 Randa M. Breikaa1,2
Randa M. Breikaa1,2 Brenda Lilly1,3*
Brenda Lilly1,3*- 1Center for Cardiovascular Research and The Heart Center, Nationwide Children's Hospital, Columbus, OH, United States
- 2Molecular, Cellular and Developmental Biology Program, The Ohio State University, Columbus, OH, United States
- 3Department of Pediatrics, The Ohio State University, Columbus, OH, United States
COVID-19 is associated with a large number of cardiovascular sequelae, including dysrhythmias, myocardial injury, myocarditis and thrombosis. The Notch pathway is one likely culprit leading to these complications due to its direct role in viral entry, inflammation and coagulation processes, all shown to be key parts of COVID-19 pathogenesis. This review highlights links between the pathophysiology of SARS-CoV2 and the Notch signaling pathway that serve as primary drivers of the cardiovascular complications seen in COVID-19 patients.
Introduction
Beginning in December 2019, the world faced a challenging nemesis presented by a member of the coronaviruses family, SARS-CoV2, later known as Coronavirus Disease 2019 or COVID-19 (1–3). First feared for its aggressive attack on the respiratory system (4, 5), it is now recognized for its severe cardiovascular complications (6–8). These range from hemodynamic instabilities, dysrhythmias, and thromboembolic events, to myocarditis, acute heart failure and cardiac arrest (9, 10). Analyzing patient data from several countries, cardiovascular disease appears in two contexts associated with COVID-19. First, studies have shown that pre-existing cardiovascular disease increases the risk of COVID-19 infection and is indeed present in a high number of cases (11–13). Second, COVID-19 patients develop cardiovascular complications during the course of the disease (14, 15). Despite a clear connection with COVID-19 and the cardiovascular system, we understand little about this relationship.
Notch signaling is a master regulator of cardiovascular function in both health and disease, and has been linked to several biological processes mediating viral infections (16, 17). A recent study by Rosa et al., characterized transcriptional signatures induced in a rhesus macaque model of SARS-CoV2 and showed an increase in Notch signaling in the lungs of the macaques (18). Another group studying human protein interactions with SARS-CoV2 using computational models, showed that proteins interacting with the 5'-region of SARS-CoV2 RNA were associated with Notch2 receptor signaling (19). The Notch pathway is also implicated in the hypoxic response and in coagulopathic processes, both of which are present in COVID-19 patients. These known roles of the Notch pathway make this signaling pathway a likely player in the COVID-19-driven cardiovascular complications.
The Beginning (Viral Entry)
The angiotensin converting enzyme 2 (ACE2) has been established to play a significant role in SARS-CoV viruses infectivity, including COVID-19, by binding to the viral spike protein and facilitating entry into the host cell (20, 21). ACE2 has distinct roles in the body, ranging from amino acid transportation and catalytic activities, to serving as functional receptors for viruses like the coronaviruses. In the heart, it is localized to cardiomyocytes, cardiac fibroblasts, epicardial adipose tissue, and the coronary vascular endothelium. In the lungs, it is expressed on the cell surface of the inner respiratory tract, protecting against lung injury. This protective effect stems from its negative regulation of the renin-angiotensin system which leads to the inhibition of the vasoconstrictive, pro-inflammatory angiotensin II (ANGII)—ANGII type 1 receptor (AT1) axis (22–24). Its unique location in both organs combined with its function make it a pivotal player in the pulmonary pathogenicity of the virus and its associated cardiovascular complications. Thus, ACE2 on one hand offers protection against injury, while on the other hand facilitates viral entry. Furthermore, upon binding of ACE2 to the viral particle, the receptor itself becomes endocytosed by the cells causing depletion of cell surface ACE2 and its mediated tissue protection (25, 26). This dilemma and the realization of the importance of ACE2 in maintaining cardiovascular homeostasis drove attempts to manipulate the ACE2/ANGII axis to mitigate virus-induced injury, while minimizing the negative effects on the protective functions of ACE2 (20, 23). One solution for this problem and an attractive target for vaccine development are the viral S-proteins, which when targeted make the enzyme unable to bind, preventing viral entry (21, 27).
Notch signaling has been known to interact with many viral particles facilitating their infectivity (Table 1). Given that Notch regulates various proliferative and differentiation events in cells, it is no surprise that the pathway is an attractive target for viruses, which are dependent on the cell cycle machinery of the cell. Those viruses tap into the Notch pathway to ensure their own survival (60–62). The first evidence that demonstrated Notch pathway-viral interactions was reported for the Epstein-Barr virus, which targets RBPJ (mouse)/CBF1 (human), the nuclear effector of Notch (28, 63). Other examples include the human papilloma virus (HPV), hepatitis B virus (HBV), and hepatitis C virus (HCV). In the case of HCV, the Notch1 receptor has been shown to facilitate nuclear localization of p65 in response to tumor necrosis factor-alpha (TNF-α) in human hepatocytes, leading to increased pathogenicity of the virus (64). Additionally, the influenza virus has been shown to block the Notch ligand Delta-like 1 (DLL1) causing a heightened inflammatory response and decreased interferon-c levels, which leads to compromised immunity against the virus. In contrast, macrophages were found to enhance their DLL1 production during the course of infection to protect against the same virus (32, 33, 65). In the case of COVID-19, an interesting enzyme that could be linking Notch and COVID-19 activation is FURIN. FURIN is a member of the protein convertases family and is both an activator and a direct target of Notch activity (66, 67). Its enzymatic activity has been proven to be exploited by a variety of bacteria and viruses, including measles, yellow fever, ebola, and avian influenza, thereby facilitating their virulence and spread (68, 69). To discern the potential role of FURIN in COVID-19, understanding the structure of the viral S-glycoprotein is important. The S-protein has two functional domains: one for receptor binding and the other for mediating fusion of the viral particle with the cell membrane. The S-protein must be cleaved by the protease to expose these fusion sequences and allow cell entry. FURIN takes on this role in coronaviruses including COVID-19 (70–72). Since Notch1 has been shown to transcriptionally induce FURIN, Notch signaling may indirectly lead to enhanced viral entry via enhanced FURIN expression (73, 74).
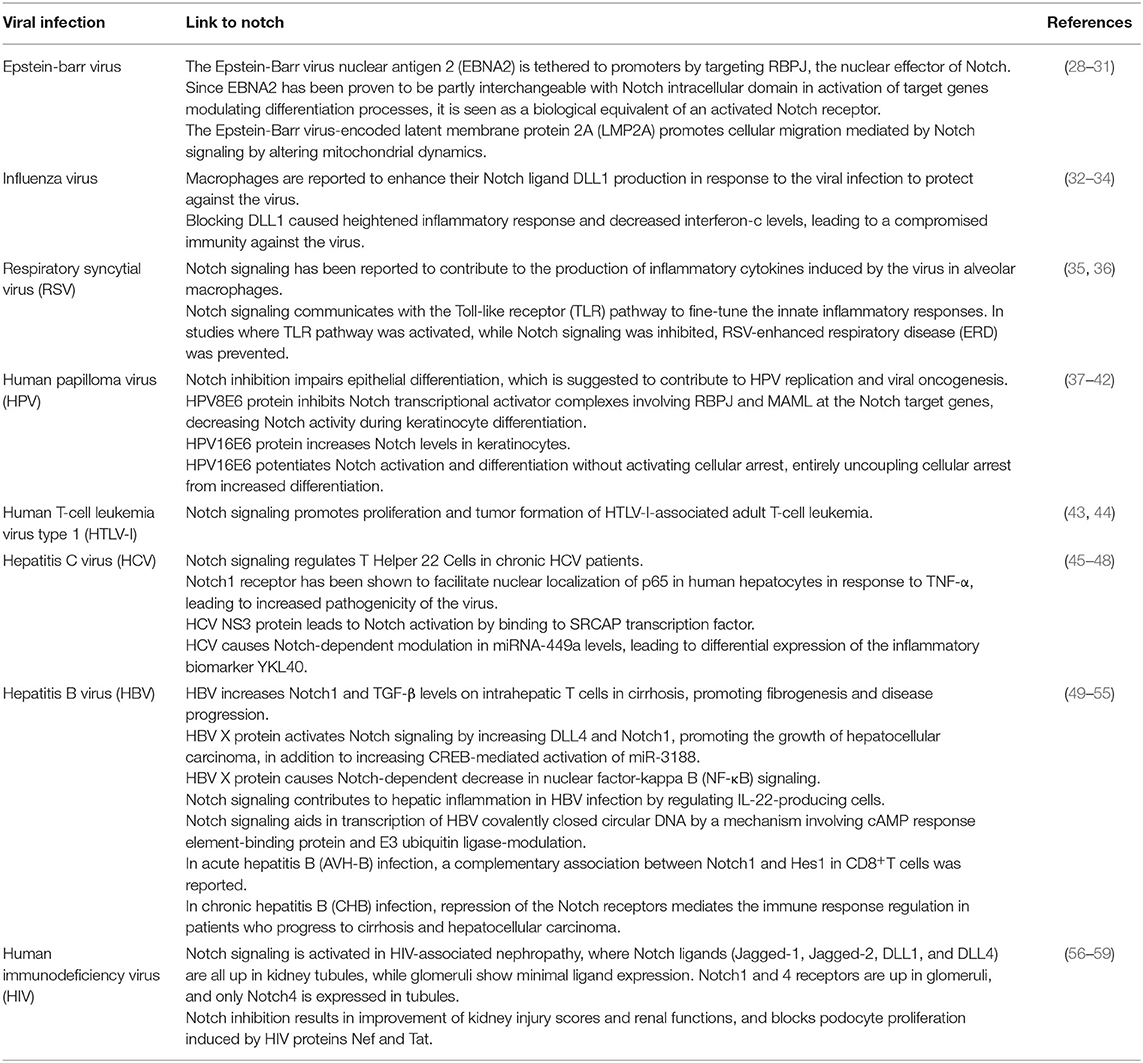
Table 1. Reported link of the Notch signaling pathway to common viral infections.
In addition to having effects on viral infectivity, interestingly both the Notch receptors and ACE2 receptor share a common mechanism of activation through cleavage by the A disintegrin and metalloproteinase (ADAM) family of enzymes, specifically ADAM17 (75, 76). ADAM17 mediates ectodomain shedding of ACE2 which can facilitate viral entry (77, 78). ADAM17 also activates the Notch signaling pathway via receptor cleavage leading to increased viral infectivity through regulation of FURIN. Therefore, Notch activity is indirectly involved in COVID-19 infectivity through FURIN induction and shared activation axis of ACE2, both of which aid in viral entry.
The Cytokine Storm
A balanced innate and adaptive host immunity is key for an effective antiviral response, including activation of T cells, macrophages, and production of various pro-inflammatory cytokines. However, in case of COVID-19, this response becomes heightened, causing a hyperinflammatory reaction known as “The Cytokine Storm Syndrome” (14, 27). The cytokine storm is one of the key factors causing cardiovascular complications in COVID-19 patients. This is attributed to the resulting inflammation-induced vascular injury, myocarditis, arrhythmia, and destabilization of coronary artery plaques leading to myocardial infarcts (79, 80). The common profile of a COVID-19 patient with cytokine storm syndrome includes elevated interleukin-6 (IL-6), IL-2 receptor, TNF-α, granulocyte-colony stimulating factor, among others. IL-6 is secreted by activated leukocytes, promotes differentiation of B lymphocytes and production of acute phase proteins, and is important for thermoregulation (14, 81).
The role of the Notch pathway in inflammation is well-documented, where it has been shown to promote the pro-inflammatory microenvironment (82–84). It is implicated in macrophage polarization and contributes to amplification of the inflammatory loop by promoting the M1 phenotype of macrophages over the M2 phenotype (17, 85). Furthermore, in macrophages, Notch1 directly binds the IL-6 promoter and activates IL-6 transcription in response to interferon-γ (81, 86). Additionally, IL-6 in turn increases the expression of the Notch ligand DLL1, amplifying the Notch signal. This works as a positive feedback loop that further drives the production of more IL-6 (87, 88). Nitric Oxide Synthase (iNOS) expression is linked to manifestation of the cytokine storm (89, 90). Direct interaction between the Notch Intracellular Domain (NICD) and TNF-α on the iNOS promoter has also been documented, indicating multiple avenues by which Notch signaling drives hyper-inflammation (91). Further, TNF-α itself has been shown to induce expression of Notch1 and Notch4, in addition to regulating NICD nuclear translocation, which leads to the activation of Notch downstream mediators (92, 93).
This interplay between Notch and pro-inflammatory processes makes the Notch pathway an attractive target for reversing inflammatory events. Indeed, genetic and pharmacological inhibition of Notch signaling was reported to ameliorate disease progression in many inflammatory disease models. These include rheumatoid arthritis, autoimmune encephalomyelitis, and several models of infectious disease (94, 95). In the case of COVID-19, the recommendation to use corticosteroids was discouraged due to controversial efficacy and reports showing exacerbation of patient symptoms. Potentially targeting the Notch pathway to specifically block the inflammatory loop re-enforced by IL-6 and TNF-α may present a viable therapy for these cases (96–98).
The Hypoxic Response
The hypoxic events in COVID-19 patients have been a mystery to medical caretakers and physicians. This is due to the fact that the patients display minimal visible distress, although clinical oxygen levels are remarkably low (99). Its presentation defies its pathophysiology, which initially led to its description of “Happy Hypoxia” (100). Hypoxia is also linked to the thrombotic events seen in these patients, which spirals quickly into more severe cardiovascular complications such as myocarditis and myocardial infarction (101–103).
The Notch pathway plays a significant role in hypoxic events (104). Notch3 is induced under hypoxic conditions in the lungs and vasculature. Notch3 deletion has been shown to protect against the development of pulmonary arterial hypertension in response to hypoxic stimulation (105, 106). Further, Notch3 was found to cooperate with the hypoxia-inducible factor-1 alpha (HIF-1α) (105, 107), a transcription factor upregulated in hypoxia and inflammatory microenvironments and a master regulator of oxygen homeostasis (108). HIF-1α also induces the expression of two of the Notch ligands, DLL4 and Jagged-1 (109–111). Another link between Notch signaling and HIF-1α is through Notch1 receptor. As mentioned previously, Notch1 receptor has been shown to promote M1 macrophage polarization and switching of macrophage metabolism to glycolysis. This is followed by induction of M1 gene transcription, coupled with an increase in mitochondrial oxidative phosphorylation and generation of reactive oxygen species (112). This in turn activates HIF-1α to induce M1 macrophage activation, in a type of positive feedback loop (111).
Additionally, enhanced Notch signaling has been linked to structural changes in air sacs in the lungs that include decreased septation of terminal alveoli, emphysematous patterns and progressive fibrotic changes (113). Furthermore, Notch3 plays a critical role in regulating alveolar epithelium and increased levels of Notch3 are associated with disruption of differentiation processes and altered lung morphology (114). Interestingly in COVID-19-associated hypoxia, the air sacs do not fill up with fluid like in pneumonia, but also show structural changes in the sacs that lead them to collapse (115, 116). Hence, Notch activation in COVID-19 patients is likely directly exacerbating the hypoxic events by cooperating with HIF-1α in addition to promoting structural defects in the air sacs.
The Coagulopathic Response
The realization that COVID-19 causes hypercoagulopathy poses more questions than answers, with studies showing severe thrombotic manifestations, while others show postmortem lung sections with extensive bleeding (117, 118). In a recent study by Boonyawa et al. a 28% incidence of venous thromboembolism was reported in COVID-19 patients in the intensive care unit (119). Another study by Klok et al. found a 31% incidence of combined deep vein thrombosis, pulmonary embolism, and arterial thrombosis in critically ill patients (120). Thus, there is an urgent need to understand the rate of bleeding and thrombotic events associated with COVID-19 coagulopathy.
Hypercoagulopathy is an important hallmark of inflammation. In fact, pro-inflammatory cytokines are directly involved in accelerating platelet hyperactivation and driving thrombotic events, while impairing crucial physiological anticoagulation pathways including antithrombin III, tissue factor pathway, and the protein C system (121, 122). The mechanisms involved in COVID-19 coagulopathy have not been fully elucidated yet, but crosstalk between the coagulation and the inflammatory systems is evident, with at least four factors seeming to contribute to this condition (123). First, the pro-inflammatory mediators such as IL-6 and IL-1β produced during the cytokine storm stimulate the production of tissue factor on immune cells. This in turn initiates the activation of the extrinsic coagulation cascade (81, 124). Secondly, those pro-inflammatory mediators directly activate the platelets themselves (125). Thirdly, a decrease in plasminogen activator coupled with an increase in plasminogen activator inhibitor suppresses the fibrinolytic system (126, 127). Lastly, the damage caused to the endothelial cells by the inflammatory reaction results in vascular homeostatic imbalances, causing accelerated local thrombotic events in addition to systemic coagulation defects. Of note is that this damaged endothelium also binds platelets more readily due to enhanced platelet-vessel wall interaction caused by the large von Willebrand factor multimers released by damaged endothelial cells (128).
Despite efforts by the scientific community to understand COVID-19-associated coagulopathy, there is still a lot to clarify regarding mechanisms involved and how to reverse the resulting homeostatic imbalances. Previous studies by Duarte et al. and Gough beautifully demonstrated a link between the Notch pathway and the coagulation pathway through fibroblast growth factor 1 (FGF1) (129, 130). These studies utilized a soluble form of the Notch ligand Jagged-1 to show the effect of Notch inhibition on FGF1 and the coagulation cascade. This link between Notch signaling and coagulation is supported by several previous findings. First, the activation of the coagulation cascade by damaged tissue generates thrombin, which activates the protease-activated receptor 1 (PAR1) and PAR1-dependent FGF1 expression and release. Released FGF1 subsequently promotes angiogenesis and induces Jagged-1 expression in the damaged tissue (131). Second, Alagille syndrome patients, who primarily have mutations in Jagged-1, show bleeding disorders (132). Consistent with this, Jagged-1 null mice show hemorrhage during their embryonic development (133, 134). Lastly, Jagged-1 was found to be the FGF1 response gene responsible for FGF1-dependent endothelial cell differentiation on fibrin matrices (131, 135). Taken together, these studies indicate that there is a Notch-dependent mechanism by which thrombin can regulate FGF1 secretion, which in turn contributes to thrombin's activity, the key protease of the coagulation cascade. Thus, through its established role in both inflammation and coagulation, Notch signaling seems likely responsible for exacerbating COVID-19-associated coagulopathy.
Endothelial Cell Involvement
The endothelium is a single layer of cells lining blood vessels, constituting a barrier between the circulation and the rest of the blood vessel wall. In addition, it is the source for several vasoreactive substances responsible for blood vessel contraction and relaxation such as endothelin and nitric oxide (136). Thus, it is a key regulator of vascular homeostasis, and damage to this layer can lead to loss of the homeostatic state and exacerbation of disease conditions. Indeed, endothelial dysfunction shifts the vascular equilibrium toward an inflammatory, pro-coagulant state (137, 138). Coordination of leukocyte trafficking in particular is critical for the inflammatory response. Under physiological conditions, the endothelial cells prevent binding and extravasation of leukocytes from the blood. However, under disease conditions such as the case in COVID-19 patients, the endothelial junctions are weakened and leaky, resulting in facilitated exit of the leukocytes from the circulation into the tissues (82, 93, 139). Interestingly, immunostaining studies have shown the confinement of Notch4 receptors on endothelial cells at the apical membrane. This localization makes Notch4 ideal for receptor/ligand communication between the endothelial and the inflammatory cells in the blood stream (140). In addition, the Notch ligand DLL4 on the endothelium has been shown to trigger a bidirectional Notch signaling between endothelial cells and monocytes (93, 141).
Several reports have linked endothelial cells to SARS-CoV2 pathology, where histological sections through hearts, kidneys and lungs showed accumulation of both inflammatory cells and viral particles within the endothelium (81, 142, 143). This COVID-19-associated endotheliitis could explain the impaired circulatory function in the various vascular beds and the clinical complications in COVID-19 patients (144–146). Furthermore, endothelial cells express the ACE2 receptor, the entry portal for the virus (147–150). This, coupled with previous reports of development of autoantibodies against endothelial cells after SARS-CoV1 infection, suggests that CoV2 infection of endothelial cells and their subsequent damage is a prominent step in the pathogenesis of COVID-19 (151, 152). Important to consider here is the discrepancy that exists between current studies, where some advocate for the endothelial cell hypothesis of COVID-19 pathology, which reinforces the idea that endothelial cells are the origin for COVID-19-associated cardiovascular impairments. In contrast, others promote a pericyte-COVID-19 hypothesis, where pericytes are the main contributors to disease progression. The studies that propose the pericyte hypothesis are based on the fact that ACE2 expression in the heart is highest in pericytes, and in the brain vasculature ACE2 is on vascular smooth muscle cells and pericytes and not on the endothelium (121, 153). Although the endothelial cell hypothesis seems to be more plausible according to consequences of endotheliitis in COVID-19 patients, these discrepancies highlight the importance of considering tissue type in disease pathology.
Conclusions
COVID-19 is associated with a large number of cardiovascular sequelae, including dysrhythmias, myocardial injury, myocarditis and thrombosis. Many of these complications seem to be linked to compromised signaling pathways in the patients, including the Notch pathway (Figure 1). Notch signaling can indirectly enhance viral entry through inducing FURIN, the protease responsible for exposing the fusion sequences of the viral S-protein. The established role of Notch signaling in both inflammation and coagulation suggests its involvement in COVID-19 cytokine storm and hypercoagulopathy, both of which are main contributors to the cardiovascular complications. Furthermore, Notch activation is known to exacerbate hypoxic events by cooperating with HIF-1α in addition to enhancing structural defects in the air sacs in the lungs, which together may contribute to enhanced lung pathology in COVID-19 patients. Lastly, the suggested role of the endothelium in COVID-19 cardiovascular impairments coupled with the specific localization of Notch4 receptors on the apical membrane of the endothelium reinforces the idea that the Notch pathway serves as a communication channel between endothelial and inflammatory cells.
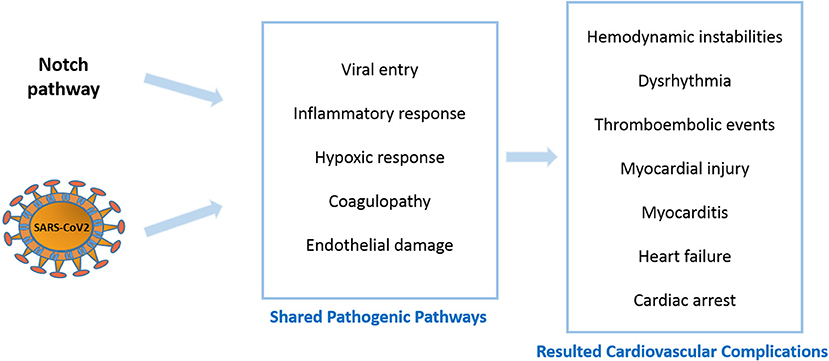
Figure 1. Notch signaling and COVID-19 shared pathogenic events and resulted cardiovascular complications.
In summary, several scenarios can be considered regarding the link between the Notch pathway and COVID-19-associated cardiovascular events. COVID-19 may act upstream to increase Notch signaling, leading to enhanced viral entry and associated pathogenic processes. Alternatively, maladaptive responses of Notch signaling due to COVID-19 infection may contribute to the enhanced inflammatory, coagulopathic, and hypoxic events. Both scenarios eventually lead to exacerbation of cardiovascular impairments in COVID-19 patients that are Notch-associated. Gamma-secretase inhibitors, which inhibit Notch receptor cleavage have been used to attenuate Notch signaling in cancer and Alzheimer disease (154, 155). These compounds, however, are associated with significant toxicity. Alternatives include Notch-specific antibodies and decoys. Antibodies allow blockade of individual Notch components, thus are not associated with complications seen with the pan inhibitors (156–158). Notch decoys also selectively block Notch receptors by a unique mechanism that involves mimicking the Notch extracellular domain of a specific Notch ligand or receptor (159, 160). Finally, uncovering new aspects of a Notch-COVID-19 relationship might help mitigate cardiac and pulmonary complications caused by the SARS-CoV family of viruses.
Author Contributions
RB wrote, edited, and conceived of topic. BL edited, wrote, and provided topic guidance. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH R01-HL132801 (BL) and NIH- 5T32-HL134616 predoctoral fellowship (RB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Masters PS. The molecular biology of coronaviruses. Adv Virus Res. (2006) 66:193–292. doi: 10.1016/S0065-3527(06)66005-3
2. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. (2020) 92:418–23. doi: 10.1002/jmv.25681
3. Cucinotta D, Vanelli M. WHO declares COVID-19 a pandemic. Acta biomed. (2020) 91:157–60. doi: 10.23750/abm.v91i1.9397
4. Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. (2020) 77:198–209. doi: 10.1111/his.14134
5. Wu J, Li X, Huang B, Su H, Li Y, Luo D, et al. Pathological changes of fatal coronavirus disease 2019 (COVID-19) in the lungs: report of 10 cases by postmortem needle autopsy. Zhonghua Bing Li Xue Za Zhi. (2020) 49:568–75. doi: 10.3760/cma.j.cn112151-20200405-00291
6. Lang JP, Wang X, Moura FA, Siddiqi HK, Morrow DA, Bohula EA. A current review of COVID-19 for the cardiovascular specialist. Am Heart J. (2020) 226:29–44. doi: 10.1016/j.ahj.2020.04.025
7. Long B, Brady WJ, Koyfman A, Gottlieb M. Cardiovascular complications in COVID-19. Am J Emerg Med. (2020) 3:1504–7. doi: 10.1016/j.ajem.2020.04.048
8. Matsushita K, Marchandot B, Jesel L, Ohlmann P, Morel O. Impact of COVID-19 on the cardiovascular system: a review. J Clin Med. (2020) 9:1407. doi: 10.3390/jcm9051407
9. Liu PP, Blet A, Smyth D, Li H. The science underlying COVID-19: implications for the cardiovascular system. Circulation. (2020) 142:68–78. doi: 10.1161/CIRCULATIONAHA.120.047549
10. Zheng YY, Ma YT. COVID-19 and the cardiovascular system. Nat Rev Cardiol. (2020) 17:259–60. doi: 10.1038/s41569-020-0360-5
11. Boureau AS, De Decker L, Berrut G, Hanon O. COVID-19 and cardiovascular diseases: viewpoint for older patients. Geriatr Psychol Neuropsychiatr Vieil. (2020) 18:141–9. doi: 10.1684/pnv.2020.0864
12. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. (2020) 395:507–13. doi: 10.1016/S0140-6736(20)30211-7
13. Ganatra S, Dani SS, Shah S, Asnani A, Neilan TG, Lenihan D, et al. Management of cardiovascular disease during coronavirus disease (COVID-19) pandemic. Trends Cardiovasc Med. (2020) 30:315–25. doi: 10.1016/j.tcm.2020.05.004
14. Ashraf O, Young M, Malik KJ, Cheema T. Systemic complications of COVID-19. Crit Care Nurs Q. (2020) 43:390–9. doi: 10.1097/CNQ.0000000000000324
15. Li G, Saguner AM, An J, Ning Y. Cardiovascular disease during the COVID-19 pandemic: think ahead, protect hearts, reduce mortality. Cardiol J. (2020) 27:616–24. doi: 10.5603/CJ.a2020.0101
16. Aster JC. In brief: notch signalling in health and disease. J Pathol. (2014) 232:1–3. doi: 10.1002/path.4291
17. Shang Y, Smith S, Hu X. Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell. (2016) 7:159–74. doi: 10.1007/s13238-016-0250-0
18. Rosa BA, Ahmed M, Singh DK, Choreño-Parra JA, Cole J, Jiménez-Álvarez LA, et al. IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. Commun Biol. (2021) 4:1–14. doi: 10.1038/s42003-021-01829-4
19. Vandelli A, Monti M, Milanetti E, Armaos A, Rupert J, Zacco E, et al. Structural analysis of SARS-CoV-2 genome and predictions of the human interactome. Nucleic Acids Res. (2020) 48:11270–83. doi: 10.1093/nar/gkaa864
20. Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, et al. Angiotensin-converting enzyme-2 (ACE2), SARS-CoV-2 and pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol. (2020) 251:228–48. doi: 10.1002/path.5471
21. Wu J, Deng W, Li S, Yang X. Advances in research on ACE2 as a receptor for 2019-nCoV. Cell Mol Life Sci. (2021). 78:531–44. doi: 10.1007/s00018-020-03611-x
22. Eguchi S, Kawai T, Scalia R, Rizzo V. Understanding angiotensin II type 1 receptor signaling in vascular pathophysiology. Hypertension. (2018) 71:804–10. doi: 10.1161/HYPERTENSIONAHA.118.10266
23. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong J-C, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res. (2020) 126:1456–74. doi: 10.1161/CIRCRESAHA.120.317015
24. Murray E, Tomaszewski M, Guzik TJ. Binding of SARS-CoV-2 and angiotensin-converting enzyme 2: clinical implications. Cardiovasc Res. (2020) 116:e87–9. doi: 10.1093/cvr/cvaa096
25. Alsibai KD. Expression of angiotensin-converting enzyme 2 and proteases in COVID-19 patients: a potential role of cellular FURIN in the pathogenesis of SARS-CoV-2. Med Hypotheses. (2020) 143:109893. doi: 10.1016/j.mehy.2020.109893
26. Wang Q, Qiu Y. Receptor utilization of angiotensin converting enzyme 2 (ACE2) indicates a narrower host range of SARS-CoV-2 than that of SARS-CoV. Transbound Emerg Dis. (2020) 1–8. doi: 10.1111/tbed.13792
27. Barillà F, Bassareo PP, Calcaterra G, Romeo F, Mehta JL. Focus on clinical practice: angiotensin-converting enzyme 2 and corona virus disease 2019: pathophysiology and clinical implications. J Cardiovasc Med (Hagerstown). (2020) 21:630–3. doi: 10.2459/JCM.0000000000001071
28. Robertson ES, Lin J, Kieff E. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ (kappa). J Virol. (1996) 70:3068–74. doi: 10.1128/JVI.70.5.3068-3074.1996
29. Hsieh J, Hayward SD. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science. (1995) 268:560–3. doi: 10.1126/science.7725102
30. Zimber-Strobl U, Strobl LJ. EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes. Semin Cancer Biol. (2001) 11:423–34. doi: 10.1006/scbi.2001.0409
31. Pal AD, Basak NP, Banerjee AS, Banerjee S. Epstein–Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis. (2014) 35:1592–601. doi: 10.1093/carcin/bgu069
32. Ito T, Allen RM, Carson Iv WF, Schaller M, Cavassani KA, Hogaboam CM, et al. The critical role of Notch ligand Delta-like 1 in the pathogenesis of influenza A virus (H1N1) infection. PLoS Pathog. (2011) 7:e1002341. doi: 10.1371/journal.ppat.1002341
33. Ito T, Matsukawa A. Notch system in influenza A/H1N1 virus infection. Inflammation and Regeneration. (2012) 32:132–136. doi: 10.2492/inflammregen.32.132
34. Yang J, Huang X, Liu Y, Zhao D, Han K, Zhang L, et al. Analysis of the microRNA expression profiles of chicken dendritic cells in response to H9N2 avian influenza virus infection. Vet Res. (2020) 51:132. doi: 10.1186/s13567-020-00856-z
35. Zhang L, Li H, Hai Y, Yin W, Li W, Zheng B, et al. CpG in combination with an inhibitor of Notch signaling suppresses formalin-inactivated respiratory syncytial virus-enhanced airway hyperresponsiveness and inflammation by inhibiting Th17 memory responses and promoting tissue-resident memory cells in lungs. J Virol. (2017) 91:e02111–16. doi: 10.1128/JVI.02111-16
36. Buonvino S, Melino S. New Consensus pattern in Spike CoV-2: potential implications in coagulation process and cell–cell fusion. Cell Death Discovery. (2020) 6:134. doi: 10.1038/s41420-020-00372-1
37. Chakrabarti O, Veeraraghavalu K, Tergaonkar V, Liu Y, Androphy EJ, Stanley MA, et al. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J Virol. (2004) 78:5934–45. doi: 10.1128/JVI.78.11.5934-5945.2004
38. Tan MJA, White EA, Sowa ME, Harper JW, Aster JC, Howley PM. Cutaneous β-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc Natl Acad Sci USA. (2012) 109:E1473–80. doi: 10.1073/pnas.1205991109
39. Meyers JM, Spangle JM, Munger K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J Virol. (2013) 87:4762–7. doi: 10.1128/JVI.02527-12
40. Vliet-Gregg PA, Hamilton JR, Katzenellenbogen RA. NFX1-123 and human papillomavirus 16E6 increase Notch expression in keratinocytes. J Virol. (2013) 87:13741–50. doi: 10.1128/JVI.02582-13
41. Das T, Zhong R, Spiotto MT. Notch signaling and human papillomavirus–associated oral tumorigenesis. Adv Exp Med Biol. (2020) 1287:105–22. doi: 10.1007/978-3-030-55031-8_8
42. Vliet-Gregg PA, Hamilton JR, Katzenellenbogen RA. Human papillomavirus 16E6 and NFX1-123 potentiate Notch signaling and differentiation without activating cellular arrest. Virology. (2015) 478:50–60. doi: 10.1016/j.virol.2015.02.002
43. Pancewicz J, Taylor JM, Datta A, Baydoun HH, Waldmann TA, Hermine O, et al. Notch signaling contributes to proliferation and tumor formation of human T-cell leukemia virus type 1–associated adult T-cell leukemia. Proc Natl Acad Sci USA. (2010) 107:16619–24. doi: 10.1073/pnas.1010722107
44. Kamdje AHN, Krampera M. Notch signaling in acute lymphoblastic leukemia: any role for stromal microenvironment? Blood. (2011) 118:6506–14. doi: 10.1182/blood-2011-08-376061
45. Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. (2007) 45:938–47. doi: 10.1002/hep.21622
46. Iwai A, Takegami T, Shiozaki T, Miyazaki T. Hepatitis C virus NS3 protein can activate the notch-signaling pathway through binding to a transcription factor, SRCAP. PLoS ONE. (2011) 6:e20718. doi: 10.1371/journal.pone.0020718
47. Jiang BC, Liu X, Liu XH, Li ZS, Zhu GZ. Notch signaling regulates circulating T helper 22 cells in patients with chronic hepatitis C. Viral Immunol. (2017) 30:522–32. doi: 10.1089/vim.2017.0007
48. Qin L, Zhou YC, Wu HJ, Zhuo Y, Wang YP, Si CY, et al. Notch signaling modulates the balance of regulatory T cells and T helper 17 cells in patients with chronic hepatitis C. DNA Cell Biol. (2017) 36:311–20. doi: 10.1089/dna.2016.3609
49. Wang F, Zhou H, Xia X, Sun Q, Wang Y, Cheng B. Activated Notch signaling is required for hepatitis B virus X protein to promote proliferation and survival of human hepatic cells. Cancer Lett. (2010) 298:64–73. doi: 10.1016/j.canlet.2010.06.003
50. Trehanpati N, Shrivastav S, Shivakumar B, Khosla R, Bhardwaj S, Chaturvedi J. Analysis of Notch and TGF-β signaling expression in different stages of disease progression during hepatitis B virus infection. Clin Transl Gastroenterol. (2012) 3:e23. doi: 10.1038/ctg.2012.17
51. Luo J, Zhou H, Wang F, Xia X, Sun Q, Wang R, et al. The hepatitis B virus X protein downregulates NF-κB signaling pathways through decreasing the Notch signaling pathway in HBx-transformed L02 cells. Int J Oncol. (2013) 42:1636–43. doi: 10.3892/ijo.2013.1842
52. Kongkavitoon P, Tangkijvanich P, Hirankarn N, Palaga T. Hepatitis B virus HBx activates Notch signaling via delta-like 4/Notch1 in hepatocellular carcinoma. PLoS ONE. (2016) 11:e0146696. doi: 10.1371/journal.pone.0146696
53. Wei X, Wang JP, Hao CQ, Yang F, Wang LX, Huang CX, et al. Notch Signaling contributes to liver inflammation by regulation of interleukin-22-producing cells in hepatitis B virus infection. Front Cell Infect Microbiol. (2016) 6:132. doi: 10.3389/fcimb.2016.00132
54. Zhou SJ, Deng YL, Liang HF, Jaoude JC, Liu FY. Hepatitis B virus X protein promotes CREB-mediated activation of miR-3188 and Notch signaling in hepatocellular carcinoma. Cell Death Differ. (2017) 24:1577–87. doi: 10.1038/cdd.2017.87
55. Wang Z, Kawaguchi K, Honda M, Hashimoto S, Shirasaki T, Okada H, et al. Notch signaling facilitates hepatitis B virus covalently closed circular DNA transcription via cAMP response element-binding protein with E3 ubiquitin ligase-modulation. Sci Rep. (2019) 9:1–12. doi: 10.1038/s41598-018-38139-5
56. Shoham N, Cohen L, Yaniv A, Gazit A. The Tat protein of the human immunodeficiency virus type 1 (HIV-1) interacts with the EGF-like repeats of the Notch proteins and the EGF precursor. Virus Res. (2003) 98:57–61. doi: 10.1016/j.virusres.2003.08.016
57. Sharma M, Callen S, Zhang D, Singhal PC, Vanden Heuvel GB, Buch S. Activation of Notch signaling pathway in HIV-associated nephropathy. AIDS. (2010) 24:2161–70. doi: 10.1097/QAD.0b013e32833dbc31
58. Fan Y, Gao X, Chen J, Liu Y, He JJ. HIV tat impairs neurogenesis through functioning as a notch ligand and activation of notch signaling pathway. J Neurosci. (2016) 36:11362–73. doi: 10.1523/JNEUROSCI.1208-16.2016
59. Yan Q, Zhao R, Shen C, Wang F, Li W, Gao J, et al. Upregulation of MicroRNA 711 mediates HIV-1 Vpr promotion of kaposi's sarcoma-associated herpesvirus latency and induction of pro-proliferation and pro-survival cytokines by targeting the Notch/NF-κB-signaling axis. J Virol. (2018) 92:e00580–18. doi: 10.1128/JVI.00580-18
60. Schaller MA, Neupane R, Rudd BD, Kunkel SL, Kallal LE, Lincoln P, et al. Notch ligand Delta-like 4 regulates disease pathogenesis during respiratory viral infections by modulating Th2 cytokines. J Exp Med. (2007) 204:2925–34. doi: 10.1084/jem.20070661
61. Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. (2012) 23:450–7. doi: 10.1016/j.semcdb.2012.01.010
62. Hayward SD. Viral interactions with the Notch pathway. Semin Cancer Biol. (2004) 14:387–96. doi: 10.1016/j.semcancer.2004.04.018
63. Kovall RA, Hendrickson WA. Crystal structure of the nuclear effector of Notch signaling, CSL bound to DNA. EMBO J. (2004) 23:3441–51. doi: 10.1038/sj.emboj.7600349
64. Sarma NJ, Tiriveedhi V, Subramanian V, Shenoy S, Crippin JS, Chapman WC, et al. Hepatitis C virus mediated changes in miRNA-449a modulates inflammatory biomarker YKL40 through components of the NOTCH signaling pathway. PLoS ONE. (2012) 7:e50826. doi: 10.1371/journal.pone.0050826
65. Duval F, Mathieu M, Labrecque N. Notch controls effector CD8+ T cell differentiation. Oncotarget. (2015) 6:21787. doi: 10.18632/oncotarget.4886
66. Ma YC, Shi C, Zhang YN, Wang G, Liu H, Jia HT, et al. The tyrosine kinase c-Src directly mediates growth factor-induced Notch-1 and Furin interaction and Notch-1 activation in pancreatic cancer cells. PLoS ONE. (2012) 7:e33414. doi: 10.1371/journal.pone.0033414
67. Qiu H, Tang X, Ma J, Shaverdashvili K, Zhang K, Bedogni B. Notch1 autoactivation via transcriptional regulation of furin, which sustains Notch1 signaling by processing Notch1-activating proteases ADAM10 and membrane type 1 matrix metalloproteinase. Mol Cell Biol. (2015) 35:3622–32. doi: 10.1128/MCB.00116-15
68. Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. (2002) 3:753–66. doi: 10.1038/nrm934
69. Braun E, Sauter D. Furin-mediated protein processing in infectious diseases and cancer. Clin Translational Immunol. (2019) 8:e1073–e1073. doi: 10.1002/cti2.1073
70. Bradding P, Richardson M, Hinks TS, Howarth PH, Choy DF, Arron JR, et al. ACE2, TMPRSS2, and furin gene expression in the airways of people with asthma—implications for COVID-19. J Allergy Clin Immunol. (2020) 146:208–11. doi: 10.1016/j.jaci.2020.05.013
71. Li X, Duan G, Zhang W, Shi J, Chen J, Chen S, et al. A furin cleavage site was discovered in the S protein of the 2019 novel coronavirus. Chinese J Bioinformatics. (2020) 18. doi: 10.12113/202002001
72. Wu C, Zheng M, Yang Y, Gu X, Yang K, Li M, et al. Furin: a potential therapeutic target for COVID-19. Iscience. (2020) 23:101642. doi: 10.1016/j.isci.2020.101642
73. Bestle D, Heindl MR, Limburg H, Van Lam Van T. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. bioRxiv [Preprint]. (2020) 3. doi: 10.1101/2020.04.15.042085
74. Xi J, Xu K, Jiang P, Lian J, Hao S, Jia H, et al. Virus strain of a mild COVID-19 patient in Hangzhou representing a new trend in SARS-CoV-2 evolution related to Furin cleavage site. medRxiv. (2020). doi: 10.1101/2020.03.10.20033944
75. Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. (2009) 29:5679–95. doi: 10.1128/MCB.00406-09
76. Parr-Sturgess CA, Rushton DJ, Parkin ET. Ectodomain shedding of the Notch ligand Jagged1 is mediated by ADAM17, but is not a lipid-raft-associated event. Biochem J. (2010) 432:283–94. doi: 10.1042/BJ20100321
77. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-α convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. (2005) 280:30113–9. doi: 10.1074/jbc.M505111200
78. Rizzo P, Dalla Sega FV, Fortini F, Marracino L, Rapezzi C, Ferrari R. COVID-19 in the heart and the lungs: could we “Notch” the inflammatory storm? Basic Res Cardiol. (2020) 115:31. doi: 10.1007/s00395-020-0791-5
79. Collins SD. Excess mortality from causes other than influenza and pneumonia during influenza epidemics. Public Health Reports. (1932) 47:2159–79. doi: 10.2307/4580606
80. Corrales-Medina VF, Madjid M, Musher DM. Role of acute infection in triggering acute coronary syndromes. Lancet Infect Dis. (2010) 10:83–92. doi: 10.1016/S1473-3099(09)70331-7
81. Zhang C, Wu Z, Li J-W, Zhao H, Wang Q. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int J Antimicrob Agents. (2020) 55:105954. doi: 10.1016/j.ijantimicag.2020.105954
82. Pabois A, Devallière J, Quillard T, Coulon F, Gérard N, Laboisse C, et al. The disintegrin and metalloproteinase ADAM10 mediates a canonical Notch-dependent regulation of IL-6 through Dll4 in human endothelial cells. Biochem Pharmacol. (2014) 91:510–21. doi: 10.1016/j.bcp.2014.08.007
83. Fazio C, Ricciardiello L. Inflammation and Notch signaling: a crosstalk with opposite effects on tumorigenesis. Cell Death Dis. (2016) 7:e2515. doi: 10.1038/cddis.2016.408
84. Lu Y, Zhang Y, Xiang X, Sharma M, Liu K, Wei J, et al. Notch signaling contributes to the expression of inflammatory cytokines induced by highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) infection in porcine alveolar macrophages. Dev Comp Immunol. (2020) 108:103690. doi: 10.1016/j.dci.2020.103690
85. Keewan E, Naser SA. The role of Notch signaling in macrophages during inflammation and infection: implication in rheumatoid arthritis? Cells. (2020) 9:111. doi: 10.3390/cells9010111
86. Wongchana W, Palaga T. Direct regulation of interleukin-6 expression by Notch signaling in macrophages. Cell Mol Immunol. (2012) 9:155–62. doi: 10.1038/cmi.2011.36
87. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. (2012) 38:904–10. doi: 10.1016/j.ctrv.2012.04.007
88. Yang Z, Guo L, Liu D, Sun L, Chen H, Deng Q, et al. Acquisition of resistance to trastuzumab in gastric cancer cells is associated with activation of IL-6/STAT3/Jagged-1/Notch positive feedback loop. Oncotarget. (2015) 6:5072. doi: 10.18632/oncotarget.3241
89. Adusumilli NC, Zhang D, Friedman JM, Friedman AJ. Harnessing nitric oxide for preventing, limiting and treating the severe pulmonary consequences of COVID-19. Nitric Oxide. (2020) 103:4–8. doi: 10.1016/j.niox.2020.07.003
90. Cespuglio R, Strekalova T, Spencer PS, Román GC, Reis J, Bouteille B, et al. SARS-CoV-2 infection and sleep disturbances: nitric oxide involvement and therapeutic opportunity. Sleep. (2021) 44:zsab009. doi: 10.1093/sleep/zsab009
91. Wang R, Li Y, Tsung A, Huang H, Du Q, Yang M, et al. iNOS promotes CD24+ CD133+ liver cancer stem cell phenotype through a TACE/ADAM17-dependent Notch signaling pathway. Proc Natl Acad Sci USA. (2018) 115:E10127–36. doi: 10.1073/pnas.1722100115
92. Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-α and its receptors by neutrophils and macrophages. J Leukoc Biol. (2007) 82:173–6. doi: 10.1189/jlb.0307193
93. Quillard T, Devallière J, Coupel S, Charreau B. Inflammation dysregulates Notch signaling in endothelial cells: implication of Notch2 and Notch4 to endothelial dysfunction. Biochem Pharmacol. (2010) 80:2032–41. doi: 10.1016/j.bcp.2010.07.010
94. Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann Rheum Dis. (2011) 70:1304–10. doi: 10.1136/ard.2010.134742
95. Liu S, Liu D, Chen C, Hamamura K, Moshaverinia A, Yang R, et al. MSC transplantation improves osteopenia via epigenetic regulation of notch signaling in lupus. Cell Metab. (2015) 22:606–18. doi: 10.1016/j.cmet.2015.08.018
96. Russell B, Moss C, Rigg A, Van Hemelrijck M. COVID-19 and treatment with NSAIDs and corticosteroids: should we be limiting their use in the clinical setting? Ecancermedicalscience. (2020) 14:1023. doi: 10.3332/ecancer.2020.1023
97. Theoharides T, Conti P. Dexamethasone for COVID-19? Not so fast. J Biol Regul Homeost Agents. (2020) 34:1241–3. doi: 10.23812/20-EDITORIAL_1-5
98. Zha L, Li S, Pan L, Tefsen B, Li Y, French N, et al. Corticosteroid treatment of patients with coronavirus disease 2019 (COVID-19). Med J Australia. (2020) 212:416–20. doi: 10.5694/mja2.50577
99. Herrmann J, Mori V, Bates JH, Suki B. Can hyperperfusion of nonaerated lung explain COVID-19 hypoxia? Res Sq. [Preprint]. (2020). doi: 10.21203/rs.3.rs-32949/v1
100. Couzin-Frankel J. The mystery of the pandemic's ‘happy hypoxia'. Science. (2020) 368:455–6. doi: 10.1126/science.368.6490.455
101. Savla JJ, Levine BD, Sadek HA. The effect of hypoxia on cardiovascular disease: friend or foe? High Alt Med Biol. (2018) 19:124–30. doi: 10.1089/ham.2018.0044
102. Kashani KB. Hypoxia in COVID-19: sign of severity or cause for poor outcomes. Mayo Clin Proc. (2020) 95:1094–6. doi: 10.1016/j.mayocp.2020.04.021
103. Thachil J. Hypoxia-an overlooked trigger for thrombosis in COVID-19 and other critically ill patients. J Thromb Haemost. (2020) 18:3109–10. doi: 10.1111/jth.15029
104. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. (2005) 9:617–28. doi: 10.1016/j.devcel.2005.09.010
105. Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. (2009) 15:1289–97. doi: 10.1038/nm.2021
106. Thistlethwaite PA, Li X, Zhang X. Notch signaling in pulmonary hypertension. Adv Exp Med Biol. (2010) 661:279–98. doi: 10.1007/978-1-60761-500-2_18
107. Liu H, Zhang W, Kennard S, Caldwell RB, Lilly B. Notch3 is critical for proper angiogenesis and mural cell investment. Circ Res. (2010) 107:860–70. doi: 10.1161/CIRCRESAHA.110.218271
108. Chun YS, Kim MS, Park JW. Oxygen-dependent and-independent regulation of HIF-1alpha. J Korean Med Sci. (2002) 17:581. doi: 10.3346/jkms.2002.17.5.581
109. Sainson RC, Harris AL. Hypoxia-regulated differentiation: let's step it up a Notch. Trends Mol Med. (2006) 12:141–3. doi: 10.1016/j.molmed.2006.02.001
110. Fung E, Tang S-MT, Canner JP, Morishige K, Arboleda-Velasquez JF, et al. Delta-like 4 induces notch signaling in macrophages. Circulation. (2007) 115:2948–56. doi: 10.1161/CIRCULATIONAHA.106.675462
111. Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, et al. Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci USA. (2008) 105:3368–73. doi: 10.1073/pnas.0711591105
112. Xu J, Chi F, Guo T, Punj V, Lee WP, French SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. (2015) 125:1579–90. doi: 10.1172/JCI76468
113. Jespersen K, Liu Z, Li C, Harding P, Sestak K, Batra R, et al. Enhanced Notch3 signaling contributes to pulmonary emphysema in a Murine Model of Marfan syndrome. Sci Rep. (2020) 10:1–11. doi: 10.1038/s41598-020-67941-3
114. Guseh JS, Bores SA, Stanger BZ, Zhou Q, Anderson WJ, Melton DA, et al. Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development. (2009) 136:1751–9. doi: 10.1242/dev.029249
115. Teo J. Early detection of silent hypoxia in COVID-19 pneumonia using Smartphone pulse oximetry. J Med Syst. (2020) 44:1–2. doi: 10.1007/s10916-020-01587-6
116. Wilkerson RG, Adler JD, Shah NG, Brown R. Silent hypoxia: a harbinger of clinical deterioration in patients with COVID-19. Am J Emerg Med. (2020) 38:2243.e5–2243.e6. doi: 10.1016/j.ajem.2020.05.044
117. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID-19 and coagulation: bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. (2020) 136:489–500. doi: 10.1182/blood.2020006520
118. Chan NC, Weitz JI. COVID-19 coagulopathy, thrombosis, and bleeding. Blood. (2020) 136:381–383. doi: 10.1182/blood.2020007335
119. Boonyawat K, Chantrathammachart P, Numthavaj P, Nanthatanti N, Phusanti S, Phuphuakrat A, et al. Incidence of thromboembolism in patients with COVID-19: a systematic review and meta-analysis. Thromb J. (2020) 18:34. doi: 10.1186/s12959-020-00248-5
120. Klok FA, Kruip M, Van Der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
121. He L, Mae MA, Sun Y, Muhl L, Nahar K, Liebanas EV, et al. Pericyte-specific vascular expression of SARS-CoV-2 receptor ACE2-implications for microvascular inflammation and hypercoagulopathy in COVID-19 patients. bioRxiv. (2020). doi: 10.1101/2020.05.11.088500
122. McGonagle D, O'donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. (2020) 2:e437–45. doi: 10.1016/S2665-9913(20)30121-1
123. Colling ME, Kanthi Y. COVID-19-associated coagulopathy: an exploration of mechanisms. Vasc Med. (2020) 25:471–8. doi: 10.1177/1358863X20932640
124. Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. Might the double X-chromosome in females be protective against SARS-CoV-2 compared to the single X-chromosome in males? Int J Mol Sci. (2020) 21:3474. doi: 10.3390/ijms21103474
125. Martín-Rojas RM, Pérez-Rus G, Delgado-Pinos VE, Domingo-González A, Regalado-Artamendi I, Alba-Urdiales N, et al. COVID-19 coagulopathy: an in-depth analysis of the coagulation system. Eur J Haematol. (2020) 105:741–50. doi: 10.1111/ejh.13501
126. Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther. (2010) 28:e72–e91. doi: 10.1111/j.1755-5922.2010.00171.x
127. Medcalf RL, Keragala CB, Myles PS. Fibrinolysis and COVID-19: a plasmin paradox. J Thromb Haemost. (2020) 18:2118–22. doi: 10.1111/jth.14960
128. O'sullivan JM, Mc Gonagle D, Ward SE, Preston RJ, O'donnell JS. Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol. (2020) 7:e553–5. doi: 10.1016/S2352-3026(20)30215-5
129. Duarte M, Kolev V, Kacer D, Mouta-Bellum C, Soldi R, Graziani I, et al. Novel cross-talk between three cardiovascular regulators: thrombin cleavage fragment of Jagged1 induces fibroblast growth factor 1 expression and release. Mol Biol Cell. (2008) 19:4863–74. doi: 10.1091/mbc.e07-12-1237
130. Gough NR. Thrombin targets notch signaling. Sci Signal. (2008) 1:ec375. doi: 10.1126/scisignal.144ec375
131. Zimrin AB, Pepper MS, Mcmahon GA, Nguyen F, Montesano R, Maciag T. An antisense oligonucleotide to the notch ligand jagged enhances fibroblast growth factor-induced angiogenesis in vitro. J Biol Chem. (1996) 271:32499–502. doi: 10.1074/jbc.271.51.32499
132. Joutel A, Tournier-Lasserve E. Notch signalling pathway and human diseases. In: Seminars in Cell and Developmental Biology. Elsevier (1998). p. 619–25. doi: 10.1006/scdb.1998.0261
133. Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. (1999) 8:723–30. doi: 10.1093/hmg/8.5.723
134. Crosnier C, Attié-Bitach T, Encha-Razavi F, Audollent S, Soudy F, Hadchouel M, et al. JAGGED1 gene expression during human embryogenesis elucidates the wide phenotypic spectrum of Alagille syndrome. Hepatology. (2000) 32:574–81. doi: 10.1053/jhep.2000.16600
135. Zimrin AB, Villeponteau B, Maciag T. Models of in vitro angiogenesis: endothelial cell differentiation on fibrin but not matrigel is transcriptionally dependent. Biochem Biophys Res Commun. (1995) 213:630–8. doi: 10.1006/bbrc.1995.2178
136. Sumpio BE, Riley JT, Dardik A. Cells in focus: endothelial cell. Int J Biochem Cell Biol. (2002) 34:1508–12. doi: 10.1016/S1357-2725(02)00075-4
137. Rubanyi GM. The role of endothelium in cardiovascular homeostasis and diseases. J Cardiovasc Pharmacol. (1993) 22:S1–14. doi: 10.1097/00005344-199322004-00002
138. Kazmi RS, Boyce S, Lwaleed BA. Homeostasis of hemostasis: the role of endothelium. Semin Thromb Hemost. (2015) 41:549–55. doi: 10.1055/s-0035-1556586
139. Quillard T, Devalliere J, Chatelais M, Coulon F, Séveno C, Romagnoli M, et al. Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS ONE. (2009) 4:e8244. doi: 10.1371/journal.pone.0008244
140. Lizama CO, Zovein AC. Polarizing pathways: balancing endothelial polarity, permeability, lumen formation. Exp Cell Res. (2013) 319:1247–54. doi: 10.1016/j.yexcr.2013.03.028
141. Pabois A, Pagie S, Gérard N, Laboisse C, Pattier S, Hulin P, et al. Notch signaling mediates crosstalk between endothelial cells and macrophages via Dll4 and IL6 in cardiac microvascular inflammation. Biochem Pharmacol. (2016) 104:95–107. doi: 10.1016/j.bcp.2016.01.016
142. Yang X, Chang Y, Wei W. Endothelial dysfunction and inflammation: immunity in rheumatoid arthritis. Mediators Inflamm. (2016) 2016:6813016. doi: 10.1155/2016/6813016
143. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
144. Fox SE Lameira FS Rinker EB Vander Heide RS Cardiac Endotheliitis and multisystem inflammatory syndrome after COVID-19. Ann Intern Med. (2020) 173:1025–7. doi: 10.7326/L20-0882
145. Endotheliitis and Endothelial Dysfunction in Patients with COVID-19: Its Role in Thrombosis and Adverse Outcomes. J Clin Med. (2020) 9:1862. doi: 10.3390/jcm9061862
146. Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. (2020) 395:1417–8. doi: 10.1016/S0140-6736(20)30937-5
147. Hamming I, Timens W, Bulthuis M, Lely A, Navis GV, Van Goor. H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203:631–7. doi: 10.1002/path.1570
148. Sluimer J, Gasc J, Hamming I, Van Goor H, Michaud A, Van Den Akker L, et al. Angiotensin-converting enzyme 2 (ACE2) expression and activity in human carotid atherosclerotic lesions. J Pathol. (2008) 215:273–79. doi: 10.1002/path.2357
149. Muus C, Luecken MD, Eraslan G, Waghray A, Heimberg G, Sikkema L, et al. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. BioRxiv. (2020). doi: 10.1101/2020.04.19.049254
150. Ratajczak MZ, Bujko K, Ciechanowicz A, Sielatycka K, Cymer M, Marlicz W, et al. SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45– precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev Reports. (2020) 16:1–2. doi: 10.1007/s12015-019-09951-x
151. Rahimi FS, Afaghi S, Tarki FE, Goudarzi K, Alamdari NM. Viral outbreaks of SARS-CoV1, SARS-CoV2, MERS-CoV, influenza H1N1, and ebola in 21st Century; a comparative review of the pathogenesis and clinical characteristics. School Med Stud J. 151. (2020). doi: 10.22037/smsj.v2i3.30455
152. Iba T, Levy JH, Levi M, Thachil J. Coagulopathy in COVID-19. J Thromb Haemost. (2020) 18:2103–9. doi: 10.1111/jth.14975
153. Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res. (2020) 116:1097–100. doi: 10.1093/cvr/cvaa078
154. Miele L, Miao H, Nickoloff B. NOTCH signaling as a novel cancer therapeutic target. Curr Cancer Drug Targets. (2006) 6:313–23. doi: 10.2174/156800906777441771
155. Woo HN, Park JS, Gwon AR, Arumugam TV, Jo DG. Alzheimer's disease and Notch signaling. Biochem Biophys Res Commun. (2009) 390:1093–7. doi: 10.1016/j.bbrc.2009.10.093
156. Li K, Li Y, Wu W, Gordon WR, Chang DW, Lu M, et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J Biol Chem. (2008) 283:8046–54. doi: 10.1074/jbc.M800170200
157. Falk R, Falk A, Dyson MR, Melidoni AN, Parthiban K, Young JL, et al. Generation of anti-Notch antibodies and their application in blocking Notch signalling in neural stem cells. Methods. (2012) 58:69–78. doi: 10.1016/j.ymeth.2012.07.008
158. Valcourt DM, Dang MN, Scully MA, Day ES. Nanoparticle-mediated co-delivery of Notch-1 antibodies and ABT-737 as a potent treatment strategy for triple-negative breast cancer. ACS Nano. (2020) 14:3378–88. doi: 10.1021/acsnano.9b09263
159. Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, et al. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discovery. (2015) 5:182–97. doi: 10.1158/2159-8290.CD-14-0650
Keywords: notch signaling pathway, COVID-19, cardiovascular, vascular biology, cardiovascular disease
Citation: Breikaa RM and Lilly B (2021) The Notch Pathway: A Link Between COVID-19 Pathophysiology and Its Cardiovascular Complications. Front. Cardiovasc. Med. 8:681948. doi: 10.3389/fcvm.2021.681948
Received: 17 March 2021; Accepted: 06 May 2021;
Published: 26 May 2021.
Edited by:
Masanori Aikawa, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Jun-ichiro Koga, Kyushu University, JapanYafeng Li, Shanxi Provincial People's Hospital, China
Copyright © 2021 Breikaa and Lilly. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brenda Lilly, YnJlbmRhLmxpbGx5QG5hdGlvbndpZGVjaGlsZHJlbnMub3Jn