 Yu Zhang†
Yu Zhang†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 26 January 2023
Sec. Cardiovascular Metabolism
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1083935
This article is part of the Research Topic Recent Advances in Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) in Heart-Related Diseases View all 5 articles
Mitochondria-associated endoplasmic reticulum membranes (MAMs) are formed by physical connections of the endoplasmic reticulum and mitochondria. Over the past decades, great breakthroughs have been made in the study of ER-mitochondria communications. It has been identified that MAM compartments are pivotal in regulating neurological function. Accumulating studies indicated that MAMs participate in the development of cardiovascular diseases. However, the specific role of MAMs in heart failure remains to be fully understood. In this article, we first summarize the structural and functional properties of MAM and MAM-associated proteins. We then focus on the roles of MAMs in myocardial infarction, cardiomyopathy and heart failure, and discuss the involvement of MAMs in disease progression and treatment. Elucidating these issues may provide important insights into therapeutic intervention of heart failure.
Mitochondria and endoplasmic reticulum (ER) are the two essential organelles which are tightly intertwined in eukaryotic cells (1). Mitochondria are the core parts of cell energy metabolism in maintaining the cellular function. Whereas ER, also known as sarcoplasmic reticulum (SR) in myocytes, participates in calcium storage, protein folding and processing, lipid metabolism (2). Mitochondria-associated ER membranes (MAMs), membranous contact sites between mitochondria and ER, bidirectionally regulates organelle physiological functions like lipid and Ca2+ homeostasis, mitochondrial dynamics, autophagy and apoptosis. Interruption of ER-mitochondria communication is a major cause of altered cellular homeostasis, which can lead to serious diseases including cancer, neurological diseases and cardiovascular diseases (CVDs) (3).
Cardiovascular diseases are the leading cause of death in the world, which consists of hypertension, acute myocardial infarction (AMI), cardiomyopathy, heart failure and other cardiac problems (4). Heart failure, the common end-stage of most cardiovascular diseases, is caused by hypertension, myocardial infarction (MI), ischemia and cardiomyopathies (5). In this article, we first describe the structural and functional properties of MAMs in cardiomyocytes, and then focus on their function in the development of heart failure. Interpretation of these issues may provide important diagnostic value and potential targets for heart failure.
Mitochondria-associated ER membranes, composed of ER subdomains placed alongside with the outer membrane of mitochondria (OMM), can fluctuate dynamically. Electron microscopy revealed a distance of approximately 10–25 nm between the ER and OMM (6). The two organelles maintain stable and dynamic communications by the protein tethers. Proteomics evaluation demonstrated that MAMs components, highly conserved among different species and different tissues, play a direct physical tethering connection role or act as modulators of the tethering complexes in MAMs (7).
The ER Ca2+ channel inositol 1,4,5-triphosphate receptors (IP3Rs) physically connect with OMM voltage-dependent anion channels (VDACs) via the cytoplasmic chaperone glucose-regulated protein 75 (GRP75), forming a tripartite complex to modulate ER-mitochondria juxtaposition (8). In mouse primary neurons, GRP75 promotes ER-mitochondria tethering and mitochondrial Ca2+, thus enhancing ATP production (9).
The ER membrane protein vesicle-associated membrane protein associated protein B (VAPB), binds to the OMM protein tyrosine phosphatase-interacting protein-51 (PTPIP51), forming VAPB-PTPIP51 tethering complex which regulates ER-mitochondria Ca2+ transmission (10). Disruption of their interaction causes MAMs dissociation, and disturbs mitochondrial Ca2+ import and ATP synthesis (11). Besides, oxysterol-binding protein-related protein 5/8 (ORP5/8), enriched at MAMs in mammalian cells, physically interacts with PTPIP51. Inhibition of ORP5/ORP8 contributes to mitochondria morphology defects and respiratory dysfunction (12).
Mitofusin2 (MFN2), mitochondrial fusion regulator, is recognized as an important constituent of MAMs. ER-resident MFN2 forms homodimer or heterodimer with either mitofusin (MFN1) or MFN2 on the OMM (13). MFN2 depletion promotes the ER-mitochondria connections and mitochondrial Ca2+ uptake from ER, indicating that MFN2 is more than a physical tether (14, 15).
During the apoptotic process, the ER-located B-cell receptor-associated protein 31 (BAP31) is associated with OMM protein, the mitochondrial fission 1 protein (Fis1), acting as another tether for MAMs to induce apoptosis (16). Besides, phosphofurin acidic cluster sorting protein 2 (PACS-2), the first MAM protein identified to be involved in MAM formation, regulates the tethering of mitochondria with ER in a BAP31-dependent manner (17).
Increasing evidence suggests that MAMs provide a platform for maintaining intracellular homeostasis and biological functions (18) (Figure 1).
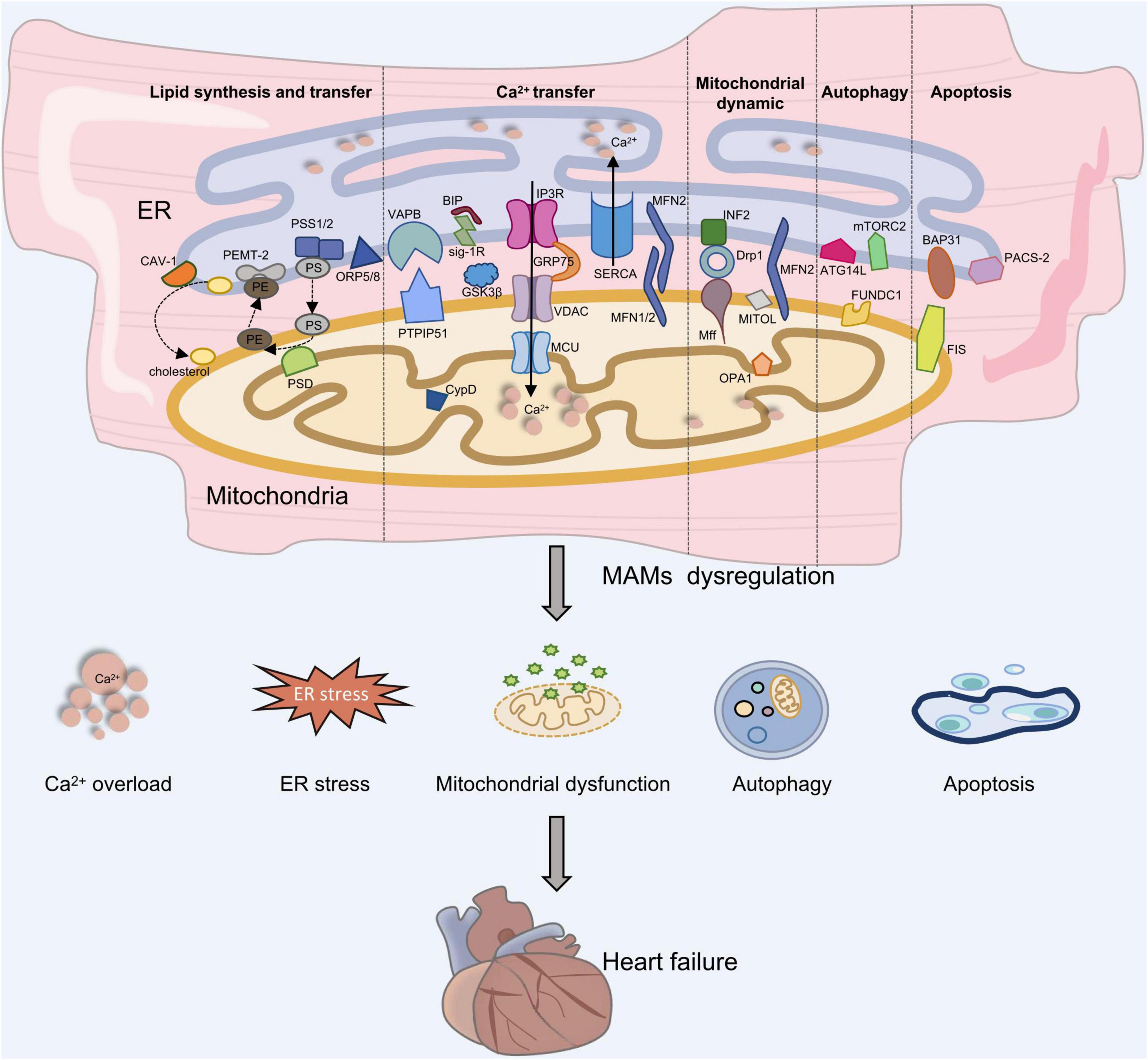
Figure 1. The role of MAMs in heart failure development. Proteins located on the ER surface, such as IP3R, VAPB, MFN2, and BAP31 interact with their counterparts on the OMM like VDAC, PTPIP51 or ORP5/8, MFN1/2, and Fis1, forming four major tethering complexes at MAMs in mammalian cells. Depletion of MAMs can lead to Ca2+ overload, ES stress, mitochondrial dysfunction, autophagy and apoptosis, which is involved in the development of heart failure.
MAMs are abundant in proteins involved in lipid metabolisms including phosphatidylserine synthase (PSS), fatty acid CoA ligase 4 (FACL4), phosphatidylethanolamine N-methyltransferase 2 (PEMT2), phosphatidylserine decarboxylase (PSD) (19), as well as in phospholipid, triacylglycerol synthesis and steroidogenesis (20). Caveolin-1 (CAV-1) is an integral component distributed abundant on MAMs, which promotes lipid and cholesterol metabolism (21).
Ca2+ is a second messenger modulating multiple cellular activities such as cell metabolism and apoptosis (22). As mentioned above, the effective Ca2+ transmission at MAMs is regulated by multiple protein complexes. IP3R1-GRP75-VDAC1 tethering complex forms a Ca2+ regulatory axis with mitochondrial calcium uniporter (MCU), which mediates Ca2+ transmission from ER to mitochondria (23). In addition, ER chaperone proteins like Sigma-1 receptor (Sig-1R) physically associate at MAM, and regulate Ca2+ transmission through IP3R3 (24).
Mitochondrial dynamics include mitochondrial fission and fusion, which are crucial for maintaining cellular homeostasis. Proteins involved in mitochondrial dynamics are enriched in MAMs (25). MFN1 and MFN2 regulate OMM fusion, while Optic atrophy 1 (OPA1) modulates IMM fusion. The mitochondrial fission process is regulated by dynamin-related protein1 (Drp1), Fis1, mitochondrial fission factor (Mff), and mitochondrial dynamic proteins of 49 and 51 kDa (MiD49/51). A recent study emphasized that MAMs are the origin sites of mitochondrial fission (26).
Many autophagic proteins are located in MAMs and autophagosomal membranes may originate from MAMs (27). Under starvation, the pre-autophagosome marker autophagy-related 14-like (ATG14L) promotes relocalization of the autophagy induction factor mammalian target of rapamycin complex 2 (mTORC2) to MAMs and initiates autophagosome formation (28). It is reported that decreased MAMs tethering complexes result in abnormal hippocampal autophagy in rats (29). Moreover, MAMs modulate apoptosis via Ca2+ regulation. Evidence showed that Ca2+ overload can induce mitochondrial permeability transition pore (mPTP) opening and apoptosis (30). BAP31-Fis1 tethering complex recruits procaspase-8, which promotes the release of Ca2+ stores in ER and causes apoptosis (16).
MI is an acute syndrome of CVD with high death rate (31). Myocardial ischemia/reperfusion (I/R) injury, a pathophysiological status after the ischemic myocardium returns to normal perfusion, is commonly deemed as a serious risk factor for coronary artery disease (32). MI is characterized by cardiac injury, myocardial cell death and abnormal cardiac function, which will lead to heart failure and death (33). Myocardial I/R injury is mainly associated with oxidative stress, ER stress and mitochondrial dysfunction (34). Among these, calcium overload is the leading cause of disordered oxidative phosphorylation and contributes to mitochondrial dysfunction (25).
Several components of ER/SR-mitochondria tethering complex participate in Ca2+ homeostasis and mediate mPTP opening and I/R damage. Cyclophilin D (CypD), encoded by Ppif, is located in the mitochondrial matrix and acts as a crucial regulator for mPTP opening and necrosis. CypD interacts synergistically with the VDAC1-Grp75-IP3R1 complex and enhances ER Ca2+ efflux into mitochondria (Table 1) (35). CypD overexpression induced mPTP opening without stimulating cell death, whereas CypD inactivation significantly reduced myocardial infarct size during I/R and ameliorated myocardial injury via impeding Ca2+ overload (36, 37). Notably, the protective effect of preconditioning was absent in Ppif–/– mice, which were more susceptible to heart failure, indicating that CypD plays a dual function in I/R (38, 39).
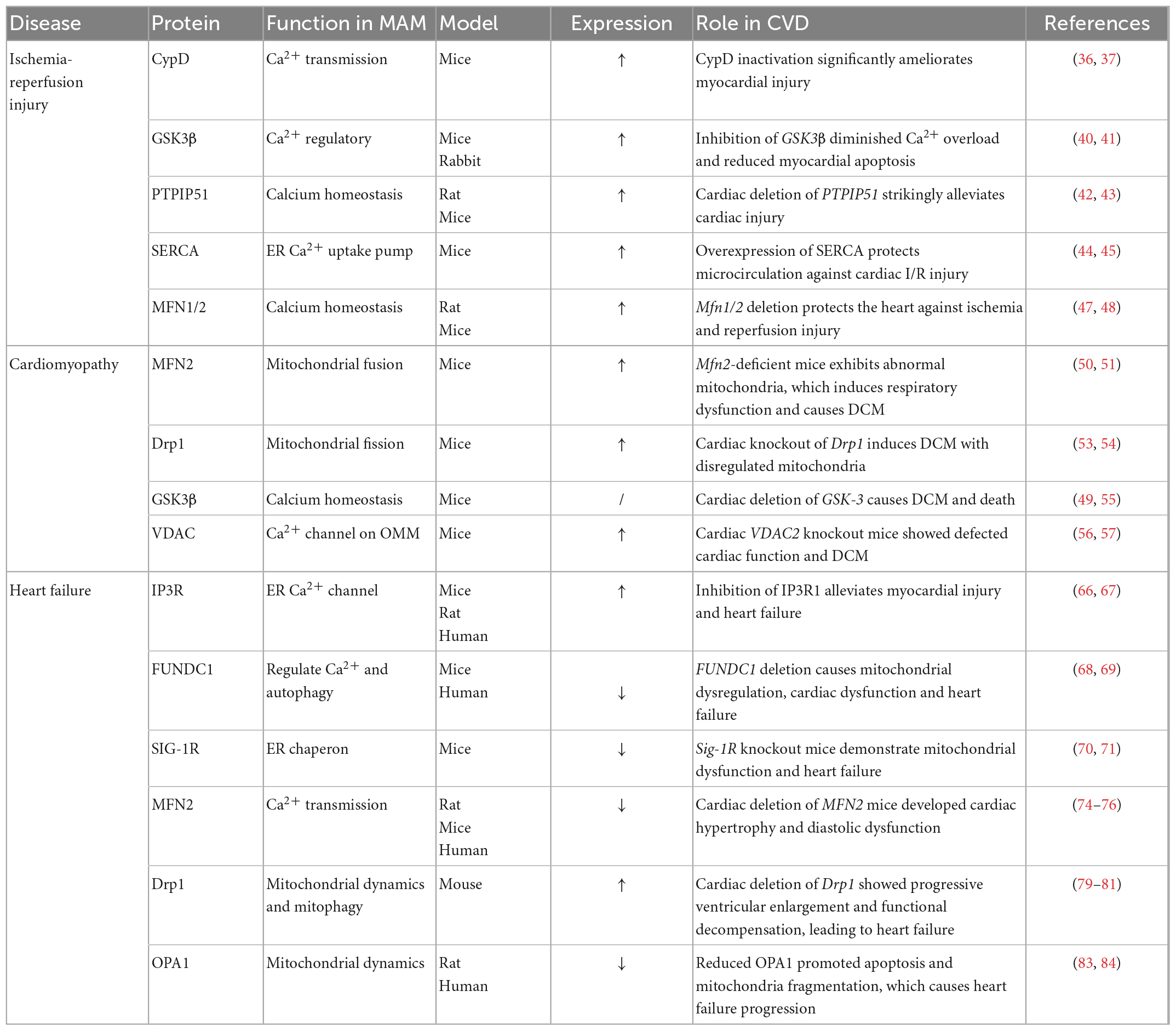
Table 1. The roles of MAMs related proteins in CVDs.
Glycogen synthase kinase-3 beta (GSK-3β), a new Ca2+ regulator located in the SR/ER, specifically interacts with the IP3R Ca2+-channeling complex and regulates Ca2+ transfer in cardiomyocytes (40). GSK-3β inhibition diminished Ca2+ overload and reduced myocardial apoptosis resulted from I/R, thereby providing cardioprotection (40). Moreover, GSK-3β inhibitors attenuated infarct size in mice and rabbits, indicating drug administration was a feasible method (41). VAPB-PTPIP51 is a widely accepted tethering complex in MAMs. PTPIP51 is markedly increased in mice I/R hearts. PTPIP51 overexpression mediated excessive mitochondrial Ca2+ uptake, but reversed by MCU inhibition, which protected cardiomyocytes against PTPIP51-mediated apoptosis (42). Cardiac knockdown of PTPIP51 strikingly alleviates cardiac injury after myocardial I/R, indicating that PTPIP51 might be a potential target for ischemic heart disease. Furthermore, downregulation of VAPB or PTPIP51 promotes autophagy by reducing mitochondrial Ca2+ levels (43).
Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), the main pump for Ca2+ uptake in the ER, regulates calcium homeostasis by interacting with calnexin. SERCA ameliorated reperfusion-induced myocardial injury by employing gene delivery strategies targeting SERCA (44, 45). Mitochondrial dynamics is crucial in myocardial I/R by regulating mPTP opening (46). MFN1-MFN2 complex is implicated in modulating mitochondrial fission and maintaining ER-mitochondria microdomain (13). Acute deletion of Mfn1 and Mfn2 prevented myocardial I/R injury and reduced infarct size (47). Mfn2 knockout hearts exhibited resistance to I/R injury, however long-term Mfn2 deletion contributed to cardiac dysfunction (47, 48).
Cardiomyopathy, myocardial disorder with abnormal cardiac muscle, can be either acquired or inherited. Hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) are the most common cardiomyopathies. Among these, DCM is the most common cause of heart failure, which is characterized by structural thinning and dilation of heart chambers with a progressively defected cardiac function (49).
It has been reported that cardiac deficiency of Mfn1 and Mfn2 exhibits progressive DCM and heart failure in succession (50). Structural and functional abnormal mitochondria were observed in Mfn2-deficient mice, which induced respiratory dysfunction and caused DCM (51). Moreover, mice with Drp1 mutation demonstrated cardiomyopathy with punctuate calcification in the heart (52). Cardiac depletion of Drp1 induced DCM after birth and rapid death in mice (53, 54). Increased mitochondrial connection, accumulated ubiquitinated proteins as well as reduced respiration was observed in Drp1 knockout cardiomyocytes. These studies indicate that Drp1 is crucial in regulating mitochondrial quality and myocardial survival.
Adult cardiac deletion of a multifunctional regulator GSK-3 contributed to severe DCM due to cell cycle dysregulation, indicating that GSK-3 is involved in maintaining cardiac homeostasis (49, 55). VDAC, the most abundant mitochondrial outer membrane protein, contains three subtypes-VDAC 1, 2, and 3 in mammalian cells (56). It has been reported that VDAC1 is upregulated in the left ventricle of HCM patients (57). VDAC1 inhibition significantly attenuated mitochondrial Ca2+ overload and protected cells from hypoxia-reoxygenation (H/O) (58). Cardiac VDAC2 knockout mice showed decreased ejection fraction and increased brain natriuretic peptide (BNP) level and cardiac fibrosis, which was consistent with DCM features (56).
Heart failure is a complicated pathophysiological syndrome of cardiac pumping failure (5). Conditions such as ischemia, pressure or volume overload, cardiac hypertrophy, cardiomyopathy, will eventually lead to heart failure (59). It is well recognized that mitochondrial function and Ca2+ homeostasis are significant in cardiac remodeling and heart failure (60–62). The cardiac rhythmicity and contraction require energy, which is driven by mitochondrial oxidative phosphorylation (63). Besides, SR, a membrane system with a high density of Ca2+-ATPases, maintains optimal calcium levels for myocardial contraction. Disruption of Ca2+ homeostasis can trigger ER stress and energy metabolism defects, affecting the development of heart failure (64, 65).
Mitochondrial Ca2+ dysregulation is involved in cardiac remodeling and heart failure. IP3R is a ligand-gated calcium channel located in the ER/SR, with isoform IP3R-2 predominant in the heart. IP3R is associated with cardiac remodeling in response to various stress that cause hypertrophy (66). The expression and activity of IP3R is enhanced under pathological conditions such as cardiac hypertrophy and heart failure. Moreover, inhibition of IP3R1 alleviates myocardial injury and heart failure (67). FUNDC1, a highly conserved OMM protein, maintains MAM formation by interacting with IP3R2 and regulates mitophagy (68). Compared with healthy group, the expression level of FUNDC1 and the number of SR-mitochondria contacts are dramatically reduced in heart failure patients. The decreased FUNDC1 level in MAMs contributed to impaired SR Ca2+ transportation to mitochondria through inhibition of IP3R2 ubiquitin-dependent degradation, resulting in perturbation of the CREB/Fis1 pathway and eventually compromising cardiac function. Besides, FUNDC1 knockout mice showed diastolic and systolic dysfunction (69). In contrast, FUNDC1 overexpression elevates both cytosolic and mitochondrial Ca2+ levels in cardiomyocytes, and lowers SR Ca2+ levels. Sig-1R disassociated from the binding immunoglobulin protein (BiP) and prolonged mitochondria Ca2+ uptake via IP3R under ER stress (70). Sig-1R regulates Ca2+ transfer into mitochondria to promote ATP production (71). Sig-1R knockout mice displayed mitochondrial dysfunction and cardiac remodeling, causing cardiac dysfunction. Besides, Fluvoxamine, possessing high Sig-1R affinity, alleviated heart failure in both mice and rat models subjected to TAC (72).
Mitochondrial dynamics is participated in cardiac hypertrophy and heart failure progression. Cardiac depletion of Mfn1/2 in mice showed impaired heart function with increased left ventricular end-diastolic volume and decreased fractional shortening (48, 73). Accumulating studies have demonstrated that MFN2 was downregulated in heart failure models induced by spontaneously hypertensive rats (SHR) or TAC (74). Consistently, MFN2 was decreased in hypertrophic cardiomyocytes induced by Angiotensin II (Ang II), accompanied by the alterations of mitochondria morphology (75). A study showed that MFN1/MFN2 double knockout mice died at the embryonic stage due to heart failure (50). Besides, cardiac deletion of MFN2 mice developed cardiac hypertrophy and moderate diastolic dysfunction (76). Conversely, MFN2 overexpression alleviated Ang-II induced cardiac hypertrophy (77). Intriguingly, sex hormones (estrogen and testosterone) can increase cardiac expression of Mfn1 and Mfn2, suggesting that further study is needed on the regulatory effect of the sex hormones, and their cardioprotective effects (78). Drp1, mitochondrial fission regulator, is upregulated in damaged cardiac tissues induced by doxorubicin. A study showed that Drp1 inhibitor will be a promising pharmacological agent, which inhibits the excessive mitochondrial fission mediated by doxorubicin and ameliorates its cardiotoxicity (79). Drp1 deficiency in adult mouse hearts showed the pathophysiological consequences of progressive ventricular enlargement and functional decompensation, resulting in heart failure (80). Drp1-dependent mitochondrial autophagy exerts a protective role in mitochondrial dysfunction and heart failure resulted from pressure overload (81). At present, microRNAs (miR) are being studied as therapeutic targets for CVDs. It is reported that miR 499 protects heart against MI by inhibiting mitochondrial fission mediated by Drp1 (82). OPA1, mediates IMM fusion and acts as a crucial regulator of morphological change in cardiac physiology. Altered OPA1 function was proposed to lead to the pathogenesis of heart failure (83). Studies have shown that protein levels of OPA1 were reduced in both rat and human heart failure models accompanied with mitochondrial fragmentation. Reduced OPA1 promoted apoptosis and mitochondria fragmentation, which may contribute to heart failure progression with progressive loss of cardiac myocytes (84). Together, mitochondrial dynamics are essential to maintain cardiac structure and function, which may act as a potential strategy to prevent myocardial hypertrophy and heart failure.
MAMs, membranous contact sites between mitochondria and ER, regulate various cellular processes including Ca2+ homeostasis, mitochondrial dynamics, autophagy and apoptosis. Heart failure is the final stage of diverse CVDs. Accumulating studies have defined the essential function of MAMs in the development of heart failure. For instance, depletion of CypD contributed to reduced myocardial infarction and ameliorated cardiac function (36). Upregulation of PTPIP51 caused cardiac injury by promoting mitochondrial Ca2+ overload and apoptosis, whereas PTPIP51 depletion significantly protected the heart from I/R injury (42). Besides, Mfn2 knockout mice developed dilated cardiomyopathy (51). Dysregulation of Drp1 in cardiomyocytes contributes to myocardial injury and heart failure (82). These studies indicate that MAMs may act as biomarkers and potential therapeutic targets in heart failure. The function of MAMs in cardiovascular diseases worth more attention due to their multifunctional. The improved understanding of MAMs integrity regulation or MAMs targets identification might provide significant therapeutic strategies for cardiovascular diseases.
XS: conceived and designed the review. YZ, JY, MZ, and YW: collected the literatures. YZ, JY, and XS: wrote the manuscript. XS, MZ, and YW: reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (82170370 and 81770381), the Natural Science Foundation of Jiangsu Province (BK20211165), and Zhishan Youth Scholar Program of SEU.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Luan Y, Ren K, Luan Y, Chen X, Yang Y. Mitochondrial dynamics: pathogenesis and therapeutic targets of vascular diseases. Front Cardiovasc Med. (2021) 8:770574. doi: 10.3389/fcvm.2021.770574
2. Yang Y, Zhou Q, Gao A, Chen L, Li L. Endoplasmic reticulum stress and focused drug discovery in cardiovascular disease. Clin Chim Acta. (2020) 504:125–37. doi: 10.1016/j.cca.2020.01.031
3. Perrone M, Caroccia N, Genovese I, Missiroli S, Modesti L, Pedriali G, et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int Rev Cell Mol Biol. (2020) 350:119–96. doi: 10.1016/bs.ircmb.2019.11.002
4. Amini M, Zayeri F, Salehi M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: results from global burden of disease study 2017. BMC Public Health. (2021) 21:401. doi: 10.1186/s12889-021-10429-0
5. Shi X, Chen R, Zhang Y, Yun J, Brand-Arzamendi K, Liu X, et al. Zebrafish heart failure models: opportunities and challenges. Amino Acids. (2018) 50:787–98. doi: 10.1007/s00726-018-2578-7
6. Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle K, et al. Structural and functional features and significance of the physical linkage between Er and mitochondria. J Cell Biol. (2006) 174:915–21. doi: 10.1083/jcb.200604016
7. Wang X, Wen Y, Dong J, Cao C, Yuan S. Systematic in-depth proteomic analysis of mitochondria-associated endoplasmic reticulum membranes in mouse and human testes. Proteomics. (2018) 18:e1700478. doi: 10.1002/pmic.201700478
8. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski M, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. (2006) 175:901–11. doi: 10.1083/jcb.200608073
9. Lee S, Wang W, Hwang J, Namgung U, Min K. Increased Er-Mitochondria tethering promotes axon regeneration. Proc Natl Acad Sci U.S.A. (2019) 116:16074–9. doi: 10.1073/pnas.1818830116
10. Gómez-Suaga P, Pérez-Nievas B, Glennon E, Lau D, Paillusson S, Mórotz G, et al. The Vapb-Ptpip51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathol Commun. (2019) 7:35. doi: 10.1186/s40478-019-0688-4
11. Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine M, et al. A -Synuclein Binds to the Er-Mitochondria Tethering Protein Vapb to Disrupt Ca(2+) Homeostasis and Mitochondrial Atp Production. Acta Neuropathol. (2017) 134:129–49. doi: 10.1007/s00401-017-1704-z
12. Galmes R, Houcine A, van Vliet A, Agostinis P, Jackson C, Giordano F. Orp5/Orp8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. (2016) 17:800–10. doi: 10.15252/embr.201541108
13. Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, et al. Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc Natl Acad Sci U.S.A. (2016) 113:11249–54. doi: 10.1073/pnas.1606786113
14. Han S, Nandy P, Austria Q, Siedlak S, Torres S, Fujioka H, et al. Mfn2 Ablation in the adult mouse hippocampus and cortex causes neuronal death. Cells. (2020) 9:116. doi: 10.3390/cells9010116
15. Mu G, Deng Y, Lu Z, Li X, Chen Y. Mir-20b suppresses mitochondrial dysfunction-mediated apoptosis to alleviate hyperoxia-induced acute lung injury by directly targeting Mfn1 and Mfn2. Acta Biochim Biophys Sin. (2021) 53:220–8. doi: 10.1093/abbs/gmaa161
16. Iwasawa R, Mahul-Mellier A, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the Mitochondria-Er Interface to establish a platform for apoptosis induction. Embo J. (2011) 30:556–68. doi: 10.1038/emboj.2010.346
17. Li C, Li L, Yang M, Zeng L, Sun L. Pacs-2: a key regulator of mitochondria-associated membranes (MAMS). Pharmacol Res. (2020) 160:105080. doi: 10.1016/j.phrs.2020.105080
18. Luan Y, Luan Y, Yuan R, Feng Q, Chen X, Yang Y. Structure and function of mitochondria-associated endoplasmic reticulum membranes (MAMS) and their role in cardiovascular diseases. Oxid Med Cell Longev. (2021) 2021:4578809. doi: 10.1155/2021/4578809
19. Anastasia I, Ilacqua N, Raimondi A, Lemieux P, Ghandehari-Alavijeh R, Faure G, et al. Mitochondria-Rough-Er Contacts in the Liver Regulate Systemic Lipid Homeostasis. Cell Rep. (2021) 34:108873. doi: 10.1016/j.celrep.2021.108873
20. Prasad M, Kaur J, Pawlak K, Bose M, Whittal R, Bose H. Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (STAR)-Voltage-Dependent Anion Channel 2 (VDAC2) Interaction. J Biol Chem. (2015) 290:2604–16. doi: 10.1074/jbc.M114.605808
21. Sala-Vila A, Navarro-Lérida I, Sánchez-Alvarez M, Bosch M, Calvo C, López J, et al. Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci Rep. (2016) 6:27351. doi: 10.1038/srep27351
22. Giorgi C, Danese A, Missiroli S, Patergnani S, Pinton P. Calcium dynamics as a machine for decoding signals. Trends Cell Biol. (2018) 28:258–73. doi: 10.1016/j.tcb.2018.01.002
23. Xu H, Guan N, Ren Y, Wei Q, Tao Y, Yang G, et al. Ip3r-Grp75-Vdac1-Mcu calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an adriamycin nephropathy rat model. BMC Nephrol. (2018) 19:140. doi: 10.1186/s12882-018-0940-3
24. Gómez-Suaga P, Bravo-San Pedro J, González-Polo R, Fuentes J, Niso-Santano M. Er-Mitochondria signaling in parkinson’s disease. Cell Death Dis. (2018) 9:337. doi: 10.1038/s41419-017-0079-3
25. Wang Y, Zhang X, Wen Y, Li S, Lu X, Xu R, et al. Endoplasmic reticulum-mitochondria contacts: a potential therapy target for cardiovascular remodeling-associated diseases. Front Cell Dev Biol. (2021) 9:774989. doi: 10.3389/fcell.2021.774989
26. Friedman J, Lackner L, West M, DiBenedetto J, Nunnari J, Voeltz G. Er tubules mark sites of mitochondrial division. Science. (2011) 334:358–62. doi: 10.1126/science.1207385
27. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at Er-mitochondria contact sites. Nature. (2013) 495:389–93. doi: 10.1038/nature11910
28. Wu X, Luo L, Kong R, Song Y, Li Q, Nice E, et al. Recent advances in autophagic machinery: a proteomic perspective. Expert Rev Proteomics. (2020) 17:561–79. doi: 10.1080/14789450.2020.1808464
29. Liu J, Wang L, Ge L, Sun W, Song Z, Lu X, et al. Lanthanum decreased Vapb-Ptpp51, Bap31-Fis1, and Mfn2-Mfn1 expression of mitochondria-associated membranes and induced abnormal autophagy in rat hippocampus. Food Chem Toxicol. (2022) 161:112831. doi: 10.1016/j.fct.2022.112831
30. Wang M, Tan J, Miao Y, Li M, Zhang Q. Role of Ca2+ and ion channels in the regulation of apoptosis under hypoxia. Histol Histopathol. (2018) 33:237–46. doi: 10.14670/hh-11-918
31. Thygesen K, Alpert J, White H, Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Universal definition of myocardial infarction. J Am Coll Cardiol. (2007) 50:2173–95. doi: 10.1016/j.jacc.2007.09.011
32. Zhou M, Yu Y, Luo X, Wang J, Lan X, Liu P, et al. Myocardial ischemia-reperfusion injury: therapeutics from a mitochondria-centric perspective. Cardiology. (2021) 146:781–92. doi: 10.1159/000518879
33. Yellon D, Hausenloy D. Myocardial reperfusion injury. N Engl J Med. (2007) 357:1121–35. doi: 10.1056/NEJMra071667
34. He J, Liu D, Zhao L, Zhou D, Rong J, Zhang L, et al. Myocardial ischemia/reperfusion injury: mechanisms of injury and implications for management (Review). Exp Ther Med. (2022) 23:430. doi: 10.3892/etm.2022.11357
35. Zhou H, Wang S, Hu S, Chen Y, Ren J. Er-Mitochondria Microdomains in Cardiac Ischemia-Reperfusion Injury: A Fresh Perspective. Front Physiol. (2018) 9:755. doi: 10.3389/fphys.2018.00755
36. Wang K, An T, Zhou L, Liu C, Zhang X, Feng C, et al. E2f1-Regulated Mir-30b Suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ. (2015) 22:743–54. doi: 10.1038/cdd.2014.165
37. Baines C, Kaiser R, Purcell N, Blair N, Osinska H, Hambleton M, et al. Loss of Cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. (2005) 434:658–62. doi: 10.1038/nature03434
38. Elrod J, Wong R, Mishra S, Vagnozzi R, Sakthievel B, Goonasekera S, et al. Cyclophilin D Controls Mitochondrial Pore-Dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest. (2010) 120:3680–7. doi: 10.1172/jci43171
39. Lim S, Davidson S, Hausenloy D, Yellon D. Preconditioning and postconditioning: the essential role of the mitochondrial permeability transition pore. Cardiovasc Res. (2007) 75:530–5. doi: 10.1016/j.cardiores.2007.04.022
40. Gomez L, Thiebaut P, Paillard M, Ducreux S, Abrial M, Crola Da Silva C. The Sr/Er-Mitochondria Calcium Crosstalk Is Regulated by Gsk3beta during reperfusion injury. Cell Death Differ. (2016) 23:313–22. doi: 10.1038/cdd.2015.101
41. Nikolaou P, Boengler K, Efentakis P, Vouvogiannopoulou K, Zoga A, Gaboriaud-Kolar N, et al. Investigating and re-evaluating the role of glycogen synthase kinase 3 beta kinase as a molecular target for cardioprotection by using novel pharmacological inhibitors. Cardiovasc Res. (2019) 115:1228–43. doi: 10.1093/cvr/cvz061
42. Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, et al. Ptpip51 regulates mouse cardiac ischemia/reperfusion through mediating the Mitochondria-Sr junction. Sci Rep. (2017) 7:45379. doi: 10.1038/srep45379
43. Gomez-Suaga P, Paillusson S, Stoica R, Noble W, Hanger D, Miller C. The Er-Mitochondria tethering Complex Vapb-Ptpip51 regulates autophagy. Curr Biol. (2017) 27:371–85. doi: 10.1016/j.cub.2016.12.038
44. Tan Y, Mui D, Toan S, Zhu P, Li R, Zhou H. Serca overexpression improves mitochondrial quality control and attenuates cardiac microvascular ischemia-reperfusion injury. Mol Ther Nucleic Acids. (2020) 22:696–707. doi: 10.1016/j.omtn.2020.09.013
45. Li C, Ma Q, Toan S, Wang J, Zhou H, Liang J. Serca Overexpression Reduces Reperfusion-Mediated Cardiac Microvascular Damage through Inhibition of the Calcium/Mcu/Mptp/Necroptosis Signaling Pathways. Redox Biol. (2020) 36:101659. doi: 10.1016/j.redox.2020.101659
46. Xu H, Cui S, Zhang Y, Ren J. Mitochondrial Ca(2+) Regulation in the Etiology of Heart Failure: Physiological and Pathophysiological Implications. Acta Pharmacol Sin. (2020) 41:1301–9. doi: 10.1038/s41401-020-0476-5
47. Hall A, Burke N, Dongworth R, Kalkhoran S, Dyson A, Vicencio J, et al. Hearts deficient in Both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. (2016) 7:e2238. doi: 10.1038/cddis.2016.139
48. Papanicolaou K, Khairallah R, Ngoh G, Chikando A, Luptak I, O’Shea K, et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. (2011) 31:1309–28. doi: 10.1128/mcb.00911-10
49. Zhou J, Ahmad F, Parikh S, Hoffman N, Rajan S, Verma V, et al. Loss of adult cardiac myocyte Gsk-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy. Circ Res. (2016) 118:1208–22. doi: 10.1161/CIRCRESAHA.116.308544
50. Chen Y, Liu Y, Dorn G II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109:1327–31. doi: 10.1161/CIRCRESAHA.111.258723
51. Chen Y, Dorn G. II. Pink1-Phosphorylated Mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. doi: 10.1126/science.1231031
52. Tian L, Neuber-Hess M, Mewburn J, Dasgupta A, Dunham-Snary K, Wu D, et al. Ischemia-Induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J Mol Med. (2017) 95:381–93. doi: 10.1007/s00109-017-1522-8
53. Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi S, et al. Parkin-Independent Mitophagy Requires Drp1 and maintains the integrity of mammalian heart and brain. Embo J. (2014) 33:2798–813. doi: 10.15252/embj.201488658
54. Tong M, Zablocki D, Sadoshima J. The Role of Drp1 in mitophagy and cell death in the heart. J Mol Cell Cardiol. (2020) 142:138–45. doi: 10.1016/j.yjmcc.2020.04.015
55. Mohamed R, Morimoto S, Ibrahim I, Zhan D, Du C, Arioka M, et al. Gsk-3beta Heterozygous knockout is cardioprotective in a knockin mouse model of familial dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. (2016) 310:H1808–15. doi: 10.1152/ajpheart.00771.2015
56. Rosenberg P. Vdac2 as a novel target for heart failure: Ca(2+) at the sarcomere, mitochondria and Sr. Cell Calcium. (2022) 104:102586. doi: 10.1016/j.ceca.2022.102586
57. Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S. Role of A -Crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. (2013) 4:e582. doi: 10.1038/cddis.2013.114
58. Paillard M, Tubbs E, Thiebaut P, Gomez L, Fauconnier J, Da Silva C, et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes from Lethal Hypoxia-Reoxygenation Injury. Circulation. (2013) 128:1555–65. doi: 10.1161/circulationaha.113.001225
59. Shi X, Verma S, Yun J, Brand-Arzamendi K, Singh K, Liu X, et al. Effect of empagliflozin on cardiac biomarkers in a zebrafish model of heart failure: clues to the empa-reg outcome trial? Mol Cell Biochem. (2017) 433:97–102. doi: 10.1007/s11010-017-3018-9
60. Brown D, Perry J, Allen M, Sabbah H, Stauffer B, Shaikh S, et al. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. (2017) 14:238–50. doi: 10.1038/nrcardio.2016.203
61. Shi X, Jiang X, Chen C, Zhang Y, Sun X. The interconnections between the microtubules and mitochondrial networks in cardiocerebrovascular diseases: implications for therapy. Pharmacol Res. (2022) 184:106452. doi: 10.1016/j.phrs.2022.106452
62. Shi X, Zhang Y, Chen R, Gong Y, Zhang M, Guan R, et al. Ndufa7 Plays a critical role in cardiac hypertrophy. J Cell Mol Med. (2020) 24:13151–62. doi: 10.1111/jcmm.15921
63. Kimball T, Vondriska T. Metabolism, epigenetics, and causal inference in heart failure. Trends Endocrinol Metab. (2020) 31:181–91. doi: 10.1016/j.tem.2019.11.009
64. Ren J, Bi Y, Sowers J, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. (2021) 18:499–521. doi: 10.1038/s41569-021-00511-w
65. Shi X, Zhang Y, Gong Y, Chen M, Brand-Arzamendi K, Liu X, et al. Zebrafish Hhatla Is Involved in Cardiac Hypertrophy. J Cell Physiol. (2021) 236:3700–9. doi: 10.1002/jcp.30106
66. Garcia M, Boehning D. Cardiac Inositol 1,4,5-Trisphosphate Receptors. Biochim Biophys Acta Mol Cell Res. (2017) 1864:907–14. doi: 10.1016/j.bbamcr.2016.11.017
67. Parys J, Vervliet T. New Insights in the Ip(3) Receptor and Its Regulation. Adv Exp Med Biol. (2020) 1131:243–70. doi: 10.1007/978-3-030-12457-1_10
68. Li G, Li J, Shao R, Zhao J, Chen M. Fundc1: a promising mitophagy regulator at the mitochondria-associated membrane for cardiovascular diseases. Front Cell Dev Biol. (2021) 9:788634. doi: 10.3389/fcell.2021.788634
69. Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, et al. Binding of Fun14 Domain Containing 1 with Inositol 1,4,5-Trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in Vivo. Circulation. (2017) 136:2248–66. doi: 10.1161/circulationaha.117.030235
70. Hayashi T, Su T. Sigma-1 receptor chaperones at the Er-Mitochondrion Interface Regulate Ca(2+) Signaling and Cell Survival. Cell. (2007) 131:596–610. doi: 10.1016/j.cell.2007.08.036
71. Abdullah C, Alam S, Aishwarya R, Miriyala S, Panchatcharam M, Bhuiyan M, et al. Cardiac dysfunction in the sigma 1 receptor knockout mouse associated with impaired mitochondrial dynamics and bioenergetics. J Am Heart Assoc. (2018) 7:e009775. doi: 10.1161/jaha.118.009775
72. Orlando R, De Martin S, Andrighetto L, Floreani M, Palatini P. Fluvoxamine pharmacokinetics in healthy elderly subjects and elderly patients with chronic heart failure. Br J Clin Pharmacol. (2010) 69:279–86. doi: 10.1111/j.1365-2125.2009.03587.x
73. Dorn G II, Song M, Walsh K. Functional Implications of Mitofusin 2-Mediated Mitochondrial-Sr Tethering. J Mol Cell Cardiol. (2015) 78:123–8. doi: 10.1016/j.yjmcc.2014.09.015
74. Fang L, Moore X, Gao X, Dart A, Lim Y, Du X. Down-Regulation of Mitofusin-2 Expression in Cardiac Hypertrophy in Vitro and in Vivo. Life Sci. (2007) 80:2154–60. doi: 10.1016/j.lfs.2007.04.003
75. Xiong W, Ma Z, An D, Liu Z, Cai W, Bai Y, et al. Mitofusin 2 Participates in Mitophagy and Mitochondrial Fusion against Angiotensin Ii-Induced Cardiomyocyte Injury. Front Physiol. (2019) 10:411. doi: 10.3389/fphys.2019.00411
76. Wang L, Qin D, Shi H, Zhang Y, Li H, Han Q. Mir-195-5p promotes cardiomyocyte hypertrophy by targeting Mfn2 and Fbxw7. Biomed Res Int. (2019) 2019:1580982. doi: 10.1155/2019/1580982
77. Yu H, Guo Y, Mi L, Wang X, Li L, Gao W. Mitofusin 2 Inhibits Angiotensin Ii-Induced Myocardial Hypertrophy. J Cardiovasc Pharmacol Ther. (2011) 16:205–11. doi: 10.1177/1074248410385683
78. Lynch S, Boyett J, Smith M, Giordano-Mooga S. Sex hormone regulation of proteins modulating mitochondrial metabolism, dynamics and inter-organellar cross talk in cardiovascular disease. Front Cell Dev Biol. (2020) 8:610516. doi: 10.3389/fcell.2020.610516
79. Deng Y, Ngo D, Holien J, Lees J, Lim S. Mitochondrial Dynamin-Related Protein Drp1: A New Player in Cardio-Oncology. Curr Oncol Rep. (2022) 24:1751–63. doi: 10.1007/s11912-022-01333-w
80. Song M, Mihara K, Chen Y, Scorrano L, Dorn G II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. (2015) 21:273–86. doi: 10.1016/j.cmet.2014.12.011
81. Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu C, et al. Drp1-Dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. (2016) 133:1249–63. doi: 10.1161/circulationaha.115.020502
82. Givvimani S, Pushpakumar S, Veeranki S, Tyagi S. Dysregulation of Mfn2 and Drp-1 proteins in heart failure. Can J Physiol Pharmacol. (2014) 92:583–91. doi: 10.1139/cjpp-2014-0060
83. Noone J, O’Gorman D, Kenny H. Opa1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol Metab. (2022) 33:710–21. doi: 10.1016/j.tem.2022.07.003
Keywords: mitochondria-associated membranes (MAMs), cardiovascular diseases, myocardial infarction, cardiomyopathy, heart failure
Citation: Zhang Y, Yao J, Zhang M, Wang Y and Shi X (2023) Mitochondria-associated endoplasmic reticulum membranes (MAMs): Possible therapeutic targets in heart failure. Front. Cardiovasc. Med. 10:1083935. doi: 10.3389/fcvm.2023.1083935
Received: 29 October 2022; Accepted: 13 January 2023;
Published: 26 January 2023.
Edited by:
Jie Ran, Shandong Normal University, ChinaCopyright © 2023 Zhang, Yao, Zhang, Wang and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xingjuan Shi,  xingjuanshi@seu.edu.cn
xingjuanshi@seu.edu.cn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.