 Myron Zaczkiewicz
Myron Zaczkiewicz Oliver Zimmermann2
Oliver Zimmermann2 Jan Torzewski
Jan Torzewski- 1Department of Cardiology, Mediclin Heart Center Lahr, Lahr, Germany
- 2Department of Cardiology, Cardiovascular Center Oberallgaeu-Kempten, Clinic Association Allgaeu, Kempten, Germany
C-reactive Protein (CRP) is synthesized in the liver. Synthesis is stimulated via the IL-1ß/IL6 pathway. CRP activates the complement system via C1q and macrophages via Fcγ receptors. Since elevated CRP plasma levels are associated with increased cardiovascular risk, CRP may play a causal role in cardiovascular disease. One approach to transfer these observations into standard medical care would be to generate hepatic CRP synthesis inhibitors and use them in controlled clinical trials. Despite huge pharmacological efforts, the search for CRP synthesis inhibitors proved to be difficult. First, the antisense oligonucleotide RNA technology, although a promising idea, has not yet led to results feasible for clinical practice. Secondly, high throughput screening assays in search for hepatic CRP inhibitors were limited by the fact that primary human hepatocytes do not adequately grow in vitro. Use of genetically engineered hepatoma cells led to the observation that cardiac glycosides are capable of inhibiting CRP synthesis. Because of patent law considerations, however, pharmaceutical companies had limited interest in further pursuing this possible path. Upstream inhibition of IL-1ß and IL-6 by antibodies has shown positive results in cardiovascular clinical trials, but because of side effects none of these antibodies has yet received FDA approval. In contrast, long-term colchicine treatment, though not being a CRP-specific approach, has recently been approved by the FDA. Taken together, there is no compelling evidence until today that hepatic CRP synthesis can specifically, effectively and safely be inhibited in vivo in human medicine. Currently, other avenues appear more promising. Here, we summarize contemporary approaches to inhibit CRP synthesis and potential goals for future clinical trials.
1 Introduction
C-reactive protein (CRP) is an acute phase protein consisting of five identical, non-covalently bound subunits and belongs to the pentraxin family (Gang et al., 2012; Du Clos, 2013). Each subunit contains a calcium-dependent phosphocholine binding site (Thompson et al., 1999). Following phosphocholine binding, a change in CRP conformation leads to C1q+ binding and, consequently, activation of the classical complement pathway (Roux et al., 1983; Volanakis, 2001; Du Clos, 2013). Unlike complement activation via antibodies, CRP-mediated complement activation is reported to result in almost complete consumption of C1, C4, C2 and (partially) C3 but only in a low activation level of terminal membrane attack complex mC5b-9 (Kaplan and Volanakis, 1974). CRP also interacts with Fcγ receptors, especially FcγRIIa, on monocytes and neutrophiles leading to opsonisation, phagocytosis and cytokine release (Bharadwaj et al., 1999; Stein et al., 2000; Manolov et al., 2004; Lu et al., 2008). From an evolutionary genetic standpoint CRP is one of the oldest and most unspecific parts of the innate human immune system being a pattern recognition molecule binding to molecules exposed during cell death and expressed on the surfaces of pathogens (Black et al., 2004)
In humans, CRP is mainly synthesized in hepatic cells through activation at the transcriptional level by a variety of pathways. A low baseline production is induced by liver specific transcription factor NF-1 (Hurlimann et al., 1966). In case of sterile or infectious danger signals (damage-associated molecular patterns, pathogen-associated molecular patterns) caused by external triggers (trauma, tissue damage, autoimmune disorders, infection), innate immune cell pattern recognition receptors (PRRs) are activated (Latz et al., 2013). A great variety of different tissue damaging patterns induce inflammasome formation, the most prominent being nucleotide oligomerization domain-, leucine-rich repeat-, and pyrin domain containing protein (3NLPR3) inflammasome. Activated inflammasomes then act as a large multimolecular signaling platform and thereby induce maturation and release of cytokines, especially pro interleukin (IL)-1ß and IL-18 (Próchnicki et al., 2016).
Pro IL-1ß is mainly a product of tissue macrophages, dendritic cells and blood monocytes and needs intracellular caspase 1 (also activated by inflammasomes) in order to be transferred into the active form IL-1ß (Martinon et al., 2009; Dinarello and van der Meer, 2013). After binding to the membrane bound IL-1 receptor (IL-1R), IL-1ß activates a cascade that results, among others, in intracellular activation of NF-kappaB, leading to cytokine and CRP induction. The same pathway and activation of NF-kappaB is also induced by IL-18 (Kaplanski, 2018). Figure 1 shows possible IL-1ß activation pathways.
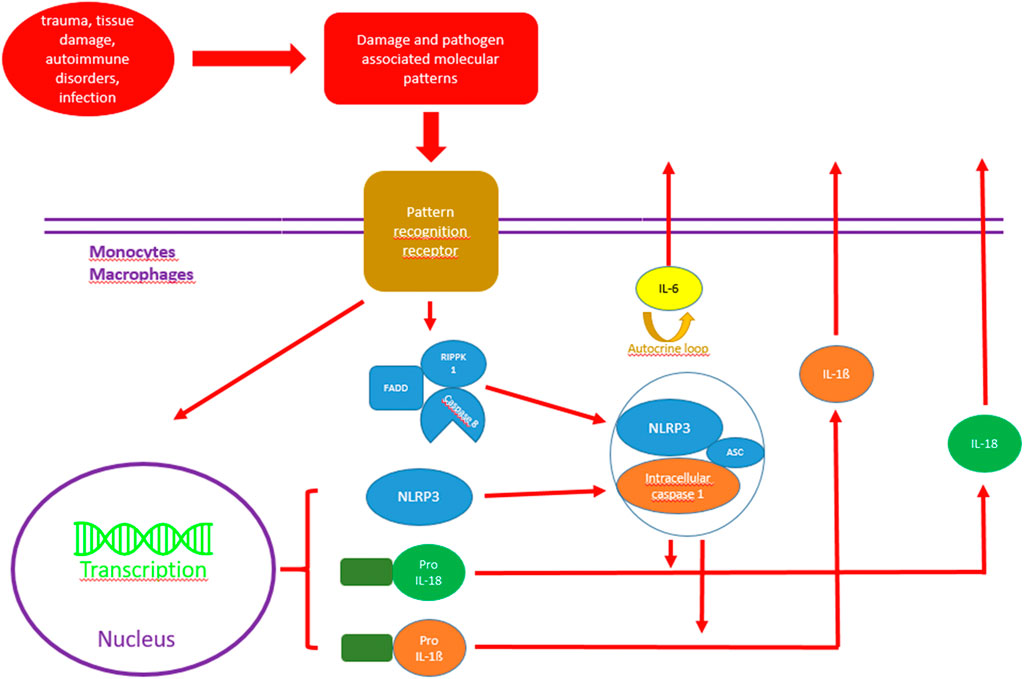
FIGURE 1. Possible activation pathways of IL-1ß and IL-18 in monocytes and macrophages. RIPK1, serine/threonine-protein kinase 1; FADD, Fas-associated protein with death domain; NLRP 3, Nod-like receptor protein family pyrin domain containing 3; Pro-IL1ß, Pro interleukin 1ß; Pro IL-18, Pro interleukin 18.
IL-6 can be found in and produced by different tissues and has many functions involving the regulation of the immune system and hematopoesis as well as inflammation and acute phase response (Tanaka et al., 2014). IL-6 is mainly produced by monocytes and macrophages (Tanaka et al., 2016). It binds to the membrane bound IL-6 receptor (IL-6R), which interacts with membrane bound glycoprotein 130 and leads to an activation of NF-kappaB and the transcription factors STAT3 and C/EBPß, all capable of upregulating CRP expression. Following a transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1 alpha, which is essential for cytokine-driven C-reactive protein gene expression, this complex binds to the CRP promoter and upregulates CRP synthesis (Zhang et al., 1996; Agrawal et al., 2001; Singh et al., 2007; Nishikawa et al., 2008; Young et al., 2008). With the simultaneous presence of interleukin-1ß (IL-1ß), hepatic CRP production is raised exponentially (Pepys and Hirschfield, 2003), potentially by triggering an autocrine IL-6 loop (Kramer et al., 2008). The latter displays the crucial role of IL-1ß in the activation of the humoral part of the innate immune system (Grebe et al., 2018).
In clinical routine, CRP has, up to the present day, predominantly been quantified in inflammatory and infectious conditions and is widely used to monitor various autoimmune diseases such as rheumatoid arthritis (Du Clos, 2013). CRP levels are also known to correlate with mortality in septic patients with multiple organ failure (Lobo et al., 2003). Elevated CRP baseline levels are associated with an increased risk for cardiovascular events, predominantly represented by stable coronary artery disease, myocardial infarction and ischemic stroke, as first reported by Ridker et al. (1997). A large meta-analysis confirmed the correlation of elevated baseline CRP levels with a higher likelihood of relevant coronary disease (i.e., patients suffering from angina on exertion or myocardial infarction), ischemic stroke and mortality (Emerging Risk Factors Collaboration et al., 2010). In addition CRP has been shown to co-localize in atherosclerotic plaques in humans as well as mice (Reynolds and Vance, 1987; Torzewski et al., 1998; Zimmermann et al., 2014). Since these findings may imply a causal role of elevated CRP plasma levels in various diseases instead of CRP just being a marker, inhibiting CRP synthesis could be a potential therapeutical target in the setting of acute infectious and autoimmune diseases as well as in cardiovascular disease. Discussions are, however, still ongoing and controversial (Torzewski, 2005; Pepys, 2008; Ridker, 2019; Jimenez and Szalai, 2021).
One way to answer the question of CRP being a worthwhile therapeutic target would be to lower plasma levels via selective CRP synthesis inhibition in the liver in the setting of the above-mentioned diseases in vivo. Consequently, this article focuses on contemporary approaches of CRP synthesis inhibition in the setting of cardiovascular disease. Substances inhibiting CRP synthesis and their potential intracellular targets are depicted in Figure 2.
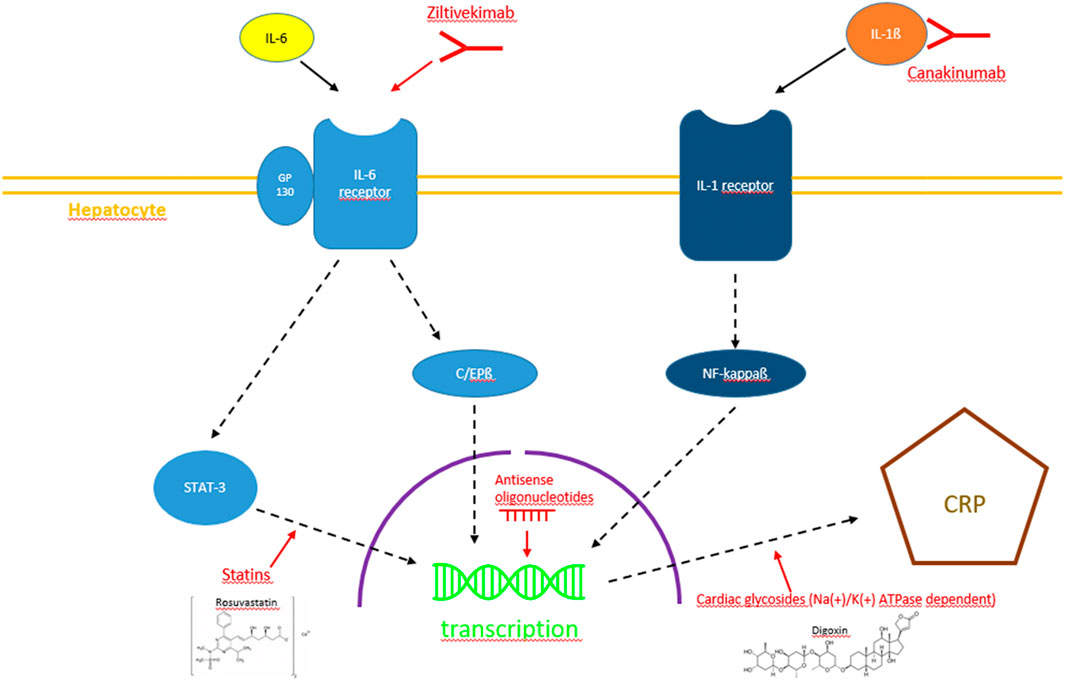
FIGURE 2. Diagram with the currently most promising pathways of CRP synthesis inhibition, i.e., IL-1ß/IL-6 inhibition by monoclonal antibodies, STAT-3 pathway interference by statins, RNA interference by antisense oligonucleotides and Na(+)/K(+)-ATPase-dependent inhibition by cardiac glycosides.
2 Natural substances capable of lowering CRP plasma levels
A variety of natural substances such as rosmarinic acid, tart cherry juice and green tea have recently been reported to have anti-inflammatory effects with the capability of reducing CRP levels (Asbaghi et al., 2019; Chai et al., 2019; Yao et al., 2019; Asbaghi et al., 2020). The mechanisms remain largely unclear. Nonetheless, in the prevention of atherosclerosis and its sequelae such substances are worth further investigation, especially for primary prevention. In Table 1 we summarize the most natural promising substances with reported effects on CRP levels.
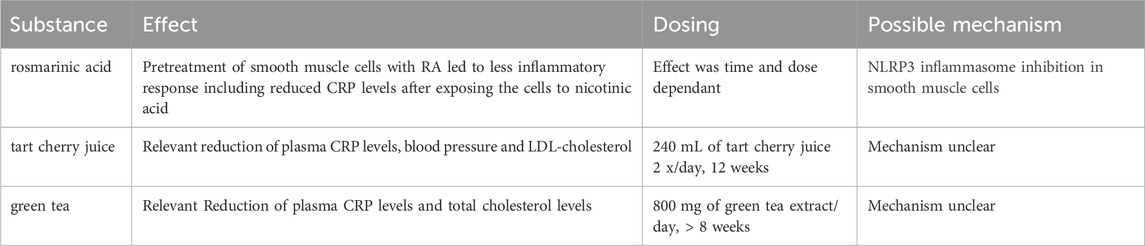
TABLE 1. Natural substances with anti-inflammatory and CRP lowering effects.
3 Reduction of CRP plasma levels via upstream inhibition of the IL-1β/IL-6/CRP pathway
The JUPITER trial (Ridker et al., 2008) showed a CRP lowering effect of 3-Hydroxy-3-Methylglutaryle-Coenzym-A-Reductase-(HMG-CoA-Reductase) inhibitors (statins) and also a reduction of cardiovascular event rates in patients with cholesterol levels of less than 130 mg/dL and CRP plasma levels of 2 mg/L or higher. These findings implied an anti-inflammatory effect of statins in addition to the reduction of cholesterol levels and underlined the importance of research on anti-inflammatory therapies in cardiovascular disease. Statins themselves have been part of the optimal medical treatment of cardiovascular disease since more than 40 years, nonetheless the mechanisms of statin-induced reduction of CRP plasma levels are not yet completely unraveled. Statins have shown to lower CRP levels in multiple ways. One reason being the reduction of LDL and oxidized LDL levels through 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibition as well as the upregulation of HDL and its key component apolipoprotein A-I. Both mechanisms lead to an indirect reduction of CRP synthesis in smooth muscle cells and macrophages in atherosclerotic plaques (Calabro et al., 2003; Arévalo-Lorido, 2016). Another potentially important mechanism involves the inhibition of protein isoprenylation, which leads to an interference with a group of intracellular signaling involving guanosine triphosphate (GTP)-binding proteins as Rho, Rac and Ras (Schönbeck and Libby, 2004). Causing a reduction of RAC-1 geranylgeranylation leads to a lower level of IL-6 induced serine phosphorylation of the transcription factor STAT3 reducing CRP synthesis (Arnaud et al., 2005).
Consecutively, Ridker et al. designed a trial which examined whether the inhibition of inflammation by IL-1β targeting without influencing cholesterol levels at all would also have an effect on cardiovascular events. The CANTOS trial (Ridker et al., 2017) used Canakinumab, a human monoclonal antibody neutralizing IL-1ß, for inhibition of the IL-1ß/IL-6/CRP pathway. The CANTOS trial indeed showed a highly relevant risk reduction for cardiovascular events in individuals with an increased risk for cardiovascular disease. Canakinumab lowered CRP, fibrinogen and IL-6 levels effectively without influencing cholesterol levels. Side effects, however, included more severe infectious diseases in the treatment group compared to the control group and thus, Canakinumab in this indication, did not receive FDA approval. Furthermore, CANTOS did not answer the question of a causal CRP involvement in cardiovascular disease but proved the vital role of inflammation once more. The COLCOT trial (Tardif et al., 2019) confirmed these results by showing reduced cardiovascular event rates due to several anti-inflammatory effects of colchicine, most notably IL-1 und IL-8 inhibition (Leung et al., 2015), in secondary disease prevention. Based on the LoDoCo2 trial (Nidorf et al., 2020), low dose Colchicine has recently received FDA approval for the secondary prevention of cardiovascular disease. The latest study investigating upstream CRP inhibition was the recently published RESCUE trial, a phase II study, which showed effective reduction of CRP levels through IL-6 inhibition via Ziltevikimab in a dose-dependent manner in patients at high cardiovascular risk and suffering from chronic kidney disease (Ridker et al., 2021). Based on these results, a large-scale randomized clinical trial has been initiated investigating the effect of Ziltevikimab on cardiovascular endpoints, results are pending. Table 2 summarizes the key results of the studies mentioned above.
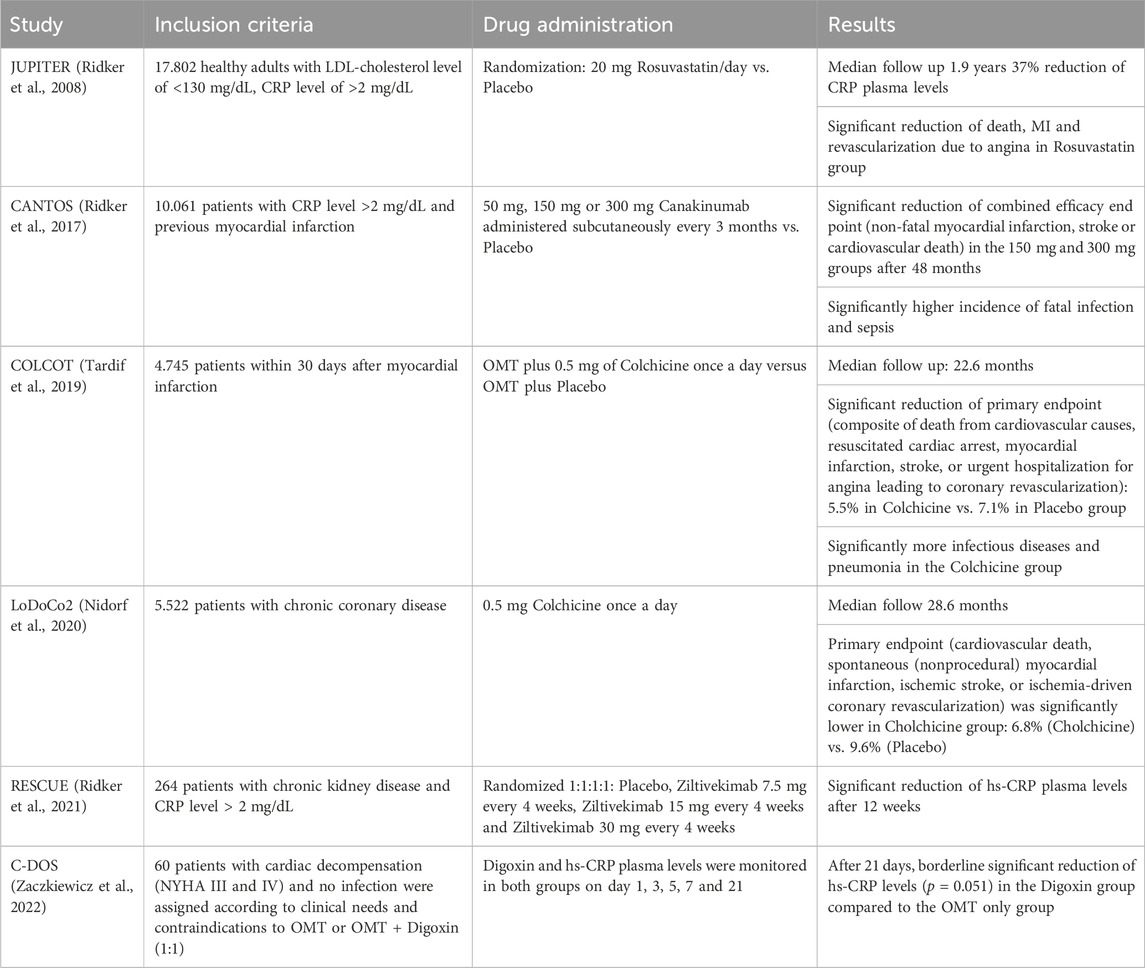
TABLE 2. Relevant clinical studies investigating reduction of CRP plasma levels.
Direct JAK inhibition to our knowledge has not yet been investigated in the context of cardiovascular disease. It has been shown though that JAK inhibitors are capable of lowering CRP levels (Pin et al., 2020). In clinical practice JAK inhibitors are mainly used to treat autoimmune disorders and strong data is available for patients with rheumathoid arthritis, which has lead to JAK inhibitors being a part of the European and American guidelines treatment algorithms for patients with rheumathoid arthritis (Fraenkel et al., 2021; Smolen et al., 2023). In this context there have been evaluations of the cardiovascular safety of JAK inhibitors showing positive effects or no relevant effects on cardiovascular events, however these were metaanalyses (Charles-Schoeman et al., 2016; Charles-Schoeman et al., 2019; Xie et al., 2019).
4 CRP synthesis inhibition via antisense oligonucleotides
Antisense oligonucleotides (ASOs) are short single-stranded nucleic acids that selectively bind to a specific part of mRNA and can be used to modify gene expression. They have to be administered parenterally and processed to ensure cellular uptake (Bennett and Swayze, 2010). Szalai et al. successfully used ASOs to inhibit CRP expression and to lower CRP plasma levels in rats and in CRP transgenic mice. ASO treatment resulted in the reduction of atherosclerotic plaque burden compared to rats and transgenic mice which were not treated with ASOs (Szalai et al., 2014). Yu et al. showed effective reduction of CRP levels with ASOs in Watanabe heritable hyperlipidemic rabbits. In contrast to Szalai et al., this study could not demonstrate any difference in atherosclerotic plaque burden between ASO-treated and -non treated Watanabe heritable hyperlipidemic rabbits (Yu et al., 2014).
The only larger clinical trial showing that ASOs can effectively inhibit CRP synthesis in humans used endotoxin to induce an acute phase reaction. An effect on the release of other acute phase proteins was not seen (Novcek et al., 2014). In 2012, another group had already reported effective reduction of CRP levels using ASOs in 8 adults (Jones et al., 2012). Since then, however, no further studies or randomized controlled trials applying ASOs in patients suffering from cardiovascular disease have been published, especially no studies investigating clinical endpoints.
5 Direct drug-induced CRP synthesis inhibition
A number of drugs affect CRP plasma levels. Most of them, however, influence CRP levels indirectly (Prasad, 2006). The search for a substance directly inhibiting CRP synthesis proved to be difficult. The main reason was that, in vitro, primary human hepatocytes (although undoubtedly being the model closest to the human liver), do not grow adequately, and limited access to adequate tissue prevents their use in high throughput screening assays (Gómez-Lechón et al., 2003). Anyway, high throughput screening assays utilizing genetically engineered hepatoma cells transfected with the human CRP promoter finally identified cardiac glycosides as potent inhibitors of in vitro CRP synthesis. It was indeed shown that cardiac glycosides inhibit the IL-1ß-/IL-6 induced expression of acute phase proteins in human hepatoma cells as well as primary human hepatocytes via a Na(+)/K(+)-ATPase-dependent pathway, which has not been completely demasked (Kolkhof et al., 2010).
Up to the present day, however, there has been no larger study investigating CRP synthesis inhibition by cardiac glycosides in humans. This is partly due to the fact that cardiac glycosides have been used in medical therapy for more than two centuries (Withering, 1785) causing relevant patent law considerations by pharmaceutical companies. Furthermore, the well-known narrow therapeutic window as well as side effects of cardiac glycosides, especially AV-blockage and ventricular arrhythmia (Hauptman and Kelly, 1999; Adams et al., 2014), limit their use in long-term medication. The single-center C-reactive protein-Digoxin Observational Study (C-DOS) provides a first piece of evidence that cardiac glycosides may indeed inhibit CRP synthesis in vivo in humans (Zaczkiewicz et al., 2022).
6 Other contemporary ways of C-reactive protein inhibition/elimination
Thiele et al. observed that in human striated muscle, human atherosclerotic plaque, and infarcted myocardium (in both rat and human myocardium), monomeric CRP (mCRP) was colocalized with inflammatory cells rather than pentameric CRP (pCRP), which is mainly found in the blood serum. These findings suggested that inhibition of the dissociation of pCRP to mCRP may prove to be a vital approach in order to reduce CRP-induced inflammation (Badimon et al., 2018, Caprio et al., 2018, Filep et al., 2023). Thiele et al. showed in 2014 that inhibiting the phopholipase A2-dependent dissociation from pCRP to mCRP via stabilization of pCRP with 1,6-bis(phosphocholine)-hexane showed significantly less inflammatory tissue damage after inducing myocardial infarction in a rat model (Thiele et al., 2014). The most recent study dealing with this issue was published by Zeller et al., in 2023. The group designed the novel substance C10M derived from phosphocholine, which is expressed in activated cells and binds to the ß-interface of pCRP. C10M was able to successfully inhibit pCRP dissociation into mCRP in vitro and in vivo in a mouse model with limb transplantation (Zeller et al., 2023). These promising results should encourage further studies following up on this approach.
A summary of CRP apheresis as a means of extracorporeal elimination of CRP has been published elsewhere (Ries et al. 2019; Torzewski et al., 2022).
7 Conclusion
In order to answer the question whether CRP plays a causal role in inflammatory processes, specific hepatic CRP synthesis inhibition may be helpful.
A promising approach seems the ASO technology. ASO technology is highly selective. Disadvantages of ASOs, however, include the need for parenteral application on the one hand and the expected high costs in case of a broader use on the other hand. No larger studies investigating the effect of CRP inhibition by ASOs on cardiovascular endpoints have been published yet.
Selective pharmacological inhibition of hepatic CRP synthesis, up to the present day, has not really been successful. Cardiac glycosides are the only substances that have been demonstrated to inhibit CRP synthesis in hepatocytes in vitro and lower CRP plasma levels in vivo in a small observational trial. Chemical modification of cardiac glycosides, pharmacological improvement and randomized controlled trials looking at cardiovascular endpoints would be necessary. This approach, however, is expensive and needs to overcome limitations like the toxicity and small therapeutic window of cardiac glycosides.
Most substantial progress has been made using upstream inhibitors, especially monoclonal IL-6 antibodies, with the first randomized controlled trial looking at cardiovascular endpoints. Potential disadvantages of this approach may be the need of parenteral application, the expected costs and the potential side effects of targeting a cytokine involved in immune regulation, hematopoesis and acute phase response. Upstream inhibition with specific IL-6 inhibitors has already reached the phase of randomized controlled clinical trials. Long-term Colchicine application for secondary prevention of cardiovascular disease has recently received FDA approval. The latter is comparatively cheap and may potentially be suitable for daily clinical practice. We summarized the current state of investigations regarding the above mentioned approaches of CRP inhibition as well as the advantages and disadvantages in Table 3.
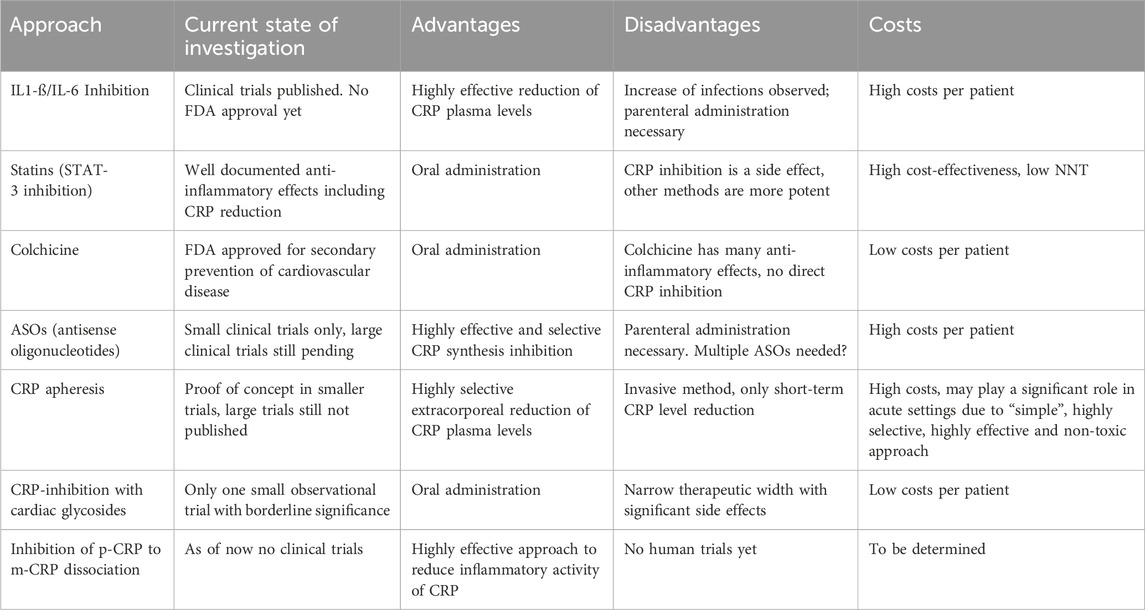
TABLE 3. Current state and advantages/disadvantages of each of the CRP-lowering approaches.
Future goals should include identifying patient populations benefiting the most from each of the specific approaches, eventually artificial intelligence could be helpful in this process. These results should be combined with considerations regarding available resources to optimize resource allocation.
Author contributions
MZ: Conceptualization, Writing–original draft. OZ: Writing–review and editing. JT: Conceptualization, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, K. F., Ghali, J. K., Herbert Patterson, J., Stough, W. G., Butler, J., Bauman, J. L., et al. (2014). A perspective on re-evaluating digoxin's role in the current management of patients with chronic systolic heart failure: targeting serum concentration to reduce hospitalization and improve safety profile. Eur. J. Heart Fail 16 (5), 483–493. doi:10.1002/ejhf.64
Agrawal, A., Cha-Molstad, H., Samols, D., and Kushner, I. (2001). Transactivation of C-reactive protein by IL-6 requires synergistic interaction of CCAAT/enhancer binding protein beta (C/EBP beta) and Rel p50. J. Immunol. 166 (4), 2378–2384. doi:10.4049/jimmunol.166.4.2378
Arévalo-Lorido, J. C. (2016). Clinical relevance for lowering C-reactive protein with statins. Ann. Med. 48 (7), 516–524. doi:10.1080/07853890.2016.1197413
Arnaud, C., Burger, F., Steffens, S., Veillard, N. R., Nguyen, T. H., Trono, D., et al. (2005). Statins reduce interleukin-6-induced C-reactive protein in human hepatocytes: new evidence for direct antiinflammatory effects of statins. Arterioscler. Thromb. Vasc. Biol. 25 (6), 1231–1236. doi:10.1161/01.ATV.0000163840.63685.0c
Asbaghi, O., Fouladvand, F., Gonzalez, M. J., Aghamohammadi, V., Choghakhori, R., and Abbasnezhad, A. (2019). The effect of green tea on C-reactive protein and biomarkers of oxidative stress in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Complement. Ther. Med. 46, 210–216. doi:10.1016/j.ctim.2019.08.019
Asbaghi, O., Fouladvand, F., Moradi, S., Ashtary-Larky, D., Choghakhori, R., and Abbasnezhad, A. (2020). Effect of green tea extract on lipid profile in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Metab. Syndr. 14 (4), 293–301. doi:10.1016/j.dsx.2020.03.018
Badimon, L., Peña, E., Arderiu, G., Padró, T., Slevin, M., Vilahur, G., et al. (2018). C-reactive protein in atherothrombosis and angiogenesis. Front. Immunol. 9, 430. doi:10.3389/fimmu.2018.00430
Bennett, C. F., and Swayze, E. E. (2010). RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 50, 259–293. doi:10.1146/annurev.pharmtox.010909.105654
Bharadwaj, D., Stein, M. P., Volzer, M., Mold, C., and Du Clos, T. W. (1999). The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J. Exp. Med. 190 (4), 585–590. doi:10.1084/jem.190.4.585
Black, S., Kushner, I., and Samols, D. (2004). C-Reactive protein. J. Biol. Chem. 279 (47), 48487–48490. doi:10.1074/jbc.R400025200
Calabró, P., Willerson, J. T., and Yeh, E. T. (2003). Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation 108 (16), 1930–1932. doi:10.1161/01.CIR.0000096055.62724.C5
Caprio, V., Badimon, L., Di Napoli, M., Fang, W. H., Ferris, G. R., Guo, B., et al. (2018). pCRP-mCRP dissociation mechanisms as potential targets for the development of small-molecule anti-inflammatory chemotherapeutics. Front. Immunol. 9, 1089. doi:10.3389/fimmu.2018.01089
Chai, S. C., Davis, K., Zhang, Z., Zha, L., and Kirschner, K. F. (2019). Effects of tart cherry juice on biomarkers of inflammation and oxidative stress in older adults. Nutrients 11 (2), 228. doi:10.3390/nu11020228
Charles-Schoeman, C., Wicker, P., Gonzalez-Gay, M. A., Boy, M., Zuckerman, A., Soma, K., et al. (2016). Cardiovascular safety findings in patients with rheumatoid arthritis treated with tofacitinib, an oral janus kinase inhibitor. Semin. Arthritis Rheum. 46, 261–271. doi:10.1016/j.semarthrit.2016.05.014
Charles-Schoeman, C., DeMasi, R., Valdez, H., Soma, K., Hwang, L. J., Boy, M. G., et al. (2019). Risk factors for major adverse cardiovascular events in phase iii and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 71, 1450–1459. doi:10.1002/art.40911
Dinarello, C. A., and van der Meer, J. W. (2013). Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 25 (6), 469–484. doi:10.1016/j.smim.2013.10.008
Du Clos, T. W. (2013). Pentraxins: structure, function, and role in inflammation. ISRN Inflamm. 2013, 379040. doi:10.1155/2013/379040
Emerging Risk Factors Collaboration Kaptoge, S., Di Angelantonio, E., Lowe, G., Pepys, M. B., Thompson, S. G., et al. (2010). C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet 375 (9709), 132–140. doi:10.1016/S0140-6736(09)61717-7
Filep, J. G. (2023). Targeting conformational changes in C-reactive protein to inhibit pro-inflammatory actions. EMBO Mol. Med. 15 (1), e17003. Epub 2022 Dec 5. PMID: 36465053; PMCID: PMC9832832. doi:10.15252/emmm.202217003
Fraenkel, L., Bathon, J. M., England, B. R., St Clair, E. W., Arayssi, T., Carandang, K., et al. (2021). 2021 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res. Hob. 73 (7), 924–939. doi:10.1002/acr.24596
Gang, T. B., Hammond, D. J., Singh, S. K., Ferguson, D. A., Mishra, V. K., and Agrawal, A. (2012). The phosphocholine-binding pocket on C-reactive protein is necessary for initial protection of mice against pneumococcal infection. J. Biol. Chem. 287 (51), 43116–43125. doi:10.1074/jbc.M112.427310
Gómez-Lechón, M. J., Donato, T., Ponsoda, X., and Castell, J. V. (2003). Human hepatic cell cultures: in vitro and in vivo drug metabolism. Altern. Lab. Anim. 31 (3), 257–265. doi:10.1177/026119290303100307
Grebe, A., Hoss, F., and Latz, E. (2018). NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 122 (12), 1722–1740. doi:10.1161/CIRCRESAHA.118.311362
Hauptman, P. J., and Kelly, R. A. (1999). Digitalis. Circulation. 99 (9), 1265–1270. doi:10.1161/01.cir.99.9.1265
Hurlimann, J., Thorbecke, G. J., and Hochwald, G. M. (1966). The liver as the site of C-reactive protein formation. J. Exp. Med. 123 (2), 365–378. doi:10.1084/jem.123.2.365
Jimenez, R. V., and Szalai, A. J. (2021). Therapeutic lowering of C-reactive protein. Front. Immunol. 11, 619564. doi:10.3389/fimmu.2020.619564
Jones, N. R., Pegues, M. A., McCrory, M. A., Singleton, W., Bethune, C., Baker, B. F., et al. (2012). A selective inhibitor of human C-reactive protein translation is efficacious in vitro and in C-reactive protein transgenic mice and humans. Mol. Ther. Nucleic Acids 1 (11), e52. doi:10.1038/mtna.2012.44
Kaplan, M. H., and Volanakis, J. E. (1974). Interaction of C-reactive protein complexes with the complement system. J. Immunol. 112 (6), 2135–2147. PMID: 4151108. doi:10.4049/jimmunol.112.6.2135
Kaplanski, G. (2018). Interleukin-18: biological properties and role in disease pathogenesis. Immunol. Rev. 281 (1), 138–153. doi:10.1111/imr.12616
Kolkhof, P., Geerts, A., Schäfer, S., and Torzewski, J. (2010). Cardiac glycosides potently inhibit C-reactive protein synthesis in human hepatocytes. Biochem. Biophys. Res. Commun. 394 (1), 233–239. doi:10.1016/j.bbrc.2010.02.177
Kramer, F., Torzewski, J., Kamenz, J., Veit, K., Hombach, V., Dedio, J., et al. (2008). Interleukin-1beta stimulates acute phase response and C-reactive protein synthesis by inducing an NFkappaB- and C/EBPbeta-dependent autocrine interleukin-6 loop. Mol. Immunol. 45 (9), 2678–2689. doi:10.1016/j.molimm.2007.12.017
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13 (6), 397–411. doi:10.1038/nri3452
Leung, Y. Y., Yao Hui, L. L., and Kraus, V. B. (2015). Colchicine--Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 45 (3), 341–350. doi:10.1016/j.semarthrit.2015.06.013
Lobo, S. M., Lobo, F. R., Bota, D. P., Lopes-Ferreira, F., Soliman, H. M., Mélot, C., et al. (2003). C-reactive protein levels correlate with mortality and organ failure in critically ill patients. Chest 123 (6), 2043–2049. doi:10.1378/chest.123.6.2043
Lu, J., Marnell, L. L., Marjon, K. D., Mold, C., Du Clos, T. W., and Sun, P. D. (2008). Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 456 (7224), 989–992. doi:10.1038/nature07468
Manolov, D. E., Röcker, C., Hombach, V., Nienhaus, G. U., and Torzewski, J. (2004). Ultrasensitive confocal fluorescence microscopy of C-reactive protein interacting with FcgammaRIIa. Arterioscler. Thromb. Vasc. Biol. 24 (12), 2372–2377. doi:10.1161/01.ATV.0000147407.17137.02
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi:10.1146/annurev.immunol.021908.132715
Nidorf, S. M., Fiolet, A. T. L., Mosterd, A., Eikelboom, J. W., Schut, A., Opstal, T. S. J., et al. (2020). Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 383 (19), 1838–1847. doi:10.1056/NEJMoa2021372
Nishikawa, T., Hagihara, K., Serada, S., Isobe, T., Matsumura, A., Song, J., et al. (2008). Transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1 alpha is essential for cytokine-driven C-reactive protein gene expression. J. Immunol. 180 (5), 3492–3501. doi:10.4049/jimmunol.180.5.3492
Noveck, R., Stroes, E. S., Flaim, J. D., Baker, B. F., Hughes, S., Graham, M. J., et al. (2014). Effects of an antisense oligonucleotide inhibitor of C-reactive protein synthesis on the endotoxin challenge response in healthy human male volunteers. J. Am. Heart Assoc. 3 (4), e001084. doi:10.1161/JAHA.114.001084
Pepys, M. B., and Hirschfield, G. M. (2003). C-reactive protein: a critical update. J. Clin. Invest. 111 (12), 1805–1812. doi:10.1172/JCI18921
Pepys, M. B. (2008). C-reactive protein is neither a marker nor a mediator of atherosclerosis. Nat. Clin. Pract. Nephrol. 4 (5), 234–235. doi:10.1038/ncpneph0778
Pin, A., Tesser, A., Pastore, S., Moressa, V., Valencic, E., Arbo, A., et al. (2020). Biological and clinical changes in a pediatric series treated with off-label JAK inhibitors. Int. J. Mol. Sci. 21 (20), 7767. doi:10.3390/ijms21207767
Prasad, K. (2006). C-reactive protein (CRP)-lowering agents. Cardiovasc Drug Rev. 24 (1), 33–50. doi:10.1111/j.1527-3466.2006.00033.x
Próchnicki, T., Mangan, M. S., and Latz, E. (2016). Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res 5, F1000. doi:10.12688/f1000research.8614.1
Reynolds, G. D., and Vance, R. P. (1987). C-reactive protein immunohistochemical localization in normal and atherosclerotic human aortas. Arch. Pathol. Lab. Med. 111 (3), 265–269.
Ridker, P. M., Cushman, M., Stampfer, M. J., Tracy, R. P., and Hennekens, C. H. (1997). Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 336 (14), 973–979. doi:10.1056/NEJM199704033361401
Ridker, P. M., Danielson, E., Fonseca, F. A., Genest, J., Gotto, A. M., Kastelein, J. J., et al. (2008). Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 359 (21), 2195–2207. doi:10.1056/NEJMoa0807646
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl. J. Med. 377 (12), 1119–1131. doi:10.1056/NEJMoa1707914
Ridker, P. M., Devalaraja, M., Baeres, F. M. M., Engelmann, M. D. M., Hovingh, G. K., Ivkovic, M., et al. (2021). IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397 (10289), 2060–2069. doi:10.1016/S0140-6736(21)00520-1
Ridker, P. M. (2019). Anticytokine agents: targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ. Res. 124 (3), 437–450. doi:10.1161/CIRCRESAHA.118.313129
Ries, W., Heigl, F., Garlichs, C., Sheriff, A., and Torzewski, J. (2019). Selective C-reactive protein-apheresis in patients. Ther. Apher. Dial. 23 (6), 570–574. doi:10.1111/1744-9987.12804
Roux, K. H., Kilpatrick, J. M., Volanakis, J. E., and Kearney, J. F. (1983). Localization of the phosphocholine-binding sites on C-reactive protein by immunoelectron microscopy. J. Immunol. 131 (5), 2411–2415. doi:10.4049/jimmunol.131.5.2411
Schönbeck, U., and Libby, P. (2004). Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation 109 (2), II18–26. doi:10.1161/01.CIR.0000129505.34151.23
Singh, P. P., Voleti, B., and Agrawal, A. (2007). A novel RBP-J kappa-dependent switch from C/EBP beta to C/EBP zeta at the C/EBP binding site on the C-reactive protein promoter. J. Immunol. 178 (11), 7302–7309. doi:10.4049/jimmunol.178.11.7302
Smolen, J. S., Landewé, R. B. M., Bergstra, S. A., Kerschbaumer, A., Sepriano, A., Aletaha, D., et al. (2023). EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann. Rheum. Dis. 82 (1), 3–18. doi:10.1136/ard-2022-223356
Stein, M. P., Mold, C., and Du Clos, T. W. (2000). C-reactive protein binding to murine leukocytes requires Fc gamma receptors. J. Immunol. 164 (3), 1514–1520. doi:10.4049/jimmunol.164.3.1514
Szalai, A. J., McCrory, M. A., Xing, D., Hage, F. G., Miller, A., Oparil, S., et al. (2014). Inhibiting C-reactive protein for the treatment of cardiovascular disease: promising evidence from rodent models. Mediat. Inflamm. 2014, 353614. doi:10.1155/2014/353614
Tanaka, T., Narazaki, M., and Kishimoto, T. (2014). IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 6 (10), a016295. doi:10.1101/cshperspect.a016295
Tanaka, T., Narazaki, M., Masuda, K., and Kishimoto, T. (2016). Regulation of IL-6 in immunity and diseases. Adv. Exp. Med. Biol. 941, 79–88. doi:10.1007/978-94-024-0921-5_4
Tardif, J. C., Kouz, S., Waters, D. D., Bertrand, O. F., Diaz, R., Maggioni, A. P., et al. (2019). Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381 (26), 2497–2505. doi:10.1056/NEJMoa1912388
Thiele, J. R., Habersberger, J., Braig, D., Schmidt, Y., Goerendt, K., Maurer, V., et al. (2014). Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: in vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation 130 (1), 35–50. doi:10.1161/CIRCULATIONAHA.113.007124
Thompson, D., Pepys, M. B., and Wood, S. P. (1999). The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 7 (2), 169–77. doi:10.1016/S0969-2126(99)80023-9
Torzewski, J., Torzewski, M., Bowyer, D. E., Fröhlich, M., Koenig, W., Waltenberger, J., et al. (1998). C-reactive protein frequently colocalizes with the terminal complement complex in the intima of early atherosclerotic lesions of human coronary arteries. Arterioscler. Thromb. Vasc. Biol. 18 (9), 1386–1392. doi:10.1161/01.atv.18.9.1386
Torzewski, J., Brunner, P., Ries, W., Garlichs, C. D., Kayser, S., Heigl, F., et al. (2022). Targeting C-reactive protein by selective apheresis in humans: pros and cons. J. Clin. Med. 11 (7), 1771. doi:10.3390/jcm11071771
Torzewski, J. (2005). C-reactive protein and atherogenesis: new insights from established animal models. Am. J. Pathol. 167 (4), 923–925. doi:10.1016/S0002-9440(10)61182-0
Volanakis, J. E. (2001). Human C-reactive protein: expression, structure, and function. Mol. Immunol. 38 (2-3), 189–197. PMID: 11532280. doi:10.1016/s0161-5890(01)00042-6
Withering, W. (1785). An account of the foxglove and some of its medical uses with practical remarks on dropsy and other diseases. London, UK: GGJ and J Robinson.
Xie, W., Huang, Y., Xiao, S., Sun, X., Fan, Y., and Zhang, Z. (2019). Impact of janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: systematic review and meta-analysis of randomised controlled trials. Ann. Rheum. Dis. 78, 1048–1054. doi:10.1136/annrheumdis-2018-214846
Yao, Y., Mao, J., Xu, S., Zhao, L., Long, L., Chen, L., et al. (2019). Rosmarinic acid inhibits nicotine induced C-reactive protein generation by inhibiting NLRP3 inflammasome activation in smooth muscle cells. J. Cell Physiol. 234 (2), 1758–1767. doi:10.1002/jcp.27046
Young, D. P., Kushner, I., and Samols, D. (2008). Binding of C/EBPbeta to the C-reactive protein (CRP) promoter in Hep3B cells is associated with transcription of CRP mRNA. J. Immunol. 181 (4), 2420–2427. doi:10.4049/jimmunol.181.4.2420
Yu, Q., Liu, Z., Waqar, A. B., Ning, B., Yang, X., Shiomi, M., et al. (2014). Effects of antisense oligonucleotides against C-reactive protein on the development of atherosclerosis in WHHL rabbits. Mediat. Inflamm. 2014, 979132. doi:10.1155/2014/979132
Zaczkiewicz, M., Kostenzer, K., Graf, M., Mayer, B., Zimmermann, O., and Torzewski, J. (2022). Cardiac glycosides lower C-reactive protein plasma levels in patients with decompensated heart failure: results from the single-center C-reactive protein-digoxin observational study (C-dos). J. Clin. Med. 11 (7), 1762. doi:10.3390/jcm11071762
Zeller, J., Cheung Tung Shing, K. S., Nero, T. L., McFadyen, J. D., Krippner, G., Bogner, B., et al. (2023). A novel phosphocholine-mimetic inhibits a pro-inflammatory conformational change in C-reactive protein. EMBO Mol. Med. 15, e16236. Epub 2022 Dec 5. PMID: 36468184. doi:10.15252/emmm.202216236
Zhang, D., Sun, M., Samols, D., and Kushner, I. (1996). STAT3 participates in transcriptional activation of the C reactive protein gene by interleukin-6. J. Biol. Chem. 271 (16), 9503–9509. doi:10.1074/jbc.271.16.9503
Keywords: cardiovascular disease, CRP synthesis, CRP inhibition, IL-6 downstream inhibition, IL-1ß down stream inhibition
Citation: Zaczkiewicz M, Zimmermann O and Torzewski J (2024) A short review on CRP synthesis inhibition in cardiovascular disease. Front. Drug Discov. 4:1338535. doi: 10.3389/fddsv.2024.1338535
Received: 14 November 2023; Accepted: 12 February 2024;
Published: 04 March 2024.
Edited by:
Stephen Geoffrey Ward, University of Bath, United KingdomReviewed by:
Werner J. Geldenhuys, West Virginia University, United StatesCopyright © 2024 Zaczkiewicz, Zimmermann and Torzewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myron Zaczkiewicz, bXlyb24uemFjemtpZXdpY3pAbWVkaWNsaW4uZGU=