 Yu Sun
Yu Sun Han Xu
Han Xu Weihan Gao2
Weihan Gao2 Xiayinan Song
Xiayinan Song Jie Li
Jie Li Xijian Liu
Xijian Liu- 1College of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2Innovation Research Institute of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
Neutrophil hyperexpression is recognized as a key prognostic factor for inflammation and is closely related to the emergence of a wide range of cardiovascular disorders. In recent years, S100 calcium binding protein A8/A9 (S100A8/A9) derived from neutrophils has attracted increasing attention as an important warning protein for cardiovascular disease. This article evaluates the utility of S100A8/A9 protein as a biomarker and therapeutic target for diagnosing cardiovascular diseases, considering its structural features, fundamental biological properties, and its multifaceted influence on cardiovascular conditions including atherosclerosis, myocardial infarction, myocardial ischemia/reperfusion injury, and heart failure.
Introduction
In recent years, the incidence rate and mortality rate of cardiovascular diseases have been increasing year by year (1). Despite the ongoing enhancements in diagnostic and treatment technologies, several shortcomings remain, such as extended diagnostic timeframes, suboptimal sensitivity and specificity of existing diagnostic markers, and, most notably, a dearth of targeted medications that are both safe and effective for particular cardiovascular conditions. Thus, the pursuit of new markers and therapeutic targets is essential for the prevention and treatment of cardiovascular diseases (2). The impact of immune system-mediated myocardial injury and the associated development of cardiovascular disease is a burgeoning area of interest in the study of disease mechanisms. Evidence has established that the innate immune response is stimulated by damage associated molecular patterns (DAMPs), leading to inflammatory processes. S100A8/A9, a component of the S100 calponin family of DAMPs, forms the S100A8/A9 heterodimer, also known as MRP8/14, and is consistently expressed (3). S100A8/A9 importance in cardiovascular diseases is underscored by its association with the extent of atherosclerosis in coronary and carotid arteries, plaque vulnerability, and its role in myocardial infarction and myocardial ischemia/reperfusion injury. Consequently, S100A8/A9 may be a promising biomarker and therapeutic target for cardiovascular conditions. The development and clinical trial approval of S100A8/A9 receptor blockers further underscore its potential (4). This review explores S100A8/A9's utility as a diagnostic tool for cardiovascular system effects and as a marker for cardiovascular events, providing a theoretical basis for the clinical treatment of cardiovascular diseases.
Overview of the S100 protein family
The S100 protein family, initially identified by Moore in 1965, comprises small molecular weight proteins (10–12 kDa) known for their solubility in a 100% neutral saturated ammonium sulfate solution and their role as calcium-regulatory proteins (5).This family now encompasses 25 members, including S100A1-16, S100G, and S100B, all of which share a significant degree of sequence and structural similarity and possess a characteristic calcium-binding motif (6). Each protein consists of two helix-loop-helix EF-hand domains surrounded by conserved hydrophobic regions at the N- and C-termini, connected by a central hinge. The N-terminus typically contains a 14-amino acid loop with low calcium affinity, whereas the C-terminus includes a high-affinity calcium-binding loop of 12 amino acids. This structural configuration facilitates a conformational change upon calcium binding, exposing hydrophobic domains that interact with various target receptors and proteins, a feature essential for the functional diversity of the S100 proteins (7). Exclusively found in vertebrates, S100 proteins are highly conserved across species. Each member of the S100 family exhibits unique tissue-specific expression patterns, determined by their respective genes (8).
Unlike calmodulin, which regulates intracellular biological activity by binding Ca2+, S100 proteins can modulate both intracellular and extracellular functions, participating in autocrine and paracrine signaling pathways (9). These proteins influence a wide array of physiological processes by interacting with cell membrane receptors such as Receptor of Advanced Glycation Endproducts (RAGE) and G Protein-Coupled Receptors (GPCRs), impacting cell proliferation, differentiation, migration, invasiveness, inflammatory responses, oxidative stress, calcium homeostasis, apoptosis, glycogen phosphorylation, and macrophage clustering (10).
Structure, origin and expression of S100a8/A9
S100A8/A9, similar to other members of the S100 protein family, is situated at a chromosomal region known for its variability, 1q21, where a distinct S100 gene cluster is formed (11). The human S100A8/A9 proteins are encoded with distinct amino acid compositions; S100A8 comprises 93 amino acids and has a molecular weight of 10.8 kDa, whereas S100A9 is made up of 113 amino acids and weighs 13.2 kDa (12). These proteins can exist as homodimers, heterodimers, and tetramers, with the homodimer being less stable, leading to a preference for the formation of S100A8/A9 heterodimer complexes, which are the predominant form of S100A8/A9 in physiological settings (13). These complexes, referred to as calprotectin, possess multiple biological characteristics (14). When Ca2+ concentration reaches a certain threshold, S100A8/A9 heterodimers form (S100A8/A9)2 tetramers, a configuration that is vital for their biological activity. Secreted mainly by immune cells such as dendritic cells, neutrophils, monocytes, and activated macrophages, these proteins participate in the pathophysiology of various inflammatory conditions by attracting leukocytes and modulating the inflammatory response to vascular injury (15) (Figure 1).
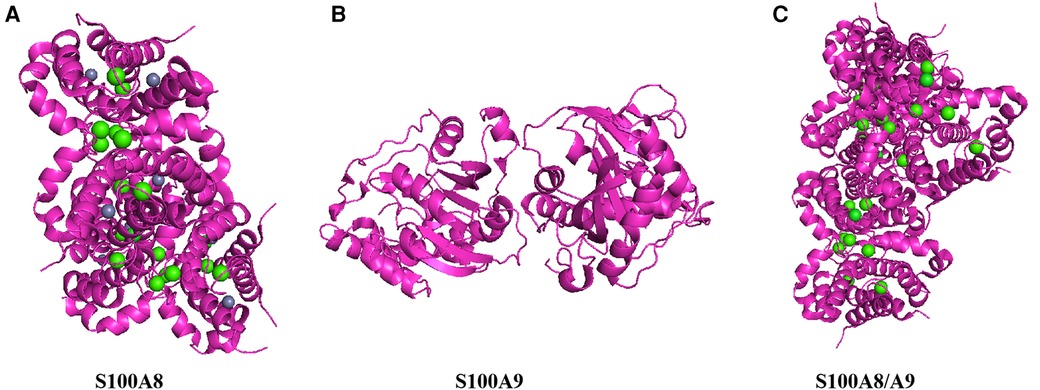
Figure 1. Structure of S100A8/A9. This figure shows the three-dimensional structure of S100A8/A9 heterodimer protein, demonstrating its complex folding and arrangement of key functional sites. (A) Color representation: Proteins are represented in magenta, displaying their alpha helix and secondary structure. Green sphere: represents the calcium ions (Ca2+) that bind to proteins, indicating that calcium binding sites are crucial for protein function. Gray sphere: may represent zinc ions (Zn2+) or other metal ions, which can stabilize protein structure and are important for its biological activity. (B) top-down perspective: This view provides a horizontal cross-section of proteins, emphasizing the internal arrangement of helices and loops. Subbase interface: The interface between S100A8 and S100A9 subunits is visible, indicating how they interact to form functional heterodimers. (C) Vertical cross-section: Provides a side view of proteins, highlighting the arrangement and spatial arrangement of alpha helices. Binding sites: The distribution of green spheres (calcium ions) throughout the structure indicates that multiple calcium binding sites are crucial for protein activity.
Biological functions of S100a8/A9
S100A8/A9 regulates phagocyte migration and exacerbates inflammation development within cells
The S100A8/A9 proteins exert their biological effects through a combination of specific expression patterns, structural alterations, distinct metal ion binding affinities, and the formation of homodimers, heterodimers, and oligomers, which are implicated in various disease states (16). Intracellular S100A8/A9 plays a pivotal role in modulating the migration of phagocytic cells, thereby facilitating the progression of inflammation. By interacting with the cytosolic components p47phox and p67phox of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, intracellular S100A8/A9 enhances the activity of NADPH oxidase, exacerbating cellular oxidative stress and contributing to pro-inflammatory effects. Phosphorylation of S100A9 regulates the p38 mitogen-activated protein kinase (MAPK) signaling pathway, leading to the formation of (S100A8/A9)2 tetramers (17). These tetramers promote microtubule polymerization and reorganization in a calcium-dependent manner, enhancing the migration of phagocytes towards the endothelium (18) Additionally, S100A8/A9 is selectively released during the interaction between phagocytes and activated endothelial cells, making it an active mediator in the development and progression of inflammation (19).
S100A8/A9 exerts significant inflammatory regulation extracellularly through multiple receptors
The biological activity of S100A8/A9 in the extracellular space is mediated by their interaction with receptors such as Toll-like receptor 4(TLR4) and the RAGE. Research indicates that the binding of S100A8/A9 to TLR4 sets off a signaling cascade that leads to the activation of NF-κB, which in turn governs cellular responses including inflammation and cell cycle progression. Studies have shown that S100A8/A9 can amplify the TLR4 response to Lipopolysaccharides (LPS) in myeloid cells, possibly by indirectly activating TLR4, which results in the induction of NO production by macrophages and intensifies inflammation. On the other hand, S100A8/A9 has been implicated in dampening acute inflammatory responses by regulating the activity of inflammatory cytokines (20).
The protein S100A8/A9 is implicated in the inflammatory response by potentially stimulating the release of inflammatory cytokines in endothelial cells through the RAGE receptor and amplifying its activation. In vitro, S100A8/A9 has been observed to trigger cell apoptosis via both caspase-dependent and -independent pathways, contributing to endothelial injury in conditions such as vasculitis and inflammatory diseases. S100A8/A9 has been shown to suppress the proliferation and differentiation of C2C12 myoblasts and induce caspase-3-mediated apoptosis. S100A8/A9 can also decrease mitochondrial membrane potential, leading to the release of mitochondrial proteins Smac/Diablo and Omi/HtrA2, and inhibit mitochondrial fission, which in turn induces cell death by tipping the balance between pro- and anti-apoptotic factors (21). Furthermore, S100A8/A9 may induce autophagy by facilitating the relocation of the mitochondrial outer membrane protein BCL2 interacting protein 3 (BNIP3) to the mitochondria and promoting Reactive Oxygen Species (ROS) production through mitochondrial-lysosomal interactions, thereby promoting apoptosis (22). In atherosclerotic lesions, S100A8/A9 is abundantly expressed in foam cells, and its release, possibly due to the upregulation of urokinase-type plasminogen activator in macrophages, may lead to endothelial cell apoptosis. Beyond its pro-inflammatory functions, S100A8/A9 also has a regulatory role in inflammation by inhibiting dendritic cell (DC) maturation and antigen presentation, which results in a reduced T cell response and prevents an overactive adaptive immune response (23). S100A8/A9 can also augment the quantity and activity of myeloid-derived suppressor cells (MDSCs), which are known for their immunosuppressive effects in various pathological states (24). This indicates that S100A8/A9 has a multifaceted role in modulating inflammation, with different molecular biological effects in a range of cell types (Figure 2).
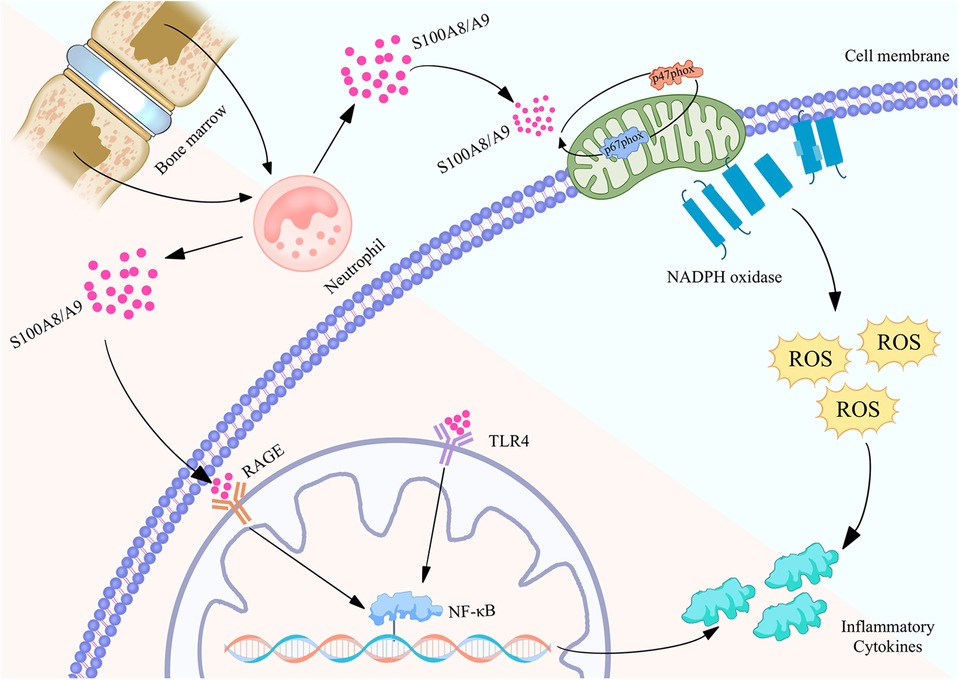
Figure 2. Biological function of S100A8/A9 proteins in neutrophils and their mechanism of regulating inflammatory action map. S100A8/A9 is released by neutrophils in the bone marrow and binds to RAGE and TLR4 receptors on the cell membrane, activating the NF - κB signaling pathway, thereby causing the expression of inflammatory factors. Meanwhile, S100A8/A9 can also activate NADPH oxidase, produce reactive oxygen species (ROS), and further promote inflammatory response. These pathways collectively promote the occurrence and development of inflammation, revealing the important role of S100A8/A9 in cardiovascular disease.
Transcriptional regulation and therapeutic development of S100a8/A9 in cardiovascular diseases
Transcriptional regulation of S100a8/A9
The transcriptional regulation of the S100A8/A9 gene is governed by multiple transcription factors, which modulate its expression in a cell-specific fashion. The S100A8/A9 gene exhibits heightened expression in human bone marrow, with mature neutrophils displaying a 40-fold increase in expression relative to monocytes (25). During inflammation in the compromised heart, specific transcription factors are implicated in the upregulation of the S100A8/A9 gene. Moreover, DNA demethylation contributes to the enhanced expression of the S100A8/A9 gene (26). This indicates a notable variation in S100A8/A9 gene expression throughout bone marrow differentiation, with its transcriptional dynamics being intricately linked to the inflammatory response mechanisms within the body. The Hoxb8 cell line is an important model system for studying the function of S100A8/A9 in myeloid cells, especially neutrophils, and can differentiate into various functional immune cell types (27). Studies have revealed that in monocytes derived from bone marrow precursor cells, S100A8/A9 mRNA levels peak during the early stages of differentiation (day 2) and subsequently decline by the later stages (days 3–5) (28). Conversely, as Hoxb8 cells differentiate into the neutrophil lineage, S100A8/A9 mRNA expression gradually increases, peaking on day 4 of culture (29). This dynamic regulation of transcription directly influences the secondary inflammatory response associated with heart failure (HF) development, thereby modulating cardiac injury and potentially decelerating the progression of cardiovascular disease.
Protein phosphorylation and S100a8/A9 regulation
Protein phosphorylation is pivotal for the intracellular activity regulation of S100A8/A9 proteins. Schenten et al. demonstrated that phosphorylated S100A8/A9 (S100A8/A9-p) induces the expression and secretion of pro-inflammatory cytokines in HL-60 cells (30). In human neutrophils, p38-MAPK acts as the upstream kinase phosphorylating threonine 113 on S100A9, regulating the translocation of S100A8/A9 proteins from the cytoplasm to the plasma membrane, thus accelerating the inflammatory response (31). Additionally, S100A8/A9 proteins in neutrophils can undergo S-nitrosylation by nitric oxide donors, resulting in S-nitrosylated S100A8 at cysteine 41 (S100A8-SNO), which exhibits anti-inflammatory properties. S100A8-SNO inhibits eukaryotic-endothelial cell interactions in the rat mesenteric microcirculation, thereby suppressing mast cell-mediated inflammation (32). These findings suggest that S100A8/A9 transcription is regulated by multiple transcription factors under varying physiological conditions, exerting both pro-inflammatory and anti-inflammatory effects through distinct mechanisms.
Therapeutic development of S100a8/A9 for cardiovascular diseases
The crucial role of S100A8/A9 protein in various cardiovascular diseases has led to the development of therapeutic tools and drugs targeting this protein. These therapeutic approaches aim to mitigate inflammation and oxidative stress by inhibiting the expression or function of S100A8/A9, thereby slowing the progression of cardiovascular diseases. Key therapeutic strategies include: RAGE Receptor Antagonists: Agents such as FPS-ZM1 reduce atherosclerosis and inflammatory responses by blocking the binding of S100A8/A9 to RAGE, thereby inhibiting NF-κB and MAPK pathways and reducing inflammatory cell infiltration and plaque formation (33). Anti-S100A8/A9 Antibodies: Specific antibodies that bind to S100A8/A9 prevent its interaction with receptors, inhibiting downstream inflammatory signaling pathways, reducing myocardial inflammation and apoptosis, and improving cardiac function (34). Small Molecule Inhibitors: Compounds like Paquinimod and ABR-238901 disrupt the interaction of S100A8/A9 with RAGE and TLR4, reducing plaque inflammation and instability, and reversing cardiac dysfunction in conditions such as sepsis (35). These inhibitors can bind directly to S100A8/A9 proteins or block their receptor interactions, reducing pro-inflammatory and pro-oxidative effects. Nanoparticle Delivery Systems: Utilizing nanoparticles or liposomes to deliver targeted anti-S100A8/A9 antibodies or small molecule inhibitors enhances drug concentration at the lesion site, thereby improving therapeutic efficacy (36). Gene Silencing Techniques: Techniques such as siRNA or CRISPR/Cas9 are employed to edit S100A8/A9 genes, reducing their expression and subsequent protein production. This approach decreases activation of NF-κB and MAPK pathways, reducing myocardial cell apoptosis and fibrosis (37). In summary, therapeutic tools and drugs targeting S100A8/A9 function through multiple mechanisms, including receptor antagonism, antibody therapy, small molecule inhibition, nanoparticle delivery, and gene silencing. These methods collectively work to attenuate the pro-inflammatory and pro-oxidative effects of S100A8/A9, offering significant potential in the treatment of cardiovascular diseases.
Significance of S100a8/A9 proteins in cardiovascular disease
S100A8/A9 regulates atherosclerotic plaque formation
S100A8/A9, as a calcium binding protein, plays an important role in the formation of atherosclerotic plaque by regulating inflammatory response and immune regulation (38, 39). Recent experimental studies have shown that S100A8/A9 activates the NF-κB signaling pathway by interacting with TLR4 and RAGE, inducing the expression of various pro-inflammatory cytokines such as tumor necrosis factor alpha TNF-α, interleukin-1β(IL-1β), and IL-6 (40). These proinflammatory factors aggravate the damage of vascular endothelial cells and promote the formation of atherosclerotic plaque (41). In addition, S100A8/A9 also plays a role in atherosclerosis by regulating oxidative stress. S100A8/A9 interacts with myeloperoxidase (MPO) to form a complex, increasing the production of reactive oxygen species (ROS) (42). ROS not only directly damages endothelial cells, but also oxidizes low-density lipoprotein (LDL), forming oxidized low-density lipoprotein (oxLDL). OxLDL is phagocytosed by macrophages to form foam cells, further promoting the formation and development of atherosclerotic plaque (43).
Other scholars explained that S100A8/A9 plays an important role in the early stage of atherosclerosis by promoting leukocyte migration and adhesion to vascular endothelial cells from the perspective of cell migration and adhesion (44). Research has shown that S100A8/A9 induces the expression of cell adhesion molecules such as Intercellular cell adhesion molecule-1(ICAM-1) and vascular cell adhesion molecule-1(VCAM-1) by binding to TLR4 and RAGE on the surface of vascular endothelial cells, enhancing the adhesion and migration of white blood cells on the arterial wall (45). At the same time, clinical studies have found that the serum S100A8/A9 level is closely related to the severity of atherosclerosis and the risk of cardiovascular events, which is expected to become a potential biomarker and therapeutic target of atherosclerosis (46). In addition, inhibiting S100A8/A9 or its signal pathway can reduce the progression of atherosclerosis, providing a new idea for the treatment of atherosclerosis (47).
In conclusion, S100A8/A9, as an important inflammatory mediator, plays a variety of roles in the occurrence and development of atherosclerosis. It promotes the formation and development of atherosclerotic plaque by regulating inflammatory response, oxidative stress, cell migration and adhesion and other mechanisms. In the future, it is expected to provide a new strategy for early diagnosis and treatment of atherosclerosis through in-depth research on the mechanism of action and clinical application value of S100A8/A9.
S100A8/A9 regulates the inflammatory response to myocardial infarction
The pathogenesis of myocardial infarction involves a sequence of two phases: an early inflammatory stage and a subsequent repair stage. Effectively managing this transition is essential for the restoration of cardiac function and the attainment of a positive prognosis in individuals with myocardial infarction (48). S100A8/A9 contributes to the regulation of both phases of myocardial infarction, with a particular impact on the migratory and differentiative behaviors of immune cells (49). The emission of myocardial inflammatory cytokines and the state of hypoxia are hallmarks of myocardial infarction and significantly contribute to the development of ischemic damage. Duet et al. have identified that myocardial ischemia or hypoxia leads to the upregulation and secretion of S100A8 and S100A9 in cardiomyocytes (50). A positive association has been observed between serum S100A8/A9 levels and neutrophil counts in patients with Acute myocardial infarction (AMI) who are under dynamic monitoring. Research has validated that blood neutrophils are the sole cell population that significantly influences the levels of circulating S100A8/A9 in humans (13). Double immunostaining analysis of infarcted hearts from AMI patients revealed that the S100A8/A9 complex was primarily co-localized with neutrophils during the early acute phase and with macrophages in the subacute phase. Sreejit using cell sorting and flow cytometry to isolate leukocytes from infarcted hearts of mice, identified neutrophils as the principal source of S100A8/A9 (51). The infiltration of neutrophils and macrophages into the infarcted myocardium was found to be the main contributor to the elevated levels of S100A8/A9 in myocardial tissue following AMI (52). In response to myocardial infarction, a surge of neutrophils expressing the alert protein S100A8/A9 rapidly infiltrates the ischemic myocardium. S100A8/A9 then interacts with TLR4 on circulating neutrophils, triggering the formation of nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammatory vesicles and promoting IL-1 secretion in a mouse model of MI (53). The concept that S100A8/A9 is an upstream regulator of MI-induced granulopoiesis is supported by a mouse model with targeted disruption of S100A9, where the deletion of the S100A9 gene or pharmacological inhibition of S100A8/A9 by ABR-215757 markedly constrained granulopoiesis, reducing the number of circulating and cardiac neutrophils and monocytes (54, 55). The binding of S100A8/A9 to TLR4 leads to the upregulation of pro-inflammatory cytokine production by inducing the nuclear translocation of NF-κB via the Toll/interleukin-1 receptor (TIR) domain adapter, either through the interferon-β-dependent pathway or the MyD88-dependent pathway, thereby amplifying the inflammatory response (56). S100A8/A9 exerts a harmful influence during the initial phase of myocardial infarction (MI), and interventions aimed at inhibiting S100A8/A9 can be advantageous for MI patients. The S100A8/A9 complex interacts with the monocyte differentiation antigen CD69, leading to the formation of a CD69-S100A8/A9 complex (29). This association downregulates signaling and transcription by enhancing the expression of cytokine signaling repressor 3 (SOCS3) and inhibiting signaling and transcription activator 3 (STAT3) signaling, while promoting the differentiation of regulatory T cells (Tregs) (57). This mechanism may induce immunosuppression in various immune cells, preventing an overactive immune response. However, overzealous inhibition of S100A8/A9 could negate this beneficial effect during the MI repair phase. Prolonged inhibition of S100A9 in ischemic mice with ABR-238901 led to progressive left ventricular remodeling and a decline in cardiac function, indicating that S100A8/A9 may have additional cardioprotective roles during the recovery phase by promoting a shift in macrophage polarization towards a reparative phenotype (4, 58). These findings advocate for a tailored therapeutic approach, suggesting short-term anti-S100A9 blockade during the early inflammatory phase post-MI, while long-term blockade could impair cardiac function recovery during the repair phase, underscoring the need for precise therapy duration determination (59).
Research has identified that female individuals with high serum S100A8/A9 protein concentrations are at a 3.8-fold greater risk for adverse vascular outcomes (60). S100A8/A9 has been recognized as a new predictor for myocardial infarction, with incremental increases in S100A8/A9 protein levels associated with a proportional rise in the risk of subsequent cardiovascular events (24). Platelet mRNA profiling has demonstrated that S100A9 mRNA is significantly elevated in ST-segment elevation myocardial infarction(STEMI) patients compared to those with stable coronary artery disease. Additionally, serum S100A8/A9 concentrations are higher in AMI patients, particularly in those with cardiac rupture (61). Katashima et al. have reported on the dynamic monitoring of S100A8/A9 levels in patients with acute-phase AMI and Unstable angina pectoris(UAP), noting that initial levels are lower in AMI patients than in those with UAP, with a significant increase in AMI patients by 3–5 days post-event. S100A8/A9 levels are also higher in patients with ischemic injury compared to those with UAP, peaking 3–5 days after the ischemic event and remaining elevated for an extended period (51). A nested case-control study on ACS patients undergoing intensive lipid-lowering therapy for 30 days post-acute cardiovascular event identified elevated S100A8/A9 in those with MI or cardiovascular death. Nonetheless, S100A8/A9 has a limited diagnostic utility, with a sensitivity of 28% for myocardial infarction in patients presenting with non-traumatic chest pain, and it does not enhance the diagnostic accuracy provided by cardiac troponin (10).
S100A8/A9 exacerbates ischemia/reperfusion (I/R) injury
Currently, an effective and standard treatment for STEMI is the reperfusion strategy (62). Reperfusion protects the ischemic heart from myocardial necrosis, but it also triggers a series of cascading responses that exacerbate and prolong post-ischemic injury (63). Inflammatory responses, oxidative stress (59), and mitochondrial dysfunction are important pathophysiological phenomena leading to alterations in cardiac structure and function after I/R injury (64). Du et al. found that S100A8/A9 was continuously expressed in mouse hearts during the early stages of myocardial ischemia/reperfusion injury (MI/RI) by time-series transcriptomics analysis, peaked at 6 h after reperfusion, and returned to baseline levels on day 7, thus clarifying that S100A8/A9 is an early mediator of I/R injury and is rapidly elevated in the early stages of MI/RI (65). Experimental Validation Findings S100A8/A9 was confirmed to be a key initiator molecule of I/R by S100A9 knockdown and overexpression experiments in animals, and in mice with a total deficiency of S100A9 after MI/RI, infarct size was significantly reduced, cardiac contractile function was improved, CM death was significantly reduced, and myocardial fibrosis was attenuated, suggesting that S100A9 increases the degree of myocardial ischemia-reperfusion injury (39, 43). Meanwhile mechanism studies revealed that S100A8/A9 down-regulated the gene expression of mitochondrial complex I subunit NDUFs by inhibiting the TLR4/ERK-mediated PGC-1α/NRF1 signaling pathway, which in turn inhibited the mitochondrial complex I function, causing mitochondrial dysfunction, leading to cardiomyocyte death, and promoting the development of MI/RI (66).
To fully understand the mechanism of action of S100A8/A9 in I/R, it is also important to clarify the cellular origin of S100A8/A9 in reperfusion injury. Experimental studies revealed that cardiac S100A8/A9 expression was significantly reduced in CXCR2-KO mice, further confirming that CXCR2+ neutrophils are the main source of S100A8/A9 secretion, which is related to the dynamic changes of CXCR2, and that cardiac neutrophil infiltration during I/R is consistent with the S100A8/A9 expression pattern. Chemokine (C-X-C motif) ligand 1 (CXCL1), a chemokine specific for CXCR2, is responsible for recruiting neutrophils expressing the chemokine receptor CXCR2 to the inflammatory microenvironment (67). In addition, mice treated with S100A9 neutralizing antibody (nAb) can observe a significant reduction in infarcted areas, increased cardiac function, and reduced myocardial fibrosis after I/R. Therefore, short-term blockade of S100A9 after I/R can effectively improve cardiac function in mice. Meanwhile, elevated serum S100A8/A9 levels 1 day after percutaneous coronary angioplasty (PCI) were found to be significantly associated with long-term adverse cardiovascular events in patients with acute myocardial infarction in the clinic (68). Targeting the signaling pathway initiated by S100A8/A9 may be a novel intervention for the treatment of MI/RI. At present, Du Jie's team has comprehensively carried out the work of early warning and intervention of S100A8/A9 after PCI for acute myocardial infarction: developed S100A8/A9 detection kit and monoclonal therapeutic antibody; and jointly carried out clinical research of “Early Warning of S100A8/A9 after PCI for Acute Myocardial Infarction”. Clinical research on S100A8/A9 in early warning after PCI for acute myocardial infarction (69).
Inhibition of S100a8/A9 slows HF development
S100A8/A9 proteins are implicated in the advancement of HF by aggravating cardiac injury through the induction of localized inflammation (70). These proteins initiate the NF-κB signaling cascade by interacting with RAGE and TLR4 on the cell membrane, which in turn activates pro-inflammatory responses in cardiomyocytes, fibroblasts, and endothelial cells of cardiac tissue, thereby intensifying HF. Myocardial hypertrophy is a pivotal aspect of HF, and S100A8/A9 may be influenced by diverse physiological conditions, leading to varied responses in the pathogenesis of myocardial hypertrophy (40). While typically low in cardiomyocytes, S100A8 expression increases in response to thyroid hormone treatment in neonatal rat cardiomyocytes (71), contributing to cardiac hypertrophy via the MyD88/NF-κB pathway (72). Conversely, S100A8/A9 mRNA and protein levels rise when cardiomyocyte hypertrophy induced by norepinephrine ceases (73), potentially mitigating hypertrophy and remodeling by reducing calmodulin neuralphosphatase-nfatc3 activation (74). The mechanisms by which endogenous S100A8/A9 proteins, stimulated by different cues, differentially influence cardiomyocyte hypertrophic responses remain unclear. However, recombinant S100A8 protein application induces inflammatory responses in human pluripotent stem cell-derived cardiomyocytes, impacting calcium transport and electrophysiological properties. CD11b+Gr1+ neutrophil-derived S100A8/A9 proteins participate in angiotensin II-induced cardiac inflammation and fibrosis, activating NF-κB signaling in cardiac fibroblasts via RAGE and amplifying chemokine and cytokine production, thus intensifying the inflammatory response and cardiac remodeling that characterize HF progression (27).
Suppression of S100A8/A9 protein expression has been shown to exhibit significant anti-inflammatory and protective effects against myocardial hypertrophy and fibrosis across various tissues, thereby mitigating the progression of heart failure (HF). In a study involving angiotensin II-infused mice, the administration of a S100A9 antibody to neutrophils was observed to protect against myocardial hypertrophy and fibrosis. This treatment also reduced the infiltration of immune cells (CD45+ leukocytes, CD45+CD11b+ monocytes, and Gr1+ neutrophils) into myocardial tissues and curtailed NF-κB-dependent proinflammatory and profibrotic gene expression (75). In rats subjected to coronary ligation, the administration of the S100A8/A9 inhibitor ABR-215757 (5 mg/kg, daily for 5 days) led to a notable decrease in fibrotic areas surrounding the myocardial infarction margins and a reduction in NF-κB p65 protein levels (69). ABR-215757 has also demonstrated protective effects against tissue fibrosis in other experimental models (76). Concurrently, researchers have developed specific antibodies and vaccines targeting the mechanotransduction of S100A8/A9 proteins within the immune system, as well as compounds that block the interaction between S100A8/A9 and TLR4 and RAGE (35). Animal studies have confirmed that inhibiting S100A8/A9 protein expression has a beneficial anti-inflammatory effect, offering a promising therapeutic target for managing the inflammatory response in HF (77).
Experimental investigations have detected a variety of immune cells, such as neutrophils, T lymphocytes, macrophages derived from monocytes, and NK cells, in the cardiac tissues of HF patients who do not exhibit significant myocardial injury, viral infection, or known immune system anomalies (78), Neutrophils, especially, have been shown to contribute to the exacerbation of HF based on both clinical and animal research (79), and in HF with preserved ejection fraction, an elevated neutrophil count is a predictor of a poor outcome (80). The detection of high neutrophil expression in myocardial biopsies from HF patients with preserved ejection fraction and in HF model rats points to intense cardiac inflammation during the disease's progression (81). The role of S100A8/A9 proteins as neutrophil mediators in the promotion of chronic cardiac injury and inflammation is not yet fully elucidated (82). Transcription profiling has demonstrated increased S100A8/A9 gene expression in neutrophils from HF patients with preserved ejection fraction, indicating a connection between neutrophil activation and systemic inflammation, as well as left ventricular diastolic dysfunction in these patients (14). Proteomic analysis of platelets from HF patients with preserved ejection fraction has identified S100A8 protein in platelets and its increased levels in plasma (83). Moreover, in elderly HF patients, high S100A8/A9 protein concentrations have been positively correlated with the levels of the inflammatory cytokines Interleukin-6 (IL-6) and Interleukin-8 (IL-8). Collectively, these observations imply that S100A8/A9 proteins may function as biomarkers in the evolution of HF (84).
Conclusions
Increasingly, research suggests that the concentrations of S100A8/A9 proteins in the bloodstream could be pivotal prognostic markers for negative cardiovascular outcomes in patients with acute and chronic heart failure, myocarditis, and thrombosis. Despite the intricate regulatory mechanisms of S100A8/A9, which involve complex transcriptional and post-translational processes and result in varied biological functions, the progress in developing potential therapeutics, such as humanized vaccines, antibodies, and inhibitors against S100A8/A9, is encouraging. These experimental findings have been successfully adapted to establish safe dosages for various immunoinflammatory diseases in clinical practice. Looking ahead, with the advancement of research technologies, the S100A8/A9 protein is expected to become a new focus for the diagnosis and treatment of cardiovascular diseases, potentially revolutionizing clinical approaches to diagnosis and therapy.
Author contributions
YS: Writing – original draft. HX: Conceptualization, Writing – original draft. WG: Writing – original draft. JD: Writing – original draft. XS: Writing – original draft. JL: Writing – review & editing. XL: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This research was supported by the grant from National Natural Science Foundation of China (82004277), Youth Natural Science Foundation of Shandong Province (ZR2020QH3090) and China Postdoctoral Funding Committee Special Funding Program (2022T150393).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fang H, Xie N, Qin L, Xia K, Fang F, Yang T. Correlation of serum calprotectin level with the range of coronary lesion in patients with acute coronary syndrome. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2014) 39(9):912–6. doi: 10.11817/j.issn.1672-7347.2014.09.008
2. Singh D, Vignat J, Lorenzoni V, Eslahi M, Ginsburg O, Lauby-Secretan B, et al. Global estimates of incidence and mortality of cervical cancer in 2020: a baseline analysis of the WHO global cervical cancer elimination initiative. Lancet Glob Health. (2023) 11(2):e197–206. doi: 10.1016/S2214-109X(22)00501-0
3. Johnson J, Jaggers RM, Gopalkrishna S, Dahdah A, Murphy AJ, Hanssen NMJ, et al. Oxidative stress in neutrophils: implications for diabetic cardiovascular complications. Antioxid Redox Signal. (2022) 36(10–12):652–66. doi: 10.1089/ars.2021.0116
4. Jakobsson G, Papareddy P, Andersson H, Mulholland M, Bhongir R, Ljungcrantz I, et al. Therapeutic S100A8/A9 blockade inhibits myocardial and systemic inflammation and mitigates sepsis-induced myocardial dysfunction. Crit Care. (2023) 27(1):374. doi: 10.1186/s13054-023-04652-x
5. Spratt DE, Barber KR, Marlatt NM, Ngo V, Macklin JA, Xiao Y, et al. A subset of calcium-binding S100 proteins show preferential heterodimerization. FEBS J. (2019) 286(10):1859–76. doi: 10.1111/febs.14775
6. Wilder PT, Varney KM, Weber DJ. Targeting S100 calcium-binding proteins with small molecule inhibitors. Methods Mol Biol. (2019) 1929:291–310. doi: 10.1007/978-1-4939-9030-6_19
7. Schafer BW, Heizmann CW. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci. (1996) 21(4):134–40. doi: 10.1016/S0968-0004(96)80167-8
8. Kazakov AS, Sofin AD, Avkhacheva NV, Deryusheva EI, Rastrygina VA, Permyakova ME, et al. Interferon-beta activity is affected by S100B protein. Int J Mol Sci. (2022) 23(4):1997. doi: 10.3390/ijms23041997
9. Liang H, Li J, Zhang K. Pathogenic role of S100 proteins in psoriasis. Front Immunol. (2023) 14:1191645. doi: 10.3389/fimmu.2023.1191645
10. Gonzalez LL, Garrie K, Turner MD. Role of S100 proteins in health and disease. Biochim Biophys Acta Mol Cell Res. (2020) 1867(6):118677. doi: 10.1016/j.bbamcr.2020.118677
11. Weisz J, Uversky VN. Zooming into the dark side of human annexin-S100 complexes: dynamic alliance of flexible partners. Int J Mol Sci. (2020) 21(16):5879. doi: 10.3390/ijms21165879
12. Charkhizadeh S, Imani M, Gheibi N, Shabaani F, Nikpajouh A, Rezvany MR. In vitro inhibitory effect of recombinant human calprotectin on Nalm6 leukemia cell line. Anticancer Agents Med Chem. (2020) 20(8):951–62. doi: 10.2174/1871520620666200331101209
13. Cotoi OS, Dunér P, Ko N, Hedblad B, Nilsson J, Björkbacka H, et al. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle-aged healthy individuals. Arterioscler Thromb Vasc Biol. (2014) 34(1):202–10. doi: 10.1161/ATVBAHA.113.302432
14. Marinkovic G, Grauen Larsen H, Yndigegn T, Szabo IA, Mares RG, de Camp L, et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur Heart J. (2019) 40(32):2713–23. doi: 10.1093/eurheartj/ehz461
15. Leukert N, Sorg C, Roth J. Molecular basis of the complex formation between the two calcium-binding proteins S100A8 (MRP8) and S100A9 (MRP14). Biol Chem. (2005) 386(5):429–34. doi: 10.1515/BC.2005.051
16. Cai Z, Xie Q, Hu T, Yao Q, Zhao J, Wu Q, et al. S100a8/A9 in myocardial infarction: a promising biomarker and therapeutic target. Front Cell Dev Biol. (2020) 8:603902. doi: 10.3389/fcell.2020.603902
17. Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and rac-2. FASEB J. (2005) 19(3):467–9. doi: 10.1096/fj.04-2377fje
18. Yi W, Zhu R, Hou X, Wu F, Feng R. Integrated analysis reveals S100a8/a9 regulates autophagy and apoptosis through the MAPK and PI3K-AKT signaling pathway in the early stage of myocardial infarction. Cells. (2022) 11(12):1911. doi: 10.3390/cells11121911
19. Sprenkeler EG, Zandstra J, van Kleef ND, Goetschalckx I, Verstegen B, Aarts CE, et al. S100a8/A9 is a marker for the release of neutrophil extracellular traps and induces neutrophil activation. Cells. (2022) 11(2):236. doi: 10.3390/cells11020236
20. Riuzzi F, Chiappalupi S, Arcuri C, Giambanco I, Sorci G, Donato R. S100 proteins in obesity: liaisons dangereuses. Cell Mol Life Sci. (2020) 77(1):129–47. doi: 10.1007/s00018-019-03257-4
21. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112(12):1796–808. doi: 10.1172/JCI200319246
22. Ghavami S, Eshragi M, Ande SR, Chazin WJ, Klonisch T, Halayko AJ, et al. S100a8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res. (2010) 20(3):314–31. doi: 10.1038/cr.2009.129
23. Petersen B, Wolf M, Austermann J, van Lent P, Foell D, Ahlmann M, et al. The alarmin Mrp8/14 as regulator of the adaptive immune response during allergic contact dermatitis. EMBO J. (2013) 32(1):100–11. doi: 10.1038/emboj.2012.309
24. Altwegg LA, Neidhart M, Hersberger M, Müller S, Eberli FR, Corti R, et al. Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J. (2007) 28(8):941–8. doi: 10.1093/eurheartj/ehm078
25. Ren X, Wen W, Fan X, Hou W, Su B, Cai P, et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. (2021) 184(7):1895–913. doi: 10.1016/j.cell.2021.01.053
26. Choi S, Liu X, Pan Z. Zinc deficiency and cellular oxidative stress: prognostic implications in cardiovascular diseases. Acta Pharmacol Sin. (2018) 39(7):1120–32. doi: 10.1038/aps.2018.25
27. Zhou Y, Hann J, Schenten V, Plançon S, Bueb JL, Tolle F, et al. Role of S100A8/A9 for cytokine secretion, revealed in neutrophils derived from ER-Hoxb8 progenitors. Int J Mol Sci. (2021) 22(16):8845. doi: 10.3390/ijms22168845
28. Jauch-Speer SL, Herrera-Rivero M, Ludwig N, De Carvalho BCV, Martens L, Wolf J, et al. C/EBPdelta-induced epigenetic changes control the dynamic gene transcription of S100a8 and S100a9. Elife. (2022) 11:e75594. doi: 10.7554/eLife.75594
29. Möller A, Jauch-Speer SL, Gandhi S, Vogl T, Roth J, Fehler O. The roles of toll-like receptor 4, CD33, CD68, CD69, or CD147/EMMPRIN for monocyte activation by the DAMP S100A8/S100A9. Front Immunol. (2023) 14:1110185. doi: 10.3389/fimmu.2023.1110185
30. Schenten V, Plançon S, Jung N, Hann J, Bueb JL, Bréchard S, et al. Secretion of the phosphorylated form of S100A9 from neutrophils is essential for the proinflammatory functions of extracellular S100A8/A9. Front Immunol. (2018) 9:447. doi: 10.3389/fimmu.2018.00447
31. Wolf M, Joseph R, Austermann J, Scharrnbeck-Davis C, Hermann S, Roth J, et al. S100a8/S100A9 integrates F-actin and microtubule dynamics to prevent uncontrolled extravasation of leukocytes. Biomedicines. (2023) 11(3):835. doi: 10.3390/biomedicines11030835
32. Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, et al. S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol. (2008) 181(8):5627–36. doi: 10.4049/jimmunol.181.8.5627
33. Jandeleit-Dahm K, Watson A, Soro-Paavonen A. The AGE/RAGE axis in diabetes-accelerated atherosclerosis. Clin Exp Pharmacol Physiol. (2008) 35(3):329–34. doi: 10.1111/j.1440-1681.2007.04875.x
34. Cesaro A, Anceriz N, Plante A, Pagé N, Tardif MR, Tessier PA. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis. PLoS One. (2012) 7(9):e45478. doi: 10.1371/journal.pone.0045478
35. Stenström M, Nyhlén HC, Törngren M, Liberg D, Sparre B, Tuvesson H, et al. Paquinimod reduces skin fibrosis in tight skin 1 mice, an experimental model of systemic sclerosis. J Dermatol Sci. (2016) 83(1):52–9. doi: 10.1016/j.jdermsci.2016.04.006
36. Stevens Barrón JC, Chapa González C, álvarez Parrilla E, De la Rosa LA. Nanoparticle-mediated delivery of flavonoids: impact on proinflammatory cytokine production: a systematic review. Biomolecules. (2023) 12(7):985. doi: 10.3390/biom12070985
37. Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, Wienbrandt AR, et al. S100a8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol. (2012) 107(2):250. doi: 10.1007/s00395-012-0250-z
38. Miyamoto S, Ueda M, Ikemoto M, Naruko T, Itoh A, Tamaki S, et al. Increased serum levels and expression of S100A8/A9 complex in infiltrated neutrophils in atherosclerotic plaque of unstable angina. Heart. (2008) 94(8):1002–7. doi: 10.1136/hrt.2007.121640
39. Averill MM, Kerkhoff C, Bornfeldt KE. S100a8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol. (2012) 32(2):223–9. doi: 10.1161/ATVBAHA.111.236927
40. Takano APC, Munhoz CD, Moriscot AS, Gupta S, Barreto-Chaves MLM. S100a8/MYD88/NF-қB: a novel pathway involved in cardiomyocyte hypertrophy driven by thyroid hormone. J Mol Med (Berl). (2017) 95(6):671–82. doi: 10.1007/s00109-017-1511-y
41. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100a8/A9 in inflammation. Front Immunol. (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
42. Björk P, Björk A, Vogl T, Stenström M, Liberg D, Olsson A, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. (2009) 7(4):e97. doi: 10.1371/journal.pbio.1000097
43. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slütter B, et al. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. (2017) 37(8):1457–61. doi: 10.1161/ATVBAHA.117.309575
44. Ionita MG, Vink A, Dijke IE, Laman JD, Peeters W, van der Kraak PH, et al. High levels of myeloid-related protein 14 in human atherosclerotic plaques correlate with the characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. (2009) 29(8):1220–7. doi: 10.1161/ATVBAHA.109.190314
45. McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, et al. S100a8 and S100A9 in human arterial wall. Implications for atherogenesis. J Biol Chem. (2005) 280(50):41521–9. doi: 10.1074/jbc.M509442200
46. Montagnana M, Danese E, Lippi G. Calprotectin and cardiovascular events. A narrative review. Clin Biochem. (2014) 47(12):996–1001. doi: 10.1016/j.clinbiochem.2014.02.021
47. Wang X, Guan M, Zhang X, Ma T, Wu M, Li Y, et al. The association between S100A8/A9 and the development of very late stent thrombosis in patients with acute myocardial infarction. Clin Appl Thromb Hemost. (2020) 26:1420625473. doi: 10.1177/1076029620943295
48. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119(1):91–112. doi: 10.1161/CIRCRESAHA.116.303577
49. Pruenster M, Vogl T, Roth J, Sperandio M. S100a8/A9: from basic science to clinical application. Pharmacol Ther. (2016) 167:120–31. doi: 10.1016/j.pharmthera.2016.07.015
50. Grebhardt S, Veltkamp C, Strobel P, Mayer D. Hypoxia and HIF-1 increase S100A8 and S100A9 expression in prostate cancer. Int J Cancer. (2012) 131(12):2785–94. doi: 10.1002/ijc.27591
51. Katashima T, Naruko T, Terasaki F, Fujita M, Otsuka K, Murakami S, et al. Enhanced expression of the S100A8/A9 complex in acute myocardial infarction patients. Circ J. (2010) 74(4):741–8. doi: 10.1253/circj.CJ-09-0564
52. Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation. (2020) 141(13):1080–94. doi: 10.1161/CIRCULATIONAHA.119.043833
53. Gupta A, Al-Tamimi AO, Halwani R, Alsaidi H, Kannan M, Ahmad F. Lipocalin-2, S100A8/A9, and cystatin C: potential predictive biomarkers of cardiovascular complications in COVID-19. Exp Biol Med (Maywood). (2022) 247(14):1205–13. doi: 10.1177/15353702221091990
54. Wu F, Zhang YT, Teng F, Li HH, Guo SB. S100a8/a9 contributes to sepsis-induced cardiomyopathy by activating ERK1/2-Drp1-mediated mitochondrial fission and respiratory dysfunction. Int Immunopharmacol. (2023) 115:109716. doi: 10.1016/j.intimp.2023.109716
55. Guo Q, Zhao Y, Li J, Liu J, Yang X, Guo X, et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe. (2021) 29(2):222–35. doi: 10.1016/j.chom.2020.12.016
56. Zheng J, Wang J, Liu H, Chen F, Wang H, Chen S, et al. Alarmins S100A8/A9 promote intervertebral disc degeneration and inflammation-related pain in a rat model through toll-like receptor-4 and activation of the NF-kappaB signaling pathway. Osteoarthritis Cartilage. (2022) 30(7):998–1011. doi: 10.1016/j.joca.2022.03.011
57. Lin CR, Wei TYW, Tsai HY, Wu YT, Wu PY, Chen ST. Glycosylation-dependent interaction between CD69 and S100A8/S100A9 complex is required for regulatory T-cell differentiation. FASEB J. (2015) 29(12):5006–17. doi: 10.1096/fj.15-273987
58. Yao Z, Zhang B, Niu G, Yan Z, Tong X, Zou Y, et al. Neutrophil infiltration characterized by upregulation of S100A8, S100A9, S100A12 and CXCR2 is associated with the co-occurrence of Crohn’s disease and peripheral artery disease. Front Immunol. (2022) 13:896645. doi: 10.3389/fimmu.2022.896645
59. Marinkovic G, Koenis DS, de Camp L, Jablonowski R, Graber N, de Waard V, et al. S100a9 links inflammation and repair in myocardial infarction. Circ Res. (2020) 127(5):664–76. doi: 10.1161/CIRCRESAHA.120.315865
60. Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. (2006) 113(19):2278–84. doi: 10.1161/CIRCULATIONAHA.105.607333
61. Silvaa EJ, Argyris PP, Zou X, Ross KF, Herzberg MC. S100a8/A9 regulates MMP-2 expression and invasion and migration by carcinoma cells. Int J Biochem Cell Biol. (2014) 55:279–87. doi: 10.1016/j.biocel.2014.09.007
62. Vogel B, Claessen BE, Arnold SV, Chan D, Cohen DJ, Giannitsis E, et al. ST-segment elevation myocardial infarction. Nat Rev Dis Primers. (2019) 5(1):39. doi: 10.1038/s41572-019-0090-3
63. Hausenloy DJ, Yellon DM. Ischaemic conditioning and reperfusion injury. Nat Rev Cardiol. (2016) 13(4):193–209. doi: 10.1038/nrcardio.2016.5
64. Walters AM, Porter GJ, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res. (2012) 111(9):1222–36. doi: 10.1161/CIRCRESAHA.112.265660
65. Schiopu A, Cotoi OS. S100a8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. (2013) 2013:828354. doi: 10.1155/2013/828354
66. Scott NR, Swanson RV, Al-Hammadi N, Domingo-Gonzalez R, Rangel-Moreno J, Kriel BA, et al. S100a8/A9 regulates CD11b expression and neutrophil recruitment during chronic tuberculosis. J Clin Invest. (2020) 130(6):3098–112. doi: 10.1172/JCI130546
67. Drummond RA, Swamydas M, Oikonomou V, Zhai B, Dambuza IM, Schaefer BC, et al. CARD9(+) microglia promote antifungal immunity via IL-1beta- and CXCL1-mediated neutrophil recruitment. Nat Immunol. (2019) 20(5):559–70. doi: 10.1038/s41590-019-0377-2
68. Ma J, Li Y, Li P, Yang X, Zhu S, Ma K, et al. S100a8/A9 as a prognostic biomarker with causal effects for post-acute myocardial infarction heart failure. Nat Commun. (2024) 15(1):2701. doi: 10.1038/s41467-024-46973-7
69. Li Y, Chen B, Yang X, Zhang C, Jiao Y, Li P, et al. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. (2019) 140(9):751–64. doi: 10.1161/CIRCULATIONAHA.118.039262
70. Boralkar KA, Kobayashi Y, Amsallem M, Ataam JA, Moneghetti KJ, Cauwenberghs N, et al. Value of neutrophil to lymphocyte ratio and its trajectory in patients hospitalized with acute heart failure and preserved ejection fraction. Am J Cardiol. (2020) 125(2):229–35. doi: 10.1016/j.amjcard.2019.10.020
71. Bai B, Xu Y, Chen H. Pathogenic roles of neutrophil-derived alarmins (S100A8/A9) in heart failure: from molecular mechanisms to therapeutic insights. Br J Pharmacol. (2023) 180(5):573–88. doi: 10.1111/bph.15998
72. Yan X, Wu L, Gao M, Yang P, Yang J, Deng Y. Omentin inhibits the resistin-induced hypertrophy of H9c2 cardiomyoblasts by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway. Exp Ther Med. (2022) 23(4):292. doi: 10.3892/etm.2022.11222
73. Hirata A, Kishida K, Nakatsuji H, Hiuge-Shimizu A, Funahashi T, Shimomura I. High serum S100A8/A9 levels and high cardiovascular complication rate in type 2 diabetics with ultrasonographic low carotid plaque density. Diabetes Res Clin Pract. (2012) 97(1):82–90. doi: 10.1016/j.diabres.2012.01.026
74. Ma L, Shi H, Li Y, Gao W, Guo J, Zhu J, et al. Hypertrophic preconditioning attenuates myocardial ischemia/reperfusion injury through the deacetylation of isocitrate dehydrogenase 2. Sci Bull (Beijing). (2021) 66(20):2099–114. doi: 10.1016/j.scib.2021.04.008
75. Wu Y, Li Y, Zhang C, Wang Y, Cui W, Li H, et al. S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II-induced cardiac inflammation and injury. Hypertension. (2014) 63(6):1241–50. doi: 10.1161/HYPERTENSIONAHA.113.02843
76. Fransén Pettersson N, Deronic A, Nilsson J, Hannibal TD, Hansen L, Schmidt-Christensen A, et al. The immunomodulatory quinoline-3-carboxamide paquinimod reverses established fibrosis in a novel mouse model for liver fibrosis. PLoS One. (2018) 13(9):e0203228. doi: 10.1371/journal.pone.0203228
77. Du L, Chen Y, Shi J, Yu X, Zhou J, Wang X, et al. Inhibition of S100A8/A9 ameliorates renal interstitial fibrosis in diabetic nephropathy. Metab Clin Exp. (2023) 144:155376. doi: 10.1016/j.metabol.2022.155376
78. Sreejit G, Latif AA, Murphy AJ, Nagareddy PR. Emerging roles of neutrophil-borne S100A8/A9 in cardiovascular inflammation. Pharmacol Res. (2020) 161:105212. doi: 10.1016/j.phrs.2020.105212
79. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17(6):327–40. doi: 10.1038/s41569-019-0326-7
80. Bai B, Yang W, Fu Y, Foon HL, Tay WT, Yang K, et al. Seipin knockout mice develop heart failure with preserved ejection fraction. JACC Basic Transl Sci. (2019) 4(8):924–37. doi: 10.1016/j.jacbts.2019.07.008
81. Bai B, Cheng M, Jiang L, Xu J, Chen H, Xu Y. High neutrophil to lymphocyte ratio and its gene signatures correlate with diastolic dysfunction in heart failure with preserved ejection fraction. Front Cardiovasc Med. (2021) 8:614757. doi: 10.3389/fcvm.2021.614757
82. Yan L, Bjork P, Butuc R, Gawdzik J, Earley J, Kim G, et al. Beneficial effects of quinoline-3-carboxamide (ABR-215757) on atherosclerotic plaque morphology in S100A12 transgenic ApoE null mice. Atherosclerosis. (2013) 228(1):69–79. doi: 10.1016/j.atherosclerosis.2013.02.023
83. Ma LP, Haugen E, Ikemoto M, Fujita M, Terasaki F, Fu M. S100a8/A9 complex as a new biomarker in prediction of mortality in elderly patients with severe heart failure. Int J Cardiol. (2012) 155(1):26–32. doi: 10.1016/j.ijcard.2011.01.082
Keywords: cardiovascular disease, S100A8/A9 protein, inflammation, neutrophil, biomarker
Citation: Sun Y, Xu H, Gao W, Deng J, Song X, Li J and Liu X (2024) S100a8/A9 proteins: critical regulators of inflammation in cardiovascular diseases. Front. Cardiovasc. Med. 11:1394137. doi: 10.3389/fcvm.2024.1394137
Received: 1 March 2024; Accepted: 24 July 2024;
Published: 8 August 2024.
Edited by:
Dragos Cretoiu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Goran Marinković, Cerba Research, NetherlandsNanako Kawaguchi, Tokyo Women's Medical University, Japan
© 2024 Sun, Xu, Gao, Deng, Song, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jie Li, MTgyNDAyNTY4MkBxcS5jb20=; Xijian Liu, NjAwMjAwNjlAc2R1dGNtLmVkdS5jbg==