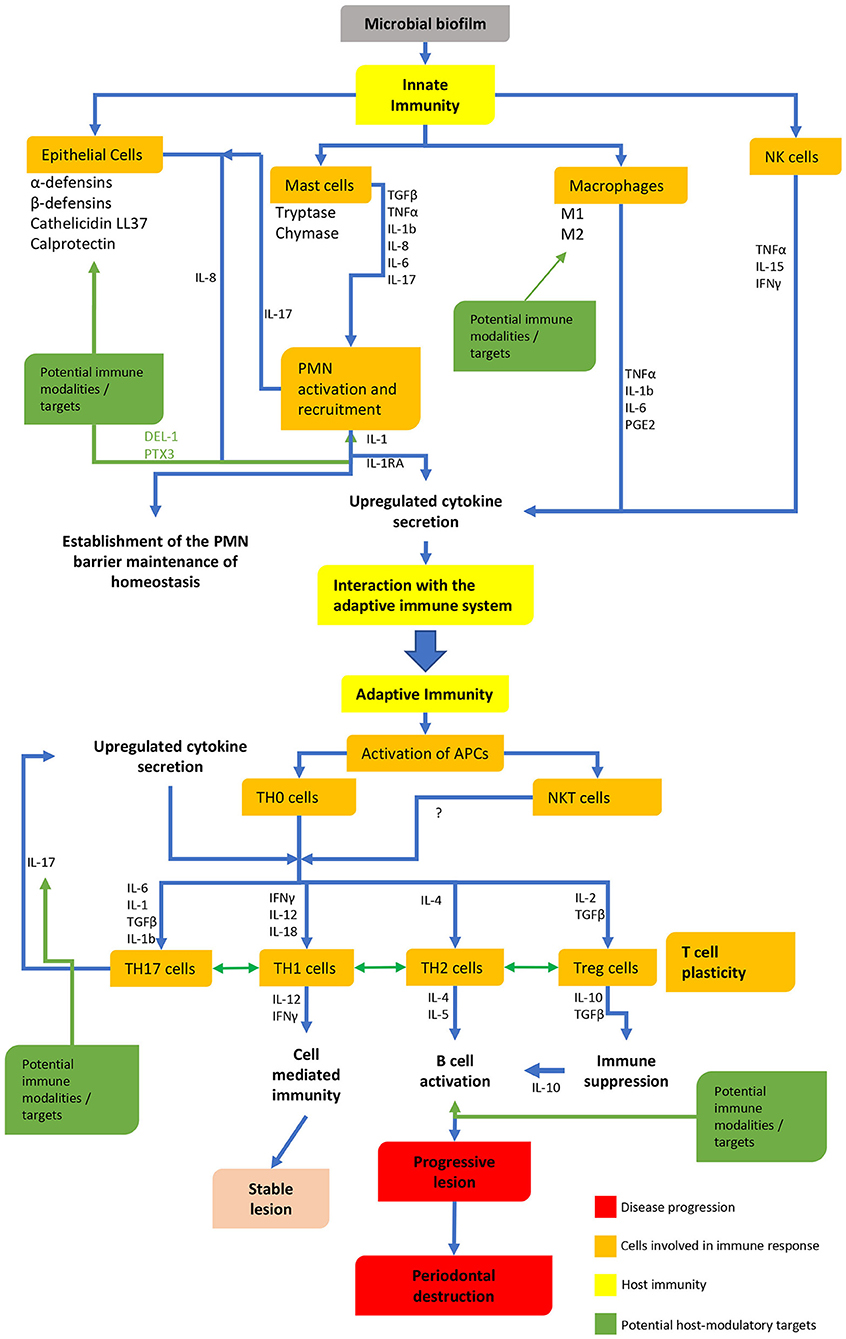
Figure 1. The innate and adaptive immune pathways, interactions and possible targets.
As a consequence, nonsurgical treatment remains the mainstay treatment modality and adjuncts are mostly limited to surgical and antibiotic therapy (11, 12). Multiple avenues of research into the possibility of host modulation therapeutics are currently being explored (13). However, recent systematic reviews have shown there is currently limited and conflicting evidence supporting the use of host modulation therapeutics as adjuncts to nonsurgical treatment (14, 15). Understanding the immunopathogenesis of periodontal disease will prove key to the development of commercially available immunomodulation therapeutics. Thus, it is important for the clinician to understand what could be modulated and why. In this context, potential targets include different immune cell populations and their sub-populations, as well as various cytokines secreted. This review, however, is not intended to be a comprehensive review of the immunopathogenesis of periodontal disease nor is it a comprehensive review of host modulation in periodontal disease, but rather the aim is to provide an overview of the role of the principal immune cell populations and their cytokines in the pathogenesis of periodontal disease and hence their potential as possible therapeutic targets.
Innate Immunity
Epithelial Cells
The epithelial cells of the gingiva not only prevent the ingress of periodontopathic bacteria and their products into the gingival tissues, but they are also important cellular components of innate immune mechanisms in the gingival sulcus. The activation of their toll-like receptors (TLRs), such as TLR-2, leads to the production of IL-8 which is a very powerful chemoattractant for polymorphonuclear neutrophils (PMNs). TLR signaling of epithelial cells and PMNs further results in the production of α- and β-defensins, the cathelicidin LL37, and calprotectin. These antimicrobial molecules not only limit the plaque microbiota within the gingival sulcus but also play a critical role in innate immunity. α-defensins activate the classical complement pathway and upregulate the production of IL-8, while β-defensins may further mediate inflammation (16). Although these molecules have long been advocated as possible therapeutic adjuvants in the control of oral infections including periodontal disease (17, 18), further research, including clinical trials, is still necessary.
Polymorphonuclear Neutrophils (PMNs)
PMNs are present at all stages of periodontal disease. They are the predominant cell in the “initial lesion” (19), and continue to migrate through the sulcular and periodontal pocket lining epithelium in gingivitis and periodontitis, respectively, where they form a barrier between the tissues and plaque. Indeed, by day 21 of an experimental gingivitis there is a four-fold increase in PMN numbers within the junctional epithelium (20). Within the gingival sulcus and periodontal pocket, they are able to control potential periodontal pathogens via reactive oxygen species, proteases and the formation of neutrophil extracellular traps (NETs) (21).
The release of tumor necrosis factor-alpha (TNF-α) and interleukin-1beta (IL-1β), from mast cells and local resident macrophages, results in the expression of adhesion molecules by endothelial cells thus mediating the adhesion and ultimate migration of PMNs into gingival tissues. PMN migration into the gingival sulcus is initially mediated by bacterially-derived chemotactic substances together with the activation of the alternate complement pathway and the formation of C5a (22). Within the gingival sulcus, PMNs produce and release a variety of cytokines, including IL-1, IL-1 receptor antagonist (IL-1RA) and high levels of IL-17, which subsequently stimulates the production of IL-8 by sulcus epithelial cells. The resultant positive feedback loop establishes a rapid flow of PMNs into the gingival sulcus via the junctional epithelium (23), allowing the formation of a PMN barrier against plaque micro-organisms (24). This barrier persists into the periodontitis lesion, likely retarding disease progression.
Paradoxically, however, PMN-mediated tissue destruction may also occur via degranulation and the release of hydrolytic enzymes, reactive oxygen species, matrix metalloproteinases and the formation of NETs (25). This destruction is confined to the tissues immediately subjacent to the pocket lining epithelium. Nevertheless, the role of PMNs in the pathogenesis of periodontitis remains to be defined. An improved understanding of PMN heterogeneity and possible subtypes (26, 27), may allow harmful sub-populations to be identified and targeted while avoiding the suppression of beneficial protective functions.
The importance of the PMN barrier function in the preservation of periodontal health is evidenced by the rapid and severe progression of periodontal destruction in conditions of PMN dysfunction (28). This may occur in complete PMN depletion [e.g. Leukocyte Adhesion Deficiency type I/LAD-I syndrome (29)], as well as in defective PMN function as seen in Papillon-Lefèvre syndrome (30) and Chediak-Higashi syndrome (31). These conditions suggest that caution must be taken when considering PMN immunomodulation, as suppression may inadvertently exacerbate disease rather than reduce it.
Developmental endothelial locus-1 (DEL-1) is an extracellular matrix protein that has been shown to interact with both the αvβ2 and αvβ3 integrins (32, 33), resulting in reduced PMN transmigration. In support of a destructive role for PMNs in periodontal disease, animal studies have shown that DEL-1-deficient mice have an accompanying increase in RANK-L and osteoclastic activity in the periodontium (34). This is in contrast to mice with combined DEL-1 and IL-17 receptor deficiency, where complete protection against bone loss was observed in the same study. In a clinical study, examining the effect of periodontal treatment an increase in DEL-1 levels in gingival crevicular fluid was observed, after scaling and root planning, in patients with active periodontitis (35). In a pre-clinical trial, the authors from the same group demonstrated that local administration of DEL-1 may prevent bone loss in non-human primates (36). Consequently, DEL-1 may be a future therapeutic target, but care must be taken before extrapolating animal studies to human disease.
In contrast, increased levels of pentraxin-3 (PTX3) have been reported in the saliva of patients with previously classified aggressive periodontitis, compared with patients with chronic periodontitis and controls (37). Pentraxins are a superfamily of proteins involved in the acute-phase response and innate immunity. Pentraxin-3 (PTX3) has been shown to bind P-selectin, which downregulates PMN recruitment via a negative feedback loop (38). The increased levels of PTX3 seen in what would be now classified as severe periodontitis—possible Stage 3 Grade C—would suggest a protective role for PMNs in periodontal disease. Current understanding of pentraxins is limited, but there may be the potential for PTX3 to be a therapeutic target in the future. As stated above care must be taken when considering PMN immunomodulation, as suppression may inadvertently exacerbate disease rather than reduce it. Clearly, phase I clinical trials involving patients with periodontal disease are essential to establish the safety of PMN immunomodulation.
Adaptive Immunity
T Cells
The importance of T cells in periodontal diseases was first suggested in seminal studies by Ivanyi and Lehner (39, 40). It is now well established that the gingivitis lesion is characterized by a predominance of T lymphocytes (41–44). The periodontitis lesion on the other hand is comprised mostly of B lymphocytes and plasma cells (45–47).
Seymour and colleagues have shown that the development of gingivitis immunologically mirrors the process involved in delayed-type hypersensitivity (DTH) (48, 49). This results in the development of a perivascular lymphocyte/macrophage infiltrate, which increases in size, leading to the typical clinical presentation of inflammation in gingivitis. In the gingivitis lesion, consistent with a lesion of DTH, the lymphocytes mainly consist of T cells with a low expression of IL-2 (<10%) and a CD4/CD8 ratio of 2:1. Langerhans cells (in oral and sulcular epithelium) are also present, increasing in quantity in tandem with lesion expansion (49). Suggestive of activation, T cells and epithelial cells express high levels of MHC class II antigens (HLADR and HLA-DQ) (49). The production of interferon gamma (IFN-γ) by activated CD4 T cells further activates PMNs and macrophages, controlling lesion progression. The persistent microbial challenge from plaque microorganisms results in a chronic but stable lesion.
Th1/Th2 Cells
It is now well established that periodontitis is dominated by large numbers of B lymphocytes (50, 51). Additionally, the proportion of B cells appears to increase with the severity of the disease (52, 53). In line with the T cell nature of gingivitis and the B cell predominance of periodontitis, it has been suggested that the stable lesion is mediated by a Th1 response, while the progressive lesion is mediated by Th2 cells (54). This model proposed that a robust innate immune response results in significant production of IL-12 (55), by both PMNs and macrophages and consequently a Th1 response. This leads to cell-mediated immunity, protective antibodies and a stable periodontal lesion. IFN-γ produced from a Th1 response enhances the phagocytic ability of PMNs and macrophages (56, 57). On the other hand, disease progression is suggested to be the result of a poor innate immune response, polyclonal B-cell activation, a resultant Th2 response and non-protective antibodies (reviewed in Gemmell et al. (58)). It is now generally accepted that both Th1 and Th2 cells are involved in the pathogenic process and a loss of homeostatic balance, shifting toward a Th2 response, is responsible for disease progression (59, 60).
A number of studies have investigated T cell cytokine profiles present and their activity during different stages of periodontal disease, with conflicting results (61–63). The ability to define the role of different cytokine profiles in the pathogenesis of periodontitis is hindered by heterogeneity in study designs and case definitions. Additionally, as periodontitis proceeds in cycles of stability and progression the ongoing inability to determine the activity of a periodontitis lesion clinically hinders a meaningful interpretation of results (64, 65). In this context and with the high level of redundancy in cytokine networks (66), careful consideration must be given if individual cytokines are to be targets for immunomodulation in periodontal disease. Nevertheless, human recombinant cytokines related to Th1 and Th2 subtypes are being examined for and have seen limited use in the treatment of malignancies with varying degrees of success (67, 68). Currently however, there are no studies investigating the role of these agents in the context of periodontitis.
In recent years, Th17 cells and the corresponding cytokine IL-17 have been suggested as possible targets for immunomodulation in the treatment of periodontitis (69). Th17 cells were identified as a distinct T cell subset which produces IL-17A (commonly referred to as IL-17) by Park et al. (70). These cells also produce IL-17F, IL-21, IL-22, and IL-26 (71, 72). Nonetheless, other cells are also capable of expressing IL-17 such as PMNs, mast cells, NKT cells and periodontal ligament cells (73–76). However, the role of Th17 cells and IL-17 in periodontitis remains enigmatic. Although IL-17 modulates bone loss, increasing osteoclastogenesis in rheumatoid arthritis (77), this has yet to be confirmed in periodontitis in humans.
There is significant controversy around the role of IL-17 in periodontitis. Animal studies have supported both a destructive (34, 69) and a protective role (78, 79). This protective role may be due to the recruitment of PMNs to inflamed gingiva (80). It has been demonstrated that P.gingivalis infection downregulates the IL-17 receptor gene in mice (81) which may explain reduced PMN entry into P.gingivalis-induced lesions in these animals (82). Utilizing IL-17RA-deficient mice, Yu et al. also showed that monoinfection with P.gingivalis reduced PMN migration, resulting in increased bone loss (79). In addition, P.gingivalis stimulation of peripheral blood mononuclear cells resulted in higher mean levels of IL-17 in gingivitis patients compared with periodontitis patients (83), further suggesting a protective role for IL-17 in periodontal disease.
Clinical studies examining the association between Th17 and IL-17 in gingival tissues, GCF, saliva and serum have shown similarly conflicting results (reviewed by Cheng et al.) (84). A comparison of gingival biopsies from both gingivitis and periodontitis patients showed that the density of CD161+ T cells, which may include a small population of IL-17 producing T-cells, was higher in periodontitis compared with gingivitis (85). In contrast, double immunofluorescence labeling has revealed that IL17+ cells were neither CD4+ nor CD8+. Instead, these cells were tryptase+, suggestive of mast cells (86, 87). This is consistent with mast cells being found to be a major source of IL-17 in a number of other lesions such as psoriasis and rheumatoid arthritis (88, 89). This has been further supported by gene expression studies on diseased human tissue in which low levels of IL-17 and low expression of IL-17 pathway genes were found (90). These results were confirmed by Parachuru et al. who demonstrated very few IL-17+ cells (<1%) in human periodontitis lesions together with down regulation of IL-17 pathway genes and low levels of IL-17 (86, 87). These findings suggest that IL-17 is not essential for periodontal bone loss in human disease, potentially playing a minor role.
T cells also have a high degree of plasticity, being able to switch from one lineage to another dependent on the antigen presenting cell (APC) and the tissue micro-environment. Th17 cells in periodontal tissues may, under the influence of IL-4, become a Th2 cell and a Th2 cell under the influence of IL-12 and dendritic APCs may become a Th1 cell (91). It has further been shown that Foxp3 positive regulatory T cells (Tregs) can become Th17 cells in autoimmune arthritis (92). Although likely not a common occurrence (90), this indicates the possibility for variation in T cell expression alongside disease progression. Predicated on the belief that Th17 and exFoxp3Th17 are pathogenic (93), some studies have advocated targeting the Th17/Treg balance. IL-35 has been shown in the treatment of ligature-induced periodontitis to reduce Th17 lymphocytes, increase Tregs and result in a benefit in terms of alveolar bone resorption (94). All-trans retinoic acid also demonstrated modulation of the Th17/Treg ratio and prevention of alveolar bone resorption (95). Further animal studies have shown that mice deficient in CD11a (LFA-1 knockout mice), with significantly higher levels of IL-23 and IL-17 than wild-type (C57BL/6) controls, experienced severe periodontal bone loss (69). When these LFA-1 knockout mice were treated with local anti-IL-17 or anti-IL-23p19 antibodies, bone loss was attenuated, while untreated mice had progressive disease. There are, however, some important differences between mouse models of periodontal bone loss and human disease. As noted above human periodontal disease tissues have low levels of IL-17 and low expression of IL-17 pathway genes (86, 87, 90) and very few IL-17-positive cells (<1%) (86). Nevertheless, Moutsopoulos et al. described significant improvement in bleeding on probing with a concurrent decrease in IL-23 and IL-17 levels after treatment with ustekinumab (IL-12/IL-23 inhibitor), in a patient with LAD-I and severe periodontitis (96). Another case report described successful resolution of acute periodontitis in five psoriatic patients, after substitution of adalimumab (TNF-alpha inhibitor) for secukinumab (IL-17 inhibitor), with no recurrence of psoriasis or periodontitis after 14 months (97). On the other hand, there is some anecdotal evidence of an exacerbation of periodontitis in patients treated with an IL-17 inhibitor. At present, the role of IL-17 in periodontitis is complex and controversial and care must be taken in extrapolating early animal studies and case reports. In view of the apparent conflicting results, further studies are clearly required to determine the role of IL-17 in the pathogenesis of human periodontal disease before its suitability as a therapeutic target can be determined.
Regulatory T Cells (Tregs)
Tregs are a subset of T cells which is closely regulated by characteristic forkhead/winged helix transcription factor (Foxp3) expression. Tregs regulate immune responses and autoimmunity (98) via both contact dependent and independent mechanisms including the release of IL-10 and TGF-β. Tregs have demonstrated suppression of other effector T cell subtypes including Th1, Th2, and Th17 (99–101).
Association studies have shown an increased number of Tregs in periodontitis lesions (87, 102, 103). Gene expression analysis by Nakajima et al. showed significantly higher Foxp3 mRNA expression in periodontitis compared with gingivitis (102). Similarly Tregs, identified with Foxp3 and other specific markers, were found with increased frequency in periodontitis patients (104). In this study Tregs increased CCR4 expression which suggested attraction to the site of inflammation by CCL17 and CCL22. Foxp3 positive cells are also significantly correlated with the B cell-Plasma cell/T cell ratio in B cell-Plasma cell dominated lesions (87) and the mean expression of STAT5A, TGFβ1 and IL10 genes has also been found to be higher in B-cell-Plasma cell predominant gingival tissues (86).
Tregs may play a key role in the pathogenesis of periodontitis. On one hand they may suppress Th1 responses and increase B cell proliferation via IL-10 production. On the other hand, animal models have supported a protective role of Treg suppression (105, 106) while Motta et al. found less Foxp3 expression in more severe periodontitis (107). Currently our understanding of Tregs in the context of periodontitis is lacking. Further complicating the situation is the already discussed plastic nature of T cells, allowing conversion between different subtypes in situ (108). Additional studies elucidating the role of these cells are clearly necessary before therapeutic targeting could be considered.
CD8 T Cells
In the progressive lesion of periodontitis there is a decrease in the CD4:CD8 ratio to 1:1 (49, 109, 110) compared with the 2:1 ratio seen in the gingivitis lesion. Despite this increased representation in the lesion, little is known about the role of CD8+ T cells in the context of periodontitis. It appears, however, they do not directly mediate the periodontal destruction in animal models (111). Currently, the potential of CD8 T-cells as host modulating targets is unknown.
Adaptive Immunity
B Cells
As stated earlier, the periodontitis lesion is dominated by B cells and plasma cells (46, 47, 112), suggesting a crucial role in disease progression.
Antibodies
The role of antibodies in periodontal disease pathogenesis is still poorly understood, however, it has been shown that oral bacteria such as P. gingivalis and F. nucleatum have polyclonal B-cell activation properties (113), which result in the production of low affinity antibodies in the periodontal tissues. In vitro studies have reported patients with severe periodontitis with high anti-P.gingivalis antibodies inhibited bone resorption, while patients with low amounts of these antibodies showed increased bone destruction (114), supporting the protective role of antibody production. On the other hand, other studies have demonstrated that higher antibody levels to subgingival plaque, positively correlated with increased periodontal bone loss, indicating a lack of protection (115).
In the context of autoimmunity, anti-collagen type I and III antibodies were observed in the gingival tissues of periodontitis patients (116) and collagen type I-specific T-cell clones were isolated from the periodontal lesions (117). It is postulated that the autoantibodies present in periodontal disease are derived from the natural antibodies and most likely play a physiological role in removal of damaged tissue and dead cells as a consequence of periodontal tissue destruction. However, there is still a possibility that the production of these antibodies may become excessive and contribute to disease progression (118).
B Cell Subsets
There are largely two types of B cells: B-1 which is subdivided into B-1a and B-1b and B-2. B-1 cells can act independent of T cells for an early antibody response of low affinity or interact with T cells and produce IgG antibodies with high affinity (119). On the other hand, B-2 cells are the traditional group of B cells that interact with T cells and develop into memory cells and plasma cells.
B-1 cells have been of particular interest due to their ability to act independently of T cells. Flow cytometry of peripheral blood has demonstrated that B-1a cells, which express the T cell marker CD5, occur in significantly larger amounts in periodontal patients compared with healthy controls (120). In addition, Berglundh et al. reported that B-1a cell proportions in peripheral blood were up to six times greater in periodontitis subjects compared with controls (121), suggesting the potential of these cells to act as a diagnostic marker for periodontitis. On the other hand, a more recent study showed similar levels of B-1a cells when comparing peripheral blood samples from patients with or without periodontitis (122). However, it is important to highlight that the case definition of periodontitis used in those studies was different. The former study included more severe periodontitis cases compared with the latter study, contributing to the contradicting observation.
More B-1a cells are also found in periodontitis tissues compared with gingivitis tissues (123) and an experimental gingivitis study in periodontitis patients also demonstrated the involvement of B-1a cells (124). Although, these studies highlight the importance of B-1a cells in the pathogenesis of periodontitis, their role in periodontitis remains to be determined. A significant aspect of B-1a cells is their ability to produce IL-10. IL-10 has both anti-inflammatory and pro-inflammatory functions, including B cell activation and as an autocrine growth factor for B-1a cells (125, 126). Although, higher levels of IL-10 have been found in gingival tissues compared with peripheral blood, its role in periodontal disease remains speculative and because of its U-shaped dose response curve its role in any lesion would be dependent upon its concentration within the tissue micro-environment. Nevertheless, transfer of IL-10 secreting B-1a cells into experimental periodontitis animals, resulted in decreased alveolar bone resorption and decreased RANKL, IL-17, TNF-α and IL-1β in the test group (127, 128). However, as pointed out earlier, care must be taken in extrapolating the results from pre-clinical animal models, when determining whether B-1a cells or indeed IL-10 offer potential therapeutic modalities.
Nevertheless, evidence supporting B cells as therapeutic targets for host modulation in periodontal disease is emerging but limited. Two cytokines, a proliferation inducing ligand (APRIL) and a B lymphocyte stimulator (BLyS), are known to be important for survival, proliferation and maturation of B cells. Abe et al. demonstrated these two cytokines to be upregulated in natural and experimental periodontal disease in humans and mice, respectively, which in turn correlated with increased numbers of B cells/plasma cells in both species (129). When APRIL and BLyS were neutralized, the number of B cells decreased in gingival tissue and inhibited bone loss in wild-type mice. Interestingly, the authors found that neutralizing either cytokine was enough to reduce bone loss.
Another group of researchers evaluated the concentrations of BLyS and APRIL in saliva and serum of patients with chronic or aggressive periodontitis compared with healthy controls (130). They found higher concentrations of BLyS in periodontitis patients compared with controls, but no statistically significant differences were found in the levels of APRIL. However, it is important to note that the levels of cytokines in the gingiva may not correlate with concentrations in the serum or saliva. Even so, the same group detected both APRIL and BLyS in gingival crevicular fluid of periodontitis patients with or without rheumatoid arthritis or osteoporosis, however, there were no comparisons with periodontally healthy controls (131). These observations indicate that B cells and its cytokines are important in the pathogenesis of periodontitis and may provide specific targets for host modulation.
Coat et al. assessed the effect of anti-B lymphocyte immunotherapy, using rituximab (an anti-CD20 monoclonal antibody), on the periodontal status of 21 subjects with rheumatoid arthritis using cross-sectional and longitudinal analyses (132). At baseline group 1, which had not received a course of rituximab, appeared to have more severe periodontitis compared with group 2, which had received at least two courses of rituximab. A follow-up of group 1, 6 months after their first rituximab treatment, found a significant decrease in probing pocket depths (PPD) and clinical attachment loss (CAL) with no changes in plaque index and which persisted for up to 4 years following the rituximab treatment. Although this study is suggestive of a therapeutic effect, further studies with larger sample sizes are nevertheless required. It must be noted that in these patients anti-TNF-α treatment had failed before rituximab therapy commenced. In this context, a study by the same group found increased gingival inflammation albeit with a reduction in attachment loss in patients after anti-TNF-α treatment (133). When comparing these two studies, it is possible that anti-B cell treatment may have provided better protection since blocking TNF-α may have led to the upregulation of a compensatory cytokine pathway.
More, recently, early phase II and III controlled clinical trials have evaluated the effects of belimumab (a recombinant human IgG monoclonal antibody), that inhibits B-cell activating factor, for the treatment of systemic lupus erythematosus (SLE) and its most common, severe, manifestation lupus nephritis (LN) (134, 135). Although the safety profile of these medications prohibits their use for periodontitis, studies evaluating the use of biologics modulating B-cell recruitment and function will provide insights into the role of B cells in periodontitis.
NK Cells
There has been suggestions that natural killer (NK) and natural killer T (NKT) cells could be potential host modulatory targets in the management of periodontal disease. However, there are no studies to date that have targeted NK or NKT cells and its effect on periodontal disease.
NK Cells in Periodontitis
There have been multiple studies demonstrating the increased levels of NK cells in relation to periodontal disease. In an experimental gingivitis group, NK cells were not present in healthy subjects and increased gradually with increasing inflammation (136). In periodontitis patients, the greatest number of NK cells were found in those lesions with the highest proportion of B cells. Fewer NK cells were found in T cell dominated lesions. Interestingly, the NK cells were also found to be in close physical association with B cells which supports the view that NK cells may regulate B cell function in vivo (137). Similarly, a immunohistochemical study of gingival specimens in humans found NK cells to have infiltrated connective tissue more greatly in severe forms of periodontitis compared with mild periodontitis (138). In addition to their cytotoxic effects (139), NK cells produce a range of cytokines including IFN-γ (140), IL-15 (141), and TNF-α (142).
The role of IFN-γ in the pathogenesis of periodontitis is controversial. Although there has been studies showing the protective role of IFN-γ (56, 57), others have demonstrated increased levels of IFN-γ in periodontitis tissues compared with healthy controls (143). This latter finding is not surprising as healthy tissue would not be expected to have high cytokine levels. Equally, the cellular source of IFN-γ in periodontitis lesions is unknown. In mice, the lack of IFN-γ has been shown to lower TNF-α and IL-1β levels and reduce the severity of the local pro-inflammatory response compared with control mice (144). When IFN-γ was reconstituted at the site of inflammation, TNF-α levels increased, and IL-10 decreased. Interestingly, it has been shown that NK and NKT cells produce the majority of IFN-γ after an E. coli lipopolysaccharide (LPS) challenge in mice spleen (145) whereas P. gingivalis LPS did not induce IFN-γ production by NK cells (146). Clearly further research is required to determine the mechanisms of how NK cells are involved in periodontal disease before they could be considered as a potential therapeutic target. Again, care must be taken before extrapolating from animal studies.
NKT Cells in Periodontitis
NKT cells were first described in 1995 where a subset of T lymphocytes were found to have similar characteristics to NK cells (147). They are directly activated by the binding of glycolipid antigens with the CD1d receptor expressed by APCs (148). The interest in NKT cells has gradually increased because they have been demonstrated to have important immunoregulatory functions through the production of pro-inflammatory and anti-inflammatory cytokines (149). However, most current research in NKT cells is in mice, with minimal studies in humans.
In vitro and in vivo studies have demonstrated a possible role of NKT cells in the pathogenesis of periodontitis. Periodontal pathogens such as P. gingivalis, A. actinomycetemcomitans, T. forsythia, and T. denticola can express glycophingolipids in their structure that have the ability to activate NKT cells (150, 151). In addition, after oral inoculation of P. gingivalis and administration of α-galatoxylceramide (α-GalCer, a synthetic glycolipid) in mice, NKT cells were found to increase systemic inflammation, RANKL production, osteoclastogenesis and alveolar bone loss. Intriguingly, when periodontitis was induced in mice without NKT cells, these mice showed less alveolar bone loss (152). In humans, gingival biopsies have shown a higher expression of NKT cells in periodontitis patients compared with gingivitis patients (151, 153). These early observations suggest a link between NKT cells and periodontitis; however, further human studies are required to strengthen this link.
NKT cells have been suggested to regulate possible autoimmunity in periodontal disease. Cross reactivity of human heat shock protein (HSP) 60 and P. gingivalis GroEL (a bacterial homolog) has been reported in periodontal disease (154, 155). In addition, it has been suggested that NKT cells may control the immune response to autoantigens such as collagen type I or HSP60 (153). However, the role of autoimmunity in periodontal disease is yet to be understood. While there is evidence showing autoimmune responses may be involved in the disease process, further studies are required to elucidate the role of autoimmunity in periodontal disease.
More recently, a subset of NKT cells called NK10 cells have been described due to its production of IL-10 (156). The ability to produce IL-10 suggests that NKT have an immunoregulatory function by increasing clonal expansion and activity of T-reg cells and M2 macrophages. This characteristic has piqued interest in its potential to be targeted for certain inflammatory diseases. Evidence has shown that NKT10 cells protect mice from autoimmune encephalomyelitis by inhibiting the pathogenicity of Th1 cells in the central nervous system (157). Furthermore, a SLE mice model study suggested that NK10 cells could have a protective role in SLE by regulating the production of autoreactive IgG by CD1d+ B cells (158). In a theoretical sense, the activation of NKT10 cells could create an anti-inflammatory environment for certain diseases, however, there have not been any studies to date attempting to understand this potential target in relation to periodontal disease.
Macrophages
Macrophages participate in the development of the gingivitis lesion. It is proposed that a strong innate immune response leads to the production of high levels of IL-12 by both PMNs and macrophages, which in turn leads to a Th1 response, cell-mediated immunity, protective antibody, and a stable periodontal lesion. The production of IFN-γ by the activated CD4 T cells further activates PMNs and macrophages. Macrophages are a non-dominant feature in periodontitis, comprising fewer than 5% of the cells (159). There have been mixed reports regarding the role of macrophages in periodontitis with some evidence suggesting that there are minimal changes in macrophage numbers when comparing advanced periodontitis and periodontal health, with little evidence of activation (160, 161). This however does not preclude their role in the pathogenic process, as quantitative measurements do not necessarily correlate to significance (66).
Macrophage Polarization
Macrophage polarization refers to an estimate of macrophage activation at a given point in space and time. Macrophages are highly plastic and may change under the influence of intrinsic factors such as epigenetics and extrinsic factors such as microbial products and cytokines which determine the tissue micro-environment (162, 163). In a simplistic point of view, two macrophage phenotypes have been identified, namely M1 and M2. The classical M1 phenotype, produces mainly pro-inflammatory cytokines (IL-6 and TNF-α), whereas M2 is thought to play a role in inflammation resolution and promotion of healing, producing comparatively higher amounts of IL-10 (164), low levels of IL-6 (165) and participating in the synthesis of specialized pro-resolving mediators (166).
While in the developing lesion most macrophages are phagocytic cells, in established gingivitis the major APC is the CD14-positive/CD83-positive dendritic cell (167), with fewer classical proinflammatory M1 macrophages compared with the alternative prohealing M2 macrophages (168). A well controlled study by Garaicoa-Pazmino et al. (168) investigated the polarization of M1 and M2 macrophages in gingivitis and in periodontitis. This study found that there were more macrophages in gingivitis samples compared with periodontitis, yet the M1:M2 ratio was similar in both lesions.
Other studies, however, have indicated an increased M1/M2 ratio in periodontitis compared with gingivitis (169, 170). Although the dichotomous model of M1/M2 polarization provides a good outline, this categorization is likely an oversimplification that describes the extremes of what is more likely a continuum of activation states (171, 172). Clarifying this phenotypic diversity during periodontitis is essential to our understanding of the role of macrophages in the pathogenic process and consequently host modulation targeting of macrophages.
Attempts to target macrophages as part of host modulation have been directed at altering the balance of macrophage polarization, suppressing M1 macrophages and the associated pro-inflammatory cytokines, which include PGE2, TNF-α, IL-1 and IL-6, as well as stimulating M2 macrophages and consequent production of anti-inflammatory factors such as IL-10 and IFN-γ (173).
Anti-cytokine therapy has been utilized to inhibit downstream M1 pro-inflammatory cytokines, although studies examining the periodontal implications of such treatment have been limited. Two such agents are infliximab (chimeric mAb to TNF-α) and etanercept (soluble form of TNF receptor). Pers et al. showed perplexingly both an increase in gingival inflammation and decreased attachment loss (133). This may indicate a transition from a progressive periodontitis to stable gingivitis. A systematic review examining the effect of anti-TNF-α therapy in rheumatoid arthritis subjects with periodontitis showed studies had produced inconsistent results (174). A more recent review however by Zamri and colleagues supported improvements in clinical parameters over time (175). Tocilizumab, a recombinant mAb which inhibits IL-6, has also been shown to produce a small statistically significant improvement in PPD and CAL after 6 months (176).
Understanding how anti-cytokine therapy may modify a patient's response to periodontal pathogens may allow us to prescribe interceptive periodontal treatment prior to anti-cytokine therapy initiation. Consistent with this are results from a study indicating an increased risk of etanercept discontinuation in patients diagnosed with periodontitis 5 years prior to or during etanercept treatment (177). Utility of anti-cytokine therapy is currently limited by cost, functional redundancy in the cytokine network and unacceptable side-effects (178). These limitations need to be addressed to make anti-cytokine therapy viable in the treatment of periodontitis.
Specialized pro-resolving mediators (SPMs) are agonists, which initiate proresolving pathways, terminating inflammation and promoting tissue homeostasis (179). As well as reversing dysbiosis and preventing PMN transmigration there is evidence that SPMs increase M2-like healing macrophages, which promotes phagocytosis of bacteria and apoptotic PMNs (180). Resolvin E1 (RvE1) has been shown in other inflammatory diseases to induce M2 macrophages (181, 182). Furthermore, macrophages from localized aggressive periodontitis patients showed reduced phagocytosis which was reversed by RvE1 (183). Consequently SPMs show promise as possible host modulation agents targeting multiple aspects of the pathogenic process including macrophage function, however the inhibition of PMN transmigration and hence the development of a PMN barrier in the gingival sulcus may represent a problem.
Mast Cells
Mast cells are resident cells of the connective tissue and contain granules rich in histamine and heparin. During periodontal disease, lipoteichoic acid and peptidoglycans from early bacterial colonizers activate complement via the ‘alternative pathway'. The production of anaphylatoxins C3a and C5a results in the release of vasoactive amines from resident mast cells and consequently leads to vascular permeability and formation of edema. In addition, mast cells can release cytokines, serine proteases, mast cell extracellular traps (184), and express MMPs (185).
Mast cells play a key role in the inflammatory process and have been found to be present in both healthy and inflamed gingival tissues (186–188). However, studies examining the proportion of mast cells within gingival tissues have been conflicting. Batista et al. found increased mast cells in chronic periodontitis/gingivitis lesions compared with clinically healthy gingival tissue (189), while Gemmell et al. found decreased numbers in chronic periodontitis tissues compared with healthy/gingivitis samples (190). Although different case definitions may have contributed to this discrepancy it is clear that mast cells play an important role not only in the initial but also the chronic stages of periodontal disease.
Many cytokines have been identified in mast cell lines or in vivo-derived mast cells, including SCF, TGF-β1, TNF-α, IL-4, IL-5, IL-6, IL-15, IL-17, bFGF, and VEGF (191). Mast cells have been reported to release a substantial amount of TNF-α without obvious stimulation, while macrophages, T and B cells contain little or no preformed TNF-α bioactivity without stimulation. In a model of acute septic peritonitis, blocking TNF-α suppressed the protective role of mast cells via reduced PMN influx (192). This study indicates the importance of mast cell derived TNF-α in inflammation and host defense. It is interesting to note that TGF-β1 released by mast cells is a potent chemoattractant for leukocytes including PMNs, monocytes and mast cells (193), and that 50% of gingival mast cells produce TGF-β1 (194).
Another possible role of mast cells within periodontal disease is the presence of serine proteases, tryptase and chymase, which have the potential to mediate breakdown of the extracellular matrix. Mast cell-derived tryptase is a potent chemoattractant for PMNs (195) promoting IL-8 secretion, increasing epithelial expression of intercellular adhesion molecule-1 (196) and inducing expression of mRNA for IL-1β and IL-8 in endothelial cells (197). Within the gingival tissues mast cells also appear to be the major source of IL-17 (86, 87) which as noted above leads to the production of IL-8 and continued PMN migration. Chymase can activate latent IL-1β (198) or MMP-9 (199). However, the biological significance of these proteases is still unknown. Small quantities of tryptase and chymase have been found in the extracellular matrix in healthy gingiva (185, 200), which may indicate periodontal lesion stability. In this context, however, it must be remembered that capability does not indicate function within the specific tissue micro-environment.
There has been limited research examining mast cells as a possible host modulation target. Jeffcoat et al. evaluated the use of iodoxamide ethyl, a mast cell release inhibitor, in beagle dogs (186). Following oral treatment with iodoxamide ethyl for 12 months, the rate of alveolar bone loss was significantly reduced compared with untreated controls. Additional benefit was observed in combination with periodontal flap surgery. Although this study suggests a possible benefit in targeting mast cell degranulation, further research is required to understand the role of mast cells in periodontitis before they can be considered a potential host modulation target.
Conclusion
In conclusion, we must address the limitations of this review. Examining components of the innate and adaptive immune response separately is artificial and ignores the complex interactions between different components and overlapping functions. This discussion has also been limited to the principal immune cells and their cytokines, and as such overlooks other cellular functions and aspects of the immune system which are also likely to play significant roles in both the protective and pathogenic responses. This review, however, highlights the current heterogeneity and deficiencies in our understanding of immunopathogenesis, which will need to be addressed when developing host modulation agents. Although immunomodulation in periodontal disease offers exciting prospects for understanding the disease and its treatment, researchers must remain cognizant of the fact that the there is a high degree of overlap, redundancy and even plasticity in the immune system (Figure 1). Furthermore, it is becoming increasingly clear that not all individuals respond in the same way, resulting in individuality of disease expression. The targeting of a single molecule or mediator, or indeed a single cell type therefore, may not result in the expected disease outcome in all individuals. Notwithstanding this caveat however, further research in all aspects of the immune system and its potential for immunomodulation in the treatment of periodontal disease is essential. In this context, the present review (summarized in Table 1) highlights that care must always be taken when extrapolating from animal studies to human trials with the principle of “do no harm” being paramount.
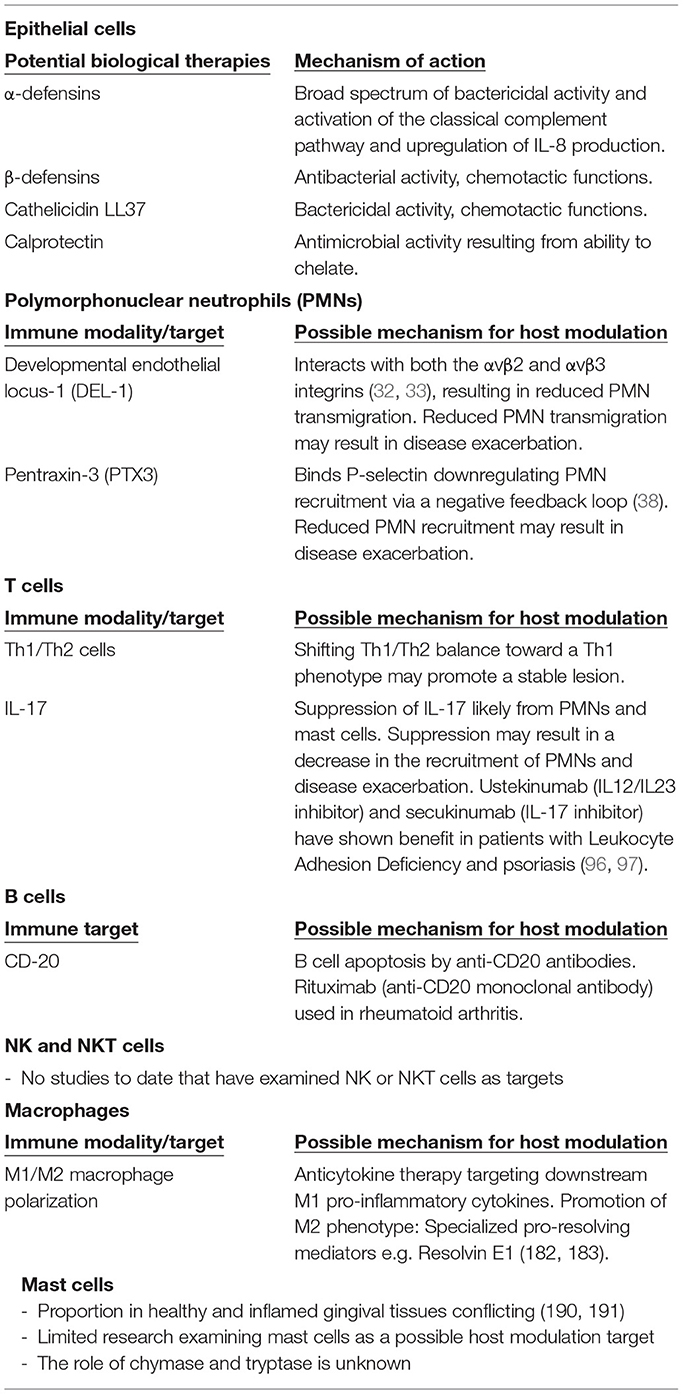
Table 1. Potential biological therapies, immune targets and their mechanisms.
Author Contributions
SQ drafted and edited the manuscript. AZ drafted the manuscript. RL was involved in concept development and editing the manuscript drafts. GS was involved in concept development and editing the manuscript drafts. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tonetti MS, Greenwell H, Kornman KS. Staging and grading of periodontitis: framework and proposal of a new classification and case definition. J Periodontol. (2018) 89:S159–S72. doi: 10.1002/jper.18-0006
2. Chen MX, Zhong YJ, Dong QQ, Wong HM, Wen YF. Global, regional, and national burden of severe periodontitis, 1990-2019: an analysis of the global burden of disease study 2019. J Clin Periodontol. (2021) 48:1165–88. doi: 10.1111/jcpe.13506
3. Vos T, Lim SS, Abbafati C, Abbas KM, Abbasi M, Abbasifard M, et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet. (2020) 396:1204–22. doi: 10.1016/s0140-6736(20)30925-9
4. Falcao A, Bullón P. A review of the influence of periodontal treatment in systemic diseases. Periodontology. (2000) 79:117–28. doi: 10.1111/prd.12249
5. Chapple ILC, Genco R. Diabetes and periodontal diseases: consensus report of the joint Efp/Aap workshop on periodontitis and systemic diseases. J Periodontol. (2013) 84:S106–12. doi: 10.1902/jop.2013.1340011
6. Seymour GJ. Importance of the host response in the periodontium. J Clin Periodontol. (1991) 18:421–6. doi: 10.1111/j.1600-051x.1991.tb02310.x
7. Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontology 2000. (2005) 38:135–87. doi: 10.1111/j.1600-0757.2005.00107.x
8. Van Dyke TE, Bartold PM, Reynolds EC. The nexus between periodontal inflammation and dysbiosis. Front Immunol. (2020) 11:00511. doi: 10.3389/fimmu.2020.00511
9. Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the Polymicrobial Synergy and Dysbiosis (Psd) model of periodontal disease etiology. Mol Oral Microbiol. (2012) 27:409–19. doi: 10.1111/j.2041-1014.2012.00663.x
10. Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. (1994) 8:263–71. doi: 10.1177/08959374940080022001
11. Heitz-Mayfield L, Trombelli L, Heitz F, Needleman I, Moles D. A systematic review of the effect of surgical debridement vs. non-surgical debridement for the treatment of chronic periodontitis. J Clin Periodontol. (2002) 29:92–102. doi: 10.1034/j.1600-051x.29.s3.5.x
12. Jepsen K, Jepsen S. Antibiotics/Antimicrobials: systemic and local administration in the therapy of mild to moderately advanced periodontitis. Periodontology 2000. (2016) 71:82–112. doi: 10.1111/prd.12121
13. Balta MG, Papathanasiou E, Blix IJ, Van Dyke TE. Host modulation and treatment of periodontal disease. J Dent Res. (2021) 100:798–809. doi: 10.1177/0022034521995157
14. Corbella S, Calciolari E, Alberti A, Donos N, Francetti L. Systematic review and meta-analysis on the adjunctive use of host immune modulators in non-surgical periodontal treatment in healthy and systemically compromised patients. Scientific Reports. (2021) 11:12125. doi: 10.1038/s41598-021-91506-7
15. Donos N, Calciolari E, Brusselaers N, Goldoni M, Bostanci N, Belibasakis GN. The adjunctive use of host modulators in non-surgical periodontal therapy. a systematic review of randomized, placebo-controlled clinical studies. J Clin Periodontol. (2020) 47:199–238. doi: 10.1111/jcpe.13232
16. Ganz T. Defensins: antimicrobial peptides of innateiImmunity. Nat Rev Immunol. (2003) 3:710–20. doi: 10.1038/nri1180
17. Komatsuzawa H, Ouhara K, Kawai T, Yamada S, Fujiwara T, Shiba H, et al. Susceptibility of periodontopathogenic and cariogenic bacteria to defensins and potential therapeutic use of defensins in oral diseases. Curr Pharm Des. (2007) 13:3084–95. doi: 10.2174/138161207782110426.
18. Chung OW, Dommisch H, Yin L, Dale AB. Expression of defensins in gingiva and their role in periodontal health and disease. Curr Pharm Des. (2007) 13:3073–83. doi: 10.2174/138161207782110435
19. Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. a summary of current work. Lab Invest. (1976) 34:235–49.
20. Lindhe J, Rylander H. Experimental gingivitis in young dogs. Eur J Oral Sci. (1975) 83:314–26. doi: 10.1111/j.1600-0722.1975.tb00444.x
21. Seymour GJ BT, Trombelli L. Pathogenesis of gingivitis and periodontitis In: Tord Berglundh WVG, Niklaus P. Lang, Mariano Sanz, (editors). Lindhe's Clinical Periodontology and Implant Dentistry. 1. 7th ed. Oxford UK: John Wiley & Sons Ltd (2022). p. 235–62.
22. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
23. Moughal NA, Adonogianaki E, Thornhill MH, Kinane DF. Endothelial cell leukocyte adhesion molecule-1 (Elam-1) and intercellular adhesion molecule-1 (Icam-1) expression in gingival tissue during health and experimentally-induced gingivitis. J Periodontal Res. (1992) 27:623–30. doi: 10.1111/j.1600-0765.1992.tb01746.x
24. Attström R. Studies on Neutrophil polymorphonuclear leukocytes at the dento-gingival junction in gingival health and disease. J Periodontal Res Suppl. (1971) 8:1–15.
25. Takahashi N, Okui T, Tabeta K, Yamazaki K. Effect of interleukin-17 on the expression of chemokines in gingival epithelial cells. Eur J Oral Sci. (2011) 119:339–44. doi: 10.1111/j.1600-0722.2011.00842.x
26. Ryder MI. Comparison of neutrophil functions in aggressive and chronic periodontitis. Periodontology 2000. (2010) 53:124–37. doi: 10.1111/j.1600-0757.2009.00327.x
27. Hirschfeld J. Neutrophil subsets in periodontal health and disease: a mini review. Front Immunol. (2020) 10:e03001. doi: 10.3389/fimmu.2019.03001
28. Khocht A, Albandar JM. Aggressive forms of periodontitis secondary to systemic disorders. Periodontology. 2000. (2014) 65:134–48. doi: 10.1111/prd.12015
29. Hanna S, Etzioni A. Leukocyte adhesion deficiencies. Ann N Y Acad Sci. (2012) 1250:50–5. doi: 10.1111/j.1749-6632.2011.06389.x
30. Roberts H, White P, Dias I, McKaig S, Veeramachaneni R, Thakker N, et al. Characterization of neutrophil function in papillon-lefèvre syndrome. J Leukoc Biol. (2016) 100:433–44. doi: 10.1189/jlb.5a1015-489r
31. Delcourt-Debruyne EMC, Boutigny HRA, Hildebrand HF. Features of severe periodontal disease in a teenager with chédiak-higashi syndrome. J Periodontol. (2000) 71:816–24. doi: 10.1902/jop.2000.71.5.816
32. Hidai C, Zupancic T, Penta K, Mikhail A, Kawana M, Quertermous EE, et al. Cloning and characterization of developmental endothelial locus-1: an embryonic endothelial cell protein that binds the alpha Vbeta 3 integrin receptor. Genes Dev. (1998) 12:21–33. doi: 10.1101/gad.12.1.21
33. Choi EY, Chavakis E, Czabanka MA, Langer HF, Fraemohs L, Economopoulou M, et al. Del-1, an Endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science. (2008) 322:1101–4. doi: 10.1126/science.1165218
34. Eskan MA, Jotwani R, Abe T, Chmelar J, Lim J-H, Liang S, et al. The leukocyte integrin antagonist Del-1 inhibits Il-17-mediated inflammatory bone loss. Nat Immunol. (2012) 13:465–73. doi: 10.1038/ni.2260
35. Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, et al. Del-1 Promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol. (2019) 20:40–9. doi: 10.1038/s41590-018-0249-1
36. Shin J, Maekawa T, Abe T, Hajishengallis E, Hosur K, Pyaram K, et al. Del-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Science Trans Medicine. (2015) 7:307ra155-307ra1. doi: 10.1126/scitranslmed.aac5380
37. Gümüş P, Nizam N, Nalbantsoy A, Özçaka Ö, Buduneli N. Saliva and serum levels of pentraxin-3 and interleukin-1β in generalized aggressive or chronic periodontitis. J Periodontol. (2014) 85:e40–e6. doi: 10.1902/jop.2013.130281
38. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of leukocyte recruitment by the long pentraxin Ptx3. Nat Immunol. (2010) 11:328–34. doi: 10.1038/ni.1854
39. Ivanyi L, Lehner T. Stimulation of lymphocyte transformation by bacterial antigens in patients with periodontal disease. Arch Oral Biol. (1970) 15:1089–96. doi: 10.1016/0003-9969(70)90121-4
40. Ivanyi L, Lehner T. Lymphocyte transformation by sonicates of dental plaque in human periodontal disease. Arch Oral Biol. (1971) 16:1117–21. doi: 10.1016/0003-9969(71)90216-0
41. Seymour GJ, Crouch MS, Powell RN, Brooks D, Beckman I, Zola H, et al. The identification of lymphoid cell subpopulations in sections of human lymphoid tissue and gingivitis in children using monoclonal antibodies. J Periodontal Res. (1982) 17:247–56. doi: 10.1111/j.1600-0765.1982.tb01151.x
42. Seymour GJ, Crouch MS, Powell RN. The phenotypic characterization of lymphoid cell subpopulations in gingivitis in children. J Periodontal Res. (1981) 16:582–92. doi: 10.1111/j.1600-0765.1981.tb02020.x
43. Longhurst P, Johnson NW, Hopps RM. Differences in lymphocyte and plasma cell densities in inflamed gingiva from adults and young children. J Periodontol. (1977) 48:705–10. doi: 10.1902/jop.1977.48.11.705
44. Seymour GJ, Powell RN, Cole KL, Aitken JF, Brooks D, Beckman I, et al. Experimental gingivitis in humans. a histochemical and immunological characterization of the lymphoid cell subpopulations. J Periodontal Res. (1983) 18:375–85. doi: 10.1111/j.1600-0765.1983.tb00373.x
45. Mackler BF, Frostad KB, Robertson PB, Levy BM. Immunoglobulin bearing lymphocytes and plasma cells in human periodontal disease. J Periodontal Res. (1977) 12:37–45. doi: 10.1111/j.1600-0765.1977.tb00107.x
46. Seymour GJ, Greenspan JS. The phenotypic characterization of lymphocyte subpopulations in established human periodontal disease. J Periodontal Res. (1979) 14:39–46. doi: 10.1111/j.1600-0765.1979.tb00216.x
47. Seymour GJ, Dockrell HM, Greenspan JS. Enzyme differentiation of lymphocyte subpopulations in sections of human lymph nodes, tonsils and periodontal disease. Clin Exp Immunol. (1978) 32:169–78.
48. Poulter LW, Seymour GJ, Duke O, Janossy G, Panayi G. Immunohistological analysis of delayed-type hypersensitivity in man. Cell Immunol. (1982) 74:358–69. doi: 10.1016/0008-8749(82)90036-3
49. Seymour GJ, Gemmell E, Walsh LJ, Powell RN. Immunohistological analysis of experimental gingivitis in humans. Clin Exp Immunol. (1988) 71:132–7.
50. Seymour GJ, Powell RN, Davies WI. Conversion of a stable T-cell lesion to a progressive B-Cell lesion in the pathogenesis of chronic inflammatory periodontal disease: an hypothesis. J Clin Periodontol. (1979) 6:267–77. doi: 10.1111/j.1600-051x.1979.tb01930.x
51. Lindhe J, Liljenberg B, Listgarten M. Some microbiological and histopathological features of periodontal disease in man. J Periodontol. (1980) 51:264–9. doi: 10.1902/jop.1980.51.5.264
52. Lappin DF, Koulouri O, Radvar M, Hodge P, Kinane DF. Relative proportions of mononuclear cell types in periodontal lesions analyzed by immunohistochemistry. J Clin Periodontol. (1999) 26:183–9. doi: 10.1034/j.1600-051x.1999.260309.x
53. Berglundh T, Wellfelt B, Liljenberg B, Lindhe J. Some local and systemic immunological features of prepubertal periodontitis. J Clin Periodontol. (2001) 28:113–20. doi: 10.1034/j.1600-051x.2001.028002113.x
54. Seymour GJ, Gemmell E, Reinhardt RA, Eastcott J, Taubman MA. Immunopathogenesis of chronic inflammatory periodontal disease: cellular and molecular mechanisms. J Periodontal Res. (1993) 28:478–86. doi: 10.1111/j.1600-0765.1993.tb02108.x
55. Orozco A, Gemmell E, Bickel M, Seymour GJ. Interleukin-1 beta, interleukin-12 and interleukin-18 levels in gingival fluid and serum of patients with gingivitis and periodontitis. Oral Microbiol Immunol. (2006) 21:256–60. doi: 10.1111/j.1399-302x.2006.00292.x
56. Ellis TN, Beaman BL. Interferon-Gamma activation of polymorphonuclear neutrophil function. Immunology. (2004) 112:2–12. doi: 10.1111/j.1365-2567.2004.01849.x
57. Hu X, Ivashkiv LB. Cross-Regulation of signaling pathways by interferon-Γ: implications for immune responses and autoimmune diseases. Immunity. (2009) 31:539–50. doi: 10.1016/j.immuni.2009.09.002
58. Gemmell E, Marshall RI, Seymour GJ. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontology 2000. (1997) 14:112–43. doi: 10.1111/j.1600-0757.1997.tb00194.x
59. Gemmell E, Yamazaki K, Seymour GJ. The role of T cells in periodontal disease: homeostasis and autoimmunity. Periodontol 2000. (2007) 43:14–40. doi: 10.1111/j.1600-0757.2006.00173.x
60. Berglundh T, Donati M. Aspects of adaptive host response in periodontitis. J Clin Periodontol. (2005) 32:87–107. doi: 10.1111/j.1600-051X.2005.00820.x
61. Sigusch B, Klinger G, Glockmann E, Simon HU. Early-Onset and adult periodontitis associated with abnormal cytokine production by activated T lymphocytes. J Periodontol. (1998) 69:1098–104. doi: 10.1902/jop.1998.69.10.1098
62. Gemmell E, Grieco DA, Cullinan MP, Westerman B, Seymour GJ. The proportion of interleukin-4, interferon-gamma and interleukin-10-positive cells in porphyromonas gingivalis -specific T-cell lines established from P. Gingivalis -Positive Subjects Oral Microbiol Immunol. (1999) 14:267–74. doi: 10.1034/j.1399-302x.1999.140501.x
63. Gemmell E, Seymour GJ. Cytokine profiles of cells extracted from humans with periodontal diseases. J Dent Res. (1998) 77:16–26. doi: 10.1177/00220345980770010101
64. Liljenberg B, Lindhe J, Berglundh T, Dahlen G, Jonsson R. Some microbiological, histopathological and immunohistochemical characteristics of progressive periodontal disease. J Clin Periodontol. (1994) 21:720–7. doi: 10.1111/j.1600-051x.1994.tb00793.x
65. Reinhardt RA, Bolton RW, McDonald TL, Dubois LM, Kaidahl WB. In Situ lymphocyte subpopulations from active vs. stable periodontal sites. J Periodontol. (1988) 59:656–70. doi: 10.1902/jop.1988.59.10.656
66. Seymour GJ, Taylor JJ. Shouts and whispers: an introduction to immunoregulation in periodontal disease. Periodontol 2000. (2004) 35:9–13. doi: 10.1111/j.0906-6713.2004.003555.x
67. Buchbinder EI, Dutcher JP, Daniels GA, Curti BD, Patel SP, Holtan SG, et al. Therapy with high-dose interleukin-2 (Hd Il-2) in metastatic melanoma and renal cell carcinoma following Pd1 or Pdl1 inhibition. J ImmunoTherapy Cancer. (2019) 7:49. doi: 10.1186/s40425-019-0522-3
68. Davar D, Ding F, Saul M, Sander C, Tarhini AA, Kirkwood JM, et al. High-Dose interleukin-2 (Hd Il-2) for advanced melanoma: a single center experience from the university of pittsburgh cancer institute. J ImmunoTherapy Cancer. (2017) 5:74. doi: 10.1186/s40425-017-0279-5
69. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local Il-17–driven inflammatory bone loss. Science Trans Med. (2014) 6:229ra40–ra40. doi: 10.1126/scitranslmed.3007696
70. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of Cd4 T cells regulates tissue inflammation by producing Interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
71. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (Il)-22 and Il-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
72. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat Immunol. (2007) 8:950–7. doi: 10.1038/ni1497
73. Park Y-D, Kim Y-S, Jung Y-M, Lee S-I, Lee Y-M, Bang J-B, et al. Porphyromonas gingivalis lipopolysaccharide regulates interleukin (Il)-17 and Il-23 expression via sirt1 modulation in human periodontal ligament cells. Cytokine. (2012) 60:284–93. doi: 10.1016/j.cyto.2012.05.021
74. Hueber AJ, Asquith DL, Miller AM, Reilly J, Kerr S, Leipe J, et al. Cutting edge: mast cells express Il-17a in rheumatoid arthritis synovium. J Immunol. (2010) 184:3336–40. doi: 10.4049/jimmunol.0903566
75. Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, et al. Cutting edge: Nkt cells constitutively express Il-23 receptor and rorγt and rapidly produce Il-17 upon receptor ligation in an Il-6-independent fashion. J Immunol. (2008) 180:5167–71. doi: 10.4049/jimmunol.180.8.5167
76. Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. Il-17, Produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: Il-15 as a possible trigger. J Immunol. (2003) 170:2106–12. doi: 10.4049/jimmunol.170.4.2106
77. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. Il-17 in Synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Inves. (1999) 103:1345–52. doi: 10.1172/jci5703
78. Settem RP, Honma K, Nakajima T, Phansopa C, Roy S, Stafford GP, et al. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol. (2013) 6:415–26. doi: 10.1038/mi.2012.85
79. Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. An essential role for Il-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires Il-17 receptor–dependent signals. Blood. (2007) 109:3794–802. doi: 10.1182/blood-2005-09-010116
80. Kelly MN, Kolls JK, Happel K, Schwartzman JD, Schwarzenberger P, Combe C, et al. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against toxoplasma gondii infection. Infect Immun. (2005) 73:617–21. doi: 10.1128/iai.73.1.617-621.2005
81. Gemmell E, Drysdale KE, Seymour GJ. Gene expression in splenic Cd4 and Cd8 cells from Balb/C mice immunized with porphyromonas gingivalis. J Periodontol. (2006) 77:622–33. doi: 10.1902/jop.2006.050211
82. Gemmell E, Bird PS, Bowman JJ, Xu L, Polak B, Walsh LJ, et al. Immunohistological study of lesions induced by porphyromonas gingivalis in a murine model. Oral Microbiol Immunol. (1997) 12:288–97. doi: 10.1111/j.1399-302x.1997.tb00393.x
83. Oda T, Yoshie H, Yamazaki K. Porphyromonas gingivalis antigen preferentially stimulates T cells to express Il-17 but not receptor activator of Nf-?b ligand in vitro. Oral Microbiol Immunol. (2003) 18:30–6. doi: 10.1034/j.1399-302x.2003.180105.x
84. Cheng W-C, Hughes FJ, Taams LS. The presence, function and regulation of Il-17 and Th17 cells in periodontitis. J Clin Periodontol. (2014) 41:541–9. doi: 10.1111/jcpe.12238
85. Thorbert-Mros S, Larsson L, Kalm J, Berglundh T. Interleukin-17–producing T cells and interleukin-17 Mrna expression in periodontitis and long-standing gingivitis lesions. J Periodontol. (2019) 90:516–21. doi: 10.1002/jper.18-0326
86. Parachuru VPB, Coates DE, Milne TJ, Rich AM, Seymour GJ. Foxp3(+) regulatory T cells, interleukin 17 and mast cells in chronic inflammatory periodontal disease. J Periodontal Res. (2018) 53:622–35. doi: 10.1111/jre.12552
87. Parachuru VPB, Coates DE, Milne TJ, Hussaini HM, Rich AM, Seymour GJ. Forkhead box P3-positive regulatory T-cells and interleukin 17-positive T-helper 17 cells in chronic inflammatory periodontal disease. J Periodontal Res. (2014) 49:817–26. doi: 10.1111/jre.12169
88. Moran EM, Heydrich R, Ng CT, Saber TP, McCormick J, Sieper J, et al. Il-17a expression is localised to both mononuclear and polymorphonuclear synovial cell infiltrates. PLoS ONE. (2011) 6:e24048. doi: 10.1371/journal.pone.0024048
89. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release Il-17 through extracellular trap formation in psoriasis. J Immunol. (2011) 187:490–500. doi: 10.4049/jimmunol.1100123
90. Okui T, Aoki Y, Ito H, Honda T, Yamazaki K. The presence of Il-17+/Foxp3+ double-positive cells in periodontitis. J Dent Res. (2012) 91:574–9. doi: 10.1177/0022034512446341
91. Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. (2010) 20:4–12. doi: 10.1038/cr.2009.138
92. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-Hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into Th17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
93. Alvarez C, Suliman S, Almarhoumi R, Vega ME, Rojas C, Monasterio G, et al. Regulatory T cell phenotype and anti-osteoclastogenic function in experimental periodontitis. Scientific Reports. (2020) 10:19018. doi: 10.1038/s41598-020-76038-w
94. Cafferata EA, Terraza-Aguirre C, Barrera R, Faúndez N, González N, Rojas C, et al. Interleukin-35 inhibits alveolar bone resorption by modulating the Th17/Treg imbalance during periodontitis. J Clin Periodontol. (2020) 47:676–88. doi: 10.1111/jcpe.13282
95. Wang L, Wang J, Jin Y, Gao H, Lin X. Oral administration of all-transretinoic acid suppresses experimental periodontitis by modulating the Th17/Treg imbalance. J Periodontol. (2014) 85:740–50. doi: 10.1902/jop.2013.130132
96. Moutsopoulos NM, Zerbe CS, Wild T, Dutzan N, Brenchley L, Dipasquale G, et al. Interleukin-12 and interleukin-23 blockade in leukocyte adhesion deficiency type 1. N Engl J Med. (2017) 376:1141–6. doi: 10.1056/nejmoa1612197
97. Brianti P, Paolino G, Mercuri SR. Successful use and safety of secukinumab in psoriatic patients with periodontitis: a valid therapeutic option. Dermatol Ther. (2020) 33:e13350. doi: 10.1111/dth.13350
98. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
99. Joller N, Lozano E, Patrick, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule tigit selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
100. Zheng Y, Chaudhry A, Kas A, Deroos P, Kim JM, Chu T-T, et al. Regulatory T-cell suppressor program co-opts transcription factor Irf4 to control Th2 responses. Nature. (2009) 458:351–6. doi: 10.1038/nature07674
101. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. Cd4 + regulatory T cells control T H 17 responses in a stat3-dependent manner. Science. (2009) 326:986–91. doi: 10.1126/science.1172702
102. Nakajima T, Ueki-Maruyama K, Oda T, Ohsawa Y, Ito H, Seymour GJ, et al. Regulatory T-cells infiltrate periodontal disease tissues. J Dent Res. (2005) 84:639–43. doi: 10.1177/154405910508400711
103. Aoyagi T, Yamazaki K, Kabasawa-Katoh Y, Nakajima T, Yamashita N, Yoshie H, et al. Elevated Ctla-4 expression on Cd4 T cells from periodontitis patients stimulated with porphyromonas gingivalis outer membrane antigen. Clinical & Experimental Immunology. (2000) 119:280–6. doi: 10.1046/j.1365-2249.2000.01126.x
104. Cardoso CR, Garlet GP, Moreira AP, Júnior WM, Rossi MA, Silva JS. Characterization of Cd4+Cd25+natural regulatory T cells in the inflammatory infiltrate of human chronic periodontitis. J Leukoc Biol. (2008) 84:311–8. doi: 10.1189/jlb.0108014
105. Garlet GP, Cardoso CR, Mariano FS, Claudino M, De Assis GF, Campanelli AP, et al. Regulatory T cells attenuate experimental periodontitis progression in mice. J Clin Periodontol. (2009) 37:591–600. doi: 10.1111/j.1600-051x.2010.01586.x
106. Alvarez C, Abdalla H, Sulliman S, Rojas P, Wu Y-C, Almarhoumi R, et al. Rve1 impacts the gingival inflammatory infiltrate by inhibiting the T cell response in experimental periodontitis. Front Immunol. (2021) 12:664756. doi: 10.3389/fimmu.2021.664756
107. Motta RJG, Almeida LY, Villafuerte KRV, Ribeiro-Silva A, León JE, Tirapelli C. Foxp3+ and Cd25+ cells are reduced in patients with stage Iv, grade C periodontitis: a comparative clinical study. J Periodontal Res. (2020) 55:374–80. doi: 10.1111/jre.12721
108. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce Cd4+Cd25–Foxp3– T cells or are self-induced to become Th17 cells in the absence oe Exogenous Tgf-?. J Immunol. (2007) 178:6725–9. doi: 10.4049/jimmunol.178.11.6725
109. Berglundh T, Liljenberg B, Lindhe J. Some cytokine profiles of T-helper cells in lesions of advanced periodontitis. J Clin Periodontol. (2002) 29:705–9. doi: 10.1034/j.1600-051x.2002.290807.x
110. Zitzmann NU, Berglundh T, Lindhe J. Inflammatory lesions in the gingiva following resective/non-resective periodontal therapy. J Clin Periodontol. (2005) 32:139–46. doi: 10.1111/j.1600-051x.2005.00649.x
111. Teng Y-TA. The role of acquired immunity and periodontal disease progression. Crit Rev Oral Biol Med. (2003) 14:237–52. doi: 10.1177/154411130301400402
112. Berglundh T, Zitzmann NU, Donati M. Are peri-implantitis lesions different from periodontitis lesions? J Clin Periodontol. (2011) (38 Suppl. 11):188–202. doi: 10.1111/j.1600-051X.2010.01672.x
113. Bick PH, Carpenter AB, Holdeman LV, Miller GA, Ranney RR, Palcanis KG, et al. Polyclonal B-cell activation induced by extracts of gram-negative bacteria isolated from periodontally diseased sites. Infect Immun. (1981) 34:43–9. doi: 10.1128/iai.34.1.43-49.1981
114. Meghji S, Henderson B, Wilson M. Higher-Titer antisera from patients with periodontal disease inhibit bacterial capsule-induced bone breakdown. J Periodontal Res. (1993) 28:115–21. doi: 10.1111/j.1600-0765.1993.tb01058.x
115. Wheeler TT, McArthur WP, Magnusson I, Marks RG, Smith J, Sarrett DC, et al. Modeling the relationship between clinical, microbiologic, and immunologic parameters and alveolar bone levels in an elderly population. J Periodontol. (1994) 65:68–78. doi: 10.1902/jop.1994.65.1.68
116. Hirsch HZ, Tarkowski A, Miller EJ, Gay S, Koopman WJ, Mestecky J. Autoimmunity to collagen in adult periodontal disease. J Oral Pathology Med. (1988) 17:456–9. doi: 10.1111/j.1600-0714.1988.tb01315.x
117. Wassenaar A, Reinhardus C, Thepen T, Abraham-Inpijn L, Kievits F. Cloning, characterization, and antigensSpecificity of T-lymphocyte subsets extracted from gingival tissue of chronic adult periodontitis patients. Infect Immun. (1995) 63:2147–53. doi: 10.1128/iai.63.6.2147-2153.1995
118. Sugawara M, Yamashita K, Yoshie H, Hara K. Detection of, and anti-collagen antibody produced by, Cd5-positive B cells in inflamed gingival tissues. J Periodontal Res. (1992) 27:489–98. doi: 10.1111/j.1600-0765.1992.tb01822.x
119. Ebeling SB, Schutte ME, Logtenberg T. The majority of human tonsillar Cd5+ B cells express somatically mutated V Kappa 4 genes. Eur J Immunol. (1993) 23:1405–8. doi: 10.1002/eji.1830230636
120. Afar B, Engel D, Clark EA. Activated lymphocyte subsets in adult periodontitis. J Periodontal Res. (1992) 27:126–33. doi: 10.1111/j.1600-0765.1992.tb01814.x
121. Berglundh T, Liljenberg B, Tarkowski A, Lindhe J. The presence of local and circulating autoreactive B cells in patients with advanced periodontitis. J Clin Periodontol. (2002) 29:281–6. doi: 10.1034/j.1600-051x.2002.290402.x
122. Demoersman J, Pochard P, Framery C, Simon Q, Boisramé S, Soueidan A, et al. B cell subset distribution is altered in patients with severe periodontitis. PLoS ONE. (2018) 13:e0192986. doi: 10.1371/journal.pone.0192986
123. Donati M, Liljenberg B, Zitzmann NU, Berglundh T. B-1a cells and plasma cells in periodontitis lesions. J Periodontal Res. (2009) 44:683–8. doi: 10.1111/j.1600-0765.2008.01178.x
124. Donati M, Liljenberg B, Zitzmann NU, Berglundh T. B-1a cells in experimental gingivitis in humans. J Periodontol. (2009) 80:1141–5. doi: 10.1902/jop.2009.080660
125. Beebe AM, Cua DJ, de Waal Malefyt R. The role of interleukin-10 in autoimmune disease: Systemic Lupus Erythematosus (Sle) and Multiple Sclerosis (Ms). Cytokine Growth Factor Rev. (2002) 13:403–12. doi: 10.1016/s1359-6101(02)00025-4
126. O'Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) Cells are the main source of B cell-derived interleukin 10. Eur J Immunol. (1992) 22:711–7. doi: 10.1002/eji.1830220314
127. Shi T, Jin Y, Miao Y, Wang Y, Zhou Y, Lin X. Il-10 secreting B cells regulate periodontal immune response during periodontitis. Odontology. (2020) 108:350–7. doi: 10.1007/s10266-019-00470-2
128. Wang Y, Yu X, Lin J, Hu Y, Zhao Q, Kawai T, et al. B10 Cells alleviate periodontal bone loss in experimental periodontitis. Infect Immun. (2017) 85:e00335–17. doi: 10.1128/IAI.00335-17
129. Abe T, Alsarhan M, Benakanakere MR, Maekawa T, Kinane DF, Cancro MP, et al. The B cell–stimulatory cytokines blys and april are elevated in human periodontitis and are required for B cell–dependent bone loss in experimental murine periodontitis. J Immunol. (2015) 195:1427–35. doi: 10.4049/jimmunol.1500496
130. Gümüş P, Nizam N, Lappin DF, Buduneli N. Saliva and serum levels of B-Cell activating factors and tumor necrosis factor-? in patients with periodontitis. J Periodontol. (2014) 85:270–80. doi: 10.1902/jop.2013.130117
131. Gümüş P, Buduneli E, Biyikoglu B, Aksu K, Saraç F, Buduneli N, et al. Gingival crevicular fluid and serum levels of april, Baff and Tnf-alpha in rheumatoid arthritis and osteoporosis patients with periodontal disease. Arch Oral Biol. (2013) 58:1302–8. doi: 10.1016/j.archoralbio.2013.07.010
132. Coat J, Demoersman J, Beuzit S, Cornec D, Devauchelle-Pensec V, Saraux A, et al. Anti-B lymphocyte immunotherapy is associated with improvement of periodontal status in subjects with rheumatoidaArthritis. J Clin Periodontol. (2015) 42:817–23. doi: 10.1111/jcpe.12433
133. Pers J-O, Saraux A, Pierre R, Youinou P. Anti–Tnf-? immunotherapy is associated with increased gingival inflammation without clinical attachment loss in subjects with rheumatoid arthritis. J Periodontol. (2008) 79:1645–51. doi: 10.1902/jop.2008.070616
134. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-Year, randomized, controlled trial of belimumab in lupus nephritis. N Eng J Med. (2020) 383:1117–28. doi: 10.1056/nejmoa2001180
135. Atisha-Fregoso Y, Malkiel S, Harris KM, Byron M, Ding L, Kanaparthi S, et al. phase Ii randomized trial of rituximab plus cyclophosphamide followed by belimumab for the treatment of lupus nephritis. Arthritis Rheumatology. (2021) 73:121–31. doi: 10.1002/art.41466
136. Wynne SE, Walsh LJ, Seymour GJ, Powell RN. In Situ demonstration of natural killer (Nk) cells in human gingival tissue. J Periodontol. (1986) 57:699–702. doi: 10.1902/jop.1986.57.11.699
137. James K, Ritchie AW. Do Natural killer cells regulate B-cell activity? Immunol Today. (1984) 5:193–4. doi: 10.1016/0167-5699(84)90219-6
138. Fujita S, Takahashi H, Okabe H, Ozaki Y, Hara Y, Kato I. Distribution of natural killer cells in periodontal diseases: an immunohistochemical study. J Periodontol. (1992) 63:686–9. doi: 10.1902/jop.1992.63.8.686
139. Kopp W. Density and localization of lymphocytes with Natural-Killer (Nk) cell activity in periodontal biopsy specimens from patients with severe periodontitis. J Clin Periodontol. (1988) 15:595–600. doi: 10.1111/j.1600-051x.1988.tb02257.x
140. Kikuchi T, Hahn CL, Tanaka S, Barbour SE, Schenkein HA, Tew JG. Dendritic cells stimulated with actinobacillus actinomycetemcomitans elicit rapid gamma interferon responses by natural killer cells. Infect Immun. (2004) 72:5089–96. doi: 10.1128/iai.72.9.5089-5096.2004
141. Takeda H, Kikuchi T, Soboku K, Okabe I, Mizutani H, Mitani A, et al. Effect of Il-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation. (2014) 37:657–69. doi: 10.1007/s10753-013-9782-0
142. Chaushu S, Wilensky A, Gur C, Shapira L, Elboim M, Halftek G, et al. Direct recognition of fusobacterium nucleatum by the Nk cell natural cytotoxicity receptor Nkp46 aggravates periodontal disease. PLoS Pathog. (2012) 8:e1002601. doi: 10.1371/journal.ppat.1002601
143. Górska R, Gregorek H, Kowalski J, Laskus-Perendyk A, Syczewska M, Madaliński K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J Clin Periodontol. (2003) 30:1046–52. doi: 10.1046/j.0303-6979.2003.00425.x
144. Houri-Haddad Y, Soskolne WA, Shai E, Palmon A, Shapira L. Interferon-Gamma deficiency attenuates local P. gingivalis-induced inflammation. J Dent Res. (2002) 81:395–8. doi: 10.1177/154405910208100608
145. Varma TK, Lin CY, Toliver-Kinsky TE, Sherwood ER. Endotoxin-Induced gamma interferon production: contributing cell types and key regulatory factors. Clin Diagn Lab Immunol. (2002) 9:530–43. doi: 10.1128/cdli.9.3.530-543.2002
146. Wang Y, Zhang W, Xu L, Jin J-O. Porphyromonas gingivalis lipopolysaccharide induced proliferation and activation of natural killer cells in vivo. Molecules. (2016) 21:1086. doi: 10.3390/molecules21081086
147. Bendelac A. Mouse Nk1+ T Cells. Curr Opin Immunol. (1995) 7:367–74. doi: 10.1016/0952-7915(95)80112-x
148. Brutkiewicz RR. Cd1d ligands: the good, the bad, and the ugly. J Immunol. (2006) 177:769. doi: 10.4049/jimmunol.177.2.769
149. Bendelac A, Savage PB, Teyton L. The biology of Nkt cells. Annu Rev Immunol. (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
150. Kinjo Y, Wu D, Kim G, Xing GW, Poles MA, Ho DD, et al. Recognition of bacterial glycosphingolipids by natural killer T cells. Nature. (2005) 434:520–5. doi: 10.1038/nature03407
151. Amanuma R, Nakajima T, Yoshie H, Yamazaki K. Increased infiltration of Cd1d and natural killer T cells in periodontal disease tissues. J Periodontal Res. (2006) 41:73–9. doi: 10.1111/j.1600-0765.2005.00837.x
152. Aoki-Nonaka Y, Nakajima T, Miyauchi S, Miyazawa H, Yamada H, Domon H, et al. Natural killer T cells mediate alveolar bone resorption and a systemic inflammatory response in response to oral infection of mice with porphyromonas gingivalis. J Periodontal Res. (2014) 49:69–76. doi: 10.1111/jre.12080
153. Yamazaki K, Ohsawa Y, Yoshie H. Elevated proportion of natural killer T cells in periodontitis lesions. Am J Pathol. (2001) 158:1391–8. doi: 10.1016/s0002-9440(10)64090-4
154. Ford P, Gemmell E, Walker P, West M, Cullinan M, Seymour G. Characterization of heat shock protein-specific T cells in atherosclerosis. Clin Diagn Lab Immunol. (2005) 12:259–67. doi: 10.1128/cdli.12.2.259-267.2005
155. Tabeta K, Yamazaki K, Hotokezaka H, Yoshie H, Hara K. Elevated humoral immune response to heat shock protein 60 (Hsp60) family in periodontitis patients. Clin Exp Immunol. (2000) 120:285–93. doi: 10.1046/j.1365-2249.2000.01216.x
156. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. Il-10-Producing Nkt10 cells are a distinct regulatory invariant Nkt cell subset. J Clin Invest. (2014) 124:3725–40. doi: 10.1172/jci72308
157. Singh AK, Wilson MT, Hong S, Olivares-Villagómez D, Du C, Stanic AK, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. (2001) 194:1801–11. doi: 10.1084/jem.194.12.1801
158. Yang JQ, Kim PJ, Singh RR. Brief treatment with Inkt cell ligand A-galactosylceramide confers a long-term protection against lupus. J Clin Immunol. (2012) 32:106–13. doi: 10.1007/s10875-011-9590-y
159. Berglundh T, Zitzmann NU, Donati M. Are peri-implantitis lesions different from periodontitis lesions? J Clin Periodontol. (2011) 38:188–202. doi: 10.1111/j.1600-051x.2010.01672.x
160. Zadeh HH, Nichols FC, Miyasaki KT. The role of the cell-mediated immune response to actinobacillus actinomycetemcomitans and porphyromonas gingivalis in periodontitis. Periodontology 2000. (1999) 20:239–88. doi: 10.1111/j.1600-0757.1999.tb00163.x
161. Chapple CC, Srivastava M, Hunter N. Failure of macrophage activation in destructive periodontal disease. J Pathol. (1998) 186:281–6. doi: 10.1002/(sici)1096-9896(1998110)186:3<281::Aid-path200>3.0.Co;2-7.
162. Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van Den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6c(High) monocytes. Cancer Res. (2010) 70:5728–39. doi: 10.1158/0008-5472.can-09-4672
163. Kratochvill F, Neale G, Haverkamp JM, Van de Velde LA, Smith AM, Kawauchi D, et al. Tnf counterbalances the emergence of M2 tumor macrophages. Cell Rep. (2015) 12:1902–14. doi: 10.1016/j.celrep.2015.08.033
164. Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. (2006) 80:1298–307. doi: 10.1189/jlb.0406249
165. Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. (2015) 185:2596–606. doi: 10.1016/j.ajpath.2015.06.001
166. Serhan CN, Yang R, Martinod K, Kasuga K, Pillai PS, Porter TF, et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med. (2009) 206:15–23. doi: 10.1084/jem.20081880
167. Gemmell E, Carter CL, Hart DNJ, Drysdale KE, Seymour GJ. Antigen-Presenting cells in human periodontal disease tissues. Oral Microbiol Immunol. (2002) 17:388–93. doi: 10.1034/j.1399-302x.2002.170609.x
168. Garaicoa-Pazmino C, Fretwurst T, Squarize CH, Berglundh T, Giannobile WV, Larsson L, et al. Characterization of macrophage polarization in periodontal disease. J Clin Periodontol. (2019) 46:830–9. doi: 10.1111/jcpe.13156
169. Yang J, Zhu Y, Duan D, Wang P, Xin Y, Bai L, et al. Enhanced activity of macrophage M1/M2 phenotypes in periodontitis. Arch Oral Biol. (2018) 96:234–42. doi: 10.1016/j.archoralbio.2017.03.006
170. Yu T, Zhao L, Huang X, Ma C, Wang Y, Zhang J, et al. Enhanced activity of the macrophage M1/M2 phenotypes and phenotypic switch to M1 in periodontal infection. J Periodontol. (2016) 87:1092–102. doi: 10.1902/jop.2016.160081
171. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. (2016) 17:34–40. doi: 10.1038/ni.3324
172. Sun X, Gao J, Meng X, Lu X, Zhang L, Chen R. Polarized macrophages in periodontitis: characteristics, function, and molecular signaling. Front Immunol. (2021) 12:763334. doi: 10.3389/fimmu.2021.763334
173. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. (2014) 26:192–7. doi: 10.1016/j.cellsig.2013.11.004
174. Han JY, Reynolds MA. Effect of Anti-Rheumatic agents on periodontal parameters and biomarkers of inflammation: a systematic review and meta-analysis. J Periodontal Implant Sci. (2012) 42:3. doi: 10.5051/jpis.2012.42.1.3
175. Zamri F, de Vries TJ. Use of Tnf inhibitors in rheumatoid arthritis and implications for the periodontal status: for the benefit of both?Front Immunol. (2020) 11:763334. doi: 10.3389/fimmu.2020.591365
176. Kobayashi T, Ito S, Kobayashi D, Kojima A, Shimada A, Narita I, et al. Interleukin-6 receptor inhibitor tocilizumab ameliorates periodontal inflammation in patients with rheumatoid arthritis and periodontitis as well as tumor necrosis factor Inhibitors. Clin Exp Dent Res. (2015) 1:63–73. doi: 10.1002/cre2.11
177. Chen HH, Chen DY, Lai KL, Chen YM, Chou YJ, Chou P, et al. Periodontitis and etanercept discontinuation risk in anti-tumor necrosis factor-naive rheumatoid arthritis patients: a nationwide population-based cohort study. J Clin Rheumatol. (2013) 19:432–8. doi: 10.1097/rhu.0000000000000041
178. Atzeni F, Sarzi-Puttini P. Anti-Cytokine antibodies for rheumatic diseases. Curr Opin Investig Drugs. (2009) 10:1204–11.
179. Van Dyke TE, Sima C. Understanding resolution of inflammation in periodontal diseases: is chronic inflammatory periodontitis a failure to resolve?Periodontology 2000. (2020) 82:205–13. doi: 10.1111/prd.12317
180. Mizraji G, Heyman O, Van Dyke TE, Wilensky A. Resolvin D2 restrains Th1 immunity and prevents alveolar bone loss in murine periodontitis. Front Immunol. (2018) 9:00785. doi: 10.3389/fimmu.2018.00785
181. Sima C, Paster B, Van Dyke TE. Function of pro-resolving lipid mediator resolvin E1 in type 2 diabetes. Crit Rev Immunol. (2018) 38:343–65. doi: 10.1615/critrevimmunol.2018026750
182. González-Périz A, Horrillo R, Ferré N, Gronert K, Dong B, Morán-Salvador E, et al. Obesity-Induced insulin resistance and hepatic steatosis are alleviated by Ω-3 fatty acids: a role for resolvins and protectins. The FASEB Journal. (2009) 23:1946–57. doi: 10.1096/fj.08-125674
183. Fredman G, Oh SF, Ayilavarapu S, Hasturk H, Serhan CN, Van Dyke TE. Impaired phagocytosis in localized aggressive periodontitis: rescue by resolvin E1. PLoS ONE. (2011) 6:e24422. doi: 10.1371/journal.pone.0024422
184. von Köckritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, et al. Phagocytosis-Independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. (2008) 111:3070–80. doi: 10.1182/blood-2007-07-104018
185. Naesse EP, Schreurs O, Helgeland K, Schenck K, Steinsvoll S. Matrix metalloproteinases and their inhibitors in gingival mast cells in persons with and without human immunodeficiency virus infection. J Periodontal Res. (2003) 38:575–82. doi: 10.1034/j.1600-0765.2003.00687.x
186. Jeffcoat MK, Williams RC, Johnson HG, Wechter WJ, Goldhabfr P. Treatment of periodontal disease in beagles with lodoxamide ethyl, an inhibitor of mast cell release. J Periodontal Res. (1985) 20:532–41. doi: 10.1111/j.1600-0765.1985.tb00837.x
187. Carranza FA. Jr., Cabrini RL. Mast cells in human gingiva. Oral Surg Oral Med Oral Pathol. (1955) 8:1093–9. doi: 10.1016/0030-4220(55)90061-x
188. Walsh LJ, Davis MF, Xu LJ, Savage NW. Relationship between mast cell degranulation and inflammation in the oral cavity. J Oral Pathol Med. (1995) 24:266–72. doi: 10.1111/j.1600-0714.1995.tb01180.x
189. Batista A, Rodini C, Lara V. Quantification of mast cells in different stages of human periodontal disease. Oral Dis. (2005) 11:249–54. doi: 10.1111/j.1601-0825.2005.01113.x
190. Gemmell E, Carter CL, Seymour GJ. Mast cells in human periodontal disease. J Dent Res. (2004) 83:384–7. doi: 10.1177/154405910408300506
191. Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol. (2014) 14:478–94. doi: 10.1038/nri3690
192. Echtenacher B, Männel DN, Hültner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. (1996) 381:75–7. doi: 10.1038/381075a0
193. Wahl SM. The Role of Transforming growth factor-beta in inflammatory processes. Immunol Res. (1991) 10:249–54. doi: 10.1007/bf02919701
194. Steinsvoll S, Halstensen TS, Schenck K. Extensive expression of Tgf-B1 in chronically-inflamed periodontal tissue. J Clin Periodontol. (1999) 26:366–73. doi: 10.1034/j.1600-051x.1999.260606.x
195. He S, Peng Q, Walls AF. Potent induction of a neutrophil and eosinophil-rich infiltrate in vivo by human mast cell tryptase: selective enhancement of eosinophil recruitment by histamine. J Immunol. (1997) 159:6216.
196. Cairns JA, Walls AF. Mast cell tryptase is a mitogen for epithelial cells. stimulation of Il-8 production and intercellular adhesion molecule-1 expression. J Immunol. (1996) 156:275–83.
197. Compton SJ, Cairns JA, Holgate ST, Walls AF. The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of Mrna for Il-1 beta and Il-8 and stimulates the selective release of Il-8 from human umbilical vein endothelial cells. J Immunol. (1998) 161:1939–46.
198. Mizutani H, Schechter N, Lazarus G, Black RA, Kupper TS. Rapid and specific conversion of precursor interleukin 1 beta (Il-1 beta) to an active Il-1 species by human mast cell chymase. J Exp Med. (1991) 174:821–5. doi: 10.1084/jem.174.4.821
199. Fang KC, Raymond WW, Blount JL, Caughey GH. Dog mast cell alpha-chymase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92 bonds of the catalytic domain. J Biol Chem. (1997) 272:25628–35. doi: 10.1074/jbc.272.41.25628
Keywords: immunopathogenesis, cell types, cytokines, immunomodulation, clinical potential
Citation: Quach SS, Zhu A, Lee RSB and Seymour GJ (2022) Immunomodulation—What to Modulate and Why? Potential Immune Targets. Front. Dent. Med. 3:883342. doi: 10.3389/fdmed.2022.883342
Received: 24 February 2022; Accepted: 11 May 2022;
Published: 03 June 2022.
Edited by:
Elena Calciolari, University of Parma, ItalyReviewed by:
Aliye Akcali, Dokuz Eylul University, TurkeyStefano Corbella, University of Milan, Italy
Copyright © 2022 Quach, Zhu, Lee and Seymour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory J. Seymour, Zy5zZXltb3VyQHVxLmVkdS5hdQ==